
Selective co-sensitization approach to increase photon conversion efficiency and electron lifetime...
6
ISSN 1463-9076 Physical Chemistry Chemical Physics www.rsc.org/pccp Volume 14 | Number 47 | 21 December 2012 | Pages 16165–16488 1463-9076(2012)14:47;1-J COMMUNICATION Grätzel, Mhaisalkar et al. A selective co-sensitization approach to increase photon conversion efficiency and electron lifetime in dye-sensitized solar cells Published on 04 October 2012. Downloaded by Nanyang Technological University on 03/07/2015 05:38:53. View Article Online / Journal Homepage / Table of Contents for this issue
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Selective co-sensitization approach to increase photon conversion efficiency and electron lifetime...

ISSN 1463-9076
Physical Chemistry Chemical Physics
www.rsc.org/pccp Volume 14 | Number 47 | 21 December 2012 | Pages 16165–16488
1463-9076(2012)14:47;1-J
COMMUNICATIONGrätzel, Mhaisalkar et al.A selective co-sensitization approach to increase photon conversion effi ciency and electron lifetime in dye-sensitized solar cells
Publ
ishe
d on
04
Oct
ober
201
2. D
ownl
oade
d by
Nan
yang
Tec
hnol
ogic
al U
nive
rsity
on
03/0
7/20
15 0
5:38
:53.
View Article Online / Journal Homepage / Table of Contents for this issue

16182 Phys. Chem. Chem. Phys., 2012, 14, 16182–16186 This journal is c the Owner Societies 2012
Cite this: Phys. Chem. Chem. Phys., 2012, 14, 16182–16186
A selective co-sensitization approach to increase photon conversion
efficiency and electron lifetime in dye-sensitized solar cellsw
Loc H. Nguyen,zab Hemant K. Mulmudi,zac Dharani Sabba,ac
Sneha A. Kulkarni,a
Sudip K. Batabyal,aKazuteru Nonomura,
abMichael Gratzel*
bdand
Subodh G. Mhaisalkar*abc
Received 23rd August 2012, Accepted 4th October 2012
DOI: 10.1039/c2cp42959d
Ruthenium-based C106 and organic D131 sensitizers have been
judicially chosen for co-sensitization due to their complementary
absorption properties and different molecular sizes. Co-sensitization
yields a higher light-harvesting efficiency as well as better dye
coverage to passivate the surface of TiO2. The co-sensitized devices
C106 + D131 showed significant enhancement in the performance
(g = 11.1%), which is a marked improvement over baseline devices
sensitized with either D131 (g = 5.6%) or C106 (g = 9.5%). The
improved performance of the co-sensitized cell is attributed to the
combined enhancement in the short circuit current, open circuit
voltage, and the fill-factor of the solar cells. Jsc is improved because
of the complementary absorption spectra and favorable energy level
alignments of both dyes; whereas, Voc is improved because of the
better surface coverage helping to reduce the recombination and
increase the electron life time. The origins of these enhancements
have been systematically studied through dye desorption, absorption
spectroscopy, and intensity modulated photovoltage spectroscopy
investigations.
Dye sensitized solar cells (DSSC) have gained much attention
from both industry and academic research during the previous
decades due to their cost effective manufacturing capability
and high photon-to-electron conversion efficiencies.1–7 The
key tunable factors to obtain a high efficiency DSSC are to
maximize the light-harvesting efficiency as well as to reduce
charge recombination.5,6 Enhancement of light-harvesting
efficiency may be achieved through incorporation of dyes that
have broad absorption bands in the near-IR of the solar
spectrum.8–17 However, a disadvantage of using a single
sensitizer with wide absorption spectra is its difficulty to
transfer photo-excited electrons from the sensitizer to the
TiO2 photoanode when the conduction band of TiO2 approaches
the LUMO of the dye.9–16 Consequently, another approach is to
co-sensitize the metal oxide photoanode with multiple sensitizers
having both complementary absorption spectra and matched
energy levels simultaneously to obtain panchromatic absorp-
tion.18,19 This approach was demonstrated in tandem device
structures.20,21 However, the method resulted in limited
improvement in the photocurrent because of the extensive loss
of incident light due to the light diffusion by the front
Pt-loaded counter-electrode. Therefore, co-sensitization strategies
were investigated to load the different dyes on a single TiO2
electrode.22–24 For example, Park et al. have reported repeated
adsorption–desorption processes mimicking the concept of the
mobile phase and the stationary phase in a column chromato-
graph, to selectively position different dyes on a single TiO2
film.24 However, the low efficiency (4.8%) obtained and the
complexity of this method limited their practical application.
Apart from these approaches, a stepwise selective dye adsorption
is a muchmore inexpensive and simpler way for co-sensitization of
multiple sensitizers on a single TiO2 film.25–27 Here, we report a
promising DSSC system using a ruthenium-based dye as a
primary sensitizer for co-sensitization via a stepwise adsorption
approach having the potential to further enhance the device
performance significantly.
The ruthenium-based C106 sensitizer, which is one of the
best dyes for DSSC in terms of device performance,28 was
chosen as the primary sensitizer due to its wide light absorp-
tion over the visible regime of the solar spectrum, high
absorption coefficient (18 500 M�1 cm�1 at ca. 550 nm) and
long hydrophobic chains with a strong steric effect reducing dye
aggregation.28–30 The organic dye D131 with its P-conjugated
indoline ring was chosen as the second sensitizer to enhance
the device performance on the C106 platform due to its small
and relatively coplanar structure, and a very high extinction
coefficient in the 400–500 nm spectral range (56 200 M�1 cm�1
at 460 nm)31 compared to C106 as well as other organic
dyes.31–34 Due to these complementary properties of the dyes,
the ruthenium-based C106 dye and organic D131 dye were used
a Energy Research Institute@NTU (ERI@N), NanyangTechnological University, Research Techno Plaza, Singapore 637553.E-mail: [email protected]; Fax: +65-6790-9081;Tel: +65-6790-4626
bCenter for Nanostructured Photosystems (CNPS), NanyangTechnological University, Research Techno Plaza, Singapore 637553
c School of Materials Science & Engineering, Nanyang TechnologicalUniversity, Singapore 639798
d Ecole Polytechnique Federale de Lausanne, Laboratory of Photonicsand Interfaces, CH-1015 Lausanne, Switzerland.E-mail: [email protected]; Tel: +41 21 69 33112,+41 21 69 33115
w Electronic supplementary information (ESI) available: Detaileddevice fabrication, photovoltaic measurements and dispersion in thevalues of J–V characteristics for individual as well as co-sensitizeddevices. See DOI: 10.1039/c2cp42959dz These authors have equally contributed.
PCCP Dynamic Article Links
www.rsc.org/pccp COMMUNICATION
Publ
ishe
d on
04
Oct
ober
201
2. D
ownl
oade
d by
Nan
yang
Tec
hnol
ogic
al U
nive
rsity
on
03/0
7/20
15 0
5:38
:53.
View Article Online
This journal is c the Owner Societies 2012 Phys. Chem. Chem. Phys., 2012, 14, 16182–16186 16183
as co-sensitizers in order to enhance light-harvesting efficiency
as well as dye coverage on the surface of TiO2. These two
factors could possibly lead to improvements not only in terms
of increased photo-excited electron injection, but also may
lower charge recombination, thus improving conversion
efficiency.
In general, there are two approaches for co-sensitization: the
cocktail approach35,36 uses a mixture of dye solutions with
certain molar ratios of the two dyes, whereas the stepwise
approach25–27,37 accomplishes sequential adsorptions of the
two sensitizers. Since the chosen primary sensitizer C106 yields
some of the highest reported device efficiencies,28–30 the objective
of co-sensitization is to further enhance device performance by
co-adsorbing the D131 sensitizer on the platform of saturated
C106 sensitized TiO2. One possible limitation of the cocktail
approach is the competition between the small, higher mobility
D13131–34 with the C106 dye, which is likely to result in
diminished adsorption of the primary sensitizer. Therefore, to
maximize the adsorption of the C106 dye, the TiO2 photoanodes
were first dipped into C106 dye solution, with the gaps in the
C106 coverage being subsequently filled with the D131 dye. The
co-sensitization route employed a stepwise fabrication approach,
which accomplishes adsorption of two dyes on the TiO2
photoanodes in a consecutive manner (details of device
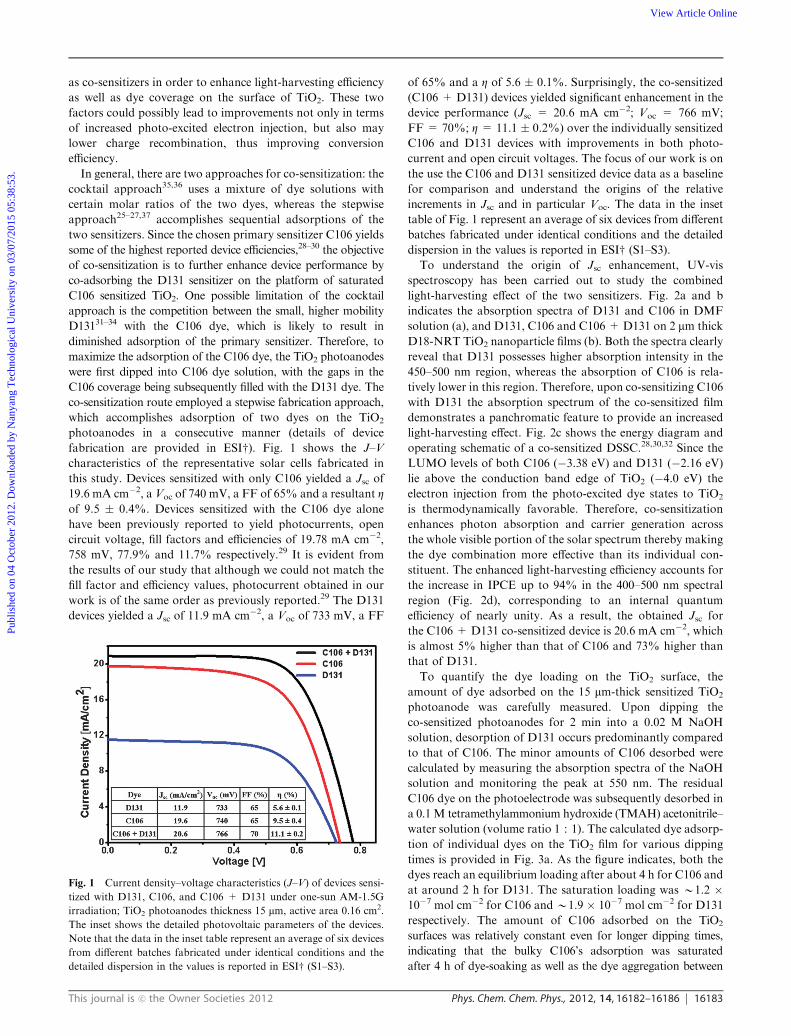
fabrication are provided in ESIw). Fig. 1 shows the J–V
characteristics of the representative solar cells fabricated in
this study. Devices sensitized with only C106 yielded a Jsc of
19.6 mA cm�2, a Voc of 740 mV, a FF of 65% and a resultant Zof 9.5 � 0.4%. Devices sensitized with the C106 dye alone
have been previously reported to yield photocurrents, open
circuit voltage, fill factors and efficiencies of 19.78 mA cm�2,
758 mV, 77.9% and 11.7% respectively.29 It is evident from
the results of our study that although we could not match the
fill factor and efficiency values, photocurrent obtained in our
work is of the same order as previously reported.29 The D131
devices yielded a Jsc of 11.9 mA cm�2, a Voc of 733 mV, a FF
of 65% and a Z of 5.6 � 0.1%. Surprisingly, the co-sensitized
(C106 + D131) devices yielded significant enhancement in the
device performance (Jsc = 20.6 mA cm�2; Voc = 766 mV;
FF = 70%; Z = 11.1 � 0.2%) over the individually sensitized
C106 and D131 devices with improvements in both photo-
current and open circuit voltages. The focus of our work is on
the use the C106 and D131 sensitized device data as a baseline
for comparison and understand the origins of the relative
increments in Jsc and in particular Voc. The data in the inset
table of Fig. 1 represent an average of six devices from different
batches fabricated under identical conditions and the detailed
dispersion in the values is reported in ESIw (S1–S3).
To understand the origin of Jsc enhancement, UV-vis
spectroscopy has been carried out to study the combined
light-harvesting effect of the two sensitizers. Fig. 2a and b
indicates the absorption spectra of D131 and C106 in DMF
solution (a), and D131, C106 and C106 +D131 on 2 mm thick
D18-NRT TiO2 nanoparticle films (b). Both the spectra clearly
reveal that D131 possesses higher absorption intensity in the
450–500 nm region, whereas the absorption of C106 is rela-
tively lower in this region. Therefore, upon co-sensitizing C106
with D131 the absorption spectrum of the co-sensitized film
demonstrates a panchromatic feature to provide an increased
light-harvesting effect. Fig. 2c shows the energy diagram and
operating schematic of a co-sensitized DSSC.28,30,32 Since the
LUMO levels of both C106 (�3.38 eV) and D131 (�2.16 eV)
lie above the conduction band edge of TiO2 (�4.0 eV) the
electron injection from the photo-excited dye states to TiO2
is thermodynamically favorable. Therefore, co-sensitization
enhances photon absorption and carrier generation across
the whole visible portion of the solar spectrum thereby making
the dye combination more effective than its individual con-
stituent. The enhanced light-harvesting efficiency accounts for
the increase in IPCE up to 94% in the 400–500 nm spectral
region (Fig. 2d), corresponding to an internal quantum
efficiency of nearly unity. As a result, the obtained Jsc for
the C106 + D131 co-sensitized device is 20.6 mA cm�2, which
is almost 5% higher than that of C106 and 73% higher than
that of D131.
To quantify the dye loading on the TiO2 surface, the
amount of dye adsorbed on the 15 mm-thick sensitized TiO2
photoanode was carefully measured. Upon dipping the
co-sensitized photoanodes for 2 min into a 0.02 M NaOH
solution, desorption of D131 occurs predominantly compared
to that of C106. The minor amounts of C106 desorbed were
calculated by measuring the absorption spectra of the NaOH
solution and monitoring the peak at 550 nm. The residual
C106 dye on the photoelectrode was subsequently desorbed in
a 0.1 M tetramethylammonium hydroxide (TMAH) acetonitrile–
water solution (volume ratio 1 : 1). The calculated dye adsorp-
tion of individual dyes on the TiO2 film for various dipping
times is provided in Fig. 3a. As the figure indicates, both the
dyes reach an equilibrium loading after about 4 h for C106 and
at around 2 h for D131. The saturation loading was B1.2 �10�7 mol cm�2 for C106 and B1.9 � 10�7 mol cm�2 for D131
respectively. The amount of C106 adsorbed on the TiO2
surfaces was relatively constant even for longer dipping times,
indicating that the bulky C106’s adsorption was saturated
after 4 h of dye-soaking as well as the dye aggregation between
Fig. 1 Current density–voltage characteristics (J–V) of devices sensi-
tized with D131, C106, and C106 + D131 under one-sun AM-1.5G
irradiation; TiO2 photoanodes thickness 15 mm, active area 0.16 cm2.
The inset shows the detailed photovoltaic parameters of the devices.
Note that the data in the inset table represent an average of six devices
from different batches fabricated under identical conditions and the
detailed dispersion in the values is reported in ESIw (S1–S3).
Publ
ishe
d on
04
Oct
ober
201
2. D
ownl
oade
d by
Nan
yang
Tec
hnol
ogic
al U
nive
rsity
on
03/0
7/20
15 0
5:38
:53.
View Article Online

16184 Phys. Chem. Chem. Phys., 2012, 14, 16182–16186 This journal is c the Owner Societies 2012
C106’s molecules during a long dye soaking process was very
unlikely, justifying the choice of 12 h dipping time in our
experiments. Fig. 3b shows the relative and total amount of dyes
loaded onto the TiO2 photoelectrode during the co-sensitization
approach. Every sample represented in Fig. 3b has been
dipped for 12 h in C106 solution before being loaded with
D131 for different dipping times. The concentration of D131
loaded onto the C106 sensitized photoanode saturates after 4 h
Fig. 2 (a) and (b) UV-Vis absorption spectra of: D131 and C106 in DMF solution (a), and D131, C106 and C106 + D131 on transparent TiO2
films (b). (c) Operating schematic of co-sensitized DSSC.28,30,32 (d) Incident-photon-to-electron conversion efficiency (IPCE) action spectra of
devices sensitized with D131, C106, and C106 + D131 under identical fabrication conditions.
Fig. 3 (a) Concentration of dye loaded onto the TiO2 photoelectrode for various dipping times of C106 and D131. The plotted values are the
average values of samples from 3 different batches. (b) Concentration of dye loaded onto co-sensitized TiO2 photoelectrodes monitored for
different D131 dipping times. All the photoanodes were prepared by dipping in a C106 solution for 12 h before varying the dipping time for the
co-sensitization of D131. (c) and (d) Schematic illustrating molecular structure and adsorption sites of C106 and D131 dyes on TiO2.
Publ
ishe
d on
04
Oct
ober
201
2. D
ownl
oade
d by
Nan
yang
Tec
hnol
ogic
al U
nive
rsity
on
03/0
7/20
15 0
5:38
:53.
View Article Online
This journal is c the Owner Societies 2012 Phys. Chem. Chem. Phys., 2012, 14, 16182–16186 16185
of dipping (in contrast to 2 h for individual sensitization).
Notably, the concentration of D131 on the co-sensitized
photoanode was only 0.9 � 10�7 mol cm�2 (vs. 1.9 �10�7 mol cm�2 for individual sensitization) indicating that
there is a lower number of binding sites left on the TiO2
surface after C106 adsorption. The total number of C106 dye
molecules on the co-sensitized samples reduces slightly (from
1.4 � 10�7 mol cm�2 to 1.2 � 10�7 mol cm�2) with increasing
dipping time due to their solubility in the D131 sensitizing
solvent (ACN, TBA). In spite of this, the total amount of dye
molecules (both C106 and D131) was significantly increased to
2.1 � 10�7 mol cm�2. The results thus reveal that the total dye
coverage has been improved by inserting small-sized D131 dye
molecules into the gaps within the C106 saturated TiO2 films,
as visualized in Fig. 3c and d. C106 is adsorbed on the TiO2
surface via its carboxylic group just as a conventional dye,28–30
whereas D131 is adsorbed via not only its carboxylic group
but also via the nitrogen in the cyano group as discussed
elsewhere.33 Consequently, the anchoring sites on the TiO2
nanocrystals are different for both dyes, thus enhancing their
adsorption–dispersion on the TiO2 surface.33 On the other
hand, D131 is expected to link perpendicularly to the TiO2
surface since the molecular structure is practically coplanar
(y anchor E0) thus giving rise to a denser packing and
coverage in tandem with C106.34
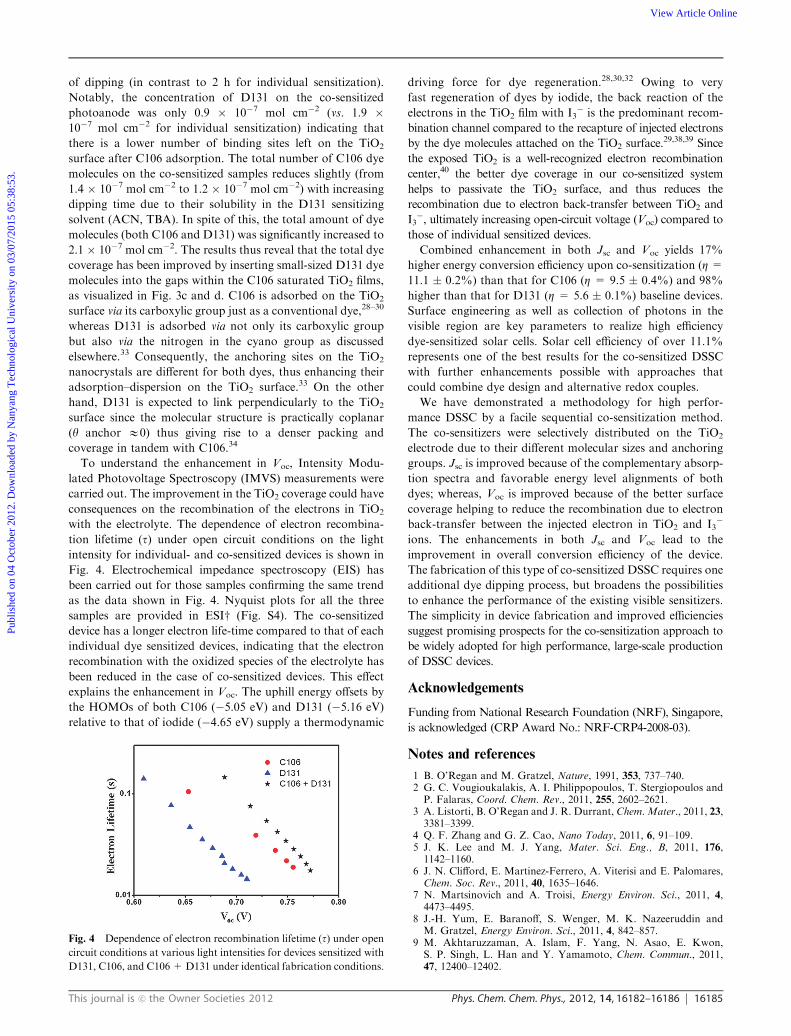
To understand the enhancement in Voc, Intensity Modu-
lated Photovoltage Spectroscopy (IMVS) measurements were
carried out. The improvement in the TiO2 coverage could have
consequences on the recombination of the electrons in TiO2
with the electrolyte. The dependence of electron recombina-
tion lifetime (t) under open circuit conditions on the light
intensity for individual- and co-sensitized devices is shown in
Fig. 4. Electrochemical impedance spectroscopy (EIS) has
been carried out for those samples confirming the same trend
as the data shown in Fig. 4. Nyquist plots for all the three
samples are provided in ESIw (Fig. S4). The co-sensitized
device has a longer electron life-time compared to that of each
individual dye sensitized devices, indicating that the electron
recombination with the oxidized species of the electrolyte has
been reduced in the case of co-sensitized devices. This effect
explains the enhancement in Voc. The uphill energy offsets by
the HOMOs of both C106 (�5.05 eV) and D131 (�5.16 eV)
relative to that of iodide (�4.65 eV) supply a thermodynamic
driving force for dye regeneration.28,30,32 Owing to very
fast regeneration of dyes by iodide, the back reaction of the
electrons in the TiO2 film with I3� is the predominant recom-
bination channel compared to the recapture of injected electrons
by the dye molecules attached on the TiO2 surface.29,38,39 Since
the exposed TiO2 is a well-recognized electron recombination
center,40 the better dye coverage in our co-sensitized system
helps to passivate the TiO2 surface, and thus reduces the
recombination due to electron back-transfer between TiO2 and
I3�, ultimately increasing open-circuit voltage (Voc) compared to
those of individual sensitized devices.
Combined enhancement in both Jsc and Voc yields 17%
higher energy conversion efficiency upon co-sensitization (Z =
11.1 � 0.2%) than that for C106 (Z = 9.5 � 0.4%) and 98%
higher than that for D131 (Z = 5.6 � 0.1%) baseline devices.
Surface engineering as well as collection of photons in the
visible region are key parameters to realize high efficiency
dye-sensitized solar cells. Solar cell efficiency of over 11.1%
represents one of the best results for the co-sensitized DSSC
with further enhancements possible with approaches that
could combine dye design and alternative redox couples.
We have demonstrated a methodology for high perfor-
mance DSSC by a facile sequential co-sensitization method.
The co-sensitizers were selectively distributed on the TiO2
electrode due to their different molecular sizes and anchoring
groups. Jsc is improved because of the complementary absorp-
tion spectra and favorable energy level alignments of both
dyes; whereas, Voc is improved because of the better surface
coverage helping to reduce the recombination due to electron
back-transfer between the injected electron in TiO2 and I3�
ions. The enhancements in both Jsc and Voc lead to the
improvement in overall conversion efficiency of the device.
The fabrication of this type of co-sensitized DSSC requires one
additional dye dipping process, but broadens the possibilities
to enhance the performance of the existing visible sensitizers.
The simplicity in device fabrication and improved efficiencies
suggest promising prospects for the co-sensitization approach to
be widely adopted for high performance, large-scale production
of DSSC devices.
Acknowledgements
Funding from National Research Foundation (NRF), Singapore,
is acknowledged (CRP Award No.: NRF-CRP4-2008-03).
Notes and references
1 B. O’Regan and M. Gratzel, Nature, 1991, 353, 737–740.2 G. C. Vougioukalakis, A. I. Philippopoulos, T. Stergiopoulos andP. Falaras, Coord. Chem. Rev., 2011, 255, 2602–2621.
3 A. Listorti, B. O’Regan and J. R. Durrant,Chem. Mater., 2011, 23,3381–3399.
4 Q. F. Zhang and G. Z. Cao, Nano Today, 2011, 6, 91–109.5 J. K. Lee and M. J. Yang, Mater. Sci. Eng., B, 2011, 176,1142–1160.
6 J. N. Clifford, E. Martinez-Ferrero, A. Viterisi and E. Palomares,Chem. Soc. Rev., 2011, 40, 1635–1646.
7 N. Martsinovich and A. Troisi, Energy Environ. Sci., 2011, 4,4473–4495.
8 J.-H. Yum, E. Baranoff, S. Wenger, M. K. Nazeeruddin andM. Gratzel, Energy Environ. Sci., 2011, 4, 842–857.
9 M. Akhtaruzzaman, A. Islam, F. Yang, N. Asao, E. Kwon,S. P. Singh, L. Han and Y. Yamamoto, Chem. Commun., 2011,47, 12400–12402.
Fig. 4 Dependence of electron recombination lifetime (t) under opencircuit conditions at various light intensities for devices sensitized with
D131, C106, and C106 +D131 under identical fabrication conditions.
Publ
ishe
d on
04
Oct
ober
201
2. D
ownl
oade
d by
Nan
yang
Tec
hnol
ogic
al U
nive
rsity
on
03/0
7/20
15 0
5:38
:53.
View Article Online

16186 Phys. Chem. Chem. Phys., 2012, 14, 16182–16186 This journal is c the Owner Societies 2012
10 A. Abbotto, F. Sauvage, C. Barolo, F. De Angelis, S. Fantacci,M. Graetzel, N. Manfredi, C. Marinzi and M. K. Nazeeruddin,Dalton Trans., 2011, 40, 234–242.
11 S. Paek, H. Choi, C. Kim, N. Cho, S. So, K. Song,M. K. Nazeeruddinand J. Ko, Chem. Commun., 2011, 47, 2874–2876.
12 Y. Liu, H. Lin, J. T. Dy, K. Tamaki, J. Nakazaki, D. Nakayama,S. Uchida, T. Kubo and H. Segawa, Chem. Commun., 2011, 47,4010–4012.
13 T. Maeda, Y. Hamamura, K. Miyanaga, N. Shima, S. Yagi andH. Nakazumi, Org. Lett., 2011, 13, 5994–5997.
14 S. Gomez Esteban, P. de la Cruz, A. Aljarilla, L. M. Arellano andF. Langa, Org. Lett., 2011, 13, 5362–5365.
15 A. Braga, S. Gimenez, I. Concina, A. Vomiero and I. n. Mora-Sero,J. Phys. Chem. Lett., 2011, 2, 454–460.
16 C. Jiao, N. Zu, K.-W. Huang, P. Wang and J. Wu, Org. Lett.,2011, 13, 3652–3655.
17 J. Warnan, F. Buchet, Y. Pellegrin, E. Blart and F. Odobel, Org.Lett., 2011, 13, 3944–3947.
18 V. P. S. Perera, P. K. D. D. P. Pitigala, M. K. I. Senevirathne andK. Tennakone, Sol. Energy Mater. Sol. Cells, 2005, 85, 91–98.
19 J.-J. Cid, J.-H. Yum, S.-R. Jang, M. K. Nazeeruddin, E. Martınez-Ferrero, E. Palomares, J. Ko, M. Gratzel and T. Torres, Angew.Chem., Int. Ed., 2007, 46, 8358–8362.
20 S.-Q. Fan, B. Fang, H. Choi, S. Paik, C. Kim, B.-S. Jeong,J.-J. Kim and J. Ko, Electrochim. Acta, 2010, 55, 4642–4646.
21 T. Yamaguchi, Y. Uchida, S. Agatsuma and H. Arakawa, Sol.Energy Mater. Sol. Cells, 2009, 93, 733–736.
22 J. N. Clifford, E. Palomares, M. K. Nazeeruddin, R. Thampi,M. Gratzel and J. R. Durrant, J. Am. Chem. Soc., 2004, 126, 5670–5671.
23 H. Choi, S. Kim, S. O. Kang, J. Ko, M.-S. Kang, J. N. Clifford,A. Forneli, E. Palomares, M. K. Nazeeruddin and M. Gratzel,Angew. Chem., Int. Ed., 2008, 47, 8259–8263.
24 K. Lee, S. W. Park, M. J. Ko, K. Kim and N.-G. Park, Nat.Mater., 2009, 8, 665–671.
25 S.-Q. Fan, C. Kim, B. Fang, K.-X. Liao, G.-J. Yang, C.-J. Li,J.-J. Kim and J. Ko, J. Phys. Chem. C, 2011, 115, 7747–7754.
26 C.-M. Lan, H.-P. Wu, T.-Y. Pan, C.-W. Chang, W.-S. Chao,C.-T. Chen, C.-L. Wang, C.-Y. Lin and E. W.-G. Diau, EnergyEnviron. Sci., 2012, 5, 6460–6464.
27 D. Kuang, P. Walter, F. Nuesch, S. Kim, J. Ko, P. Comte,S. M. Zakeeruddin, M. K. Nazeeruddin and M. Gratzel, Langmuir,2007, 23, 10906–10909.
28 M. Wang, J. Liu, N.-L. Cevey-Ha, S.-J. Moon, P. Liska,R. Humphry-Baker, J.-E. Moser, C. Gratzel, P. Wang,S. M. Zakeeruddin and M. Gratzel, Nano Today, 2010, 5, 169–174.
29 Q. Yu, Y. Wang, Z. Yi, N. Zu, J. Zhang, M. Zhang and P. Wang,ACS Nano, 2010, 4, 6032–6038.
30 Y. Cao, Y. Bai, Q. Yu, Y. Cheng, S. Liu, D. Shi, F. Gao andP. Wang, J. Phys. Chem. C, 2009, 113, 6290–6297.
31 M. Matsui, M. Kotani, Y. Kubota, K. Funabiki, J. Jin,T. Yoshida, S. Higashijima and H. Miura, Dyes Pigm., 2011, 91,145–152.
32 J. Y. Kim, Y. H. Kim and Y. S. Kim, Curr. Appl. Phys., 2011, 11,S117–S121.
33 R. Y. Ogura, S. Nakane, M. Morooka, M. Orihashi, Y. Suzukiand K. Noda, Appl. Phys. Lett., 2009, 94, 073308–073303.
34 T. Le Bahers, T. Pauporte, G. Scalmani, C. Adamo and I. Ciofini,Phys. Chem. Chem. Phys., 2009, 11, 11276–11284.
35 Y. Chen, Z. Zeng, C. Li, W. Wang, X. Wang and B. Zhang,New J.Chem., 2005, 29, 773–776.
36 J.-H. Yum, S.-R. Jang, P. Walter, T. Geiger, F. Nuesch, S. Kim,J. Ko, M. Gratzel and M. K. Nazeeruddin, Chem. Commun., 2007,4680–4682.
37 J. N. Clifford, A. Forneli, H. Chen, T. Torres, S. Tan andE. Palomares, J. Mater. Chem., 2011, 21, 1693–1696.
38 M. Wang, P. Chen, R. Humphry-Baker, S. M. Zakeeruddin andM. Gratzel, ChemPhysChem, 2009, 10, 290–299.
39 B. A. Gregg, F. Pichot, S. Ferrere and C. L. Fields, J. Phys. Chem. B,2001, 105, 1422–1429.
40 S. A. Haque, Y. Tachibana, R. L. Willis, J. E. Moser, M. Gratzel,D. R. Klug and J. R. Durrant, J. Phys. Chem. B, 1999, 104,538–547.
Publ
ishe
d on
04
Oct
ober
201
2. D
ownl
oade
d by
Nan
yang
Tec
hnol
ogic
al U
nive
rsity
on
03/0
7/20
15 0
5:38
:53.
View Article Online




















