
Siamese-Twin Porphyrin: A Pyrazole-Based Expanded Porphyrin Providing a Bimetallic Cavity
Upload
independentCategory
view
1download
0
Bioorganic & Medicinal Chemistry Letters 22 (2012) 6896–6902
Contents lists available at SciVerse ScienceDirect
Bioorganic & Medicinal Chemistry Letters
journal homepage: www.elsevier .com/ locate/bmcl
Synthesis and biological evaluation of new phenothiazine derivatives bearinga pyrazole unit as protein farnesyltransferase inhibitors
Lavinia Baciu-Atudosie a,�, Alina Ghinet a,b,c,�, Amaury Farce b,d, Joëlle Dubois e, Dalila Belei a, Elena Bîcu a,⇑a Department of Organic Chemistry, ‘Al. I. Cuza’ University of Iasi, Faculty of Chemistry, Bd. Carol I nr. 11, 700506 Iasi, Romaniab Univ Lille Nord de France, F-59000 Lille, Francec UCLille, EA 4481 (GRIIOT), Laboratoire de Pharmacochimie, HEI, 13 rue de Toul, F-59046 Lille, Franced Institut de Chimie Pharmaceutique Albert Lespagnol, EA GRIIOT (4481), IFR114, 3 Rue du Pr Laguesse, B.P. 83, F-59006 Lille, Francee Institut de Chimie des Substances Naturelles, UPR2301 CNRS, Centre de Recherche de Gif, Avenue de la Terrasse, F-91198 Gif-sur-Yvette Cedex, France
a r t i c l e i n f o
Article history:Received 4 August 2012Revised 7 September 2012Accepted 10 September 2012Available online 17 September 2012
Keywords:Farnesyltransferase inhibitorPhenothiazinePyrazolePyrazolineAcylhydrazoneCytostatic agent
0960-894X/$ - see front matter � 2012 Elsevier Ltd.http://dx.doi.org/10.1016/j.bmcl.2012.09.030
⇑ Corresponding author.E-mail address: [email protected] (E. Bîcu).
� These authors have made equal contributions.
a b s t r a c t
A new family of protein farnesyltransferase inhibitors, based on a phenothiazine scaffold, was designedand synthesized. The biological evaluation of these products showed that compounds 28 and 30 were themost active, with protein farnesyltransferase inhibition potencies in the low micromolar range. Com-pounds were also evaluated for their antiproliferative activity on a NCI-60 cancer cell line panel. Inden-opyrazole 30 exhibited the most potent in vitro cytostatic activity inhibiting the growth of HCT-116, LOXIMVI and SK-MEL-5 cell lines.
� 2012 Elsevier Ltd. All rights reserved.
The farnesyltransferase (FTase) has generated much attentionas an important target for the conception of new anticancer agentswith reduced toxicity.1 It is a heterodimeric metalloenzyme thatbelongs to the prenyltransferase protein family and catalyzes thetransfer of a prenyl unit (C15) from farnesylpyrophosphate (FPP)to the free thiol group of the C cysteine found in the terminalCa1a2X motif (were a1, a2 are aliphatic amino acids and X a serine,a methionine, an alanine or a glutamine)2 of a group of membranesmall G-proteins such as lamin A and lamin B, RhoB or RhoE. Thisfarnesylation is critical for membrane binding and the biologicalfunction of G-proteins.3 The protein farnesylation is an importantpost-translational modification occurring on several cell signalingproteins for example oncogenic Ras proteins.4 Inhibition of proteinfarnesyltransferase prevents membrane localization of Ras, andthus constitutes a valid target for the conception of new cytostaticanticancer drugs.5 Hence, many farnesyltransferase inhibitors(FTIs) demonstrated antitumor efficacy in preclinical human xeno-graft models and diverse molecules (compounds I–III, Fig. 1) haveundergone clinical trials.
All rights reserved.
A recent biological screening of the chemical library of our Or-ganic Chemistry Department (Iasi) allowed us to discover new FTIshits, heretofore unencountered, such as compound IV (Fig. 1), bear-ing a phenothiazine unit.6 On the other hand, the investigationrealized by our team of the steric tolerance of the A2 binding siteof farnesyltransferase revealed that the ferrocene unit was awell-tolerated bulky group (compound V, Fig. 1).7 Molecular mod-elling7 learned us that the phenothiazine moiety can be placed inthe same A2 binding site as the bulky ferrocene unit. We theninvestigated the possibility of obtaining phenothiazine-containingFTIs able to coordinate with the zinc atom of the protein via chelat-ing groups and to interact with the A2 binding site via the bulkyphenothiazine unit. Then, we decided to perform a preliminarySAR study on the phenothiazine derivatives bearing a pyrazole unitwith some structural modifications of the scaffold of compound VI(Fig. 1).
Heterocyclic compounds with a pyrazole skeleton take a signif-icant place in medicinal chemistry. They have been investigated fortheir potential antitumor activity8–12 as potent inhibitors of thehistone deacetylase (HDAC),13 as inhibitors of type I TGF-b recep-tor,14 as inhibitors of tubulin polymerization,15 as androgenreceptor (AR) antagonists,16 or as inhibitors of ERK kinase.17 Thepyrazole pattern is also found in the structure of antimicrobial11

N
N
N
Cl
O
NH2
ClTipifarnib I22
NO
NH2NO
N
Br
Cl
Br
N
N
NC
NH
N
SO2
S
BMS-214662 III24
IV6
Br
O
NOO
NNN
S
Lonafarnib II23
YXN
S
NN
VI
Fe2+
O OMeOMe
OMe-
-
V7
R
X = (CH2)2, CH2, COY = CO, CH2R = Me, C6H5
bulky unit
bulky unit bulky unit
chelating groups
IC50 (bovine FTase) = 0.65 nM22c IC50 (bovine FTase) = 8.3 nM22c IC50 (human FTase) = 1.4 nM24
IC50 (human FTase) = 2.2 µM7IC50 (human FTase) = 1.3 µM6
Figure 1. Structure of known FTIs I–III,22–24 of FTIs synthesized by our team IV–V,6,7 and of target compounds VI.
L. Baciu-Atudosie et al. / Bioorg. Med. Chem. Lett. 22 (2012) 6896–6902 6897
and antifungal agents,18 and molecules with activity againstHepatitis C Virus (HCV).12 It provided also anti-inflammatorypotential.19
The phenothiazine unit represents an important pharmaco-phore. Phenothiazine derivatives show significant antitumoral,antimicrobial, antimalarial, antipsychotic and numerous other bio-logical activities.20 The association of the phenothiazine with thepyrazole ring resulted recently in the synthesis of new agents withantimicrobial potential.21
To the best of our knowledge, phenothiazines containing a pyr-azole ring have not encountered in the field of FTIs. In order toidentify new FTIs with original skeleton, we report here our designand synthesis of a series of phenothiazine based FTIs with pyrazoleunit (compounds VI, Fig. 1).
We have recently described the optimization of the synthesis of10-(hydrazinoacetyl)-10H-phenothiazine 3.25 The condensation ofacylhydrazines 1,26 227 and alkylhydrazine 3 with acyclic 1,3-dike-tones 4–6 afforded pyrazoles 7–10, 12, 14, 17 and 18 (35–93%yields) and/or 5-hydroxy-4,5-dihydropyrazoles 11, 13, 15 and 16
N
S
XYNH
NH2
+ R
O
R
O
1 2
N
S
XYN
NOHR
R
1: X = (CH2)2, Y = CO2: X = CH2, Y = CO3: X = CO, Y = CH2
1
2
4: R1 = CH3, R2 = CH35: R1 = CH3, R2 = C6H56: R1 = CO2C2H5, R2 = C6H5
(i)
11: X = (CH2)2, Y = CO, R1 = C13: X = CH2, Y = CO, R1 = CH315: X = (CH2)2, Y = CO, R1 = C16: X = CH2, Y = CO, R1 = CO
Scheme 1. Reactions of acylhydrazines 1, 2 and alkylhydrazine 3 with acyclic 1,3-dike
(25–67% yields) (Scheme 1 and Table 1). The formation of5-hydroxypyrazolines by reacting 1,3-diketones with hydrazidederivatives are largely described in the literature.28 Note that treat-ment of alkylhydrazine 3 with 4-aryl-2,4-dioxoester 6 led to theformation of two isomers (pyrazoles 17 and 18) in 55% and 30%yield, respectively.
The importance of the nature of the pyrazole ring was consid-ered and structural variations were obtained by reacting hydrazines1–3 with cyclic di- and triketones 19–21 (Scheme 2) in order to givetetrahydroindazoles and indenopyrazoles. Thus, reaction of acy-lhydrazines 1, 2 and alkylhydrazine 3 with cyclic diketone 19 orwith triketone 20 led to 25–65% of 4,5,6,7-tetrahydroindazolederivatives 22–27. When the reactions were conducted with acy-lhydrazines 1–2 and triketone 21, the hydrazones 28–29 were iso-lated in good yields (77% and 79% respectively). Condensation of thealkylhydrazine 3 with triketone 21 in DMA at 100 �C afforded thecyclized indenopyrazole 30 as unique product, in 71% yield.
To complete this preliminary study, we were interested onortho-aminocyanopyrazoles and their derivatives as inhibitors of
and/orN
S
XYN
N
R
R
1
2
H3, R2 = C6H5, 38%, R2 = C6H5, 25%O2C2H5, R2 = C6H5, 64%
2C2H5, R2 = C6H5, 67%
7: X = (CH2)2, Y = CO, R1 = CH3, R2 = CH3, 93%8: X = CH2, Y = CO, R1 = CH3, R2 = CH3, 65%9: X = CO, Y = CH2, R1 = CH3, R2 = CH3, 82%10: X = (CH2)2, Y = CO, R1 = CH3, R2 = C6H5, 56%12: X = CH2, Y = CO, R1 = CH3, R2 = C6H5, 35%14: X = CO, Y = CH2, R1 = CH3, R2 = C6H5, 71%17: X = CO, Y = CH2, R1 = CO2C2H5, R2 = C6H5, 55%18: X = CO, Y = CH2, R1 = C6H5, R2 = CO2C2H5, 30%
tones 4–6. Conditions: (i) absolute EtOH (reflux) or DMA (100 �C), 3–8 h, 25–90%.

Table 1Structure and inhibitory activities of the tested compounds on human FTase
N
S
XYR
Product no X Y R % Inhibition of FTase IC50a (lM ± SD)
9 CO CH2
NN
Me
Me
22.4 n.d.
12 CH2 CO
NN
Me
C6H5
0 n.d.
14 CO CH2
NN
Me
C6H5
0 n.d.
15 (CH2)2 CO
NN
CO2Et
OH C6H5
22.8 n.d.
16 CH2 CO
NN
CO2Et
OH C6H5
30.7 n.d.
17 CO CH2
NN
C6H5
CO2Et
0 n.d.
18 CO CH2
NN
C6H5
EtO2C
0 n.d.
22 (CH2)2 CO
NN
Me
0 n.d.
23 CH2 CO
NN
Me
0 n.d.
24 CO CH2
NN
Me
0 n.d.
25 (CH2)2 CO
NN
Me O
19.5 n.d.
26 CH2 CO
NN
Me O
0 n.d.
27 CO CH2
NN
Me O
46.6 n.d.
28 (CH2)2 CO NNH
Me O
OH
95.5 3.73 ± 0.46
30 CO CH2
NN
MeO
67.7 3.35 ± 0.28
6898 L. Baciu-Atudosie et al. / Bioorg. Med. Chem. Lett. 22 (2012) 6896–6902
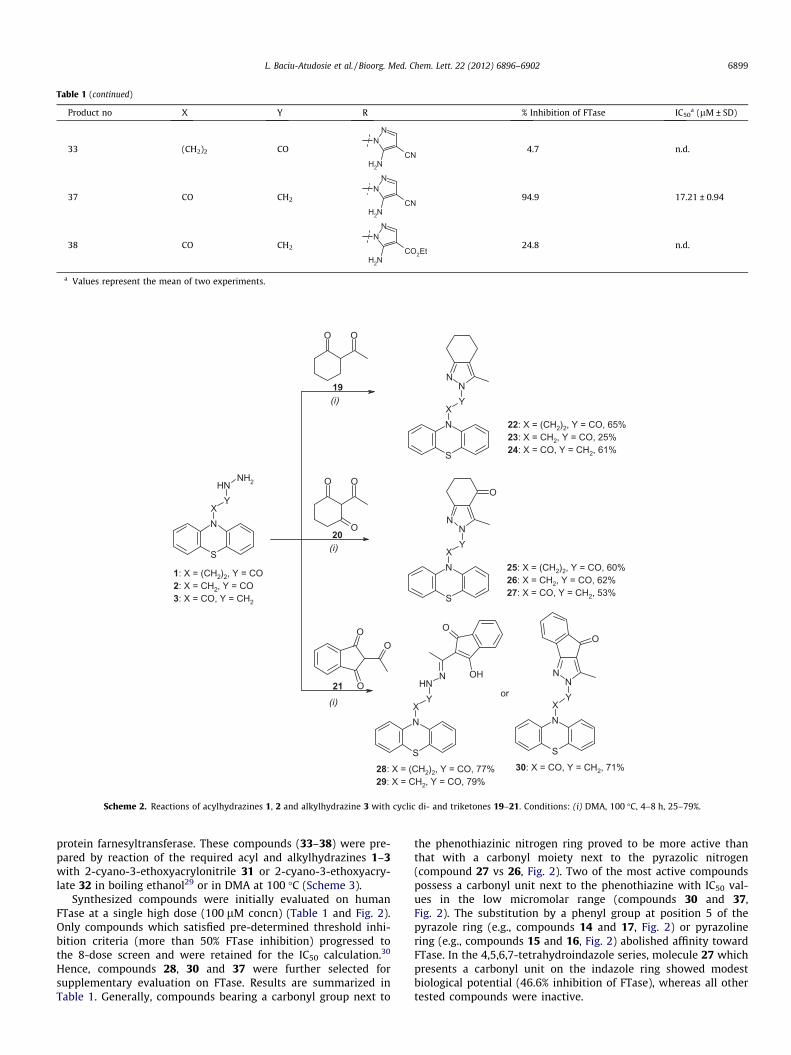
Table 1 (continued)
Product no X Y R % Inhibition of FTase IC50a (lM ± SD)
33 (CH2)2 CO
NN
H2NCN
4.7 n.d.
37 CO CH2
NN
H2NCN
94.9 17.21 ± 0.94
38 CO CH2
NN
NH2
CO2Et24.8 n.d.
a Values represent the mean of two experiments.
N
S
XYNH
NH2
O O
O O
O
N
S
XYN
N
N
S
XYN
N
O
OO
O
N
S
XYNH
N OH
O
N
S
XYN
N
O
(i)
(i)
(i)
19
20
1: X = (CH2)2, Y = CO2: X = CH2, Y = CO3: X = CO, Y = CH2
22: X = (CH2)2, Y = CO, 65%23: X = CH2, Y = CO, 25%24: X = CO, Y = CH2, 61%
25: X = (CH2)2, Y = CO, 60%26: X = CH2, Y = CO, 62%27: X = CO, Y = CH2, 53%
21
28: X = (CH2)2, Y = CO, 77%29: X = CH2, Y = CO, 79%
or
30: X = CO, Y = CH2, 71%
Scheme 2. Reactions of acylhydrazines 1, 2 and alkylhydrazine 3 with cyclic di- and triketones 19–21. Conditions: (i) DMA, 100 �C, 4–8 h, 25–79%.
L. Baciu-Atudosie et al. / Bioorg. Med. Chem. Lett. 22 (2012) 6896–6902 6899
protein farnesyltransferase. These compounds (33–38) were pre-pared by reaction of the required acyl and alkylhydrazines 1–3with 2-cyano-3-ethoxyacrylonitrile 31 or 2-cyano-3-ethoxyacry-late 32 in boiling ethanol29 or in DMA at 100 �C (Scheme 3).
Synthesized compounds were initially evaluated on humanFTase at a single high dose (100 lM concn) (Table 1 and Fig. 2).Only compounds which satisfied pre-determined threshold inhi-bition criteria (more than 50% FTase inhibition) progressed tothe 8-dose screen and were retained for the IC50 calculation.30
Hence, compounds 28, 30 and 37 were further selected forsupplementary evaluation on FTase. Results are summarized inTable 1. Generally, compounds bearing a carbonyl group next to
the phenothiazinic nitrogen ring proved to be more active thanthat with a carbonyl moiety next to the pyrazolic nitrogen(compound 27 vs 26, Fig. 2). Two of the most active compoundspossess a carbonyl unit next to the phenothiazine with IC50 val-ues in the low micromolar range (compounds 30 and 37,Fig. 2). The substitution by a phenyl group at position 5 of thepyrazole ring (e.g., compounds 14 and 17, Fig. 2) or pyrazolinering (e.g., compounds 15 and 16, Fig. 2) abolished affinity towardFTase. In the 4,5,6,7-tetrahydroindazole series, molecule 27 whichpresents a carbonyl unit on the indazole ring showed modestbiological potential (46.6% inhibition of FTase), whereas all othertested compounds were inactive.

Table 2In vitro percentage growth inhibition (GI%) caused by the selected compounds against some tumor cell lines in the single-dose assaya
Cell type Cell line GI% at 10 lM
12 22 24 30 37 38
Leukemia CCRF-CEM —b 12.8 6.5 12.1 — —HL-60(TB) 31.5 19.6 — — 46.2 83.2MOLT-4 9.6 — 20.1 25.4 n.d.c 7.6RPMI-8226 — — 16.2 — — —
CNS cancer SF-295 — — — 35.8 13.1 6.7SNB-19 — — — 35.7 — —U251 9.2 — 10.2 24.8 — —
Colon cancer HCT-166 — 12.4 8.4 55.7 — —HCT-15 — — 8.0 23.0 — 25.4HT29 — — — 17.7 — 13.2SW-620 — — — 23.8 — —
Non-Small Cell EKVX 5.0 8.3 — 39.9 — 5.3Lung Cancer NCI-H226 7.2 12.8 — 25.3 — 8.1
HOP-92 14.0 31.6 n.d. — 12.5 —NCI-H522 — 9.1 15.2 9.6 — 31.9
Melanoma LOX IMVI — — — 54.8 — —SK-MEL-5 — — — 51.4 — 6.9UACC-62 — 7.1 13.2 31.2 — 6.3
Renal Cancer ACHN — — — 29.7 — —SN12C 7.2 — 5.6 34.8 — —A498 17.3 n.d. n.d. 19.4 14.0 11.8CAKI-1 16.2 — 8.1 38.3 — 16.0UO-31 9.0 — 10.0 29.5 6.4 13.3
Ovarian Cancer OVCAR-3 — — — 29.7 — —OVCAR-8 6.3 — — 28.2 5.5 —NCI/ADR-RES — 8.2 6.2 33.5 — —
Prostate Cancer PC-3 13.0 13.0 n.d. 31.0 9.9 21.3Breast Cancer MDA-MB-231/ATCC — — — 27.8 — —
MDA-MB-468 — 11.2 — 37.6 11.7 15.3
a Data obtained from NCI’s in vitro disease-oriented human tumor cell screen at 10 lM concentration.b Inactive.c Not determined.
N
S
XYNH
NH2
+N
S
XYN
N
W
NH2
1: X = (CH2)2, Y = CO2: X = CH2, Y = CO3: X = CO, Y = CH2
31: W1 = W2 = CN32: W1 = CN, W2 = CO2C2H5
(i)
W
W
EtO
33: X = (CH2)2, Y = CO, W2 = CN, 52%34: X = (CH2)2, Y = CO, W2 = CO2C2H5, 25%35: X = CH2, Y = CO, W2 = CN, 74%36: X = CH2, Y = CO, W2 = CO2C2H5, 57%37: X = CO, Y = CH2, W2 = CN, 76%38: X = CO, Y = CH2, W2 = CO2C2H5, 73%
1
2
2
Scheme 3. Reactions of acylhydrazines 1, 2 and alkylhydrazine 3 with a,b-unsaturated b-disubstituted ethers 31, 32. Conditions: (i) EtOH, reflux, except for compound 35(DMA, 100 �C), 1–8 h, 25–76%.
Figure 2. Screening of synthesized compounds on human FTase at a 100 lM concentration.
6900 L. Baciu-Atudosie et al. / Bioorg. Med. Chem. Lett. 22 (2012) 6896–6902

Figure 3. Structure and docking of new phenothiazines in the active site of FTase: (a) compound 28; (b) compound 30.
L. Baciu-Atudosie et al. / Bioorg. Med. Chem. Lett. 22 (2012) 6896–6902 6901
The six derivatives 12, 22, 24, 30, 37 and 38 were selected byNCI for screening against 60 human tumor cell lines.31 The repre-sentative results are summarized in Table 2. Indenopyrazole 30exhibited the most interesting cell growth inhibition among thetested compounds (55.7% inhibition on HCT-166, 54.8% inhibitionon LOX IMVI and 51.4% inhibition on SK-MEL-5 cell lines at10 lM concn). Compound 37 showed decreased growth inhibitorypotential compared to pyrazole 30, conserving an increased activ-ity only on the HL-60(TB) leukemia cell line (46.2% inhibition).Molecules 12, 22 and 24, which were inactive in the fluores-cence-based FTase assay proved also to be inactive on cell growthinhibition in the single-dose assay (Table 2). Pyrazole 38 proved tobe the most interesting, as it showed selective cell growth inhibi-tion on leukemia cells at a 10 lM concentration (83% inhibitionof HL-60(TB), Table 2).
The compound docking onto 1LD7 binding site32 was realizedwith GOLD5.1 (CCDC 2011), using the standard parameters, and asphere of 10 Å around the extracted cocrystallized ligand. The bestsolution of the docking run was defined as the most frequent con-formation among the 30 conformations generated.
Hydrazone 28 and pyrazole 30 (Fig. 3) possess the same pheno-thiazine unit; both behave in the same way with the indene/pyra-zole unit in front of the zinc atom of the enzyme and thephenothiazine group deep in the pocket. They don’t form anyhydrogen bonds, however, but may be stabilized by p-p interac-tions which are stronger for compound 30, for which the angle be-tween the aromatic group and the Trp sidechain is of 80�,compared to a less favorable angle of about 60� and longer distancefor compound 28.
The second carbonyl function in the structure of compounds25–27 facilitates the placement of these compounds with their car-bonyl toward the zinc atom with its electrons readily able to formfavorable interaction. There are nonetheless less potent than com-pound 30 that has both a carbonyl and a supplementary aromaticring next to the zinc atom, therefore displaying a stronger affinityfor the enzyme. This translates in a moderate activity, while com-pounds 22–24 are utterly lacking any.
The difference between the two series of compounds (11, 13, 15and 16 vs 7–10, 12, 14, 17 and 18) is the addition of a hydroxyl onthe former, giving again a stronger possibility of interaction withthe zinc. However, it is noteworthy that the most active com-pounds are those two bearing an ester group on the pyrazole ring(compounds 15 and 16, Table 1). We can hypothesize that it is infact the acidic moiety that is relevant for their higher activity; a
free carboxylic acid group could be more amenable to the zinc che-lation. Compound 18 is hindered by the position of the phenylgroup on its pyrazole unit, rendering it unable to reach the zincand explaining its lack of activity. On the contrary, compound 9may be involved in a fairly stable stacking, explaining its activity.
In the series of compounds 25–27 (Table 1), compound 25 dis-plays a large array of divers conformations, with only a few, high-est ranked solutions, pointing both their carbonyls toward the zincatom. This is in clear relation with its low activity. The less flexiblecompound 26 also displays a large number of conformations, butnone of them is able to interact with the metal of the enzyme. Thisis most probably due to the fact that both its carbonyls are close tothe methyl group on the pyrazole moiety, rendering them unableto interact and leaving only the tricyclic head as an anchor point.On the contrary, compound 27 has two major conformations; thehighest scored putting its carbonyl in front of the zinc while thelargest cluster of solutions behaves as compound 24, lying in thepocket on a perpendicular plane when compared to compound30. It is enough to give it a relatively good activity, in contrast tocompound 26. We can safely assume on the basis of compounds24 and 27 that the same holds true for the remaining compounds:22 and 23 both lacks the terminal carbonyl and their carbonyl lin-ker is shielded by the methyl, giving them a very low affinity andtherefore, a lack of activity.
In conclusion, we have synthesized a new series of phenothia-zines bearing a pyrazole unit with protein farnesyltransferase inhi-bition potencies in the low micromolar range.
Synthesized compounds were also evaluated for their antipro-liferative activity on the NCI-60 cell line panel. Compound 30exhibited the most potent in vitro cytostatic activity: inhibitionof the growth of HCT-116, LOX IMVI, and SK-MEL-5 cell lines,and pyrazole 38 showed selectivity against leukemia HL-60(TB)cell lines.
The best FTIs hits issued from this study will be evaluated in thefuture against other biological targets.
Acknowledgements
The authors gratefully acknowledge the ’Programul OperationalSectorial Dezvoltarea Resurselor Umane 2007–2013’ (project POS-DRU/88/1.5/S/47646) and the CommScie (project POSDRU/89/1.5/S/63663) for financial support and the NCI (National Cancer Insti-tute) for the biological evaluation of compounds on their 60-cellpanel.

6902 L. Baciu-Atudosie et al. / Bioorg. Med. Chem. Lett. 22 (2012) 6896–6902
Supplementary data
Supplementary data (synthesis details and physico-chemicalcharacterization for all new compounds are available) associatedwith this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bmcl.2012.09.030.
References and notes
1. (a) Ohkanda, J.; Knowles, D. B.; Blaskovich, M. A.; Sebti, S. M.; Hamilton, A. D.Curr. Top. Med. Chem. 2002, 2, 303; (b) Haluska, P.; Dy, G. K.; Adjei, A. A. Eur. J.Cancer 2002, 38, 1685.
2. (a) Handb, Zhang H. Metalloproteins 2004, 3, 37; (b) Park, H. W.; Beese, L. Curr.Opin. Struct. Biol. 1997, 7, 873.
3. Asoh, K.; Kohchi, M.; Hyoudoh, I.; Ohtsuka, T.; Masubuchi, M.; Kawasaki, K.;Ebiike, H.; Shiratori, Y.; Fukami, T. A.; Kondoh, O.; Tsukaguchi, T.; Ishii, N.; Aoki,Y.; Shimma, N.; Sakaitani, M. Bioorg. Med. Chem. Lett. 2009, 19, 1753. andreferences cited therein.
4. Russo, P.; Loprevite, M.; Cesario, A.; Ardizzoni, A. Curr. Med. Chem. AnticancerAgents 2004, 4, 123.
5. (a) Bell, I. M. J. Med. Chem. 1869, 2004, 47; (b) Singh, S. B.; Lingham, R. B. Curr.Opin. Drug Discov. Devel. 2002, 5, 225.
6. Belei, D.; Dumea, C.; Samson, A.; Farce, A.; Dubois, J.; Bîcu, E.; Ghinet, A. Bioorg.Med. Chem. Lett. 2012, 22, 4517.
7. Ghinet, A.; Rigo, B.; Dubois, J.; Farce, A.; Hénichart, J.-P.; Gautret, P. Med. Chem.Commun. 2012, 3, 1147.
8. Zhang, D.; Wang, G.; Zhao, G.; Xu, W.; Huo, L. Eur. J. Med. Chem. 2011, 46, 5868.9. Insuasty, B.; Tigreros, A.; Orozco, F.; Quiroga, J.; Abonía, R.; Nogueras, M.;
Sanchez, A.; Cobo, J. Bioorg. Med. Chem. 2010, 18, 4965.10. Farag, A. M.; Mayhoub, A. S.; Barakat, S. E.; Bayomi, A. H. Bioorg. Med. Chem.
2008, 16, 881.11. El-borai, M. A.; Rizk, H. F.; Abd-Aal, M. F.; El-Deeb, I. Y. Eur. J. Med. Chem. 2012,
48, 92.12. Riyadh, S. M.; Farghaly, T. A.; Abdallah, M. A.; Abdalla, M. M.; Abd El-Aziz, M. R.
Eur. J. Med. Chem. 2010, 45, 1042.13. Li, X.; Liu, J.-L.; Yang, X.-H.; Lu, X.; Zhao, T.-T.; Gong, H.-B.; Zhu, H.-L. Bioorg.
Med. Chem. 2012, 20, 4430.14. Franchini, M. C.; Bonini, B. F.; Camaggi, C. M.; Gentili, D.; Pession, A.; Rani, M.;
Strocchi, E. Eur. J. Med. Chem. 2010, 45, 2024.15. Balbi, A.; Anzaldi, M.; Macciò, C.; Aiello, C.; Mazzei, M.; Gangemi, R.;
Castagnola, P.; Miele, M.; Rosano, C.; Viale, M. Eur. J. Med. Chem. 2011, 46, 5293.16. Yamamoto, S.; Tomita, N.; Suzuki, Y.; Suzaki, T.; Kaku, T.; Hara, T.; Yamaoka,
M.; Kanzaki, N.; Hasuoka, A.; Baba, A.; Ito, M. Bioorg. Med. Chem. 2012, 20, 2338.17. Choi, W.-K.; El-Gamal, M. I.; Choi, H. S.; Baek, D.; Oh, C.-H. Eur. J. Med. Chem.
2011, 46, 5754.18. Zhao, Y.; Wang, G.; Dong, W.; Shi, Y.; Li, B.; Wang, S.; Li, Z. ARKIVOC 2010, ii, 16.19. Longhin Bosquesi, P.; Ferreira Melo, T. R.; Oliveira Vizioli, E.; dos Santos, J. L.;
Chung, M. C. Pharmaceuticals 2011, 4, 1450.20. Pluta, K.; Morak-Młodawska, B.; Jelen, M. Eur. J. Med. Chem. 2011, 46, 3179.21. Bansode, T. N.; Ansari, R. M.; Gawale, Y. K. J. Pharm. Res. 2011, 4, 1141.22. (a) Santucci, R.; Mackley, P. A.; Sebti, S.; Alsina, M. Cancer Control 2003, 10, 384;
(b) Rao, S.; Cunningham, D.; de Gramont, A.; Scheithauer, W.; Smakal, M.;Humblet, Y.; Kourteva, G.; Iveson, T.; Andre, T.; Dostalova, J.; Illes, A.; Belly, R.;Perez-Ruixo, J. J.; Park, Y. C.; Palmer, P. A. J. Clin. Oncol. 2004, 22, 3950; (c) Li, Q.;Li, T.; Woods, K. W.; Gu, W.-Z.; Cohen, J.; Stoll, V. S.; Galicia, T.; Hutchins, C.;Frost, D.; Rosenberg, S. H.; Sham, H. L. Bioorg. Med. Chem. Lett. 2005, 15, 2918.
23. Khuri, F. R.; Glisson, B. S.; Kim, E. S.; Statkevich, P.; Thall, P. F.; Meyers, M. L.;Herbst, R. S.; Munden, R. F.; Tendler, C.; Zhu, Y.; Bangert, S.; Thompson, E.; Lu,C.; Wang, X. M.; Shin, D. M.; Kies, M. S.; Papadimitrakopoulou, V.; Fossella, F.V.; Kirschmeier, P.; Bishop, W. R.; Hong, W. K. Clin. Cancer Res. 2004, 10, 2968.
24. Hunt, J. T.; Ding, C. Z.; Batorsky, R.; Bednarz, M.; Bhide, R.; Cho, Y.; Chong, S.;Chao, S.; Gullo-Brown, J.; Guo, P.; Kim, S. H.; Lee, F. Y.; Leftheris, K.; Miller, A.;Mitt, T.; Patel, M.; Penhallow, B. A.; Ricca, C.; Rose, W. C.; Schmidt, R.;Slusarchyk, W. A.; Vite, G.; Manne, V. J. Med. Chem. 2000, 43, 3587.
25. Baciu-Atudosie, L.; Ghinet, A.; Belei, D.; Gautret, P.; Rigo, B.; Bîcu, E. TetrahedronLett. Accepted manuscript, doi: http://dx.doi.org/10.1016/j.tetlet.2012.08.152.
26. Godefroi, E. F.; Wittle, E. L. J. Org. Chem. 1956, 21, 1163.27. (a) Hogale, M. B.; Deshmukh, D. S. J. Indian Chem. Soc. 1989, 66, 212–213; (b)
Srivastava, S. D.; Kohli, P. Proc. Natl. Acad. Sci., India, Sect. B 2007, 77, 199.28. (a) de Sousa, G. F.; Garcia, E.; Gatto, C. C.; Resck, I. S.; Deflon, V. M. J. Mol. Struct.
2010, 981, 46; (b) Zhao, Y.; Bacher, A.; Illarionov, B.; Fischer, M.; Georg, G.; Ye,Q.-Z.; Fanwick, P. E.; Franzblau, S. G.; Wan, B.; Cushman, M. J. Org. Chem. 2009,74, 5297; (c) Joshi, K. C.; Bohra, R.; Joshi, B. S. Inorg. Chem. 1992, 31, 598.
29. (a) Gupta, S.; Rodrigues, L. M.; Esteves, A. P.; Oliveira-Campos, A. M. F.;Nascimento, M. S. J.; Nazareth, N.; Cidade, H.; Neves, M. P.; Fernandes, E.; Pinto,M.; Cerqueira, N. M. F. S. A.; Brás, N. Eur. J. Med. Chem. 2008, 43, 771; (b)Oliveira-Campos, A. M. F.; Salaheldin, A. M.; Rodrigues, L. M. ARKIVOC 2007, xvi,92.
30. Farnesyltransferase assay: Assays were realized in 96-well plates, prepared witha Biomek NKMC and a Biomek 3000 from Beckman Coulter and read on aWallac Victor fluorimeter from Perkin-Elmer. Per well, 20 lL of farnesylpyrophosphate (10 lM) was added to 180 lL of a solution containing 2 lL ofvaried concentrations of potential inhibitors (dissolved in DMSO) and 178 lL ofa solution composed by 10 lL of partially purified recombinant human FTase(1.5 mg/mL) and 1.0 mL of Dansyl-GCVLS peptide (in the following buffer:5.6 mM DTT, 5.6 mM MgCl2, 12 lM ZnCl2 and 0.2% (w/v) octyl-ß-D-glucopyranoside, 52 mM Tris/HCl, pH 7.5). Fluorescence was recorded for15 min (0.7 s per well, 20 repeats) at 30 �C with an excitation filter at 340 nmand an emission filter of 486 nm. Each measurement was reproduced induplicate. The kinetic experiments were realized under the same conditions,either with FPP as varied substrate with a constant concentration of Dns-GCVLS of 2.5 lM, or with Dns-GCVLS as varied substrate with a constantconcentration of FPP of 10 lM. Non linear regressions were performed withKaleidaGraph 4.03 software. Coudray, L.; de Figueiredo, R. M.; Duez, S.; Cortial,S.; Dubois, J. J. Enz. Inh. Med. Chem. 2009, 24, 972.
31. Cell proliferation assay: Six compounds were tested against a panel of 60 humancancer cell lines at the National Cancer Institute, Bethesda, MD. Thecytotoxicity studies were conducted using a 48 h exposure protocol usingthe sulforhodamine B assay (a) Boyd, R. B. The NCI in vitro anticancer drugdiscovery screen. In Anticancer Drug Development Guide: Preclinical Screening,Clinical Trials, and Approval; Teicher, B., Ed.; Humana Press Inc. Totowa: NJ,1997; p 23; (b) Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.;Vistica, D.; Warren, J. T.; Bokesh, H.; Kenney, S.; Boyd, M. R. J. Natl. Cancer Inst.1990, 82, 1107.
32. Molecular modelling: Based on previous studies, 1LD7 (Bell, I. M.; Gallicchio, S.N.; Abrams, M.; Beese, L. S.; Beshore, D. C.; Bhimnathwala, H.; Bogusky, M. J.;Buser, C. A.; Culberson, J. C.; Davide, J.; Ellis-Hutchings, M.; Fernandes, C.;Gibbs, J. B.; Graham, S. L.; Hamilton, K. A.; Hartman, G. D.; Heimbrook, D. C.;Homnick, C. F.; Huber, H. E.; Huff, J. R.; Kassahun, K.; Koblan, K. S.; Kohl, N. E.;Lobell, R. B.; Lynch Jr., J. J.; Robinson, R.; Rodrigues, A. D.; Taylor, J. S.; Walsh, E.S.; Williams, T. M.; Zartman, C. B. J. Med. Chem. 2002, 45, 2388) was chosen dueto the structural similarity of the cocrystallized ((20S)-19,20,22,23-tetrahydro-19-oxo-5H,21H–18,20-ethano-12,14-etheno-6,10-methenobenz[D]imidazo[4,3-L][1,6,9,13]oxatriazacyclonoadecosine-9-carbonitrile) ligand with the compounds related in this work. The referenceligand and water molecules were removed, then the cocrystallized inhibitorwas repositioned into the binding site defined as a 10 Å sphere around itsposition in the crystal, needing a distance constraint to maintain proximity tothe zinc. This constraint was later withdrawn as metal atoms were directlytaken into account by newer versions docking software and the studiedcompounds became more remote from the cocrystallized inhibitor. Theprotonation state of the protein and ligands was found to be compatiblewith a pH of 7.5 and the geometry of the ligands optimized using Gasteiger-Hückel partial charges, with a formal charge of +2 applied to iron atoms and adielectric constant set to 4. After applying a formal charge of +2 on thefarnesyltransferase zinc, metal atoms were handled by GOLD with the defaultgeometry corresponding to their coordination. The 30 docking solutions wereranked based on a consensus scoring by GoldScore and X Score, then, visuallyinspected to assess the consistency of the best ranking conformation and selectthe most inhibitory clusters or conformation, when several were found. Millet,R.; Domarkas, J.; Houssin, R.; Gilleron, P.; Goossens, J.-F.; Chavatte, P.; Logé, C.;Pommery, N.; Pommery, J.; Hénichart, J.-P. J. Med. Chem. 2004, 47, 6812.
Copyright © 2022 FDOKUMEN













![4-[(2,4-Difluorophenyl)hydrazinylidene]-3-methyl-5-oxo-4,5-dihydro-1 H -pyrazole-1-carbothioamide](https://static.fdokumen.com/doc/165x107/63448600596bdb97a9087f01/4-24-difluorophenylhydrazinylidene-3-methyl-5-oxo-45-dihydro-1-h-pyrazole-1-carbothioamide.jpg)






