
Segmental dynamics in nanophase separated comb-like polymers with long side chains
6
Segmental dynamics in nanophase separated comb-like polymers with long side chains E. Laredo a, * , M. Grimau b , A. Bello a , F. Lo ´ pez-Carrasquero c a Physics Department, Universidad Simo ´ n Bolı ´var, Apartado 89000, Caracas 1080 A, Venezuela b Materials Science Department, Universidad Simo ´ n Bolı ´var, Apartado 89000, Caracas 1080 A, Venezuela c Chemistry Department, Facultad de Ciencias, Universidad de Los Andes, Me ´rida 5101 A, Venezuela Available online 25 September 2007 Abstract Poly(a-n-alkyl b-L-aspartate)s, PnAALAs, are comb-like polymers with rod-like helical conformations which present crystallinity for long alkyl side chains (n > 10). Dielectric spectroscopy experiments for PnAALAs (10 6 n 6 18) show the existence at low temperatures of two secondary modes corresponding to localized motions of the carbonyl group. At higher temperatures two cooperative mobilities coexist. The first corresponds to the segmental mobility of the amorphous side chains and has the characteristic of a polyethylene-like dynamic glass transition common to various series of nanophase segregated side chain polymers with different morphologies. A second segmental mode is attributed to the restricted rotational motion of the rigid rods which is hindered when the spacing between rods decreases. An extension to the results found for other polymer series with shorter lateral chains is presented, showing the tendency to approximate amorphous polyethylene dynamics in these confined nanodomains of increasing sizes. Ó 2007 Elsevier B.V. All rights reserved. PACS: 64.70.Pf; 77.22.Gm; 77.84.Jd; 64.70.Nd Keywords: Dielectric properties, relaxation, electric modulus; Glass transition; Nanocrystals; Polymers and organics; Long range order 1. Introduction Poly(a-n-alkyl b-L-aspartate)s, PnAALAs, where n is the number of carbons in the alkyl group, are nylon-3 deriva- tives which repeating unit is represented in Fig. 1. These materials with variable side chain lengths have been thor- oughly characterized [1,2] and shown to form helical struc- tures. The most frequent conformation are right handed helices with an axial period of 2 nm, which are stabilized by the intramolecular H-bond existing between the amino group of the i residue and the carbonyl group of the i +3 residue. The PnAALAs are another example of a family of comb-like polymers with incompatible main chains and flexible paraffinic side chains of variable lengths such as the poly(n-alkyl acrylates), PnAA, the poly(n-alkyl methacrylates), PnAMA, the poly(n-alkyl itaconates), PnAI, poly(n-alkyl glutamates), PnALG, and the hairy rods polyimides. Recently, Beiner and Huth [3] have studied the nano- phase separation on size domains from 1 to 5 nm for PnAA (n 6 10) with incompatible main and side chains and shown that it is a general phenomenon in amorphous comb-like polymers with short to medium length alkyl branches. Also, they showed that important dynamic aspects are main chain independent as they occur in self- assembled alkyl nanodomains. The effect of this self con- finement on the cooperative motions of the families of amorphous side chain polymers is the existence of a poly- ethylene-like glass transition within the nanodomains and which is shown to have similar features for all the comb- like polymers with alkyl side chains compiled in that work. In the PnAALAs WAXS, SAXS combined with calori- metric, DSC, experiments [2] have shown that for n P 12 0022-3093/$ - see front matter Ó 2007 Elsevier B.V. All rights reserved. doi:10.1016/j.jnoncrysol.2007.03.032 * Corresponding author. Tel.: +58 212 906 3536; fax: +58 212 906 3527. E-mail address: [email protected] (E. Laredo). www.elsevier.com/locate/jnoncrysol Available online at www.sciencedirect.com Journal of Non-Crystalline Solids 353 (2007) 4324–4329
Transcript of Segmental dynamics in nanophase separated comb-like polymers with long side chains

Available online at www.sciencedirect.com
www.elsevier.com/locate/jnoncrysol
Journal of Non-Crystalline Solids 353 (2007) 4324–4329
Segmental dynamics in nanophase separated comb-like polymerswith long side chains
E. Laredo a,*, M. Grimau b, A. Bello a, F. Lopez-Carrasquero c
a Physics Department, Universidad Simon Bolıvar, Apartado 89000, Caracas 1080 A, Venezuelab Materials Science Department, Universidad Simon Bolıvar, Apartado 89000, Caracas 1080 A, Venezuela
c Chemistry Department, Facultad de Ciencias, Universidad de Los Andes, Merida 5101 A, Venezuela
Available online 25 September 2007
Abstract
Poly(a-n-alkyl b-L-aspartate)s, PnAALAs, are comb-like polymers with rod-like helical conformations which present crystallinity forlong alkyl side chains (n > 10). Dielectric spectroscopy experiments for PnAALAs (10 6 n 6 18) show the existence at low temperaturesof two secondary modes corresponding to localized motions of the carbonyl group. At higher temperatures two cooperative mobilitiescoexist. The first corresponds to the segmental mobility of the amorphous side chains and has the characteristic of a polyethylene-likedynamic glass transition common to various series of nanophase segregated side chain polymers with different morphologies. A secondsegmental mode is attributed to the restricted rotational motion of the rigid rods which is hindered when the spacing between rodsdecreases. An extension to the results found for other polymer series with shorter lateral chains is presented, showing the tendency toapproximate amorphous polyethylene dynamics in these confined nanodomains of increasing sizes.� 2007 Elsevier B.V. All rights reserved.
PACS: 64.70.Pf; 77.22.Gm; 77.84.Jd; 64.70.Nd
Keywords: Dielectric properties, relaxation, electric modulus; Glass transition; Nanocrystals; Polymers and organics; Long range order
1. Introduction
Poly(a-n-alkyl b-L-aspartate)s, PnAALAs, where n is thenumber of carbons in the alkyl group, are nylon-3 deriva-tives which repeating unit is represented in Fig. 1. Thesematerials with variable side chain lengths have been thor-oughly characterized [1,2] and shown to form helical struc-tures. The most frequent conformation are right handedhelices with an axial period of �2 nm, which are stabilizedby the intramolecular H-bond existing between the aminogroup of the i residue and the carbonyl group of the i + 3residue. The PnAALAs are another example of a familyof comb-like polymers with incompatible main chainsand flexible paraffinic side chains of variable lengths suchas the poly(n-alkyl acrylates), PnAA, the poly(n-alkyl
0022-3093/$ - see front matter � 2007 Elsevier B.V. All rights reserved.
doi:10.1016/j.jnoncrysol.2007.03.032
* Corresponding author. Tel.: +58 212 906 3536; fax: +58 212 906 3527.E-mail address: [email protected] (E. Laredo).
methacrylates), PnAMA, the poly(n-alkyl itaconates),PnAI, poly(n-alkyl glutamates), PnALG, and the hairyrods polyimides.
Recently, Beiner and Huth [3] have studied the nano-phase separation on size domains from 1 to 5 nm for PnAA(n 6 10) with incompatible main and side chains andshown that it is a general phenomenon in amorphouscomb-like polymers with short to medium length alkylbranches. Also, they showed that important dynamicaspects are main chain independent as they occur in self-assembled alkyl nanodomains. The effect of this self con-finement on the cooperative motions of the families ofamorphous side chain polymers is the existence of a poly-ethylene-like glass transition within the nanodomains andwhich is shown to have similar features for all the comb-like polymers with alkyl side chains compiled in that work.
In the PnAALAs WAXS, SAXS combined with calori-metric, DSC, experiments [2] have shown that for n P 12

HNC
COORn
H
CH2
CO
Fig. 1. Chemical Structure of the poly(a-n-alkyl b-L-aspartate)s repeatingunit, Rn=–(CH2)n�1–CH3.
E. Laredo et al. / Journal of Non-Crystalline Solids 353 (2007) 4324–4329 4325
the long linear alkyl chains tend to form two-dimensionalarrangements with rows of 13/4 helices separated by a par-tially crystallized paraffinic nanophase. This lamellarordered arrangement, phase A, exists for temperaturesT < T1, and T1 being an increasing function of n. This ther-mal transition is associated to the fusion of the paraffiniccrystallites and causes a contraction of the interlayer dis-tance, L0. At temperatures between T1 and T2 phase Bexists and it is presumably similar to phase A but with sidechains in a liquid state. Phase C exists above T2 where thelayered structure is not observed anymore and L0 is furtherreduced as shown in Table 1. No evidence of a glass tran-sition was found by DSC [2].
In the present study of long side chains PnAALAs,n P 10, dielectric spectroscopy (DS) was used in order tofollow the local and segmental dynamics in the whole tem-perature range. At high temperatures multiple cooperativemodes are found which show the coexistence of polyethyl-ene-like segmental dynamics in the alkyl nanodomains witha second cooperative mobility that could be assigned to arestricted rotational diffusion of the rods in a liquid med-ium formed by the molten side chains [4]. The objectiveof this work is to compare the results obtained in thesemi-crystalline PnAALAs to those obtained on the nano-phase separated PnAAs, PnAMAs and PnAIs which areamorphous and present common trends in the variationof their relaxation characteristics.
2. Experimental
2.1. Materials
The five PnAALAs studied here were synthesized bynon-assisted anionic ring polymerization of the opticallypure (S)-4-alkoxycarbonyl-2-azetidinones [5,6]. The molec-ular weights are about 105 g/mol and the products are sta-ble at temperatures below 473 K. Films of PnAALA with10 6 n 6 18 were obtained by evaporation of chloroformsolutions and dried under vacuum. The average thicknesswas about 50 lm.
Table 1Transition temperatures and L0 for the PnAALA [2]
N T1 (K) T2 (K) L0A (nm) L0B (nm) L0C (nm)
12 258 323 2.5 2.4 2.314 271 335 2.6 2.5 2.216 307–321 366 2.9 2.7 2.418 327–337 390 3.1 2.8 2.3
2.2. Dielectric spectroscopy
The DS experiments were carried out in a NovocontrolConcept Twelve Spectrometer by heating the sample from133 to 358 K by 2 K steps. The temperature stability wasbetter than 0.1 K. The 35 frequencies chosen here rangedfrom 10�2 to 3.16 · 106 Hz. The relative error on thee00ðxÞ determinations is less than 3%.
The analysis of the dynamic spectra was performed bydecomposing the experimental isothermal trace of theimaginary part e00ðxÞ into up to three relaxation modeswith Cole–Cole distributions plus a conductivity term.The fit of the experimental data is made to the expression
e00ðxÞ ¼X3
k¼1
Dekðx�skÞak sinðpak=2Þ½1þ 2ðx�skÞak cosðpak=2Þ þ ðx�skÞ2ak �
þ r0
e0x
� �;
ð1Þwhere e0 denotes the permittivity of free space. Each modeis characterized by the width, ak, the mean relaxation time,�sk, of the Cole–Cole distribution and the dielectric strength,Dek . Such a procedure yields the relaxation maps present-ing the variation of log �xk ¼ � log �sk vs. 1/T. The curvesare fitted to Arrhenius (linear dependences) or to a Vo-gel–Fulcher–Tammann (VFT) expression
sVFTðT Þ ¼ s0VFT exp½EVFT=kðT � T 0Þ�: ð2Þ
The three parameters s0VFT, EVFT and T0 can be deducedfor each cooperative mode. Cole–Cole distribution was cho-sen instead of Havriliak–Negami function as it has beenshown that for semi-crystalline polymers the local and seg-mental modes are best described by symmetric distributions.Recent works on in situ crystallization of polycarbonate [7],polyethylene terephtalate [8] and PEEK [9] have shown thatthe asymmetric parameter of the HN distributions tends to1 as the crystallization process advances.
3. Results
The experimental loss tangent isochrones are repre-sented in Fig. 2(a) and (b) as tan(d) vs. temperature for100 Hz and 10 kHz for the five n values of PnAALA’s stud-ied here. The shift of the spectrum to higher temperature asn increases is observed. The low temperature region ismade of wide peaks which translation with n is weak; acareful observation of these peaks indicates the presenceof two modes, labelled c1 and c2. Around room tempera-ture are found the most intense relaxations, aPE and a,which are also broad and shift considerably with n as seenin both isochrones. At 100 Hz the conductivity contribu-tion is already significant and overlaps the a mode; forP18AALA this second peak is apparent in Fig. 2(a). Forthe isochrone at 10 kHz, the two processes overlap butcan still be distinguished in the data for P12AALA andP18AALA. The multicomponent character of these peaksneeds to be taken into account when determining the relax-ation parameters of each mode.
100 Hz
tan(
δ )
10-3
10-2
10-1
10 kHz
T (K)
150 200 250 300 350
tan(
δ )
10-4
10-3
10-2
a
b
γ1
γ2
γ1
γ2
αPE
αPE
α
α
Fig. 2. Variation with temperature of tan(d) for (a) m = 100 Hz, (b)m = 10 kHz for the PnAALAs: h, n = 18; � n = 16; 4, n = 14; d, n = 12;5, n = 10.
307 K
log(ν /Hz)
-3 -2 -1 0 1 2 3 4 5 6 7
ε"(x
10-3
)
0
2
4
6
8
10
150 K
ε"(x
10-3
)
0
1
2
3
4
α
αPE
γ1
γ2
a
b
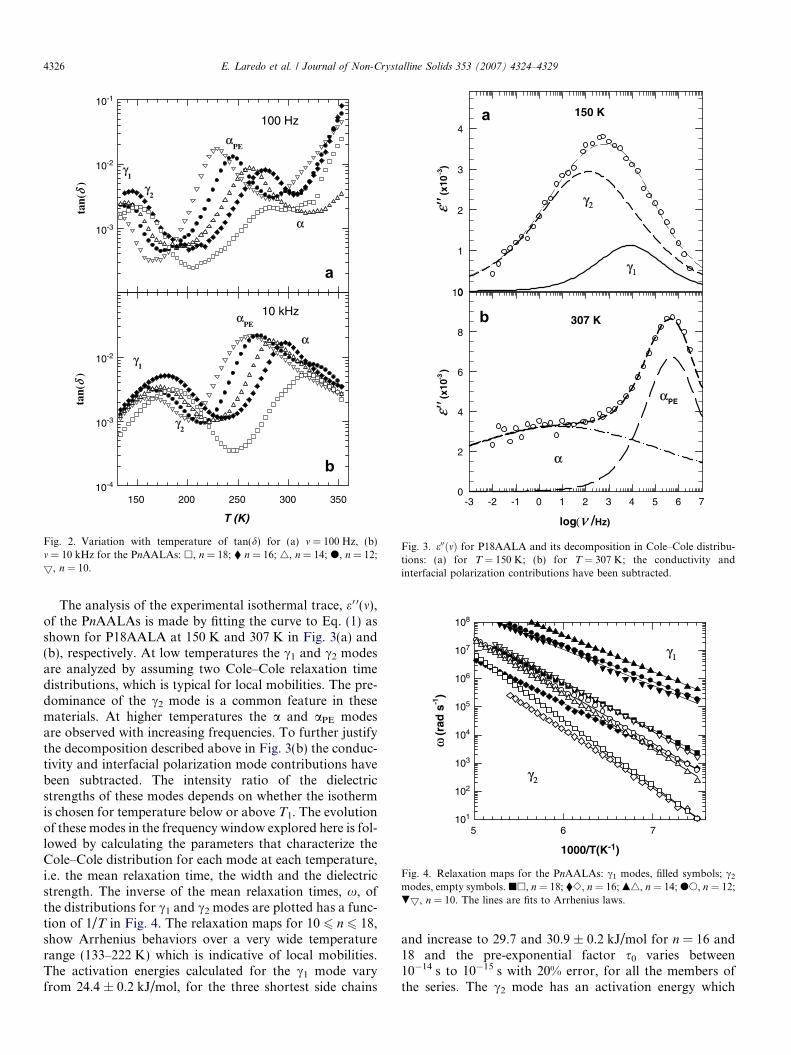
Fig. 3. e00ðmÞ for P18AALA and its decomposition in Cole–Cole distribu-tions: (a) for T = 150 K; (b) for T = 307 K; the conductivity andinterfacial polarization contributions have been subtracted.
1000/T(K-1)
5
ω (r
ad s
-1)
101
102
103
104
105
106
107
108
γ1
γ2
76
Fig. 4. Relaxation maps for the PnAALAs: c1 modes, filled symbols; c2
modes, empty symbols. jh, n = 18; �e, n = 16; m4, n = 14; ds, n = 12;.5, n = 10. The lines are fits to Arrhenius laws.
4326 E. Laredo et al. / Journal of Non-Crystalline Solids 353 (2007) 4324–4329
The analysis of the experimental isothermal trace, e 0 0(m),of the PnAALAs is made by fitting the curve to Eq. (1) asshown for P18AALA at 150 K and 307 K in Fig. 3(a) and(b), respectively. At low temperatures the c1 and c2 modesare analyzed by assuming two Cole–Cole relaxation timedistributions, which is typical for local mobilities. The pre-dominance of the c2 mode is a common feature in thesematerials. At higher temperatures the a and aPE modesare observed with increasing frequencies. To further justifythe decomposition described above in Fig. 3(b) the conduc-tivity and interfacial polarization mode contributions havebeen subtracted. The intensity ratio of the dielectricstrengths of these modes depends on whether the isothermis chosen for temperature below or above T1. The evolutionof these modes in the frequency window explored here is fol-lowed by calculating the parameters that characterize theCole–Cole distribution for each mode at each temperature,i.e. the mean relaxation time, the width and the dielectricstrength. The inverse of the mean relaxation times, x, ofthe distributions for c1 and c2 modes are plotted has a func-tion of 1/T in Fig. 4. The relaxation maps for 10 6 n 6 18,show Arrhenius behaviors over a very wide temperaturerange (133–222 K) which is indicative of local mobilities.The activation energies calculated for the c1 mode varyfrom 24.4 ± 0.2 kJ/mol, for the three shortest side chains
and increase to 29.7 and 30.9 ± 0.2 kJ/mol for n = 16 and18 and the pre-exponential factor s0 varies between10�14 s to 10�15 s with 20% error, for all the members ofthe series. The c2 mode has an activation energy which

E. Laredo et al. / Journal of Non-Crystalline Solids 353 (2007) 4324–4329 4327
slowly increases with n from 32.9 ± 0.2 kJ/mol to44.3 ± 0.2 kJ/mol, while the pre-exponential factor is nearlyconstant with a value within 10�16.0±0.3 s for the first fourmembers of the series and equal to 10�18.40±0.01 s forn = 18. The weak variation of these parameters when n var-ies confirms the short range motions at the origin of thesemodes. More detailed information on these values and var-iation with temperature and n of these parameters can befound in our previous work [4]. Also, the variation withtemperature of the shape and strength of these modes isnot significantly different as n varies as expected for veryshort range motions [4]. The variation with 1/T of theinverse of the mean relaxation times for the high tempera-ture modes, saPE
and sa, extracted from the best fits, arereported in the relaxation map plotted in Fig. 5(a) and(b), respectively. The lines represent the fit to a VTF depen-dence from which the relaxation parameters are calculated.Clear differences in the VTF parameters that define bothcooperative modes are evident as n increases. The variationof the mean xa and xaPE
as a function of 1/T, besides beinglocated at increasing temperatures, show several crossoversamong VTF plots which affects the fragility indexes of thesamples as discussed below. It is worth noting that the relax-ation time variation for the two cooperative modes is insen-sitive to the structural changes that occur in PnAALAs at
1000/T (K-1)
3 5
ωα (r
ad s
10-2
10-1
100
101
102
103
104
105
106
ωα
PE (r
ad s
-1)
-1)
10-1
100
101
102
103
104
105
106
107
αα
αPE
a
b
4
Fig. 5. Relaxation maps for the PnAALAs: (a) aPE mode; (b) a mode:jh, n = 18; �e, n = 16; m4, n = 14; ds, n = 12; .5, n = 10. The linesare fits to VTF laws.
the transitions temperatures T1 and T2. This indicates thatthe relaxation kinetics are not perturbed by the changes inthe dipolar population involved or in the changes in con-straints on the mobile phases responsible for these modes.On the contrary both the shape and dielectric strength ofthe cooperative modes are strongly affected as the tempera-ture increases above T1 and T2 showing the influence of thestructural evolution. The dielectric strengths show oppositebehaviors for the two cooperative modes except for n = 10where both of them slowly decrease with temperature. Forn = 12, 16, 18, DeaPE
increases drastically above the firstphase transition, T > T1, where the side chains order isdestroyed and the number of orientable dipoles grows asthe number of amorphous chain increases. Simultaneously,the strength of the a mode decreases above T1 for the twolongest side chains and above T2 for n = 12,14. At hightemperatures the a mode intensity becomes insignificant.More details might be found in Ref. [4].
4. Discussion
The deconvolution of the relaxation spectra used here isin agreement with similar analysis proposed for other poly-mer series having incompatible main and side chain parts.Side chains formed by alkyl groups with independentdynamics have been reported in PnALG [10,11], PnAMAand PnAA [3]. In the case of polyglutamates two localmodes have been reported, as found here in PnAALAs.Also, in all these polymers independent segmental relaxa-tion processes have been associated to the relaxationsdynamics of the main and the side chains.
The secondary relaxations with low activation energiesare attributed to the local motions of the carbonyl groupof the side chains. The lowest temperature mode, c1, isoriginated by the reorientation of short molecular segmentsof the side chains located between the main chain and thecrystallized side chains or in the amorphous alkyl chains.The higher temperature secondary mode, c2, is then attrib-uted to the motion of longer side chain segments accompa-nying the reorientation of the carbonyl group, possiblylocal motion in whole amorphous side chains which arealready present below T1. The activation energies reportedfor the low temperature mode in the poly(n-alkyl gluta-mates) with very short side chains n 6 3 are much higherthan our values for the PnAALA showing strong con-straints on the local motions of the PnALG [12].
The presence of a rigid main chain – the a-helix – inpolyglutamates and polyaspartates as compared to theamorphous main chain in acrylic and methacrylic polymersresults in an inversion of the intensities of the two cooper-ative processes present in all these polymers. From therelaxation maps shown in Fig. 5(a) the variation of thetemperature, Tx, corresponding to x = 10 rad/s, can bedetermined for the aPE mode; its variation with n has beenplotted in Fig. 6(a) (filled triangles) together with the com-pilation by Beiner and Huth [3] which includes the pointscorresponding to the glass transition temperature estimated

0 2 4 6 8 10 12 14 1 8
Tω
(ºC
)
-100
0
100
200
Aspartates
Imides
ItaconatesMetacrylatesAcrylates
α
αPE
n
mω(α
PE)
00 2 4 6 8 10 12 14 16 18
40
80
a
b
Fig. 6. (a) Variation of the relaxation temperature (x = 10 rad/s) for theaPE mode (filled symbols) and the a mode (empty symbols) as a functionof the number of carbons in the lateral chain of the PnAAs (jh),PnAMAs (ds), Polyimides (�e), PnAIs ( ) from Fig. 2(c) in Ref. [3].m4, PnAALAs results. (b) Fragility index, mx, of the aPE process forPnAAs, PnAMAs and PnAALAs. Error bars are smaller than symbol sizesand not shown.
1000/T (K -1)4.0 4.5 5.0 5.5 6.0
log
(ω
/rad
s-1
)
0
2
4
6
P10AA
P5AA
P8AA
P4AA
P6AA
P10AALAP12AALAP14AALAP16AALAP18AALA
Fig. 7. Comparison of the segmental dynamics of the side chains in thePnAAs (filled symbols) (Fig. 2(b) in Ref. [3]) and the PnAALAs (emptysymbols).
4328 E. Laredo et al. / Journal of Non-Crystalline Solids 353 (2007) 4324–4329
for amorphous PE [13] and for the c relaxation of semi-crystalline polyethylene which is taken as its glass transi-tion temperature [14,15]. It is worth noting that the generaltrend found here fits nicely the findings of purely amor-phous alkyl nanodomains existing in PnAAs, PnAMAs,4 6 n 6 12, PnAI, n 6 11, and hairy rods polyimides forn 6 16. This result indicates that the dynamics of the sidechains in the PnAALAs as in the other families representedin this figure, are independent of the main chain structureand conformation and of the existence of crystalline paraf-fin-like lamellae. Another example of this situation hasbeen found by Yildirim et al. [16] in discotic liquid crystalswhere a hindered glass transition of the alkyl chains con-strained by the columnar discotic structure exists. Thecomparison of the variation of the Tx for aPE, occurringin nanodomains with the values of the glass transition insemi-crystalline and amorphous PE, seems to point to thesimilarity between the confinements imposed either by thereduction of the alkyl nanodomains sizes or by the size ofthe amorphous regions in semi-crystalline PE. The compa-rable values for n = 16 with amorphous PE might be anindication for a finite CRR size in polyethylene.
The fragility indexes mx(aPE) plotted in Fig. 6(b) for theaPE in PnAALAs (x = 10 rad/s) are slightly higher thanthose found for the PnAAs and PnAMAs with strongerconfinements. For n P 10 the fragility is above 40 andincreases with n showing a cooperativity enhancement;for n = 18 lateral chains are still crystallized at Tx and thisadditional confinement on the amorphous side chainsresults in a lower fragility index. The addition of the datafrom the PnAALAs to this figure extends the knowledgeof the dynamics characteristics in self-assembled nanodo-mains with sizes ranging from 2 to 3 nm where the confine-ment effect on amorphous chains is very weak even in thepresence of side chain crystallinity.
The effect of the nanophase confinement on the dynam-ics of the aPE mode is shown in the relaxation maps plottedin Fig. 7 for the PnAA series measured by Beiner and Huth[3] where the present results on the PnAALAs have beenadded. Again one can observe that the variation of therelaxation times of the amorphous nanoconfined alkylchains in the PnAALAs, follows the general trend of thePnAAs: (i) for strong confinements, PnAAs 4 6 n 6 10,the dependence is nearly Arrhenius and the alkyl nanodo-mains are much smaller than the characteristic length ofthe CRR introduced by Adam and Gibbs [17]; (ii) as thesize of the alkyl nanodomains increases there is a strongto fragile transition in PnAA [3] and in this weak confine-ment the VTF dependence is recovered.
A second mode, labelled a, shows a VFT behavior fors�1(1/T) as seen in Fig. 5(b), an its origin differs from thatof the series compiled previously [3] where the a mode iscaused by the segmental dynamics of the amorphous mainchains. Here, the ratio Dea=DeaPE
decreases drasticallyabove T1 for n = 16,18 and above T2 for n = 12,14 [4].The increase in DeaPE
above T1 in the heating runs, due tothe fusion of the side chains observed here is analogousto the strength reduction of the high temperature modeobserved by Hempel et al. [18] in P16AMA, where the sidechains crystallize, when cooling the sample, as a conse-quence of the variation in the number of orientable dipoles.In poly(c-benzyl-L-glutamate) the a mode has been attrib-uted to a restricted rotational fluctuation of the rigid rods,i.e. a chopstick-like motion, within a cone with an apexangle h0 [10,19]. According to the model of Wang and Pec-ora [20] the rotational diffusion occurs in the cage formedby neighboring rods which size in the PnAALAs decreasesabove the phase transitions at T1 and T2 (see Table 1). Thecontraction necessary to inhibit the chopstick motion forthe shortest side chains is only reached above T2. Recently,Papadopoulos et al. [11] have proposed a different interpre-tation for the a relaxation process in the oligoPBLG. Basedon the variation of the relaxation parameters with increas-ing low molecular weights, they suggest that it couldbe assigned to amorphous peptide segments found in

E. Laredo et al. / Journal of Non-Crystalline Solids 353 (2007) 4324–4329 4329
amorphous-like defects within and at the end of the helices,or in a small fraction of amorphous chains. In the discoticliquid crystals the existence of a second cooperative mobil-ity attributed to intracolumnar fluctuations of rotationaldisorder has also been reported [16].
5. Conclusions
Results of dielectric spectroscopy measurements on thePnAALAs with long side chains have clarified the charac-teristics of the various mobilities originated by the existenceof nanophase segregation in these comb-like polymers withrod-like main chains and a fraction of long side chains par-tially crystallized for n > 10. This layered biphasic structurehas two cooperative mobilities. In the PnAALAs the coop-erative dynamics are affected by the combined effect of theconfinement due to the size of the nanodomains and to thepresence of a lamellar structure. The aPE segmental mode isa polyethylene-like dynamic glass transition completelyconsistent with the variation with n 6 12 in other seriesof nanophase segregated materials which are wholly amor-phous. Their shorter lateral chains length result in theoccurrence of much stronger constraints for the side chaindynamics. The crossover from less cooperative chaindynamic to a bulk-like one is observed as n increasesregardless of the main chain structure and the crystallinityof the material. The fragility index variation for the PnAAand PnAMA is expanded here for longer paraffinic chains,confirming the strong to fragile transition as n increases.
Acknowledgments
We thank FONACIT (Project G2005-000776) for finan-cial support of this work. Thanks are due to Freddy San-chez for his help with the data analysis.
References
[1] J.M. Fernandez-Santin, J. Aymami, A. Rodriguez-Galan, S. Munoz-Guerra, J.A. Subirana, Nature (London) 311 (1984) 53.
[2] F. Lopez-Carrasquero, S. Montserrat, A. Martinez de Ilarduya, S.Munoz-Guerra, Macromolecules 28 (1995) 5535.
[3] M. Beiner, H. Huth, Nat. Mater. 2 (2003) 595.[4] M. Grimau, E. Laredo, F. Sanchez, F. Lopez-Carrasquero, M.E.
Baez, A. Bello, Eur. Phys. J. E 15 (2004) 383.[5] F. Lopez-Carrasquero, M. Garcıa-Alvarez, S. Munoz-Guerra, Poly-
mer 35 (1994) 4502.[6] M. Garcıa-Alvarez, F. Lopez-Carrasquero, E. Tort, A. Rodrıguez-
Galan, S. Munoz-Guerra, Synth. Commun. 24 (1994) 745.[7] E. Laredo, M. Grimau, P. Barriola, A. Bello, A.J. Muller, Polymer 46
(2005) 6532.[8] K. Fukao, Y. Miyamoto, J. Non-Cryst. Solids 212 (1997) 208.[9] A. Nogales, T. Ezquerra, J.M. Garcia, F.J. Balta-Calleja, J. Chem.
Phys. 115 (2001) 3804.[10] A. Schmidt, S. Lehmann, M. Georgelin, G. Katana, K. Mathauer, F.
Kremer, K. Schmidt-Rohr, C. Boeffel, G. Wegner, W. Knoll,Macromolecules 28 (1995) 5487.
[11] P. Papadopoulos, G. Floudas, H.A. Klok, I. Schnell, T. Pakula,Biomacromolecules 5 (2004) 81.
[12] M. Kakizaki, H. Nakayama, T. Hideshima, Polym. J. 17 (1986) 141.[13] U. Gaur, B. Wunderlich, Macromolecules 13 (1980) 445.[14] E. Laredo, N. Suarez, A. Bello, L. Marquez, J. Polym. Sci., Part B:
Polym. Phys. Ed. 34 (1999) 641.[15] E. Laredo, N. Suarez, A. Bello, B. Rojas, M.A. Gomez, J.M.G.
Fatou, Polymer 40 (1999) 3038.[16] Z. Yildirim, M. Wubbenhorst, E. Mendes, S.J. Picken, I. Paraschiv,
A.T.M. Marcelis, H. Zuilhof, E.J.R. Sudholter, J. Non-Cryst. Solids351 (2005) 2622.
[17] G. Adam, J.H. Gibbs, J. Chem. Phys. 43 (1965) 139.[18] E. Hempel, H. Huth, M. Beiner, Thermochim. Acta 403 (2003) 105.[19] L. Hartmann, F. Kremer, T. Kratzmuller, H.G. Braun, IEEE Trans.
Dielect. Elect. Insul. 8 (2001) 390.[20] C.C. Wang, R. Pecora, J. Chem. Phys. 72 (1980) 5333.




















