
ROS-independent preconditioning in neurons via activation of mitoKATP channels by BMS-191095
14
ROS-independent preconditioning in neurons via activation of mitoK ATP channels by BMS-191095 Tama ´s Ga ´spa ´r 1 , James A Snipes 1 , Anna R Busija 1 , Be ´la Kis 1 , Ferenc Domoki 1,2 , Ferenc Bari 2 and David W Busija 1 1 Department of Physiology and Pharmacology, Wake Forest University Health Sciences, Winston-Salem, North Carolina, USA; 2 Department of Physiology, Faculty of Medicine, University of Szeged, Szeged, Hungary Previously, we have shown that the selective mitochondrial ATP-sensitive potassium (mitoK ATP ) channel opener BMS-191095 (BMS) induces neuronal preconditioning (PC); however, the exact mechanism of BMS-induced neuroprotection remains unclear. In this study, we have identified key components of the cascade resulting in delayed neuronal PC with BMS using isolated rat brain mitochondria and primary cultures of rat cortical neurons. BMS depolarized isolated mitochondria without an increase in reactive oxygen species (ROS) generation and induced rapid phosphorylation of Akt and glycogen synthase kinase-3b. Long-term (3 days) treatment of neurons with BMS resulted in sustained mitochondrial depolarization, decreased basal ROS generation, and elevated ATP levels. This treatment also elicited almost complete protection against glutamate excitotoxicity, which could be abolished using the phosphoinositide 3-kinase (PI3K) inhibitor wortmannin, but not with the superoxide dismutase (SOD) mimetic M40401. Long-term BMS treatment induced a PI3K-dependent increase in the expression and activity of catalase without affecting manganese SOD and copper/zinc-dependent SOD. Finally, the catalase inhibitor 3-aminotriazole dose- dependently antagonized the neuroprotective effect of BMS-induced PC. In summary, BMS depolarizes mitochondria without ROS generation, activates the PI3K–Akt pathway, improves ATP content, and increases catalase expression. These mechanisms appear to play important roles in the neuroprotective effect of BMS. Journal of Cerebral Blood Flow & Metabolism (2008) 28, 1090–1103; doi:10.1038/sj.jcbfm.9600611; published online 30 January 2008 Keywords: mitochondria; mitoK ATP ; neuronal culture; neuroprotection; phosphoinositide 3-kinase; reactive oxygen species Introduction Stroke is a leading cause of death and permanent disability worldwide. Therefore, strategies to pre- vent or decrease the damage caused by prolonged ischemia are of great importance. One of the promising protective approaches is preconditioning (PC). This ubiquitous natural phenomenon was first described in the brain more than 40 years ago by Dahl and Balfour (1964) and was defined by Murry et al (1986) as an increased tolerance induced by a subtoxic stimulus (e.g., a short period of anoxia) against a subsequent, otherwise lethal insult such as prolonged ischemia. Preconditioning can be in- duced by several different approaches, including the selective pharmacological activation of the ATP- sensitive potassium (K ATP ) channel in the inner mitochondrial membrane (mitoK ATP ; reviewed by Busija et al, 2004). Diazoxide (DZ) is the proto- typical activator of the potassium channel and it has been used in most pharmacological PC studies. However, DZ is known to have effects unrelated to the mitoK ATP channels, such as the inhibition of the mitochondrial electron transport chain (Andrukhiv et al, 2006; Kis et al, 2003; Minners et al, 2007). Furthermore, in higher doses, DZ also opens plasma membrane K ATP channels (Garlid et al, 1996). There- fore, new and more selective activators are needed to further explore the protection mediated by mitoK ATP channels. BMS-191095 (BMS) is a novel opener of the mitoK ATP channels and it has shown itself to be more potent and selective than DZ Received 3 October 2007; revised 11 December 2007; accepted 26 December 2007; published online 30 January 2008 Correspondence: Dr T Ga ´spa ´r, Department of Physiology and Pharmacology, Wake Forest University Health Sciences, Medical Center Boulevard, Winston-Salem, NC 27157-1010, USA. E-mail: [email protected] This work was supported by the National Institutes of Health Grants (HL-077731, HL-030260, and HL-065380), YF Wu Research and Education Fund, WFUSM Venture Fund, KG Phillips Fund for the Prevention and Treatment of Heart Disease, and WFUSM Interim Funding for DW Busija and partly by the Hungarian Science Fund (OTKA K63401 and IN69967) for F Bari. Journal of Cerebral Blood Flow & Metabolism (2008) 28, 1090–1103 & 2008 ISCBFM All rights reserved 0271-678X/08 $30.00 www.jcbfm.com
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of ROS-independent preconditioning in neurons via activation of mitoKATP channels by BMS-191095

ROS-independent preconditioning in neurons viaactivation of mitoKATP channels by BMS-191095
Tamas Gaspar1, James A Snipes1, Anna R Busija1, Bela Kis1, Ferenc Domoki1,2, Ferenc Bari2
and David W Busija1
1Department of Physiology and Pharmacology, Wake Forest University Health Sciences, Winston-Salem,North Carolina, USA; 2Department of Physiology, Faculty of Medicine, University of Szeged, Szeged, Hungary
Previously, we have shown that the selective mitochondrial ATP-sensitive potassium (mitoKATP)channel opener BMS-191095 (BMS) induces neuronal preconditioning (PC); however, the exactmechanism of BMS-induced neuroprotection remains unclear. In this study, we have identified keycomponents of the cascade resulting in delayed neuronal PC with BMS using isolated rat brainmitochondria and primary cultures of rat cortical neurons. BMS depolarized isolated mitochondriawithout an increase in reactive oxygen species (ROS) generation and induced rapid phosphorylationof Akt and glycogen synthase kinase-3b. Long-term (3 days) treatment of neurons with BMS resultedin sustained mitochondrial depolarization, decreased basal ROS generation, and elevated ATPlevels. This treatment also elicited almost complete protection against glutamate excitotoxicity,which could be abolished using the phosphoinositide 3-kinase (PI3K) inhibitor wortmannin, but notwith the superoxide dismutase (SOD) mimetic M40401. Long-term BMS treatment induced aPI3K-dependent increase in the expression and activity of catalase without affecting manganeseSOD and copper/zinc-dependent SOD. Finally, the catalase inhibitor 3-aminotriazole dose-dependently antagonized the neuroprotective effect of BMS-induced PC. In summary, BMSdepolarizes mitochondria without ROS generation, activates the PI3K–Akt pathway, improves ATPcontent, and increases catalase expression. These mechanisms appear to play important roles inthe neuroprotective effect of BMS.Journal of Cerebral Blood Flow & Metabolism (2008) 28, 1090–1103; doi:10.1038/sj.jcbfm.9600611; published online30 January 2008
Keywords: mitochondria; mitoKATP; neuronal culture; neuroprotection; phosphoinositide 3-kinase; reactive oxygenspecies
Introduction
Stroke is a leading cause of death and permanentdisability worldwide. Therefore, strategies to pre-vent or decrease the damage caused by prolongedischemia are of great importance. One of thepromising protective approaches is preconditioning(PC). This ubiquitous natural phenomenon was firstdescribed in the brain more than 40 years ago byDahl and Balfour (1964) and was defined by Murry
et al (1986) as an increased tolerance induced by asubtoxic stimulus (e.g., a short period of anoxia)against a subsequent, otherwise lethal insult such asprolonged ischemia. Preconditioning can be in-duced by several different approaches, includingthe selective pharmacological activation of the ATP-sensitive potassium (KATP) channel in the innermitochondrial membrane (mitoKATP; reviewed byBusija et al, 2004). Diazoxide (DZ) is the proto-typical activator of the potassium channel and it hasbeen used in most pharmacological PC studies.However, DZ is known to have effects unrelated tothe mitoKATP channels, such as the inhibition of themitochondrial electron transport chain (Andrukhivet al, 2006; Kis et al, 2003; Minners et al, 2007).Furthermore, in higher doses, DZ also opens plasmamembrane KATP channels (Garlid et al, 1996). There-fore, new and more selective activators are neededto further explore the protection mediated bymitoKATP channels. BMS-191095 (BMS) is a novelopener of the mitoKATP channels and it has shownitself to be more potent and selective than DZ
Received 3 October 2007; revised 11 December 2007; accepted 26December 2007; published online 30 January 2008
Correspondence: Dr T Gaspar, Department of Physiology andPharmacology, Wake Forest University Health Sciences, MedicalCenter Boulevard, Winston-Salem, NC 27157-1010, USA.E-mail: [email protected]
This work was supported by the National Institutes of Health
Grants (HL-077731, HL-030260, and HL-065380), YF Wu Research
and Education Fund, WFUSM Venture Fund, KG Phillips Fund
for the Prevention and Treatment of Heart Disease, and WFUSM
Interim Funding for DW Busija and partly by the Hungarian
Science Fund (OTKA K63401 and IN69967) for F Bari.
Journal of Cerebral Blood Flow & Metabolism (2008) 28, 1090–1103& 2008 ISCBFM All rights reserved 0271-678X/08 $30.00
www.jcbfm.com

(Grover et al, 2001; Mayanagi et al, 2007). Theeffective dose of this compound for PC has novasoactivity and does not influence whole-cell K +
currents or plasma membrane potential. In previousreports from our laboratory, we have shown thatacute and delayed PC with BMS protected ratcortical neuronal cultures in vitro against toxicstimuli (Kis et al, 2004) and induced delayedtolerance against transient middle cerebral arteryocclusion in vivo (Mayanagi et al, 2007). In addition,BMS PC abolished the reactive oxygen species(ROS) surge that was induced by glutamate exposure(Kis et al, 2004). We have also reported that theapplication of BMS depolarized isolated piglet brainmitochondria without induction of ROS generation(Busija et al, 2005). BMS was also shown to induceboth acute and delayed cardioprotection against leftanterior descending coronary artery occlusion in therat heart (Gross et al, 2007; Wang et al, 2007b) by theactivation of the phosphoinositide 3-kinase (PI3K)signaling pathway. Despite these results, the exactmechanism of BMS-induced neuroprotection hasnot yet been clarified. Therefore, further studies areneeded to explore the details of immediate anddelayed PC induction by BMS.
The purpose of this study was to identify keycomponents of the cytoprotective cascade activatedby BMS, ultimately resulting in delayed neuronalPC. We analyzed the immediate and long-termeffects of BMS on isolated rat mitochondria andcultured rat cortical neurons, respectively. Further-more, we examined the role of the PI3K signalingpathway and the expression of several cytoprotec-tive proteins in the mediation of BMS-induceddelayed neuronal PC. Through these experiments,we have also shown that the PC due to activation ofmitoKATP channels by BMS is independent of ROSproduction.
Materials and methods
Materials
Cell culture plastics were purchased from Becton Dickinson(San Jose, CA, USA). Dulbecco’s modified Eagle medium,Neurobasal medium, B27 Supplement, 2-mercaptoethanol,and horse serum were obtained from Gibco BRL (GrandIsland, NY, USA). Percoll was purchased from AmershamBiosciences (Uppsala, Sweden), dispase I from Roche(Mannheim, Germany), and M40401 from MetaphorePharmaceuticals (St Louis, MO, USA). CellTiter-GloLuminescent Assay, CellTiter 96 AQueous One SolutionAssay, and SV Total RNA Isolation Kit were procured fromPromega (Madison, WI, USA). Hydroethidine (HEt),tetramethylrhodamine ethyl ester (TMRE), and AmplexRed Catalase Assay Kit were purchased from MolecularProbes (Eugene, OR, USA). The SOD Assay Kit waspurchased from Fluka (Buchs, Switzerland). Taqman GeneExpression Assays for catalase (assay identification no.Rn00560930_m1), manganese-dependent superoxide dis-mutase (MnSOD; Rn00566942_g1), and b-actin (4352340E)
were procured from Applied Biosystems (Foster City, CA,USA). Antibodies were obtained from the followingsources: anti-glial fibrillary acidic protein antibody fromChemicon (Temecula, CA, USA); anti-microtubule-asso-ciated protein-2, monoclonal anti-MnSOD, monoclonalprotein kinase B (PKB)a/Akt antibodies from BectonDickinson; polyclonal anti-catalase, and polyclonal anti-copper and zinc-dependent SOD antibodies from Calbio-chem (San Diego, CA, USA); polyclonal anti-pS473 Aktantibody from Promega; polyclonal anti-phospho-glycogensynthase kinase 3a/b (Gsk3a/b) (Ser 21/9) antibody andmonoclonal anti-Gsk3b antibody from Cell SignalingTechnology (Danvers, MA, USA); and anti-rabbit IgG andanti-mouse IgG from Jackson ImmunoResearch (WestGrove, PA, USA). All other chemicals were from Sigma(St Louis, MO, USA).
Primary Rat Cortical Neuronal Culture
Timed pregnant Sprague–Dawley (SD) rats were obtainedfrom Harlan (Indianapolis, IN, USA) and were maintainedand used in compliance with the principles set forth bythe Animal Care and Use Committee of Wake ForestUniversity Health Sciences. Primary rat cortical neuronswere isolated from E18 SD fetuses as described in Kis et al(2003). The cells were plated at a density of 2� 105 cells/cm2 onto poly-D-lysine-coated glass coverslips for confocalmicroscopic analysis, whereas 106 cells/cm2 were placedonto poly-D-lysine-coated plates or dishes for the otherexperiments in plating medium, which consisted of 60%Dulbecco’s modified Eagle medium, 20% Ham’s F-12Nutrient Mixture, 20% horse serum, and L-glutamine(0.5 mmol/L). After cell attachment, the plating mediumwas replaced with Neurobasal medium supplementedwith B27 (2%), L-glutamine (0.5 mmol/L), 2-mercaptoetha-nol (55 mmol/L), and KCl (25 mmol/L). Positive immunos-taining for microtubule-associated protein-2 and negativeimmunostaining for glial fibrillary acidic protein verifiedthat the cultures consisted of more than 98% of neuronson day 7 in vitro. Experiments were carried out on 7- to9-day-old cultures, during which period neurons ex-pressed N-methyl-D-aspartate, a-amino-3-hydroxy-5-methylisoxazole-4-propionate, and kainate receptors andwere vulnerable to glucose deprivation (Mattson et al,1991, 1993).
Treatment with BMS
The time line of the experiments is shown in Figure 1A.To induce delayed PC, 7-day-old neuronal cultureswere treated with BMS (30 mmol/L) in the regular culturemedium once a day for three consecutive days. The doseof BMS was based on that used in previous studiesfrom our laboratory in which the dose-dependency ofBMS-induced neuroprotection was examined (Kis et al,2004). In some of the experiments, the cells were cotreatedwith BMS (30 mmol/L) and the putative selective mitoKATP
blocker 5-hydroxydecanoic acid (5HD), the protein kinaseC (PKC) inhibitor chelerythrine (Chel), the SOD mimeticM40401, the large conductance Ca2 +-activated K + (KCa)
ROS-independent preconditioning by BMS-191095T Gaspar et al
1091
Journal of Cerebral Blood Flow & Metabolism (2008) 28, 1090–1103
channel antagonist paxilline (Pax), or the PI3K inhibitorwortmannin (Wrt). At the end of the treatments, all drugswere washed out thoroughly with phosphate-bufferedsaline (PBS), and the cells were then maintained in theregular cell culture medium.
Glutamate Excitotoxicity
Cell cultures in 96-well plates were exposed to glutamate(200 mmol/L) 24 h after the initiation of the last BMStreatment in the regular culture medium for 60 mins at371C in the 5% CO2 incubator. Afterward, the cells wererinsed and returned to the 5% CO2 incubator in regularculture medium. In some experiments, different concen-trations of the catalase inhibitor 3-aminotriazole (3AT; 1,10, 100 mmol/L) were applied to the cultures during andfor 3 h after glutamate excitotoxicity.
Quantification of Neuronal Survival
The cell viability in neuronal cultures was determined24 h after the neurotoxic insults using the tetrazolium-based CellTiter 96 AQueous One Solution Assay, asdescribed previously (Kis et al, 2003). Absorbance at492 nm was measured with a microplate reader (FLUOstarOPTIMA, BMG Labtech GmbH, Offenburg, Germany).Comparisons were always made in the same manner,between sister cultures exposed to the neurotoxic stimu-lus on the same day, and cell viability was expressed asa percentage of the corresponding control culture (un-treated and not exposed to the lethal insult) as follows: %viability = (absorbanceSAMPLE�absorbanceBACKGROUND)� 100/(absorbanceCONTROL�absorbanceBACKGROUND).
Isolation of Rat Brain Mitochondria
Brain mitochondria were isolated using a discontinuousPercoll gradient as described previously, with somemodifications (Rajapakse et al, 2001; Sims, 1990). Allthe instruments and buffers were kept ice-cold duringthe procedure. Male SD rats (280 to 320 g; Harlan) wereoveranesthetized with isoflurane and decapitated. Thebrain, without the cerebellum, was removed, weighed,and then homogenized in mitochondrial isolation buffer(MIB; sucrose, 250 mmol/L; K+-EDTA (ethylenediaminete-traacetic acid), 0.5 mmol/L; Tris-HCl (pH 7.4), 10 mmol/L;bovine serum albumin, 1%) using Dounce homogenizers
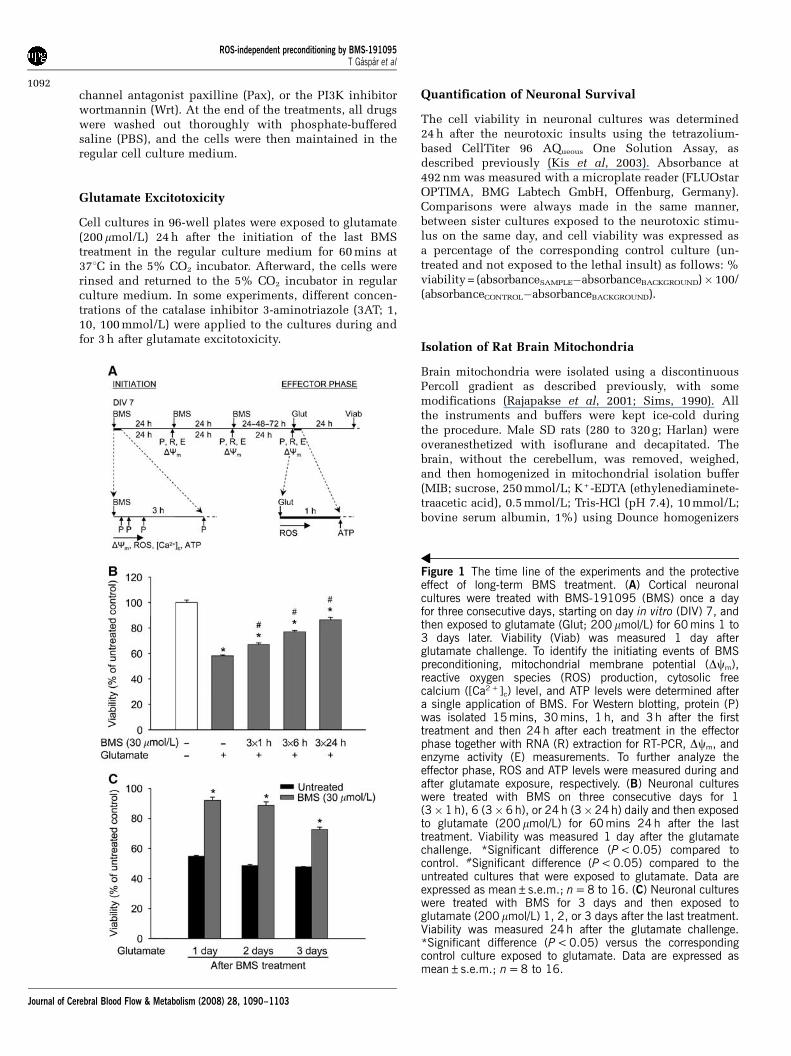
Figure 1 The time line of the experiments and the protectiveeffect of long-term BMS treatment. (A) Cortical neuronalcultures were treated with BMS-191095 (BMS) once a dayfor three consecutive days, starting on day in vitro (DIV) 7, andthen exposed to glutamate (Glut; 200 mmol/L) for 60 mins 1 to3 days later. Viability (Viab) was measured 1 day afterglutamate challenge. To identify the initiating events of BMSpreconditioning, mitochondrial membrane potential (Dcm),reactive oxygen species (ROS) production, cytosolic freecalcium ([Ca2 +]c) level, and ATP levels were determined aftera single application of BMS. For Western blotting, protein (P)was isolated 15 mins, 30 mins, 1 h, and 3 h after the firsttreatment and then 24 h after each treatment in the effectorphase together with RNA (R) extraction for RT-PCR, Dcm, andenzyme activity (E) measurements. To further analyze theeffector phase, ROS and ATP levels were measured during andafter glutamate exposure, respectively. (B) Neuronal cultureswere treated with BMS on three consecutive days for 1(3�1 h), 6 (3�6 h), or 24 h (3�24 h) daily and then exposedto glutamate (200 mmol/L) for 60 mins 24 h after the lasttreatment. Viability was measured 1 day after the glutamatechallenge. *Significant difference (P < 0.05) compared tocontrol. #Significant difference (P < 0.05) compared to theuntreated cultures that were exposed to glutamate. Data areexpressed as mean±s.e.m.; n = 8 to 16. (C) Neuronal cultureswere treated with BMS for 3 days and then exposed toglutamate (200 mmol/L) 1, 2, or 3 days after the last treatment.Viability was measured 24 h after the glutamate challenge.*Significant difference (P < 0.05) versus the correspondingcontrol culture exposed to glutamate. Data are expressed asmean±s.e.m.; n = 8 to 16.
ROS-independent preconditioning by BMS-191095T Gaspar et al
1092
Journal of Cerebral Blood Flow & Metabolism (2008) 28, 1090–1103

and glass pestles (Kontes Glass Co., Vineland, NJ, USA).The homogenate was centrifuged for 3 mins at 500g andthen the supernatant was collected and resuspended inequal amounts of 24% Percoll in MIB. This suspensionwas layered onto a discontinuous Percoll gradient (24%and 40% in MIB). The gradient was centrifuged for 5 minsat 28,000g, and the layer between the 24% and 40% Percollsuspensions containing the purified mitochondria wascollected. The preparation was washed in MIB andcentrifuged for 12 mins at 15,000g. For fluorescent platereader measurements, the protein content was determinedand then equal amounts of mitochondria were transferredin MIB into black-walled, clear-bottomed 96-well plates.
Analysis of Reactive Oxygen Species Formation
Reactive oxygen species generation was assessed inblack-walled 96-well plates with HEt using the samemicroplate reader used for the viability measurements(lex = 510, lem = 590 nm). Isolated mitochondria werewashed and then loaded with HEt (5mmol/L) and BMS(10 to 40 mmol/L) or DZ (500 mmol/L) in MIB 1 min beforethe assay. Hydroethidine fluorescence in each well wasmeasured at 371C every minute for 30 mins. Data wereexpressed as a percentage of the starting intensity of theuntreated control: % HEt fluorescenceSAMPLE = (HEtfluorescenceSAMPLE�HEt fluorescenceBACKGROUND)� 100/(HEtfluorescenceCONTROL AT TIME 0�HEt fluorescenceBACKGROUND).
To assess the delayed effects of BMS treatment, neuronalcultures were treated with BMS for 3 days. On the dayafter the last treatment, the cells were loaded with HEt(5mmol/L) and subjected to glutamate (200mmol/L). Theintensity of HEt fluorescence was measured at 371C everyminute for 30 mins. The change of HEt fluorescence(steepness of the curves) was calculated using the follow-ing equation: HEt fluorescenceSAMPLE in relative fluores-cent units/min (DRFU/min) = (HEt fluorescenceSAMPLE at30 mins�HEt fluorescenceSAMPLE at 0 min)/30.
Determination of Mitochondrial Membrane Potential
The changes of mitochondrial membrane potential (Dcm)were analyzed using the Dcm-sensitive dye TMRE.Measurements were performed in PBS containing1 mg/mL glucose (neurons) or in MIB (mitochondria) at371C. To measure immediate changes, isolated mitochon-dria in MIB were loaded with BMS (10 to 40 mmol/L) orwith DZ (500 mmol/L) and TMRE (0.5 mmol/L) for 20 minsat 41C in the dark after which the drugs were washed outthoroughly. To analyze the delayed effects of BMS on Dcm,neuronal cultures in black-walled, clear-bottomed 96-wellplates were treated with BMS (30mmol/L) and 5HD (1,5 mmol/L) once a day for 1 to 3 days. Twenty-four hoursafter the last treatment, the cells were loaded withTMRE (0.5 mmol/L) at 371C in a 5% CO2 incubator. After20 mins, the neurons were washed with PBS, and TMREfluorescence was measured in each well using thesame microplate reader, which was used for viabilitymeasurements (lex = 510 nm, lem = 590 nm). Data wereexpressed as a percentage of the intensity of the untreated
control culture: % TMRE fluorescenceSAMPLE = (TMREfluorescenceSAMPLE�TMRE fluorescenceBACKGROUND)� 100/(TMRE fluorescenceCONTROL�TMRE fluorescenceBACKGROUND).
Monitoring Free Cytosolic Calcium Levels
Changes of free cytosolic calcium levels ([Ca2 + ]c) weremonitored using the Ca indicator dye Fluo-4 AM inglucose containing (1 mg/ml) PBS. Neuronal cultures wereloaded with 2 mmol/L Fluo-4 AM and 1 mmol/L PluronicF-127 in PBS in the dark for 60 mins at room temperature(211C) and then washed three times. BMS (30 mmol/L) wasmixed with the buffer, and confocal images of cellularFluo-4 AM fluorescence (lex = 488 nm, lem = 520 nm) wereacquired using a laser scanning microscope (LSM 510;Zeiss, Jena, Germany) with a � 63 water immersionobjective (Zeiss). Images were recorded every 20 secs for10 mins, and the average pixel intensity of individual cellbodies was determined using the software supplied by themanufacturer (Zeiss). Data were expressed as a percentageof the starting intensity of the untreated control culture: %Fluo-4 fluorescenceSAMPLE = (Fluo-4 fluorescenceSAMPLE�Fluo-4 fluorescenceBACKGROUND)� 100/(Fluo-4 fluorescenceCONTROL
AT TIME 0�Fluo-4 fluorescenceBACKGROUND).
ATP Assay
The ATP level of neurons was measured with the glow-type CellTiter-Glo Luminescent Assay, as directed by themanufacturer. Cortical neurons cultured in opaque-walled96-well plates were equilibrated to room temperature(211C) for 30 mins. CellTiter-Glo was added to each well,and the plates were incubated at room temperature for10 mins to stabilize the luminescent signal, which wasthen measured with a FLUOstar OPTIMA microplatereader. An ATP standard curve was generated for eachmeasurement to calculate the ATP contents of wells.
Western Blotting for Catalase, Manganese-DependentSuperoxide Dismutase, Copper and Zinc-DependentSuperoxide Dismutase, Total and Phosphorylated Akt,and Glycogen Synthase Kinase-3b
Cultured cells were washed twice in ice-cold PBS andthen harvested by scraping in ice-cold Nonidet P40 lysisbuffer supplemented with proteinase inhibitors (1 mg/mLaprotinin, 50 mg/mL phenylmethylsulfonyl fluoride,and 1 mg/mL leupeptin) and a phosphatase inhibitorcocktail (1 mmol/L EDTA, 1 mmol/L sodium orthovana-date, 10 mg/mL benzamidine, 1 mmol/L sodium pyropho-sphate, and 1 mmol/L sodium fluoride). Equal amounts ofprotein for each sample were separated by 10% sodiumdodecyl sulfate-polyacrylamide gel electrophoresis andtransferred onto a polyvinylidine difluoride sheet (Poly-screen PVDF; Perkin Elmer Life Sciences, Boston, MA,USA). Membranes were incubated in a blocking buffer(Tris-buffered saline, 0.1% Tween 20, and 5% skimmedmilk powder) for 1 h at room temperature after which theblots were incubated with monoclonal anti-MnSOD
ROS-independent preconditioning by BMS-191095T Gaspar et al
1093
Journal of Cerebral Blood Flow & Metabolism (2008) 28, 1090–1103

(1:1,000), polyclonal anti-copper and zinc-dependentsuperoxide dismutase (CuZnSOD) (1:1,000), polyclonalanti-catalase (1:4,000), monoclonal anti-Akt (1:1,000),polyclonal anti-pS473 Akt (1:2,500), monoclonal anti-Gsk3b (1:1,000), and polyclonal anti-phospho-Gsk3a/b(1:1,000) antibodies overnight at 41C. The membraneswere then washed three times in Tris-buffered saline with0.1% Tween 20 and incubated for 1 h in the blockingbuffer with anti-rabbit IgG (1:50,000) or anti-mouse IgG(1:5,000) conjugated to horseradish peroxidase. The finalreaction products were visualized using enhanced chemi-luminescence (SuperSignal West Pico; Pierce, Rockford,IL, USA) and recorded on X-ray film.
Real-Time Reverse Transcription-PCR for Catalaseand Manganese-Dependent Superoxide Dismutase
RNA was isolated from neurons using the SV Total RNAIsolation Kit as directed by the manufacturer. The RNAwas incubated with DNase to eliminate any residualDNA that might amplify during PCR. Real-time quantita-tive reverse transcription-PCR (RT-PCR) experiments wereperformed in the ABI/Prism 7000 Sequence DetectionSystem (Applied Biosystems). From each sample, 25 pgtotal RNA was reverse transcribed and amplified usingQuantiTect Probe RT-PCR Kit (Qiagen, Valencia, CA, USA)and cycle program as follows: 30 mins at 501C, 15 mins at951C, and then 40 cycles each at 941C for 15 secs and 601Cfor 60 secs. All amplifications were conducted with thePre-Developed TaqMan Gene Expression Assays (AppliedBiosystems). All reactions were performed in triplicatewith b-actin serving as an internal control. Relativequantification of the mRNA expression levels of targetgenes was calculated using the 2�DDCT method (Schmittgenet al, 2000).
Catalase Assay
Catalase activity was assessed with the Amplex RedCatalase Activity Assay Kit. Neurons on 96-well plateswere pretreated with BMS (30mmol/L) for 1, 2, or 3 daysand then, 24 h after the last treatment, the plates wererinsed three times with PBS and enzyme activity wasmeasured with the same microplate reader that was usedfor other fluorescent measurements (lex = 555 nm andlem = 590 nm). A standard curve was generated for eachmeasurement to calculate the catalase activity of wells.Data were expressed as a percentage of the activity of theuntreated control culture using the following equation: %catalase activitySAMPLE = catalase activitySAMPLE� 100/catalase activityCONTROL.
Enzyme Activity Assay for Manganese-DependentSuperoxide Dismutase
Manganese-dependent superoxide dismutase activitywas measured using the tetrazolium-based SOD AssayKit as directed by the manufacturer. Neurons on 96-wellplates were pretreated with BMS (30 mmol/L) for 1, 2, or 3
days, then 24 h after the last treatment, the cells wereincubated with NaCN (5 mmol/L) for 10 mins to inhibitCuZnSOD activity. Subsequently, the plates were rinsedthree times with PBS and enzyme activity was measuredwith the same microplate reader that was used forthe viability measurements (labs = 460 nm). Data wereexpressed as a percentage of the activity of the untreatedcontrol culture using the following equations:
SOD activity SAMPLE¼ f½ðabsorbancePOSITIVE CONTROL
�absorbanceNEGATIVE CONTROLÞ�ðabsorbanceSAMPLE
�absorbanceBACKGROUNDÞ�=ðabsorbancePOSITIVE CONTROL
�absorbanceNEGATIVE CONTROLÞg�100
and
%SOD activitySAMPLE
¼ SOD activitySAMPLE�100=SOD activityCONTROL
Statistical Analysis
Statistical analyses were performed with SigmaStat (SPSS,Chicago, IL, USA). Data are presented as means±s.e.m.Differences between groups were assessed by one-wayanalysis of variance followed by Tukey comparison tests.A value of P < 0.05 was considered to be statisticallysignificant.
Results
Long-Term Treatment with BMS Induced ProlongedProtection against Glutamate Excitotoxicity
Treatment of neurons with BMS during glutamateexposure did not provide protection (viability:untreated, 64.00%±0.83%; BMS 30 mmol/L,65.4%±0.70%, data expressed as mean±s.e.m.,n = 8 to 16). One, six, and twenty-four hours dailytreatments for 3 days with 30 mmol/L BMS inducedincreasing tolerance against glutamate excitotoxicitywith the best protection after the longest dailytreatment (Figure 1B). Therefore, the 24-h protocolwas chosen as a long-term treatment paradigmfor the further experiments. This paradigm didnot influence the viability of quiescent neurons(untreated, 100.00%±1.40%; BMS 30 mmol/L,102.99%±0.69%, data expressed as mean±s.e.m.,n = 32 in each group), but the protection againstglutamate exposure was still apparent 3 days afterthe last treatment (Figure 1C).
BMS Depolarized Mitochondria
Exposure of isolated rat brain mitochondria toincreasing concentrations of BMS resulted inan immediate dose-dependent depolarization,as shown by the decrease of TMRE fluorescenceintensity (Figure 2A). DZ (500 mmol/L) was usedas a positive control.
ROS-independent preconditioning by BMS-191095T Gaspar et al
1094
Journal of Cerebral Blood Flow & Metabolism (2008) 28, 1090–1103
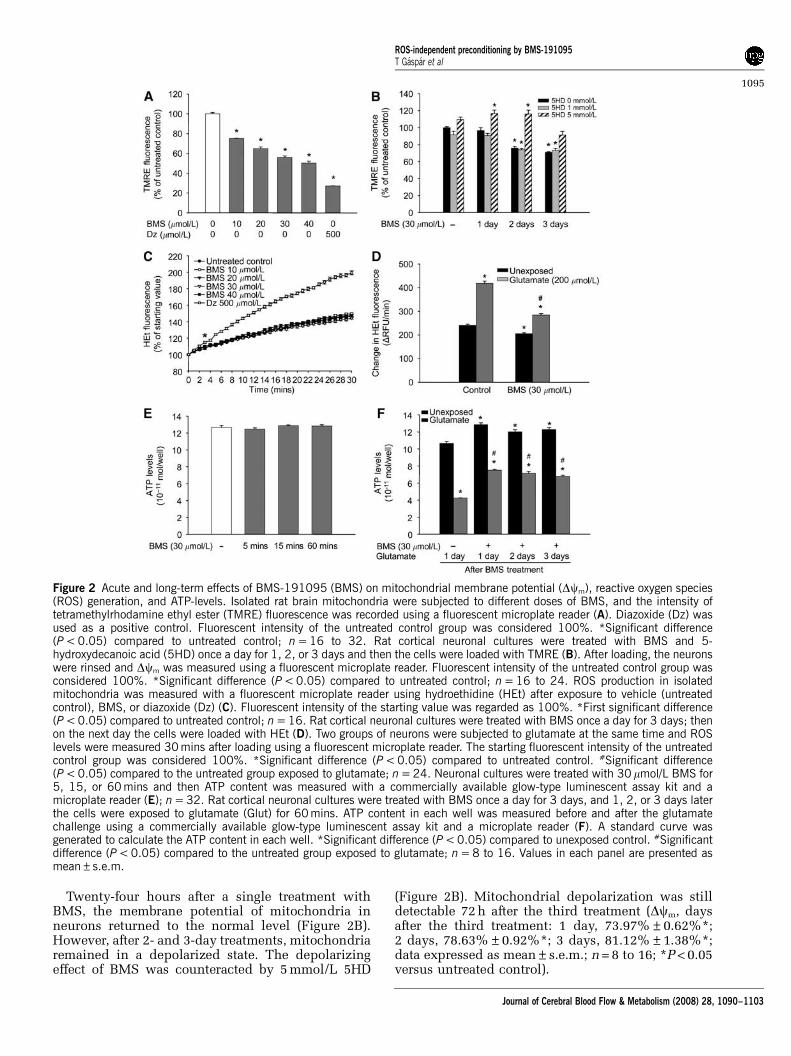
Twenty-four hours after a single treatment withBMS, the membrane potential of mitochondria inneurons returned to the normal level (Figure 2B).However, after 2- and 3-day treatments, mitochondriaremained in a depolarized state. The depolarizingeffect of BMS was counteracted by 5 mmol/L 5HD
(Figure 2B). Mitochondrial depolarization was stilldetectable 72 h after the third treatment (Dcm, daysafter the third treatment: 1 day, 73.97%±0.62%*;2 days, 78.63%±0.92%*; 3 days, 81.12%±1.38%*;data expressed as mean±s.e.m.; n = 8 to 16; *P < 0.05versus untreated control).
Figure 2 Acute and long-term effects of BMS-191095 (BMS) on mitochondrial membrane potential (Dcm), reactive oxygen species(ROS) generation, and ATP-levels. Isolated rat brain mitochondria were subjected to different doses of BMS, and the intensity oftetramethylrhodamine ethyl ester (TMRE) fluorescence was recorded using a fluorescent microplate reader (A). Diazoxide (Dz) wasused as a positive control. Fluorescent intensity of the untreated control group was considered 100%. *Significant difference(P < 0.05) compared to untreated control; n = 16 to 32. Rat cortical neuronal cultures were treated with BMS and 5-hydroxydecanoic acid (5HD) once a day for 1, 2, or 3 days and then the cells were loaded with TMRE (B). After loading, the neuronswere rinsed and Dcm was measured using a fluorescent microplate reader. Fluorescent intensity of the untreated control group wasconsidered 100%. *Significant difference (P < 0.05) compared to untreated control; n = 16 to 24. ROS production in isolatedmitochondria was measured with a fluorescent microplate reader using hydroethidine (HEt) after exposure to vehicle (untreatedcontrol), BMS, or diazoxide (Dz) (C). Fluorescent intensity of the starting value was regarded as 100%. *First significant difference(P < 0.05) compared to untreated control; n = 16. Rat cortical neuronal cultures were treated with BMS once a day for 3 days; thenon the next day the cells were loaded with HEt (D). Two groups of neurons were subjected to glutamate at the same time and ROSlevels were measured 30 mins after loading using a fluorescent microplate reader. The starting fluorescent intensity of the untreatedcontrol group was considered 100%. *Significant difference (P < 0.05) compared to untreated control. #Significant difference(P < 0.05) compared to the untreated group exposed to glutamate; n = 24. Neuronal cultures were treated with 30 mmol/L BMS for5, 15, or 60 mins and then ATP content was measured with a commercially available glow-type luminescent assay kit and amicroplate reader (E); n = 32. Rat cortical neuronal cultures were treated with BMS once a day for 3 days, and 1, 2, or 3 days laterthe cells were exposed to glutamate (Glut) for 60 mins. ATP content in each well was measured before and after the glutamatechallenge using a commercially available glow-type luminescent assay kit and a microplate reader (F). A standard curve wasgenerated to calculate the ATP content in each well. *Significant difference (P < 0.05) compared to unexposed control. #Significantdifference (P < 0.05) compared to the untreated group exposed to glutamate; n = 8 to 16. Values in each panel are presented asmean±s.e.m.
ROS-independent preconditioning by BMS-191095T Gaspar et al
1095
Journal of Cerebral Blood Flow & Metabolism (2008) 28, 1090–1103

The Effect of BMS on Reactive Oxygen SpeciesGeneration
The moderately increasing baseline curve of HEtfluorescence showed continuous basal ROS genera-tion in mitochondria (Figure 2C). Administration ofincreasing doses of BMS (10 to 40 mmol/L) did notchange the kinetics of ROS production of mitochon-dria. DZ (500 mmol/L), which is known to induceROS generation, was used as a positive control toshow the viability of mitochondrial preparations.
Compared with the untreated control group, long-term BMS treatment resulted in significantlyreduced basal ROS generation rate (Figure 2D) incultured neurons. Exposure to glutamate (200 mmol/L, 30 mins) induced a ROS surge in both untreatedand BMS-treated neurons. However, although therate of ROS generation doubled in control neurons,the increase in the BMS-treated group was markedlyless (Figure 2D).
BMS Treatment Improved ATP Homeostasis andUtilization in Neurons
Treatment with BMS (30 mmol/L) for 5, 15, or60 mins did not change ATP levels of neurons(Figure 2E). Conversely, neurons treated with BMSfor three consecutive days showed significantlyhigher ATP content than control cells. Moreover,increased ATP content of BMS-treated cells waspreserved for 3 days (Figure 2F). The ATP content ofcontrol groups showed a significant decrease afterglutamate challenge (200 mmol/L, 60 mins). In theBMS-treated group, the decrease was markedly lessand BMS-treated neurons contained almost twice asmuch ATP as the control neurons at the end of the60-min glutamate exposure.
BMS Application Induced an Increase in the FreeCytosolic Ca2 + Level of Neurons
In quiescent cells, the intensity of Fluo-4 fluores-cence did not change over time (Figure 3). Admin-istration of 30 mmol/L BMS resulted in anapproximate two-fold increase in the signal. Tenminutes later, at the end of the measurement,however, [Ca2 + ]c of neurons almost reached theresting level (Figure 3). A similar response in [Ca2 +]c
levels was seen using DZ and a more robust increasewith the KCa channel opener NS1619 (data notshown).
BMS Increased Akt and Glycogen Synthase Kinase-3bPhosphorylation
Western blot analysis with the anti-pS473 Akt anti-body revealed increased phosphorylation of Aktwithin 15 mins after the initiation of BMS treatment(Figure 4A). However, 3 h after the beginning ofthe incubation, the level of phosphorylated Akt
returned to the baseline. Phosphorylation (i.e.,inhibition) of Gsk3b showed a significant increase15 mins after BMS treatment, and it reached its peakat 30 mins. At the end of the measurement (3 h afterthe application of BMS), the level of the phosphory-lated protein was still very high (Figure 4B). Oneday after a single treatment with BMS, phosphoryla-tion levels of both Akt and Gsk3b returned tobaseline, and no further changes were detected24 h after 3-day treatments, at the time of glutamatechallenge (data not shown).
Phosphoinositide 3-Kinase Inhibition did notAntagonize Mitochondrial Depolarization
To determine whether PI3K activation is a cause or aconsequence of the mitoKATP opening effect of BMS,we loaded neuronal cultures with TMRE in thepresence of BMS (30 mmol/L) and Wrt (30, 100 nmol/L) for 20 mins. Wrt did not change Dcm and did notalter the depolarizing effect of BMS (Dcm: control,100.0%±1.65%; Wrt 30 nmol/L, 100.9%±1.09%,Wrt 100 nmol/L, 101.4%±1.34%, BMS 30 mmol/L,65.6%±0.94%*; BMS 30 mmol/L +Wrt 30 nmol/L,63.9%±0.82%*; BMS 30 mmol/L +Wrt 100 nmol/L,66.4%±0.89%*; data expressed as mean±s.e.m.;n = 16 in each group; *P < 0.05 versus control).
Wortmannin, but not M40401, and PaxillineAntagonized the Neuroprotective Effect of BMS
The PI3K inhibitor Wrt (10 to 100 nmol/L) had noeffect on the survival of neurons when used once aday for 3 days (Figure 5A). Furthermore, Wrt did notinfluence cell survival after exposure to glutamate
Figure 3 Administration of BMS-191095 (BMS) inducestransient increase in cytosolic free Ca2 + [Ca2 +]c levels inneurons. Neuronal cultures on poly-D-lysine-coated glasscoverslips were loaded with Fluo-4 and then the changes offree cellular calcium levels were analyzed with confocal laserscanning microscopy. Fluo-4 fluorescence was recorded inresting cells and after the application of 30 mmol/L BMS.Fluorescent intensity of the starting value of the untreatedcontrol group was considered 100%. *First significantdifference (P < 0.05) compared to untreated control; data areshown as mean±s.e.m.; n = 37 to 49.
ROS-independent preconditioning by BMS-191095T Gaspar et al
1096
Journal of Cerebral Blood Flow & Metabolism (2008) 28, 1090–1103
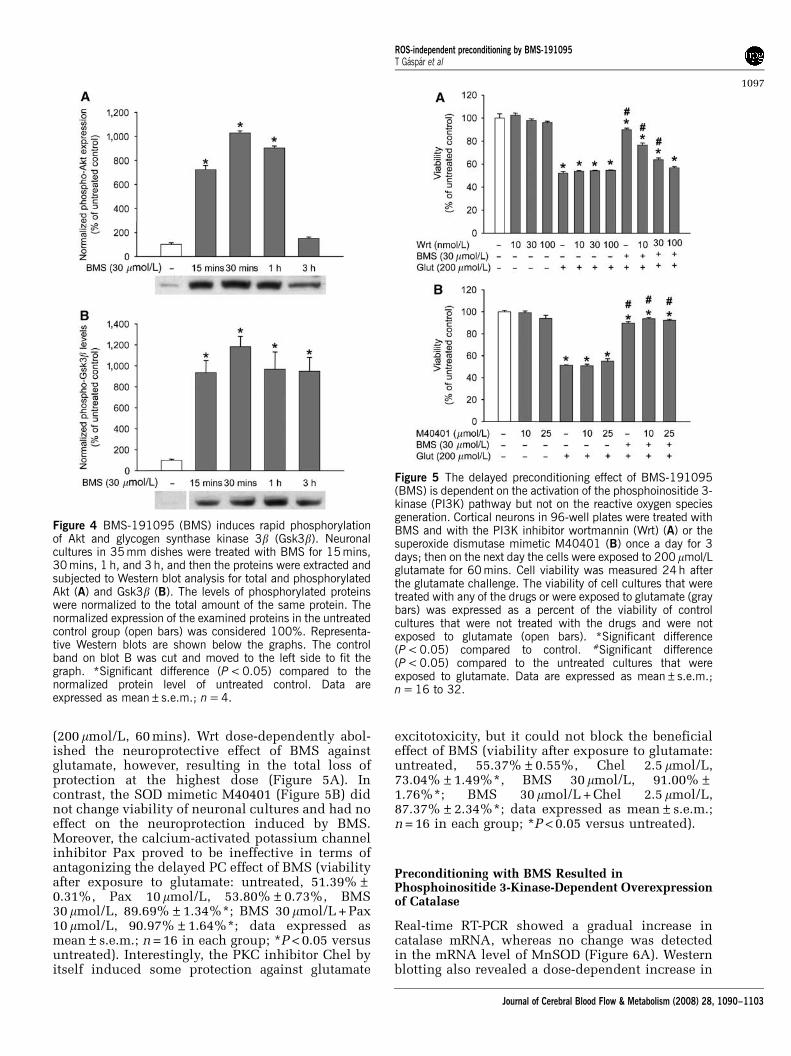
(200 mmol/L, 60 mins). Wrt dose-dependently abol-ished the neuroprotective effect of BMS againstglutamate, however, resulting in the total loss ofprotection at the highest dose (Figure 5A). Incontrast, the SOD mimetic M40401 (Figure 5B) didnot change viability of neuronal cultures and had noeffect on the neuroprotection induced by BMS.Moreover, the calcium-activated potassium channelinhibitor Pax proved to be ineffective in terms ofantagonizing the delayed PC effect of BMS (viabilityafter exposure to glutamate: untreated, 51.39%±0.31%, Pax 10mmol/L, 53.80%±0.73%, BMS30 mmol/L, 89.69%±1.34%*; BMS 30 mmol/L + Pax10 mmol/L, 90.97%±1.64%*; data expressed asmean±s.e.m.; n = 16 in each group; *P < 0.05 versusuntreated). Interestingly, the PKC inhibitor Chel byitself induced some protection against glutamate
excitotoxicity, but it could not block the beneficialeffect of BMS (viability after exposure to glutamate:untreated, 55.37%±0.55%, Chel 2.5 mmol/L,73.04%±1.49%*, BMS 30 mmol/L, 91.00%±1.76%*; BMS 30 mmol/L + Chel 2.5 mmol/L,87.37%±2.34%*; data expressed as mean±s.e.m.;n = 16 in each group; *P < 0.05 versus untreated).
Preconditioning with BMS Resulted inPhosphoinositide 3-Kinase-Dependent Overexpressionof Catalase
Real-time RT-PCR showed a gradual increase incatalase mRNA, whereas no change was detectedin the mRNA level of MnSOD (Figure 6A). Westernblotting also revealed a dose-dependent increase in
Figure 4 BMS-191095 (BMS) induces rapid phosphorylationof Akt and glycogen synthase kinase 3b (Gsk3b). Neuronalcultures in 35 mm dishes were treated with BMS for 15 mins,30 mins, 1 h, and 3 h, and then the proteins were extracted andsubjected to Western blot analysis for total and phosphorylatedAkt (A) and Gsk3b (B). The levels of phosphorylated proteinswere normalized to the total amount of the same protein. Thenormalized expression of the examined proteins in the untreatedcontrol group (open bars) was considered 100%. Representa-tive Western blots are shown below the graphs. The controlband on blot B was cut and moved to the left side to fit thegraph. *Significant difference (P < 0.05) compared to thenormalized protein level of untreated control. Data areexpressed as mean±s.e.m.; n = 4.
Figure 5 The delayed preconditioning effect of BMS-191095(BMS) is dependent on the activation of the phosphoinositide 3-kinase (PI3K) pathway but not on the reactive oxygen speciesgeneration. Cortical neurons in 96-well plates were treated withBMS and with the PI3K inhibitor wortmannin (Wrt) (A) or thesuperoxide dismutase mimetic M40401 (B) once a day for 3days; then on the next day the cells were exposed to 200 mmol/Lglutamate for 60 mins. Cell viability was measured 24 h afterthe glutamate challenge. The viability of cell cultures that weretreated with any of the drugs or were exposed to glutamate (graybars) was expressed as a percent of the viability of controlcultures that were not treated with the drugs and were notexposed to glutamate (open bars). *Significant difference(P < 0.05) compared to control. #Significant difference(P < 0.05) compared to the untreated cultures that wereexposed to glutamate. Data are expressed as mean±s.e.m.;n = 16 to 32.
ROS-independent preconditioning by BMS-191095T Gaspar et al
1097
Journal of Cerebral Blood Flow & Metabolism (2008) 28, 1090–1103

the expression of catalase after 1, 2, and 3 days ofBMS treatment (Figure 6B). The same treatment,however, did not induce any changes in the proteinlevels of MnSOD and CuZnSOD (data not shown).Similar to its mRNA and protein levels, the activityof catalase increased as well (untreated control,100.00%±3.73%; BMS 30 mmol/L for 3 days,169.23%±2.67%*, data expressed as mean±s.e.m.;n = 24 in each group; *P < 0.05 versus untreatedcontrol). In contrast, enzymatic activity of MnSODwas not altered by BMS treatment (data not shown).Finally, the overexpression of catalase could be
antagonized by cotreatment of neurons with30mmol/L BMS and 100 nmol/L Wrt for 3 days(Figure 6C).
The Catalase Inhibitor 3AT Dose-DependentlyAntagonized the Protective Effect of BMS
In untreated cells, the high doses of 3AT that wereused had no toxic effect on neuronal survival(Figure 6D). In addition, 3AT did not influence cellsurvival after exposure to glutamate (200 mmol/L) for
Figure 6 Delayed preconditioning with BMS-191095 (BMS) increases the expression and activity of catalase through thephosphoinositide 3-kinase (PI3K) pathway. Cultured neurons in 35 mm dishes were treated with BMS once a day for 1, 2, or 3 days,and then total RNA was isolated and subjected to real-time reverse transcription-PCR for catalase and manganese-dependentsuperoxide dismutase (MnSOD) (A). *Significant difference (P < 0.05) compared to the mRNA level of untreated control. Data areexpressed as mean±s.e.m.; n = 5; CT, threshold cycle. Another set of neurons in 35 mm dishes was treated using the same protocol,and then the extracted protein was subjected to Western blot analysis for catalase (B). The intensity of bands was normalized to thatof b-actin. The normalized level of catalase in the untreated control group (open bars) was considered 100%. A representativeWestern blot is shown below the graph. *Significant difference (P < 0.05) compared to the normalized protein level of untreatedcontrol. Data are expressed as mean±s.e.m.; n = 4. A third subset of neurons was treated with BMS and the phosphoinositide3-kinase inhibitor wortmannin (Wrt) once a day for 3 days after which catalase protein levels were examined by Western blotting(C). The intensity of bands was normalized to that of b-actin. The normalized level of the examined protein in the untreated controlgroup (open bars) was considered 100%. A representative Western blot is shown below the graph. *Significant difference (P < 0.05)compared to untreated control. Data are expressed as mean±s.e.m.; cells from two individual cultures; n = 4. Rat cortical neuronalcultures were treated with BMS-191095 once a day for 3 days and then exposed to glutamate for 60 mins (D). The cultures werecotreated with different concentrations of the catalase inhibitor 3-aminotriazole (3AT) during and for 3 h after glutamateexcitotoxicity. The viability of cell cultures that were treated with any of the drugs or were exposed to glutamate (gray bars) wasexpressed as a percent of the viability of control cultures that were not treated with the drugs and were not exposed to glutamate(open bar). *Significant difference (P < 0.05) compared to untreated control, not exposed to glutamate. #Significant difference(P < 0.05) compared to the untreated group, exposed to glutamate. zSignificant difference (P < 0.05) compared to BMS treatedgroup, exposed to glutamate. Data are expressed as mean±s.e.m.; n = 16.
ROS-independent preconditioning by BMS-191095T Gaspar et al
1098
Journal of Cerebral Blood Flow & Metabolism (2008) 28, 1090–1103

60 mins. However, 3AT dose-dependently decreasedthe neuroprotective effect of BMS. The maximalreduction in the protection was observed using10 mmol/L 3AT, which is known to effectivelyinhibit catalase, and the highest dose (100 mmol/L)had no additional effect on cell survival.
Discussion
Several important findings of our study are asfollows: (1) the selective mitoKATP channel openerBMS-191095 dose-dependently depolarized isolatedrat brain mitochondria without an increase in ROSgeneration; (2) long-term BMS treatment inducedprolonged protection along with sustained mito-chondrial depolarization, reduced basal ROS gen-eration, and improved ATP levels in culturedneurons; and (3) BMS activated the PI3K signal-transduction pathway that led to the phosphoryla-tion of Akt and Gsk3b, increased expression andactivity of catalase, and ultimately elicited neuro-protection.
Since the first report on the presence of a smallconductance ATP-sensitive potassium channel inthe inner mitochondrial membrane by Inoue et al(1991), the selective pharmacological activation ofmitoKATP channels became a major target of bothimmediate and delayed PC studies. However, theexact steps of PC after mitoKATP activation are stillunclear. The role of mitoKATP channels in ROSgeneration is controversial. ROS released by mito-chondria and acting as second messengers couldprovide a link between mitoKATP opening and theactivation of cytoprotective pathways. However,most of the pharmacological studies on the mitoKATP
channel showing ROS generation were performedusing DZ, which was shown to inhibit the mito-chondrial electron transport chain at Complex II(succinate dehydrogenase) resulting in excessiveROS release (Kis et al, 2003; Minners et al, 2007).Others have shown that DZ may also increasemitochondrial ROS generation by inhibiting Com-plex I (Andrukhiv et al, 2006). Indeed, ROS seem toplay a pivotal role in the tolerance-inducing effect ofDZ, as the neuroprotection could be completelyblocked with an SOD mimetic if administeredduring the PC phase (Kis et al, 2003). Conversely,Facundo et al (2006) tested the redox effects ofmitoKATP activation and found no evidence ofincreased mitochondrial ROS generation by openingof the mitoKATP channels. Furthermore, Ferranti et al(2003) showed that mitochondria depolarized by K+
entry generate less ROS. To clarify this issue, studiesusing more selective activators of the mitoKATP
channels such as BMS are needed. In our experi-ments, comparable to our previous studies onisolated piglet brain mitochondria (Busija et al,2005) and on cultured neurons (Mayanagi et al,2007), BMS induced dose-dependent depolarizationof the inner membrane of isolated rat brain
mitochondria without ROS generation. Hence, weprovide strong evidence that mitochondrial ROSgeneration is independent from the activation of themitoKATP channels. Consequently, it is likely thatROS generation is a nonspecific side effect of someless selective mitoKATP openers. It is worth mention-ing, however, that ROS produced during PC isknown to activate cytoprotective pathways, whichmay act synergistically with mitoKATP activationinduced by nonselective openers. Additionally, ROSmay directly or indirectly modulate the activation ofmitoKATP channels.
We have also shown that long-term treatment ofcultured neurons with BMS leads to sustainedmitochondrial depolarization and a parallel reduc-tion in basal ROS production. Furthermore, thiseffect was reversed by the putatively selectivemitoKATP blocker 5HD. These results are in agree-ment with previous studies from our laboratory inwhich we showed that at least 5 mmol/L 5HD isneeded to sufficiently antagonize the effects of BMSand that doses of 1 mmol/L or less are ineffective(Kis et al, 2004; Mayanagi et al, 2007). The exactmechanism of sustained depolarization is unknown.Nevertheless, in a previous study, we found asimilar phenomenon after a different PC stimulus,namely, transient energy deprivation (Gaspar et al,2006). Therefore, it seems that sustained depolariza-tion of mitochondria may play a crucial role indelayed PC induced by various stimuli.
K+ influx through mitoKATP channels mightpreserve mitochondrial integrity by maintainingmatrix volume and reducing Ca2 + accumulation(Busija et al, 2004; Domoki et al, 2004). This isparticularly important during an ischemic orchemical challenge and allows the rapid restorationof normal ATP production by maintaining theoptimal configuration of respiratory complexes.Our results appear to support this hypothesis, aswe found higher basal levels of ATP after long-termBMS treatment despite sustained mitochondrialdepolarization. This may seem controversial; never-theless, a similar trend in Dcm and ATP content wasobserved in neuronal cultures after delayed PC withtransient energy deprivation (Gaspar et al, 2006).Moreover, BMS-preconditioned neurons better pre-served ATP during a glutamate challenge and,consequently had a better survival rate. In ourexperiments, neuronal viability was assessed witha probe that measures the reducing capacity of cellsthat is primarily dependent on mitochondrialNADH generation. Thus, increased ATP contentand preserved viability in BMS-treated neurons alsoindicate preserved mitochondrial integrity.
To further explore the immediate effects of BMS,we performed Western blot analysis of two keycomponents of the PI3K signaling pathway, namely,Akt (also known as protein kinase B) and Gsk3b, andfound that both enzymes were phosphorylatedwithin 15 mins of the initiation of BMS treatment.However, whereas activity of Akt returned to
ROS-independent preconditioning by BMS-191095T Gaspar et al
1099
Journal of Cerebral Blood Flow & Metabolism (2008) 28, 1090–1103

baseline within 3 h, Gsk3b remained highly phos-phorylated until the end of the measurement (3 h).Akt is a serine/threonine protein kinase with highsequence homology to both protein kinase A andPKC; its role in neuronal survival was recentlyreviewed by Brunet et al (2001). Among manyothers, active Akt phosphorylates Bad (Kamadaet al, 2007) and Gsk3b (Grimes and Jope, 2001) andthereby induces antiapoptotic effects. Intracerebro-ventricular infusion of the PI3K inhibitor Wrt beforeischemic PC was shown to inhibit both Aktphosphorylation and protection against bilateralcommon carotid artery occlusion in the Mongoliangerbil model (Yano et al, 2001). Glycogen synthasekinase 3b is a critical component of a vast number ofcellular signaling pathways (for a detailed reviewsee Grimes and Jope, 2001). As one of the centraltargets of the PI3K–Akt pathway, Gsk3b is also a keymodulator of apoptosis. The enzyme is constitu-tively active and is primarily regulated throughinhibition by phosphorylation. Liang and Chuang(2007) showed that inhibition of either isoforms ofGsk3 completely blocks glutamate excitotoxicityand protects rat cortical neuronal cultures. Further-more, Kajta et al (2007) reported that pharmacolo-gical inhibition of Gsk3b enhanced theneuroprotective effect of the selective estrogenreceptor modulator genistein against glutamatechallenge. These results support our findings withBMS concerning inhibition of Gsk3b and almostcomplete protection against glutamate excitotoxi-city. However, it seems that Gsk3b is not theultimate effector of long-term BMS treatment butrather an important component of the cytoprotectivecascade, as its phosphorylation level did not differfrom the control at the time of glutamate exposure.
The activation of the PI3K pathway is usuallylinked to cell surface receptors such as receptortyrosine kinases and G-protein-coupled receptors(Brunet et al, 2001; Oudit et al, 2004). This mightsuggest that the effects of BMS are also mediated bybinding to such a receptor. We are unaware,however, of any targets other than the mitoKATP
channel. Moreover, Wrt was unable to block themitochondrial depolarizing effect of BMS, suggest-ing that the opening of mitoKATP channels isupstream to PI3K activation. The proposed mechan-ism of mitoKATP activation is depicted in Figure 7.A potential retrograde signaling link between theactivation of mitoKATP channels and the PI3K path-way could be the production of ROS by mitochon-dria. However, our results clearly show that BMSopens mitoKATP channels without ROS generation.Calcium is one of the possible links betweenmitochondria and cytosolic signaling pathways,and it was shown to be related to the PI3K cascadeby several mechanisms. First, the small GTP-binding protein p21ras that is upstream to PI3K isactivated by Ca2 + (Finkbeiner and Greenberg, 1996).Second, the Ca2 +/calmodulin-dependent proteinkinase (CaMK) II may activate PI3K, and the
calmodulin-binding sequence in PI3K may facilitatethe association of calmodulin with PI3K (Wang et al,2007a). Third, Akt was shown to be phosphorylatedby CaMK kinase (Yano et al, 2005). Finally, Bicklerand Fahlman (2004) showed that moderate increasesin [Ca2 +]c induced protection against a subsequentlethal stress in hippocampal slice cultures by theactivation of cytoprotective kinases, including Akt.An elevation of [Ca2 +]c was also observed after thedissipation of Dcm, and this led to the activation ofCa2 +-sensitive signaling pathways (Biswas et al,2005). Consistent with these observations, we havealso found that the application of BMS resulted in a
Figure 7 A schematic diagram illustrating the activation ofsignaling pathways of preconditioning after mitochondrial ATP-sensitive potassium (mitoKATP) channel opening. Activation ofmitoKATP channels by BMS-191095 induces the dissipation ofthe mitochondrial membrane potential (Dcm) without anelevation in reactive oxygen species generation. Mitochondrialdepolarization causes an increase in cytosolic free calcium([Ca2 +]c) levels, inhibits phosphatase and tensin homologuedeleted on chromosome 10 (PTEN), and may induce therelease of lipid second messengers (LSM) from the mitochon-drial membrane. These effects give rise to the activation ofphosphoinositide 3-kinase (PI3K), Akt, protein kinase C (PKC),inhibition of glycogen synthase kinase 3b (Gsk3b), and to theactivation of NF-Y that is involved in the transcription ofcatalase. Solid arrows indicate activation and broken arrowsshow putative activation; whiskered lines indicate inhibition;gray ellipses indicate components of the signaling pathway thatwere discussed in this study. Although catalase upregulationprovides the bulk of the neuroprotection, it seems likely that theactivation of these pathways also elicit other beneficial cellularchanges. 5HD, 5-hydroxydecanoic acid; Chel, chelerythrine;PI3K, phosphoinositide 3-kinase; Wrt, wortmannin.
ROS-independent preconditioning by BMS-191095T Gaspar et al
1100
Journal of Cerebral Blood Flow & Metabolism (2008) 28, 1090–1103

mild and transient increase in [Ca2 +]c and asubsequent phosphorylation of Akt and Gsk3b.Another possible ROS-independent mitochondrialmechanism of Akt activation could be the inhibitionby increased NADH/NADPH ratio of phosphataseand tensin homologue deleted on chromosome 10(PTEN) that dephosphorylates Akt (Pelicano et al,2006). Finally, Shang et al (2005) found thatmitochondrial depolarization, independently ofROS formation, led to the dephosphorylation ofdeath-associated protein kinase through the activa-tion of a class III PI3K in a neuroblastoma cell line.The authors proposed the role of lipid secondmessengers in the activation of PI3K released fromthe mitochondrial membrane during depolarization.
Next, we tested the delayed effects of BMS PC andfound that Wrt dose-dependently and completelyantagonized the neuroprotective effect of BMS,providing evidence on the crucial role of the PI3Ksignaling pathway in the initiation and/or mediationof PC. Conversely, the SOD mimetic M40401 had noeffect on the protection, showing that indeed ROSdid not have a role in the initiation of PC by BMS.The KCa channel inhibitor Pax was also ineffective,proving again the selectivity of BMS. Interestingly,subtoxic doses of the PKC inhibitor Chel by itselfinduced protection against glutamate challenge.Although the beneficial role of PKC activation(especially that of its e isoform) in PC was shownin various models, our results are in agreement withprevious reports showing that in neurons theinhibition or downregulation of PKC confers protec-tion against ischemia and other insults (Favaronet al, 1990). Additionally, Reshef et al (1997)reported that both activation and inhibition of PKCelicit neuroprotection. These authors concludedthat differential activation (or inhibition) of PKCisoforms might be responsible for the protection.Surprisingly, cotreatment with Chel did not signifi-cantly change the viability of BMS-preconditionedneurons exposed to glutamate. Nevertheless, thisfinding may be explained as the parallel actions ofChel: PC by PKC inhibition, and antagonizing theeffects of BMS. Additionally, the activation of PKCwas shown to be secondary to that of PI3K (Tonget al, 2000); this may explain the complete inhibi-tion of PC by Wrt. Finally, several isoforms of PKCwere reported to inactivate Gsk3b (Grimes and Jope,2001).
We also examined the effects of long-term BMStreatment on the expression of several cytoprotec-tive proteins and found that the expression ofcatalase increased at both transcriptional and trans-lational levels. A similar, although not entirelyparallel, increase in its enzymatic activity was alsofound. Furthermore, Wrt abolished catalase over-expression showing that catalase was activated bythe PI3K pathway. Finally, to examine whetherelevated catalase levels play any role in theneuroprotective effect of BMS, we performed viabi-lity experiments in which the catalase inhibitor 3AT
was used to antagonize the protective effect of BMSagainst glutamate challenge and found a partial anddose-dependent inhibition of the protection. Theexpression of catalase is regulated both transcrip-tionally and posttranscriptionally. Our resultsindicate that BMS-induced catalase overexpressionwas, at least in part, due to transcriptional activa-tion. Luo and Rando (2003) reported that in mice thecatalase expression is regulated by the CCAAT-binding factor NF-Y. In addition, Lee et al (2005)showed that Akt upregulates the DNA-bindingactivity of NF-Y. Furthermore, the presence ofNF-Y was reported in several neural cell lines.Thus, NF-Y may provide a link between Aktactivation and catalase overexpression. Althoughincreased levels of catalase provided much of theprotection against neuronal cell death during gluta-mate exposure, it is likely that other factors alsoparticipate in the mediation of BMS-induced PC.
In conclusion, our results provide novel compo-nents to the mechanism of action of delayed PCinduced by long-term BMS treatment showing thatactivation of mitoKATP channels is independent ofROS generation. Long-term treatment with BMSresults in sustained mitochondrial depolarization,reduced basal ROS formation, and improved ATPhomeostasis. Together with the activation of thePI3K–Akt–Gsk3b axis and upregulation of catalase,these effects play important roles in BMS-inducedneuroprotection.
Acknowledgements
We gratefully thank Nancy Busija, MA, for criticalreading of the paper. BMS-191095 was a generousgift from Bristol-Myers Squibb (Princeton, NJ, USA).
References
Andrukhiv A, Costa AD, West IC, Garlid KD (2006)Opening mitoKATP increases superoxide generationfrom complex I of the electron transport chain. Am JPhysiol Heart Circ Physiol 291:H2067–74
Bickler PE, Fahlman CS (2004) Moderate increases inintracellular calcium activate neuroprotective signalsin hippocampal neurons. Neuroscience 127:673–83
Biswas G, Guha M, Avadhani NG (2005) Mitochondria-to-nucleus stress signaling in mammalian cells: nature ofnuclear gene targets, transcription regulation, andinduced resistance to apoptosis. Gene 354:132–9
Brunet A, Datta SR, Greenberg ME (2001) Transcription-dependent and -independent control of neuronalsurvival by the PI3K–Akt signaling pathway. Curr OpinNeurobiol 11:297–305
Busija DW, Katakam P, Rajapakse NC, Kis B, Grover G,Domoki F, Bari F (2005) Effects of ATP-sensitivepotassium channel activators diazoxide and BMS-191095 on membrane potential and reactive oxygenspecies production in isolated piglet mitochondria.Brain Res Bull 66:85–90
ROS-independent preconditioning by BMS-191095T Gaspar et al
1101
Journal of Cerebral Blood Flow & Metabolism (2008) 28, 1090–1103

Busija DW, Lacza Z, Rajapakse N, Shimizu K, Kis B, Bari F,Domoki F, Horiguchi T (2004) Targeting mitochondrialATP-sensitive potassium channels—a novel approachto neuroprotection. Brain Res Brain Res Rev 46:282–94
Dahl NA, Balfour WM (1964) Prolonged anoxic survivaldue to anoxia pre-exposure: brain ATP, lactate, andpyruvate. Am J Physiol 207:452–6
Domoki F, Bari F, Nagy K, Busija DW, Siklos L (2004)Diazoxide prevents mitochondrial swelling and Ca2+accumulation in CA1 pyramidal cells after cerebralischemia in newborn pigs. Brain Res 1019:97–104
Facundo HT, Carreira RS, de Paula JG, Santos CC, FerrantiR, Laurindo FR, Kowaltowski AJ (2006) Ischemicpreconditioning requires increases in reactive oxygenrelease independent of mitochondrial K+ channelactivity. Free Radic Biol Med 40:469–79
Favaron M, Manev H, Siman R, Bertolino M, Szekely AM,DeErausquin G, Guidotti A, Costa E (1990) Down-regulation of protein kinase C protects cerebellargranule neurons in primary culture from glutamate-induced neuronal death. Proc Natl Acad Sci USA87:1983–7
Ferranti R, da Silva MM, Kowaltowski AJ (2003) Mito-chondrial ATP-sensitive K+ channel opening decreasesreactive oxygen species generation. FEBS Lett 536:51–5
Finkbeiner S, Greenberg ME (1996) Ca(2+)-dependentroutes to Ras: mechanisms for neuronal survival,differentiation, and plasticity? Neuron 16:233–6
Garlid KD, Paucek P, Yarov-Yarovoy V, Sun X, SchindlerPA (1996) The mitochondrial KATP channel as areceptor for potassium channel openers. J Biol Chem271:8796–9
Gaspar T, Kis B, Snipes JA, Lenzser G, Mayanagi K, Bari F,Busija DW (2006) Transient glucose and amino aciddeprivation induces delayed preconditioning incultured rat cortical neurons. J Neurochem 98:555–65
Grimes CA, Jope RS (2001) The multifaceted roles ofglycogen synthase kinase 3beta in cellular signaling.Prog Neurobiol 65:391–426
Gross ER, Hsu AK, Gross GJ (2007) GSK3beta inhibitionand K(ATP) channel opening mediate acute opioid-induced cardioprotection at reperfusion. Basic ResCardiol 102:341–9
Grover GJ, D’Alonzo AJ, Garlid KD, Bajgar R, Lodge NJ,Sleph PG, Darbenzio RB, Hess TA, Smith MA, PaucekP, Atwal KS (2001) Pharmacologic characterization ofBMS-191095, a mitochondrial K(ATP) opener with noperipheral vasodilator or cardiac action potential short-ening activity. J Pharmacol Exp Ther 297:1184–92
Inoue I, Nagase H, Kishi K, Higuti T (1991) ATP-sensitiveK+ channel in the mitochondrial inner membrane.Nature 352:244–7
Kajta M, Domin H, Grynkiewicz G, Lason W (2007)Genistein inhibits glutamate-induced apoptotic pro-cesses in primary neuronal cell cultures: an involve-ment of aryl hydrocarbon receptor and estrogenreceptor/glycogen synthase kinase-3beta intracellularsignaling pathway. Neuroscience 145:592–604
Kamada H, Nito C, Endo H, Chan PH (2007) Bad as aconverging signaling molecule between survival PI3-K/Akt and death JNK in neurons after transient focalcerebral ischemia in rats. J Cereb Blood Flow Metab27:521–33
Kis B, Nagy K, Snipes JA, Rajapakse NC, Horiguchi T,Grover GJ, Busija DW (2004) The mitochondrial K(ATP)channel opener BMS-191095 induces neuronalpreconditioning. Neuroreport 15:345–9
Kis B, Rajapakse NC, Snipes JA, Nagy K, Horiguchi T,Busija DW (2003) Diazoxide induces delayedpre-conditioning in cultured rat cortical neurons.J Neurochem 87:969–80
Lee SR, Park JH, Park EK, Chung CH, Kang SS, Bang OS(2005) Akt-induced promotion of cell-cycle progressionat G2/M phase involves upregulation of NF-Y bindingactivity in PC12 cells. J Cell Physiol 205:270–7
Liang MH, Chuang DM (2007) Regulation and function ofglycogen synthase kinase-3 isoforms in neuronalsurvival. J Biol Chem 282:3904–17
Luo D, Rando TA (2003) The regulation of catalase geneexpression in mouse muscle cells is dependent on theCCAAT-binding factor NF-Y. Biochem Biophys ResCommun 303:609–18
Mattson MP, Wang H, Michaelis EK (1991) Developmentalexpression, compartmentalization, and possible role inexcitotoxicity of a putative NMDA receptor protein incultured hippocampal neurons. Brain Res 565:94–108
Mattson MP, Zhang Y, Bose S (1993) Growth factorsprevent mitochondrial dysfunction, loss of calciumhomeostasis, and cell injury, but not ATP depletion inhippocampal neurons deprived of glucose. Exp Neurol121:1–13
Mayanagi K, Gaspar T, Katakam PV, Kis B, Busija DW(2007) The mitochondrial K(ATP) channel openerBMS-191095 reduces neuronal damage after transientfocal cerebral ischemia in rats. J Cereb Blood FlowMetab 27:348–55
Minners J, Lacerda L, Yellon DM, Opie LH, McLeod CJ,Sack MN (2007) Diazoxide-induced respiratoryinhibition—a putative mitochondrial K(ATP) channelindependent mechanism of pharmacological precondi-tioning. Mol Cell Biochem 294:11–8
Murry CE, Jennings RB, Reimer KA (1986) Precondition-ing with ischemia: a delay of lethal cell injury inischemic myocardium. Circulation 74:1124–36
Oudit GY, Sun H, Kerfant BG, Crackower MA, PenningerJM, Backx PH (2004) The role of phosphoinositide-3kinase and PTEN in cardiovascular physiology anddisease. J Mol Cell Cardiol 37:449–71
Pelicano H, Xu RH, Du M, Feng L, Sasaki R, Carew JS, Hu Y,Ramdas L, Hu L, Keating MJ, Zhang W, Plunkett W,Huang P (2006) Mitochondrial respiration defects incancer cells cause activation of Akt survival pathwaythrough a redox-mediated mechanism. J Cell Biol175:913–23
Rajapakse N, Shimizu K, Payne M, Busija D (2001)Isolation and characterization of intact mitochondriafrom neonatal rat brain. Brain Res Brain Res Protoc8:176–83
Reshef A, Sperling O, Zoref-Shani E (1997) Activation andinhibition of protein kinase C protect rat neuronalcultures against ischemia–reperfusion insult. NeurosciLett 238:37–40
Schmittgen TD, Zakrajsek BA, Mills AG, Gorn V, Singer MJ,Reed MW (2000) Quantitative reverse transcription-polymerase chain reaction to study mRNA decay:comparison of endpoint and real-time methods. AnalBiochem 285:194–204
Shang T, Joseph J, Hillard CJ, Kalyanaraman B (2005)Death-associated protein kinase as a sensor ofmitochondrial membrane potential: role of lysosomein mitochondrial toxin-induced cell death. J Biol Chem280:34644–53
Sims NR (1990) Rapid isolation of metabolically activemitochondria from rat brain and subregions using
ROS-independent preconditioning by BMS-191095T Gaspar et al
1102
Journal of Cerebral Blood Flow & Metabolism (2008) 28, 1090–1103

Percoll density gradient centrifugation. J Neurochem55:698–707
Tong H, Chen W, Steenbergen C, Murphy E (2000) Ischemicpreconditioning activates phosphatidylinositol-3-kinaseupstream of protein kinase C. Circ Res 87:309–15
Wang JQ, Fibuch EE, Mao L (2007a) Regulation ofmitogen-activated protein kinases by glutamate recep-tors. J Neurochem 100:1–11
Wang Y, Ahmad N, Wang B, Ashraf M (2007b) Chronicpreconditioning: a novel approach for cardiac protection.Am J Physiol Heart Circ Physiol 292:H2300–5
Yano S, Morioka M, Fukunaga K, Kawano T, Hara T, Kai Y,Hamada J, Miyamoto E, Ushio Y (2001) Activationof Akt/protein kinase B contributes to inductionof ischemic tolerance in the CA1 subfield ofgerbil hippocampus. J Cereb Blood Flow Metab 21:351–60
Yano S, Morioka M, Kuratsu J, Fukunaga K (2005)Functional proteins involved in regulation of intracel-lular Ca(2+) for drug development: role of calcium/calmodulin-dependent protein kinases in ischemicneuronal death. J Pharmacol Sci 97:351–4
ROS-independent preconditioning by BMS-191095T Gaspar et al
1103
Journal of Cerebral Blood Flow & Metabolism (2008) 28, 1090–1103




















