
Comparison ofthe methods available for assessing cytotoxicity
Upload
independentCategory
view
2download
0
RPR-115135, A FARNESYLTRANSFERASE INHIBITOR, INCREASES5-FU- CYTOTOXICITY IN TEN HUMAN COLON CANCER CELL LINES:ROLE OF P53Patrizia RUSSO
1*, Davide MALACARNE1, Carla FALUGI
2, Sonya TROMBINO1 and Patrick M. O’CONNOR
3
1Laboratory Experimental Oncology, Molecular Pathology Section, National Institute for Research on Cancer, Genova, Italy2Department of Experimental, Environmental and Applied Biology (DIBISAA) University of Genova, Genova, Italy3Laboratory of Molecular Pharmacology, Division Basic Sciences, National Cancer Institute, National Institutes of Health, BethesdaMD, USA
A new non peptidic farnesyltransferase inhibitor,RPR-115135, in combination with 5-FU was studied in 10human colon cancer cell lines (HCT-116, RKO, DLD-1, Colo-320, LoVo, SW-620, HT-29, HCT-15, Colo-205 and KM-12)carrying several mutations but well characterized for p53 andRas status. We found that there was a slight tendency (notstatistically significant) for the p53 inactivated cells to be lesssensitive to 5-FU after 6 days continuous treatment. Simul-taneous administration of RPR-115135 and 5-FU, at subtoxicconcentrations, resulted in a synergistic enhancement of5-FU cytotoxicity in the p53 wildtype cells (HCT-116, RKO,DLD-1, Colo-320, LoVo). In the p53 mutated cells (SW-620,HT-29, HCT-15, Colo-205, KM-12) the effect was very com-plicated. In HCT-15 the combination resulted in antagonism,in KM-12 in antagonism or in synergy (at different concen-trations) and in SW-620, HT-29 and Colo-205 cells in synergybut only when 5-FU was administered at high concentrations.Growth inhibition could be accounted for on the basis of aspecific cell cycle arrest phenotype (G2-M arrest), as assayedby flow cytometry, only in the p53 functioning cell lines. Thecombination RPR-115135 � 5-FU increases apoptotic eventsonly in these cell lines. In the mutated cell lines no majoralterations on cell cycle arrest phenotype and no induction ofapoptosis was observed. Although RPR-115135 can potenti-ate the effect of 5-FU in cells in which p53 function is dis-rupted, these data suggest strongly that RPR-115135 signifi-cantly enhances the efficacy of 5-FU only when p53 isfunctioning.© 2002 Wiley-Liss, Inc.
Key words: farnesyltransferase; apoptosis; 5-FU; cell cycle; p53
The wealth of new knowledge, concerning the molecular andbiochemical pathways required for neoplastic transformation, hasprovided important insights into the clinical behavior of colorectalcancer. It is now widely accepted that the multistep carcinogenicprocess that is involved in colon cancer is driven by mutationalevents that ultimately give the cancer cells a growth advantage.1Some of these genetic alterations are prognostic factors in colo-rectal cancer.2 Molecular alterations also may have the potential topredict survival after chemotherapy.2,3 Advances in the molecularunderstanding of normal and pathological disturbed cellular sig-naling networks may have profound impact on the efficacy ofchemotherapy. It has been suggested that modifications of p53,that influence a cell’s tendency to apoptosis, may play a significantrole in modifying response to radiation and chemotherapy. Cyto-toxicity of 5-FU in cell lines of the NCI anticancer drug screeningprogram correlated with their p53 status.4,5 Disruption in p53, bytargeted homologous recombination, rendered human colon cancercell lines strikingly resistant to the effects of 5-FU. Althoughalteration of p53 is a plausible predictive factor discrepant resultsin patients entered in same clinical trials (adjuvant chemotherapywith 5-FU regimen) are reported.2,3
5-FU cytotoxicity is determined by either thymidine deprivation(inhibition of TS) or by RNA incorporation. Data strongly6,7
supports the hypothesis that cell death in intestinal epithelia or inhuman epithelial colon cancer cell lines requires 5-FU metabolitesto be incorporated into RNA. Cell death therefore occurs by
apoptosis and is p53-dependent. The effects on 5-FU sensitivitywere observed both in vitro and in vivo, were independent ofp21waf-1, and appeared to be the result of alterations in RNA, ratherthan DNA, metabolism.8
Recent insights into the biology of colon cancer have spurreddevelopment of new drugs [i.e., new folate-based TS inhibitors(Tomudex, AG337 or LY231514), inhibitors of glycinamide ribo-nucleotide formyltransferase (AG2034, Lemetrexol), Topoisomer-ase I inhibitors (CPT11) or farnesyltransferase inhibitors (R11577,SCH66336)].9,10 Farnesylation is catalyzed by FTase, which isresponsible for catalyzing farnesylation of several cellular proteinsby transfer a C-15 farnesyl moiety from FPP. Studies have shownthat farnesylation of Ras is the obligatory, first step in a series ofpost-translational modifications that lead to the membrane associ-ation that determine the switch from an inactive to an activeRas-GTP bound form. Upon receiving a signal input, Ras-GTPacts as a molecular switch that turns on downstream effectors.11–15
When farnesylation is blocked, the function of Ras protein isseverely impaired because of the inability of the nonfarnesylatedprotein to anchor to the membrane. All mutant Ras protein in-volved in human cancer are modified exclusively by FTase,13 socompounds that specifically inhibit FTase were sought as strategyto block the activity of oncogenic Ras in cancer cells.12–15
Activating point mutations of Ras genes occur in approximately30% of human cancers, with certain malignancies having a par-ticular high incidence. The incidence of K-Ras mutations in pan-creatic cancers approaches 90%, whereas mutations in K- Ras aredetected in 50% colorectal carcinomas and nonsmall cell lung
Abbreviations: ANT, antagonism; CI, combination index; CPT11,camptotecin-11; DMSO, dimethyl sulfoxide; 5-FU, 5-fluorouracil; FPP,farnesyl pyrophosphate; FTase, farnesyltransferase; FTI, farnesyltrans-ferase inhibitor; IC, inhibitory concentration; mu, mutated; SYN, synergy;TS, thymidylate synthetase; wt, wildtype.
Grant sponsor: Fondazione Italiana per la Ricerca sul Cancro (FIRC),Milan, Italy; Grant sponsor: European Community, Ispra, Italy; Grantnumber: TENDER 2000/S 118-076796.
Dr. O’Connor’s current address is: Oncology Research Division, PfizerGlobal Research & Development Inc., La Jolla, CA 92121, USA.
*Correspondence to: Molecular Pathology Section, Laboratory of Ex-perimental Oncology, National Institute for Research on Cancer, LargoRosanna Benzi 10, I-16132 Genova, Italy.Fax: �39-010-5600217. E-mail: [email protected].
Received 27 November 2001; Revised 1 February 2002, 22 March 2002;Accepted 2 April 2002
DOI 10.1002/ijc.10461Published online 28 May 2002 in Wiley InterScience (www.interscience.
wiley.com).
Int. J. Cancer: 100, 266–275 (2002)© 2002 Wiley-Liss, Inc.
Publication of the International Union Against Cancer

cancers. Hematologic malignancies harbor N-Ras mutations.12 H-Ras transformed cells appeared to be more sensitive to FTIs thanthose transformed by either K-Ras or N-Ras.12–15 This observationwas initially disappointing in terms of human cancers, becauseK-Ras mutations12 occurred more frequently in human malignan-cies than H-Ras mutations. Nevertheless, human cancer cell linesharboring K-Ras mutations are sensitive to FTIs.12–15 This sensi-tivity is explained by the peculiarity of K-Ras4B to have a 10–20-fold greater affinity to FTase necessitating much higher levelsto inhibit the post-translational processing of this Ras isotype.16
Although it has been demonstrated clearly that FTIs inhibit Rasfarnesylation, it is uncertain whether the antiproliferative effects ofthese compounds result exclusively from effects on Ras. Gera-nylgeranylated forms of K-Ras and N-Ras (not H-Ras), which arecapable of transforming cells, are observed in cells treated withFTIs.14 Despite this alternative prenylation pathway, FTIs interferewith the proliferation of many tumor cells expressing activatedK-Ras in vitro and in vivo.12–16 Several cell types that lack Rasmutations are also sensitive to FTIs in vivo and in vitro. This effectcan be, partially, related to overexpression of normal Ras (thatcauses deregulation of wildtype Ras isoform) as a common featureof some human cancers (i.e., glioblastoma and breast cancer).17,18
High grade human astrocytomas do not harbor oncogenic/acti-vating Ras mutations, however Ras activation occurs in thesetumors, attributable in part to overexpression of receptor tyrosinekinases. They are, however, sensitive to FTIs. Recent findingssupport the hypothesis that Ras is a major therapeutic target ofFTIs in human astrocytomas.19
Moreover, no clear correlation between Ras mutation status andFTIs sensitivity has been observed by several investigators, andthere remains no consensus as to the relevant targets(s) [i.e., RhoB,centromere associated CENP-E and CENP-F12–15] of FTIs that canexplain the mosaic pharmacology.
There are, however, a growing number of FTIs in varying stagesof preclinical and clinical development.12,20,21
RPR-115135 is a new and selective inhibitor of FTase in vitro.Starting from RPR-104213, a hit uncovered through SPA assayusing FTase and H-Ras related peptide as substrate, AventisPharma developed chemical strategies to prepare 4,9-ethano-2,3,3a,4,9,9a-hexahydro-1H-benzo(f)isoindole derivatives modi-fied at position 2 and 3a. Compounds were evaluated for theinhibition of the farnesylation of purified H-Ras protein (Gly12)obtained by recombinant E. coli in TCA assay, which lead to theselection of RPR-115135 for further evaluation. This compoundexhibited a specific FTase inhibition on H-Ras and K-Ras inenzymatic tests (TCA and related-peptide scintillation) [farnesy-lation of Lamin B (IC50 � 70 nM) or H-Ras (IC50 � 300 nM) orK-Ras (IC50 � 38 nM)] unaffecting the closely related enzymegeranylgeranyltransferase I (IC50 � 6,000 nM).22,23. Kinetic ex-periments showed that this compound inhibits FTase in a FPP-competitive way.22
RPR-115135 potently inhibited cloning of several human tumorcell lines, independently of Ras mutations (wt in breast carcinomaMCF-7), however, there was a tendency for cell lines harboringK-Ras mutations [such as colon HCT-116 or SW-620, lung H460or A549 and pancreas MiaPaCa-2] or expressing high levels ofK-Ras proteins [such as ovary OVCAR-3 or Pa-1] to be moresensitive to RPR-115135.22–26 The inhibitory effect was p53-independent (several lines with different p53 status and HCT-116cells transfected with p53 negative dominant mutant).22–26
The growth inhibition induced by RPR-115135 was independentof the cell cycle status of HCT-116 cells (synchronized cells).Addition of RPR-115135 only to cells arrested in G0–G1, however,resulted in a strong enhancement of G0–G1 arrest, independently ofp53 and p21waf1.23
Interestingly, RPR-115135 inhibited cell cloning in an isogeniccell line model system consisting of human normal breastMCF10A cells stably transfected with either c-Ha-Ras or c-erbB-2
or both H-Ras �c-erbB-2 that do not contain ras mutation (exceptthe H-Ras cells). This might be explained, in part, by the action ofoncogenes or autocrine factors that lie upstream in the Ras signaltransduction pathway. The untransfected cells, however, were 10times less sensitive than H-Ras transfected cells.24
RPR-115135 inhibited the growth of normal human T lympho-cytes (induced by PHA/IL2) however, the concentration to achievethe IC50 was 30-fold higher than that in cells harboring oncogenicforms of Ras [H, K or N-Ras24,26].
Moreover, RPR-115135 induced a significant increase in thefrequency of micronuclei (only in tumors cells) in a complexmanner, dependent upon the complement of mutations in cellregulatory genes and this activity was independent of Ras geno-type.24
We recently studied the new non-peptidomimetic FTase inhib-itor, RPR-115135, in combination with 5-FU in an isogenic cellline model system consisting of human colon cancer HCT-116cells. HCT-116 cells were transfected with an empty controlpCMV vector or with a dominant-negative mutated p53 trans-gene.25 Because the only difference among these cell lines is theabsence or presence of a single gene, the interpretation of resultsis particularly straightforward and is uncomplicated by the over-expression of exogenous genetic elements. We found that, relativeto control transfectants, there was a slight tendency for the p53inactivated cells to be less sensitive to 5-FU after 6 days contin-uous treatment. Simultaneous administration of RPR-115135 and5-FU, at subtoxic concentrations, resulted in a synergistic enhance-ment of 5-FU cytotoxicity, especially in the p53 wt clone. Growthinhibition could be accounted for on the basis of a specific cellcycle arrest phenotype (G2-M arrest in p53 functioning and Sarrest in mutated clones). The combination RPR-115135 � 5-FUincreases strongly apoptotic events only in the p53 wt clone. Theseresults suggest that p53 has a profound influence not only on theresponse to 5-FU but also in modulating RPR-115135-enhanced5-FU cytotoxicity.
Our present study was undertaken to investigate the possibleeffects of RPR-115135 used in combination with 5-FU in 10human colon cancer cell lines, carrying several mutations but wellcharacterized for p53 and Ras status.
MATERIALS AND METHODS
Chemical treatmentsRPR-115135 (C31H29NO4, m.w. � 479.58) (Fig. 1) is produced
by Aventis Pharma (Centre de Recherches de Vitry Alfortville,France).22 It was prepared as a 1 mM stock solution in DMSO andaliquots were stored at �20°C until needed. 5-FU (Fig. 1) waspurchased by Sigma (St. Louis, MO).
Cell cultureHuman colon cancer cell lines HCT-116, HT-29, SW-620,
DLD-1, KM-12, Colo-205, LoVo, HCT-15, RKO and Colo-320(Table I) were obtained by NCI anticancer drug screening (Be-
FIGURE 1 – Chemical structure of RPR-115135 and 5-FU.
2675-FU CYTOTOXICITY

thesda, MD) or by ATCC (Rockville, MD). They were grown inRPMI 1640 (Gibco BRL, Grand Island, NY) supplemented with5% heat inactivated fetal bovine serum (Gibco BRL). Cell countswere determined using a Coulter Counter with Channelyzer attach-ment to monitor cell size (Coulter Electronics, Hialeah, FL). Cellmembranes integrity was determined by trypan blue dye exclusionassay.
Cell cytotoxicityCells were plated in log phase into 96-multiwells plates (250
cells/well) with 190 �l of complete medium for 24 hr and thentreated with various concentrations of RPR-115135 or 5-FU [from0.01 to 10 �M (10 �l)] for 6 days. At the end of the incubationtime (6 days) 40 �l of MTS tetrazolium solution (CellTiter 96�AQueous One Solution Cell Proliferation Assay, Promega, MadisonWI) was added for 2 hr and then absorbance was read at 490 nmwith a 96-well plate reader.
The IC50 was calculated as the drug concentration that inhibitscell growth to 50% of the control cells. IC50 values were estimatedfitting the data with a non linear regression to the dose-effectmodel derived by Chou and Talalay27,28):
fa/fu � �D/Dm�m (1)
where D is the dose of the drug, Dm is the IC50, fa is the fractionaffected by the dose, fu is the fraction unaffected and m is acoefficient that determines the sigmoidicity of the curve.
Flow cytometryCells were plated in log phase in T75 flakes (2,700 cells/cm2) in
complete medium for 24 hr, then treated over 6 days with RPR-115135 in combination with 5-FU and then counted before flowcytometry. Samples were prepared for flow cytometry essentiallyas described previously.4,25 Briefly, cells were washed with 1�phosphate buffered saline pH 7.4 and then fixed with ice-cold 70%ethanol. Samples were washed with 1� PBS and stained withpropidium iodide 6 �g/ml (Sigma) containing RNase 2 �g/ml(Sigma) for 30 min at 37°C. Cell cycle analysis was performedusing a Becton Dickinson Fluorescence-activated cell analyzer andCell Quest version 1.2 software (Becton Dickinson Immunocito-metry Systems, Mansfield, MA). For each sample at least 15,000cells were analyzed and quantitation of the cell cycle distributionwas performed using the ModFit LT Version 1.01 software (VeritySoftware House Inc., Topsham, ME).
Morphological analysisMorphological assessment was determined by staining cells
with 4,6-diamidino-2-phenylindole (DAPI), 105 cells were seededon a slide 24 hr before treatment, after six days treatment slideswere washed twice in PBS, fixed for 5 min at room temperature in
95% ethanol, air dried and then dipped for 5 min in 0.1 �gDAPI/ml in a methanol solution (1,000 cells were scored for eachslide).
RESULTS
We have shown recently26 that 5-FU-induced cytotoxicity after2 days treatment was p53-independent. In contrast when cells wereexposed to 5-FU for 6 days (continuous treatment) there was atendency for p53 wt cells to be more sensitive than mutated clones.Accordingly to these observations ten human colon cancer celllines carrying different mutations (Table I) were exposed to 5-FUfor 6 days. Comparison between the 2 groups [IC50 values for wtp53 cells (HCT-116, RKO, DLD-1, Colo-320, LoVo) and mutatedones (SW-620, HT-29, HCT-15, Colo-205, KM-12)] yields differ-ences that do not reach statistical significance with both parametric(Student’s t-test) and non-parametric (Mann-Whitney test) statistic(Fig. 2).
The same panel of cells was exposed to RPR-115135 for 6 days,as reported in previous experiments,23–26 cell sensitivity was bothp53 (Fig. 2) and Ras-independent.
Different cell lines were exposed to subtoxic concentrations ofRPR-115135 [0.03, 0.1 or 0.3 �M] simultaneously to differentsubtoxic concentrations of 5-FU [0.03, 0.1 or 1.0 �M] for six dayscontinuous exposure (Fig. 3a, b). For each drug combination theCI and the statistical significance were calculated. Table II showsthese results. In all the p53 wt cells (HCT-116, RKO, DLD-1,Colo-320, LoVo) a strong synergic effect (C.I.: �0.3) was ob-served. Synergy was obtained combining very low subtoxic con-centrations of 5-FU (0.1–0.3 �M) and RPR-115135 (0.03–0.1�M).
In the mutated cells (SW-620, HT-29, HCT-15, Colo-205, KM-12) the effect was very complicated. In one line (HCT-15) thecombination resulted in antagonism, in KM-12 in antagonism andin synergy [at different drug concentrations] and finally in SW-620, HT-29 and Colo-205 cells in synergy, but in a larger range ofconcentrations (0.03–1.0 �M for 5-FU and 0.1–0.3 �M for RPR-115135) than in the wt cell lines (Table II).
The concentration of 0.1 �M both for 5-FU, RPR-115135 andfor the combination was chosen to investigate time course, cellcycle alteration and induction of apoptosis. In MTS assay thisconcentration was synergic (statistically significant) in all wt cells(HCT-116, LoVo, RKO, DLD-1, Colo-320). In mutated ones it
FIGURE 2 – IC50 values [�M] obtained for 5-FU or RPR-115135 in10 human colon cancer cell lines. Cells were treated for 6 dayscontinuous exposure.
TABLE I – STATUS OF THE P53 AND K-RAS GENE IN HUMAN COLONCANCER CELL LINES USED IN THE PRESENT STUDIES
Cell line p53 gene status Ras gene status
HCT-116 wt1 mu2
LoVo wt3 mu4
RKO wt5 wt6
DLD-1 wt7 mu7
Colo-320 wt8 wt9
SW-620 mu1 mu2
HT-29 mu1 wt2
HCT-15 mu1 mu2
Colo-205 mu1 wt2
KM-12 mu1 wt2
1According to O’Connor et al.4.–2In HCT-116 mutation at residue13 [(DGGE analysis) sequence GAC (Asp � D)], in SW-620 atresidue 12 [GTT (Val), in HCT-15 at residue 13 [GAC (ASP)],according to Koo et al.40.–3According to Donald et al.41.–4 Accordingto Nagase et al.42.–5According to Petak et al.43.–6According toLengyel et al.44.–7According to Rajesh et al.45.–8According to Zhenget al.31.–9According to Di Paolo et al.46.
268 RUSSO ET AL.
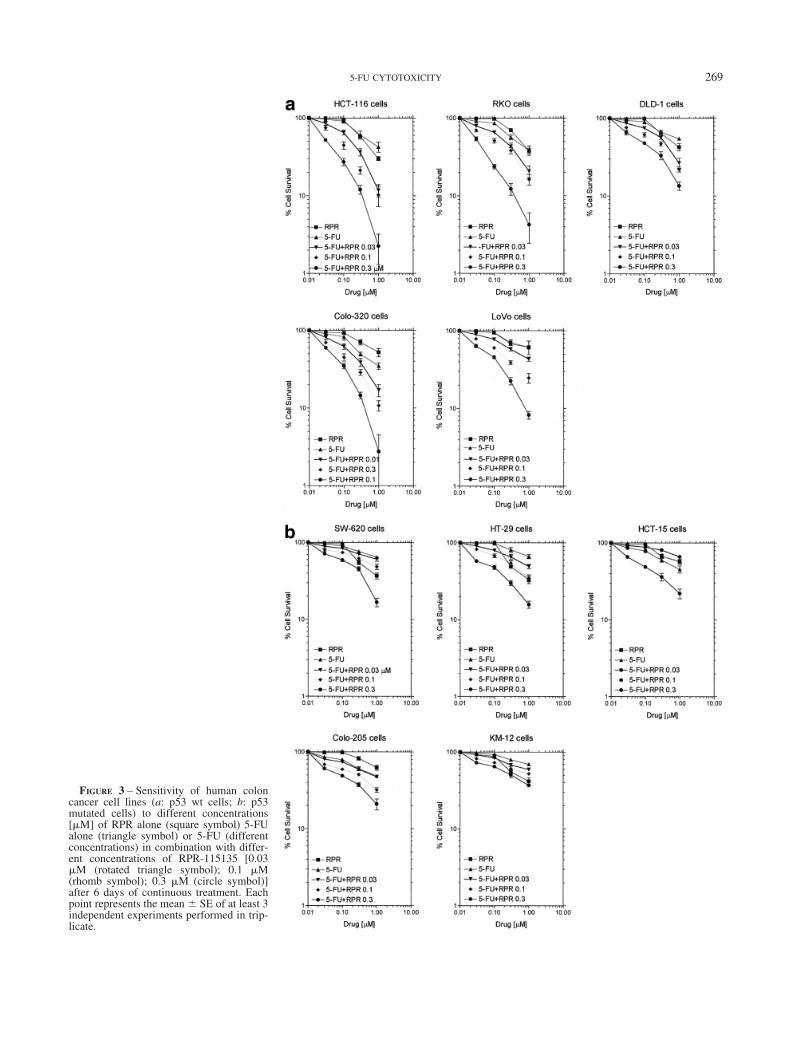
FIGURE 3 – Sensitivity of human coloncancer cell lines (a: p53 wt cells; b: p53mutated cells) to different concentrations[�M] of RPR alone (square symbol) 5-FUalone (triangle symbol) or 5-FU (differentconcentrations) in combination with differ-ent concentrations of RPR-115135 [0.03�M (rotated triangle symbol); 0.1 �M(rhomb symbol); 0.3 �M (circle symbol)]after 6 days of continuous treatment. Eachpoint represents the mean SE of at least 3independent experiments performed in trip-licate.
2695-FU CYTOTOXICITY

was moderately synergic but not statistically significant in SW-620(C.I. � 0.743) and HT-29 cells (C.I. � 0.478), antagonistic (sta-tistically significant) in HCT-15 and KM-12 cells and synergistic(statistically significant) only in Colo-205 (Table II).
Time-course experiments confirmed that, when nontoxic con-centrations of 5-FU and RPR-115135 (Fig. 4a) were given togethera great synergistic effect in growth inhibitory activity was ob-served in p53 wt cell lines. In the mutated cell lines this effect wasless apparent (Fig. 4b).
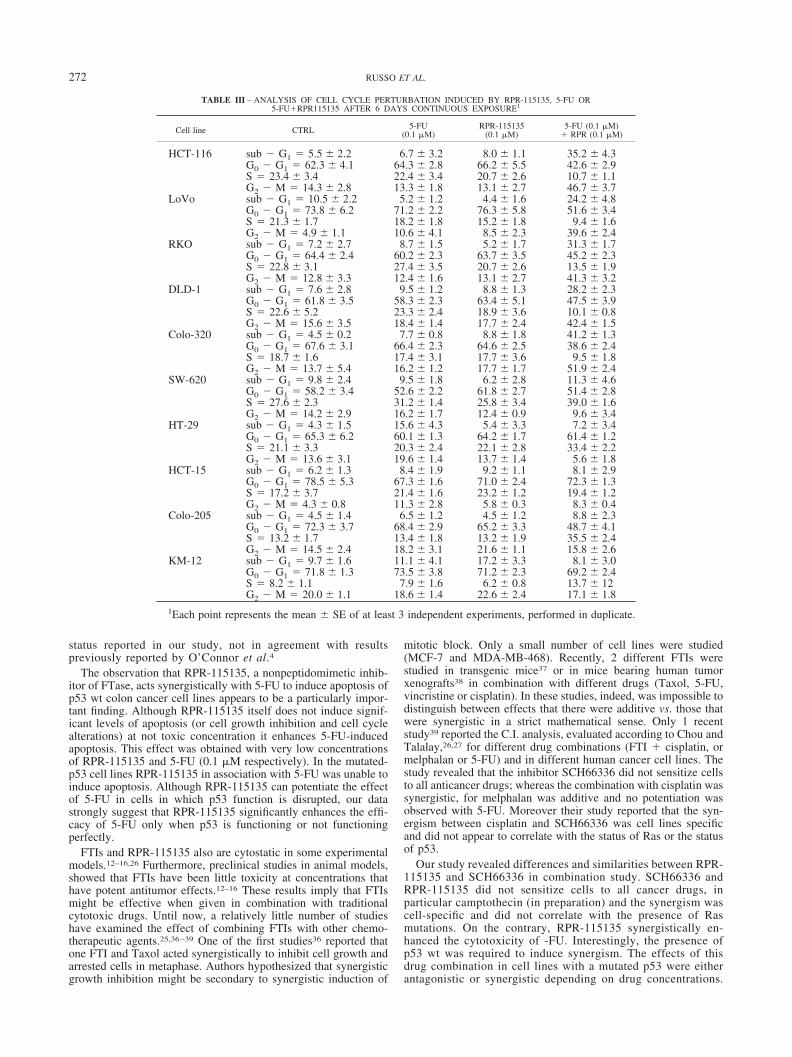
The cell cycle was analyzed over 6 days of continuous treatment(Table III, Fig. 5). Growth inhibition could be accounted for on thebasis of a specific cell cycle arrest phenotype, as assayed by flowcytometry only in the p53 wt cell lines (Table III). Treatment withRPR-115135, or with 5-FU, at concentration equal to 0.1 �M, didnot produce any apparent alteration in the composition of the cellcycle in overall cell lines (Table III). These data are in agreementwith previous both for 5-FU26 and for RPR-115135, obtained alsoat higher concentrations (10 �M).23 The combination 5-FU �RPR-115135 induced a G2-M redistribution (p � 0.002 Student’st-test) in the wt p53 cell lines. It is also possible, however, that thedrug combination can result in apoptosis of G1 population andeventual disappearance from the population. A dual G1 and G2effect is also possible In the mutated cells the combination induceda tendency to S increase only in 2 cell lines (HT-29, Colo-205),whereas in 3 (SW-620, HCT-15, KM-12) no major alteration in thecomposition of the cell cycle was observed (Table III and Fig. 5).
Flow cytometry in the p53 wt cell lines indicated the presence ofa sub-G1 population only when cells were treated with the com-bination 5-FU � RPR-115135 (Table III).
In mutated cells the sub-G1 population was not significant(Table III).
To further support the apoptotic induction, revealed by flowcytometry (sub-G1 population), nuclei of treated cells were ob-served for typical morphological apoptotic changes after DAPIstaining. Table IV shows that in the wt p53 cell lines a stronginduction of apoptotic nuclei were observed only for the combi-nation 5-FU � RPR-115135. Table IV shows that in the mutatedcell lines no induction of apoptotic figures was present in both5-FU, RPR-115135 and in the combination treated cells.
DISCUSSION
The objective of our study was to test the ability of RPR-115135, a new non peptidomimetic FTI, to increase the cyto-
toxic activity of 5-FU and to correlate the possible interac-tive effect with the status of p53 gene. To accomplish this 10human colon cancer cell lines were analyzed. We have shownpreviously that RPR-115135 enhances the antiproliferativeeffects of 5-FU in the human colon cancer HCT-116 iso-genic cell system. These results indicated that RPR-115135enhances 5-FU-induced apoptosis26 only when p53 wt ispresent. We demonstrate the ability of RPR-115135, at not toxicconcentrations, to strongly increase 5-FU cytotoxicity in humancolon cancer cell lines wt for p53 (HCT-116, RKO, DLD-1,Colo-320, LoVo). The enhanced growth inhibition, in thesecells, could be accounted for on the basis of a specific cell cyclearrest phenotype (G2-M arrest) and on severe induction ofapoptosis.
In p53 mutated cell lines (SW-620, HT-29, HCT-15, Colo-205,KM-12) subtoxic concentrations of RPR-115135 in combinationwith 5-FU were antagonist or synergistic, but did not influence thecell cycle and did not induce apoptosis.
Despite being p53 wildtype HCT-116 cells are not prone toapoptosis induced by radiation,29,30 5-FU26 or serum depriva-tion.23 probably due to the fact that HCT116 cells have lost thenormal functions of the gene products of p14ARF and p16INK4.31
DLD-1 and LoVo cells (p53 wt), also, showed extensive andpartial methylation, respectively, at the p14ARF locus and conse-quently p14ARF mRNA were expressed at extremely low (DLD-1)or intermediate (LoVo) levels.31 The hypermethylation of CpGisland results in gene silencing32 and often p14ARF is hypermethy-lated in colon carcinoma.31–33
The INK4A/ARF locus, on human chromosome 9p21, con-sists of two overlapping genes that encode two unrelated pro-teins p16INK4A and p14ARF. ARF may act upstream of p53promoting MDM2 degradation and thus preventing the MDM2-mediated neutralization of p53.34,35 Alterations in ARF func-tion would result in overexpression of MDM-2 and functionalinactivation of p53 and finally might compromise anticancertherapy.34,35 Consequently, these observations predict that dis-ruption or silencing of ARF locus could contribute to chemore-sistance in human tumors. Furthermore, these data imply that asubstantial number of p53 “normal” tumors would be misclas-sified (e.g., those harboring ARF mutations or methylation) andmay explain why some studies fail to correlate p53 mutationswith adverse clinical features. These observations can alsoexplain the discrepancies between 5-FU sensitivity and p53
TABLE II – ESTIMATED COMBINATION INDEX (CI) VALUES AS DEFINED BY CHOW AND TALALAY28,29
IN ORDER TO EVALUATE THE EFFECT OF THE COMBINATION 5�FU�RPR�115135 IN 10 HUMANCOLON CANCER CELL LINES1
Cell line Drug concentrations2
5-FU�RPR (�M)Statistical significant
CISign of theinteraction 95% CI3
HCT-116 0.1 � 0.1 0.191 SYN (0.076–0.693)LoVo 0.1 � 0.1 0.182 SYN (0.105–0.416)RKO 0.1 � 0.03 0.480 SYN (0.304–0.920)DLD-1 0.1 � 0.3 0.280 SYN (0.139–0.861)Colo-320 0.1 � 0.1 0.098 SYN (0.042–0.261)SW-620 1.0 � 0.3 0.103 SYN (0.034–0.308)HT-29 0.3 � 0.3 0.283 SYN (0.093–0.777)HCT-15 0.3 � 0.03 5.883 ANT (2.045–39.212)Colo-205 0.03 � 0.1 0.285 SYN (0.177–0.567)KM-12 0.03 � 0.3 1.776 ANT (1.006–4.225)
1.0 � 0.1 0.582 SYN (0.417–0.875)1Data were obtained from Figure 3. To analyze the interactions between the 2 agents, for each
tested combination of the 2 drugs CI values were calculated as proposed by Chou,47 for mutuallyexclusive drugs: CI � (D)1/(Dx)1 � (D)2/(Dx)2
. Where Dx represent the estimated dose of the drugalone capable of producing the same effect of the combined drugs, as estimated from the median-effect equation [1]. In outline CI near to 1 indicates additivity, CI�1 indicates antagonism, CI�1indicates synergism (CI�.03 strong synergism).–2Lowest drug concentration at which the effectarises.–3 To extrapolate confidence limits (i.e. statistical significance) for CI values we conducted aparametric bootstrapping where we assumed the distribution of parameters (m and Dm) as formulatedin equation [1] to be approximately gaussian with estimated mean and asymptotic standard error ascalculated using non-linear regression commands of SPSS software.
270 RUSSO ET AL.
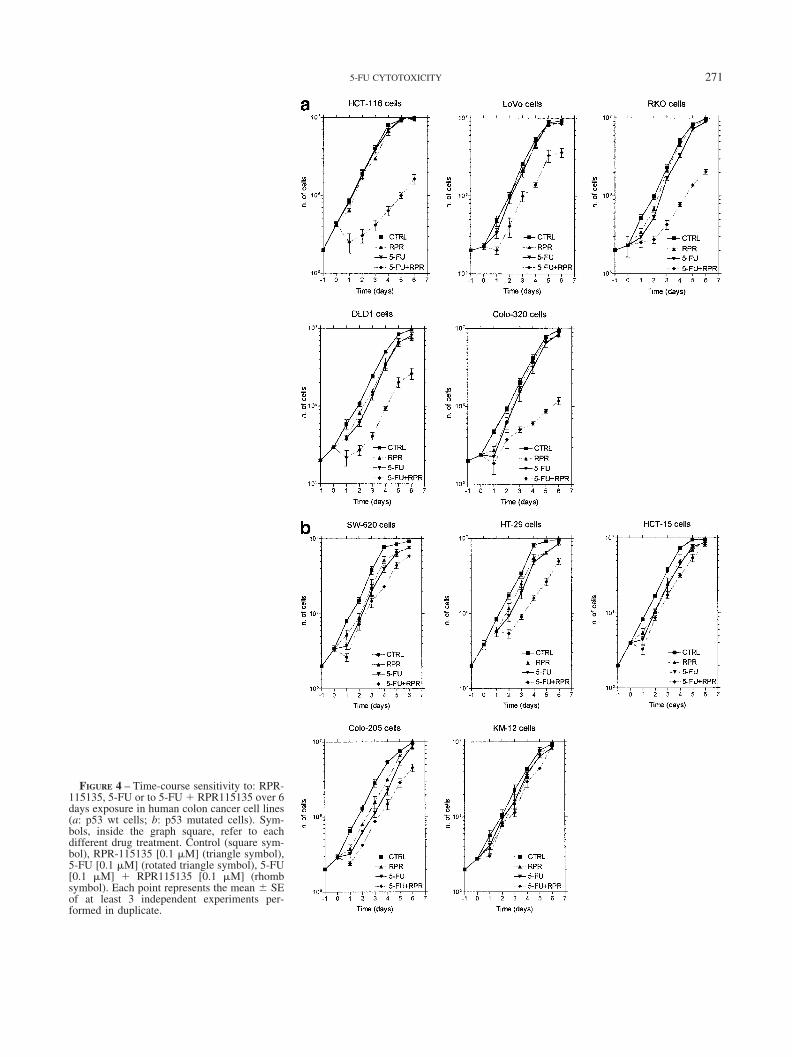
FIGURE 4 – Time-course sensitivity to: RPR-115135, 5-FU or to 5-FU � RPR115135 over 6days exposure in human colon cancer cell lines(a: p53 wt cells; b: p53 mutated cells). Sym-bols, inside the graph square, refer to eachdifferent drug treatment. Control (square sym-bol), RPR-115135 [0.1 �M] (triangle symbol),5-FU [0.1 �M] (rotated triangle symbol), 5-FU[0.1 �M] � RPR115135 [0.1 �M] (rhombsymbol). Each point represents the mean SEof at least 3 independent experiments per-formed in duplicate.
2715-FU CYTOTOXICITY
status reported in our study, not in agreement with resultspreviously reported by O’Connor et al.4
The observation that RPR-115135, a nonpeptidomimetic inhib-itor of FTase, acts synergistically with 5-FU to induce apoptosis ofp53 wt colon cancer cell lines appears to be a particularly impor-tant finding. Although RPR-115135 itself does not induce signif-icant levels of apoptosis (or cell growth inhibition and cell cyclealterations) at not toxic concentration it enhances 5-FU-inducedapoptosis. This effect was obtained with very low concentrationsof RPR-115135 and 5-FU (0.1 �M respectively). In the mutated-p53 cell lines RPR-115135 in association with 5-FU was unable toinduce apoptosis. Although RPR-115135 can potentiate the effectof 5-FU in cells in which p53 function is disrupted, our datastrongly suggest that RPR-115135 significantly enhances the effi-cacy of 5-FU only when p53 is functioning or not functioningperfectly.
FTIs and RPR-115135 also are cytostatic in some experimentalmodels.12–16,26 Furthermore, preclinical studies in animal models,showed that FTIs have been little toxicity at concentrations thathave potent antitumor effects.12–16 These results imply that FTIsmight be effective when given in combination with traditionalcytotoxic drugs. Until now, a relatively little number of studieshave examined the effect of combining FTIs with other chemo-therapeutic agents.25,36–39 One of the first studies36 reported thatone FTI and Taxol acted synergistically to inhibit cell growth andarrested cells in metaphase. Authors hypothesized that synergisticgrowth inhibition might be secondary to synergistic induction of
mitotic block. Only a small number of cell lines were studied(MCF-7 and MDA-MB-468). Recently, 2 different FTIs werestudied in transgenic mice37 or in mice bearing human tumorxenografts38 in combination with different drugs (Taxol, 5-FU,vincristine or cisplatin). In these studies, indeed, was impossible todistinguish between effects that there were additive vs. those thatwere synergistic in a strict mathematical sense. Only 1 recentstudy39 reported the C.I. analysis, evaluated according to Chou andTalalay,26,27 for different drug combinations (FTI � cisplatin, ormelphalan or 5-FU) and in different human cancer cell lines. Thestudy revealed that the inhibitor SCH66336 did not sensitize cellsto all anticancer drugs; whereas the combination with cisplatin wassynergistic, for melphalan was additive and no potentiation wasobserved with 5-FU. Moreover their study reported that the syn-ergism between cisplatin and SCH66336 was cell lines specificand did not appear to correlate with the status of Ras or the statusof p53.
Our study revealed differences and similarities between RPR-115135 and SCH66336 in combination study. SCH66336 andRPR-115135 did not sensitize cells to all cancer drugs, inparticular camptothecin (in preparation) and the synergism wascell-specific and did not correlate with the presence of Rasmutations. On the contrary, RPR-115135 synergistically en-hanced the cytotoxicity of -FU. Interestingly, the presence ofp53 wt was required to induce synergism. The effects of thisdrug combination in cell lines with a mutated p53 were eitherantagonistic or synergistic depending on drug concentrations.
TABLE III – ANALYSIS OF CELL CYCLE PERTURBATION INDUCED BY RPR-115135, 5-FU OR5-FU�RPR115135 AFTER 6 DAYS CONTINUOUS EXPOSURE1
Cell line CTRL 5-FU(0.1 �M)
RPR-115135(0.1 �M)
5-FU (0.1 �M)� RPR (0.1 �M)
HCT-116 sub � G1 � 5.5 2.2 6.7 3.2 8.0 1.1 35.2 4.3G0 � G1 � 62.3 4.1 64.3 2.8 66.2 5.5 42.6 2.9S � 23.4 3.4 22.4 3.4 20.7 2.6 10.7 1.1G2 � M � 14.3 2.8 13.3 1.8 13.1 2.7 46.7 3.7
LoVo sub � G1 � 10.5 2.2 5.2 1.2 4.4 1.6 24.2 4.8G0 � G1 � 73.8 6.2 71.2 2.2 76.3 5.8 51.6 3.4S � 21.3 1.7 18.2 1.8 15.2 1.8 9.4 1.6G2 � M � 4.9 1.1 10.6 4.1 8.5 2.3 39.6 2.4
RKO sub � G1 � 7.2 2.7 8.7 1.5 5.2 1.7 31.3 1.7G0 � G1 � 64.4 2.4 60.2 2.3 63.7 3.5 45.2 2.3S � 22.8 3.1 27.4 3.5 20.7 2.6 13.5 1.9G2 � M � 12.8 3.3 12.4 1.6 13.1 2.7 41.3 3.2
DLD-1 sub � G1 � 7.6 2.8 9.5 1.2 8.8 1.3 28.2 2.3G0 � G1 � 61.8 3.5 58.3 2.3 63.4 5.1 47.5 3.9S � 22.6 5.2 23.3 2.4 18.9 3.6 10.1 0.8G2 � M � 15.6 3.5 18.4 1.4 17.7 2.4 42.4 1.5
Colo-320 sub � G1 � 4.5 0.2 7.7 0.8 8.8 1.8 41.2 1.3G0 � G1 � 67.6 3.1 66.4 2.3 64.6 2.5 38.6 2.4S � 18.7 1.6 17.4 3.1 17.7 3.6 9.5 1.8G2 � M � 13.7 5.4 16.2 1.2 17.7 1.7 51.9 2.4
SW-620 sub � G1 � 9.8 2.4 9.5 1.8 6.2 2.8 11.3 4.6G0 � G1 � 58.2 3.4 52.6 2.2 61.8 2.7 51.4 2.8S � 27.6 2.3 31.2 1.4 25.8 3.4 39.0 1.6G2 � M � 14.2 2.9 16.2 1.7 12.4 0.9 9.6 3.4
HT-29 sub � G1 � 4.3 1.5 15.6 4.3 5.4 3.3 7.2 3.4G0 � G1 � 65.3 6.2 60.1 1.3 64.2 1.7 61.4 1.2S � 21.1 3.3 20.3 2.4 22.1 2.8 33.4 2.2G2 � M � 13.6 3.1 19.6 1.4 13.7 1.4 5.6 1.8
HCT-15 sub � G1 � 6.2 1.3 8.4 1.9 9.2 1.1 8.1 2.9G0 � G1 � 78.5 5.3 67.3 1.6 71.0 2.4 72.3 1.3S � 17.2 3.7 21.4 1.6 23.2 1.2 19.4 1.2G2 � M � 4.3 0.8 11.3 2.8 5.8 0.3 8.3 0.4
Colo-205 sub � G1 � 4.5 1.4 6.5 1.2 4.5 1.2 8.8 2.3G0 � G1 � 72.3 3.7 68.4 2.9 65.2 3.3 48.7 4.1S � 13.2 1.7 13.4 1.8 13.2 1.9 35.5 2.4G2 � M � 14.5 2.4 18.2 3.1 21.6 1.1 15.8 2.6
KM-12 sub � G1 � 9.7 1.6 11.1 4.1 17.2 3.3 8.1 3.0G0 � G1 � 71.8 1.3 73.5 3.8 71.2 2.3 69.2 2.4S � 8.2 1.1 7.9 1.6 6.2 0.8 13.7 12G2 � M � 20.0 1.1 18.6 1.4 22.6 2.4 17.1 1.8
1Each point represents the mean SE of at least 3 independent experiments, performed in duplicate.
272 RUSSO ET AL.
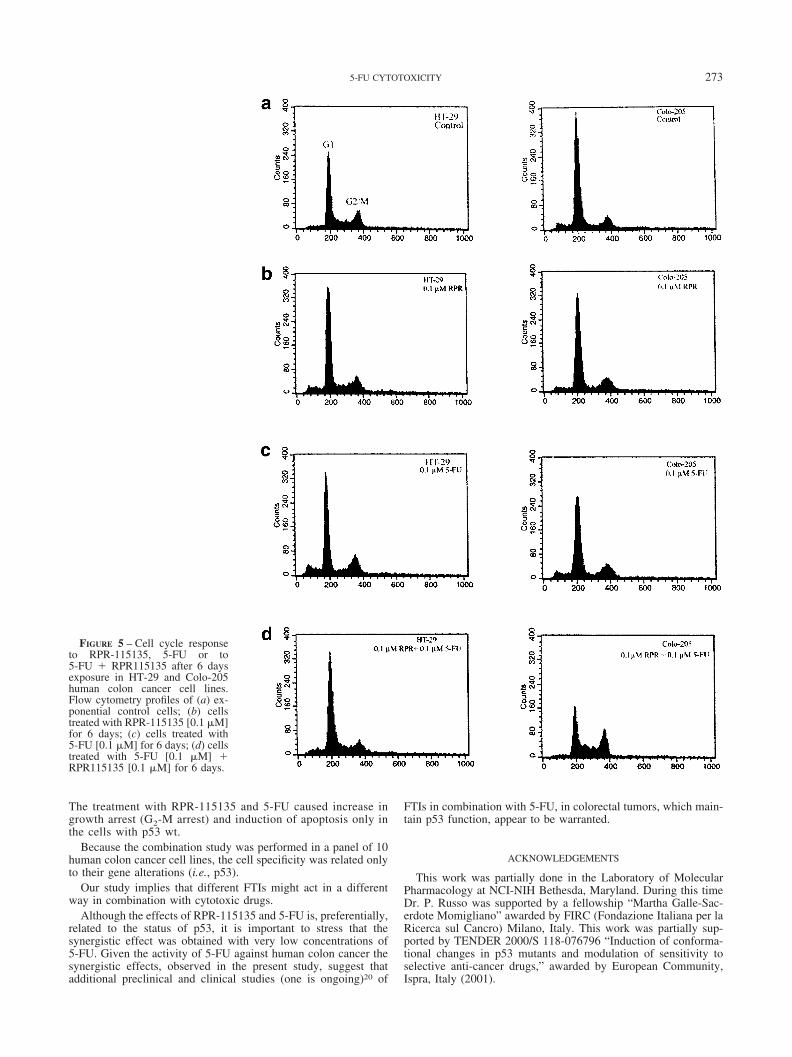
The treatment with RPR-115135 and 5-FU caused increase ingrowth arrest (G2-M arrest) and induction of apoptosis only inthe cells with p53 wt.
Because the combination study was performed in a panel of 10human colon cancer cell lines, the cell specificity was related onlyto their gene alterations (i.e., p53).
Our study implies that different FTIs might act in a differentway in combination with cytotoxic drugs.
Although the effects of RPR-115135 and 5-FU is, preferentially,related to the status of p53, it is important to stress that thesynergistic effect was obtained with very low concentrations of5-FU. Given the activity of 5-FU against human colon cancer thesynergistic effects, observed in the present study, suggest thatadditional preclinical and clinical studies (one is ongoing)20 of
FTIs in combination with 5-FU, in colorectal tumors, which main-tain p53 function, appear to be warranted.
ACKNOWLEDGEMENTS
This work was partially done in the Laboratory of MolecularPharmacology at NCI-NIH Bethesda, Maryland. During this timeDr. P. Russo was supported by a fellowship “Martha Galle-Sac-erdote Momigliano” awarded by FIRC (Fondazione Italiana per laRicerca sul Cancro) Milano, Italy. This work was partially sup-ported by TENDER 2000/S 118-076796 “Induction of conforma-tional changes in p53 mutants and modulation of sensitivity toselective anti-cancer drugs,” awarded by European Community,Ispra, Italy (2001).
FIGURE 5 – Cell cycle responseto RPR-115135, 5-FU or to5-FU � RPR115135 after 6 daysexposure in HT-29 and Colo-205human colon cancer cell lines.Flow cytometry profiles of (a) ex-ponential control cells; (b) cellstreated with RPR-115135 [0.1 �M]for 6 days; (c) cells treated with5-FU [0.1 �M] for 6 days; (d) cellstreated with 5-FU [0.1 �M] �RPR115135 [0.1 �M] for 6 days.
2735-FU CYTOTOXICITY

REFERENCES
1. Midgley R, Kerr D. Colorectal cancer. Lancet 1999;353:391–9.2. Watanabe T, Wu T-T, Catalano PJ, Ueki T, Satriano R, Haller DG,
Benson AB 3rd, Hamilton Sr. Molecular predictors of survival afteradjuvant chemotherapy for colon cancer. N Engl J Med 2001;344:1196–206.
3. Ahnen DJ, Feigl P, Quan G, Fenoglio-Preiser C, Lovato LC, Bunn PAJr, Stemmermann G, Wells JD, McDonald JS, Meyskens FL Jr. Ki-rasmutation and p53 overexpression predict the clinical behavior ofcolorectal cancer: a Southwest Oncology Group Study. Cancer Res1998;58:1149–58.
4. O’Connor PM, Jackman J, Bae I, Myers TG, Fan S, Motho M,Scudiero DA, Monks A, Sausville EA, Weinstein JN, Friend S,Fornace AJ Jr, Kohn KW. Characterization of the p53 tumor suppres-sor pathway in cell lines of the National Cancer Anticancer DrugScreen and correlations with the growth inhibitory potency of 123anticancer agents. Cancer Res 1997;57:4285–300.
5. Grem JL, Danenberg KD, Behan K, Parr A, Young L, Danenberg PV,Nguyen D, Drake J, Monks A, Allegra CJ. Thymidine kinase, thymi-dylate Synthase, and dihydropyrimidine dehydrogenase profiles ofcell lines of the National cancer Institute’s anticancer drug screen.Clin Cancer Res 2001;7:999–1009.
6. Pritchard DM, Watson AJM, Potten CS, Jackman AL, Hickman JA.Inhibition by uridine but not thymidine of p53-dependent intestinalapoptosis initiated by 5-fluorouracil: evidence for the involvement ofRNA perturbation. Proc Natl Acad Sci USA 1997;94:1795–9.
7. Pritchard DM, Potten CS, Hickman JA. The relationships betweenp53-dependent apoptosis, inhibition of proliferation, and 5-fluoroura-cil-induced histopathology in murine intestinal epithelia. Cancer Res1998;58:5453–65.
8. Bunz F, Hwang PM, Torrance C, Waldman T, Zhang Y, Dillehay L,Williams J, Lengauer C, Kinzler KW, Vogelstein B. Disruption of p53in human cancer cells alters the responses to therapeutic agents. J ClinInvest 1999;104:263–9.
9. Rustum YM, Cao S. New drugs in therapy of colorectal cancer:preclinical studies. Semin Oncol 1999;26:612–20.
10. Sobrero A, Kerr D, Glimelius B, Van Cutsem E, Milano G, PritchardDM, Rougier P, Aapro M. New directions in the treatment of colo-rectal cancer: a look to the future. Eur J Cancer 2000;236:559–66.
11. Katz ME, McCormick F. Signal transduction from multiple Raseffectors. Current Opin Gen Dev 1997;7:75–9.
12. Adjei AA. Blocking oncogenic Ras signaling for cancer therapy.J Natl Cancer Inst 2001;93:1062–74.
13. Gibbs JB, Oliff A, Kohl NE. Farnesyltransferase inhibitors, ras re-search yields a potential cancer therapeutic. Cell 1994;77:175–8.
14. Sebti SM, Hamilton AD. Farnesyltransferase and geranyltransferase Iinhibitors in cancer therapy: important mechanistic and bench tobedside issues. Expert Opin Investig Drugs 2000;9:2767–-82.
15. Cox AD. Farnesyltransferase inhibitors. Potential role in the treatmentof cancer. Drugs 2001;61:723–32.
16. Zhang FL, Kirschmeier P, Carr D, James L, Bond RW, Wang L,Patton R, Windsor WT, Syto R, Zhang R, Bishop WR. Characteriza-tion of Ha-ras, N-ras, Ki-Ras4A, and Ki-Ras4B as in vitro substratesfor farnesyl protein transferase and geranylgeranyl protein transferasetype I. J Biol Chem 1997;272:10232–9.
17. Bredel M, Pollack IF. The p21-Ras signal transduction pathway andgrowth regulation in human high-grade gliomas. Brain Res Brain ResRev 1999;29:232–49.
18. Miyakis S, Sourvinos G, Spandidos DA. Differential expression andmutation of the ras family genes in human breast cancer. BiochemBiophys Res Commun 1998;251:609–12.
19. Feldkamp MM, Lau N, Roncari L, Guha A. Isotype-specific Ras.
GTP-levels predict the efficacy of farnesyl transferase inhibitorsagainst human astrocytomas regardless of Ras mutational status. Can-cer Res 2001;61:4425–31.
20. Hahn SM, Bernhard E, McKenna WG. Farnesyltransferase inhibitors.Semin Oncol 2001;28(Suppl):86–93.
21. Karp JE, Kaufmann SH, Adjei AA, Lancet JE, Wright JJ, End DW.Current status of clinical trials of farnesyltransferase inhibitors. CurrOpin Oncol 2001;13:470–6.
22. Giraud E, Luttmann C, Lavelle F, Riou JF, Mailliet P, Laoui A.Multivariate data analysis using D-optimal designs, partial leastsquares, and response surface modelling: a directional approach forthe analysis of farnesyltransferase inhibitors. J Med Chem 2000;43:1807–16.
23. Russo P, Ottoboni C, Crippa A, Riou JF, O’Connor PM. RPR-115135,a new non peptidomimetic farnesyltransferase inhibitor, inducesG0/G1 arrest only in serum starved cells. Int J Oncol. 2001; 18:855–62.
24. Ottoboni C, Crippa A, Falugi C, Russo P. Induction of micronuclei bya new non-peptidic mimetic farnesyltransferase inhibitor RPR-115135: role of gene mutations. Mutagenesis. 2001;16 423–30.
25. Russo P, Ottoboni C, Malacarne D, Crippa A, Riou JF, O’Connor PM.Nonpeptidomimetic Farnesyltransferase Inhibitor RPR-115135 In-creases Cytotoxicity of 5-Fluorouracil: Role of p53. J Pharmacol ExpTher. 2002;300:220–6.
26. Falugi C, Arzani D, Trombino S, Russo P. c-myc down regulationinduces apoptosis in human cancer cell lines exposed to RPR-115135,a non peptidomimetic farnesyltransferase inhibitor. Cell Death Diff, inpress.
27. Chow T-C, Talalay P. A simple generalized equation for the analysisof multiple inhibitions of Michaelis-Menten kinetic systems. J BiolChem 1977;252:6438–42.
28. Chow T-C, Talalay P. Generalized equations for the analysis ofinhibitors of Michaelis-Menten and higher order kinetic systems withtwo or more mutually exclusive and nonexclusive inhibitors. EurJ Biochem 1981;115:207–16.
29. Fan S, Chang JK, Smith ML, Duba D, Fornace AI Jr, O’Connor PM.Cell lacking CIP1/WAF1 genes exhibit preferential sensitivity tocisplatin and nitrogen mustard. Oncogene 1997;14:2127–36.
30. Fan S, Cherney B, Reinhold W, Rucker K, O’Connor PM. Disruptionof p53 function in immortalized human cells does not affect survivalor apoptosis after Taxol or vincristine treatment. Clin Cancer Res1998;4:1047–54.
31. Zheng S, Chen P, McMillan A, Lafuente A, Lafuente MJ, Ballesta A,Trias M, Wiencke JK. Correlations of partial and extensive methyl-ation at the p14(ARF) locus with reduced mRNA expression incolorectal cancer cell lines and clinicopathological features in primarytumors. Carcinogenesis 2000;21:2057–64.
32. Magdinier F, Wolffe AP. Selective association of the methyl-CpGbinding protein MBD2 with the silent p14/p16 locus in human neo-plasia. Proc Natl Acad Science USA 2001;98:4990–5.
33. Esteller M, Corn PG, Baylin SB, Herman JG. A gene hypermethyl-ation profile of human cancer. Cancer Res 2001;61:3225–9.
34. Schmitt CA, McCurrach ME, de Stanchina E, Wallace-Brodeur RR,Lowe SW. INK4a/ARF-mutations accelerate lymphomagenesis andpromote chemoresistance by disabling p53. Genes Dev 1999;13:2670–7.
35. Lloyd AC. p53: only ARF the story. Nat Cell Biol 2000;2:E48–50.36. Moasser MM, Sepp-Lorenzino L, Kohl NE, Oliff A, Balog A, Su DS,
Danishefsky SJ, Rosen N. Farnesyl transferase inhibitors cause en-hanced mitotic sensitivity to Taxol and epothilones. Proc Natl AcadSci USA 1998;95:1369–74.
TABLE IV – PERCENTAGE OF APOPTOTIC CELLS AFTER 6 DAYS CONTINUOUS TREATMENT1
Cell line CTRL 5-FU(0.1 �M)
RPR-115135(0.1 �M)
5-FU (0.1 �M)� RPR (0.1 �M)
HCT-116 8.8 1.2 10.5 0.8 12.0 1.7 68.4 3.4LoVo 14.3 2.7 8.2 1.7 2.5 1.1 54.7 3.2RKO 10.2 5.5 9.8 1.8 11.3 1.4 71.6 2.4DLD-1 4.8 0.8 7.8 1.4 9.5 1.8 48.7 3.3Colo-320 6.5 1.1 19.2 1.4 7.9 1.2 81.6 7.7SW-620 11.4 4.3 12.5 3.3 9.2 1.2 21.7 1.4HT-29 22.3 2.7 25.0 2.8 16.4 1.4 29.2 2.7HCT-15 8.4 17 10.4 1.7 11.2 2.3 15.6 2.2Colo-205 8.3 0.8 7.5 1.4 9.9 2.7 14.2 1.8KM-12 13.5 1.2 9.4 2.1 19.4 2.3 9.7 0.811,000 cells were scored for each DAPI stained slide. Each point represents the mean SE of at least
3 independent experiments, performed in duplicate.
274 RUSSO ET AL.

37. Sun J, Blaskovich MA, Knowles D, Qian Y, Ohkanda J, Bailey RD,Hamilton AD, Sebti SM. Antitumor efficacy of a novel class ofnon-thiol-containing peptidomimetic inhibitors of farnesyltransferaseand geranylgeranyltransferase I: combination therapy with the cyto-toxic agents cisplatin, Taxol and gemcitabine. Cancer Res 1999;59:4919–26.
38. Liu M, Bryant MS, Chen J, Lee S, Yaremko B, Lipari P, MalkowskiM, Ferrari E, Nielsen L, Prioli N, Dell J, Sinha D, et al. Antitumoractivity of SCH 66336, an orally bioavailable tricyclic inhibitor offarnesyl protein transferase, in human tumor xenograft models andwap-ras transgenic mice. Cancer Res 1998;58:4947–56.
39. Adjei AA, Davis JN, Bruzek LM, Erlichman C, Kaufmann SH.Synergy of the protein farnesyltransferase inhibitor SCH66336 andcisplatin in human cancer cell lines. Clin Cancer Res 2001;7:1438–45.
40. Koo HM, Monks A, Mikheev A, Rubistein LV, Gray-Goodrich M,McWilliams MJ, Alvord WG, Oie HK, Gazdar AF, Paull KD, ZarblH, Vande Woude GF. Enhanced sensitivity to 1-�-D-arabinofurano-sylcytosine and topoisomerase II inhibitors in tumor cell lines har-boring activated ras oncogens. Cancer Res 1996;56:5211–6.
41. Donald SP, Sun XY, Hu CA, Yu J, Mei JM, Valle D, Phang JM.Proline oxidase, encoded by p53-induced gene-6, catalyzes the gen-eration of proline-dependent reactive oxygen species. Cancer Res2001;61:1810–5.
42. Nagase T, Kawata S, Tamura S, Matsuda Y, Inui Y, Yamasaki E,
Ishiguro H, Ito T, Miyagawa J, Mitsui H, Yamamoto K, Kinoshita M,et al. Manumycin and gliotoxin derivative KT7595 block Ras farne-sylation and cell growth but do not disturb lamin farnesylation andlocalization in human tumour cells. Br J Cancer 1997;76:1101–10.
43. Petak I, Tillman DM, Houghton JA. p53 dependence of Fas inductionand acute apoptosis in response to 5-fluorouracil-leucovorin in humancolon carcinoma cell lines. Clin Cancer Res 2000;6:4432–41.
44. Lengyel E, Wang H, Gum R, Simon C, Wang Y, Boyd D. Elevatedurokinase-type plasminogen activator receptor expression in a coloncancer cell line is due to a constitutively activated extracellularsignal-regulated kinase-1-dependent signaling cascade. Oncogene1997;14:2563–73.
45. Rajesh D, Schell K, Verma AK. Ras mutation, irrespective of cell typeand p53 status, determines a cell’s destiny to undergo apoptosis byokadaic acid, an inhibitor of protein phosphatase 1 and 2A. MolPharmacol 1999;56:515–25.
46. Di Paolo A, Danesi R, Nardini D, Bocci G, Innocenti F, Fogli S,Barachini S, Marchetti A, Bevilacqua G, Del Tacca M. Manumycininhibits ras signal transduction pathway and induces apoptosis inCOLO-320-DM human colon tumour cells. Br J Cancer 2000;82:905–12.
47. Chou T-C. The median-effect principle and the combination index forquantitation of synergism and antagonism. In: Chou T-C, Rideout DC,eds. Synergism and antagonism in chemotherapy. San Diego: Aca-demic Press, 1991. 61–102.
2755-FU CYTOTOXICITY
Copyright © 2022 FDOKUMEN




















