
Semiempirical and density functional theory molecular modeling of brown coal chars with iron species...
12
Semiempirical and Density Functional Theory Molecular Modeling of Brown Coal Chars with Iron Species and H 2 , CO Formation George Domazetis,* Monthida Raoarun, and Bruce D. James Chemistry Department, La Trobe UniVersity, Victoria, 3086, Australia ReceiVed March 13, 2007. ReVised Manuscript ReceiVed May 15, 2007 This paper is an overview of the calculations performed on various molecular models of char containing iron species, developed from a brown coal model with iron complexes, to mimic pyrolysis over 200-700 °C. The semiempirical (SE) optimization of four char models and density functional theory (DFT) calculations on the fourth smaller model were assessed on the basis of calculated heats of formation, total energy, bond lengths and angles, and partial charges. The formation of hydrogen was examined via H abstraction from [O-H] and [C-H] groups by iron clusters in char to form the hydride, followed by the dihydride, and accompanied by [C-O-Fe] and [C-Fe] bond formation. Single-point self-consistent field DFT and SE calculations for relevant structures containing iron clusters indicated that H abstraction and hydrogen formation were energetically favored. Two routes were examined for CO formation: via the adsorption of FeO and Fe 2 O on graphite followed by a loss of CO and via decomposition of the newly formed [C-O-Fe] group. The latter was shown to be the likely route. A concerted reaction for hydride formation during CO formation has been discussed and a reaction scheme for H 2 and CO suggested; the chemistry for H 2 and CO from catalytic steam gasification is briefly discussed. Introduction Studies of metal-mediated pyrolysis and the gasification of low-rank coals require the characterization of inorganic species added to the macromolecular structure of the coals and their forms in char. To this end, we have systematically studied the addition and characterization of inorganic complexes to low- rank coals, particularly iron hydroxyl complexes, and have examined the decomposition reactions involving coal oxygen functional groups coordinated to iron hydroxyl complexes during low-temperature pyrolysis. 1-4 Semiempirical (SE) molecular modeling and experimental methods have shown that the chemistry during low-temperature pyrolysis includes the trans- formation of the iron hydroxyl complexes into Fe(III)/Fe(II) oxides and ultimately into Fe(0). The present work extends our effort to include SE and density functional theory (DFT) molecular modeling of char containing iron complexes and examines the formation of H 2 and CO from models of char that contain iron species. The formation of CO 2 and CO during the initial stages of pyrolysis involves the thermal breakdown of coal oxygen functional ligands coordinated to the inorganic complex. As the temperature increases, the amount of CO increases during pyrolysis, and significant amounts of H 2 O, H 2 , and CH 4 are observed. These events can precede gasification chemistry (e.g., char gasification with O 2 or H 2 O) and include the transformation of the iron hydroxyl into various Fe(III)/(II) and Fe(0) com- plexes. Such iron species may subsequently be involved in catalytic activity. The modeling approach adopted in our studies commenced with a molecular model of brown coal, and this was transformed into a char model by removing functional groups to mimic the loss of oxygen functional groups during pyrolysis and also the transformation of iron hydroxyl species into iron oxides and reduced iron species. Calculated data obtained from the transformation of the model of coal into char have been compared with experimental data, that is, elemental composition, percentage weight loss, the ratio CO 2 /CO measured during pyrolysis at given temperatures, iron hydroxyl species, and the various Fe(III)/Fe(II) oxides and Fe(0) moieties, over the temperature range 200-700 °C, as discussed previously. 4 The molecular modeling theory used in our studies has been SE with the PM5 Hamiltonian (SE-PM5) for large molecule models and DFT for smaller char models with iron clusters Fe m O n (n ) 1-4 and m ) 0-3), and also a char/graphite model containing the CtC group, reported by a number of workers for comparison with our char models. 5-9 The approach in this work has been to use relative changes in the calculated heat of * Corresponding author. Tel.: 61 3 9479 2811. Fax: 61 3 9479 1399. E-mail: [email protected]. (1) Domazetis, G.; Raoarun, M.; James, B. D. Studies of Mono- and Poly-nuclear Iron Hydroxyl Complexes in Brown Coal. Energy Fuels 2005, 19, 1047-1055. (2) Domazetis, G.; Liesegang, J.; James, B. D. Studies of inorganics added to low-rank coals for catalytic gasification. Fuel Process. Technol. 2005, 86, 463-486. (3) Domazetis, G.; James, B. D. Molecular Models of Low Rank Coals Incorporating Metal Containing Species. Org. Geochem. 2006, 37, 244- 259. (4) Domazetis, G.; Raoarun, M.; James, B. D. Low-Temperature Pyrolysis of Brown Coal and Brown Coal Containing Iron Hydroxyl Complexes. Energy Fuels 2006, 20, 1997-2007. (5) Zhu, Z.; Lu, G. Q.; Finnerty, J.; Yang, R. T. Electronic structure methods applied to gas-carbon reactions. Carbon 2003, 41, 635-658. (6) Sendt, K.; Haynes, B. S. Density functional study of the chemisorption of O2 on the armchair surface of graphite. Proc. Combust. Inst. 2005, 30, 2141-2149. (7) Haynes. B. S. A turnover model for carbon reactivity I. Development. Combust. Flame 2001, 126, 1421-1432. (8) Ma, X.; Wang, Q.; Cis, L-Q.; Cermignani, W.; Schobert, H. H.; Pantano, C. G. Semi-empirical Studies on Electronic Structures of a Boron- doped Graphene Layer - Implications on the Oxidation Mechanism. Carbon 1997, 35, 1517-1525. (9) Chan, N.; Yang, R. T. Ab initio Molecular Orbital Calculations on Graphite: Selection of Molecular Systems and Model Chemistry. Carbon 1998, 36, 1061-1070. 2531 Energy & Fuels 2007, 21, 2531-2542 10.1021/ef070129v CCC: $37.00 © 2007 American Chemical Society Published on Web 07/14/2007
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Semiempirical and density functional theory molecular modeling of brown coal chars with iron species...

Semiempirical and Density Functional Theory Molecular Modelingof Brown Coal Chars with Iron Species and H2, CO Formation
George Domazetis,* Monthida Raoarun, and Bruce D. James
Chemistry Department, La Trobe UniVersity, Victoria, 3086, Australia
ReceiVed March 13, 2007. ReVised Manuscript ReceiVed May 15, 2007
This paper is an overview of the calculations performed on various molecular models of char containingiron species, developed from a brown coal model with iron complexes, to mimic pyrolysis over 200-700°C.The semiempirical (SE) optimization of four char models and density functional theory (DFT) calculations onthe fourth smaller model were assessed on the basis of calculated heats of formation, total energy, bond lengthsand angles, and partial charges. The formation of hydrogen was examined via H abstraction from [O-H] and[C-H] groups by iron clusters in char to form the hydride, followed by the dihydride, and accompanied by[C-O-Fe] and [C-Fe] bond formation. Single-point self-consistent field DFT and SE calculations for relevantstructures containing iron clusters indicated that H abstraction and hydrogen formation were energeticallyfavored. Two routes were examined for CO formation: via the adsorption of FeO and Fe2O on graphite followedby a loss of CO and via decomposition of the newly formed [C-O-Fe] group. The latter was shown to be thelikely route. A concerted reaction for hydride formation during CO formation has been discussed and a reactionscheme for H2 and CO suggested; the chemistry for H2 and CO from catalytic steam gasification is brieflydiscussed.
Introduction
Studies of metal-mediated pyrolysis and the gasification oflow-rank coals require the characterization of inorganic speciesadded to the macromolecular structure of the coals and theirforms in char. To this end, we have systematically studied theaddition and characterization of inorganic complexes to low-rank coals, particularly iron hydroxyl complexes, and haveexamined the decomposition reactions involving coal oxygenfunctional groups coordinated to iron hydroxyl complexes duringlow-temperature pyrolysis.1-4 Semiempirical (SE) molecularmodeling and experimental methods have shown that thechemistry during low-temperature pyrolysis includes the trans-formation of the iron hydroxyl complexes into Fe(III)/Fe(II)oxides and ultimately into Fe(0). The present work extends oureffort to include SE and density functional theory (DFT)molecular modeling of char containing iron complexes andexamines the formation of H2 and CO from models of char thatcontain iron species.
The formation of CO2 and CO during the initial stages ofpyrolysis involves the thermal breakdown of coal oxygenfunctional ligands coordinated to the inorganic complex. As thetemperature increases, the amount of CO increases duringpyrolysis, and significant amounts of H2O, H2, and CH4 are
observed. These events can precede gasification chemistry (e.g.,char gasification with O2 or H2O) and include the transformationof the iron hydroxyl into various Fe(III)/(II) and Fe(0) com-plexes. Such iron species may subsequently be involved incatalytic activity. The modeling approach adopted in our studiescommenced with a molecular model of brown coal, and thiswas transformed into a char model by removing functionalgroups to mimic the loss of oxygen functional groups duringpyrolysis and also the transformation of iron hydroxyl speciesinto iron oxides and reduced iron species. Calculated dataobtained from the transformation of the model of coal into charhave been compared with experimental data, that is, elementalcomposition, percentage weight loss, the ratio CO2/CO measuredduring pyrolysis at given temperatures, iron hydroxyl species,and the various Fe(III)/Fe(II) oxides and Fe(0) moieties, overthe temperature range 200-700 °C, as discussed previously.4
The molecular modeling theory used in our studies has beenSE with the PM5 Hamiltonian (SE-PM5) for large moleculemodels and DFT for smaller char models with iron clustersFemOn (n ) 1-4 andm) 0-3), and also a char/graphite modelcontaining the CtC group, reported by a number of workersfor comparison with our char models.5-9 The approach in thiswork has been to use relative changes in the calculated heat of
* Corresponding author. Tel.: 61 3 9479 2811. Fax: 61 3 9479 1399.E-mail: [email protected].
(1) Domazetis, G.; Raoarun, M.; James, B. D. Studies of Mono- andPoly-nuclear Iron Hydroxyl Complexes in Brown Coal.Energy Fuels2005,19, 1047-1055.
(2) Domazetis, G.; Liesegang, J.; James, B. D. Studies of inorganicsadded to low-rank coals for catalytic gasification.Fuel Process. Technol.2005, 86, 463-486.
(3) Domazetis, G.; James, B. D. Molecular Models of Low Rank CoalsIncorporating Metal Containing Species.Org. Geochem.2006, 37, 244-259.
(4) Domazetis, G.; Raoarun, M.; James, B. D. Low-Temperature Pyrolysisof Brown Coal and Brown Coal Containing Iron Hydroxyl Complexes.Energy Fuels2006, 20, 1997-2007.
(5) Zhu, Z.; Lu, G. Q.; Finnerty, J.; Yang, R. T. Electronic structuremethods applied to gas-carbon reactions.Carbon2003, 41, 635-658.
(6) Sendt, K.; Haynes, B. S. Density functional study of the chemisorptionof O2 on the armchair surface of graphite.Proc. Combust. Inst.2005, 30,2141-2149.
(7) Haynes. B. S. A turnover model for carbon reactivity I. Development.Combust. Flame2001, 126, 1421-1432.
(8) Ma, X.; Wang, Q.; Cis, L-Q.; Cermignani, W.; Schobert, H. H.;Pantano, C. G. Semi-empirical Studies on Electronic Structures of a Boron-doped Graphene Layer- Implications on the Oxidation Mechanism.Carbon1997, 35, 1517-1525.
(9) Chan, N.; Yang, R. T. Ab initio Molecular Orbital Calculations onGraphite: Selection of Molecular Systems and Model Chemistry.Carbon1998, 36, 1061-1070.
2531Energy & Fuels2007,21, 2531-2542
10.1021/ef070129v CCC: $37.00 © 2007 American Chemical SocietyPublished on Web 07/14/2007

formation of the compound from its elements in their standardstate (∆Hf), partial charges of char and iron species, bondlengths, and angles as a basis for assessing the molecularstructures in reaction sequences for hydrogen and carbonmonoxide formation. In this paper, we discuss (i) the modelingof brown coal char formation, (ii) molecular models of charcontaining various iron species, and (iii) reaction routes for H2
and CO formation from char containing iron clusters. Thechemistry of H2 and CO formation from catalytic steamgasification is also briefly discussed.
Computer Molecular Models
Molecular models of low-rank coals have been discussedpreviously, and the brown coal molecule, discussed below, wasmodified into molecular structures of brown coal char by removingfunctional groups, to mimic the loss of carboxyl, carbonyl, ether,and hydroxyl groups, observed during the pyrolysis of brown coal.3,4
The same coal model with iron hydroxyl complexes was alsotransformed in a similar manner into molecular structures of browncoal char with iron oxide, but in this case, the carboxyl and hydroxylgroups coordinated to the iron centers were lost as CO2 and CO,and the hydroxyl groups within the iron complex were lost as watermolecules;4 for example, [Fe6(OH)14]4+ in the initial coal modellost (OH) groups as H2O and changed into [(Fe3+)4(Fe2+)2(O2-)8]for the char model. The weight percent loss, elemental analysis,and CO2/CO ratio were calculated for each of the resulting charmolecular models and compared with experimental data for charsamples from pyrolysis at particular temperatures; the models,experimental data, and loss of functional groups have been discussedpreviously.1,2,4,10
Molecular structures were optimized using molecular mechanics,single-point self-consistent field calculations using the PM5 Hamil-tonian (1scf-PM5), semiempirical calculations using the PM5Hamiltonian (SE-PM5), single-point self-consistent field calcula-tions using DFT (1scf-DFT), and DFT. Calculations for smallmolecules were performed with the Fujitsu CAChe ab initio 5.04software package and for large molecules with MOPAC200211a atthe Australian Partnership for Advanced Computing NationalFacility (APAC-NF). DFT calculations of small molecules wereperformed with the CAChe 5.04 DGauss 4.1/UC-4.1 program, usingBecke ‘88: Perdew & Wang ‘91 theory, with a triple-ú-valenceuncontracted 63321/531/41 Gaussian basis set (DZVP, A1), and aLi-Rn pseudopotential which includes relativistic effects for heavyatoms.11b DFT calculations were also carried out at the VictorianPartnership for Advanced Computing facility (VPAC) using theSchrodinger Jaguar package at the B3LYP level of theory (exactHartree-Fock, Slater local exchange functional, Becke’s 1988nonlocal gradient correction; the correlation is the Vosko-Wilk-Nusair local functional and Lee-Yang-Parr local and nonlocalfunctionals) and using lacvp** or lacvp3** basis sets, which includeeffective core potentials (ECPs) for Fe developed at Los AlamosNational Laboratory; the basis set is valence-only, containing thehighest s and p shells for main group atoms and the highest s, p,and d shells for transition metals including the outermost coreorbitals; the 631G basis set developed by Pople and co-workerswas used for atoms not described by ECPs. Polarization functionswere used on all atoms except for transition metals, and the effectivecore potentials include one-electron mass-velocity and relativisticcorrections.12aThe geometries of three smaller molecular structuresof char, and char with the cluster Fe3, were optimized using DFT-
B3LYP and lacvp**; in this case, the very large computer resourcesrequired for this task necessitated limited medium-accuracy com-putations.
Our goal has been to develop specific molecular models thatcould be used to examine reactions leading to H2 and CO formationfrom char containing iron complexes, and also the reactions of thesemolecules with H2O, as models for catalytic steam gasification;consequently, an exhaustive study of all configurations, electronicstates, and transition species, for all models of chars with inorganicspecies, is outside the scope of our work. Conformational analysesfor particular structures were initially performed using CAChe 5.04Cornflex and the Monte Carlo multiple minimum method withSchrodinger’s MacroModel 9.1.12b Details have been provided inthe discussion of particular conformations of molecules and theirimpact on the calculated data.
SE geometric optimization using the default options in MOPACwas obtained for molecular models of brown coal and brown coalchars. Calculations for structures of char containing iron complexeswere complicated, however, by multielectron configuration interac-tions (MECI), and such calculations for the large structures requiredexcessively large computer wall time. Consequently, only a fewsmaller models containing iron complexes were examined usingMECI, and for these molecules, the singlet state was the lowest-energy configuration, with singlet and triplet states separated by 1to 2 eV. It is noted that the structures formed during pyrolysis areof chemically reactive systems at elevated temperatures, and it isobvious that SE results of structures optimized to a ground statecan only be used to provide a relative indication of energeticallyfavored geometries. It was observed that a number of molecularstructures used to examine hydrogen and carbon monoxide reactionswere relatively stable molecules, and thus the calculated∆Hf, andthe total energy for these, may be used as a relative measure ofenergetically favored structures provided the data were for the samemolecule that had undergone a simple internal transformation. Ourapproach has been simplified in this way: we have also used dataobtained from 1scf-PM5, 1scf-DFT, and SE calculations, to ensurethat the relative comparison was reasonable. The default optionfor structure optimization in MOPAC was the Baker’s EigenFollowing geometry optimization method; results from the Broy-den-Fletcher-Goldfarb-Shanno method were occasionally usedfor larger structures.
A number of options were examined in the SE treatment,including relaxing the default conditions for geometry optimizationand removing safety checks in MOPAC. In virtually all molecularmodels containing iron complexes, removing safety checks oftenresulted in a catastrophic failure, while in other cases, the structurewas optimized with very short distances between coal oxygenfunctional groups, and also between organic functional groups andiron complexes. This behavior was not observed for the organicmolecular models of coal and char, but only these models containingiron species; additionally, this behavior was not observed whensimilar molecular models of char, but without hydroxyl or phenolicgroups, and containing the same iron complexes, were optimized.It is likely, therefore, that the behavior observed for these structuresstemmed from the SE treatment of hydrogen bonds and coordinationbonds formed in these models. The structures that were optimizedusing DFT did not display this behavior. SE results were obtainedfor these difficult structures by modifying the options available inMOPAC that vary the minimum allowed ratio for energy change.In minimum energy searches, it is usually desirable that the energydecreases in each iteration; MOPAC searches for lower energychanges for rigid systems by stipulating values for a maximum-(RMAX) and minimum- (RMIN) allowed ratio for energy change.In cases where the optimization may terminate before the stationarypoint has been reached, the trust radius may be set to a low value,or set to zero, and RMIN may be set to a negative value to allowthe program to continue searching for a stationary point with stepsthat allow the energy to increase as well as decrease.
The data obtained from SE calculations include∆Hf, total energy,bond lengths, bond angles, bond orders, partial charges, andcontributions ofσ andπ components to bonding with iron clusters.
(10) Domazetis, G.; Raoarun, M.; James, B. D.; Liesegang, J.; Pigram,P. J.; Brack. N.; Glaisher, R. Analytical and Characterization Studies ofOrganic and Inorganic Species in Brown Coal.Energy Fuels2006, 20,1556-1564.
(11) (a) Stewart, J. J. P.MOPAC 2002, version 2.5.2; Fujitsu Ltd: Tokyo,Japan. (b)CAChe ab initio, version 5.04; Fujitsu Ltd: Tokyo, Japan, 2000-2002; Oxford Molecular Ltd: Oxford, U.K., 1989-2000.
(12) (a) Jaguar, version 6.5; (b) MacroModel, version 9.1; Schro¨dinger,LLC: New York, 2005.
2532 Energy & Fuels, Vol. 21, No. 5, 2007 Domazetis et al.

Three SE calculations were carried out: (i) using default optionsin MOPAC, (ii) without MECI capabilities, and (iii) stipulatinglower values for the trust radius (including zero) and a negativevalue for RMIN. MOPAC provides Wyberg indices as bond ordersthat mirror the simple ideas of single, double, and triple bonds;bond orders of less than 0.2 are indicative of “no bond”; the bondsmatrix is split intoσ-π-δ components, and the net charges, orpartial charge, on each atom from MOPAC were the Coulsoncharges, while for specific models, Mulliken populations and partialcharges were also computed. DFT and 1scf-DFT data included totalenergy, bond lengths and angles, Mulliken atomic charges, and bondorder, and if needed, Mayer total atomic valence and bond order.
Results and Discussion
Char Formation. The molecular model of brown coal wastransformed into the various char molecular models by progres-sively performing the following steps: (a) loss of carboxylgroups to form CO2; (b) loss of carboxyl groups to form CO;(c) loss of carbonyl groups to form CO; (d) loss of hydroxylgroups from phenol functional groups to form CO and H2O,and associated reactions; and (e) loss of “links” comprising esterand aliphatic groups to form CO, H2O, H2, and hydrocarbons(e.g., CH4, CH2dCH2).
The changes made to the coal molecular model, progressingfrom step a through to step e, were consistent with the pyrolysischemistry observed at progressively higher temperatures. Thecalculated data from each of the transformations of the coalmolecular model usually compared well with the experimentaldata obtained from the pyrolysis of acid-washed brown coal.4
Coal molecular models containing iron hydroxyl species,however, were lower in total oxygen content compared to thoseamounts measured for these char samples. This was due to thesimplifying assumptions adopted for these models, in that allhydroxyl and coordinated water molecules were removedinstantly as water molecules, resulting in a lower total oxygencontent for the molecular model. Experimentally, however, atthe relatively low temperatures of these studies, it is likely someiron hydroxyl groups, and water molecules strongly coordinatedto iron hydroxyl, remain in the char, and this would provide alarger amount of total oxygen in the sample. The transformationsof the molecular model of brown coal containing iron hydroxylcomplexes, into char models with iron oxide species, alsofollowed steps a to e, but the carboxyl and hydroxyl groupsbound to the iron complex were initially lost.
In cases when functional groups that linked units in the 3Dstructure were removed, the resulting char molecular modelsfragmented, and these molecular structures were optimized assingle char models consisting of the resulting large molecularfragments. The molecular modeling results are for three chars,with and without iron hydroxyl/oxide complexes, and arecompared to chars obtained experimentally from the pyrolysisof brown coal at temperatures of∼300, ∼450, and∼600-700°C. Above 700°C, our interest is in oxygen/steam catalyticgasification chemistry, which will be discussed in detail insubsequent publications.
Molecular Models. The properties of the brown coal mo-lecular model, a molecular formula (MF) of [C263H239NO90] anda formula weight (FW) of 4853.734, are similar to propertiesreported as typical for brown coal; that is, molecular model:Car/Ctot ) 0.6 and Har/Htot ) 0.2 (ar) aromatic, tot) total);elemental composition: C 65.1%, H 5.0%, N 0.3%, O 29.7%;distribution of oxygen: carboxyl O(COOH) ) 21%, phenolic O(OH)
) 34%, methoxy O(O-CH3) ) 7%, methyl, ether, and aliphatichydroxy O(R-OH) ) ∼4% and 18%, carbonyl O(RC)O) ) 11%,other) 5%. The measured values vary considerably due to the
heterogeneous nature of brown coal:10,13Car/Ctot ) 0.57-0.65;Har/Htot ) ∼0.3; elemental composition: C 67.8%, H 4.9%, N0.61%, O 26.4%. The distribution of oxygen is as follows:O(COOH) 17-23%; O(OH) 35-38%; O(O-CH3) ∼12%; O(R-OH)
∼4%; O(RC)O) ∼23%.The SE calculated∆Hf for the optimized coal molecular
model structure was-3434.7 kcal (total energy-60598.2 eV).As discussed previously, structures of brown coal molecularmodels in which carboxyl groups formed monodentate coordi-nation bonds with iron hydroxyl complexes, containing watermolecules coordinated to iron, provided a lower energy thanthose for the brown coal molecular model;3 for example, the1scf-PM5∆Hf forthebrowncoal/ironcomplex{coal[COO-]7[(Fe3-Fe4)(OH)14(15H2O)]7+} was -4229.7 kcal (total energy of-71052.2 eV).
The char molecular model incorporating pyrolysis steps aand b of brown coal is shown in Figure 1 (labeled Char1; MF) C249H239NO65; FW ) 4285.595). The calculated data arecomparable to those for char samples obtained at∼300°C: thatis, the calculated weight loss on a dry basis is 12 wt %; themeasured weight loss (dry basis) from acid-washed brown coalwas 14 wt %; char model elemental composition: C 69.8%, H5.6%, O 24.3%, N 0.3%; elemental analysis of the char: C70.1%, H 4.1%, O 25.4%, N 0.3%; calculated CO2/CO ratio3.7:1; experimental CO2/CO ratio 3.3:1. The structure wasoptimized (SE) with a∆Hf of -2300.2 kcal and a total energyof -51930.6 eV; the phenyl groups formed a disorderedarrangement with some two-phenyl groups adopting a parallelarrangement separated by 3.5-4 Å, which is similar to valuesobtained from X-ray diffraction (XRD) studies of char.14-16
The decarboxylation reactions of the iron hydroxyl complexesin low-temperature pyrolysis include the formation of carbonato,µ-oxo-iron, and ultimately Fe(II) and Fe(0) complexes.4 Thechemistry for oxides and reduced iron species has been modeledby using iron oxide species containing six, five, and four ironcenters, to resemble structures reported for Fe2O3, Fe3O4, and
(13) Verheyen, T. V.; Perry, G. J.The Science of Victorian Brown Coal;Durie, R. A., Ed.; Butterworth Heimann: London, 1991; p 280.
(14) Wertz, D. L. Interlayer Structural Models of Beulah Zap LigniteBased on Its Wide Angle X-ray Scattering.Energy Fuels1999, 13, 513-517.
(15) Sahajwalla, L. L. V.; Kong, C.; Harris, D. Quantitative X-raydiffraction analysis and its application to various coals.Carbon2001, 39,1821-1833.
(16) Feng, B.; Bhatia, S. K.; Barry, J. C. Variation of the CrystallineStructure of Coal Char during Gasification.Energy Fuels2003, 17, 744-754.
Figure 1. Molecular model of Char1.
Molecular Modeling of Brown Coal Chars Energy & Fuels, Vol. 21, No. 5, 20072533

FeO;17 additionally, a number of FemOn and Fem clusters, whichhave been studied using DFT methods,18-22 have also beenplaced within the char to model reduced and Fe(0) iron speciesformed in pyrolysis at higher temperatures (discussed below).
The char molecular model originally shown in Figure 1, butnow containing the [Fe6O8] moiety, is shown in Figure 2; MF) [C249H239NO73Fe6], FW ) 4748.67; calculated weight loss15 wt %; elemental composition: C 62.8%, H 5.4%, O 24.5%,Fe 7.0%; calculated CO2/CO ratio 3.7:1. The measured elemen-tal composition of the char sample was as follows: C 56.7%,H 3.8%, O 30.2%, ash (mainly iron oxide) 8.4%; experimentalweight loss 16-20%; measured CO2/CO ratio 4:1. As notedpreviously, the calculated composition differs from the experi-mental data mainly because of the lower amount of oxygenpresent in the molecular model, due to the simplifying assump-tion of instantaneous conversion of the iron hydroxyl complexinto the iron oxide moiety.
1scf-PM5 data for Char1 with the various iron oxides wereas follows: [Fe3O2] ∆Hf ) -1585.22 kcal (total energy-53659.30 eV); [Fe3O4] ∆Hf ) -1703.43 kcal (total energy-54216.58 eV); [Fe6O8] ∆Hf ) -1599.5 kcal (total energy-56523.97 eV). 1scf-PM5 calculations were performed for themodel [C249H239NO73Fe6] using the MECI routine with 27configurations; in these, the lowest level was the singlet,separated from the triplet by∼1.3 eV. SE optimization usingdefault settings in MOPAC produced a structure with very shortOH‚‚‚H bonds, but calculations using the various alternateoptions available in MOPAC provided the following values of∆Hf: Char1[Fe3O4], [C249H239NO69Fe3] ) -2124.7 kcal (total
energy-54 234.84 eV); Char1[Fe6O8], [C249H239NO73Fe6] )-2371.2 kcal (total energy-56 556.76 eV) (∆Hf for this modelwithout the default MECI routine was-2366.2 kcal, total energywas-56557.22 eV). The data show that these char molecularmodels were destabilized by the larger size of the iron oxidesbut were stabilized by the additional coordination bonds betweencoal oxygen functional groups with the iron centers. Significantionic interactions were indicated by the increase in partialcharges on the iron oxide, and the bond orders were consistentwith the partially covalent and partially ionic character of themolecule. The iron oxide [(Fe3+)4(Fe2+)2(O2-)8] core consistedof four Fe(III) and two Fe(II) centers, with eight O atoms, andthe remaining coordination sites were filled by neighboringhydroxyl ligands. The Fe(III) centers were octahedral and theFe(II) centers tetrahedral; distortions in the octahedral andtetrahedral sites were observed, as discussed for iron hydroxylin coal models.3 The calculated bond orders for the iron atomsin Char1[Fe6O8] varied from 1 to 0.4; partial charges on Fe(+) were 0.4, 0.5, 0.6, 0.7, 0.9, 0.9, and on the O bound to iron(-) were 0.8, 0.8, 0.7, 0.7, 0.7, 0.7, 0.7, and 0.6. Although partialcharges on two iron centers were higher, none reflected theformal charges on the iron centers; the results were consistentwith the significant ionic character of the hydrophilic molecule.Strong H‚‚‚OH interactions were evidenced by the shorterdistances between some of the coal-OH ligands.
A molecular model of Char1 was also formed with an ironoxide moiety resembling Wu¨stite, [(Fe5O4)]4+ (tetrahedral Fecenters,µ-oxo bonds, and four Fe-OR oxidative bonds in the{coal[(Fe5O4)(OR)4]} macromolecule). 1scf-PM5 results for thismolecule were similar to those for the structure shown in Figure2, but SE results indicated that this structure may be lessenergetically favored than{Char1[Fe6O8]}, shown in Figure 2.The Fe-O bond orders varied between 1.1 and 0.8. Significantionic character was evidenced by the partial charges on ironcenters of (+) 0.5-0.6 and+0.9; partial charges on oxygenbound to iron centers were (-) 0.6-0.7, and the ligand-oxygenbound to the iron centers [R-O] had values of-0.6. Thismolecular model was further modified into four similar mol-ecules, by varying the formal oxidation states of the Fe, to reflectthe reduction of the iron centers during pyrolysis; that is, theiron moieties were formally [FeIV(FeII)4(O2-)4]4+, [(FeII)4(O2-)2]4+,[(FeII)5(O2-)4]2+, and [FeIV(O2-)4(FeI)4]. Similar ∆Hf valueswere obtained for these structures, and the calculated partialcharges did not reflect the formal oxidation states indicated onthe iron moieties. The steric crowding observed for the structurein Figure 2 was not observed for these models. SE results forthe molecular models were as follows:{Char1[FeIV(FeII)4-(O2-)4]4+} ∆Hf ) -2203.5 kcal (total energy-54 990.14 eV);{Char1[FeIV(O2-)4FeI
4]} ∆Hf ) -2213.68 kcal (total energy-55 043.77 eV);{Char1[(FeII)4(O2-)2]4+} ∆Hf ) -2150.17kcal (total energy-54 033.14 eV). The structure for Char1[Fe4O2], however, contained bonds between Fe centers andtwo C atoms of a phenyl group (bond orders 0.8) with bondlengths typical for single Fe-C bonds; the partial charges onthese carbon atoms were more negative than those on neighbor-ing oxygen atoms.
Data for a molecular model of char formed using steps a tod (labeled Char2) resembling char at 350-450 °C were asfollows: SE∆Hf ) -1228.57 kcal (total energy) -43298.4eV); MF ) [C222H182N2O48]; FW ) 3645.864; elementalcomposition: C 73.14%, H 5.03%, N 0.77%, O 21.06%; weightloss 25 wt %; CO2/CO ratio 1:1.5. The measured weight lossat this temperature range was 20-25 wt %, and the CO2/COratio was 1:1.9. Although significant ring condensation was not
(17) Cornell, R. M.; Schwertmann, U.The Iron Oxides; VCH: Weinheim,Germany, 1996.
(18) Gutsev, G. L.; Bauschlicher, C. W., Jr. Structural and electronicproperties of iron monoxide clusters FemO and FemO- (m ) 2-6) Acombined photoelectron spectroscopy and density functional theory study.J. Chem. Phys.2003, 119, 11135-11145.
(19) Gutsev, G. L.; Mochena, M. D.; Bauschlicher, C. W., Jr. Structureand Properties of Fe4 with Different Coverage by C and CO.J. Phys. Chem.A 2004, 108, 11409-11418.
(20) Knickelbein, M. B.; Koretsky, G. M.; Jackson. K. A.; Pederson,M. R.; Hajnal, Z. Hydrogenated and deuterated iron clusters: Infraredspectra and density functional calculations.J. Chem. Phys.1998, 109,10692-10700.
(21) Wang, Q.; Sun, Q.; Sakurai, M.; Yu, J. Z.; Gu, B. L.; Sumiyama,K.; Kawazoe, Y. Geometry and electronic structure of magic iron oxideclusters.Phys. ReV. B: Condens. Matter Mater. Phys.1999, 59, 12672-12677.
(22) Castro, M.; Salahub. D. R. Theoretical study of the structure andbinding of iron clusters: Fen (n< 5). Phys. ReV. B: Condens. Matter Mater.Phys.1993, 47, 10955-10958.
Figure 2. Model of {Char1[FeB6BOB8B]}, showing OH f Fecoordination bonds (purple atoms) Fe, red atoms) O).
2534 Energy & Fuels, Vol. 21, No. 5, 2007 Domazetis et al.

invoked in forming this structure, the proportion of aromaticgroups had increased dramatically, and the proportion of twophenyl rings in the molecule had also increased. About 85% ofall carbons were aromatic, and a number of CdC bonds wereformed from the elimination of functional groups, for example,RCH2-CH2-COOH f RCHdCH2 + CO + H2O
The char model developed after all of steps a through e wereperformed, mimicking char at 600-700 °C, provided aweight loss of 48 wt % (measured weight loss 45-50 wt % at∼600 °C). This char model was designated Char3: MF)[C177H151NO14]; FW ) 2516.1; elemental composition: C84.5%, H 6.0%, O 8.9%, N 0.6%. All of the carboxyl groups,over half of the phenolic groups, and most of the carbonyl andether groups had been lost. The latter also acted as links for the3-D macromolecule, and as a result, breaking these linksdisrupted the 3-D structure. The pyrolysis chemistry includesthe formation of carbon- and oxygen-centered radicals throughhydrogen abstraction and the recombination of H radicals intoH2.4 XRD indicates that the iron oxides in the char at 600-700°C were reduced mainly to Fe(0), and consequently, these weremodeled by using the iron clusters [Fe2O], [Fe3O], [Fe3], [Fe4O],and [Fe4]. Iron clusters have been studied by numerousworkers.18-23 The larger-sized iron cluster in this char desta-bilized the molecule, consistent with the trend discussedpreviously for char/iron oxide models. The formation of OHfFecoordination bonds between the iron clusters and hydroxylgroups in the char molecule also caused changes to that regionof the macroligand. The∆Hf values for Char3 were as follows:1scf-PM5 ) 51.5 kcal and SE) -87.4 kcal (total energy-27756.2 eV). 1scf-PM5∆Hf values for Char3 with thefollowing iron clusters were as follows: [Fe3O] ) 371.6 kcal,[Fe4O] ) 532.6 kcal, and [Fe4] 573.4 kcal. The SE∆Hf valuesfor Char3 with iron clusters were as follows: [Fe3O] ) 145.8kcal, [Fe4O] ) 370.8 kcal, and [Fe4] ) 576.1 kcal. Data for the{Char3[Fe3O]} molecule were as follows: Fe-O bond orders0.8 and 0.9, bond lengths 1.88 Å and 1.92 Å, and the third Feat 2.77 Å from the cluster oxygen with a bond order< 0.1;Fe-Fe bond orders were 2.7, 1.2, and 0.5 and bond lengthswere 1.74, 2.03, and 2.26 Å; one coordinate bond was typical(Fe r OH bond length of 2.06 Å, bond order 0.4), but thesecond Fe‚‚‚OH distance was 2.20 Å and the bond order was<0.2, indicating only a weak electrostatic interaction. The lengthof a hydrogen bond between the cluster and a nearby hydroxylgroup (Fe3O‚‚‚HO-R) was 2.08 Å. The partial charges in thecluster were (Coulson) Fe+0.3, +0.2, +0.2; O -0.8 and, oncoordinated oxygen,-0.4; Mulliken partial charges on Fe were+0.6,+0.4, and+0.4; the Fe centers with lower partial chargeswere situated near C-H groups, with Fe‚‚‚H distances of 1.78and 2.70 Å. SE treatment of this model using default MOPACsettings and also calculations without MECI provided∆Hf
values that differed by< 2 kcal.DFT treatment of{Char3[Fe3O]} provided a structure that
was less compacted than that obtained from SE, although bothstructures contain a similar number of hydrogen bonds betweenhydroxyl groups. The DFT-optimized structure was more “open”and contained fewer phenyl groups in parallel arrangementscompared to the SE-optimized structure; the distances betweenparallel phenyl groups were 4.4 Å for DFT and 4.3 Å for SE.1scf-DFT data for{Char3[Fe3O]} included partial charges on
Fe of+0.1,-0.1, and+0.1, and on the cluster O of-0.7; theFer OH coordination bonds lengths were typical at 2.27, 2.25,and 2.29 Å.
SE results for{Char3[Fe4]} included [R-HOf(Fe)4] bondorders at 0.2-0.3, partial charges on Fe∼ 0, and Fe-Fe bondlengths of 2.33-2.35 Å. The Fer OH coordination bonds were2.06 and 2.17 Å. Bond lengths have been reported for the Fe3Oand Fe4 clusters as follows: Fe-Fe, 2.20-2.25 Å, and for Fe-O, 1.80-1.83 Å, while Fe-O bond lengths in iron oxides are1.95 Å (R-Fe2O3), 1.89 Å (Fe3O4), and 2.15 Å (Fe-O); Fe-Febond lengths for Fe4(CO) have been reported to vary from 2.26to 2.55 Å.18-24
The Formation of H2 and CO from Char with IronSpecies.CO2 and CO are the major products from the pyrolysisof brown coal at relatively low temperatures; with an increasein temperature, the amount of CO increases, and significantamounts of H2, H2O, and hydrocarbons also begin to form. Inthe absence of inorganic species, the formation of char andgaseous products at higher temperatures is mainly via radicalchemistry, but for chars containing inorganic species (such asthe iron species discussed here), metal-mediated chemistrypredominates. The formation of reduced iron species has beenshown to take place via carbonato andµ-oxo- complexes, butreactions involving radicals and reduced iron complexes havealso been postulated.4
It is impractical to provide exhaustive modeling encompassingall possible iron oxides and iron clusters of the form Fem andFemOn (wheren and m are up to 20) and the concentrationsthat may be contemplated in the char models. The data fromthree molecular models of char with some iron oxides have beencompared to experimental data, and additionally, chemistry willnow be examined dealing with hydrogen and carbon monoxideformation at 600-700 °C. Reaction routes for the breakdownof carboxyl groups into CO2 and CO have been discussedpreviously, including the abstraction of hydrogen from functionalgroups;4 hydrogen abstraction from hydroxyl groups in closeproximity, or coordinated to iron species, would be expectedto occur as the temperature increases. Hydrogen abstraction fromFer OH groups may lead to the oxidative formation of [-C-O-Fe] bonds and iron hydrides and ultimately yields H2; thebreakdown of the newly formed [-C-O-Fe] bonds wouldyield CO. An alternate route for CO formation has also beenconsidered, using a simpler model of char containing a CtCgraphite edge, as a comparison for CO formation after thechemisorption of FeO or Fe2O.
Hydrogen Abstraction and H2 Formation. The formationof hydrides in iron clusters has been discussed theoretically anddemonstrated experimentally, and hydride formation has beenidentified from reactions between [Fem]+ clusters and meth-ane.20,25 Hydrogen abstraction and transfer reactions involvingaliphatic or aromatic hydrocarbons are important in understand-ing the formation of oil, gas, and coal, and in understandingthe chemistry of gasification and combustion; ab initio studieshave been reported for small organic molecules.26 Dehydroge-
(23) Dieguez, O.; Alemany, M. M. G.; Rey, C.; Pablo Ordejo´n, P.;Gallego, L. J. Density-functional calculations of the structures, bindingenergies, and magnetic moments of Fe clusters with 2 to 17 atoms.Phys.ReV. B: Condens. Matter Mater. Phys.2001, 63, 205407-205413.
(24) Wu, Z. Y.; Gota, S.; Jollet, F.; Pollak, M.; Gautier-Soyer, M.; Natoli,C. R. Characterization of iron oxides by x-ray absorption at the oxygen Kedge using a full multiple-scattering approach.Phys. ReV. B: Condens.Matter Mater. Phys.1997, 55, 2570-2577.
(25) Liyanage, R.; Zhang, X.-G.; Armentrout, P. B. Activation of methaneby size-selected iron cluster cations, Fem
+ (m ) 2-15): Cluster-CHx (x )0-3) bond energies and reaction mechanisms.J. Chem. Phys.2001, 115,9747-9763.
(26) Schimmel, P. H.; Paul, J. A.; Ruttink, J. A.; de Jong, B. H.W. S. Ab Initio Calculations on Hydroaromatics: Hydrogen Abstractionand Dissociation Reaction Pathways.J. Phys. Chem. B1999, 103, 10506-10516.
Molecular Modeling of Brown Coal Chars Energy & Fuels, Vol. 21, No. 5, 20072535

nation of ethylene by iron, nickel, and cobalt clusters and theformation of hydrogen have been studied experimentally, andcatalyzed hydrogen transfer by nickel has also been demon-strated.27,28 Hydride formation is modeled in our studies via Habstraction by iron clusters in char. Although hydrogen abstrac-tion could involve any of the C-H or O-H groups in char, itis more likely to commence by abstraction from hydroxyl groupscoordinated to the Fe centers, followed by oxidative formationof the O-Fe bond and the iron-hydride complex. Theplausibility of these reactions has been based on relative changesin calculated energies of the relevant molecular structure from1scf-PM5, 1scf-DFT, and SE calculations.
Hydrogen abstraction for the model{Char3[Fe3]}, andformation of the hydride{Char3[(C-O-Fe)(Fe2O)(H)]}, in-dicated that this structure was energetically favored relative to{Char3[Fe3]}; however, additional H abstraction followed byH2 desorption resulted in a neutral overall relative change to∆Hf. Hydrogen abstraction for the model{Char3[Fe3O]},followed by the migration of H to the iron cluster oxygen (e.g.,in Fe3O) to form the hydroxide{Char3[-O-Fe2(Fe-OH)]} wasexothermic, indicating that this may compete with hydrideformation. Additional H abstraction for this model, to form H2Owith the [Fe3OH] cluster, was significantly endothermic; theseresults indicate that the iron-cluster-hydride complex wasenergetically favored, and the formation of H2 was more likelythan that of H2O. SE values obtained for the model{Char3-[(C-O)2(Fe3O)]} (after a loss of H2) using default settings ofMOPAC gave relative changes to the∆Hf values of between9.1 and 15.3 kcal. The overall relative change in∆Hf beforeand after the loss of H2 was between-130 and-140 kcal.The iron cluster after the loss of H2 had “opened up” with oneFe-Fe bond distance at 1.76 Å, and the other at>3 Å; theiron species had formed two Fe-O bonds (1.93 and 1.96 Å),with Fe-O-Fe bond lengths of 1.90 and 1.76 Å, and one Fer OH coordination bond length of 2.32 Å. Short Fe‚‚‚Hdistances were also observed in this structure.
The large size of the [Char3/iron cluster] model requiredextremely large computer resources for DFT calculations, andconsequently, a smaller char model was developed by reducingthe size of the [Char3] model into the smaller, similar molecularmodel, labeled CharD: MF) [C144H119NO10]; FW ) 2023.49;elemental composition: C 85.6%, H 5.9%, O 7.9%, N 0.7%.The [Fe3] cluster was added to this to form the molecular model[CharD(Fe3)]: MF ) [C144H119NFe3O10]; elemental composi-tion: C 78.9%, H 5.5%, Fe 7.7%, O 7.3%, N 0.6%. Optimiza-
tion of the CharD molecular structure, in which the internalarrangements of phenyl groups were changed relative to eachother, provided SE∆Hf values of 13.0 and 15.0 kcal, while acompacted configuration which maximized OH‚‚‚H interactions(terminated at a trust radius of<0.0001) gave a∆Hf value of12.0 kcal. DFT geometry optimization of this model provideda structure with normal C-C, C-H, and C-O-H bond lengthsand angles. The model CharD with the Fe3 cluster, [CharD-(Fe3)], was used for detailed studies of H2 and CO formation,and DFT calculations were performed for this model and thetwo molecular models that resulted after H2 elimination (bondlengths listed in Table 1). The shape and size of the Fe3 clusterwere consistent with the reported ground state and Fe-Fe bondlengths obtained using DFT.22,23
The changes in∆Hf from configurations of this model arisingfrom the placement of the Fe3 cluster in [CharD(Fe3)], in whichthe Fer OH coordination bonds lengths were 2.3 and 2.4 Å,respectively (similar to values obtained for models discussedabove), provided SE∆Hf values that differed by only 9 kcal;changes in the position of the Fe3 moiety that resulted in FerOH coordination bond lengths of up to 2.8 Å gave a differenceof 90 kcal to∆Hf. The large difference in∆Hf was due to thechanges in the configurations of the organic molecule resultingfrom the different position of [Fe3] and the longer Fer OHbond lengths. SE optimization of a structure in which the Fe3
cluster was placed in the least sterically crowded area of CharD,and which maximized Fe‚‚‚C interactions with neighboringphenyl carbons, provided the lowest∆Hf value; this structurewas rejected, however, as such a configuration would haverequired an instantaneous appearance of the Fe3 cluster in thechar, without the prior iron-hydroxyl and iron-oxide trans-formations discussed for pyrolysis.
Inspection of the DFT-optimized structures of CharD and{CharD[Fe3]} revealed that the Fe3 cluster had caused asignificant change to the relative positions of all major groups(compared with the arrangements in [CharD]), including thehydroxyl and phenyl groups; fewer phenyl groups assumed localparallel arrangements, and the hydrogen bonds present in theCharD molecule were not observed in{CharD[Fe3]}. The twoHOfFe coordination bond distances in the DFT-optimizedstructure were typical for iron-hydroxyl complexes. Thiscontrasted with the SE results discussed previously, in whichvariable Fer OH and OH‚‚‚H bond distances and the differentenergies were observed. The data highlight the differenttreatment by SE and DFT of hydrogen bonds and coordinationbonds in these structures. Lengthy DFT optimization of thegeometry of the three structures in Table 1 revealed significantchanges in the relative positions of the organic fragments ofthe overall structure, but the iron moiety remained bonded tothe respective functional groups (Fe-O, OHf Fe, and Fe-C)
(27) Ichihashi, M.; Hanmura, T.; Kondow, T. How many metal atomsare needed to dehydrogenate an ethylene molecule on metal clusters?:Correlation between reactivity and electronic structures of Fen
+, Con+, and
Nin+. J. Chem. Phys.2006, 125, 133404-133409.(28) Le Page, M. D.; James, B. R. Nickel bromide as a hydrogen transfer
catalyst.Chem. Commun.2000, 1647-1648.
Table 1. Bond Lengths for Three Models Using SE, 1scf-DFT and DFT
bond lengths (Å)
model method Fe-Fe Fe-O Fe-C
CharD[(CO)-Fe3(-C)] SE 2.34, 2.34, 2.37 1.91, 2.17 1.951scf-DFT 2.34, 2.34, 2.35 1.90, 2.08 1.95DFT 2.37, 2.12, 2.22 1.86, 2.03 1.85
CharD[(OC)2(Fe3)] SE 1.76, 1.76, 2.01 1.95, 2.09SEa 1.73, 1.79, 1.99 2.00, 2.101scf-DFT 2.34, 2.35, 2.35 1.92, 1.92DFT 2.24, 2.22, 2.50 1.76, 1.85 2.33, 2.01
CharD[Fe3] SE 1.73, 1.73, 1.73 2.44, 2.481scf-DFT 2.34, 2.34, 2.34 2.17, 2.17DFT 2.24, 2.11, 2.16 2.06, 2.11
a Using other options available in MOPAC (see text).
2536 Energy & Fuels, Vol. 21, No. 5, 2007 Domazetis et al.
with relatively small variations in the bond lengths ((0.02 Å).These results may indicate a lower impact of hydrogen bondsin DFT calculations, compared to SE calculations. For example,SE calculations for similar configurations of{CharD[(O-C)2-(Fe3)]}, formed after the loss of 2H from the coordinated FerOH groups, agreed within 0.2 kcal, but if this model wascompacted to maximized hydrogen bonding,∆Hf was lowerby -25 kcal. The compacted model gave different Fe-Fe bondlengths and different distances of iron atoms to the neighboringhydrogen atoms of the OH and CH groups.
The formation and loss of H2 from [CharD(Fe3)] may occur(1) through the cleavage of O-H and C-H groups, to form{CharD(C-O)-Fe3(-C)]}, with [Fe-O-C], Fe r OH, and[Fe-C-] bonds and (2) by the cleavage of two O-H groups,to form {CharD[-O-C)2(Fe3)]} containing two (-C-O-Fe)bonds. The structures resulting from 1 and 2 were examinedusing 1scf-PM5, SE, and DFT (bond lengths are shown in Table1). SE data for 1,{CharD[(C-O)-Fe3(-C)]}, indicated a weakFer OH coordination (bond order 0.2), and Fe-O and Fe-Cbonds (bond orders of 0.6 and 0.8). The Fe-Fe bond ordersfor the Fe3 cluster (2.0, 2.4, and 1.2) are consistent with theshorter Fe-Fe bond lengths, and the partial charges on Fe were+(0.3, 0.4, 0.2), on the (Fe-O) oxygen they were-(0.6, 0.3),and on the (Fe-C) carbon was-0.4; these values were similarfor {CharD[-O-C)2(Fe3)]} and significantly larger than thoseobtained for the model{CharD[Fe3]}. Shorter Fe‚‚‚H distanceswere also present in this structure, including one (OH‚‚‚Fe)distance of 1.96 Å, while the Fe-O bond lengths are typicalfor structures of iron with oxygen ligands.1,3 The Fe-Fe bondlengths obtained from the DFT geometry, however, are typicalfor iron clusters, and may be compared to reported values fromDFT calculations for the following: Fe-Fe) 2.14-2.26 Å inthe Fe3, Fe-C ) 1.61-1.93 Å in FeCn, and Fe-O ) 1.80-1.85 Å in FemO clusters.18-23 The DFT-optimized structure{CharD[-O-C)2(Fe3)]} also contained Fe-C distances indica-tive of significant interactions between the iron cluster andneighboring phenyl groups, as shown by the Fe‚‚‚C distancesin Table 1. The importance of maintaining a similar configu-ration for this model may be highlighted by comparing the SE∆Hf values of two molecules, which were identical in all aspectsinitially, with the exception of the internal configuration of thosephenyl groups situated at a considerable distance from the Fe3
cluster that differed by 200 kcal. As a result, every effort hasbeen made to maintain a similar configuration for the modelsused to obtain relative∆Hf values for H abstraction and theloss of H2, and also for the formation of CO by the loss of COfrom the [Fe-O-C] group, as discussed before.
1scf-PM5 and SE calculations for the hydride formed bycleavage of the C-OH group coordinated to Fe in{CharD-[FeB3B]} showed that the hydride structure was energeticallyfavored, as was the dihydride by abstraction of another H bycleavage of either O-H or C-H bonds. The partial charges inthe iron cluster were initially zero in{CharD[Fe3]}, and theseincreased to+(0.6-0.4) for the dihydride complex; the partialcharges on the O, C, and H groups, associated with the Fe clusterwere-0.6, -0.5, and-0.2. The bond orders were consistentwith the single bonds Fe-O and Fe-C. The bond order for theFe‚‚‚OH group indicated a very weak coordination bond; theFe-H distances were 1.62, 1.62, and 1.51 Å, with one Hassociated with two Fe centers; one Fe-Fe bond length was2.31 Å, but two Fe-Fe distances (1.87 and 1.77 Å) were shorterthan the Fe-Fe bonds reported for iron clusters and also shorterthan the Fe-Fe bond lengths obtained from the DFT-optimizedstructures of individual Fem and FemOn clusters (m ) 2-4, n )
1-2).18-23 ∆Hf for the formation of the iron dihydride complexwas exothermic; the particular configuration of the molecularstructure, in which two H’s are situated on either side of theFe3 cluster, encountered a relatively large energy barrier to theformation and loss of the H2 molecule. The thermochemistryfor the{CharD[Fe3]} model, however, differs significantly fromthat for [CharD] (without the iron cluster), for which Habstraction and H2 formation from [CharD] was very endot-hermic.
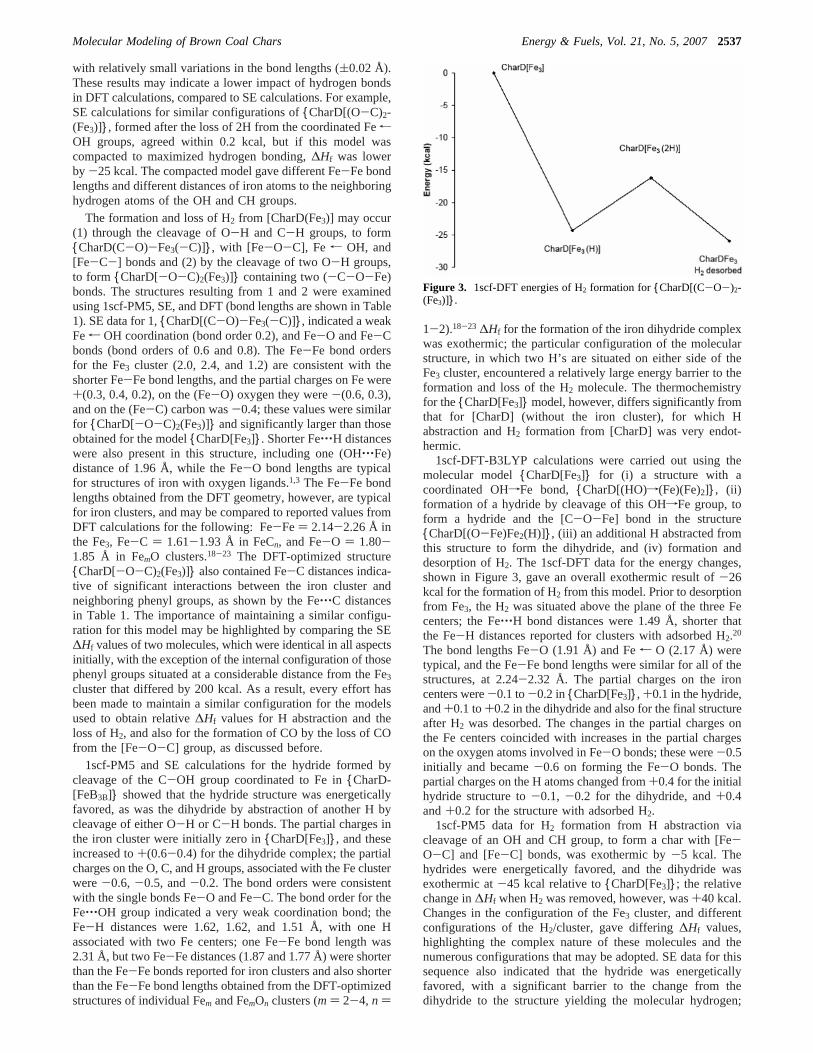
1scf-DFT-B3LYP calculations were carried out using themolecular model{CharD[Fe3]} for (i) a structure with acoordinated OHfFe bond, {CharD[(HO)f(Fe)(Fe)2]}, (ii)formation of a hydride by cleavage of this OHfFe group, toform a hydride and the [C-O-Fe] bond in the structure{CharD[(O-Fe)Fe2(H)]}, (iii) an additional H abstracted fromthis structure to form the dihydride, and (iv) formation anddesorption of H2. The 1scf-DFT data for the energy changes,shown in Figure 3, gave an overall exothermic result of-26kcal for the formation of H2 from this model. Prior to desorptionfrom Fe3, the H2 was situated above the plane of the three Fecenters; the Fe‚‚‚H bond distances were 1.49 Å, shorter thatthe Fe-H distances reported for clusters with adsorbed H2.20
The bond lengths Fe-O (1.91 Å) and Fer O (2.17 Å) weretypical, and the Fe-Fe bond lengths were similar for all of thestructures, at 2.24-2.32 Å. The partial charges on the ironcenters were-0.1 to-0.2 in{CharD[Fe3]}, +0.1 in the hydride,and+0.1 to+0.2 in the dihydride and also for the final structureafter H2 was desorbed. The changes in the partial charges onthe Fe centers coincided with increases in the partial chargeson the oxygen atoms involved in Fe-O bonds; these were-0.5initially and became-0.6 on forming the Fe-O bonds. Thepartial charges on the H atoms changed from+0.4 for the initialhydride structure to-0.1, -0.2 for the dihydride, and+0.4and+0.2 for the structure with adsorbed H2.
1scf-PM5 data for H2 formation from H abstraction viacleavage of an OH and CH group, to form a char with [Fe-O-C] and [Fe-C] bonds, was exothermic by-5 kcal. Thehydrides were energetically favored, and the dihydride wasexothermic at-45 kcal relative to{CharD[Fe3]}; the relativechange in∆Hf when H2 was removed, however, was+40 kcal.Changes in the configuration of the Fe3 cluster, and differentconfigurations of the H2/cluster, gave differing∆Hf values,highlighting the complex nature of these molecules and thenumerous configurations that may be adopted. SE data for thissequence also indicated that the hydride was energeticallyfavored, with a significant barrier to the change from thedihydride to the structure yielding the molecular hydrogen;
Figure 3. 1scf-DFT energies of H2 formation for{CharD[(C-O-)2-(Fe3)]}.
Molecular Modeling of Brown Coal Chars Energy & Fuels, Vol. 21, No. 5, 20072537
the SE∆Hf data for the formation and desorption of H2 for{CharD[Fe3]}, however, gave an overall exothermic value of-99 kcal.
While these calculations indicate that hydrogen formation viathe Fe3 cluster hydride may be energetically favored, the resultsare contingent on a well-defined structure used to commencethe reaction sequence, and also on the assumption that a suitablysized cluster, such as Fe3, may form in the char. However, anFe-hydride molecular structure may also be invoked in aconcerted reaction leading to H2 and CO formation. Forexample, SE data of the dihydride structure, in which the [-C-O-Fe] group was modified into an intermediate structure usedfor CO formation, provided a lower∆Hf than the previousdihydride structure. It is difficult, therefore, to simply postulatea discrete reaction path for H2 formation; the present resultsshow that H abstraction by the iron cluster is energeticallyfavored and also indicate the types of reactions that yield H2
from brown coal char containing iron complexes.CO Formation Using a CtC Graphite Edge with FeO
and Fe2O. DFT calculations have been reported on theformation of CO from reactions of O2 or H2O with char (orgraphite) models containing the CtC edge.6-10 Here, weexamine a similar mechanism for CO formation from the char/graphite model [C22H10] with the iron species FeO and Fe2O;we then compare these results with the mechanism in whichCO is eliminated from the [Fe-O-C] bond formed after Habstraction. The CtC graphite edge is assumed to form afterthe loss of H from two adjacent C-H groups as H2; this hasbeen shown to be very endothermic. The char model [C22H10]is a flat structure and does not contain oxygen functional groupsobserved in brown coal char and thus cannot account for stericinteractions resulting from the various bonds between char [OH]groups and the iron species, nor for hydrogen bonds. However,this is a smaller and simpler molecular model, and it is easierto examine the adsorption of the FeO or FemO clusters followedby the loss of CO; the structures for this sequence also providesome comparison to those invoked for CO formation from the[Fe-O-C] group formed after H abstraction.
The molecular model with FeO adsorbed onto the CtC groupis shown in Figure 4i. SE optimization of this structure withMECI provided nine configurations, with the triplet at 2.58 eVhigher energy compared to the ground state singlet. Thesequence of structures examined was adsorption of FeO on CtC, followed by the formation of a CdO group coordinated toFe, and ultimately the loss of CO. The relative changes in theenergies for this sequence followed a similar trend using 1scf-PM5, 1scf-DFT-B88-PW91, and 1scf-DFT-B3LYP.
The 1scf-B3LYP results for this sequence gave an overallexothermic value of-20 kcal/mol for the formation of CO
(Figure 5). The change in the energy for the structure in whichonly one atom was adsorbed to the char edge (b) was-30 kcal,but the value was-120 kcal for the structure (c) shown inFigure 4i, in which both O and Fe had adsorbed; this moleculewas almost identical in energy to one with a structure containinga [CdOfFe] coordination bond, shown in Figure 5. The lossof CO from this molecular model involves the movement ofCO above the plane of the char molecule in Figure 4ii, untilthe Fe atom had occupied the vacated site in the char model,and the CO molecule was situated on the other side of the Featom, as illustrated in Figure 4iii. The energy for the loss ofCO in [char(Fe)CO] designated e in Figure 5, was 30 kcal/mol, but this varied by∼10 kcal (illustrated by two values inthe Figure 5) depending on the configuration adopted for COto the Fe, shown in Figure 4iii. Desorption energies for CO forvarious models of char have been reported at 33.0-54.9 kcal/mol.29 An alternate structure containing a five-member carbonring, with the Fe bound to the apex pentyl carbon (analogousto the pentyl structure discussed for the reaction mechanismfor char and O2),6 was calculated with a similar energy to thatof the structure in Figure 4iii after the loss of CO.
1scf-DFT data for this graphite/char model with the Fe2Ocluster indicated a greater affinity for the Fe2O cluster to theCtC edge, and a larger energy barrier to the loss of CO fromthe [Char(Fe2)] complex, compared to FeO. This sequenceincluded a molecular structure in which the [CdO] group wascoordinated to one iron center [FerOdC], while both Fe atomsformed Fe‚‚‚C bonds, as shown in Figure 6i; the loss of CO
(29) Montoya, A.; Truong, T.-T. T.; Fanor Mondrago´n, F.; Thanh, N.;Truong, T. N. CO Desorption from Oxygen Species on CarbonaceousSurface: 1. Effects of the Local Structure of the Active Site and the SurfaceCoverage.J. Phys. Chem. A2001, 105, 6757-6764.
Figure 4. Structure in the formation of CO: (i) FeO adsorbed to graphite edge, (ii) Fe displacing CO, and (iii) Fe in graphite plane, prior to lossof CO.
Figure 5. Energy changes (1scf-B3LYP) for CO formation fromCharFeO.
2538 Energy & Fuels, Vol. 21, No. 5, 2007 Domazetis et al.

gave the structure in Figure 6ii; the Fe2 may assume either theconfiguration shown or an “end-on” one in which only one Feinteracts with the carbon atoms.
The loss of CO involved the Fe2 unit moving above the charplane, and the CO below. The loss of CO from this molecularmodel was exothermic by-150 kcal, due to the stability of theiron complex shown in Figure 6ii. The large difference betweenthe energies of the [Char-FeO] and [Char-Fe2O] models wasdue to the additional Fe-C bond formed in the latter. Althoughthese calculations show that the adsorption of FeO and Fe2Oon CtC is energetically favored, it is doubtful that such a charedge is readily available for adsorption by iron oxide clustersat 600-700 °C.
An attempt was made to use this simple model of char inassessing steric crowding introduced by the relatively larger Feclusters, by using a molecule consisting of two and four parallellayers of this char. The molecular model consisting of twoparallel char molecules, with the FeO placed adjacent to thetwo CtC edges, was optimized using SE into a flat structurewith each char molecule on either side of the FeO, Fe-C, andO-C bonds formed with each of the CtC edges; all Fe bondsand the two graphite molecules were situated in the same plane.DFT treatment of this molecular model resulted in both carbonsof the CtC edges bonding to Fe, to give a V-shaped molecularconfiguration with four Fe-C bond lengths of 1.86 Å. Themodel consisting of four parallel char structures, with FeO (andalso Fe3O) adsorbed on the CtC edges, was optimized into adisordered arrangement, in which bonds had formed by the Feand O atoms with the available CtC edges. These results showthat the iron species caused a major disruption to the originalparallel arrangement of the molecular structure. As an alterna-tive, [Char3] was modified to contain a CtC edge, andcalculations were performed for the{Char3[FeO]}, {Char3-[Fe3O2]}, and {Char3[Fe4O]} models. 1scf-PM5 calculationsprovided similar results, consisting of a large exothermic stepfor the adsorption of the iron cluster to the CtC edge, whilethe loss of CO required changes to the configuration of themolecular structures, to allow CO to move above and away fromthe iron cluster. The desorption of CO required+32 kcal for{Char3[FeO]}, but for{Char3[Fe3O2]}, conformational changesgave a total of+80 kcal for the loss of CO. 1scf-PM5 data for{Char3[Fe4O]} with the CtC edge gave-90 kcal for theadsorption of Fe4O and+60 kcal for the desorption of CO, withthe resulting molecule containing the [(Fe4)-C] bond. Theenergy required to form a CtC edge, however, is much greaterthan that required for H abstraction by an iron cluster; thus, theenergy required to form the CtC edge by breaking two adjacentC-H groups on a phenyl group in [Char3] (to yield H2) wascalculated at+150 kcal (SE) and+129 kcal (1scf-DFT).
CO Formation after H Abstraction. The formation of COby decomposition of the [C-O-Fe] group (formed after H
abstraction) was initially modeled using [Char3(Fem)] (m )1-5). After formation of the [Fe-O-C] group, the Fe movescloser to the phenyl group, the (CO) moves above the plane ofthe phenyl group, and the Fe below, creating two Fe‚‚‚C bondswith the adjacent carbon atoms; the sequence may be comparedto that illustrated in Figure 4ii and iii, and also Figure 6. 1scf-PM5 and SE calculations for [Char3(Fe4)] provided a structurein which the iron atom in the cluster was 1.9 and 2.2 Å fromthe (CdO) atoms. The (CdO) group had moved above the Fe4
cluster, analogous to that in Figure 4ii. Detailed 1scf-PM5, SE,and 1scf-DFT calculations were carried out using the smallermolecular model [CharD(Fem)]. 1scf-DFT calculations wererestricted to simplified schemes for H2 and CO formation.
Hydrogen abstraction, H2 formation, and oxidative formationof the [C-O-(Fe3)] bond, followed by the loss of CO (limitedby the available OH groups in the char) may be schematicallywritten as
The formation of the [Fe-O-C] groups resulting from Habstraction has been discussed. On the basis of the relative heatsof formation, the structure containing two [Fe-O-C] bonds(yielding H2) would be energetically favored; however, therelative energies of the monohydride structures indicated thatthe structure containing the [Fe-C] bond is also energeticallyfavored. Consequently, both structures were examined for theformation of CO, on the assumption that CO formation may beconsidered as a discrete reaction sequence. 1scf-PM5 treatmentof {CharD[(C-O-Fe)2Fe]}, in which CO is displaced by themovement of Fe to the phenyl group, forming two Fe-C bonds,shown in Figure 7i, gave a relative increase in∆Hf of +80kcal. For the structure shown in Figure 7ii, in which the COmoved away from the phenyl group,∆Hf decreased by-32kcal relative to the Figure 7i structure. The subsequentmovement of CO parallel to the cluster, then to an “end-on”(CdO‚‚‚Fe) prior to desorption, shown in Figure 7iii, gave∆Hf
values that varied by+7 and+32 kcal, respectively. The energychange for the loss of CO from the structure in Figure 7iii, intothat in Figure 7iv, was-54 kcal. This treatment provided anoverall endothermic result (relative to{CharD[(C-O-Fe)2Fe]})of between+1 and+36 kcal.
The results of energy changes from 1scf-DFT calculationson this sequence of molecular models for CO formation andloss, commencing with the molecular model formed after H2
loss, are shown in Figure 8. This sequence contained anadditional configuration, in which CO was bound “parallel” tothe Fe cluster. The overall result for the formation and loss ofCO was exothermic by-11 kcal. However, it is necessary topoint out that the data in Figure 8 would include an exothermic-144 kcal, if the sequence were to commence with a structureincluding hydride formation but prior to H2 loss; this againhighlights the differences between a discrete reaction schemeand a concerted reaction scheme in which a hydride had formedduring the sequence of structures leading to the loss of CO.
The structures typifying concerted chemistry are shown inFigure 9; Figure 9i is one example that illustrates an [Fe‚‚‚(CO)] intermediate at the beginning of the CO formationsequence, in which the [Fe-H] bond has resulted from Habstraction from an OH group coordinated to the Fe3 cluster.This iron-hydride achieved a lower energy because the iron
Figure 6. Structure of graphite with (i) adsorbed Fe2O and (ii) afterCO loss.
{Char(C-OH)(Fe3)} f {Char(OH)(C-O-)(Fe3)(H)} f
{Char(C-O-)2(Fe3)(2H)} f {Char(C-O-)2(Fe3)} +H2 f {Char(C-O-)(Fe3)(CO)} f
{Char(C-O-)(Fe3)} + CO
Molecular Modeling of Brown Coal Chars Energy & Fuels, Vol. 21, No. 5, 20072539
cluster moved closer to the OH group, and also closer to theadjacent C atoms of the phenyl group, causing the CO groupto move above the plane of the phenyl group.
The second example is shown in Figure 9ii in which themolecule was further stabilized byπ-π bonding between theiron-phenyl group with additional Fe‚‚‚H interactions betweenFe3 and two nearby C-H groups; SE optimization for thisstructure gave the lowest heat of formation of all hydridesconsidered in this study. Finally, Figure 9iii is an example of ahydride structure formed after the loss of CO from the [Fe3]cluster. This structure was optimized to a lower energy when ahydride formed by the cleavage of a nearby C-H group, theFe of the [Fe3] cluster forming the [Fe-C] bond and anotherFe atom occupying the space vacated by the CO group. Thethird Fe retained the [Fe-O-C] bonding from the previous Habstraction. The Fe atom involved in the cleavage of the C-Hbond had also formed a coordination bond with an OH groupin the vicinity (this OH group may participate in further H
abstraction to yield H2). SE data for this structure were asfollows: Bond lengths Fe-Fe ) 1.96, 1.79, 2.48 Å; Fe-H )1.63, 1.62 Å; Fe-C ) 1.92, 1.94, 1.92, 1.88 Å; Fe-O ) 2.03,2.22 Å. Partial charges Fe (+) 0.7, 0.7, 0.6; C (-) 0.6, 0.5, 0.5;O (-) 0.6, 0.3; H (-) 0.16 Bond orders: Fe-Fe ) 1.4, 0.2,2.2; Fe-O ) 0.5; Fer(OH) ) 0.3; Fe-H ) 0.5, 0.4; Fe-C )0.7, 0.9, 0.9, 0.7. The bond order between one pair of Fe-Featoms involved in hydride formation had reduced to 0.2 and abond distance of 2.48 Å, indicating a weakening and lengtheningof this Fe-Fe bond in the cluster, while for the other pair, theFe-Fe bond order had increased to 2.2 and a smaller bondlength of 1.79 Å.
These results further illustrate the difficulty encountered intreating the reactions for H abstraction and H2 loss and thosefor CO loss as two separate and discrete events. Our attemptsat modeling concerted reaction sequences provided a range of∆Hf values for the loss of CO; these models may commencewith H abstraction, and subsequent intermediates would lead
Figure 7. Sequence of molecular model structures used for CO formation: (i) Fe and CO(Ph), (ii) Fe(CO), (iii) CO end-on, and (iv) CO removed.
Figure 8. 1scf-DFT energies for CO formation for the molecule [CharD(C-O)2(Fe3)].
Figure 9. Structures typifying concerted chemistry of hydride and CO formation.
2540 Energy & Fuels, Vol. 21, No. 5, 2007 Domazetis et al.

to H2 formation and CO formation. The difficulty in arriving ata value of the relative changes in∆Hf for the formation of CO,however, was further compounded by the differing structuresobtained at the end of such a reaction sequence. SE values ofthe relative ∆Hf changes for CO formation for a reactionsequence commencing with the molecular model{CharD[-C-O-(Fe3)-C)]} included a decrease of-120 kcal, and a finalexothermic value of-95 kcal, with hydride structures giving arelative decrease in∆Hf to -172 kcal. Changes in theconfiguration of the Fe3 cluster, particularly the distance betweenthe third Fe to the H-C group of an adjacent phenyl group,also provided different∆Hf values. All data, however, showthat the loss of CO and H abstraction from char functionalgroups by iron clusters was energetically favored.
Chemistry of H2 and CO Formation and Catalytic SteamGasification. The modeling results have indicated that H2 andCO may form via metal-mediated chemistry by cleavage of theO-H and C-H groups in char. Hydride/CO structures discussedfor the formation of both H2 and CO may continue until eitherall OH and CH groups were eliminated or the iron moietybecame situated at a relatively large distance from such groups.The chemistry can be summarized in the reaction schemedisplayed in Figure 10, which outlines the reaction routesdiscussed for H2 and CO formation from char containing thecluster Fe3.
Although three reaction routes are shown, the number wouldbe much greater if the scheme were to include all of the possibleiron complexes that may form during the pyrolysis of browncoal containing various iron-hydroxyl complexes. It is likelythat some iron species may be more reactive than others. AsH2 and CO are lost, the molecular size of the char decreases,and this is designated by the models Char*(Fe3), Char#(Fe3),and Charq(Fe3). The formation of the iron-hydride complexby H abstraction is postulated as the initial step in this reactionscheme; the scheme does not include chemistry leading to theformation of other products, such as methane, nor does it includeradical chemistry observed as the temperature increases.
The chemistry of H2 and CO formation may be extended tothe catalytic gasification of char with H2O as follows: duringpyrolysis, reactions yielding CO2, CO, and H2 would provide{Char[Fem]} complexes with Fe-C and Fe-O bonds andFe‚‚‚OH interactions. The partial charges on the iron clustersin these complexes increase to values observed for Fe(III)/(II)oxides in char. These atoms would act as sites for the adsorptionof H2O via the oxygen atom, which contains an appreciablenegative partial charge; this step would be followed by H
abstraction from H2O, to yield an iron-hydride complex, andan additional [HO-C] functional group.
The catalytic gasification of char containing iron species(designated Fem) with H2O may be schematically written as
Initial results for the model [CharD/Fe3], after the loss of H2and CO, indicated that the adsorption of H2O to the Fe3 clusterwas energetically favored; a structure then formed with anadditional abstraction of H from a nearby C-H group. Thisparticular molecular model contained an H2OfFe distancetypical for these coordination bonds and a distorted six-coordinate Fe(III) center, with the Fe‚‚‚Fe distance of 3.1 Åsimilar to that observed in iron complexes containing coordi-nated bonds, such as iron-hydroxyl complexes in coal.2,3 Theother two Fe centers had formed distorted tetrahedral structures,and the H atom was situated between these two tetrahedral Fecenters, with Fe-H bond lengths of 1.8 and 1.6 Å; the clusteralso formed two [Fe-O-C-] bonds. The partial charges onall Fe centers had increased to+(0.9, 0.8, 0.6), and theσ-πandπ-π contribution to the bonding in the six-coordinated Fecenter and ligands had increased significantly. This modelfurther emphasizes the importance of iron-hydride complexesin the concerted chemistry discussed; the abstraction of H froma H2O molecule to form the dihydride-iron complex wouldyield H2 and a new [C-O-H] group; as discussed previously,the decomposition of one of the [C-O-Fe] bonds in thecomplex would yield CO.
A molecular model was also optimized in which the H2Omolecule provided a short Fe-H distance, and an [OH-C]organic group, indicating a direct cleavage of the H-(OH) watermolecule by the iron cluster, may also occur. Molecularmodeling is continuing on catalytic steam gasification involvingthe transition species Fe, Ni, and Co.
Experimental data of pyrolysis at 700-800°C of brown coalchar containing iron species yielded H2, CO, CH4, and CO2, asdiscussed previously. Initial results of steam gasification of thesame char/iron sample at 700-800 °C yielded a gas streamconsisting of significantly larger amounts of H2, and CH4, withhigher ratios of CO to CO2. These results are consistent withthe catalytic steam gasification of char, and some additionalcatalysis of the water-gas-shift reaction between steam and COto yield H2 and CO2.30 Almost complete conversion of theorganic substrate had occurred over a relatively short period,indicating effective catalysis of char/steam gasification. Ourexperimental work, and molecular modeling, is continuing onthe nature of the catalytically active species present in the chars,aimed at maximizing the production of hydrogen from catalyticsteam gasification of brown coal.
Conclusions
Molecular models of char containing iron oxides, and reducediron species, have been developed with similar properties tothose of char/iron samples obtained experimentally. The mo-lecular structures of these char models were optimized usingSE techniques, and a smaller but similar molecular model using
(30) Rhodes, C.; Hutchings, G. J.; Ward, A. M. Water-gas shiftreactions: finding the mechanistic boundary.Catal. Today1995, 23, 43-58.
Figure 10. Reaction routes for H2 and CO from{Char[Fe3]}.
{Char[(C-Fe)(H)(Fem-1)]} + H2O f
{Char[(C-Fe)(H)(Fem-1 H2O)]} f
{Char[(Fem) - (HO-C)]} + H2 f
{Char#[(Fem-1)(Fe)(H)]} + CO
Molecular Modeling of Brown Coal Chars Energy & Fuels, Vol. 21, No. 5, 20072541

DFT. The computer molecular modeling provided insights intothe chemical transformations during the pyrolysis of brown coalcontaining iron-hydroxide complexes, particularly on changesto the iron species during the pyrolysis of brown coal containingiron-hydroxyl species. Molecular models of char containingiron clusters were also used to examine reactions that yieldhydrogen and carbon monoxide. The modeling of structures usedto examine the chemistry of H2 and CO formation, have shownthe following:
1. Char/iron-hydride complexes were energetically favoredwhen they contained iron clusters such as FeO, Fe2O, Fe3O2,Fe3O, Fe3, and Fe4.
2. Hydrogen was formed by the abstraction of H via cleavageof C-H and O-H groups in char by iron clusters; a dihydride-iron complex formed, followed by a loss of H2, and theformation of [Fe-O-C-] bonds.
3. The decomposition of [Fe-O-C-] bonds yielded CO andFe-C bonds, formed when Fe from the iron cluster fitted intothe space vacated by the CO.
4. The relative changes in the energies of the molecules indiscrete reactions for H2 and CO formation show that ironcomplexes in char provided lower-energy reaction routes.
5. A detailed examination of the various molecular complexesfor the formation of H2 and CO from brown coal char containingiron complexes indicated that the chemistry was more likely tooccur via concerted reactions, involving iron-hydride com-plexes during CO formation.
Initial examination of the adsorption of H2O on char/ironmodels, formed after pyrolytic formation of H2 and CO, showthat the adsorption of H2O on iron clusters was energeticallyfavored, indicating that the resulting iron/char complexes couldact as sites for the catalytic steam gasification of low-rank char.
Acknowledgment. This work was supported by grants andcomputer facilities from VPAC and the provision of computer timeby VPAC at APAC-NF. Rheinbraun GBT supplied samples ofGerman brown coal, and Loy Yang Open Cut Mine suppliedsamples of Victorian brown coal.
EF070129V
2542 Energy & Fuels, Vol. 21, No. 5, 2007 Domazetis et al.




















