
Rôle du monoxyde d'azote dans la résistance à l'insuline ...
208
GENEVIÈVE PILON RÔLE DU MONOXYDE D'AZOTE DANS LA RÉSISTANCE À L'INSULINE ASSOCIÉE À L'INFLAMMATION Thèse présentée à la Faculté des études supérieures de l'Université Laval dans le cadre du programme de doctorat en physiologie-endocrinologie pour l'obtention du grade de Philosophioe I)octor ( Ph]). ) © Geneviève Pilon, 2008 F ACUL TÉ DE MÉDECINE UNIVERSITÉ LA V AL QUÉBEC 2008
-
Upload
khangminh22 -
Category
Documents
-
view
12 -
download
0
Transcript of Rôle du monoxyde d'azote dans la résistance à l'insuline ...

GENEVIÈVE PILON
RÔLE DU MONOXYDE D'AZOTE DANS LA RÉSISTANCE À L'INSULINE ASSOCIÉE À
L'INFLAMMATION
Thèse présentée à la Faculté des études supérieures de l'Université Laval
dans le cadre du programme de doctorat en physiologie-endocrinologie pour l' obtention du grade de Philosophiœ I)octor (Ph]).)
© Geneviève Pilon, 2008
F ACUL TÉ DE MÉDECINE UNIVERSITÉ LA V AL
QUÉBEC
2008

Résumé
La résistance à l' insuline du muscle squelettique se situe au niveau des étapes subséquentes
à la liaison de l' hormone à son récepteur. L'origine de la résis'tance à l' insuline n est pas
identifiée avec précision, bien que l'on sache qu'une association de plusieurs facteurs en
soit responsable. Parmi ces facteurs de risque, nous avons identifié la synthase inductible
du monoxyde d' azote (NO). Cette enzyme (iN OS) a la particularité de produire de fortes
concentrations de NO e'n réponse à divers stimuli inflammatoires. Les études présentées
dans cette thèse perniettent de mieux comprendre l' implication de cette enzyme dans la
résistance à l' insuline ainsi que les mécanismes d'action via lesquels le NO inhibe 1 action
de l' insuline.
Dans la première étude, nous avons confirmé dans un modèle cellulaire de muscles la
capacité de iNOS à causer un défaut du transport du glucose en réponse à l'insuline. Nous
avons toutefois également observé que les adipocytes ne développaient pas ce défaut suite à
l' induction de l' enzyme. Dans une seconde étude, nous avons cherché à comprendre le
mécanisme par lequel la production de NO pouvait être reliée à la résistance à l' insuline.
Nous avons montré que la génération d'un oxydant découlant de la synthèse de NO, le
peroxynitrite (ONOO-), causait la nitration de tyrosine de la molécule IRS-l et ainsi
interfère dans la cascade des étapes de signalisation de l' insuline. Dans une troisième étude,
nous avons démontré que l'incubation de muscles in vitro causait des altérations dans la
captation de glucose via l' induction de iNOS causée par le procédé d' incubation.
Finalement, dans une quatrième étude, nous proposons un mécanisme d'inhibition de iNOS
pouvant être mis à profit dans la recherche de cibles thérapeutiques contre la résistance à
l' insuline. Nous avons démontré que l'activati.on de la kinase sensible à l'AMP (AMPK)
réduisait l' induction de iNOS, tant au niveau cellulaire (myocytes, adipocytes et
macrophages) que dans les muscles et le tissu adipeux de rats traités à la lipopolysaccharide
(LPS). L' ensemble de nos études précise l'implication de iNOS dans la résistance à
l' insuline associée à l'inflammation et oriente la recherche vers de nouvelles pistes et un
certain nombre d'applications thérapeutiques.

Avant-Propos
Chapitre 1 : Ce manuscrit a été publié dans la revue Hormone and Metabolic Research. Pilon, G. ; Penfornis, P.; Marette, A. Nitric Oxide Production by Adipocytes: A Role in the Pathogenesis of Insulin Resistance? Horm Metab Res 2000; 32: 480-484 © Georg Thierne Verlag Stuttgart. New York Pour cette étude, j ' ai effectué la majorité des expériences en collaboration avec Patrice Penfornis. De plus, j ' ai rédigé le premier jet de l ' article.
Chapitre 2 : Ce manuscrit est en préparation. Pour cette étude, j ' ai effectué la majorité des
expériences. De plus, j ' ai rédigé le premier jet de l ' article. Il est à noter que Patrice
Dallaire, Mylène Perreault et Sonia Kapur ont aussi aidé aux expérimentations.
Chapitre 3 : Ce manuscrit est en préparation. Pour cette étude, j ' ai effectué la majorité des expériences avec l ' aide de Kathleen Lemieux. De plus, j ' ai rédigé le premier jet de l ' article.
Chapitre 4 : Ce manuscrit a été publié dans la revue Journal of Biological Chemistry (2004
May 14;279(20):20767-74). Pour cette étude, j ' ai effectué la grande majorité des
expériences avec l ' aide de Patrice Dallaire. De plus, j ' ai rédigé le premier j et de l ' article.

IV
Reinerciements
Je tiens à remercier le Dr André Marette pour sa disponibilité, son amitié et la confiance
qu' il m ' a toujours accordée. André fut un directeur de recherche extraordinaire. Je tiens à le
remercier par-dessus tout de m'avoir transmis son amour de la science. Mille fois merci !
Merci au Dr Claude H Côté. Claude fut présent tout au long de mes années d' études. Grâce
à ses encouragements, ses conseils tant scientifiques que personnels mes dernières années
d'apprentissage furent encore plus riches. Je veux également remercier les autres membres
de mon jury de thèse, soit Dr. Pascale Mauriège et Dr. André Carpentier.
Merci à tous mes collègues du laboratoire qui ont pris une place immense dans ma vie
durant toutes ces années. Ma reconnaissance va tout particulièrement à Bruno Marcotte qui
m'a beaucoup appris. Merci à Phillip James White pour sa générosité et à mes amis Marie
Julie Dubois et Frédéric Tremblay avec qui j ' ai développé une amitié qui débordera, j ' en
suis certaine, ces années d'études.
Un merci bien particulier à ma famille. À mes grandes sœurs qui m'ont toujours
encouragée. À ma mère, pour sa patience et sa persévérance, sans lesquelles je n' aurais
jamais étudié aussi longtemps. Merci à mon père qui a toujours voulu le meilleur pour moi
et m'a touj ours incitée à aller plus loin.
Merci enfin aux organismes subventionnaires qui ont rendu mes études possibles: le Fonds
FCAR-FRSQ-Santé, les Instituts de Recherche en Santé du Canada et Diabète Québec.

À mes parents et mes sœurs

Table des matières
Résumé ................................................................................................................................... ii Avant-Propos .................................................................... .................................................... iii Liste des abréviations ..................................................... ...................................................... vii Introduction ........................................................................................................................... 11
1. Diabète de type 2 .................................. ........................................................................ Il 2. Rôles et mécanismes d ' action de l ' insuline .................................................................. 12
2.1 L' insuline ................................................................................................................ 12 2.2 Le récepteur de l ' insuline (IR) ............... : ................................................................ 13 2.3 Les substrats du récepteur à l' insuline (IRS) .......................................................... 14 2.4 La phosphatidylinosito13-kinase (PI 3-Kinase) ..................................................... 15 2.5 L'AktIPKB ....................................................................................... ....................... 18 2.6 Les PKC atypiques .................................................................................................. 20 2.7 Les transporteurs de glucose: GLUT ............................................................. ......... 21 2.8 La résistance à l'insuline ......................................................................................... 26 2.9 Autres facteurs adipocytaires importants dans la sensibilité à l'insuline ................ 33
3. L' AMPK ....................................................................................................................... 37 3.1 La structure de l' AMPK ......................................................................................... 3 7 3.2 L' activation de l' AMPK .................................................. : ...................................... 39 3.3 Les modulateurs physiologiques ............................................................................. 41 3.4 Les modulateurs chimiques .......................................................................... .......... 43 3.5 Rôles de l 'AMPK ........................................................ ............................................. 45
4. Le monoxyde d' azote .................................................................................................... 50 4.1 La famille des NOS ................................................................................................. 51 4.2 La structure des NOS ............... ~ .............................................................................. 52 4.3 eNOS ....................................................................................................................... 53 4.4 nNOS ....................................................................................................................... 55 4.5 MtNOS .................................................................................................................... 57 4.6 iNOS ....................................................................................................................... 58 4.7 La résistance à l'insuline induite par la S-nitrosylation .......................................... 67 4.8 Le ONOO- ............................................................................................................... 68 4.9 L'origine du O2- •••••••.••••••••••••••••••••.••••••••••.••.••••...•••••••••••••••••••••••••••••.•••••••••••••••••••••• 69 4.10 La nitrotyrosination ............................................................................................... 69
5. Buts de l'étude .................................................................................................................. 73 Chapitre 1. La production de monoxyde d'azote par les adipocytes : un rôle dans la pathogenèse de la résistance à l' insuline? ........................................................................... 74 Chapitre 2. L ' induction de iNOS cause la résistance à l'insuline par un mécanisme dépendant du peroxynitritre par la nitration d' IRS-1 dans le muscle squelettique ............... 92 Chapitre 3. Une incubation prolongée du muscle épitrocléaris cause l'induction de iNOS et des altérations du transport de glucose ............................................................................... 116 Chapitre 4. L ' inhibition de la synthase de NO inductible par les activateurs de la kinase activée par l'AMP (AMPK): un nouveau mécanisme anti-inflammatoire ......................... 137 Conclusion .......................................................................................................................... 165 Bibliographie ...................................................................................................................... 178

Liste des abréviations
ACC: ADP: AGEs: AMP: AMPK: API: . ATF2 : ATP: BAEC:
Acetyl-CoA Carboxylase Adenosine diphosphate Advanced glycation end products Adenosine monophsphate Kinase dépendante de l ' AMP Acti vator protein 1 Activating transcription factor 2 Adénosine triphosphate Bovine aorta endothelial cells
BH4 : Tertrahudrobiopterine BMI : Body mass index CaMKK : Ca2+/calmodulin-dependent protein kinase kinase CEACAM-1 : Carcinoembryonic antigen-related cell adhesion molecule cGMP : Cyclic guanidine monophosphate ChREBP : Carbohydrate response element binding prote in CPT -1 : Camityl plamitotransférase-1 CRP : C-reactive protein DAG : Diacylglycerol DNA-PK: DNA-dependent protein kinase EDL : Extensor digitorium longus EDRF : Endothelium-derived relaxing factor ERK : Extracellular signal-regulated kinase F AD : Flavine adenine nucleotide F AD : Flavine adénine dinucléotide FF A : Free fatty acid FMN: Flavine mononucleotide FOXO-l : Forkhead box class 0-1 G6Pase: GAPDH: GDE: GEF: Ghrelin: GLUT: GM3 : Grb10 : GSK3: GTP: HMG-Coa: HNF4: IFN: IGF: IKK:
G 1 ucose-6-phosphatase Glyceraldhéhyde phosphate déshydrogénase Glycogen-debranching enzyme GLUT 4 enhancer factor Growth hormone releasing peptide Transporteur de glucose Gangliosides Growth-factor-receptor-bound protein 10 Glycogen Synthase Kinase-3 Guanosine triphosphate 3-Hydroxy-3-methyl glutaryl-Coenzyme A Hepatic nuclear factor 4 alpha Interféron F acteur de croissance de l'insuline Kinase de iKB

IL-I et IL-6 : ILK: IR: IRAK: IRF : IRS: iKB : JAKISTAT: JIPI : JNK: KSRP: LDL: L-NAME: LPA: LPS: MAPK: MEF2: mTOR: NADH: NAPIIO: NEMO: NFKB NMDAR: NO: N02 :
N03 :
NOS: NRFl: O2- :
ONOO- : PCI: PDK: PDZ: PECAM: PEPCK: PH: PI3K: PIKfyve: PIP3 : PKA: PKB/AKT: PKC: PPAR: PPRE: PTEN: PTP: RNS:
Interleukines-l et 6 Integrin-linked kinase Récepteur de l' insuline IL-l receptor-associated kinase Interferon response factor Substrat du récepteur à l' insuline inhibiteur de NFKB Janus kinase-signal transducer and activator of transcription JNK-interacting protein-l c-Jun NH(2)-terminal kinase KR-type splicing regulatory protein lipoprotéine de faible densité NG-nitro-L-arginine methyl ester Lysophosphatidic acid Lipopolysaccharide Mitogen activated protein kinase Myocyte enhancer factor 2 Mammalian Target of Rapamycine ~-nicotinamide adenine dinucleotide phopshate NOS-associated protein-ll 0 kDa . Nuclear factor kappa B essential modulator Nuclear factor kappa B N -methy 1-D-aspartate receptor Monoxyde d'azote Nitrite Nitrate
. Monoxyde d'az9te synthase N uclear respiratory factor 1 Superxoxyde Peroxynitrite Plasma glycoprotein-l Kinase dépendante des phosphoinositides PSD/Discs-large/ZO-l Platelet endothelial cell adhesion molecule Phosphoenolpyruvate carboxylase Pleckstrin homology do main Phosphatidylinositol 3-kinase Phosphoinositide kinase for five position containing a Fyve finger Phosphoinositols (3,4,5) triphosphates Protéine kinase A Protéine kinase B Protéine kinase C Peroxisome proliferator activated receptor PP AR response element Phosphatase and tensin homologue Phosphotyrosine binding domain Reactive nitrogen species
Vll1

IX
ROS: Reactive oxygen species SH2 domain : Src homology domain SHIP2 : SHP-I : siRNA: SNFI: SNP: SOCS: SREBPlc: STAT: TAKI: TLR: TNFa: TORC2:
Tubules-T: ZMP: ZTP:
SH2 domain-containing inositol phosphatase Src homology domain 2-containing tyrosine phosphatase Small interfering RNA Sucrose non-fermenting 1 Sodium nitroprusside Supressors of cutokine signlaing . Sterol-regulatory element binding pro teins 1 c Signal transducer and activator of transcription Transforming growth factor-beta-activated kinase T 0 ll-like receptors Tumor necrosis factor a cAMP response element-binding protein (CREB) coactivator transducer of regulated CREB activity 2 Tubules transversaux 5-amino-4-imidazolecarboxamide riboside monophosphate 5 -amino-4-imidazo lecarboxamide ri boside triphosphate

Liste des figures
Introduction Figure 1. Éléments de signalisation activés via le récepteur de l' insuline 25
Chapitre 1 Figure 1: iNOS' expression in white and brown adipose tissues 88 Figure 2: Effect of the iNOS inhibitor 1400W on NO production and glucose uptake in cytokines/LPS-treated 3T3-L1 white and T37i brown adi pocytes 89 Figure 3: Proposed role for adipose iNOS induction in skeletal muscle insulin resistance in obesity and inflammation 90
Chapitre 2 . Figure 1: Effect of aminoguanidine on iNOS induced insulin resistance in muscle 110 Figure 2: iNOS disruption protects mice against insulin resistance induced by LP S treatment 111 Figure 3: Effect of ONOO- and iNOS induction on basal and insulin stimulated glucose transport in L6 muscle cells 112 Figure 4: Effect of iNOS and ONOO- on IRS-1 associated PI 3-kinase activity in L6 myocytes 113 Figure 5: Effect ofONOO- and iNOS induction on IRS-1 tyrosine nitration and phosphorylation 114
Chapitre 3 Figure 1: Long-terrn incubation of muscle modulates NOS expression 132 Figure 2: Time course ofiNOS and GLUT1 induction 133 Figure 3: iNOS induction causes insulin resistance in incubated muscle 134 Figure 4: NO donor increases GLUT1 expression in L6 muscle cells 135
Chapitre 4 Figure 1: Effects of AMPK activators on NO production and iNOS expression in L6 myocytes 159 Figure 2: Effects of cytokines/LPS and AMPK activators on IRS-1 associated PI 3-kinase activity in L6 myocytes 160 Figure 3: Effects of AMPK activators on NO production and iNOS expression in 3T3-L1 adipocytes and macrophages 161 Figure 4: Effect of AICAR and metforrnin on iNOS in skeletal muscle and adipose tissue in vivo 162 Figure 5: siRNA-mediated reduction in AMPK expression reverses the inhibitory effects of AMPK activators on iNOS expression and activity 163

Introduction
Le diabète est une affection chronique résultant de l'incapacité du corps à produire
suffisamment d'insuline ou à lui répondre comme il se doit. L'insuline sécrétée par le
pancréas permet l'utilisation ou l' entreposage du glucose en circulation. Sans insuline, ou
sans son signal, les celhiles du corps -principalement celles des muscles, des tissus
adipeux et du foie - ne répondent plus adéquatement aux besoins de l'organisme causant
une augmentation du glucose en circulation. Une glycémie toujours élevée provoque la
dysfonction et la défaillance de divers organes comme les reins, les yeux, les nerfs, le cœur
et les vaisseaux sanguins.
Les études de cette thèse visent à mieux comprendre la résistance du muscle à l' insuline
observée dans le diabète de type 2.
1. Diabète de type 2
Le diabète de type 2 fait habituellement son apparition après l'âge de 40 ans et s' attaque
principalement aux gens présentant un surplus de poids. En perdant du poids, en faisant de
l'exercice ou en prenant des médicaments, beaucoup de patients surmontent la résistance à
l'insuline. Certains doivent toutefois finalement s'en remettre à des injections quotidiennes
d'insuline.
La maladie se développe souvent de la façon suivante: un état d' insulinorésistance est
d' abord observé. Même si l'insuline peut se fixer nonnalement à son .récepteur, certains
mécanismes empêchent l'insuline de transporter le glucose en circulation à l'intérieur des
cellules. Les personnes touchées produisent alors des quantités variables, voir normales ou
élevées, d'insuline. Au début, ces quantités suffisent pour surmonter cette résistance.
À la longue, le pancréas devient incapable de produire suffisamment d'insuline pour
surmonter la résistance. Ce deuxième stade entraînera donc une élévation anormale de la
glycémie après un repas: c'est ce qu'on appelle l'hyperglycémie postprandiale. Le cycle de

12
. l'hyperglycémie finit par entraver encore davantage le fonctionnement des cellules bêta du
pancréas, ce qui interrompt complètement la production d'insuline et provoque l'apparition
du diabète, comme en témoigne l'hypergly cémie à jeun ; une glycémie qui demeure élevée
durant presque toute la période où la personne diabétique est à jeun.
2. Rôles et mécanismes d'action de l'insuline
2.1 L'insuline
L' insuline est une petite hormone de 51 acides aminés produite par les cellules p du
pancréas ayant un rôle décisif sur le transport du glucose et son métabolisme, la synthèse
protéique et la régulation de plusieurs gènes. Ces effets sont régulés par un système
complexe de signalisation intracellulaire impliquant une cascade d' événements de
phosphory lation à partir de son récepteur - ci -après identifié par IR.
À la suite de la prise alimentaire, la quantité de glucose en circulation augmente et
provoque la production d' insuline par les cellules fJ du pancréas. Plus précisément, le
glucose métabolisé augmentera la quantité" d'ATP disponible, élevant ainsi le rapport
ATP/ADP. Ce rapport modifié occasionnera la fermeture des canaux potassique (K+) de la
cellule, augmentant sa charge positive. Cette modification de la charge causera la
dépolarisation des cellules, ouvrant les canaux calcium. L'augmentation de calcium
intracellulaire causera l'exocytose de l'insuline [1]. L' insuline en circulation permet alors
l' entrée du glucose au muscle et au tissu adipeux et inhibe la lipolyse de ce dernier. Au
niveau du foie, l' hormone stimule la synthèse de glycogène et inhibe la production de
glucose, contrecarrant ainsi les processus de gluconéogenèse et de glycogénolyse. Bref,
l'insuline défavorise les actions dites cataboliques au profit de l'entreposage du glucose.
Ensuite, pour occasionner ses effets métaboliques, l'insuline doit lier le IR. Cette liaison
provoque son autophosphorylation permettant ainsi son activité tyrosine kinase. Le IR

13
servira ensuite de point d ' ancrage pour, entre autres, les substrats du récepteur de l' insuline,
les protéines IRS. Une fois associés, les IRS sont phosphorylés à leur tour permettant alors
le recrutement de la sous-unité régulatrice de la phosphatidylinositol 3-kinase (PI 3-Kinase)
: la p85. Cette dernière active la sous-unité catalytique de la même enzyme: la pllO. La PI
3-Kinase, comme son nom l' indique, phosphoryle et transforme à la membrane les
phosphatidylinositols PIC 4,5)P2 en PI(3 ,4,5)P3. Ces derniers vont permettre l'activation de
la 3-phosphoinositide-dependent kinase-l (PDK-l /2) qui phosphoryle les enzymes
Akt/PKB et les PKC-Ç/À. Ces kinases conduiront à la translocation des transporteurs de
glucose GLUT4 qui réaliseront l' étape ultime: la captation du glucose. Les prochaines
sections reprennent en détails les processus moléculaires impliqués dans cette cascade
signalétique.
2.2 Le récepteur de l'insuline (IR)
La signalisation de l' insuline menant au transport de glucose de l'extérieur vers l ' intérieur
de la cellule débute alors par le contact de l' insuline à son récepteur. Le récepteur de
l' insuline est un hétérotétramère composé de deux sous-unités -a et deux ~ liées entre
elles par des liens disulfures. La sous-unité a possède une section extracellulaire et lie ainsi
l'insuline circulante, alors que la sous-unité {3 est transmembranaire et permettra la
transmission du signal intracellulaire. La liaison de l' insuline provoquera un changement
de structure du récepteur rapprochant les sous-unités a à proximité l' une de l' autre. Ce
changement de conformation permettra l' autophosphorylation des sous-unités {3. Une fois
activé, le IR pourra phosphoryler plusieurs cibles sous-jacentes dont les substrats du
récepteur à l'insuline (IRS). Le IR doit absolument être phosphorylé sur son résidu tyrosine
Y960 dans le domaine juxta membranaire pour permettre son association avec IRS-l
contenant un domaine de liaison à la phosphotyrosine (PTB) [2]. Afin d'assurer un fin
control du IR, plusieurs mécanismes assurent sa régulation négative. D'abord, la protéine
tyrosine phosphatase PTP-lB interagit directement avec le récepteur afin de diminuer son
activité [3]. D'autres protéines interagissent directement avec le récepteur, empêchant son
interaction avec ses substrats ou induisant la dégradation du récepteur. C'est le cas des

14
suppressor of cytokine signaling (SOCS-l , SOCS-6 et SOCS-3), du growth-factor
receptor-bound protein 10 (Grb 1 0) et de la plasma glycoprotein-l (PC 1) [4] [5] [6].
2.3 Les substrats du récepteur à l'insuline (IRS)
Parmi les six IRS connus, IRS-1 fut le premier à avoir été identifié [7] . Alors qu'IRS-1 et
IRS-2 sont largement distribués, IRS-3 se situe au niveau du cerveau et dans le tissu
adipeux. IRS-4 pour sa part est particulièrement exprimé dans les tissus embryonnaires
alors qu'IRS-5 et IRS-6, sont peu exprimés, et semblent jouer un rôle mineur dans la
signalisation de l'insuline [8] [9] [10] [11] [12]. Les IRS peuvent être phosphorylés sur de
nombreux résidus sérine et thréonine (possiblement une cinquantaine) ainsi que sur des
résidus tyrosine. Même si l'ensemble des fonctions de ces phosphorylations n'est pas tout à
fait comprise, on sait que généralement les phosphorylations sur tyrosine permettent la
continuité du signal par l'insuline, alors que les modifications sur sérine et thréonine
peuvent provoquer des effets surtout négatifs. Le domaine pleckstrin homology (PH) des
IRS leur permettent d' être localisés au niveau de la membrane à proximité du récepteur de
l' insuline, tandis que par leur domaine PTB, IRS-1 et IRS-2 peuvent interagir avec le
récepteur de l' insuline. Ainsi activés, les IRS peuvent relayer la cascade de phosphorylation
par l'activation d'une enzyme clé: la PI 3-Kinase. Le recrutement de cette kinase se fait par
l' association de son domaine SH2 [13]. Du côté d' IRS-1 , les sites Tyr 608 et 628 (chez le
rat) représenteraient des sites majeurs d'ancrage [14]. Malgré l'expression ubiquitaire
d ' IRS-1 et d ' IRS-2, le rôle d'IRS-1 serait de permettre les ' fonctions métaboliques de
l' i'nsuline au muscle squelettique, au tissu adipeux et, dans une moindre mesure, au foie,
alors que les rôles d' IRS-2 s' exerceraient davantage au niveau du foie et du pancréas [15]
[16]. Dans les myocytes L6, où IRS-1 et IRS-2 sont exprimés, des études utilisant la
technologie des ARN interférant, ont montré qu' IRS-l aurait un rôle majeur sur le transport
du glucose stimulé par l'insuline alors qu'une réduction importante d' IRS-2 serait sans
impact [17].

15
L'exposition prolongée à l'insuline, à certaines cytokines et aux acides gras libres active des
serine/thréonine kinases qui occasionnent la phosphorylation sur sérine d'IRS-l. Les
kinases suivantes ont été impliquées: extracellular signal - regulated kinase (ERK), la S6
kinase, la c-Jun-N-terminal kinase (JNK), la PKC theta, l'AMPK, les salt-induced kinases
(SIKI et SAK2), l ' IL-l receptor-associated kinase (IRAK) et NF-kB [18] [19] [20] [21]
[22] [23] [24] [25] [26]. Finalement, la dégradation ou l'inhibition de la traduction des IRS
exercent aussi un contrôle sur le signal conduit par l'insuline [27] [28].
2.4 La phosphatidylinositol3-kinase (PI3-Kinase)
2.4.1 Son fonctionnement et ses composantes
Découlant de l' association des IRS à la sous-unité régulatrice p85 de la PI 3-Kinase, la
sous-unité catalytique plI 0 est à son tour recrutée. Il en résulte une activation de la PI 3-
kinase. Cette kinase est essentielle à l ' exercice de plusieurs fonctions métaboliques de
l' insuline comme la translocation des transporteurs de glucose GLUT4, la synthèse de
glycogène, l' inhibition de la lipolyse et la stimulation de la synthèse protéique [29] [30]
[31] [32].
Plusieurs enzymes possèdent une activité PI 3-kinase, c'est-à-dire capables de phosphoryler
en position D-3 l' anneau inositol de phospholipides membranaires [33] [34]. Ces enzymes
regroupées dans la grande famille des PI 3-kinases sont cependant plutôt hétérogènes et ont
donc été regroupées en sous catégories sur la base de leur ressemblance, de leur séquence,
et de leur fonction.
La classe 1, composée d'une sous-unité dite régulatrice nommée p85 et d 'une autre dite
catalytique nommée plI 0, se subdivise en classes lA et lB. Panni toutes ces fonnes de PI
3-kinase, seules celles de la classe lA pennettent la translocation des transporteurs de
glucose GLUT4, l' étape ultime pennettant l ' entrée du glucose dans la cellule stimulée par
l' insuline.

16
La sous-unité p85 sert d' adaptateur pour lier la sous-unité catalytique pllO. La p85 possède
un domaine SH3 et deux domaines SH2. Ces derniers vont permettre la liaison de peptides
contenant un motif composé d'une méthionine en position + 1 et +3 en amont d'une
tyrosine phosphorylée [35] [36] [37]. Cette capacité à lier ce motif particulier lui permet de
s' associer à IRS-l [38][39]. Dans la classe lA des PI 3-kinases, trois formes de sous-unités
régulatrices ont été caractérisées à ce jour: la p85a, la p85~ et la forme tronquée p55y [36]
[39]. La raison d' être des différentes sous-unités n 'est pas encore bien définie. En effet, la .
délétion des sous-unités p85a et p85~ cause des effets surprenants puisqu' il en résulte une
plus grande sensibilité à l' insuline. Cette conséquence inattendue pourrait s' expliquer par le
fait qu'en situation normale, la sous-unité p85 est plus abondante que la plIO, on retrouve
donc la p85 sous forme de monomère et aussi associée à la plI O. En effet, la sous-unité
p1l0 est rarement retrouvée non associée à sa partie régulatrice puisque seule elle est
rapidement dégradée [40]. Dans cette situation existe alors une compétition entre les deux
formes pour lier IRS-l. En réduisant l' expression de la p85, le nombre d 'hétérodimères
composés de la p85 et de la plI 0 sera alors favorisé [41] [42] [43]. Cependant, cet effet
d'un débalancement de la sous-unité p85 ne peut pas simplement s'expliquer par un
désordre stœchiométrique puisque la surexpression des sous-unités p55a ou p50a ne cause
pas cette augmentation de la sensibilité à l'insuline malgré leur effet semblable sur la plI 0
[44]. D'un autre côté, la sous-unité p85 peut causer l'activation de JNK par un mécanisme
encore nébuleux mais indépendant de l'activité PI 3-kinase. Étant donné que JNK peut
moduler négativement la sensibilité à l'insuline (comme nous le verrons plus loin), il est
concevable qu'une diminution de la p85 puisse augmenter la sensibilité à l'insuline en
diminuant ainsi l' activation de la JNK [43]. .
Trois versions de la sous-unité catalytique de la PI 3-kinase de la classe lA sont connues:
la plI Oa, la p Il O~ et la plI 08. Cependant, seules les formes a et fJ seraient impliquées
dans les rôles de signalisation de l'insuline puisqu'elles sont exprimées de façon
ubiquitaire, alors que la forme 8 n'est pas exprimée dans les tissus' sensibles à l'insuline
[45] [46] [47].

17
Il est clairement établi que la classe 1 A est nécessaire à l' exercice de plusieurs fonctions
métaboliques de l' insuline. Cependant, il existe aussi certaines hypothèses suggérant
qu' elle n'est pas seule en cause. Par exemple, l' introduction du produit de la PI 3-kinase
soit PI(3 ,4,5)P3 --dans une cellule n ' est pas suffisante pour permettre le transport de
glucose [48]. Des études réalisées à l' aide d'un inhibiteur de PI 3-kinase -la wortmannine
- montrent, par exemple, que deux processus semblent réguler la translocation de GLUT4,
puisque seule une partie de la translocation des GLUT4 est sensible à l' inhibiteur [49] [50].
Les autres classes de PI 3-kinases ne semblent pas en mesure de produire des PI(3 ,4,5)P3 in
vivo. Elles peuvent cependant produire le PI(3)P [51] .[52] [53] [54] [55]. Il est suggéré que
le PI(3)P est aussi important pour le transport du glucose stimulé par l' insuline. Une
diminution de PI(3)P par une phosphatase spécifique aux lipides, la myotubularine,
diminue le transport du glucose par l'insuline [5-6]. Une stimulation à l'insuline cause une
augmentation des niveaux de PI(3)P dans la cellule, mais son origine n' est pas encore tout à
fait claire. Comme cette augmentation n'est pas sensible à la wortmannine, on pourrait
croire que la production de PI(3)P provient de la PI3 K -C2a : une forme résistante à
l' inhibiteur [57] [58].
2.4.2 Les mécanismes inhibant la PI 3-kinase
Comme la PI 3 .. kinase joue plusieurs rôles physiologiques importants, son activation doit
être finement contrôlée. Son inhibition s'effectue par plusieurs mécanismes. La protéine
tyrosine phosphatase PTP-l B, par exemple, permet de déphosphoryler le récepteur à
l' insuline et ses substrats ayant ainsi pour conséquence, l'inhibition de la PI 3-kinase. Il fut
proposé que l'insuline activait elle-même la PTP-1B afin de limiter son propre signal
[59][60]. Il existe par ailleurs des phosphatases ciblant directement les PI(3 ,4,5)P3 tels que
SH2 domain-containing inositol phosphatase (SHIP2) et phosphatase and tensin
homologue (PTEN), contrecarrant ainsi l'effet de l'activation de la PI 3-kinase. Si le rôle de
PTEN fut clairement démontré pour inhiber la translocation de GLUT4, celui de SHIP2 est

18
plus controversé. En effet, même si les souris SHIP2 hétérozygottes ' sont plus sensibles à
l'insuUne, des études in vitro à l'aide de petits ARN interférant (siRNA) dirigés contre
SHIP2 n'ont pas été en mesure d'augmenter la sensibilité à l'insuline d'adipocytes 3T3-L1
[61] [62]. De plus, récemment, un nouveau modèle de souris hétérozygotes de SHIP2 n'a
pas démontré une amélioration de la sensibilité à l'insuline. Cependant, sous une diète riche
en graisses, une diminution de l'expression de SHIP2 fut démontrée bénéfique [63]. D'un
autre côté, des études de notre laboratoire ont montré l' impact négatif de la sre homology
domain 2-eontaining tyrosine phosphatase (SHP-1) sur la signalisation de l' insuline au
niveau du muscle et au foie par l'utilisation de souris invalidées pour cette phosphatase. De
plus, cette étude a permis de mettre en lumière un rôle de la phosphatase dans la clairance
hépatique de l'insuline par son interaction avec la protéine Careinoembryonie antigen~
related cel! adhesion moleeule (CEACAM-1) [64].
L' activité de la PI 3-kinase peut être aussi directement inhibée par sa phosphorylation. La
phosphorylation sur la sous-unité p85 sur sérine 608 inhibe l' activité catalytique de plI Oa
et serait le résultat d'un mécanisme de rétroaction de l'insuline [65] [66]. La PI 3-kinase
pourrait aussi phosphoryler IRS-1 sur des résidus sérine dans le même but, celui de
contrôler l'action de l'insuline, du moins dans un modèle cellulaire in vitro [67]. De la
même façon, la p110p peut être phosphorylée et être ainsi inhibée in vitro ; mais la
pertinence de ce mécanisme in vivo doit encore être démontrée [68].
L' importance de la PI 3-kinase pour les effets métaboliques de l' insuline est donc
indéniable, mais les processus qui la régissent sont très complexes et demandent à être
mIeux connus.
2.5 L'Akt/PKB
L'activation de la PI 3-kinase mènera à la mise en action d'une sérine/thréonine kinase
nommée Akt ou PKB. Ce processus est réalisé en deux étapes. D'abord, la production de
PI(3 ,4)P2 et PI(3 ,4,5)P3 est nécessaire pour l'attachement de l'Akt à la membrane

19
plasmique par l' intermédiaire de son domaine PH N-terminal. Cette translocation lui
permettra de se positionner à proximité d'une kinase qui pourra la phosphoryler et ainsi
l' activer: laphosphoinositide-dependent kinase-l (PDK-1) [69].
Trois formes d' Akt sont actuellement identifiées : Akt 1, Akt2 et Akt3 (aussi nommées
PKBu, PKBp et PKBy, respectivement) [70]. Leur expression est ubiquitaire et en absence
de stimulation, elles sont situées dans le cytosol. Une fois au niveau de la membrane,
l' interaction entre le domaine PH et ~es lipides membranaires permet un changement
conformationnel de l ' Akt, démasquant ainsi le site Thr308 et la rendant apte à être
phosphorylée par PDK-1. Afin d ' être complètement active, l' Akt doit aussi être
phosphorylée sur le résidu Ser4 73. La kinase responsable de cette dernière phosphorylation
n'est pas encore clairement identifiée. Cependant, la mitogen-activated protein kinase
activated protein kinase 2 (MAPKAPK2) , l'integrin-linked kinase (ILK) , la DNA-dependent
protein kinase (DNA-PK) et le complex rictor-mTOR complex 2, peuvent phosphoryler Akt
sur ce résidu sérine in vitro; il pourrait donc s'agir de candidats potentiellement
responsables de la phosphorylation in vivo [71] [72] [73] [74] [75].
En plus de ces deux sites de phosphorylation bien décrits, l'Akt serait aussi phosphorylée de
façon constitutive sur la sérine 124 et la thréonine 450 et l'activité de la kinase pourrait
aussi être régulée par sa phosphorylation sur tyrosine Y315 et Y326 [76].
Plus récemment, une étude a suggéré la présence de deux nouveaux sites de
phosphorylation responsables de l'activité de l'Akt : la thréonine 72 et la serine 246. Ces
sites seraient phosphorylés par l'Akt elle-même par autophosphorylation et seraient
responsables de l'activité de base de la kinase mais pourraient aussi être stimulés par des
facteurs externes [77].
Akt aura par la suite plusieurs fonctions. Le premier rôle physiologique de l'Akt à avoir été
rapporté fut la synthèse de glycogène par la phosphorylation et l'inactivation de la
glycogène synthase kinase 3 (GSK3). Lorsque cette dernière est inactivée, elle lève une
inhibition sur la glycogène synthase (GS) permettant ainsi son activation [78]. L'Akt a

20
aussi pour cible le facteur FOXO-l. Une fois modifié par la kinase, ce facteur de
transcription est exclus du noyau; il en découle qu' il ne peut plus permettre l'expression
des gènes de la phosphoenolpyruvate carboxylase (PEPCK) et la glucose-6-phosphatase
(G6Pase), deux enzymes importantes dans la gluconéogenèse [79].
Par ailleurs, le lien subséquent entre l'Akt et la translocation de GLUT4, quoique toujours
un peu nébuleux, se clarifie peu à peu [80] [81]. D'abord, l'AS 160 ou (Fre-2/Bub2/Cdc16)
domain fami/y, member 4 (TBC 1 D4), 'qui serait le substrat le plus abondant de l' Akt, a un
poids moléculaire de 160kDa et possède une activité Rab GTP-ase. Cette protéine serait
impliquée dans la translocation des transporteurs de glucose GLUT4. L'AS 160 est
phosphorylée en réponse à l'insuline sur cinq sites ayant le motif ciblé de l'Akt [82]. En
condition basale, l'AS160 pourrait retenir les vésicules GLUT4 dans la cellule. Cependant,
lorsque l'insuline occasionne la phosphorylation de l' ~S 160 par l' intermédiaire de l' Akt,
cette dernière se dissocierait des vésicules GLUT4 permettant leur exocytose [83]. Un
défaut d'expression de l'AS160 ne serait pas en cause chez les diabétiques de type 2 mais
la phosphorylation de la protéine en réponse à l'insuline serait par contre réduite. De plus,
comme AS 160 est une protéine possédant une activité Rab GTP-ase, plusieurs membres de
la famille des Rab pourraient être impliqués dans le processus de translocation des GLUT4 ·
[84] [85]. Il est à noter que la contraction musculaire occasionne aussi la phosphorylation de
l'AS160 et permettrait ainsi la translocation de GLUT4 et le transport du glucose [86].
Finalement, phosphoinositide kinase for five position containing a Fyve finger (PIKfyve)
et Synip seraient aussi activées par l'insuline en fonction de l'Akt et pourraient aussi être
impliquées dans la translocation de GLUT4 [87] [88].
2.6 Les PKC atypiques
Parmi les trois classes de protein kinase C (PKC) existantes - conventionnelles (u, p, y),
nouvelles (8, E, v, 0) et atypiques (À, Ç) -, seules les dernières sont activées par l'insuline.
Ces PKC sont qualifiées d'atypiques puisque, contrairement aux formes classiques, elles

21
n'ont pas besoin de diacylglycérol ni de calcium pour leur activation [89] [90]. Elles sont
cependant, sensibles à la présence de PI(3 ,4,5)P3, produit de la PI 3-kinase [91] .
L' interaction entre les PI (3 ,4,5)P3 et les PKC atypiques occasionnera un changement de
conformation, ce qui les rendra sensibles à la phosphorylation par PDKI.
Tout comme l'Akt, les PKC atypiques semblent importantes pour permettre la translocation
des GLUT4 par l' insuline et rendre possible le transport du glucose. Plusieurs études
témoignent de leur importance dans ce processus. D'abord, la surexpression d'une forme
constitutivement active ou sauvage des PKC,,) Ç dans des cellules de muscles ou de tissu
adipeux mime l'action de l'insuline sur le transport de glucose alors que la présence de
dominant négatif produit l'effet contraire [92] [93]. Une autre étude montre que
l'expression de PKCÂ rétablit le transport du glucose stimulé par l' insuline dans des
cellules invalidées pour cette kinase. À l' opposé, une diminution de l' expression des deux
formes atypiques à l' aide de petits ARN interférant dans des adipocytes 3T3-L1 n' a pas
suffi à produire un défaut de stimulation de transport de glucose par l' insuline [94]. Il est à
noter cependant, qu'un défaut d'activation de PKC atypique chez l'humain diabétique de
type 2 de même que chez l'humain obèse intolérant au glucose a été observé [95] [96] [97].
Il faudra donc chercher encore avant d' établir la contribution exacte des PKC atypiques
dans le transport du glucose stimulé par l'insuline.
2.7 Les transporteurs de glucose: GLUT
L'un des objectifs ultimes de cette cascade de signalisation provoquée par l'insuline est de
permettre l 'entrée du glucose dans les cellules.
La membrane lipidique des cellules étant imperméable aux molécules comme les hydrates
de carbone, le transport du glucose doit s' effectuer par l' intermédiaire de transporteurs
particuliers, les GLUT. Jusqu'à maintenant, quatorze membres de la grande famille des
transporteurs de glucose ont été caractérisés. Ces protéines sont le produit de gènes
différents possédant des caractéristiques particulières au niveau de leur localisation

22
tissulaire et cellulaire, de leur affinité pour le glucose et de leur .expression en réponse à
certains facteurs particuliers. Grâce à leur diversité et à leurs caractéristiques, les GLUT
pennettent un fin contrôle de l'homéostasie glucidique. La grande famille des GLUT est
subdivisée en trois classes. La classe 1 regroupe les isofonnes les plus caractérisés :
GLUT1 , GLUT2, GLUT3 et GLUT4. La classe II inclut le transporteur de fructose
GLUT5, ainsi que trois autres protéines semblables: GLUT7, GLUT9 et GLUT11. La
classe III, enfin, est composée de GLUT6, GLUT8, GLUT10, GLUT12 et HMIT
(cotransporteur de proton/myoinositol) [98].
GLUTI fut identifié à l ' origine dans les érythrocytes, mais on sait aujourd hui qu' il est
exprimé de façon constitutive dans la plupart des tissus. Cet isofonne est responsable du
transport basal de glucose. GLUT2 est exprimé dans les cellules épithéliales de l'intestin,
aux reins, dans les hépatocytes et dans la membrane plasmique des cellules insulino
sécrétrices du pancréas et dans certaines régions de l'hypothalamus. La localisation
tissulaire de GLUT2 laisse supposer que ce transporteur joue un rôle majeur dans le
transport trans-épithélial du glucose au niveau de l'intestin et du rein [99]. Au niveau des
cellules f3 du pancréas, il pennet un équilibre rapide du glucose entre les espaces extra et
intracellulaires contrôlant ainsi la sécrétion de l'insuline [100]. Le rôle hypothalamique de
GLUT2 n'est pas connu avec précision. Il pourrait cependant intervenir dans les
mécanismes de détection des neurones sensibles au glucose contrôlant la satiété [101]. Au
niveau du foie, GLUT2 est nécessaire aussi bien au captage du glucose qu'à son
relâchement dans le sang au cours du jeûne. L'absence de GLUT2 induit chez la souris un
phénotype diabétique [102]. GLUT3 pour sa part est le principal transporteur de glucose
dans les neurones [103]. GLUT4 est présent dans le muscle et le tissu adipeux et est
responsable du transport de glucose en situation postprandiale et lors de l'exercice [104].
Comme les isofonnes 1 et 4 sont les principales responsables du transport de glucose basal
et en réponse à l'insuline au muscle, j'approfondirai davantage leurs caractéristiques et leur
régulation.

----------,
23
2.7.1 GLUTI
Le transporteur de glucose GLUT1 fut le premier à être cloné. Même si GLUT1 est
responsable du transport basal de glucose, son expression et son activité peuvent varier.
GLUT1 peut être considéré comme une protéine de stress. Il est en effet conce~able qu' en
situation stressante les cellules aient rapidement besoin de glucose et que GLUT1 permette
de répondre à ce besoin. Dans des cellules de muscle L8 en culture, la présence
d' ionophore de calcium, de 2-mercaptoethanol ou de tunicamycine, cause une forte
augmentation de l' expression de GLUT1 [105]. GLUT1 peut aussi être induit par les
concentrations environnantes de glucose. Ainsi, l' expression de GLUT1 augmente dans des
hépatocytes en culture maintenus dans un milieu à concentration de glucose sous la
normale et au foie de rats à jeun [106]. Le même phénomène est observable dans une
culture de cellules de muscles L6 en absence de glucose pour une période prolongée [107].
La dénervation de certains muscles peut aussi augmenter l' expression de ce transporteur
[108]. La présence de NO peut contribuer à l'induction de GLUT1 dans différents tissus.
Dans des cellules humaines endothéliales de veine ombilicale (HUVEC), la présence de
deux donneurs de NO différents, SNP et DETA, augmente l' expression de GLUT1 [109].
De plus, une étude de notre laboratoire a démontré que l' ajout de cytokines et de
l' endotoxine LPS dans le milieu de culture de cellules de muscles L6 augmentait
l ' expression de GLUT1 et que la présence d'un inhibiteur de NOS bloquait cette induction,
suggérant l' implication du NO dans cet effet [110]. De plus, l'hypoxie, le choc osmotique
. et certains éléments tels le facteur de croissance endothéliale vasculaire (VEGF) et le TNF a
peuvent causer une augmentation de l'expression de GLUT1 [111] [112] [113] [114].
Finalement, le stress oxydatif pourrait aussi induire l'expression de GLUT1. En effet, le
stress oxydatif est reconnu pour moduler certains facteurs de transcription tel que l' AP l , un
facteur impliqué dans la régulation de ce transporteur de glucose [115]. Des études
réalisées dans les myocytes L6 de même que dans des adipocytes 3T3-L1 démontrent qu'un
stress oxydatif imposé pendant dix-huit heures par le H202 cause d'ailleurs l'induction de
GLUT1 [113].
Il semble par ailleurs que l'activité intrinsèque des GLUT1 puisse être modifiée par un
mécanisme impliquant l'AMPK (Les rôles de cette kinase seront discutés ultérieurement).

24
En effet, dans des cellules épithéliales de foie de rat (clone 9 ceUs) , le transport basal de
glucose par GLUTI est augmenté lors d'un stress osmotique par un mécanisme mal défini,
mais qui ferait en sorte que le transporteur de glucose serait démasqué et, ainsi, plus apte à
lier le glucose [116].
2.7.2 GLUT 4
En présence d' insuline, le glucose pénètre dans le muscle et le tissu adipeux à l' aide des
transporteurs GLUT4. Pour cela, les transporteurs GLUT4 doivent rapidement gagner la
surface des cellules. Au niveau du muscle squelettique, il a été démontré qu'en plus de l~
membrane plasmique, l'insuline induisait la translocation de GLUT4 au niveau d'un second
compartiment de surface, les tubules-T [117] [118]. Ces transporteurs sont en lent transit
continuel entre le cytoplasme et la membrane plasmique et ce processus est accéléré en
présence d ' insuline [119]. D'un autre côté, l' exercice va aussi permettre l' entrée de glucose
par le déplacement des transporteurs GLUT4 à la membrane plasmique et aux t-tubules
[120][121]. Dans le cytosol, les transporteurs de glucose GLUT4 se retrouvent associés à
des vésicules et la production de ces vésicules enrichies de GLUT4 est augmentée par la
présence d' insuline [122].
Pour que cette translocation des GLUT4 ait lieu dans les cellules musculaires, les filaments
d 'actine de .la cellule doivent se réorganiser en réponse à l'insuline. En présence d' agents
pharmacologiques bloquant ce processus de remodelage, la translocation des GLUT4 est
presque totalement abolie dans les cellules de muscles et en partie diminuée dans les
adipocytes [123] [81][124] [125] [126] [127] [128]. Cependant, tel qu'abordé dans la
section du rôle de la PI 3-kinase dans l'action de l'insuline, la simple translocation des
GLUT4 ne semble pas suffisante pour permettre l'entrée du glucose dans la cellule. En
effet, la présence de PI(3,4,5)P3 permet la translocation des GLUT4 à la membrane mais
cela ne suffit pas à provoquer la réponse normale de l' insuline [129] [130] [131]. De plus,
dans des cellules de muscles, la présence d'une faible concentration de wortmannine, un
inhibiteur de la PI 3-kinase, inhibe davantage le transport du glucose que la translocation
des GLUT4. Pour inhiber les deux processus, il faut utiliser une dose plus importante de

25
l' inhibiteur [132]. L' étape subséquente pourrait être la dissociation d'une protéine
inhibitrice associée à GLUT4 ou un changement conformationnel. Il fut proposé que,
malgré la translocation du transporteur en présence de PI(3 ,4,5)P3 ajouté de façon
artificielle, sa section C-terminale ne serait pas insérée dans la membrane - comme c' est
le cas à la suite d'une stimulation par l' insuline [129].
Grâce à des inhibiteurs pharmacologiques, certains résultats suggèrent que l' activité des
transporteurs serait sous le contrôle de la voie p38 MAPK. Des études récentes, réalisées à
l' aide de la technologie d ' interférence à l 'ARN, n'ont cependant pas permis de confmner la
responsabilité de cette avenue de signalisation [133] [134].
Les résultats probants de plusieurs rech~rches démontrent que les GLUT 4 seraient
déterminants dans la sensibilité à l'insuline. Dans le muscle, la résistance à l'insuline serait
principalement due à l'incapacité de recruter les GLUT4 à la membrane en réponse à
l'insuline, alors qu'au niveau du tissu adipeux, l'express,ion réduite des transporteurs serait
plutôt en cause. De plus, des souris invalidées pour l'expression de GLUT4 spécifiquement
dans le tissu adipeux deviennent résistantes à l'insuline au niveau muscles et au niveau foie
indiquant l'importance de ce transporteur de glucose dans la sensibilité à l'insuline [135].
Dans le même ordre d' idée, l' absence de GLUT4 spécifiquement dans le muscle cause,
chez la souris, la résistance à l' insuline dans ce tissu [136] [137].

26
GlUT
Glycogen synthestSJ PP1
Gen~SSion
Figure 1 : Représentation schématique de certains éléments de signalisation intracellulaire
activés via le récepteur de l' insuline et impliqués dans la translocation de GLUT4.
Adapapté de la page internet du Dr Arun Srinivasan .
http://arunbt.googlepages. com/pi3k.jpg
2.8 La résistance à l'insuline
Lorsque la résistance à l ' insuline s'installe, l'hormone ne peut agir aux tissus normalement
sensibles à l'hormone qui sont" résistants" à son action. Ceci oblige le pancréas à produire
de plus en plus d' insuline pour compenser. Le pancréas finira ultimement par se fatiguer et
par ne plus fournir assez d' insuline entraînant alors l ' apparition du diabète de type 2 [138].
La source de la résistance à l'insuline n'est pas identifiée avec précision, mais on devine
qu'une combinaison de facteurs génétiques et environnementaux est en cause. L'adiposité
est définitivement un facteur associé à la résistance à l'insuline. Il est bien établi que
l'accumulation excessive de tissu adipeux mène à la sécrétion de cytokines et des

27
hormones; les adipokines. À une époque où la nourriture se faisait rare, quand il fallait
chasser pour se nourrir et souffrir de longues périodes de jeûne, il était utile - voire
nécessaire - de mettre en réserve les graisses. Mais, aujourd'hui, alors que la noUrriture
est généralement disponible et l'exercice, la dépense d' énergie, moins répandus, l'obésité
est un tueur redoutable.
2.8.1 Rôle de l'inflammation dans la résistance à l'insuline
La survie des organismes dépend largement de l'habileté qu' ils ont à se défendre contre les
infections au moyen de leur système immunitaire; mais ce même processus, lorsqu' il est
mal contrôlé, peut conduire à la résistance à l'insuline. L'obésité peut désormais être
considérée comme une maladie inflammatoire. En effet, plusieurs études d'expression de
gènes démontrent, une nette induction de différents facteurs inflammatoires et de stress
dans le tissu adipeux de modèles d'animaux obèses [139] [140] [141].
2.8.1.1 Les cytokines pro-inflammatoires
Une augrpentation de la concentration de tumor necrosis factor a (TNFa) en circulation
peut, de différentes manières, causer la résistance à l'insuline. ' On a d'abord observé
l'augmentation de cette cytokine dans le tissu adipeux du rongeur au moyen de différents
modèles d'obésité [142] [143]. La présence de TNFa fut ensuite retracée dans le tissu
adipeux et le muscle de sujets humains obèses [144] [145] [146]. Bien que les adipocytes
en soit produisent du TNFa, on sait aujourd'hui que les macrophages infiltrant le tissu
adipeux d ' animaux obèses seraient les grands responsables de cette production massive
[147]. Le lien entre la TNFa et la résistance à l'insuline fut établi lorsque l'on a observé que
l'administration de TNF a à des animaux ou à des cellules musculaires en culture causait la
résistance à l'insuline. Plus convaincant encore est le fait que l'expression transgénique
d ' une forme non fonctionnelle du TNFa ou de son récepteur chez les souris obèses leur
procure une protection contre la résistance à l'insuline [144] [148].

28
Le TNFa est aussi en mesure de causer la phosphorylation d'IRS-l sur la sérine 307 (sérine
312 chez l'humain). Cette modification d'IRS-l est associée à une réduction de la
phosphorylation sur les résidus tyrosine d'IRS-l , ce qui diminue à son tour son association
avec IR [149]. En plus d'être associé à la phosphorylation de la sérine 307 d'IRS-l , le
TNFa peut causer la phosphorylation sur la sérine 632 d'IRS-l qui se trouve à proximité du
site de liaison de ' la PI 3-kinase. Le fait que cette phosphorylation soit sensible à la
rapamycine - un inhibiteur de mTOR (mammalian target of rapamycin) - sous-entend
que cette voie est concernée dans cet effet du TNFa [150]. Une étude récente montre que
les facteurs myosin le (Myolc) et nuclear factor kappa B essential modulator (NEMO)
seraient aussi essentiels pour l'effet du TNFa sur la phosphorylation sur sérine 307 d IRS-l
[151]. Le TNFa peut aussi diminuer l'expression d'IRS-l et l'expression des GLUT4 [152]
[153]. Finalement, un nouveau mécanisme détériorant le métabolisme de glucose par
l'action de cette cytokine fut récemment- établi. Dans les circonstances de l ' étude, la
présence de TNFa causait l'induction de la phosphatase 2C inhibant l'activation de l'AMPK
causant la résistance à l'insuline [154]. L'importance de cette kinase sera discutée
ultérieurement.
Cette cYtokine peut aussi être indirectement responsable de la résistance à l'insuline. Étant
un facteur inflammatoire, le TNFa peut, par exemple, conduire à la production d'autres
cytokines telles que l'interleukine-6 (IL-6) [155]. Le TNFa provoque également la lipolyse
des adipocytes, ce qui a pour effet d ' augmenter la quantité d'acides gras libres en
circulation et altérer la sensibilité à l'insuline [156]. Le rôle de ces facteurs dans la
résistance à l'insuline sera examiné ultérieurement dans cette section.
Le TNF a contribue encore à la résistance à l'insuline par la production de céramides. Il
active la sphingomyélinase, ce qui dégrade les sphingolipides et a, à son tour, pour effet de
produire des céramides [157]. Cette production de céramides serait associée à la
phosphorylation d'IRS-l sur sérine. Les céramides peuvent aussi causer la résistance à
l'insuline en interférant avec la translocation de l'Akt à la membrane plasmique et par la
déphosphorylation d'Akt par l'activation de la protéine phosphatase 2A [158] [159] [160]. Il

29
faut noter, incidemment, que la quantité de céramides retrouvée au niveau des muscles de
patients obèses est deux fois plus élevée que celle observée chez des sujets normaux [161].
Le TNFa peut aussi causer la résistance à l'insuline, du moins dans les adipocytes 3T3-L1 ,
en augmentant la présence de gangliosides (GM3). Les GM3 se situent dans des
microdomaines de la membrane plasmique et sont reconnus pour causer la résistance à
l'insuline dans les adipocytes [162]. Des souris génétiquement modifiées, incapables de
produire de GM3 , sont plus sensibles à l'insuline et sont protégées contre la résistance à
l'hormone induite par une diète riche « en gras» [163]. Il est aussi important de noter que le
TNF-u est un facteur contribuant à l' induction de iNOS, un joueur majeur dans la résistance
à l' insuline [164][165]. Les effets de cette enzyme et de son produit; le NO seront discutés
en détail dans une section ultérieure. Finalement, le TNFa bloque la production
d'adiponectine, une adipokine favorisant la sensibilité à l'insuline [166].
En dépit de, l'implication du TNFa dans la résistance à l'insuline, son rôle dans cette
pathologie chez l'humain reste controversé puisque la neutralisation de cette cytokine chez
des sujets diabétiques n'a pas permis d'améliorer la sensibilité à l'insuline et ce d'autant plus
que certaines différences distinguent le TNF a du rongeur et de l'humain [167]. En effet,
contrairement à ce qui est observé chez le rongeur, le TNFu ne serait pas relâché dans la
circulation à partir du tissu adipeux chez l'humain [168].
Outre le TNF -u, plusieurs autres cytokines pro-inflammatoires seraient impliquées dans la
résistance à l ' insuline c' est le cas de l'IL-1 et l'interféron-y qui sont surexprimés dans le
tissu adipeux de sujets humains obèses [144]. Des études chez l'humain ont aussi montré
que les cellules du tissu adipeux pouvaient aussi produire de l'IL-6. La présence de l ' IL-6
est augmentée avec l'indice de masse corporelle et le degré de résistance à l'insuline [169].
Cette cytokine a des effets directs sur la signalisation de l'insuline dans les adipocytes et les
hépatocytes [170]. L'IL-6 diminuerait l'activité d'IRS-1 , possiblement par l'activation de
supressor of cytokines signaling (SOCS-3) [171]. Comme c'est le cas pour le TNFa, l'IL-6
aurait des effets indirects sur la sensibilité à l'insuline par la stimulation de la lipolyse et la
libération d'acides gras [172]. C'est le cas chez l'humain où l'administration d'IL-6

30
augmente la lipolyse et inhibe le métabolisme du glucose [173]. Cela dit, les effets de l ' IL-
6 sur le métabolisme sont d ' autant plus complexes que les souris invalidées pour le gène de
l ' IL-6 deviennent obèses. Cette cytokine agirait au niveau central afin de contrôler la
dépense énergétique [174]. De plus, chez l 'humain, les niveaux d ' IL-6 dans le liquide
cérébrospinal sont inversement proportionnels au degré d ' obésité [175]. Le rôle de l' IL-6
dans la résistance à l'insuline demande donc à être mieux compris.
Plusieurs autres facteurs inflammatoires sont induits par l'obésité. Parmi les autres facteurs
inflammatoires possiblement impliqués dans la résistance à l' insuline, on a identifié l'acide
sialic et la protéine C-réactive [176] [177]. L ' apeline, une adipokine, est également induite
dans différents modèles d ' obésité ainsi qu'en réponse au TNFa dans les adipocytes. Le rôle
de cette dernière dans la sensibilité à l' insuline n ' a cependant pas clairement défini pour le
moment [1 78].
2.8.1.4 Le rôle des macrophages dans la résistance à l'insuline
Le tissu adipeux blanc se compose principalement d'adipocytes ; mais on y retrouve aussi
d'autres types de cellules, dont les macrophages. Le nombre de macrophages retrouvé dans
le tissu adipeux blanc est proportionnel à l'adiposité et à la grosseur des adipocytes ; tant
chez l'humain que chez les rongeurs [179] [141]. Bien qu' il y ait de nombreuses similitudes . entre les macrophages et les adipocytes - les préadipocytes pouvant même se différ·encier
en macrophages - , l'augmentation du nombre de macrophages associée à l'obésité provient
de la moelle osseuse, il s'agit donc de macrophages infiltrants [180]. Cette accumulation de
macrophages pourrait être impliquée dans la résistance à l'insuline et ce d'autant plus
qu'elle est la 'source majeure de TNFa et serait responsable d'environ 50 % de la
production d'IL-6 [141]. Une étude récente a suggéré que la présence des macrophages
dans le tissu adipeux serait cyclique et aurait pour but la réorganisation du tissu adipeux en
débarassant celui-ci des adipocytes morts; trop gros, laissant place à de nouveaux
adipocytes plus petits. Encore dans cette étude, le lien entre la résistance à l'insuline et la

31
présence des macrophages est souligné puisque le moment fort de leur occupation du tissu
adipeux correspond aux périodes de résistance à l ' insuline plus marquée de l' animal [181].
2.8.2 Les mécanismes moléculaires de la résistance à l'insuline
Les facteurs impliqués dans la résistance à l'insuline iront activer certaines VOles
contrecarrant la signalisation de l'insuline à divers niveaux.
2.8.2.1 La sérine/thréonine kinase c-jun N-terminal kinase: JNK
Puisque la phosphorylation d'IRS-1 sur les résidus sérine cause souvent une diminution de
son activité, beaucoup de recherches se sont intéressées à des kinases capables d' agir sur
ces résidus. On a par exemple cherché à établir le rôle que pourrait jouer la sérine/thréonine
kinase c-jun N-terminal kinase (JNK) dans la résistance à l'insuline en regard de l'obésité.
On a ainsi observé que l'activité JNK était anormalement élevée dans le foie, le muscle et le
tissu adipeux de modèles murins diabétiques de type 2. Des souris invalidées pour le gène
JNK sont par ailleurs protégées contre l'obésité et la résistance à l'insuline induite par une
diète riche en lipides [26]. La résistance à l'insuline associée à l'activation de JNK serait
due à la phosphorylation de IRS-1 sur la sérine 307 [182]. Cependant, puisque ces souris
sont aussi résistantes à l'obésité, il a été difficile de déterminer si JNK est vraiment en cause
dans la sensibilité à l'insuline observée ou si le responsable de cet effet ne serait pas plutôt
la résistance au gain de poids. Des études récentes sont cependant venues confirmer le rôle
de JNK dans la résistance à l' insuline. On a pu démontrer en effet que la surexpression d'un
adénovirus dominant négatif contre JNK au niveau du foie de souris obèses et résistantes à
l'insuline améliore clairement leur sensibilité à l'hormone en supprimant la production de
glucose hépatique et ce, sans modifier l'adiposité [183]. De plus, JNK doit s'associer à une
protéine no'mmée JNK-interacting protein-l (lIP 1) pour avoir une activité normale. Les
souris n'exprimant pas le gène lIP1 sont donc, elles aussi, plus sensibles à l'insuline [184J.

32
Finalement, une récente étude a démontré que l' absence de JNK 1 spécifiquement des les
cellules hématopoïétiques protégeait contre la résistance à l ' insuline induite par une diète
riche en «gras» sans modifier l' adiposité [185]. Dans un même ordre d' idée, il est
intéressant de noter que la voie JNK serait importante dans l' activation par les acides gras
libres des macrophages infiltrant le tissu adipeux d' animaux soumis à un régime riche en
« gras » [186] .
. 2.8.2.2 La sérine/thréonine kinase :IKKp
La kinase IKK~ est responsable de la phosphorylation de IKB qui permet l'activation du
facteur de transcription NF -KB. Cette kinase aurait aussi un rôle à jouer dans la résistance à
l'insuline puisqu' il a été démontré que les souris hétérozygotes pour IKK~ sont
partiellement protégées contre la résistance à l'insuline induite par une infusion de lipides,
une diète riche en « gras» ou par une modification génétique les rendant obèses [187]
[136]. De plus, une étude a démontré que des souris invalidées pour le gène IKK~
spécifiquement dans les cellules myéloïdes étaient complètement protégées contre la
résistance à l' insuline associée à une diète riche en « gras », à l' obésité ou au vieillissement
[188]. IKK~ pourrait avoir cet impact sur la sensibilité à l'insuline en activant le facteur de
transcription NF-KB, permettant ainsi la production d'autres facteurs inflammatoires [189]
[190].
2.8.2.3 Stress du réticulum endoplasmique : ER stress
On a récemment émis l' hypothèse que l'obésité surchargeait les capacités du réticulum
endoplasmique, ce qui y causerait un stress, le ER stress. Ce stress cellulaire contribuerait à
la résistance à l'insuline en activant des voies inflammatoires telles que JNK [191]. Le ER
stress ~st causé par une accumulation au réticulum endoplasinique de protéines mal formées
et une protéine chaperone, la ORP150, permettrait de réduire ce stress. Dans un modèle
murin diabétique de type 2, la surexpression de ORP150 améliore d'ailleurs la sensibilité à

33
l'insuline au niveau du foie et du muscle [192]. Il fut aussi proposé que les inhibiteurs de
protéases utilisés contre le HIV causaient comme effet secondaire la résistance à l' insuline
en surchargeant le reticulum endoplasmique de protéines qui auraient dues être dégradées
[193]. Dans des cellules adipeuses 3T3-L1 , une augmentation du ER stress fut aussi associé
à une induction de iNOS ce qui pourrait contribuer à la résistance à l' insuline (le rôle de
iNOS dans la résistance à l' insuline sera discuté ultérieurement) [194].
2.8.2.4 Suppressors of cytokine signaling (SOCS)
Les SOCS sont activés par des cytokines dont IL-l , IL-6, TNFa et IFNy. Elles ont pour
objectif de limiter la suractivation des récepteurs des cytokines mais sont également
étudiées en regard de leur éventuelle implication dans la résistance à l'insuline associée à
l'inflammation [195]. On a ainsi pu établir que les SOCS-1 ,3 et 6 peuvent interagir avec
IRS-1 , bloquant ainsi sa tyrosine phosphorylation et son association avec la PI 3-kinase
[196] [197] [198]. L'interaction des SOCS avec IRS-1 et IRS-2 peut aussi avoir pour effet
de les diriger vers les protéosomes afin de les dégrader [199] [4].
2.9 Autres facteurs adipocytaires importants dans la sensibilité à l'insuline
2.9.1 Adiponectine
L'adiponectine, aussi connue sous le nom de Acrp30 et AdipoQ, est réduite dans le ,diabète
de type 2 [200]. Une réduction des niveaux d'adiponectine chez les individus en santé est
d'ailleurs une prédisposition à la . résistance à l'insuline [201]. Plusieurs études montrent
qu'une diminution de l'adiponectine est associée à une augmentation de l'indice de masse
corporelle et à une augmentation du taux de glucose, d'insuline et de triglycérides sanguins

34
tant chez l'humain que chez le rongeur [202]. Cette hormone, exclusivement sécrétée par
les adipocytes, est formée d'une section collagène et d'un trimère globulaire.
L'adiponectine se retrouve en différents complexes. Dans le sérum, l'adiponectine peut être
exprimée sous la forme d'un dimère de trimère (la forme de bas poids moléculaire LMW
camplex) ou sous celle, plus large, de 12 à 18 sous-unités [203] [204]. On comprend
aujourd'hui que le rapport de ces deux formes est plus important que leur quantité
respective pour prédire la sensibilité à l'insuline. L'adiponectine favorisera la sensibilité à
l'insuline par différents moyens. Cette adipokine diminue l'expression de deux enzymes
clés de la production de glucose par le foie : la glucose-6-phosphatase (G6Pase) et la
phosphoenolpyruvate carboxykinase (PEPCK). L'adiponectine augmente le transport de
glucose dans le muscle et l'oxydation des acides gras. Elle favorise l'expression d'UCP2 et
d'UCP3 ; ce qui a pour effet d' augmenter la dépense énergétique [205].
2.9.2 La résistine
La résistine fut d'abord décrite comme une adipokine puisqu'elle fut isolée d'adipocytes.
Alors que chez les rongeurs, la principale source de résistine est l'adipocyte, ce sont plutôt
des macrophages dans la circulation sanguine qui en sont responsables chez l'humain[206].
La résistine tient son nom du fait que chez la souris, son injection cause une intolérance au
glucose et une diminution de la sensibilité à l'insuline. D'ailleurs, l'administration
d'anticorps neutralisant l'hormone réduit l'hyperglycémie de souris induite par une diète
riche « en gras» [207]. Le foie serait une cible déterminante dans la résistance à l'insuline
induite chez l'animal par la présence de ce facteur [208]. Chez l'humain, par contre, le rôle
de la résistine dans la résistance à l'insuline est ambigu.
2.9.3 La leptine
La leptine est une hormone majeure dans le métabolisme glucidique et lipidique. Il n'y a
pas actuellement de preuve d'un effet direct de la leptine sur la signalisation de l'insuline,

35
mais plusieurs des effets de l'hormone profitent indirectement à la sensibilité à l'insuline. La
leptine permet, par exemple, de réduire l'appétit, d'augmenter la dépense énergétique et de
stimuler l'oxydation des lipides au niveau du foie et du muscle. Les souris dont le gène de
la leptine (ob/ob) ou du récepteur de la leptine (db/db) est éliminé deviennent obèses. Ces
modèles sont très utiles pour la recherche sur le métabolisme. Chez ces animaux déficients
en leptine, on observe une susceptibilité aux infections, suggérant que l'hormone joue aussi
un rôle anti-inflammatoire [209].
2.9.4 Les acides gras libres
Dans les cas d'obésité, il y aura accumulation excessive de « gras» dans le tissu adipeux,
mais aussi dans le muscle squelettique et le foie, et une augmentation d'acides gras libres en
circulation. La concentration d'acides gras libres est directement liée à l'hyperglycémie, la
résistance à l'insuline au niveau du muscle et le risque de développer le diabète de type 2
[210].
À l'origine, le groupe de Randle expliqua que la résistance à l'insuline induite par les acides
gras libres provenait de la compétition entre l'utilisation du glucose et des acides gras par la
cellule. Leur hypothèse avait pour prémices le fait qu'en situation d'abondance d'acides
gras, la cellule les oxyde préférentiellement et cause une accumulation de citrate, ce qui a
pour effet d' inhiber la phosphofructokinase (PFK) et de causer, par voie de conséquence,
une accumulation de glucose-6-phosphate dans la cellule [211]. Randle et son groupe
proposèrent donc que l'accumulation de ce glucose intracellulaire causerait une inhibition
de l'entrée supplémentaire de glucose. Ce mécanisme de résistance à l'insuline fut revu par
le groupe de Shulman [212] [213]. Cette équipe démontra que les acides gras libres chez
l'humain causaient une diminution de la synthèse de glycogène au niveau du muscle
squelettique en raison d'une réduction de la quantité de glucose-6-phosphate dans la
cellule. On a donc compris qu'une diminution de l'entrée de glucose dans la cellule, plutôt
que son accumulation, était à l'origine du défaut induit par les acides gras libres [214].

36
Au ruveau du muscle squelettique, différents mécanismes expliquent la résistance à
l'insuline induite par les acides gras libres. Comme les PKCs (mis à part les formes
atypiques) sont activées par les lipides, plus particulièrement par le diacylglycérol (DAG),
ces kinases furent suspectées de jouer un rôle dans la résistance à l'insuline induite par les
acides gras libres. On a établi que l'accumulation de DAG par une augmentation
plasmatique d'acides gras libres activait la PKCpII et PKC8 chez l'humain et la PKC theta
chez le rat [215] [216]. L'activation de ces PKC pourrait causer une phosphorylation sur les
résidus sérine et thréonine des substrats du récepteur à l'insuline [217].
On a aussi démontré que la présence d'acides gras libres causait l'activation de NF -KB via la
dégradation de IKB pouvant ainsi induire des gènes inflammatoires impliqués dans la
résistance à l'insuline [218]. On sait aussi que les acides gras libres augmentent l'expression
de GLUTI et diminuent celle de GLUT4 contribuant à la diminution de la réponse à
l'insuline [219].
Dans une étude récente, des adipocytes 3T3-Ll traités avec un mélange d'acides gras
présentaient une diminution de la translocation de GLUT4 en réponse à l'insuline. Cette
résistance à l'insuline par la présence d'acides gras libres était accompagnée d'une activation
de JNK, d'IKKp, SOCS3, d'une augmentation de TNFa et d'une diminution de la sécrétion
d'adiponectine dans le milieu de culture [220].
Il faut aussi dire, finalement, que les acides gras libres inhibent la capacité de l'insuline à
supprimer la production de glucose par le foie. Le mécanisme de cette résistance dans le cas
du foie n'est pas encore déterminé, mais puisque les acides gras libres peuvent y activer la
PKC8, celle-ci pourrait bien être en cause - comme elle l'est, dans les mêmes
circonstances, dans le cas du muscle [221].

~-~ --------~-----
37
3. L'AMPK
La kinase activée par l'AMP (AMPK) fut d' abord perçue comme un simple détecteur et un
régulateur d' énergie à l' intérieur de la cellule. Une réduction en ATP intracellulaire active
en effet l' AMPK dans le but de régénérer et de conserver l' énergie de la cellule. On sait
maintenant que l' AMPK contrôle la balance énergétique de tout l'organisme en répondant
aux signaux hormonaux et nutritionnels des tissus périphériques et du système nerveux
central. D'ailleurs, l' orthologue de l'AMPK chez la levure nommée SNFI est activé en
réponse au stress causé par l' absence de glucose [222].
L'AMPK chez le mammifère fut découverte, simultanément, par deux groupes. En 1973,
Beg et ses collaborateurs découvraient qu'une enzyme phosphorylait et inhibait la 3-
hydroxy-3-methylglutaryl-coenzyme A reductase (HMG-Coa reductase) réduisant ainsi la
synthèse de cholestérol [223]. Pendant ce temps, Carlson et Kim observaient que l 'acetyl
CoA carboxylase (ACC) était phosphorylée et inhibée par une kinase non identifiée [224].
Plus tard, au milieu des années 80, le groupe de Carling purifiait cette enzyme qui, en
présence d'AMP, phosphorylait l'ACC avant de découvrir que cette kinase était aussi
responsable de la phosphorylation de l'HMG-Coa réductase. On la rebaptisa la kinase
stimulée par l' AMP - (AMPK) [225] -[226].
3.1 La structure de l'AMPK
L'AMPK est un hétérotrimère composé d'une sous-unité catalytique a et de deux sous
unités non catalytiques P et y. Les sous~unités sont exprimées sous différents isoformes : la
sous-unité a est exprimée sous les formes a 1 et a2, la sous-unitép en pl et p2 et la sous
unité y en yI , y2 et y3. Les différents agencements d' isoformes pourraient servir à répondre
à différents stimuli selon les tissus, mais la raison fondamentale de toute cette diversité
n' est pas encore connue. L' AMPK est exprimée de façon ubiquitaire même si les différents
isoformes des sous-unités varient selon les tissus étudiés [227] [228].

38
La sous-unité a est de 62 kDa et contient le domaine sérine/thréonine kinase dans la partie
N-terminale. Son activation permet de phosphoryler des protéines sur une séquence
revêtant des caractéristiques particulières. La séquence protéique des protéines ciblées par
l'AMPK contient un résidu hydrophobique en position -5 et +4 du site de phosphorylation
et au moins 1 résidu basique entre - 1 et --4. Par contre, cette séquence dite consensus ne se
retrouve pas dans toutes les cibles de l' AMPK [229]. La sous-unité a est phosphorylée en
position N-terminale sur le résidu thréonine 172 (Thr-172) et permet ainsi son activité. À
l'opposé, deux résidus sérines (ser-485) et (ser-491) peuvent être phosphorylés sur les sous
unités al et a2 respectivement et réduisent ainsi l'activité de l'AMPK. Il est aussi proposé
que l'AMPK pourrait s'autophosphoryler sur ces sites pour limiter son propre signal [230].
Les deux isoformes a 1 et a2 sont présentes dans le cytosol mais certaines études indiquent
que la sous-unité a2 se localise aussi dans le noyau, suggérant un rôle pour la transcription
de gène [231] [232]. D'ailleurs, l' AMPK fut impliquée dans la régulation des facteurs de
transcription TORC2, HNF4, SREBP1c et ChREBP [233] [234] [235]. Les sous-unités al
et a2 sont exprimées dans tous les tissus, mais elles auraient des impacts différents sur les
fonctions de l'AMPK. Par exemple, l' hétérotrimère composé de la sous-unité a2 serait plus
dépendant de l'AMP et essentiel à l'exercice, même s' il est présent en quantité semblable à
a 1. Pour sa part, a 1 serait recruté lors de stress cellulaires plus forts , par exemple en
situation d'hypoxie ou d ' intense activité physique [236]. Les souris AMPKa1-Ko ont une
homéostasie normale du glucose alors que les AMPKa2-KO sont moins sensibles à
l' insuline dans les muscles et ont un défaut de production d'insuline. On doit cependant
noter que les défauts de ces souris ne s' expliquent pas par un problème de transport de
glucose intrinsèque aux muscles causé par l'absence de sous-unité a2, puisque les muscles
isolés de ces souris répondent normalement à l' insuline. Ce défaut s'expliquerait plutôt par
une production accrue de catécholamines causant ainsi indirectement une résistance à
l' insuline dans le muscle [237].
La sous-unité P est une protéine de 30kDa. Celle qui est exprimée sous la forme pl peut
être phosphorylée et myristosylée, bien que les causes de ces modifications post-

39
transcriptionelles ne soient pas clairement identifiées [238]. Dans le muscle, la sous-unité
p2 est le plus exprimée. La sous-unité P lie tant les sous-unités a que y et pourrait donc
servir de joint entre les sous-unités. Cette sous-unité pourrait aussi réguler l' activité de
l'AMPK en fonction du glycogène puisqu'elle possède un site de liaison à cet effet [239].
Une étude récente a montré que la sous-unité pl interagit avec la glycogen-debranching
enzyme (ODE), une enzyme responsable de la dégradation du glycogène. La raison de
l'association entre l'AMPK et le glycogène n' est pas clairement établie, mais elle
diminuerait selon toute vraisemblance l' activité de l'AMPK [240]. Il n ' existe pas pour le
moment de modèle de modification génétique pour l' isoforme p ,ce qui en fait la sous-unité
la moins connue des trois.
Finalement, la sous-unité y est une protéine de 37 kDa essentielle à l' activité catalytique de
la sous-unité a. Cette sous-unité permet à l' AMPK d'être régulée par l' AMP et l'A TP. En
effet, c'est sur y que se lient les nucléotides sur deux paires de séquences nommées
cystathionine b-synthase [241]. La sous-unité y 3, exprimée uniquemènt au muscle revêt un
intérêt particulier puisqu' elle permettrait éventuellement de cibler l' action d'un
médicament spécifiquement sur ce tissu. Cet isofome peut aussi être induit par
l' entraînement physique spécifiquement dans le quadriceps rouge [242]. L' isoforme y 3
serait déterminant dans le métabolisme du glycogène basé sur un modèle de mutation
génétique connu chez le cochon : Hampshire pig, où l'isoforme y 3 est mutée et où le
glycogène s' accumule de façon anormale au niveau du muscle. En effet, cette mutation
activerait en premier lieu l'AMPK, augmentant le transport de glucose et la synthèse de
glycogène au niveau du muscle causant ainsi une rétroinhibition de l' activité de l' AMPK
qui est sensible à la présence de glycogène [243].
3.2 L'activation de l'AMPK
L'activation de l'AMPK requiert absolument la phosphorylation de sa sous-unité a sur le
résidu thréonine (Thr 172) par les AMPKK [244] [245]. Une interaction directe avec l'AMP
permet à l 'AMPK d'être davantage phosphorylé par les AMPKK. Un deuxième niveau de

40
contrôle par l' AMP est de rendre l' AMPK moins sensible à sa déphosphorylation par les
phosphatases PP2AC et PP2C. D'un autre côté, la présence en fortes concentrations d'ATP
antagonise les effets de l' AMP, procurant ainsi une régulation fine à cette kinase [246].
L' identité de la ou des kinases responsables de la phosphorylation de l'AMPK était jusqu'à
récemment inconnue. C'est grâce à l'analogie faite avec son orthologue SNF-1 qu'une
première AMPKK fut identifiée. Chez la levure, les kinases responsables de l'activation de
SNF-1 se nomment PaK1p, Tos3p et Elm1p. Chez les mammifères, la première kinase étudiée
- à cause de sa ressemblance avec les kinases de SNF-1 - fut la Ca2+/calmodulin
dependent protein kinase kinase (CaMKK) [247]. Cette kinase ne fut pas d'abord considérée
comme la kinase de l'AMPK puisqu'on observait que l'activation de l'AMPK était
indépendante du calcium et que la CaMKK extraite in vitro de cerveau de cochon n'arrivait
pas à activer de façon non équivoque l'AMPK in vitro. On a conclu de ces observations que la
CaMKK ne jouait pas un rôle physiologique significatif dans l'activation de l'AMPK. On a
cependant identifié récemment une autre kinase ayant de fortes similitudes avec la kinase
responsable de l'activation de snfl chez la levure. Cette kinase, présente chez les mammifères,
est LKB 1. Il fut clairement établi que la LKB 1 activait la phosphorylation de l' AMPK. La
stimulation de l'AMPK n'était pas observable dans des cellules Hela n'ayant pas de LKB1 ou
dans des cellules dont le gène LKB 1 avait été éliminé. Cependant, il semblait y avoir une autre
AMPKK en action puisqu'une certaine activation de base était observable même en l'absence
de LKB1 [248].
Par ailleurs on a revu, récemment, le rôle de la CaMKK. Différents groupes ont observé que
l'AMPK pouvait être activée en l'absence de LKB1 , que l'augmentation de Ca2+
intracellulaire menait à l'activation de l' AMPK, que l'expression de la CamKK augmentait
l' activation de l'AMPK, que l' inhibition pharmacologique ou génétique de la CamKK
réduisait l'activation de l'AMPK et , finalement, que l'AMPK in vitro était un substrat de la
CaMKK [249]. Il est à noter que les deux formes de la CaMKK- a et p- peuvent agir au
niveau de AMPKK [250]. Finalement, la transforming growth factor-beta-activated kinase
(T AK1), pourrait aussi causer la phosphorylation et l'activation de l'AMPK [251].

41
3.3 Les modulateurs physiologiques
Plusieurs phénomènes physiologiques conduisent à l' activation de l'AMPK. Puisqu'une des
fonctions majeures de. l'AMPK est de percevoir une baisse d'énergie intracellulaire et de la
corriger, les événements causant une élévation du rapport AMP/ATP et créatine/créatine
phosphate mèneront à son activation [252]. Une autre étude a proposé que le rapport
NADINADH, active l'AMPK. Dans ce cas, la présence en fortes concentrations de NAD
activait la kinase alors que l' ajout de NADH avait l'effet contraire [253].
La contraction musculaire qui cause une augmentation de ces rapports s' est donc
rapidement imposée comme l' un des activateurs clés de cette kinase. Les groupes de
Winder et de Ruderman démontrent que la contraction musculaire, lors de l' exercice ou
d'une décharge électrique, causent l'activation de l'AMPK au niveau du muscle [254]
[255] [256]. Par contre, l'exercice active aussi l'AMPK au foie et au tissu adipeux, ce qui
pourrait s'expliquer par une production d'IL-6 par le muscle. On a en effet suggéré que
cette cytokine jouerait un rôle dans l' activation de l' AMPK lors de la contraction du muscle
; en réponse à l' exercice, on a incidemment observé moins d'activité AMPK chez des
souris invalidées pour le gène de l' IL-6 [257].
Les facteurs de stress cellulaire - l 'hypoxie, une carence en glucose, les chocs osmotiques
et thermiques et l'ischémie - permettent aussi l'activation de l'AMPK [258] [259]. On
peut concevoir que ces facteurs recruteront l' AMPK pour rétablir un débalancement
cellulaire, donc à petite échelle.
La fonction de l'AMPK s'est récemment vue attribuer une fonction beaucoup plus globale;
elle répondrait à des besoins physiologiques élargis à tout l'organisme. Il fut démontré que
le contrôle de la prise alimentaire au niveau ducerveau est en partie régulé par les
modulations de l' AMPK dans l'hypothalamus. Ainsi, la leptine, qui inhibe la
consommation de nourriture, agirait en empêchant l'activation de l' AMPK, et à l'opposé, la
growth hormone releasing peptide (grhelin) et l'activation des récepteurs aux canabinoïdes
augmenteraient la satiété en activant l'AMPK [260]. Par contre, la leptine présente une

42
complexité particulière puisque, contrairement à son action au cerveau, elle active l'AMPK
des muscles et du foie [261]. Ce double effet se retrouve aussi dans le cas de l' acide u
lipoique, un anti-oxydant naturel qui diminue la prise alimentaire inhibant l'AMPK au
cerveau tout en l' activant au niveau du muscle [262] [263]. Cette différence de modulation
selon les tissus pourrait s' expliquer par la présence de la kinase responsable de la
phosphorylation de l ' AMPK. Il a en effet, été proposé que l' AMPKK responsable de
l' activation de l'AMPK au cerveau pourrait être la CaMKKb ; tandis que LKB-1 serait,
elle, responsable de son activation aux tissus périphériques [264].
L'AMPK contrôlerait ainsi la prise alimentaire par l' augmentation de l' expression des
neuropeptides orexigéniques NPY (neuropeptide Y) et AgRP (agouti related prote in) et
l'inhibition des facteurs anorexigéniques (POMC (proopiomélanocortine) et CART
(cocain-and amphetamine-regulated transcript). La mise en évidence d'une hyperphagie
couplée à une activation de l' AMPK hypothalamique chez des souris rendues diabétiques
par injection à la streptozotocine conforte également l' idée selon laquelle l'AMPK pourrait
constituer une cible thérapeutique intéressante dans la prise en charge des troubles du
comportement alimentaire [265].
L' adiponectine, une autre hormone déterminante dans l'homéostasie glucidique et
lipidique, produit pour sa part ses effets positifs par l' activation de l'AMPK en périphérie
[205]. Pendant, qu' au contraire, une hormone, proposée pour nuire à la sensibilité à
l' insuline, la résistine, diminuerait la phosphorylation de l' AMPK au niveau du foie et dans
les cellules de muscle en culture (L6) [266].
Bien que le mécanisme ne semble pas jouer ou être présent dans tous les tissus, d'autres
études proposent une modulation de l'AMPK par une augmentation des niveaux d'AMP
cyclique. Le groupe de Denton établit ce lien en 1998 ; le groupe de Bimbaum, en 2003.
Dans l'étude de Bimbaum, des adipocytes 3T3-Ll traités à la forskoline qui active la"
production de l'AMPc, de même qu'un traitement à l'analogue de l'AMPc, le Sp-cAMP,
activent l' AMPK. Ces résultats furent appuyés par une étude qui rapporte elle aussi l' effet

43
de la forskolin et d'un analogue de l'AMPc, le Bt2cAMP, sur l' activation de l'AMPK sur
des adipocytes isolés [267] [268].
Tout récemment, un nouveau mode de régulation de l'AMPK fut proposé. En effet, un
groupe de recherche a observé que l'AMPK pouvait être modifiée et activée par la
glycosylation causée par la suractivation chronique de la voie des hexosamine. Cette voie
étant sollicitée lors d'apports importants en glucose va à l'encontre du rôle habituellement
octroyé à l'AMPK. Cependant, ces observations furent réalisées sur les adipocytes où le
rôle de l'AMPK semble différer de celui des myocytes [269].
Finalement, les deux hormones clés du métabolisme du glucose, l' insuline et le glucagon,
exercent aussi certains de leurs effets au moyen de l' AMPK. D' abord, l' AMPK est
suspectée d'effectuer certains des effets cataboliques du glucagon. L' AMPK inhiberait la
synthèse protéique au niveau du foie en inhibant la voie mTOR par l'activation de la voie
LKB1/AMPK [270]. L' insuline, pour sa part, inhiberait l 'AMPK dans l' hypothalamus et
ainsi inhiberait la prise alimentaire et favoriserait l' utilisation du glucose au détriment des
acides gras au niveau cœur par l'inhibition de l'AMPK par la phosphorylation sur les
résidus sérine de la sous-unité a (485/491) [271] [272] [273].
3.4 Les modulateurs chimiques
L'activateur chimique par excellence de l' AMPK est sans aucun doute le 5' aminoimidazole-4-
carboxamide 1 ~-D-ribofuranoside ou AICAR. Le AICAR est transporté dans la cellule par
des transporteurs de nucléosides, avant d'être transformé en ZMP par l' adénosine kinase. Le
ZMP étant un analogue de l'AMP, il permet l'activation de l'AMPK [274]. Certains ont
cependant, questionné la spécificité du AICAR puisque, mimant l' AMP, il peut activer
d' autres kinases qui lui sont sensibles, produisant ainsi des effets non reliés à l'AMPK.
Récemment par exemple, une étude a montré que l'inhibition de la synthèse de
phosphatidylcholine (PC) par un traitement au AICAR s'effectuait de façon indépendante de
l'activation de l'AMPK puisque tant l'expression d'un dominant négatif que la présence d'un
adenovirus surexprimant la forme active de l'AMPK ne modulaient la production de PC [24].

- - - ----~~~~~----,
44
Un extrait de lilac, était déjà utilisé à l'époque médiévale pour ses propriétés médicinales
contre ce qui fut plus tard identifié comme le diabète de type 2. On sait aujourd'hui que
l ' ingrédient actif de la plante est la biguanidine, commercialisé sous le nom de metformine
ou glucophage [275]. La metformine est maintenant largement utilisée pour traiter le
diabète de type2. Par contre, les mécanismes d' action de la metformin sont demeurés
obscurs jusqu'à ce que Zhou et ses collaborateurs démontrent l' implication de l' activation
de l' AMPK dans les effets bénéfiques de la metformine. Ils ont montré que, dans des
hépatocytes ou des muscles isolés, la metformine activait l' AMPK en provoquant
l ' inhibition de l' ACC, donc l' oxydation des acides gras, et une suppression de SREBP-l :
un facteur clé dans la lipogenèse. De plus, grâce à un inhibiteur de l' AMPK, le compound
C, ils montrèrent que la metformine nécessitait l' activation de l' AMPK pour produire ces
effets bénéfiques [235].
Les thiazolidinediones (TZDs), qui sont eux aussi largement utilisés pour le traitement du
diabète de type2, activent l'AMPK [276]. Les TZDs lient et activent les récepteurs
nucléaires PP ARy, permettant ainsi la transcription de gènes cibles. Par contre, le
mécanisme par lequel les TZDs activent l' AMPK est encore nébuleux. Il a été suggéré que
la metformin et les TZDs agissaient sur, l' AMPK en inhibant le complexe 1 de la chaîne
respiratoire [277] [278]. D'autre part, les TZD augmentent les niveaux d' adiponectine et
diminuent ceux de la résistine en circulation ; ces mécanismes pourraient donc aussi
contribuer à l' activation de l' AMPK. Enfin, puisque l'activateur naturel des pp ARy (non
TZD) : la PGJ2 active l'AMPK, il est permis de croire que la mobilisation des récepteurs
nucléaires joue un rôle dans l' activation de l'AMPK [165]. On a récemment démontré, dans
le même ordre d' idées, que l' activation des PPAR8 causait aussi l'activation de l'AMPK.
Cette dernière découverte vient renforcer le potentiel antidiabétique de l' AMPK, puisque
l' activation des PP AR8 est maintenant considérée comme une cible prometteuse pour le
traitement du diabète de type 2 [279].

45
3.5 Rôles de l'AMPK
3.5.1 La conservation de l'énergie
Le rôle par excellence de l'AMPK est sans contredit la conservation de l' énergie
intracellulaire. Étant donné que le processus de synthèse de protéine est énergivore, il est
peu surprenant d' y voir l 'AMPK exercer son contrôle [280] [281]. D'abord, l 'AMPK peut
phosphoryler et activer la tuberine (TSC2) qui pourra ainsi s'associer avec l'hamartin
, (TSC1). Une fois activé, ce complexe sert de GTPase en causant une augmentation des
niveaux de Rheb-GDP diminuant l'activité d'une voie qui contrôle la synthèse protéique:
mTOR/S6K1. Il a aussi été démontré que l'AMPK peut directement phosphoryler et inhiber
mTOR [282]. Un deuxième niveau de contrôle de la synthèse protéique s' effectue par la
phosphorylation et l' inhibition du facteur d' élongation eucaryote eEF2 [280]. L' activation
de l'AMPK causera la phosphorylation de eEF2 sur son résidu thréonine 56 (Thr-56). Ainsi
modifié, le facteur de traduction bloquera la synthèse protéique en empêchant l'avancement
des ribosomes sur l' ARN messager. La phosphorylation de eEF2 ne se ferait pas
directement par l'AMPK ; l'AMPK activerait plutôt la kinase de eEF2, en la phosphorylant
[283]. Finalement, l'AMPK peut réduire la stabilité de 'certains ARNm en diminuant la
disponibilité de la protéine stabilisatrice HUR. Cette dernière lie les ARNm dans le noyau
et permet son transport au niveau du cytoplasme pour sa traduction éventuelle. Plusieurs
études révèlent que l' activation de l' AMPK réduit les niveaux cytoplasmiques de HUR,
altérant ainsi la synthèse de protéines ciblées [284] [285] [286].
L'AMPK régule aussi négativement la synthèse de glycogène. Ce processus étant lui aussi
énergivore, l'AMPK ira inhiber, par sa phosphorylation sur le résidu ser7, de la glycogène
synthase (GS) [236]. Par ailleurs, la présence de glycogène, signifiant l' abondance
énergétique au niveau muscle, inhibe l' activation de l'AMPK par la contraction ou par la
présence de AICAR [287]. La synthèse de cholestérol au moyen de la HMG-Coa reductase
est un autre processus régulé négativement par l'AMPK qui demande aussi beaucoup
d' énergie.

46
3.5.2 La production d'énergie
L'activation de l'AMPK ne se limite pas à réduire les processus consommant l'énergie. Elle
déclenchera plusieurs processus afm d'augmenter la régénération d'énergie. Un premier
mécanisme, bien établi, est l'augmentation de l'entrée de glucose dans la cellule. En effet, un
des paramètres très étudié de l'activation de l' AMPK fut l'augmentation du transport de
glucose dans le muscle. En fait, plusieurs d'études furent menées afin de déterminer si
l'augmentation du transport du glucose au niveau du muscle par la stimulation de l'AMPK ne
correspondrait pas au mécanisme par lequel l'exercice permet l' entrée de glucose,
indépendamment des voies de signalisation de l'insuline. En effet, lors de la contraction
musculaire, le muscle utilise l' ATP et la créatine phosphate disponible et déséquilibre les
ratios AMP/ATP et crétine/créatine phosphate; autant de conditions nécessaires à l'activation
de l'AMPK. On sait en outre que le AICAR stimule le transport du glucose au muscle, et cela,
de façon additive à l' insuline, comme le fait l'exercice. Comme c' est le cas durant l'exercice,
le transport du glucose stimulé par le AICAR n'est pas, ici non plus, sensible à l' inhibition de
la PI 3-kinase. Cependant, les expériences réalisées sur les modèles d' animaux transgéniques
remettent en question l'hypothèse selon laquelle l' AMPK serait bel et bien la kinase
responsable du transport du glucose stimulé par la contraction. Une première étude a montré
que l' inactivation de la sous-unité a2 agissant comme dominant négatif de l'AMPK empêche
le transport du glucose stimulé par le AICAR et 1 'hypoxie mais réduit le transport stimulé par
la contraction de seulement 30 à 40 % [288]. Jorgensen et collaborateurs ont par ailleurs
rapporté que le transport de glucose stimulé par la contraction n'est pas modifié, ni dans des
souris invalidées pour la sous-unité al ou a2 de l'AMPK. Une étude récente vient de montrer
que des souris dont la sous-unité a2 de l'AMPK a été éliminée au muscle ne peuvent pas
stimuler le transport de glucose par le AICAR ou par la roténone qui inhibe le complexe l de
la chaîne respiratoire mais, qu'à l'opposé, le transport du glucose stimulé par la contraction ou
par un choc osmotique créé par le sorbitol n'est pas affecté non plus par l' inactivation de la
sous-unité a2. Comme la sous-unité a2 qui est activée par l'exercice et que les auteurs n'ont
pas observé de compensation par la sous-unité al , ils concluent que le transport du glucose au
muscle ne peut être stimulé que de trois façons : par un mécanisme dépendant de l' insuline,

47
par un mécanisme dépendant de l'AMPK ou par un indépendant de l' insuline et de l' AMPK
[289].
L' activation de l'AMPK va permettre l' augmentation du transport du glucose par trois
mécanismes différents: le premier étant la translocation des transporteurs de glucose
GLUT4 à la surface cellulaire, le second par l' augmentation de l'expression de ces
transporteurs par les facteurs de transcription GLUT4 enhancer factor (GEF) et myocyte
enhancer factor 2 (MEF2) et, enfin, par l' augmentation de l' activité intrinsèque de ces
mêmes transporteurs [290] [291] [292] [293] [294]. L'AMPK pourrait utiliser sa capacité à
stimuler la map kinase p38 pour permettre l ' activité intrinsèque des GLUT4 [295] [296].
Le mécanisme par lequel la p38 est activée au moyen de l'AMPK pourrait faire intervenir
la protéine T AB 1. T AB 1 semble au moins importante au cœur, puisque la phosphorylation
de la p38 nécessite la formation d'un complexe avec l' AMPK et la protéine T AB 1 [297].
Cependant, le rôle de la p38 dans l' activité intrinsèque de GLUT4 demande encore à être
éclairci [134].
Tel que mentionné précédemment, on a par ailleurs montré que le AICAR augmentait
l' activité par le démasquage des transporteurs de glucose GLUTI dans des cellules du foie
- les clones 9 cells - et que cet effet était médié par l' activation de l' AMPK [298] [116].
Une autre façon de s'assurer de l'approvisionnement suffisant en énergie par l'AMPK consiste
en l'oxydation des acides gras libres. L'AMPK active ce processus par l' inhibition de l' acétyl
CoA carboxylase (ACC). En effet, l'AMPK inhibe cette enzyme par sa phosphorylation.
Considérant que l'ACC produit le malonyl-CoA qui bloque l'entrée et l'oxydation des acides
gras libres par l'obstruction de la carnityl plamitotransférase (CPT -1) aux mitochondries,
l' inhibition de l'ACC provoquera l'utilisation des acides gras. Ainsi, l'augmentation de
l'oxydation des acides gras par la leptine et par l' adiponectine' serait directement reliée à
l' activation de l'AMPK. Il fut démontré à cet effet que la présence d'un dominant négatif de
l'AMPK dans des cèllules de muscle H-2Kb empêche l'utilisation des acides gras par la
leptine [299]. Pour sa part, le groupe de Kadowaki a démontré que la présence d'un dominant

48
négatif de l'AMPK au niveau du muscle empêche l'adiponectine d'oxyder les acides gras dans
les myocytes C2C12 [205].
3.5.3 Rôle anti-inflammatoire
Quelques données récentes supportent le rôle anti-inflammatoire de l'AMPK. Le groupe de
Singh fut le premier à l' établir. Il a montré que l' activation de L'AMPK par le AICAR inhibe
la production de cytokines (TNFa, IL-1 p et IL-6) de même que l' induction de iNOS à la suite
d'un traitement à la LPS dans des astrocytes de rat en culture primaire, dans des cellules de la
microglie, de même que dans des macrophages péritonéaux. L'AMPK semblait bel et bien
responsable de ces effets puisque la présence d'un dominant négatif contre l 'AMPK
empêchait les effets du AICAR de se produire. Plus récemment, le même groupe a montré que
le AICAR pouvait servir d 'agent anti-inflammatoire en réduisant la production de certaines
cytokines dans le cas d'une maladie du système nerveux central nommée sclérose en plaque
[300] [301]. On a aussi établi que l ' inhibition de l'expression protéique de iNOS était reliée à
une réduction de la stabilité de l' ARNm engendrée par l' activation de l' AMPK [286]. Plus
récemment, la capacité de l'AMPK à réduire l'expression de la cytokine IL-2 dans des cellules
T fut aussi établie [302]. De plus, la metfonnine empêcherait certaines cytokines d'activer la
voie NfKB par l'action de l'AMPK [303]. Une autre étude a, pour sa part, démontré que
l' activation de l'AMPK dans l'hypothalamus renversait l' anorexie causée par une tumeur
possiblement en inhibant la production de molécules pro-inflammatoires [304]. Il fut aussi
rapporté que dans des cellules épithéliales de bronches cette fois, que l' activation de l' AMPK
permettait un meilleur contrôle de l' inflammation [305]. Finalement, la nicotine jourait un
certain rôle anti-inflammatoire par l' activation de l'AMPK dans les macrophages [306].
3.5.4 Les autres rôles bénéfiques de l'AMPK
Les rôles de l' AMPK qui viennent d' être mentionnés sont tous utiles pour empêcher le
développement du syndrome métabolique. À la longue liste de ces bénéfices, on peut ajouter

49
la synthèse de mitochondries par l' activation du facteur de transcription nuclear respiratory
factor 1 (NRF-1). Une plus grande concentration de mitochondries augmenterait la dépense
énergétique, contribuant ainsi à un meilleur contrôle du poids corporel [307].
L'AMPK inhibe aussi la néoglucogenèse hépatique en phosphorylant et en inhibant le facteur
de transcription TORC2 qui ne pourra plus activer les gènes responsables de cette production
de glucose [308]. Plusieurs auteurs ont finalement suggéré que l'AMPK, stimulé par un
environnement à faible concentration de glucose, inhibe l' exocytose de l' insuline par les
cellules fJ du pancréas, contrôlant ainsi l'hyperinsulinémie associée à la résistance à 1 insuline
[309] [231]. Aussi, une production modérée de NO par eNOS est reconnue pour dilater les
vaisseaux sanguins et contribuer ainsi à l'accessibilité de l' insuline aux tissus. Le groupe de
Kemp fut le premier à démontrer que l'AMPK pouvait activer eNOS en présence de Ca2+
calmoduline et ainsi permettre son activation [310]. Depuis, différents groupes ont observé des
effets semblables, bien que ce ne soit pas le cas pour tous les types de cellules ; dans les
cellules rénales, par exemple, l'activation de l' AMPK ne cause pas l'activation de eNOS par
sa phosphorylation [311] [312].

50
4. Le monoxyde d'azote
En 1987, les docteurs Murad, Ignarro et Furghgott firent une découverte majeure en
révélant un nouveau système biologique, ce qui leur valut un prix Nobel. Ils découvrirent
un gaz libéré par les cellules endothéliales qui se manifeste de multiples façons dans tout
l ' organisme: le monoxyde d' azote (NO).
Dans le système vasculaire, le NO maintient un tonus vasodilatateur nécessaire pour la
régulation de la circulation sanguine et le contrôle de la pression artérielle ; il inhibe
l ' agrégation et l ' adhésion des plaquettes, empêche l' adhésion des leucocytes et module la
prolifération des cellules musculaires lisses.
Dans le système nerveux central, il agit comme un neuromédiateur..Il est associé à diverses
fonctions physiologiques ; par exemple, la formation de la mémoire, la coordination entre
l' activité neuronale et le débit sanguin et la modulation de la douleur.
Le NO est, en outre, produit en grandes quantités dans les manifestations de défense de
l 'organisme. Il contribue à protéger contre les cellules tumorales, les bactéries et les virus.
Ainsi libéré, le NO peut cependant aussi favoriser le développement de pathologies comme
le choc septique et certaines formes d ' inflammation aiguë ou chronique.
Le NO est donc un médiateur biologique ubiquitaire dont les fonctions sont tantôt
physiologiques, tantôt pathophysiologiques.
En raison de son étendue d'action, de ses effets bénéfiques autant que de ses effets néfastes
et de sa càpacité à interagir avec une multitude de cibles, le NO mérite grandement son titre
de molécule de l' année attribué en 1992 par la revue Science. Et pourtant, le NO est l 'une
des plus vieilles molécules connues. La génération de NO dans l' atmosphère est née de la
réaction entre l' azote et l ' oxygène à des températures de 1 000 degrés Celsius. Pour cette
raison, la production cellulaire de NO fut longtemps perçue commé impossible.

51
Sans rien lui enlever, il faut aussi réaliser qu'une partie des effets du NO n'est pas produite
par la molécule elle-même, mais bien par ses produits de dégradation ou encore le produit
de ses interactions avec d'autres molécules chimiques ou protéiques. En milieux aqueux et
oxygéné par exemple, le NO est rapidement oxydé en nitrite (N02) et en nitrate (N03) ; il
activera aussi la production de cyclic guanidine monophosphate cGMP par l'activation de
la guanylyl cyclase ; il pourra devenir un oxydant puissant par son association avec le
superoxyde (02-) pour former le peroxynitrite (ONOO-), modifiant ainsi des protéines par
leur nitration. Le NO peut aussi interagir avec les domaines S-H de protéines leur causant
une modification nommée s-nitrosation. En milieu biologique, sa grande capacité à difuser
l' amènera souvent à sortir de la cellule qui l' a produite pour atteindre ses cibles. Il s'agit
donc d'une molécule simple aux effets complexes [313] [314].
4.1 La famille des NOS
Les synthétases de NO (NOS) sont les enzymes responsables de la synthèse du NO
catalysée par l' oxydation de la L-arginine en NO et en citrulline. Trois isoformes de NOS
distinctes ont été identifiées à ce jour: eNOS caractérisée initialement dans les cellules
endothéliales vasculaires, nNOS découverte dans les neurones et iNOS, une forme
inductible, découverte dans les macrophages. Les NOS sont codées par 3 gènes distincts:
NOSI (nNOS), NOS2 (iNOS) et NOS3 (eNOS) présents, respectivement, sur les
chromosomes 7, 12 et 17, et numérotés selon leur ordre de découverte. L'organisation
intron/exon des gènes NOS humains présente des similitudes structurales, mais les
mécanismes de régulation sont distincts - particulièrement dans le cas de iNOS. Les 3
isoformes sont distribuées dans un grand nombre de tissus et de cellules et il est maintenant
établi qu'une même cellule peut exprimer plusieurs isoformes de NOS. Les noms de ces
enzymes sont donc trompeurs, malgré qu'ils soient déjà consacrés par l'usage. Les enzymes
nNOS et eNOS sont dites constitutives, exprimées de façon basale et générant de faibles
quantités de NO ; elles sont ~econnues pour leur rôle bénéfique. À l'opposé, iNOS est peu
ou pas exprimée à l'état basal, mais elle est fortement induite en présence de stimuli

52
inflammatoires. Les quantités de NO générées par iNOS sont beaucoup plus importantes
que celles produites par les NOS constitutives. Cette dernière caractéristique en fait une
enzyme à « double tranchant» puisqu'elle peut exercer un rôle bénéfique pour combattre
un envahisseur viral ou bactérien, mais elle peut aussi avoir des conséquences délétères, si
son expression est mal contrôlée [315] [316] [317] [318j [319] [320].
4.2 La structure des NOS
On dit souvent que les NOS doivent s' autodimériser pour être actives. Il faudrait plutôt dire
que les NOS doivent être sous forme de tétramères puisqu'elles ont toutes besoin d' être
associées à deux calmodulines (CaMs). La structure simîlaire des trois enzymes suggère un
ancêtre commun. En effet, les NOS possèdent toutes un domaine oxygénase dans leur
section N -terminale et un domaine réductase en position C-terminale.
Si l' on regarde de près le domaine oxygénase, on remarque qu' il contient les sites de
liaison des cofacteurs essentiels suivants : le (6R)-5,6,7,8-tétrahydrobioptérine (BH4),
l 'hème, et la L-arginine. Pour sa part, le domaine réductase lie la. flavine adénine
mononucléotide (F AD), la flavine mononucléotide FMN et la nicotinamide adénine
~inucléotide phosphate (NADPH) [321] [322] [323]. Les NOS doivent obligatoirement être
associées en homodimères pour être activées. Le site de liaison qui permet cette interaction
déterminante se trouve dans le domaine oxygénase pour iNOS, alors que le domaine
réductase serait aussi concerné dans le cas des deux NOS constitutives. Les co-facteurs
BH4, hème et la L-arginine contribuent tous à l' association et à la stabilisation du dimère
[324] [325].
L'oxydation de la L-arginine qui permet la production de NO et de citrulline par les NOS
est réalisée en deux étapes. D'abord, la NADPH se fixe au domaine réductase par lequel il
est oxydé. Les deux électrons ainsi libérés transitent par la FAD, puis la FMN, avant
d' atteindre le domaine oxygénasè où ~e trouve le site de liaison du BH4 et de l'hème. Ce
flux d' électrons est rendu possible par la présence de la c~lmoduline. Le substrat, la L
arginine, sera hydroxylé par l'hème en présence de BH4 en Nw-hydroarginine (NOHA)

53
grâce aux électrons provenant de la NADPH et de l' oxygène moléculaire. Ensuite, le
composé intermédiaire sera oxydé à son tour pour produire le NO et la citrulline [326].
4.3 eNOS
4.3.1 La modulation de eNOS
eNOS est une protéine de 133kDa possédant un site de myristosylation et un site de
palmitosylation la distinguant des autres NOS. eNOS est d'abord myristosylée ce qui lui
permet d' être dirigée vers l' appareil de Golgi où elle sera palmitosylée ; elle pourra alors
s' intégrer à la membrane intérieure des structures nommées caveolea. Il s' agit de petites
invaginations de la membrane plasmique. À l' intérieur de ces structures; eNOS interagit
avec la calvéoline inhibant son action. L' augmentation de calcium intracellulaire cause la
dissociation du complexe eNOS/calvéolin permettant son actiyation [327].
Lorsqu' eNOS n' est plus palmitosylée, elle peut se déplacer au niveau cytoplasme où elle
pourra être activée ou inhibée par sa phosphorylation sur différents sites par des kinases
telles que l ' Akt/PKB, la PKA et l'AMPK [328] [310]. L'AMPK, l ' AkT/PKB et PKCa sont
connues pour phosphoryler eNOS sur le résidu sérine 1177 ou 1179, selon l' espèce. Cette
phosphorylation servirait à activer l' enzyme. Par contre, le groupe de Cohen a rapporté que
l' activation de l'AMPK par le ONOO- causait la phosphoryla~ion de eNOS sur ser1179
produisant un découplage de l' enzyme et augmentant la production de O2- au détriment de
celle du NO [329]. eNOS peut aussi être modulé négativement par sa phosphorylation sur
son résidu thréonine 497 ou 495 [330].
Plusieurs facteurs peuvent moduler eNOS. Au niveau de la transcription par exemple, le
TNFa peut déstabiliser l'ARNm de eNOS. Le mécanisme d ' action de cette cytokine sur
eNOS n'est pas complètement défini, mais on sait que le traitement au TNFa de cellules
endothéliales BAEC cause la formation d'un complexe de ribonucléoprotéines non
identifiées sur la séquence 3 '-untranslated region (3 ' -UTR) de eNOS réduisant la stabilité

54
de l' enzyme [331]. La lipopolysaccharide (LPS), seule ou en combinaison avec des
cytokines, peut aussi causer une diminution de l' expression de l'ARNm de eNOS [332]. À
l' opposé, le TGF-B1 est reconnu pour stabiliser l' expression de l'ARNm de eNOS dans des
cellules endothéliales HUVEC par un mécanisme impliquant la liaison du facteur de
transcription SMAD2 au site 3' -UTR de l'enzyme [333]. Le NO est incidemment lui-même
reconnu pour inhiber l' activité de eNOS. En effet, le NO peut lier le groupement hème de
l' enzyme et diminuer significativement sa production [334].
4.3.2 Les rôles d'eNOS.
Le premier rôle du NO généré par eNOS est sans contredit le contrôl~ de la pression
artérielle. Il fut, en effet, d' abord identifié comme étant l' endothelium-qerived rel~ing
factor EDRF découvert par Furchgott & Zawadski 1981. L'utilisation d'un inhibiteur
semblable à la L-arginine, le L-NMMA, augmente la pression artérielle in vivo et in vitro.
Plus récemment, ces résultats ont été confirmés chez des souris invalidées pour le gène
eNOS. Ces souris sont hypertendues, mais l'administration de NO exogène sous forme de
NaN02 (nitrite de sodium) permet le rétablissement d'une pression artérielle normale [335].
L'acétylcholine, la bradikinine; et le stress de cisaillement sont des stimuli qui activent des
récepteurs à la surface des cellules endothéliales augmentant alors le flux de calcium
intracellulaire. eNOS ainsi activée par l' augmentation de calcium dans la cellule génère du
NO qui diffuse jusqu' aux cellules musculaires lisses où il cause la dilatation des vaisseaux
par la production de cGMP par l' activation de la guanylate cyclase [336] [337] [338]. D'un
autre côté, l' administration exogène de NO par des drogues telles que la nitroglycérine ou
le sodium nitroprusside cause aussi la relaxation par la production de cGMP.
Comme. l' hypertension artérielle et la résistance à l'insuline sont souvent associées, le
groupe de Scherrer a cherché à comprendre si l' absence de eNOS pouvait contribuer à la
résistance de l 'hormone. En utilisant des souris eNOS( -/-), ils ont observé que ces souris
étaient bien sûr hypertendues, mais qu' elles étaient également hyperinsulinémiques et que

55
le transport du glucose à l ' intérieur du muscle était réduit de 40% comparativement aux
souris contrôles. De plus, différents groupes ont expliqué l ' association d'un polymorphisme
du gène eNOS à un défaut de la sensibilité à l' insuline. Le bon fonctionnement de eNOS
pourrait donc avoir un impact déterminant sur la sensibilité à l' insuline même si son rôle
n' est pas encore totalement défini [339] [340] {341].
La production du NO par eNOS peut aussi être associée à l' athérosclérose qui s' établit par
la formation de plaques d 'athérome. En inhibant l' adhésion des leukocytes et des
monocytes, en diminuant l' oxydation des lipoprotéine de f aible densité (LDL) et en
inhibant la synthèse des cytokines, le NO généré par l ' endothélium protège contre la
maladie. Une étude démontre d'ailleurs une susceptibilité des souris eNOS-/- à développer
des lésions athéroscérotiques [342].
Certains ont par ailleurs suggéré que eNOS pouvait exercer un rôle pro-inflammatoire. En
l' absence de l'enzyme, la LPS induit en effet moins efficacement iNOS et sa capacité à
causer une production de TNF a est réduite dans des cellules endothéliales et dans des
cardiomyocytes. Il est aussi rapporté que, dans la vessie de rats traités à la LPS, eNOS était
rapidement phosphorylé sur serl179 et qu' ainsi eNOS contribuait à la réponse
inflammatoire [343] [344] [345] [346].
4.4 nNOS
4.4.1 Modulation de nNOS
Malgré son appellation, la forme neuronale des NOS se retrouve aussi dans des tissus autres
que le cerveau; par exemple, dans le placenta, les testicules, la rate, les reins, les poumons
et le muscle squelettique. nNOS, une protéine de 160kDa, fut d'abord identifiée dans les
neurones. Elle se distingue de ses deux « enzymes-sœurs» par son domaine PSD/Discs
large/ZO-l (PDZ) positionné en N-terminal. Cette région lie des domaines PDZ d' autres
protéines, tels que la post synaptic density protein (PSD-95 et la PSD-93) La PSD-95 fait le

1
1
l' 1
56
pont entre nNOS et le récepteur de N-methy l-D-aspartate (NMDA). Ce motif PDZ permet
aussi à nNOS retrouvée au niveau du muscle de lier la protéine syntrophine, formant un
complexe avec la dystrophine [347].
nNOSa (la forme complète) peut subir un épissage alternatif de son ARNm permettant
l'expression de plusieurs variants (nNOSp, nNOSy, nNOSfJ, et nNOS-2). Les fonctions
précises de tous ces dérivés sont peu caractérisées [348]. On remarque par contre que les
formes nNOSp et nNOSy se retrouvent sous forme libre ; ils ne possèdent pas de domaine
PDZ et ne sont pas associées aux membranes [349].
nNOSfJ, se retrouve au niveau du cœur et est la forme prédominante du muscle' squelettique
[350]. La fonction de nNOS-2 est peu définie, mais elle pourrait agir comme dominant
négatif. En effet, nNOS-2 ne possède pas la région liant la L-arginine ; donc, cette forme de
l' enzyme est incapable de produire du NO. Cependant, l' enzyme présente tout de même
une activité NADPH diaphorase qui peut lui permettre de produire du O2-. Certains auteurs
ont émis l 'hypothèse que cette production de O2 pourrait contribuer à certains désordes
neuronaux causés par son interaction avec le NO, expliqués par la production de ONOO-
, [351].
Tout comme eNOS, l' activité de nNOS est dépendante du calcium. Elle est inhibée par la
présence de NO et peut aussi l'être en s'associant à la cavéoline. nNOS est aussi modulée par
sa phosphorylation mais ces modifications seraient dans l' ensemble inhibitrices. Par exemple,
la calmodulin kinase 1 et II (CaMKI et CaMKII) phosphorylent nNOS sur son résidu sérine
741 , ce qui inhibe son activité [352]. Sa phosphorylation sur le résidu thréonine 1296 cause
aussi son inhibition. D'un autre côté, sa déphosphorylation par les phosphatase 2A (PP2A) ou
SH2 domain-containingproteinphosphatase 1 (SHP-1) active l' enzyme [353] [354].
La -régulation de l' expression de nNOS en situation inflammatoire varie selon les tissus.
Dans le coeur de rats injectés à la LPS, l' expression de nNOS est réduite, alors qu'une autre
étude montre l' effet contraire dans l'utérus [355] [356]. Par ailleurs, l' induction de iNOS
dans les artères de porcs inhibe l'activité de nNOS [357].

57
4.4.2 ~ôles du NO généré par nNOS
Un des rôles importants de nNOS se situe au cerveau. Par son domaine PDZ, nNOS active
les récepteurs N-methyl-Daspartate type glutamate (NMDAR) par la liaison de la PSD-95
[358]. D'un autre côté, nNOS peut inhiber la transmission synaptique par la S-nitrosylation
des récepteurs NMDAR [359] (le processus de S-nitrosylation sera expliqué plus loin). Au
cerveau, nNOS joue un rôle dans le comportement, l' apprentissage et la sensibilité face à la
douleur. Par contre, la production de NO au cerveau doit être très bien régulée puisqu'une
trop grande production du gaz est associée à la maladie de Parkinson et à la maladie de
d'Huntington ; elle est aussi en cause dans l' étiologie des migraines [360].
Au muscle squelettique, la forme épissée nNOSJ.! est aussi très importante. Lors de la
contraction, le NO est libéré par le muscle et dilate les vaisseaux sanguins adjacents
permettant un apport sanguin adéquat. nN OS J.! pourrait pallier à certaines dystrophies
musculaires puisque l' expression d'un transgène de l' enzyme améliore le phénotype
d' animaux malades [361]. nNOSJ.! est aussi en cause dans la force et le développement du
muscle, dans le contrôle de l 'homéostasie du contenu en sodium du corps en régulant la
réabsorption aux reins ; elle est indispensable à l'exercice des fonctions sexuelles mâles et
au contrôle du péristaltisme de l ' intestin [362] [363]. Finalement, nNOS peut jouer un rôle
dans la sécrétion de l' insuline par les cellules f3 du pancréas [364].
4.5 MtNOS
L' existence d'une quatrième forme de NOS soulève actuellement bien des débats. Il s' agit
d'une forme mitochondriale nommée mtNOS. L' expression d'une forme mitochondriale de
NOS fut observée par différents groupes - presque simultanément -, il y a environ une
dizaine d 'années [365] [366]. Mais depuis, on a très peu appris sur ce sujet controversé.

58
L' identification d'une enzyme NOS propre aux mitochondries est rendue difficile d' abord
en raison des méthodes d' isolation des mitochondries qui ne permettent pas une pureté
absolue des organelles ; sans compter qu' eNOS possède des sites d' ancrage aux
membranes (palmitoyl et myristoyl) pouvant contribuer à la contamination des
mitochondries. Un autre argument de poids pour les détracteurs de l' existence des mtNOS
vient du fait que chaque NOS est codé par un gène distinct et que l' analyse extensive du
génome eukaryote n' a pas permis l' identification d'une autre séquence de NOS. Ce qui
signifie que mtNOS proviendrait d'une modification d'une des isoformes de NOS actuelles.
Cependant, aucune des NOS existantes ne possède de séquence la dirigeant vers les
mitochondries. D ' un autre côté, le NO semble avoir un rôle non équivoque dans la
biogenèse des mitochondries et qu' en présenc~ de calcium, mtNOS produirait du ONOO
[367]. La production de ONOO- par mtNOS conduirait à la nitration de protéines associées
à une dysfonction mitochondriale [368]. Ces organelles sont par ailleurs de plus en plus
mises en cause pour leur rôle potentiel dans la résistance à l' insuline [213]. Il serait donc
intéressant de mieux comprendre les mécanismes régulant la production de reactive oxygen
species (ROS) et reactive nitrogen species RNS par les mitochondries.
Finalement, il est à noter que le NO peut être produit de façon non enzymatique à partir de
nitrites en situation réductrice dans un environnement à bas pH. Cependant, l' importance
physiologique de ce NO et son rôle ne sont pas encore clairement définis [369].
4.6 iNOS
iNOS est une protéine de 130kDa qui, contrairement à eNOS et nNOS est peu ou pas
exprimée en situation basale. Elle a une activité indépendante de la présence de calcium étant
toujours fortement liée à la calmoduline [370]. Cependant, en présence de signaux
inflammatoires, iNOS est induite et génère des quantités de NO de loin supérieures aux autres
formes de NOS. Même si cette forme de iNOS fut découverte dans les macrophages, on sait 1
aujourd'hui que iNOS peut être induite dans plusieurs tissus, dont le muscle et les tissus
adipeux [371] [372].

59
4.6.1 Facteurs et mécanismes responsables de l'induction de iNOS
Les facteurs inflammatoires induisant iNOS sont nombreux. Il y a, bien sûr, les cytokines
pro-inflammatoires: TNFa, IL-lp , IL-6, INF-y. L ' endotoxine LPS est aussi un 'activateur
de iNOS très étudié. Parmi les signaux qui mènent à l' induction de l ' ARNm de iNOS on
compte la voie NF-KB et la voie JAKISTAT [372]. Par ailleurs, la voie des mitogen
activated protein kinases (MAPK) va conduire à l ' activation d'autres facteurs de
transcription impliqués dans l' induction de iNOS, soit activator protein 1 (API),
l'acti"yating transcription factor 2 (ATF2), et d' autres facteurs de transcription de la famille
des Ets [373] [374] [375] [376] [377]. Dans les macrophages, les récepteurs toll-like
receptors (TLR4) et CD14 sont très importants pour permettre l' induction de iNOS. CD14
est le récepteur permettant la liaison de la LPS qui rendra alors possible l' activation de la
voie NF-KB [378]. Cette voie est un mécanisme déterminant de l' induction de iNOS. NF
KB est présent dans le cytosol et est inhibé lorsqu' il est associé IKB. Lorsque le complexe
est phosphorylé par IKB kinase (IKK) en présence de certains facteurs, NF-KB est libéré et
se dirige au noyau où il activera le promoteur de gènes tels qu' iNOS. Des études récentes
ont aussi démontré la présence des (TLR2) et (TLR4) dans le muscle, le foie et le tissu
adipeux, pouvant ainsi contribuer à l' induction de iNOS dans ces tissus[379] [380]. L' IFNy,
pour sa part, active la voix JAKISTAT qui mène à la synthèse du facteur de transcription
interferon response factor (IRF -1) stimulant à son tour la transcription de iNOS.
Dans la plupart des études in vitro, seule une combinaison de plusieurs facteurs
inflammatoires peut mener à une induction soutenue de iNOS. Donc, son expression est
relativement contrôlée et ne répond pas au premier stimuli. La région du promoteur de
iNOS contient d' ailleurs plusieurs éléments de liaison tels que NF-KB, c-juniFos, C/EBT,
CREB et STAT. On peut deviner que plus d'un site doit être stimulé pour qu' il y ait
induction de iNOS [381] [382] [383]. Par exemple, il existe une synergie entre les actions
de la LPS et de l ' IFNy, puisque IRF -1 et NF -KB interagissent au niveau du promoteur
causant une transformation dans la conformation de ce dernier, ce qui a pour effet de
l' activer davantage [384]. À l'opposé, la grande quantité de NO générée par iNOS inhibe sa
propre production en inhibant la voie NF-KB [382].
~I

60
Du côté traductionnel, la stabilité de l' ARNm détermine la quantité de protéine iNOS
produite. Un des mécanismes connus de stabilisation de iNOS implique la protéine HUR.
Cette nucléoprotéine lie une région 3'-UTR de iNOS et, ce faisant, maintient l'ARNm
disponible pour la traduction [286]. De plus, la protéine tristetraproline (TTP) aurait aussi
un rôle à jouer dans la stabilité de l' ARNm de iNOS pour sa traduction par un mécanisme
impliquant vraisemblablement JNK [385]. À l' opposé, la protéine KH-type splicing
regulatory protein (KSRP) se lie à la même région de l' ARNm de iNOS, mais diminue sa
stabilité [386].
L' inhibition de la dimérisation de iNOS est un autre mécanisme par lequel iNOS est
contrôlée. En effet, comme iNOS doit être exprimée sous forme d 'homodimères pour
produire le NO, des agents tels que la Kalirine protègent des effets néfastes de iNOS au
moyen de ce mécanisme. De la même façon, une protéine produite par les macrophages
NOS-associated protein-ll 0 kDa (NAP 110) contrôle la production de NO [387] [388].
La production de NO par iNOS peut aussi dépendre de la disponibilité du substrat, la L
arginine [389]. Comme pour les deux autres NOS, la phosphorylation de l ' enzyme pourrait
modifier son activité. En effet, sa phosphorylation en présence de LPS dans les
macrophages concorderait avec son activité [390]. Des études chez l' humain semblent aussi
suggérer que certains polymorphismes du gène iNOS sont associés à une plus grande
activité de l'enzyme puisque les gens présentant cette variation du gène seraient plus aptes
à se défendre contre le virus de la malaria [391].
Finalement, tout comme nNOS, iNOS peut aussi subir un épissage alternatif. Trois
variantes sont possibles. La raison d'être de ces enzymes n'est pas clairement définie, mais
elles ne présenteraient plus d'activité NO synthase et conserveraient une activité NADPH
diaphorase qui leur permettait de produire du superoxyde (02-) [392] [393].

,--------------_. --- --- - ~-~----
61
4.6.2 Autres Facteurs induisant iNOS
Mis à part les cytokines pro-inflammatoires, plusieurs autres facteurs d'origines diverses
peuvent contribuer à ·l ' induction de iNOS. Parmi eux, plusieurs sont augmentés dans
l' obésité ou associés à la résistance à l'insuline tels que les acides gras libres (AGL), les
céramides, les advanced glycation end products (AGEs), la leptine, la C-reactive protein
(CRP), les gangliosides, l 'homocystéine et une concentration de glucose circulante élevée
[394][395] [396] [397] [398] [399] [400] [401].
L' insuline qui est présente en plus fortes concentrations au début de l'établissement de la
résistance à celle ci, a des effets différents selon les tissus. Ainsi, des études de notre
laboratoire ont démontré que l'insuline augmentait l'expression de iNOS dans les adipocytes
en culture alors que l'effet opposé était observé dans les myotubes (Penfornis et Maretle
résultats non-publiés) [164]. L'effet inducteur de l'insuline a aussi été rapporté dans des
cellules d'îlots pancréatiques isolés en culture et dans des ostéoblastes. L' insuline pourrait
également actualiser la capacité d' IL-1 f3 à induire iNOS dans des cellules glomérulaires [402].
À l' opposé, dans les leucocytes de rats rendus diabétiques de type 1 à la suite d'un traitement à
l' alloxane (donc, qui ne produisent plus d' insuline), on observe une induction de iNOS.
Cependant, si les animaux sont préalablement traités à l' insuline, cette modulation de
l'enzyme n'est plus observée [403]. Bien entendu, in vivo, comme l' insuline a de multiples
actions, on ne peut pas déduire une relation directe. Cependant, ces études démontrent que
l' insuline et iNOS sont reliés d'une façon ou d'une autre et l'on suppose que les mécanismes
complexes qui sous-tendent cette interaction peuvent l'être davantage dans les cas d'obésité où
plusieurs autres facteurs modulent iNOS et où la résistance à l' insuline s' installe.
La concentration de glucose est aussi augmentée lorsque le pancréas ne parvient plus à
compenser pour la résistance à l' insuline et cela peut contribuer ainsi à l' induction de
iNOS. Ainsi, dans des cellules mésengliales de rats, une forte concentration de glucose
(30mM) potentialise l' induction d'iNOS en présence de LPS et d'IFNy [404]. Ces résultats

62
confirment ceux d'une étude précédente du groupe de Shanna [405]. Des résultats
similaires furent observés dans les cellules épithéliales humaines où la région de NF-KB
était activée en présence d' une forte concentration de glucose [406].
Par ailleurs, dans les kératinocytes où iNOS joue un rôle dans la réparation des blessures et
protège contre les infections, une forte concentration de glucose activerait iNOS après une
journée d' exposition, mais l ' inhiber~it après 10 jours en raison d'une inhibition de NF-KB
[407]. Une inhibition de iNOS fut aussi observée dans des cellules endothéliales bovine
aorta endothelial ceUs (BAEC) et dans des cellules musculaires lisses vasculaires (VSMC)
en présence d'une concentration élevée de glucose [408].
Le NO lui-même exercerait un effet biphasique sur sa transcription. À de faibles
concentrations, le gaz active NF-KB et augmente l'expression de iNOS. À de fortes
concentrations, l' effet opposé est observé prévenant une surproduction de NO [409] [410].
4.6.3.1 Le contrôle de iNOS par les Peroxisomes Proliferator Activated Receptors (PPAR)
Les peroxisome proliferator activated receptors r (PP ARy) sont des récepteurs nucléaires
contrôlant notamment des gènes impliqués dans la différentiation des adipocytes et inhibant
certains mécanismes pro-inflammatoires. L'étude des PPARy a suscité beaucoup d' intérêt
parce qu'une catégorie de drogues, les thiazolidinediones (TZDs), est reconnue pour lier
ces récepteurs et avoir des effets bénéfiques sur la sensibilité à l' insuline.
L'activation des PPARy cause une inhibition de iNOS, mais le mécanisme exact de
régulation n' a pas encore été bien identifié. Même si de nombreuses études montrent que
les agonistes de PPARy inhibent iNOS le mécanisme d'action en cause reste encore mal
connu.
La prostaglandine 15-deoxy-delta 12,14-prostaglandin J2 (15dPGJ2) serait un ligand
endogène potentiel pour l 'activation PPARy, mais l'importance des PPARy dans l' inhibition

63
de iNOS est loin d'avoir été démontrée. Par exemple, dans les astrocytes, les effets anti
inflammatoires de la 1SdPGJ2 ne seraient pas dépendants des PP ARy puisque la
surexpression d'un dominant négatif de PP ARr ou d'un inhibiteur pharmacologique de
PPARy (le GW9662), n' empêche pas la 1SdPGJ2 d' inhiber l'expression de iNOS. La
1SdPGJ2 utiliserait plutôt l' inhibition de l'activité IKK pour permettre ses actions anti
inflammatoires [411]. Dans le même ordre d' idées, la 1SdPGJ2 dans des cellules de
pancréas RinmSF empêche, d'une part, la dégradation de Iill en présence de IL-1 et,
d' autre part, l' activation de STAT en présence d'IFNy, mais là encore, la présence d'un
dominant négatif ou du GW9662 n' altère en rien les activités de la 1SdPGJ2 sur iNOS
[412]. Certaines études récentes suggèrent plutôt que le véritable ligand endogène des
PP ARy serait la lysophosphatidic acid (LP A) qui traverse les membranes cellulaires et
nucléaires et qui lie les PPARy [413]. Même si la LPA peut inhiber iNOS, son mécanisme
d' action exact est encore inconnu. La LP A est un produit lipidique généré par les cellules
endothéliales, les plaquettes et les fibroblastes des tissus endommagés, et qui contribuerait
à leur réparation - possiblement en contrôlant l' expression de iNOS [414].
Par ailleurs, le groupe de Gilkeson a suggéré que les PP ARy ne sont pas impliqués dans
l' inhibition de iNOS par les agonistes synthétiques tels que le GW34784SX, la
rosiglitazone et la pioglitazone. En effet, dans des macrophages RA W 264,7, où la présence
de PPARy est peu ou pas détectée, les agonistes de PPARy inhibent tout de même iNOS.
De plus, pour être convaincus de l' absence de PPARy fonctionnels dans leur modèle, les
auteurs ont utilisé un dominant négatif de PPARy. Même en présence de ce dominant
négatif, les TZDs inhibent iNOS de la même façon que dans les cellules contrôles. Dans la
même étude, les auteurs utilisent aussi des macrophages provenant de souris dont le gène
de PP ARy avait été préalablement détruit dans ces cellules précisément au moyen d'un
système Cre/Lox. Même en absence de PPARy, les TZDs étaient en mesure d' inhiber
iNOS, de la même façonque dans les cellules exprimant les PPARy. Les auteurs en
concluent que les PPARy ne sont pas nécessaires à l'inhibition de iNOS dans les
macrophages. Cependant, dans leur modèle Cre/Lox, une plus grande induction d'iNOS en
présence d' interféron et de LPS fut observée, suggérant que même si les TZDs ne semblent

64
pas utiliser ces récepteurs pour inhiber iNOS dans les macrophages, leur présence semble
avoir un rôle à jouer sur la modulation endogène de l' enzyme [415]. Cela pourrait
s'expliquer par les récentes publications démontrant un rôle des pp AR Y dans la polarisation
des macrophages. En effet, il fût observé par deux groupes différents, que l' absence de
PPARy dans les macrophages augmentait l' expression de facteurs inflammatoires
occasionnant la résistance à l' insuline au foie, au muscle et au tissu adipeux [416] [417].
Le groupe de Nowling a aussi identifié un élément de réponse aux PPAR: (PPRE) dans le
promoteur du gène iNOS. Par contre, cet élément ne serait pas impliqué dans l'inhibition de
iNOS par les agonistes PP ARy [418]. Finalement, comme les TZD à plus fortes
concentrations peuvent aussi activer la forme 8 des PP AR, il fut suggéré que leurs effets
observés sur iNOS concernent plutôt ces récepteurs. Il fut démontré en effet qu'iNQS était
inhibé par la présence d'un agoniste de PPAR8, le GW0742 et que, tout comme dans
l 'étude précédente, l' absence de PPARy dans des macrophages n 'empêchait pas un TZD (la
rosiglitazone) d'avoir des effets sur iNOS. Cependant, dans cette étude, l' inhibition de
iNOS par la rosiglitazone était plus efficace en présence de PPARy que dans le modèle KO.
Ces résultats suggèrent donc que l' inhibition de iNOS implique des mécanismes
dépendants et indépendants des PPARy [419].
D'autres observations suggèrent également que des PPARy exercent un rôle déterminant
dans l'inhibition de iNOS. Les résultats d'une étude réalisée dans les poumons de souris
injectées à la LPS suggèrent que la rosiglitazone inhibe iNOS par l' intermédiaire des
PPARy puisqu'en présence de BADGE, un inhibiteur de PPARy, le TZD est moins efficace
[420]. Il a aussi été montré que l' expression d'un dominant négatif des PPARy empêche
iNOS d'être inhibé par la 15dPGJ2 dans des myocytes [421].
Finalement, deux études distinctes vont à contre-courant en suggérant que dans des cellules
rénales ou de rétines, la présence d' activateurs de PPARy contribue à l ' induction d' iNOS.
Ces résultats contradictoires avec les autres études mentionnées ci-haut pourraient
s' expliquer par le type cellulaire étudié [422] [423].

65
4.6.3.2 L'inhibition de iNOS par l'activité physique
L' activité physique aurait une incidence sur l' expression de iNOS. Des études montrent
qu'un entraînement régulier permet de réduire considérablement l' expression de iNOS au
niveau du muscle squelettique. Dans ces études; iNOS était présent chez ces patients à la
suite d'une crise cardiaque et l' activité physique permettait une inhibition de l' enzyme
[424] [425]. Dans un autre modèle, où iNOS est induit chez les lapins par une diète
hypercholestérolémique, un · programme d' exercice régulier permet aussi d' inhiber
l' expression de iNOS dans les aortes [426]. Plus récemment, une étude du groupe de Saad a
montré qu' une seule période d ' exercice intense chez des rats rendus obèses par une diète
riche en gras, inhibait iNOS dans le muscle. Les auteurs ont aussi observé que la s
nitrosation d' éléments de signalisation de l ' insuline était bloquée par l'activité physique,
pouvant donc ainsi contribuer à la sensibilité à l' insuline [427].
4.6.4 iNOS et l'obésité
Il est maintenant acquis que l' obésité est une condition associée à une inflammation
modérée mais chronique. Le surplus de tissu adipeux produit des cytokines et des hormones
qui touchent l' ensemble des tissus avoisinants. Comme iNOS ·répond aux signaux
inflammatoires, il n ' est pas surprenant qu' il soit induit dans des cas de surcharges
pondérales importantes.
La première recherche faisant mention d'une association entre l' obésité et une induction de
iNOS remonte à plus de 10 ans. Dans cette étude, les auteurs observèrent une induction de
iNOS dans l' hypothalamus de souris obèses ( ob/ob) [428]. Des études plus récentes
montrent que dans ce modèle d'obésité, iNOS est aussi induit dans le muscle squelettique,
dans le tissu adipeux et au foie [429] [430]. Dans un autre modèle, chez le rat Zucker cette
fois, l ' induction de iNOS est observ~e au cœur [431]. Une étude réalisée dans notre
laboratoire montre pour sa part que l' obésité induite par une diète riche en « gras» est aussi
associée à une induction de iNOS dans le muscle squelettique et le tissu adipeux [432].

66
Cette association entre iNOS et l' obésité est aussi observée chez l 'humain. Une étude
réalisée chez trente femmes obèses a révélé une induction de iNOS dans leur tissu adipeux
[433]. De plus, une analyse du plasma de 363 adolescents montre que la concentration des
produits dérivés du NO (N02-/N03-) était près de 14 fois plus élevée chez les sujets obèses
(BMI >30 kg/m2) et 4,2 fois plus élevée chez les sujets présentant un surplus de poids
(BMI >25kg/m2) que chez les sujets normaux lai~sant supposé l'induction de iNOS [434] . 1
Plus récemment, une étude réalisée chez des patients obèses ayant subit une chirurgie
gastrique afin de perdre du poids s'est avérée très révélatrice. En effet, il fut observé que la
perte de poids induite par la chirurgie était associée à une nette réduction des concentrations
de nitrite et de nitrates en circulation laissant supposer une réduction de iNOS [435]. Il Y
aurait donc une relation certaine entre l' expression de iNOS et l' obésité.
4.6.9 iNOS et la résistance à l'insuline
Considérant le lien de causalité entre l' obésité et la résistance à l' insuline, et puisque
l' obésité semble souvent associée à une induction de iNOS, on a établi un lien entre iNOS
et la pathologie. Des études de notre laboratoire ont démontré que dans des myocytes en
culture, la présence d' IFNy, de TNFa et de LPS induit iNOS et augmente le transport de
glucose basal, mais cause un défaut dans le transport de glucose stimulé par l ' insuline.
iNOS semble dans ce cas bel et bien responsable de l'effet produit puisque la présence d'un
inhibiteur de NOS, le L-NAME, empêche cette manifestation [110]. Dans une autre étude,
Perreault et Marette ont démontré que l' absence du gène iNOS dans des souris
transgéniques protège contre la résistance à l ' insuline induite par une diète riche en « gras »
[432]. Ces résultats furent d'ailleurs confirmés par le groupe de Keamey quelques années
plus tard [436].
Chez l'humain, il a été démontré que les niveaux plasmatiques de nitrites et de nitrates
étaient plus élevés chez des patients diabétiques de type 2 que chez des sujets contrôles,
suggérant l'induction de iNOS. En effet, une étude a montré que chez quatorze patients
diabétiques de type 2, comparés à douze volontaires en bonne santé, l' expression de iNOS
était augmentée au niveau muscle squelettique [437]. Une autre étude, de polymorphisme

67
cette fois , montre une variante dans le promoteur de iNOS chez des patients diabétiques de
type 2, ce qui pourrait laisser croire à une modulation d ' activité de iNOS chez les
personnes diabétiques [438].
Perreault et Marette ont établi que iNOS cause la résistance à l'insuline au muscle
squelettique en créant une défaillance de la signalisation de l ' insuline au niveau de la PI 3-
kinase et de l 'Akt, une voie déterminante des effets métaboliques de l ' insuline [432]. Plus
récelllIllent, une étude a montré que l'exposition à un donneur de NO ou la transfection de
iNOS réduisait l'expression de la protéine IRS-1 dans les cellules musculaires en culture
primaire. Le donneur de NO augmente l'ubiquitination d'IRS-l , et les inhibiteurs du
protéasome bloquent la réduction de l'expression d'IRS-1 induite par le donneur de NO.
L'expression de iNOS est augmentée dans le muscle squelettique des souris obèses (ob/ob)
par rapport aux souris de type sauvage et la délétion du gène iNOS ou le traitement aux
inhibiteurs de iNOS améliore la diminution . de l'expression d'IRS-1 dans le muscle
squelettique de ces souris. Ces données proposent donc que iNOS réduit l'expression d'IRS-
1 dans le muscle squelettique par la dégradation d' IRS-1 et qu' il pourrait donc contribuer à
l'insulino-résistance associée à l' obésité [429]. Kaneki et Fuj imito ont pour leur part
observé les effets de iNOS induit au foie dans les souris obèses (ob/ob). Ils ont déterminé
que iNOS jouait un rôle dans l'hyperglycémie à jeun des souris obèses puisque la présence
d ' un inhibiteur de l' enzyme réduisait ce défaut. Cette étude révèle aussi que les souris
obèses traitées avec l'inhibiteur de iNOS présentaient une plus grande expression d ' IRS-1
et d' IRS-2 au foie et que la présence d'iNOS ou du donneur de NO dans des hépatocytes en
culture diminuait l' expression des deux substrats du récepteur à l'insuline [430].
4.7 La résistance à l'insuline induite par la S-nitrosylation
La S-nitrosylation est une modification post-traductionnelle réversible résultant de la
réaction du NO et d'un résidu cystéine d'un motif particulier d'une protéine en présence
d' 02- [439]. Cette modification peut altérer l ' activité de certaines enzymes telle H-ras, c
Jun N-terminal kinase-1 (JNK1), NF-KB et nNOS [440] [441] [442] [359].

68
Des études ont établi un rapprochement entre la S-nitrosylation induite par iNOS dans
l'obésité et la résistance à l' insuline. Dans une première étude, les auteurs obsérvèrent qu'un
donneur de NO dans un muscle isolé causait la S-nitrosylation du récepteur à l' insuline (IR),
de son substrat (IRS-I) et de l'Akt. L' expression d'IRS-I était aussi réduite par le traitement.
Ils utilisèrent ensuite deux modèles d'obésité associés à l' induction de iNOS, soit une diète
riche en ~< gras» et un modèle d'obésité génétique (ob/ob). Dans ces modèles, les auteurs ont
aussi observé la S-nitrosylation de IR, d' IRS-I et d'Akt et une diminution de la sensibilité à
l' insuline. L' inhibition de iNOS a permis de rétablir la signalisation par l' insuline [443]. Dans
une autre étude, les auteurs ont identifié le résidu cystéine d'Akt ciblé par la S-nitrosylation en
présence de iNOS [444].
D'un autre côté, une étude récente propose un rôle protecteur de la S-nitrosylation contre les
effets néfastes de l'hyperglycémie associée à la résistance à l' insuline. En effet, les auteurs
suggèrent qu'une forte concentration de glucose dans des cellules endothéliales, réduit la S
nitrosylation de eNOS et pourrait ainsi contribuer au dysfonctionnement de l' endothélium
chez les patients résistants à l'insuline. De plus, les auteurs de cette études observent que NF
KB est inhibé par la S-nitrosylation et suggèrent que le NO utilise donc ce mécanisme pour
réguler le facteur de transcription [445].
4.8 Le ONOO-
Le NO réagit très rapidement avec le superoxide (02-) pour former le ONOO- : le
peroxynitrite. Comme ces deux molécules sont souvent produites au site d' inflammation où
iNOS est induit par les macrophages et/ou par le tissu touché, le ONOO- est alors formé
très rapidement [446]. Bien que ni le NO ni le O2- ne soit des oxydants puissants, le
ONOO- est très réactif et peut attaquer plusieurs types de protéines par son action
oxydante [ 447].

69
4.9 L'origine du O2-
Le (02-) peut avoir différentes origines. Il peut d' abord être formé à partir des trois enzymes
NOS. En faibles concentrations de certains co-facteurs et substrats, les enzymes peuvent se
découpler et produire du (02-). Le découplage d' une enzyme fait référence à la perte d'un
électron qui devrait normalement être utilisé pour générer un produit à partir de son
substrat. L' oxygène peut alors servir d' accepteur d ' électrons et ainsi produire le (0 2-) . Dans
le cas des NOS, lorsque les niveaux de L-arginine et de BH4 sont sous-maximales, le
transfert d' électrons peut être déficient et produire du (02-) [448] [449]. Il est ainsi
intéressant de noter que les macrophages sont reconnus pour migrer aux endroits où la L
arginine est limitée,. ce qui peut contribuer à leur production de ONOO- [450]. Le NO peut
aussi inhiber la cytochrome c oxydase et ainsi inhiber la chaîne respiratoire mitochondriale,
menant alors à la production d' (02-) [451]. Plusieurs autres enzymes telles que la NADPH
oxydase, la cyclooxygenase, la lipoxygénase, et la xanthine oxydase peuvent aussi
contribuer à la génération de (02-) [452] [453].
4.10 La nitrotyrosination
La nitrotyrosination est une modification posttraductionelle sur un résidu tyrosine. Ce
processus peut faire intervenir plusieurs molécules telles que N02-, N02+, HONO et le
ONOO-. Le nitrite N02- qui est le produit majeur de dégradation du NO peut devenir un
agent de nitration. L' acidification du N02- en HN02 par des péroxydases peut mener à la
formation de N02CI un agent nitratant [454][455]. Ainsi donc, il fut proposé que ce
mécanisme de nitration était plus important que celui impliquant le NO in vivo chez la
souris. En effet, l'injection du parasite T spiralis augmente la présence de nitrotyrosine
dans des souris iNOS ,.../- au même titre que dans les souris sauvages, alors que l'induction
de l'expression de la myeloperoxidase (MPO) est pour sa part augmentée [456]. Cependant,
une autre étude a démontré que les macrophages n'exprimant pas iNOS pouvaient
effectivement produire une réaction oxydative en réponse à des cytokines, mais que la

70
quantité de nitrotyrosine était grandement réduite [457]. Chez l 'humain, la nitration de
résidus tyrosine sur des protéines particulières est retrouvée dans plus de cinquante
maladies [458].
La nitration est un processus sélectif qui ne touche pas tous les résidus tyrosine. Il faut
d' abord que le résidu tyrosine soit dans un environnement pauvre en cystéine, à proximité
d'une charge négative, dans une région bien exposée et de préférence dans une structure en
forme de boucle [459] [460].
Le rôle physiologique de la nitration pourrait être un signal de transduction dans la réponse
immunitaire. En effet, les protéines nitratées pourraient alors servir à la formation
d' antigènes pour la production d' anticorps, de facteur chémoattractant ou de signal pour
que les macrophages procèdent à la phagocytose. Il fut aussi démontré que la nitration peut
occasionner la dégradation de protéines par la chymotrypsine en particulier [461]. Il fut
aussi postulé que la nitration pouvait altérer la conformation, la structure et l'activité
catalytique de protéines [462]. Par ailleurs, comme la nitration est à la base de plusieurs
protéines dans différents tissus, cette modification pourrait avoir un rôle physiologique en
l' absence d' inflammation. Aussi, certains ont-ils suggéré que la nitration pouvait jouer un
rôle dans la transmission synaptique et dans l'ovulation [463].
4.10.1 Effets de la nitrotyrosination sur la signalisation cellulaire
La phosphorylation sur tyrosine est l'un des processus ' les plus reconnus ' dans la
signalisation cellulaire. Les tyrosines kinase catalysent le transfert d'un groupement
phosphate à un groupe hydroxyle d'un résidu tyrosine. Cette réaction est réversible en
présence de phosphatases. Le processus de phosphorylation sur tyrosine est sélectif. Par
exemple, une séquence particulière d'acides aminés entourant le résidu tyrosine et une
conformation adéquate de la protéine lui confèrent sa susceptibilité à être phosphorylée. De
la même façon, la nitration sur tyrosine demande un environnement particulier et est lui
aussi réversible par la présence de dénitrase [464]. Certains suggèrent que les deux
processus de modification sur tyrosine requièrent les mêmes exigences [460]. On suppose

71
que la nitration sur tyrosine empêche la phosphorylation de ce même résidu. En effet, il est
postulé que la nitration de résidus tyrosine fige l' enzyme cible dans une conformation
inactive [465] [466]. Par exemple, la molécule d' adhésion platelet endothelial cel! adhesion
molecule (PECAM-1) ne peut plus être modulée par sa phosphorylation à la suite de la
nitration sur le résidu tyrosine 686 [467]. Beaucoup d' autres enzymes telles que la
glutamine synthase, la prostacyclin synthase, la tyrosine hydrolase et la Ca-ATPase du
reticulum sarcoplasmique peuvent être inactivées ainsi [459] [468] [469] [470].
4.10.2 La nitration et la résistance à l'insuline
Il semble exister un lien entre la présence de nitrotyrosine et le diabète de type 2. Dans une
étude sur quarante sujets diabétiques type 2, comparés à 35 sujets en bonne santé, la
présence de nitrotyrosine fut observée dans tous les plasmas des sujets malades et non
détectée chez les contrôles [471]. Cette présence de nitrotyrosine pourrait connaître son
origine dans le déséquilibre des niveaux de glucose observés chez les diabétiques. En effet,
une forte concentration de glucose semble induire la nitration dans certains tissus et
certaines cellules en culture. Ainsi la perfusion de cœur de rat avec une solution à forte
teneur en glucose augmente la production d' 02- de 300 %, augmente de 40 % la production
de NO causée par une induction de iNOS et permet la détection de nitrotyrosine [472]. De
la même façon, des cellules endothéliales en culture (HAECs) exposées à des
concentrations de glucose au-dessus des normales cause la nitration de certaines protéines
[473]. Finalement, une étude chez des sujets humains en bonne santé a montré qu'une
augmentation du glucose sanguin à 15 mmollL durant deux heures augmentait la présence
de nitrotyrosine dans le plasma [474].
L' insuline qui possède des résidus tyrosine importants pour son interaction avec son
récepteur pourrait elle même être la cible de nitration. Différentes études indiquent que
l' insuline peut être nitratée sur des résidus tyrosine causant ainsi une réduction de sa
capacité à lier son récepteur [475] [476]. Il est à noter par ailleurs, qu' il a été suggéré que le
peroxynitrite inhiberait l' activation des PPARy par leur nitration dans les macrophages. Tel
que mentionné plus haut, ces derniers ont des rôles bénéfiques contre la résistance à

72
l' insuline donc leur inactivation par leur nitration pourrait contribuer au développement de
la pathologie [477].

73
5. Buts de l'étude
Les tissus adipeux étant sensibles à l'insuline, nous avons voulu dans une première étude
déterminer si les tissus adipeux, blanc et brun, étaient aussi la cible des effets néfastes de
iNOS sur la sensibilité à l'insuline au même titre que nous l'avions établi pour le muscle
squelettique. Cette étude fut publiée dans la revue Hormone and Metabolic Research
(2000 ; 32 : 480-484). Je suis première auteure de cet article.
Afin d'approfondir les mécanismes impliqués dans la résistance à l'insuline observée dans
le muscle squelettique en présence de iNOS, nous nous sommes questionnés sur
l'implication de la formation du ONOO- dans ce phénomène. Manuscrit en préparation. Je
suis première auteure de cet article.
Dans un troisième temps, nous avons déterminé si l'induction de iNOS pouvait contribuer
aux désordes métaboliques observés lors de l'incubation prolongée de muscles in vitro.
Cette étude est particulièrement déterminante pour l'interprétation des données obtenues par
quiconque utilisant cette technique pour répondre à ses objectifs scientifiques. Manuscrit en
préparation. Je suis première auteure de cet article.
Finalement, nous avons évalué si les effets sensibilisateurs à l' insuline de drogues activant
l'AMPK n'impliqueraient pas leurs capacités à inhiber l'expression de iNOS. Cette étude
fut publiée dans la revue Journal of Biological Chemistry 2004 May 14,·279(20):20767-74.
Je suis première auteure de cet article.

Chapitre 1. La production de monoxyde d'azote par les adipocytes : un rôle dans la pathogenèse de la résistance à l'insuline?
Cette étude compare les effets d' iNOS sur le muscle et le tissu adipeux en ce qui a trait à la
sensibilité à l' insuline au niveau du transport de glucose. Nous avons déjà rapporté que
l' induction d' iNOS au niveau du muscle cause une réduction de la capacité de l' insuline à
permettre le transport du glucose. Les conséquences de l' induction de l' enzyme sur la
sensibilité à l' insuline dans les adipocytes restaient à déterminer. Nous utilisons donc dans
cette étude, un modèle d'adipocytes blancs (3T3-Ll) et bruns (T37i) dans lesquels iNOS
est induit par un mélange de cytokines (TNFa et IFNy) accompagné de LPS. Le rôle
d' iNOS dans les effets observés fut déterminé par l' utilisation d'un inhibiteur spécifique:
le 1400W. Contrairement à ce que nous avions précédemment observé au niveau du
muscle, nous avons constaté que dans les adipocytes en culture la résistance à l' insuline
induite par la présence de cytokines et de LPS, n' impliquait pas iNOS puisque son
inhibition par le 1400W ne prévenait pas le défaut de transport du glucose stimulé par
l' insuline. Nous proposons finalement que, même si la présence d'iNOS dans le tissu
adipeux n' est pas dommageable pour ce dernier, il n' en demeure pas moins que son
induction pourrait contribuer à la résistance à l'insuline du muscle qui lui est affecté par la
présence de monoxyde d'azote.

75
Nitric oxide production by adipocytes : a role in the pathogenesis of insulin
resistance ?
Geneviève Pilon, Patrice Penfomis and André Marette*
Department of Physiology, Lipid Research Unit, Laval University Hospital Center, Québec,
G1V 4G2, Canada . .
Short title: NO and glucose metabolism in adipocytes.
* To whom correspondence should be adressed at:
Departement ofPhysiology & Lipid Research Unit
Laval University Hospital Research Center
2705 Laurier Boulevard, RC 9502
Sainte-Foy, Québec, G 1 V 4G2, Canada
Fax: (418) 654-2176
E-mail: [email protected]

76
INTRODUCTION
The radical gas nitric oxide (NO) has been shown to participate in an ever-growing
number of cellular processes. NO can be recognized as an intracellular second messenger, a
paracrin~ substance for regulation ofneighboring cells, and a neurotransmitter [see (1 , 2)].
NO is synthetized from L-arginine by a family of enzymes known as NO synthases (NOS).
There are at least three different isotypes of NOS enzymes: the endothelial type (eNOS or
NOS III), the neuronal type (nNOS or NOS 1), and the inducible type (iNOS or NOS II).
eNOS is mainly expressed in endothelial cells and catalyzes the synthesis of NO which
mediates vasorelaxation of the underlying smooth muscle. nNOS is highly expressed in the
central nervous system where NO serves as a messenger for neurons, much like a
neurotransmitter. Unlike its constitutive Ca2+-dependent eNOS and nNOS counterparts,
iNOS is mainly regulated at the expression level and its activity is in large part independent
from Ca2+. In most species, iNOS is expressed to very low levels under normal conditions.
However, its cellular expression is markedly stimulated by bacterial endotoxins or
inflammatory cytokines (2, 3). When induced, this isoform generates high concentrations of
NO as compared to eNOS or nNOS. In macrophages, sustained production of NO enables
cytotoxic activity against invading microorganisms. Although this high-output NO pathway
probably evolved to protect the host from infection, there is also evidence that it can cause
deleterious effects to other normal host cells which conf ers to iNOS the
protective/destructive duality inherent in every other major component of the immune
response. For example, the marked NO production that is consequent to iNOS expression in
many tissues in endotoxic shock is believed to cause hypotension, organ injury and
dysfunction [reviewed in (3)].
NOS expression in adipose tissue
Whereas iNOS expression in macrophages is well documented, we and others have
recently reported that the enzyme can also be induced by LPS and cytokines in many other
tissues including classical targets of insulin action such as the liver, skeletal muscle and
white and brown adipose tissues (4, 5, 6, 7, 8). The effects of LPS and proinflammatory

77
cytokines on iNOS induction in white and brown adipose tissues are depicted in Figure 1.
Low but detectable expression of iNOS mRNA and prote in can be foùnd in adipose tissues
. of normal rats (Figure lA-B). A single injection ofbacteriallipopolysaccharide (LPS) in rat
(a model of endotoxic shock) markedly increased iNOS expression in white (epididymal
and perirenal) and interscapular brown adipose tissues, raising iNOS enzymatic activity by
10-20-fold (Figure lB).
We have also used adipose cell lines to investigate the regulation of iNOS
expression by LPS and preinflammatory cytokines. We used 3T3-Ll adipocytes as a model
of white fat cells and T37i adipocytes as a model of brown adipose cells. The T37i fat cell
line was derived from a mouse hibemoma (9) and present typical features of brown
adipocytes (e.g. uncoupling protein-l expression; see (10)). As shown in Figure le, iNOS
cellular expression and activity can be directly increased by incubating cultured white and
brown adipocytes with a mixture of cytokines and LPS. We have previously reported that
neither LPS alone nor individual cytokine can increase iNOS-mediated NO production in
3T3-Ll adipocytes (8) and we have obtained similar findings with T37i brown adipocytes
(Penfomis and Marette, unpublished observations). This indicates that iNOS induction in
endotoxin-challenged rats is mediated by a complex network of interactions between
inflammatory cytokines and endotoxin at the level of the adipocyte.
1t has recently been reported that eNOS is also expressed in hum an adipose tissue
(11). Although adipose tissue eNOS may be explained by the presence of endothelial cells
in the tissue extracts, the fact that the enzyme could also be detected in isolated adipose
cells strongly suggest that eNOS is also expressed in adipocytes. However, we could not
detect eNOS in either 3T3-Ll white adipocytes (8) or T37i brown adipocytes (Penfomis
and Marette, unpublished observations). The neuronal isoform nNOS was not detected in
adipose tissues of either rat or humans (8, Il).
Biologica~ role of NOS in adipocytes
NO and lipid metabolism

78
There is sorne evidence that NO can regulate adipocyte lipolysis. 1ndeed, Gaudiot et
al. (12) reported that NO-releasing compounds inhibits catecholamine-stimulated lipolysis
in rat fat cells. The NO-dependent inhibition of catecholamine-stimulated lipolysis has been
recently confirmed in isolated human fat cells (13). On the other hand, in the absence of
catecholamines, NO donors can also increase basallipolysis (12). Whether NO is mediating
the known stimulatory effects of cytokines (such as TNF -u) on lipolysis remains to be
tested. 1t has also been speculated that eNOS-mediated NO production may decrease
lipolysis in human fat cells (11). 'Clearly, more studies are needed to clarify the precise role
of NO in modulating fat celllipolysis.
There is also evidence that NO regulates lipoprotein metabolism in brown
adipocytes. One study reported that TNF-u-induced inhibition of lipoprotein lipase (LPL)
activity was mediated by iNOS induction and NO production in brown adipocytes (14).
More recent studies from Deshaies and colleagues further suggest that iNOS induction in
skeletal muscle and brown fat of LPS-challenged rats is also mediated by iNOS activity
(Picard, Perreault, Kapur, Marette, and Deshaies, unpublished data). 1ndeed, they found
that LPS-induced LPL inhibition in these tissues was blocked by the specific iNOS
inhibitor aminoguanidine (AGN) and was prevented in iNOS knockout mice. 1ntriguingly,
although LPS also blunted LPL activity in white adipose tissue, this effect could not be
abrogated by AGN treatment or by targeted disruption of iNOS in mice, therefore
suggesting that NO exerts tissue-specific modulation of LPL activity in endotoxemia.
NO and glucose metabolism
1nsulin is believed to enhance glucose disposaI in peripheral tissues at least in part by
increasing muscle perfusion by vasodilatory mechanisms [reviewed in (15)]. The
stimulatory effect of insulin on tissue blood flow has been reported to be dependent on
endothelium NOS (eN OS). T~e implication of eNOS in insulin-mediated glucose disposaI
is also supported by our previous observation that in vivo administration of the NOS
inhibitor L-NAME significantly reduced insulin-stimulated glucose uptake by individual
skeletal muscles and white and brown adipose tissues (16). A role for eNOS in mediating

79
insulin-dependent glucose disposaI is also supported by the finding that mi ce with targeted
disruption in eNOS gene develop insulin resistance (17). Thus, eNOS-mediated NO
production in the microvasculature of muscle and adipose tissues appears to be important
for optimal glucose metabolism by these tissues
On the other hand, overproduction of NO by. iNOS induction may also directly
affect glucose metabolism in fat and muscle cells. Thus, we have previously demonstrated
that LPS and cytokines modulate glucose metabolism by inducing iNOS expression in
skeletal muscle and cultured L6 myocytes (6, 18). In L6 myocytes, chronic treatment with
cytokines and LPS caused a large increase in basal glucose transport but also completely
inhibited the ability of insulin to stimulate glucose transport. Both effects were largely
abrogated by the non-specific NOS inhibitor L-NAME (18). On the other hand, the
potential effect of adipose NO production on glucose metabolism has not yet been
investigated. As shown in Figure 1 C, the 3T3-L1 and T37i celllines represent valuable in
vitro models to study the impact of iNOS induction and NO production on white and brown
adipocyte glucose metabolism, respectively. Thus, we have investigated the potential role
of NO in mediating changes in glucose uptake in cytokines/LPS-treated adipocytes.
To test the implication of NO in cytokine/LPS-treated adipocytes, we used the
recently described and selective iNOS inhibitor 1400W (19). As shown in ' Figure 2A,
addition of 1400W to cytokines/LPS-treated cells totally abrogated iNOS activity, as
measured by nitrite production over 24 hrS, in 3T3-Ll white adipocytes. The inhibitor also
largely, but not completely, inhibited iNOS-mediated NO production in T37i brown
adipocytes (Fig. 2B). Cytokines/LPS markedly increased (3-4-fold) basal glucose transport
in both 3T3-L1 white adipocytes and T37i brown fat cells. However, iNOS inhibition with
1400W failed to prevent the increase in basal glucose uptake in white adipocytes (Fig. 2C)
and only partially inhibited this effect in brown adipocytes (Fig. 2D). These data indicate
that the elevated basal glucose uptake is entirely or mostly explained by NO-independent
pathways in cytokine/LPS-treated white and brown fat cells, respectively. The
cytokines/LPS mixture also markedly reduced the acute stimulatory effect of insulin on
glucose uptake in both white and brown adipocytes (Fig. 2E and F). Addition of 1400W

,------------~~--- - - -- ~
80
failed to reverse insulin resistance in both cell types. This is in marked contrast with our
observations that iNOS inhibition with L-NAME (18) or 1400W (Pilon and Marette,
unpublished data) reduced ~oth the basal increase in glucose uptake and the insulin
resistance induced by cytokines/LPS exposure in skeletal muscle cells.
These results indicate that NO modulates glucose metabolism in a cell type-specific
manner. In this regard, it is interesting to note that a similar difference in NO action was
observed for the effect of endotoxin challenge on LPL activity in skeletal muscle and
brown adipose tissue versus white adipose tissue. The mechanisms behind this selective
action of NO in different insulin target cells remain to be clarified.
Adipocyte iNOS and insulin resistance.
What is the physiological role of iNOS in adipose tissue? We have previously
suggested that iNOS induction in adipose tissues may play a role in the local immune
defense during inflammatory processes [see (8)]. White and brown fat are dispersed
throughout the body and are widely distributed along and even within maj or organs such as
the intestine, liver, kidney, blood vessels and skeletal muscle. It is therefore likely to
represent a major source of local NO generation in inflammatory settings. Such a link
between the immune system and adipose tissue is consistent with previous observations
that adipsin, the initial and rate-limiting enzyme of the alternative pathway of complement
activation (20), is mainly expressed in adipose tissue and that fat cells are a major site of
expression and synthesîs of immunoregulatory molecules such as the cytokines TNF -u and
leptin, the product of the ob gene T see (21) for a review].
On the other hand, iNOS-derived NO is also known as a double-edged sword since
it can also be detrimental to the host in a variety of inflammatory conditions. For example,
the marked NO production that is consequent to iNOS expression in many tissues in
endotoxic shock is believed to cause hypotension, organ injury and dysfunction [see(3)].
Our previous work indicates that another deleterious effect of endotoxemia that appears to
be related to iNOS induction is the development of muscle insulin resistance (6). Indeed,
both LPS challenge in vivo and treatment with cytokines in vitro caused insulin resistance

81
for glucose uptake in skeletal muscle. These effects can be mainly abrogated by the NOS
inhibition with L-NAME (18) or the selecctive iNOS inhibitor aminoguanidine (Perreault,
Kapur and Marette, unpublished data).
On the other hand, the present data suggest that NO is not an autocrine modulator of
glucose metabolism in white adipose cells since iNOS inhibition with ,1400W failed to
restore insulin action in both white and brown adipocytes chronically exposed to cytokines
and LPS. However, as proposed in Figure 3, it may be possible that NO exerts sorne
paracrine modulation of skeletal muscle glucose metabolism. Indeed, NO is a low
molecular weight, highly lipophilic molecule, and can diffuse rapidly to neighboring cells.
It is therefore conceivable that the marked iNOS induction and NO production by local
adipose cells contributes to muscle insulin resistance in endotoxemia. In this model, NO
may inhibit insulin action only when its levels are abnormally increased, i.e. when iNOS
expression is induced. In this situation, NO may react more readily with 02- to form the
potent oxidant · peroxynitrite (ONOO-) and override the cell natural defense against
oxidation (antioxidant enzymes and oxidant scavengers). This double-edged sword effects
of NO is well documented for other systems (e.g. cardiovascular, central nervous), and can
be compared to the protective/deleterious nature of adipose tissue TNF-a, which may
provide local immune protection but also inhibits insulin action in skeletal muscle of obese
animaIs (22, 23).
It is also possible that iNOS induction in adipocytes contribute to obesity-linked
insulin resistance. Indeed, since skeletal muscle is infiltrated with adipose tissue in obese
subjects, elevated NO production by adipocytes may thus contribute to the development of
insulin resistance in obesity. In line with this hypothesis, we have recently found that iNOS
expression is induced in both skeletal muscle and white adipose tissue of high-fat fed rats
and mice and that iNOS knockout mice are protected from the development of insulin
resistance in high-fat diet-induced obesity (Perreault and Marette, unpublished
observations ).

82
Conclusions
The studies reported and the new data presented in this minireview article present indicate
that iNOS expression is inducible in classical targets of insulin action, namely skeletal
muscle, white and brown adipose tissues. Our results also indicate that NO is not the
mediator of cytokine/LPS-mediated insulin resistance in white and brown fat cells,
respectively. Howev'er, iNOS induction in adipose tissue could contribute to insulin
resistance through a paracrine modulation of muscle insulin-stimulated glucose uptake.
Future studies will be aimed at testing whether selective iNOS invalidation in adipose
tissue of obese animaIs could reduce or even prevent the development of insulin resistance,
hypertriglyceridemia and associated cardiovascular complications.

83
Acknowledgments
We wish to thank Dr Amira Klip (Hospital for Sick Children, Toronto, Canada) and Dr
Marc Lombès (INSERM U478, Université Bichat, Paris, France) for providing the 3T3-Ll
and_T37i adipose cell lines, respectively. We also wish to thank Drs Yves Deshaies and
Claude'H. Côté for critical reading ofthis review.
The work presented in this review was supported by a grant from the Canadian Institute for
Health Research and by scholarships from the Fonds de la Recherche en Santé du Québec
to A. Maretle
G. Pilon was supported by studentship from Energy Metabolism Research Center
(CREME), Laval university, Québec.
P. Penfomis was supported by Doctoral studentship from the Canadian Institute for Health
Research.

84
References
1. Murad, F. Nitric oxide signaling: would you believe that a simple free radical could be a
second messenger, autacoid, paracrine substance, neurotransmitter, and hormone? Recent
Prog Honn Res 1998, 53: 43-59; discussion 59-60.
2. Moncada, S, RMJ Palmer, and EA Higgs. Nitric oxide: physiology, pathophysiology and
phannacology. Pharmacol Rev 1991 , 43: 109-142.
3. Nathan, C. lnducible nitric oxide synthase: what difference does it make? J Clin lnvest
1997, 100:2417-23.
4. Annane, D, S Sanquer, V Sebille, A Faye, D Djuranovic, JC Raphael, P Gajdos, and E
Bellissant. Compartmentalised inducible nitric-oxide synthase activity in septic shock.
Lancet 2000, 355: 1143-8.
5. Liu, S, lM Adcock, RW Old, PJ Bames, and TW Evans. Lipopolysaccharide treatment in
vivo induces widespread tissue expression of inducible nitric oxide synthase mRNA.
Biochem Biophys Res Commun 1993, 196: 1208-13.
6. Kapur, S, S Bedard, B Marcotte, CH Cote, and A Marette. Expression of nitric oxide
synthase in skeletal muscle: a novel role for nitric oxide as a modulator of insulin action.
Diabetes 1997, 46: 1691-700.
7. Ribière, C, AM Jaubert, N Gaudiot, D Sabourault, ML Marcus, JL Boucher, D Denis
Henriot, and Y Giudicelli. White adipose tissue nitric oxide synthase: a potential source for
NO production. Biochem Biophys Res Commun 1996, 222: 706-12.
8. Kapur, S, B Marcotte, and A Marette. Mechanism of adipose tissue iNOS induction in
endotoxemia. Am J Physiol 1999, 276: E635-41.

85
9. Zennaro, MC, D Le Menuet, S Viengchareun, F Walker, D Ricquier, and M Lombes.
Hibemoma development in transgenic mice identifies brown adipose tissue as a novel
target ofaldosterone action. J Clin Invest 1998, 101: 1254-60.
10. Penfornis, P, S Viengchareun, D Le Menuet, F Cluzeaud, MC Zennaro, and M Lombes.
The mineralocorticoid receptor mediates aldosterone-induced differentiation of T37i cells
into brown adipocytes. Am J Physiol Endocrinol Metab 2000, 279: E386-94.
Il. Elizalde, M, M Ryden, V van Harmelen, P Eneroth, H Gyllenhammar, C Holm, S
Ramel, A Olund, P Amer, and K Andersson. Expression of nitric oxide synthases in
subcutaneous adipose tissue of nonobese and obese humans [In Process Citation] . J Lipid
Res 2000, 41: 1244-51.
12. Gaudiot, N, AM Jaubert, E Charbonnier, D Sabourault, D Lacasa, Y Giudicelli, and C
Ribiere. Modulation of white adipose tissue lipolysis by nitric oxide. J Biol Chem 1998,
273: 13475-81.
13. Andersson, K, N Gaudiot, C Ribiere, M Elizalde, Y Giudicelli, and P Amer. A nitric
oxide-mediated mechanism regulates lipolysis in hum an adipose tissue in vivo. Br J
Pharmacol 1999, 126: 1639-45.
14. Uchida, Y, F Tsukahara, K Ohba, A Ogawa, K Irie, E Fujii, T Yoshimoto, T Yoshioka,
and T Muraki. Nitric oxide mediates down regulation of lipoprotein lipase activity induced
by tumor necrosis factor-alpha in brown adipocytes. Eur J Pharmacol 1997, 335: 235-43.
15. Baron, AD. The coupling of glucose metabolism and perfusion in hum an skeletal
muscle. The potential role of Endothelium-derived nitric oxide. Diabetes 1996, 45 (Suppl.
1): SI05-S109.
16. Roy, D, M Perreault, and A Marette. Insulin stimulation of glucose uptake in skeletal
muscles and adipose tissues in vivo is NO dependent. Am J Physiol 1998,274: E692-9.

86
17. Shankar, RR, y WU, HQ Shen, JS Zhu, and AD Baron. Mice with gene disruption of
both endothelial and neuronal nitric oxide synthase exhibit insulin resistance. Diabetes
2000, 49: 684-7.
18. Bédard, S, B Marcotte, and A Marette. Cytokines modulate glucose transport in skeletal
muscle by inducing the expression of inducible nitric oxide synthase. Biochem J 1997,
325: 487-93.
19. Garvey, EP, JA Oplinger, ES Furfine, RJ Kiff, F Laszlo, BJ Whittle, and RG Knowles.
1400W is a slow, tight binding, and highly selective inhibitor of inducible nitric-oxide
synthase in vitro and in vivo. J Biol Chem 1997, 272: 4959-63.
20. Rosen, BS, KS Cook, J Yaglom, DL Groves, JE Volanakis, D Damm, T White, and BM
Spiegelman. Adipsin and complement factor D activity: an immune-related defect in
obesity. Science 1989, 244: 1483-7.
21. Spiegelman, BM, and JS Flier. Adipogenesis and obesity: rounding out the big picture.
Cell 1996, 87: 377-89.
22. Hotamisligil, GS, P Amer, JF Caro, RL Atkinson, and BM Spiegelman. Increased
adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin
resistance. J Clin Invest 1995, 95: 2409-15.
23. Hotamisligil, GS, and BM Spiegelman. Tumor necrosis factor alpha: a key component
of the obesity-diabetes link. Diabetes 1994, 43: 1271-8.

,----------------- ~ ~~
87
Legends to figures
Figure 1: iNOS expression in white and brown adipose tissues
A) LPS-induced expression of iNOS synthase (iNOS) mRNA ln white epididymal
(EW AT), perirenal (PW AT), and interscapular brown adipose tissue (BAT). Rats were
treated for 4 hours with LPS or vehicle, RNA was isolated and iNOS mRNA was measured
by RT-PCR, as in ref. (18). B) Effects ofLPS on iNOS prote in levels and iNOS activity in
white and brown adipose tissues. Rats were treated or not with LPS for 4 hours and tissue
iNOS protein levels and iNOS activity were determined by Western blotting with an iNOS-
specific antibody " and by the 3H-citrulline assay, respectively, as described in (6 8). C)
Effects of cytokines/LPS (interferon-y 200 U/ml, TNF-a lOng/ml and LPS 10Jlg/ml) on
iNOS protein levels in T37i brown adipocytes and in 3T3-Ll white adipocytes. Cells were
treated with cytokines/LPS for 24 hours and iNOS protein levels were detected by Western
blotting. Nitrite production was measured by the Griess reaction, as in (6). AlI data
represent means ± SE ofthree individual experiments. *P<O.Ol vs control values.
Figure 2 : Effect of the iNOS inhibitor 1400W on NO production and glucose uptake
in cytokines/LPS-treated 3T3-Ll white and T37i brown adipocytes.
Cells were treated or not for 24 ho urs with interferon-y (200 U/ml, lnM), TNF-a (lOng/ml),
LPS "(1 o Jlg/ml) ± l400W (0.1 mM), and A) NO production, B) basal glucose uptake, and
C), insulin-mediated glucose uptake was measured. NO production was determined by
measuring the accumulation of nitrite in the medium by the Griess reaction. Cellular
glucose uptake was measured by use of the glucose analogue 2-[3H]-2-deoxy-D-glucose as
described in (18). When present, insulin (100 nM) was added 30 min before uptake
measurements. Data represent means ± SD of 2 (T37i) and 3 (3T3-Ll) individual
experiments. *P<O.Ol vs control values. +P<O.Ol vs cytokines/LPS values.
Figure 3: Proposed role for adipose iNOS induction in skeletal muscle insulin
resistance in obesity and inflammation.

88
Increased concentrations of proinflammatory cytokines (and perhaps other mediators)
induce iNOS expression in skeletal muscle and adipocytes. NO inhibits insulin-stimulated
glucose uptake in skeletal muscle (6, 18) but not in adipose cells (results of the present
study). However, marked NO production by local adipocytes may affect insulin action in
muscle cells through a paracrine action, especially in obesity where muscle tissue is
infiltrated with fat cells.

Figure. 1
A LP8 :
B
c
+
EWAT
o COî1t'ol • LPS
EWAT
Basal Cyto IlPS
313 -ll vil ite adipocytes
+
FWAT
PW"AT
irJOS
500 c
'S 400 e :; 300 e ~2OJ o z 2 100 =l
+
"'-- îN08
~ GAPDH
BAT
BAT
Basal CytoA...PS
137i b rom ooipocytes
iN0 8
89
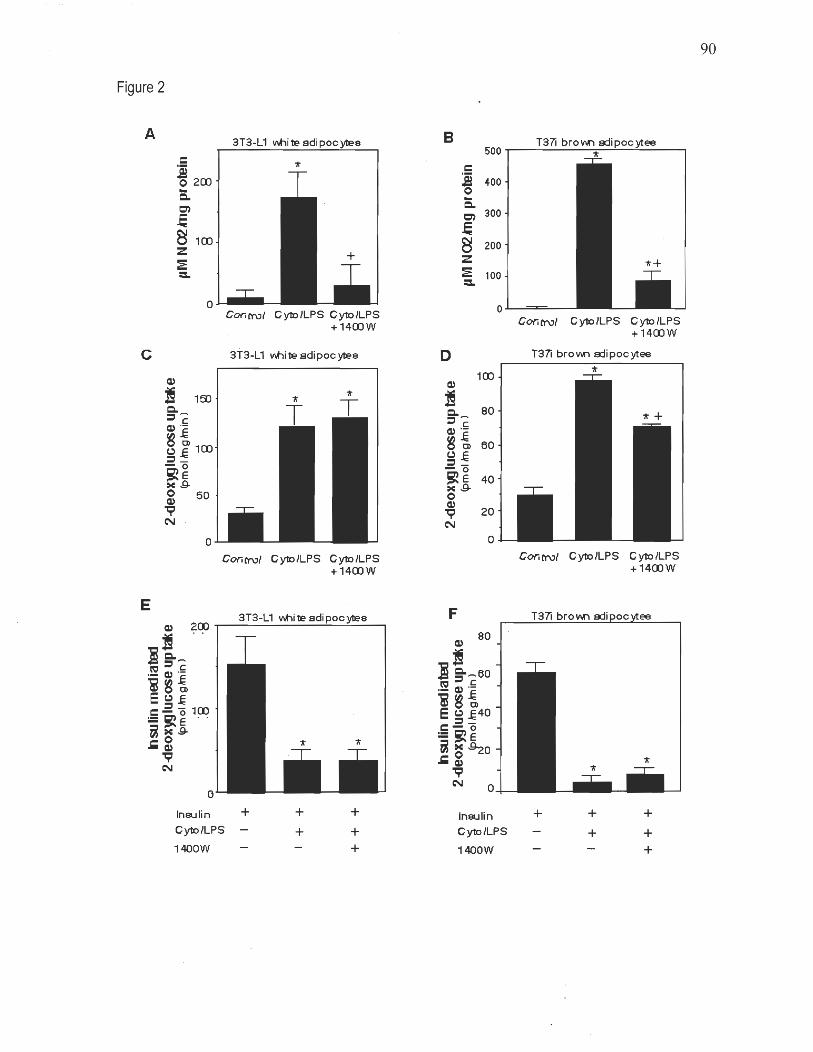
Figure 2
A
c:
~2oo Q. CT)
.e ~1oo Z :E =-
3T3-L 1 V\hi te adi poe e
O~---
c
o
E
Cor. rrul C yto ILPS C yto ILPS + 1400W
3T3-L 1 V\hi te adi poe ytee
7;
T
Cor. rrul C yto ILPS C yto ILPS + 1400W
3T3-L 1 V\hi te adi poe}'tee Cl) 200~------------~~--~
! j §o-l'tl ,.., .~ .- -~ i~o> E"~ c: -E, 0 100 = ~E .. ~)C-&). .. g
'ÇJ C'\I
o Inaulin
Cyto/LPS
1400W
+ + +
+
+ +
90
B T37i bro W""l aji poe ytee 500
c: j 400 0 -Q.
cr') 300
.e ~ 200 Z :E =- 100
0 Cor.rrul Cyto/LPS Cyto/LPS
+ 1400W
D T37i bro W"l aji poe ytee
100 Cl)
! §o- 80
c: Cl) .-
~t 60 ,,~
~--0 ~E 40 )C-&).
0 Cl)
'ÇJ 20 C'\I
0
Cor.trul Cyto/LPS Cyto/LPS + 1400W
F T37i bro W""l aji ee
Cl) 80
! j §o-60 l'tl Cl) .~ .- ~
i~o> E " ~40 ~-c: - 0
= ~E ~ )C 9.z0 .. g
'ÇJ C'\I 0
Inaulin + + + Cyto/LPS + + 1400W +

Figure 3
Obesity Inftammation
\ 1 t proinftammatory cytokines
ether mediaors ?
Skeletal Muscle
1 NO~
I."suu" resista"ce 1
Adipoc.yles
91

92
Chapitre 2. L'induction de iNOS cause la résistance à l'insuline par un mécanisme dépendant du peroxynitritre par la nitration d'IRS-l dans le muscle squelettique.
La synthase de NO inductible (iNOS) est connue pour produire de fortes concentrations de
monoxyde d' azote (NO) et est impliquée dans la défense immunitaire par ses activités anti
tumorales et antimicrobiennes. Cependant, une expression incontrôlée de iNOS a été
associée à la pathogenèse de différentes maladies. En particulier, il fut rapporté récemment
que l' induction de iNOS avait un rôle à jouer dans la résistance à l' insuline du muscle
associée à l' obésité. Dans la présente étude, nous avons démontré qu'un traitement à la LPS
chez le rat induisait iNOS et causait un défaut du transport du glucose stimulé par l' insuline
due à une diminution de l' activité PI 3-kinase associée à IRS-l. Nous avons trouvé que le
NO est un joueur majeur dans cet effet puisqu'un inhibiteur de iNOS ; l' aminoguanidine
rétablissait l' action de l'insuline. De plus, les souris invalidées pour le gène iNOS étaient
protégées contre la résistance à l'insuline induite par l' injection de LPS. L'exposition-de
cellules musculaires en culture à des cytokines et à la LPS causait aussi l' induction de
iNOS et la résistance à l'insuline par un mécanisme dépendant de l' activité PI 3-kinase
associée à IRS-l. Cependant, un inhibiteur spécifique de iNOS le 1400W ou l' agent
neutralisant de ONOO-, epigallocatechin gallate (EGCG) empêchaient tous deux la
résistance à l'insuline causée par le' traitement. De plus, nous avons observé que
l' exposition de ces mêmes cellules musculaires au ONOO- directement reproduisait l ' effet
inhibiteur des cytokines/LPS sur l'action de l'insuline au niveau du transport du glucose et
sur l' activité PI 3-kinase. Finalement, l' induction de iNOS ou le traitement direct à l'aide
du ONOO- causent la nitration d' IRS-l sur des résidus tyrosine causant ainsi une capacité
réduite d'IRS-l d'être phosphorylée sur ces tyrosines par l'action de l'insuline. Nos
résultats suggèrent donc que le ONOO- est le principal médiateur de la résistance à
l' insuline suivant l'induction de iNOS.
~----------------------------------------------------------------------------- ---- - ----

93
iNOS induction causes insulin resistance by a peroxynitrite-dependent tyrosine nitration of IRS-1 in skeletal muscle
Geneviève Pilon, Patrice Dallaire, Mylène Perreault, Sonia Kapur, and André Marette*
Department of Anatomy and Physiology, and Lipid Research Unit, Laval University Hospital Research Center, Sainte-Foy, Québec, G 1 V 4G2, Canada.
RUNNING TITLE: Peroxynitrite causes insulin resistance in muscle.
* Address correspondence to: Dr. André Marette Department ofPhysiology & Lipid Research Unit Laval University Hospital Research Center 2705, Làurier Blvd Ste-Foy, (Québec), Canada, G1V 4G2 Tel: (418) 656-4141 ext. 47908 Fax: (418) 654-2176 E-mail: [email protected]

94
Abstract
Inducible nitric oxide synthase (iNOS), an enzyme known to produce large amount of nitric
oxide (NO), is implicated in immune defense through its antitumor and its antimicrobial
activities. However, marked expression of iNOS has also been associated with the
pathogenesis of various diseases. Indeed, we have previously reported evidence for a
detrimental role of iNOS in obesity-linked insulin resistance in muscle. In the present
study, we show that LPS treatment in rats induces iNOS and causes a defect in insulin
stimulated glucose uptake in muscle due to a decrease in IRS-l associated PI 3-kinase
activity. Importantly, the insulin resistant effect of LPS could be blunted by the iNOS
inhibitor aminoguanidine or by targeted di~ruption of iNOS. Exposure of cultured muscle
cells to cytokines and LPS also induced iNOS and caused insulin resistance through
impairment of IRS-l associated PI 3-kinase activity. Interestingly, both the specific iNOS
inhibitor, 1400W, and the ONOO- scavenger, epigallocatechin gallate (EGCG), prevented
the cytokine/LPS induced insulin resistance in these cells. Moreover, we found that
exposure of muscle cells to authentic ONOO- reproduced the inhibitory effect of
cytokines/LPS on insulin mediated glucose transport and PI 3-kinase activity. Imp o rtantl y ,
both iNOS induction and ONOO- treatment caused nitration of IRS-l on tyrosine residues
which was linked to a reduced capacity of IRS-l to undergo insulin dependent tyrosine
phosphorylation. These results suggest that ONOO- is an important mediator of muscle
insulin resistance following iNOS induction and that this oxidant derivative inhibits insulin
action via nitration of IRS-l and inhibition of its associated PI 3-kinase activity.

95
INTRODUCTION
The highly reactive gas nitric oxide (NO) has been shown to participate in a large variety of
physlological processes such as neuronal transmission, kidney function, vascular relaxation
and immune modulation [1]. There are at least three different isoforms of the NO synthase
(NOS) enzymes: the endothelial type eNOS, the neuronal type nNOS and the inducible
type iNOS. Unlike the other members of the NOS family which are constitutively
expressed Ca2+-dependent enzymes, iNOS must be induced by inflammatory cytokines
and/or bacterial endotoxins. Once induced, iNOS then generates NO at a much higher rate
and for a longer period oftime than the other two NOS enzymes.
Even though iNOS has an important role to play in host defense, there is much evidence
that the mass production of NO by this enzyme also has a deleterious impact on healthy
host cells [2]. Indeed, it is weIl accepted that uncontrolled iNOS induction is involved in
the etiology of multiple disease states including Alzheimer' s disease, apoptosis/necrosis of
pancreatic Islets, hypotension, inflammatory bowel disease, and rheumatoid arthritis [3] [4]
[5] . Along with these findings our lab has shown that iNOS induction leads to disturbances
in glucose metabolism and is inyolved in the pathogenesis of obesity-linked insulin
resistance. However, this is not surprising since it is now weIl established that obesity is a
chronic inflammatory disorder characterized by increased expression of inflammatory
cytokines su ch as TNF-a, interferon-y, IL-6, IL-l~ that are known to induce iNOS
expression[ 6] . Indeed, iNOS induction has been observed in aorta, skeletal muscle and
adipose tissue of high-fat fed mice, in the heart of diabetic Zucker rats and in skeletal
muscle and platelets of type 2 diabetic subjécts [7] [8] [9] [10]. We have also previously
shown that iNOS knockout mice are protected against insulin resistance induced by high
fat feeding; and these results have 'been confirmeçl recently by Keamey' s group [7] [8].
Persistent high levels of NO in the cellular environment can lead to the production of
oxidative metabolites. Protein S-nitrosylation is one such form of oxidative modification
that has been proposed to play an important role in the pathogenesis of iNOS induced
insulin rësistance. Indeed, S-nitrosylation is elevated in patients with type 2 diabetes and

96
increased S-nitrosylation of the insulin receptor, insulin receptor substrate-1 and AKT have
been reported in skeletal muscle of obese, diabetic mice [11][12]. Alternatively, due to its
unpaired electron, NO may also act as a free radical within the cellular environrnent and
will readily react with superoxide O2- to form peroxynitrite (ONOO-). This powerful
oxidant has been reported to modulate cellular functions through nitration of tyrosine
residues and the detection of prote in tyrosine nitration is used as a footprint of peroxynitrite
formation. It is noteworthy that nitrated proteins have been detected in at least 50 human
diseases such as: Alzheimer disease, rheumatoid arthritis, glaucoma, asthma and
inflarnmatory bowel disease [13]. What is more it has been shown that nitration of tyrosine
residues decreases their subsequent phosphorylation capacity and different groups have
reported that protein tyrosine nitration is found at higher levels in type 2 diabetics [14] [15]
[16]. Thus, it is therefore conceivable that ONOO- could impair insulin action through
nitration of key residues on proteins involved . in insulin signaling. The aim of this study
was to determine the role of ONOO- in iNOS-mediated insulin resistance and to clarify the
target of its action within the insulin signaling pathway.
Materials and Methods
Interferon-y and TNF-a were purchased from Research Diagnostics Inc. , (Flanders, NJ),
and RD systems (Minneapolis, MN), respectively. 1400W and polyclonal nitrotyrosine
antibody were from Biomol Research Laboratories Inc. AlI cell culture solutions and
supplements were purchased from Life Technologies, Inc. except for fetal bovine serum,
which was purchased from Wisent (St-Bruno, QC, Canada). Reagents for SDS
polyacrylamide gel electrophoresis and immunoblotting were from Bio-Rad. ECL and [2-
3H]Deoxyglucose were from PerkinElmer Life Sciences. [y_32P]ATP, protein A- and G
Sepharose, anti-mouse or anti-rabbit immunoglobulin G conjugated ta horseradish
peroxidase were purchased from Amersham Pharmacia Biotech (Baie d'Urfé, QC, Canada).
Polyclonal antibodies against IRS-1 (raised against 20 C-terminal amino acids (C-20)),
were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). The antibody IRS-1 for
the western blot was from Cell Signaling (Beverly, MA). Polyclonal iNOS antibody was
purchased from Upstate Biotechnology and eNOS and nNOS antibodies were from

97
transduction (San Jose, CA). PI 3-kinase substrate (Crosstide) and monoclonal antibody
against phosphotyrosine were obtained from Upstate Biotechnology (Lake Placid, NY).
Human insulin was obtained from Eli Lilly (Toronto, ON, Canada). L-a -
phosphatidylinositol was from Avanti Polar Lipids (Alabaster, AL). Oxalate-treated TLC
si li ca gel H plates were obtained from Analtech (Newark, DE). AIl other chemicals were
from Sigma (St.Louis, MO).
Cell culture
L6 myoblasts were grown in a-MEM (10% FBS) containing 1 % antibiotic/antimycotic
(100 000 units/ml penicillin, 100 mg/ml streptomycin, 0.25 mg/ml amphotericin) and
differentiated into myotubes in a-MEM (2% FBS). iNOS induction by cytokines and LPS
in L6 myotubes was the result of 24 hours incubation in presence of TNF-a (lOng/ml),
IFN-y (200U/ml) and LPS (lOJlg/ml). The accumulation of nitrite in the incubation medium
was used as an index of NO production. Nitrite was determined spectrophotometrically as
described previously [17].
AnimaIs
This study was approved by the Animal Care and Handling Committee of Laval University.
~ale Sprague-Dawley rats (200- 250 g) purchased from Charles River (Montréal, Canada)
were used in these studies. Rats were pre-treated with a single intraperitoneal injection of
aminoguanidine or saline (vehicle) 30 min prior to LPS (20 mg/kg, BW) or saline .
treatment. AnimaIs were sacrificed 6 h later, and muscles tissues were removed and stored
at -80 oC until further processing. Breeding pairs of Nos2-/- mice were a gift from C.
Nathan and J.S.Mudgett. The mice have been subsequently bred under specific pathogen
free conditions at the Animal Facility of Laval University hospital research center. Mice
were given free access to a standard low-fat diet (Purina Rodent Chow, Madison,
Wisconsin). AnimaIs were sacrificed 6 h after LPS treatment.
Tyrosine phosphorylation of the insulin receptor and IRS.
Cell or tissue lysates (500ug of prote in) were immunoprecipitated with 2 Jlg of anti
phosphotyrosine (4GlO) coupled to prote in A-Sepharose ovemight at 4°C. The immune

98
complex was washed three times in PBS (pH 7.4) containing 1 % NP-40 and 2 rnmol/l
Na3 V04 , resuspended in Laemmli buffer, and boiled for 5 min. Proteins were resolved on
SDS-PAGE (6% gel) and processed for Western blot analysis.
PI 3-Kinase Activity
Cell or tissue lysates (500 J.lg) were immunoprecipitated with 2 J.lg of anti-IRS-1 coupled to
protein A-Sepharose overnight at 4°C. PI 3-kinase activity was determined as described
previously [18].
Measurement of2-Deoxyglucose uptake
Measurement of 2-Deoxyglucose uptake was performed as previously described [17]. In
brief, cells were incubated for 8 min in HEPES-buffered saline containing 10 J.lM unlabeled
2-deoxyglucose and 10 J.lM D-2-deoxy-e H]glucose (0.5 J.lCi/ml). The reaction was
terminated by washing three times with ice-cold 0.9% NaCI (w/v). Cell-associated
radioactivity was determined by lysing the cells with 0.05 N NaOH, followed by liquid
scintillation counting. Glucose transport in isolated soleus and EDL muscles was measured
by use of the glucose analogue 2-eH]2-deoxy-D-glucose as described [19].
Nitrotyrosine measurement
Cell lysates (500J.lg of prote in) were immunoprecipitated with 2 J.lg of anti-nitrotyrosine
coupled to prote in G-Sepharose overnight at 4°C. The immune complex was washed three
times in PBS (pH 7.4) containing 1% NP-40 and 2 mmol/l Na3V04, resuspended in
Laemmli buffer, and boiled for 5 min. Proteins were resolved on SDS-PAGE (7,5% gel)
and processed for Western blot analysis.
Measurement of Plasma Nitrite and Nitrate
Nitrite and nitrate levels in plasma were measured by fluorometry. Briefly, blood was
collected in tubes containing EDTA and centrifuged for 10 min at 3200 x g to obtain
plasma. Plasma was then centrifuged at 5000 x g (4 OC) overnight in an Ultrafree®
Microcentrifuge 10,000 NMWL filter unit. Nitrate was reduced to nitrite using nitrate

99
reductase and the NADPH regenerating system. The fluorescence was measured at ex of
360 nm and em of 450 nm. NOS activity was performed as described previously [19].
Data Analysis
All data are presented as means ± SE. The effects of the treatments were compared by an
ANOV A followed by Pisher's post hoc test. Differences were considered to be statistically
significant at p<0.05.

100
RESULTS
iNOS inhibition prevents insu/in resistance in skeletal muscle of LPS-treated rats.
To study the role of iNOS in insulin resistance, we treated rats with LPS with or without
the iNOS inhibitor aminoguanidine. LPS was found to induce iNOS as reflected by
N02/N03 accumulation in the plasma (figure lA). As expected, pretreatment with
aminoguanidine partially (lOOmg/kg) or totally (150mg/kg) blocked NO production and
iNOS protein expression (figure lB). However, the two constitutive NOS enzymes, eNOS
and nNOS, were not affected by the aminoguanidine treatment. We next investigated the
influence of iNOS induction on insulin sensitivity ex vivo by measuring glucose uptake in
isolated soleus muscle. Here we observed that insulin-stimulated glucose uptake was
reduced in LPS-treated animaIs compared to the control group (figure 1 C). Importantly the
aminoguanidine treatment restored insulin stimulated glucose uptake in LPS treated
animaIs in a dose dependant manner such that the lower dose of aminoguanidine' partially
improved insulin sensitivity while the higher dose fully restored muscle insulin action
(figure 1C). Similar results were obtained in EDL muscle (data not shown). We had
previously reported that iNOS induction causes a defect in IRS-1 associated PI 3-kinase, a
key enzyme involved in insulin signaling [20] [7]. Therefore, we determined whether the
activity of this enzyme was altered in LPS-treated rats. Here we observed that LPS
treatment decreased the ability of insulin to stimulate PI 3-kinase activity while iNOS
inhibition with aminoguanidine fully restored insulin' s action on PI 3-kinase (figure 1 D).
Aminoguanidine treatment alone had no effect on insulin sensitivity (data not shown).
Targeted disruption of iNOS protects mice against LPS induced insu/in resistance.
We next used a genetic model of iNOS disruption to further demonstrate the role of iNOS
in LPS-induced insulin resistance. As mentioned above, we have previously reported that
mice lacking iNOS are protected against insulin resistance induced by obesity [7]. To

101
demonstrate the role of iNOS in endotoxemia induced insulin resistance, we challenged
iNOS knockout mi ce with LPS. As expected, iNOS induction and NO production by LPS
treatment were abrogated in these genetically modified mice (figure 2 A, B). As observed
after the pharmacological inhibition of iNOS with aminoguanidine, we found that iNOS
gene deletion protected mi ce against LPS-induced insuliri resistance. Indeed, we found that
insulin-mediated 2-DG glucose uptake was normalized in the soleus of iNOS-/- mice that
. had been treated with LPS compared to saline treated iNOS -/- mice (figure 2C). LPS
induced inhibition of muscle insulin-stimulated PI 3-kinase was also significantly
abrogated by iNOS deletion (figure 2D). Similar results were obtained in EDL muscle (data
not shown).
iNOS inhibits insulin-stimulated glucose uptake via the production of ONOO-
We next test whether iNOS-mediated insulin resistance could be reproduced in vitro.
Cytokine/ LPS exposure markedly induced NO production as detected by the accumulation
of nitrite in the culture medium and this was completely inhibited by co-incubation with the
iNOS inhibitor 1400W (ctr: 0.03 +/- 0.07 f.lM, cyto/LPS: 9.18 +/- 0.62 f.lM,
cyto/LPS+ 1400W: 0.0 +/- 0.06 f.lM). As depicted in Figure 3A, cytokine/LPS treatment
. increased basal glucose transport. This effect was, in major part, due to chronic iNOS
induction since 1400W greatly prevented this augmentation. In contrast to the basal state,
cytokine/LPS treatment caused a significant decrease in insulin-stimulated glucose
transport as shown in Figure 3B. As expected, this defect was reversed by iNOS inhibition.
These results clearly support a role for iNOS in cytokine/LPS-induced insulin resistance.
To determine the role of ONOO- in this process, we first used the peroxynitrite scavenger
epigallocathechin gallate (EGCG). As observed with iNOS inhibition, the neutralization of
ONOO- blunted the increase of basal glucose transport and reversed the deleterious effect
of iNOS on insulin-stimulated glucose uptake (Figure 3C and 3D). Next, we treated cells
directly with authentic ONOO-. As observed for iNOS induction, ONOO- treatment
increased basal glucose transport and decreased insulin-stimulated glucose uptake (Figure
3E and 3F). These data support a role for ONOO- in the deleterious effect of iNOS on
insulin-stimulated glucose transport.

102
iNOS inhibits insulin-stimulated PI 3-kinase activity via the production of ONOO- in
myocytes.
Following on from the observed defect in insulin stimulated PI 3-kinase activity in LPS
treated animaIs, we investigated the involvement of ONOO- in this defect in the cellular
model. As shown in figure 4, cytokine/ LPS treatment impaired insulin signaling as
revealed by a '" 50% reduction in the ability of insulin to stimulate IRS-1-associated PI 3-
kinase activity as compared to control cells (figure 4A). However, 1400W restored insulin
stimulated PI 3-kinase activity. We then used EGCG to look at the role of ONOO- in iNOS
mediated insulin resistance at the level of PI 3-kinase. As for glucose transport the ONOO
scavenger significantly prevented the inhibition caused by cytokines/LPS treatment (figure
4B). Furthermore exposing cells to ONOO- per se also reduced the ability of insulin to
stimulate IRS-1 associated PI 3-kinase activity (figure ' 4C). Importantly both the iNOS
inhibitor and EGCG failed to modulate PI 3-kinase in the absence or in the presence of
insulin (data not shown) and no effect of cytokines/LPS or ONOO- was observed on basal
PI 3-kinase activity. These results support a role for ONOO- in the iNOS-induced
impairment of insulin-stimulated PI 3-Kinase activity in skeletal muscle.
ONOO- causes nitration of tyrosine on IRS-l
Since a major reaction of ONOO- with proteins is the formation of nitrotyrosine, we looked
at this modification on the insulin receptor substrate-1, a critical element for insulin action.
To evaluate specifically the nitration of IRS-1 we first immunoprecipitated aIl nitrated
proteins following treatment of L6 muscle cells with ONOO- or cytokine/LPS and then we
looked at whether IRS-1 was nitrated using an antibody against IRS-1. We observed that
increasing doses of ONOO- (Fig. SA) as weIl as cytokine/LPS (Fig.SB) treatment increases
the tyrosine nitration of IRS-1. Furthermore, we observed that iNOS inhibition by 1400W
in cytokine/LPS treated cells blunted the tyrosine nitration of IRS-1 (figure SB). We then
confirmed these results by performing the reverse experiment; where we

103
immunoprecipitated IRS-1 and confirmed its tyrosine nitration by western blotting (figure 1
5C and 5D). Since our results reveal a .role for ONOO- in iNOS induced insulin resistance,
we asked whether nitration of IRS-1 on tyrosine residues couIC! decrease the ability of
insulin to stimulate IRS-l tyrosine phosphorylation. To answer this question we measured
IRS-1 tyrosine phosphorylation of cells treated with insulin in the absence or in the
presence of ONOO-. Here we found that ONOO- treatment caused a decrease in the
capacity of insulin to induce tyrosine phosphorylation of IRS-1 (figure 5E).
DISCUSSION
Until now our understanding of the molecular mechanisms by which iNOS mediates
insulin resistance in skeletal muscle was limited to NO mediated S-nitrosylation[12] [21].
Herein we show for the first time that the radical NO oxidation product ONOO- also
contributes to the iNOS mediated insulin signaling defects in skeletal muscle via tyrosine

104
nitration of IRS-1. This post translational modification appears to impede insulin signal
transduction by blocking tyrosine phosphorylation of IRS-1 and thereby limiting its
interaction with PI 3 -Kinase.
Our report of a negative role for elevated tyrosine nitration in skeletal muscle exposed to
cytokines and LPS or authentic ONOO- is in line with observations in other tissues and
fields. Indeed, ONOO- mediated tyrosine nitration has been shown to negatively modulate
the function of several other enzymes such as glutamine synthase, prostacyclin synthase,
cytochrome P450, tyrosine hydrolase and sarcoplasmic reticulum Ca-ATPase [22] [23] [24]
[25] [26] . Furthemore tyrosine nitration has previously been implicated in the diminution
of insulin stimulated PI 3-kinase activity in other tissues. Indeed, in endothelial cells,
authentic ONOO- or high glucose concentration was found to cause protein nitration
associated with a weaker PI 3-kinase activity. While, in 3T3-L1 adipocytes, it has been
reported that the presence of ONOO- is associated with IRS-1 tyrosine nitration and a
subsequent decline in insulin stimulated glucose transport. A defect in PI 3-kinase activity
has also been reported in ONOO- treated macrophages [27] [28]. However, in the latter
work, it was shown that the failure in PI 3-kinase activity resulted from a defect in the
association between the p85 and plI 0 subunits of the kinase rather than IRS-1
nitrotyrosination as demonstrated by our work. Since insulin mediates its action through
phosphorylation of specific tyrosine residues on IRS-1 , we can assume that tyrosine
alteration likely - affects this manner of signal transduction. As with most other post
translational modifications tyrosine nitration is a selective process and not aIl tyrosine
residues present in proteins are nitrated. To date no specific consensus sequence or motif
has been identified for prote in tyrosine nitration. However, it is known that the presence of
one or more acidic residues and a relative paucity of cysteine, methionine and basic amino
acids are required for tyrosine nitration to occur [13]. After searching the sequence of IRS-
1 we have identified one tyrosine residue that respoIids to these critieria, the tyrosine at
position 895 in rat or 896 in humans. Further experiments will be needed to determine
whether this key tyrosine is the target of ONOO- nitration in skeletal muscle.

105
In conclusion, we and others have previously shown that iNOS can be induced in muscle
and adipose tissues in obesity [8] [7] [29] [30]. Furthermore, it has also been proposed that
obesity is associated with an increase in infiltration of macrophages which highly express
iNOS [31]. We thus propose here that iNOS induction in obesity can cause insulin
resistance in muscle via tyrosine modification of IRS-1 following ONOO- formation. A
better understanding of the mechanism linking iNOS to insulin resistance will provide
means to develop tools to treat this ever growing disease.

106
LEGENDS TO FIGURES
Figure 1. Effect of aminoguanidine on iNOS induced insulin resistance in muscle in vivo.
Rats were injected i.p with LPS 20mg/kg with or without aminoguanidine (100 or
l50mg/kg) and sacrificed 6 hours later. A) Nitrite accumulation in plasma was measured by
fluorometry. B) NOS enzyme protein expression was measured by western blot in soleus
muséle. C) Effect of LPS with or without aminoguanidine was measured on basal or insulin
stimulated glucose transport in strips of soleus muscle. D) IRS-l associated PI 3-kinase
activity was measured in muscle of LPS +/- aminoguanidine (100mg/kg) treated animaIs.
Insulin (3 ,8U/kg i.v) was, injected 4 minutes prior to sacrifice.
Figure 2. iNOS disruption protects mlce against insulin resistance induced by LPS
treatment. Wild type (WT) or iNOS-/- mice were treated with saline or LPS A) nitrite
accumulation in plasma was measured by fluorometry. iNOS Protein expression is
represented in panel B). Basal and insulin stimulated 2-DG uptake is represented in panel
C) and insulin induced IRS-l-associated PI 3-kinase activity is represented in panel D).
Figure3. Effect of·ONOO- and iNOS induction on basal and insulin stimulated glucose
transport in L6 muscle cells. Glucose transport was measured as described under "Materials
and Methods" following these treatments. A) The effect of cyto/LPS on basal and insulin
stimulated glucose uptake was measured in L6 muscle cells treated with TNF-a (lOng/ml),
IFN-y (200units/ml) and LPS (lOJlg/ml) for 24 hours, +/- l400W ( expressed as % offold
change relative to insulin alone) (B). (C) The effect of EGCG (lOJlM) on basal glucose
transport in the presence of cytokines/L~S(D) The effect of EGCG(lOuM) on insulin
stimulated glucose transport in the presence of cytokines/LPS. (E) Basal glucose transport
in the presence of ONOO- is represented F) Insulin-stimulated glucose transport in the
presence ofONOO-.
Figure 4. Effect of iNOS and ONOO- on IRS-1-associated PI 3-kinase activity in L6
myocytes. PI 3-kinase was measured in anti-IRS-I immonoprecipitates of L6 cells treated
--------------------------------------------------------- ---------------------- - --- - - ----

107
with A) (cyto/LPS) with TNF-a (lOng/ml), IFN-y (200 units/ml) and LPS (lOJlg/ml) for 24
hours, +/- iNOS inhibitor l400W (O, lmM), B) +/- EOCG (lOJlM) or C) in cells directly
treated for 1 hour with ONOO- . Quantification of 32p incorporated into PI 3-phosphate (PI
3P) was expressed relative to insulin control value. A representative autoradiograph is
shown at the top of the figure.
Figure5. Effect of ONOO- and iNOS induction on IRS-l tyrosine nitration and
phosphorylation. A) and B) Protein lysates from cells treated with increasing doses of
ONOO- or cytokines +/- the iNOS inhibitor 1400W (O.lmM) were immunoprecipitated
with anti-nitrotyrosine antibody and then immunoblotted with anti-IRS-l antibody. C) and
D) Cells lysates were immunoprecipitated with anti-IRS-l antibody and then
immunoblorted with anti-nitrotyrosine antibody. E) L6 cells were serum deprived for 4
hours and then treated with or without ONOO- for 1 hour. Cells were then stimulated with
100nM insulin for the last 5 minutes of the treatment. Celllysates were immunoprecipitated
with anti-phosphotyrosine antibody (4010) and then immunoblorted using the IRS-l
antibody.

108
References
1. Murad, F., Nitric oxide signaling: would you believe that a simple free radical could
be a second messenger, autacoid, paracrine substance, neurotransmitter, and hormone?
Recent Prog Horm Res, 1998.53: p. 43-59; discussion 59-60.
2. 'Moncada, S., R.M. Palmer, and E.A. Higgs, Nitric oxide: physiology,
pathophysiology, andpharmacology. Pharmacol Rev, 1991.43(2): p. 109-42.
3. Suarez-Pinzon, W.L. and A. Rabinovitch, Approaches to type 1 diabetes prevention
by intervention in cytokine in1munoregulatory circuits. Int J Exp Diabetes Res, 2001. 2(1):
p.3-17.
4. Strunk, V., et al., Selective iNOS inhibition prevents hypotension in septic rats while
preserving endothelium-dependent vasodilation. Anesth Analg, 2001.92(3): p. 681-7.
5. 'Fernandez-Vizarra, P., et al., Expression of nitric oxide system in clinically
evaluated cases of Alzheimer's disease. Neurobiol Dis, 2004. 15(2): p. 287-305.
6. Marette, A., Pathogenic role of inflammatory cytokines in obesity: from insulin
resistance to diabetes mellitus. Nestle Nutr Workshop Ser Clin Perform Programme, 2004.
9: p. 141-50; discussion 151-3.
7. Perreault, M. and A. Marette, Targeted disruption of inducible nitric oxide synthase
protects against obesity-linked insulin resistance in muscle. Nat Med, 2001.7(10): p. 1138-
43.
8. Noronha, B.T., et al., Inducible nitric oxide synthase has divergent effects on
vascular and metabolic function in obesity. Diabetes, 2005. 54(4): p. 1082-9.
9. Shimabukuro, M., et al., Role of nitric oxide in obesity-induced beta cell disease. J
Clin Invest, 1997. 100(2): p. 290-5.
10. Torres, S.H., et al., Inflammation and nitric oxide production in skeletal muscle of
type 2 diabetic patients. J Endocrinol, 2004. 181(3): p. 419-27.
Il. Padron, J., et al., Enhancement of S-nitrosylation in glycosylated hemoglobin.
Biochem Biophys Res Commun, 2000. 271(1): p. 217-21.
12. Yasukawa, T., et al., S-nitrosylation-dependent inactivation of A kt/prote in kinase B
in insulin resistance. J Biol Chem, 2005. 280(9): p. 7511-8.

109
13. Greenacre, S.A. and H. Ischiropoulos, Tyrosine nitration: localisation,
quantification, consequences for prote in function and signal transduction. Free Radic Res,
2001.34(6): p. 541-81.
14. Gow, A.J. , et al. , Effects of peroxynitrite-induced protein modifications on tyrosine
phosphorylation and degradation. FEBS Lett, 1996. 385(1-2): p. 63-6.
15. Randriamboavonjy, V. , et al. , Platelet sarcoplasmic endoplasmic reticulum Ca2+-
ATPase and mu-calpain activity are altered in type 2 diabetes mellitus and restored by
ros iglitazo ne. Circulation, 2008. 117(1): p. 52-60.
16. Ceriello, A. , et al. , Detection of nitrotyrosine in the diabetic plasma: evidence of
oxidative stress. Diabetologia, 2001. 44(7): p. 834-8.
17. Bedard, S., B. Marcotte, and A. Marette, Cytokines modulate glucose transport in
skeletal muscle by inducing the expression of inducible nitric oxide synthase. Biochem J,
1997. 325 ( Pt 2): p. 487-93.
18. Tremblay, F. and A. Marette, Amino acid and insulin signaling via the mTORlp70
S6 kinase pathway. A negative feedback mechanism leading to insu lin resistance in skeletal
muscle cells. J Biol Chem, 2001. 276(41): p. 38052-60.
19. Kapur, S., et al. , Expression ofnitric oxide synthase in skelet~l muscle: a novel role
for nitric oxide as a modulator of insulin action. Diabetes, 1997. 46(11): p. 1691-700.
20. Pilon, G., P. Dallaire, and A. Marette, Inhibition of inducible nitric-oxide synthase
by activators of AMP-activated protein kinase: a new mechanism of action of insulin
sensitizing drugs. J Biol Chem, 2004.279(20): p. 20767-74.
21. Carvalho-Filho, M.A., et al. , Targeted disruption ofiNOSprevents LPS-induced S
nitrosation of IRbeta/IRS-l and Akt and insulin resistance in muscle of mice. Am J Physiol
Endocrinol Metab, 2006.291(3): p. E476-82.
22. Tien, M., et al. , Peroxynitrite-mediated modification of proteins at physiological
carbon dioxide concentration: pH dependence of carbonyl formation, tyrosine nitration,
and methionine oxidation. Proc Nad Acad Sci USA, 1999.96(14): p. 7809-14.
23. Nie, H. , et al. , Endothelial nitric oxide synthase-dependent tyrosine nitration of
prostacyclin synthase in diabetes in vivo. Diabetes, 2006. 55(11): p. 3133-41.

110
24. Lin, H.L. , et al. , Peroxynitrite inactivation of human cytochrome P450s 2B6 and
2Ei: heme modification and site-specifie nitrotyrosine formation. Chem Res Toxicol, 2007.
20(11): p. 1612-22.
25. Gorg, B. , et al. , Reversible inhibition of mammalian glutamine synthetase by
tyrosine nitration. FEBS Lett, 2007.581(1): p. 84-90.
26. Xu, S. , et al. , Detection of sequence-specific tyrosine nitration of manganese SOD
and SERCA in cardiovascular disease and aging. Am J Physiol Heart Circ Physiol, 2006.
290(6): p. H2220-7.
27. Nomiyama, T., et al. , Reduction of insulin-stimulated glucose uptake by ·
peroxynitrite is concurrent with tyrosine nitration of insulin receptor substrate-l. Biochem
Biophys Res Commun, 2004.320(3): p. 639-47.
28. Hellberg, C.B., S.E. Boggs, and E.G. Lapetina, Phosphatidylinositol 3-kinase is a
targetfor prote in tyrosine nitration. Biochem Biophys Res Commun, 1998.252(2): p. 313-
7.
29. Engeli, S., et al., Regulation of the nitric oxide system in human adipose tissue. J
Lipid Res, 2004. 45(9): p. 1640-8.
30. Fujimoto, M., et al., A role for iNOS in fasting hyperglycemia and impaired insulin
signaling in the liver of obese diabetic mice. Diabetes, 2005.54(5): p. 1340-8.
31. Weisberg, S.P., et al. , Obesity is associated with macrophage accumulation in
adipose tissue. J Clin Invest, 2003. 112(12): p. 1796-808.
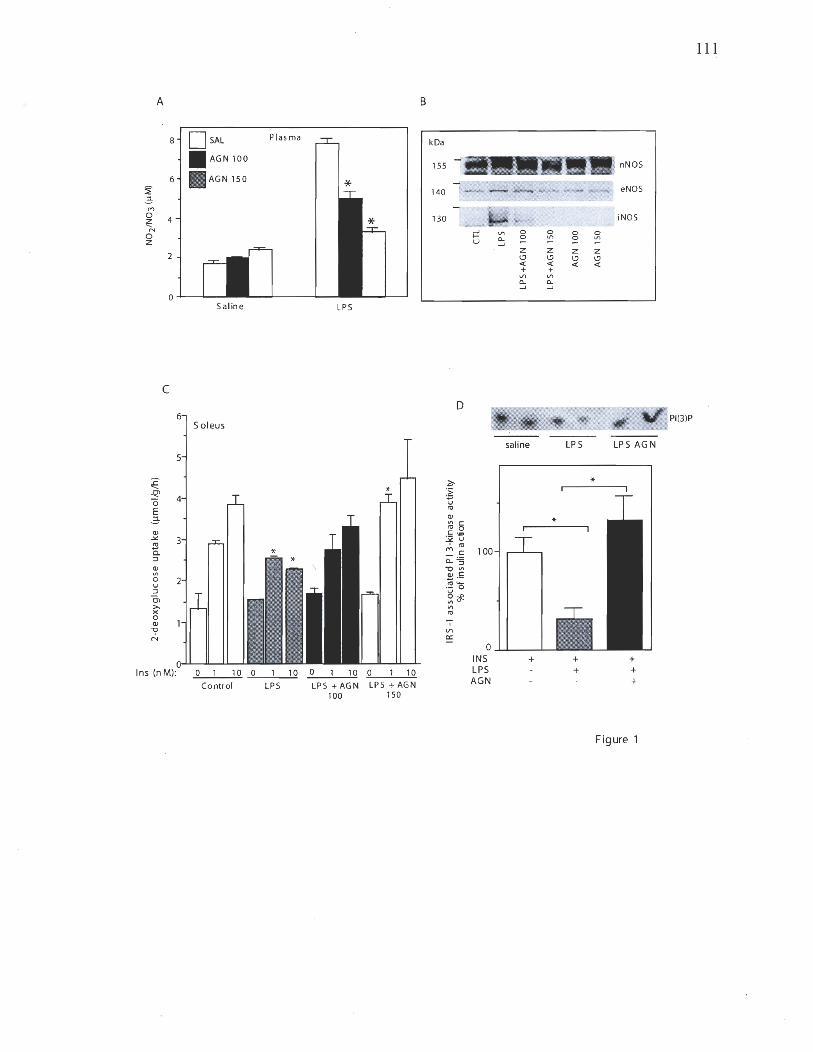
111
A B
8 DSAL Plasma kOa
• AGN 100 155 nNO S
6 . AGN 150
* ~ 140 eNOS ~
CV')
0 4 ~ 130 i NOS
N ....J 0 V'l 0 0 0 5;
z .... a.. 0 ~ ~ U -J
2 z z z Z l:J l:J l:J l:J « « « « + + Vl V'l a.. a.. -J -J
0 Saline LPS
C
D
Soleus
saline LP 5 LP 5 AG N
? p '* ~ * ':;:
:;:; 0 v E l1J
.=; <1> Vl c::
Q) l1J 0
~ 3 .E 'P
.œ ~v
1 l1J
c.. ~c:: 100 :::J o..=S <1> VI
"0 VI
0 ~ .E v 11J ......
~ ,- 0 v
Dl ~~ >- Vl x 0
l1J
Q) '7' '? V'l N c:::
0 INS + + +
Ins (nM): 0 10 0 1 10 0 1 10 0 10 LPS + + Control LPS LPS +AGN LP5 + AGN AGN +
100 150
Figure 1

A
C
~ ~ (5 E 2-Q)
~ a. :::> ~ Cl N
50
i 3- 40
30 !V'l
o .~
N 20 o z
10
o
20 soleus
15
10
·5
0
Insu lin +
SAL LPS
iNOS +/+
B
iNOS -1-
+ + + SAL LPS
iNOS -/-
112
kDa
155 nNOS
140 eNOS
130 i NOS
iNOS +/+ iNOS -/-
D PI(3)P
CTL LPS LPS
è iNOS +/+ iNOS -/-
:~ 120 v
ra Q)
V') c .~ .~ 100 ..:.:: v M ra - .~ 80 CL::; -C <Il
~ .~ ra-.-'v 0 60
~~ <Il ra 40 "7 V'l
~ 20
0
al LPS LPS
iNOS+/+ iNOS-/-
Figure 2

113
A B
3 120
* 100 0 s:::::::
Q.J ~ <lJ 0 ~ c 2 ~ .p
80 1'0 0 ,rs u nU 0.1'0 :::l ~
::J .~ 60 Q.J
~ > \.9"'5 o 0
o .~ , ""0
40 N -
N g ~ 0
20
0 0
cytolLP S + + cyto/LP S + + 1400W + + 1400W + +
insulin + + + +
C D
3 120
* 100 "0 s:::::::
<lJ.Q Q.J!J ~ .....
80 .:::t:. c 2 ru v 1'0 0 ..... ro E..u §-.~ ::J ~ 60 Q.J
\.9"'5 ~ > o 0 o.~ l ""0 40 N - N g ~ 0
20
0 0 cyto/LPS + + cyto/LP S + + EGCG + + EGCG + + insulin + + + +
E F
3 120
* 100
"0 c: (l).Q
* Q.J ~ 2 .,Y. ..... 8 .:::t:. c ,rsu 1'0 0 E..u 0.1ï:I ::J ~ ::J.~ (LI
\.9"'5 1..9 > o 0 o .~ 1 "'0
N -N g ~ 0
0 ctr 300 500 0 300 500
o NOO- (IlM) ins + ONOO- (IlM)
Figure 3

" \oC C
ro ~
~. ~ IRS -l -associated PI 3- kinase activity S. 0 (% of insulin-treated cells) 5' q
o
+ 1
1 + 1 H
IV o
.f:>.. (j\ o 0
(Xl o
o IV o 0
1*
n
t
++ .J 11 4 ./ 1
..... ~
9 -0
5 ' m.Q ~C).-+ _no 5' C)?
+
" V1
+
+ 1 +
1 + +
+ + +
co
IRS-l -associated PI 3-kinase activity (% ofinsulin -treated cells)
o IV o
.f:>..
o (j\ o
(Xl
o o IV o 0
1. 1 ITHa , 1
' ' '''''''''@''' I ~
.9 -0
:5""'" n VI ~ '< coS 5'~~
LA
+ 1 1
+
+ 1 +
1 + +
+ + +
»
1 R 5 -1 -a 5 s ocia ted PI 3- kinas e activity (% ofinsulin-treated (ells)
IV .f:>..
o o 0
g .
Pt
(j\ o
,
(Xl o
...1
o IV o 0
II I
1 1· 1 . ,
L " ~ "
~
~
~

A
c V1 o .'!: '+=J C ~ ::1
.t:: ~ C ~
ro '7 .~ Vl-e ~ ro
C
CTR 0.25 0.5
12
10
8
6
4
2
C V1 .Q :'!: .... c ~ ::1
.~ ~ C '-
ro .- '-1 ....
Vl 15 a:: '-_ ro
ONOO - (mM )
0 .25 0.5
ONOO-(mM )
eTR 0. 5
ONOO-(mM} 6
5
4
3
2
o -L...I. __ ----'-_
e T R
E
0.5 1
ONOO-(mM )
, Vl
Sf
'0 c .g 10 lU
>(;
.s::: Q.
Ô .s::: c.. Q)
.~
ô z- o
ONOOins
IRS -1
IR S-1
I ~ ~~::]I,
+
B
o
eTR eYTO eYTO 1400W
4~--------------~
o ~-----'--CTR
eTR
8
6
2
CYTO
eYTa
CYTO 1400W
eYTa 1400W
0-..------'--
eTR eYTa eYTa 1400W
·iiit _ I@ IRS-' IP: PY lB : IRS -l
+ + +
IRS- l IP : ntyr lB: IRS -l
IRS -l IP: IRS -l lB: n-tyr
11 5
Figure 5

Chapitre 3. Une incubation prolongée du muscle épitrocléaris cause l'induction de iNOS et des altérations du transport de glucose.
Le but de la présente étude était d' éval:uer l' implication de la synthase de NO inductible
(iNOS) dans les limitations observées lors de l' incubation prolongée d'un muscle. Des
épitrocléaris de rats furent incubés ex vivo pendant 10 heures et l' induction de iNOS fut
évaluée. Nous avons observé que le processus d' incubation causait l' induction de l'ARNm
de iNOS, de la protéine et l ' accumulation de nitrites et nitrates dans le milieu d 'incubation.
L' incubation était aussi associée à une augmentation de l' activité de iNOS tandis que
l' activité des NOS constitutives (cNOS) était réduite. Puisque iNOS est connu pour affecter
le métabolisme du glucose dans le muscle, nous avons vérifié l' impact de cette production
de NO dans ce modèle. L' induction de iNOS était corrélée à l' induction de l' expression du
transporteur de gl~cose 1 (GLUT1). De plus, la présence d'un inhibiteur spécifique de
iNOS; le 1400W dans le milieu d' incubation a bloqué l'augmentation de l' expression de
GLUTl. Nous avons par ailleurs observé que la présence d'un donneur de NO (NOC-15)
dans un modèle de cellules musculaires en culture (L6) avait des effets similaires sur
l' induction de GLUT1, supportant ainsi le rôle du NO sur l'expression de ce transporteur de
glucose. Nous avons observé que l'incubation des muscles causait la résistance à l'insuline
au niveau du transport du glucose et que cet effet était renversé par la présence du 1400W.
Ce manque d' effet de l' insuline était relié à la réduction de la stimulation de l' activité PI 3-
kinase associée à IRS-1 dans le muscle incubé. Ces résultats suggèrent donc que
l' incubation à long terme de muscle cause l'induction de iNOS induisant des altérations
dans l' utilisation du glucose. Cette étude souligne une fois de plus l'effet délétère de iNOS
sur la sensibilité à l'insuline, et questionne les limites des études utilisant l' incubation des
muscles pour une période prolongée.

117
Long-term incubation of epitrochlearis muscle causes iNOS induction and nitric
oxide-dependent alteratio:Ds in glucose uptake
Geneviève Pilon, Kathleen Lemieux, and André Marette*
Department ofPhysiology, Lipid Research Unit, Laval University Hospital Center, Québec,
G 1 V 402, Canada.
* To whom correspondence should be addressed at:
Department ofPhysiology & Lipid Research Unit
Laval University Hospital Research Center
2705 Laurier Boulevard, RC 9502
Sainte-Foy, Québec, G 1 V 4G2, Canada
Fax: (418) 654-2176

118
Abstract
The aim of the present study was to evaluate sorne of the limitations of prolonged skeletal
muscle incubation related to the modulation of inducible nitric oxide synthase (iN OS).
iNOS expression was evaluated in rat epitrochlearis following a 10 hour incubation period.
Rere we observed that the incubation process leads to iNOS mRNA and protein induction,
as well as nitrite and nitrate accumulation in the incubation medium. As expected these
changes were associated with an increase in iNOS activity, however in contrast to iNOS the
activity of constitutive NOS (cNOS) was reduced. Since iNOS is known to disturb glucose
metabolism in muscle, we investigated the impact of high NO output in the incubation
model on the expression of glucose transporter 1 (GLUT1) and insulin action. iNOS
induction in the incubation model was associated with an increase in GLUT1 expression
importantly the presence of 1400W (a specific iNOS inhibitor) in the incubation medium,
prevented this effect of long-term skeletal muscle incubation. In L6 skeletal muscle cells,
the NO donor NOC-15 had similar effects on GLUT1 expression, adding further support to
a role for NO in the induction of this glucose transporter. We also observed that long-term
muscle incubation impaired insulin stimulated glucose transport and this was reversed by
the presence of 1400W. This defect in insulin action was related to a reduced insulin
stimulated IRS-1 associated PI 3-kinase activity in incubated muscle. Taken together, these
results suggest that long-term muscle incubation causes iNOS induction and thereby leads
to alterations in both basal and insulin stimulated glucose utilization. Rence, these findings
provide further support for a deleterious role of iNOS on insulin sensitivity. Our data also
call for caution when using long-term muscle incubation for metabolic studies.

119
Introduction
The radical gas nitric oxide (NO) is an essential player in an ever-growing number of
cellular processes. This diffusible and reactive molecular messenger is fundamental for the
regulation of vasodilatation, neurotransmission and immune responses. The synthesis of
NO is generated by the oxidation of nitrogen atoms from L-arginine by at least three
isoforms of nitric oxide synthase (NOS): the endothelial type (eNOS or NOS3), the
neuronal type (nNOS or NOSl) and the inducible type (iNOS or NOS2) [1]. While eNOS
and nNOS are expressed constitutively and transiently activated by an increase in
intracellular Ca2+, iNOS activity is relatively calcium independent and its prolonged
activation plays an important role in host defense [2]. 1ndeed, iNOS is expressed at very
low levels under normal conditions. However, inflammatory cytokines and bacterial
endotoxins markedly stimulate iNOS expression [3]. Although this mechanism evolved to
protect the host from bacterial or viral invaders, it is well accepted that an uncontrolled
iNOS induction is directlY involved in the etiology of many diseases. 1ndeed, iNOS has
been associated with the necrosis of pancreatic islets, hypotension, Huntington' s disease,
Alzheimer' s disease, rheumatoid arthritis, and inflammatory bowel disease.
Among all deleterious effects of marked iNOS induction, we have clearly demonstrated its
role in glucose metabolism and insulin sensitivity [4] [5]. 1ndeed, we have shown that
iNOS knockout micewere protected against insulin resistance induced by high-fat feeding
and these results have been confirmed recently by the group of Kearney [6] [7]. 1t has also
been reported that treatment with the iNOS inhibitor L-N1L reverses fasting hyperglycemia
and restores hyperinsulinemia in ob/ob mice [8] [8]. We have also demonstrated, that iNOS
induction by cytokines and LPS in L6 muscle cells, causes insulin resistance by blunting
insulin-induced 1RS-1 associated P13-kinase activity, a key signaling event in insulin's
metabolic action [5]. This link between iNOS and insulin resistance is not surprising since
iNOS induction has been detected in different tissues from humans and animal models of

120
both diabetes and obesity. lndeed, iNOS has been detected in aorta and skeletal muscle of
high-fat fed mice, in skeletal muscle, liver and adipose tissue of ob/ob mice, in heart of
diabetic Zucker rats and in skeletal muscle and platelets from type 2 diabetic subj ects [9]
[7] [10].
The incubation of isolated skeletal muscle in vitro lS a useful technique for the
understanding of the tissue response to specific stimuli without the influence of other
organs and hormonal factors. However this method has its limitations and disadvantages.
Several groups have observed that long-term muscle incubation was associated with
disturbance of glucose metabolism [11][12]. However the cellular mechanism behind this
defect has been left unanswered. In the present study, we show that prolonged muscle
incubation gives rise to iNOS induction causing major perturbations in the regulation of
glucose uptake. Our findings point out a limitation for long-term muscle incubation and
confirm a role for iNOS in insulin resistance.

121
Experimental procedures
AnimaIs
This study was approved by the Animal Care and Handling Committee of Laval University.
Male Sprague-Dawley rats (175-200 g) purchased from Charles River (Montreal, Qc,
Canada) were maintained on a 12 h dark and light schedule, and fed ad libitum with Purina
rat chow. '
Muscle sample preparation
Rats were anesthetised by a ketamine/xylazine injection after over-night fasting. The
epitrochlearis muscles were rapidly isolated and immediately flash frozen in liquid nitrogen
or preincubated for 30 min in 6 ml of Krebs-Ringer bicarbonate (KRB) buffer, pH 7.4,
containing 2 mM sodium pyruvate (KRB-P). Muscles were then incubated for the
appropriate times (up to 12 hours) in the KRB buffer supplement containing aIl amino
acids and the incubation medium was changed every two hours. After the appropriate
incubation period, the muscles were frozen in liquid nitrogen or kept for in vitro glucose
transport.
Materials
1400W was from Biomol Research Laboratories (Plymouth, PA). ADP-Sepharose 4B
beads were from Amersham Biosciences, and the Renaissance enhanced
chemiluminescence kit was from PerkinElmer Life Sciences (Boston, MA). Monoclonal
antibodies against iNOS and eNOS and the polyclonal anti,body against nNOS were
obtained from Transduction Laboratories (Mississauga, Canada). IRS-l antibody (C-20)
was obtained from Santa Cruz Biotechnology (Santa Cruz, CA). NOC-15 was from Alexis
(San Diego, Calif. , USA). [3H]-I-arginine was from Amersham (OakviIle, Ont. , Canada)
and Dowex AG50W-X8 from Bio-Rad (Mississauga, Ont. , Canada). a-Minim~ essential
medium (a-MEM), fetal bovine serum and other tissue-culture products were obtained from
Gibco BRL. Human insulin (Humulin R) was purchased from Eli Lilly. 2,3-
diaminonaphthotriazole (DAN) was purchased from Aldrich chemicals (Milwaukee, WI) ,
and aIl others products were from Sigma Chemicals (St. Louis, MO).,

122
.Cell Culture
A line of L6 skeletal muscle cells (kind gift of Dr. Amira Klip, Hospital for Sick Children,
Toronto, ON, Canada) clonally selected for high fusion potential were used in the present
study. The L6 cellline was derived from neonatal rat thigh skeletal muscle cells and retains
many morphological, biochemical, and metabolic characteristics of skeletal muscle. Cells
were grown and maintained in monolayer culture in oc-MEM containing 2% (v/v) fetal
bovine serum and 1 % (v/v) antibiotic/antimycotic solution (10000 units/ml penicillin,
10000 J.lg/ml streptomycin and 25 J-lg/ml amphotericin B) in an atmosphere of 5% CO2 at
37 oC.
Glucose transport assay
2-Deoxyglucose uptake was deterrnined as previously described [4]. Briefly, L6 myotubes
were incubated with or without NOC-15 for 1h, followed by the addition of insulin 100nM
(or medium alone) for an additional 30 min. Cells were rinsed once with glucose-free
Hepes-buffered saline solution, pH 7.4 (140 mM NaCI, 20 mM Hepes/Na, 5 mM KCI, 2.5
mM MgS04 and 1 mM CaCb), and were subsequently incubated for 8 min with la JlM 2-
deoxy-D-glucose containing 0.3 J-lCi/ml 2-deoxy-D-eH]glucose in the same buffer. After
the incubation in transport medium, cells were rinsed three times with ice-cold saline
solution, and then disrupted by adding 50 mM NaOH. Cell-associated radioactivity was
deterrnined by scintillation counting. Protein concentrations were deterrnined by the
bicinchoninic acid method (Pierce), and the results were expressed in pmol/min per mg.
Glucose uptake values were corrected for non-carrier-mediated transport by measuring
hexose uptake in the presence of la J-lM cytochalasin B.
In vitro glucose uptake in isolated muscle.
Glucose transport in isolated epitrochlearis muscles was measured uSlng the glucose
analogue 2-eH]2-deoxy-d-glucose as described in [13].

123
Immunoblotting
Protein extraction was prepared using (20 mM Tris (pH 7.5), 150 mM NaCI, 10% glycerol,
1% Triton X-100, 1 mM EDTA, 2 mM Na3V04, 1 mM phenylmethylsulfonylfluoride, 10
f.!g/ml aprotinin, and 10 f.!g/ml leupeptin), and insoluble material was removed by
centrifugation at 12,000 x g. For western blotting, prote in extracts were resolved by SDS
P AGE and transferred to a polyvinylidene difluoride (PVDF) membrane. Blots were
blocked with 5% non-fat dry milk in 10 mM Tris (pH 7.5), 150 mM NaCI, and 0.05%
Tween 20; incubated with primary antibody; and then il?-cubated with a secondary
horseradish-conjugated antibody. Immunoreactive bands were visualized by enhanced
chemiluminescence.
RNA analysis
Total cellular RNA was isolated uSlng guanidinium thiocyanate-phenol-chloroform
extraction and iNOS, eNOS, nNOS, GLUT4 and GLUT1 mRNAs were measured by semi
quantitative RT-PCR as described previously [14][4].
PI 3-Kinase Activity
500 f.!g of muscle lysates were immunoprecipitated with 2 f.!g of anti-IRS-1 coupled to
protein A-Sepharose overnight at 4 oC and the IRS-1 associated PI 3-kinase activity was
measured as described previously [15].
Nitric oxide synthase activity
Nitric oxide synthase activity was quantified by the conversion of [3H]-I-arginine to [3H]-I
citrulline as described previously in [4] [6].
Nitrite and nitrate measurements
Nitrite and nitrate levels in plasma and incubation medium were measured by fluorometry.
Nitrate was reduced to nitrite using nitrate reductase and the NADPH regenerating system
as described previously [16]. Nitrite levels were determined by measuring the fluorescence
of 2,3-diaminonaphthotriazole DAN at Àex of 360 nm and :.\em of 450 nm.

124
Data Analysis- All data are presented as means ± S.E. The effects of the treatments were
compared by an analysis of variance followed by Fisher's post hoc test. Differences were
considered to be statistically significant at p < 0.05
Results
Long-term incubation of epitrochlearis muscle modulates NOS expression
Since muscle incubation has been shown to promote insulin resistance and we have
previously demonstrated that iNOS induction causes insulin resistance in skeletal muscle,
we speculated that muscle incubation may lead to induction of the iNOS enzyme. Rat
epitrochlearis muscles were isolated and frozen immediately or incubated for 10 hours (as
indicated on the figures) and th en frozen. In figure 1 we show that, without any treatment,
long-term muscle incubation leads to robust iNOS mRNA and protein induction (Fig. lA,
~). In contrast, the two calcium dependent NOS, eNOS and nNOS, were down-regulated
following incubation of the muscle. This alteration in NOS expression was associated with
shifts in NOS activity. Indeed, cNOS activity was dramatically reduced by long-term
skeletal muscle incubation while iNOS activity was up-regulated in muscle following 10
hours of incubation (Fig. 1 C). We also measured nitrite accumulation, a stable product of
NO degradation, in the incubation medium. In accordance with the iNOS expression and
activity data we observed that nitrite concentration was about 4 times higher in long-term
incubated muscle medium compared to the frozen muscle control, indicating that the iNOS
in these tissues was functional (Fig. ID). These results suggest that long-term incubation of
skeletal muscle enhances iNOS mRNA and protein expression along with enzyme activity
at the expense of cNOS.
Time course ofiNOS and GLUTl induction
To further characterize iNOS modulation by the muscle incubation, we looked at the time
course of iNOS induction. Muscles were incubated and frozen every hour for up to 12
hours. In muscles incubated for 2 and 3 hours, a slight iNOS prote in induction was
observed followed by a stronger induction at 2:4 hours (fig.2 A). We have previously

125
reported that iNOS induction by cytokines and LPS increased the expression of the glucose
trarisporter-l (GLUTl), leading to increased basal glucose transport [4]. We thus next
investigated whether GLUTI was modulated by iNOS induction in incubated
epitrochlearis. Here we observed that GLUTI expression was actually induced by the
incubation and the time course of its expression closely followed the time course ·of iNOS
expression (Fig.2B).
iNOS induction causes insulin resistance in incubated muscle
We next sought to determine whether iNOS induction leads to insulin resistance in these
long-term incubated muscles. We measured basal and insulin-stimulated glucose transport
in muscle incubated for 10 hours in the presence or in the absence of 1400W, a specific
iNOS inhibitor. Insulin failed to stimulate glucose transport after long-term incubation in
untreated muscles (Figure 3A). However, insulin significantly enhanced glucose transport
in the presence of 1400W. The effect of 1400W on insulin stimulated glucose transport
appeared to be explained in most part by normalization of basal glucose transport
potentially through "inhibition of GLUTI induction. To directly test this hypothesis, we
looked at the expression of GLUTI and GLUT4 in these muscles. GLUTI expression was
induced by long-term skeletal muscle incubation but this response was repressed by the
presence of the iNOS inhibitor (Fig. 3C). Importantly, no change in GLUT4 expression was
observed in incubated muscle treated with or without 1400W (Fig. 3D). To further
investigate the mèchanism by which iNOS caused the defect in insulin signal transmission,
we next assessed IRS-l associated PI 3-kinase activity, an essential step in the stimulation
of glucose transport by insuline No effect of incubation was observed on basal PI 3-kinase
activity; however, the potent effect of insulin on IRS-l associated PI 3-kinase activity was
markedly impaired following long-term skeletal muscle incubation (Fig 3E).
NO donor increases GLUTl expression in L6 muscle ceUs
To clarify the link between high NO output and GLUTI expression, we tested the effect of
NOC-15 (a chemical NO donor) in L6 muscle cells. Increasing doses ofNOC-15 caused a
dose dependent augmentation of GLUTI mRNA expression (Fig. 4A). The induction of
GLUTI mRNA corresponded with GLUTI prote in expression (Fig. 4B). As in incubated

126
muscle, no effect of the NO donor was observed on GLUT4 mRNA expression in this
cellular model. To further test the role of NO in mediating insulin resistance, we looked at
the effect of NOC-15 on basal and insulin stimulated glucose uptake in these cells. L6
myocytes were incubated with or without increasing doses ofNOC-15 for 24h, followed by
measurement of glucose uptake. Basal glucose uptake was augmented by the NO donor
while insulin-stimulated glucose uptake was reduced (Fig. 4C and 4D). These results are
consistent with our observations in incubated muscles and confirm the implication of iNOS
derived NO in the perturbations of glucose metabolism during long-term incubation of
muscles ex vivo.
Discussion
We have previously reported a causal link between iNOS induction and skeletal muscle
insulin resistance. In effect, we have shown that cytokines and LPS induce iNOS in
myocytes and in rat muscle, impairing insulin stimulated glucose transport [5] [13].
Furthermore, at the genetic level, we demonstrated that targeted disruption of iNOS
prevents the development of muscle insulin resistance associated with obesity caused by
high fat feeding [6]. In the present study, we consolidated this relationship in a new model
of iNOS induction. Here we observed that, long-term skeletal muscle incubation induces
iNOS, giving rise to alterations in glucose uptake. Under these conditions we observed that
basal glucose uptake was increased whereas insulin mediated glucose transport was
hindered. These perturbations in glucose utilization appear to be linked to increased
GLUTI expression and an impairment of insulin to stimulate IRS-1-associated PI 3-kinase
activity. We already reported that in L6 muscle cells, iNOS induction by cytokines and LPS
treatment leads ta insulin resistance by causing a defect ln the activation of IRS-1
as~ociated PI 3-kinase [5]. Furthermore insulin mediated PI 3-kinase activation was
reduced in skeletal muscle of wild type high-fat-fed obese mice, but remained normal in
high-fat fed obese iNOS-I- mice [6]. Consistent with these observations, we observed here
that iNOS mediated insulin resistance is caused by the inhibition of IRS-1 associated PI 3-
kinase activity in incubated muscles.

127
Whereas insulin-stimulated glucose uptake was reduced, basal glucose uptake was
increased in our model. This augmentation of basal glucose transport in incubated muscle is
in accordance with a previous study [12]. It has been proposed that the increase in basal
glucose uptake in incubated muscle could be due to a greater amount of glucose reaching
the cells by diffusion compared to the amount of glucose available in vivo by capillaries
within the muscle [12]. On the other hand, different groups have also reported that GLUT1
is induced in stressful conditions such as oxidative stress, denervation, hyperosmolarity,
glucose deprivation and hypoxia which could likely explain the elevated basal glucos~
uptake.
ln stressful situations, where energy is critical for survival, it is likely that GLUT1 is
mobilized to fulfill essential functions and ensure host defense. It is noteworthy that these
stress situations have a~so been linked to insulin resistance [17]. We can consider the
incubation process as stressful since the muscle is denervated and isolated from its natural
environment. Accordingly, it has been demonstrated that situations such as crush injuries,
shear stress or oxidative stress, also lead to iNOS induction [18] [19] [20]. In these
circumstances, cellular damage or stress may induce cytokine expression leading to iNOS
expression. Interestingly, it has been shown that heat shock protein 70 (HSP70), which is
produced during cellular stress, enhances cytokine-induced iNOS expression by activating
p38 MAP kinase [21]. In our model, muscle denervation could cause enough stress to
increase cytokine expression or activate stress-signaling pathways leading to iNOS
induction. Importantly, high NO concentrations are known to activate GLUT1 in different
models. Indeed, we have already reported that cytokines and LPS treatment increase
glucose transport in L6 muscle cells via GLUT1 induction [4]. Furthermore, the NO donors
soduim nitroprusside (SNP) or diethylenetriamine (DETA) induce GLUT1 in HUVEC cells
[22]. However, to our knowledge, NO's potential role as an inducer of GLUT1 in intact
muscle has never been investigated. Here, we provide novel complementary evidence that
iNOS induction leads to GLUT1 overexpression since the iNOS inhibitor 1400W blunted
both GLUT1 protein expression and the increase in basal glucose transport in intact skeletal
muscle that had been incubated long-terme

128
We also observed a decreased expreSSIon and activity of cNOS following long-term
skeletal muscle incubation. This downregulation could be explained by the effect of high
NO output from iNOS. Indeed, sorne studies have reported that NO inhibits NOS activity
and that nNOS and eNOS are more sen~itive than iNOS to the inhibitory action of NO [23].
Our finding of reduced expression of eNOS and nNOS is also consistent with previous
observations that iNOS induction in endothelial cells, reduces the expression of eNOS [24].
It is thus possible that this mechanism has evolved to temporarily inhibit cNOS in an effort
to keep cofactors available for iNOS during infection.
While previous studies on denervated and incubated muscles have reported a decrease in
GLUT4 expression, we could not explain the reduction in insulin sensitivity seen in our
study by changes in the expression of this glucose transporter [25] [12]. This apparent
discrepancy is likely explained by the composition of the muscle fiber type. Following
denervation, Handberg et al. reported a much greater GLUT4 decrement in oxidative red
muscle (type 1) compared to (type lIB) glycolytic white muscle [26]. Thus, the fact that
epitrochlearis is a fast twitch glycolytic muscle could explain why we did not observe a
reduction ofGLUT4 expression in our study.
In summary, the present study demonstrates that long-term incubation causes iNOS
induction in skeletal muscle leading to altered basal and insulin stimulated glucose
metabolism. The incubation dependant iNOS expression caused an increase in basal
glucose transport through elevated GLUT1 expression and decreased insulin-stimulated PI
3-kinase activity leading to defective insulin stimulated glucose transport. These results
may possibly provide a molecular basis for the alterations in glucose utilization that
manifest in patients suffering from denervated muscle following injuries. Our findings also
provide important considerations for those planning studies with incubated muscle
preparations.

129
LEGENDS TO FIGURES
Figure 1. Long-term incubation of epitrochlearis muscle modulates NOS expression. Rat
epitrochlearis muscles were isolated and frozen immediately or incubated for 10h before
being frozen. A) RT-PCR of iNOS, eNOS, nNOS and GAPDH rnRNA is shown. B)
immunoblot of iNOS, eNOS and nNOS protein is presented C) NOS activity in n0t:!
incubated (CTL) or incubated muscle (INC). D) Measurement of nitrite production in the
incubation medium.
Figure 2. Time-course of iNOS induction. Rat epitrochlearis muscles were incubated and
frozen each hour for up to 12 hours. A) Representative immunoblot of iNOS prote in B)
Representative RT-PCR ofiNOS and GLUTI mRNA and quantification.
Figure 3. Incubation causes alterations in glucose metabolism. A) Basal and insulin
stimulated glucose transport were measured in incubated muscle for 10 hours in the
presence or in the absence of 1400W, a specific iNOS inhibitor. B) Insulin stimulated
glucose transport is expressed as fold stimulation over basal. C) and D) show GLUTI and
GLUT4 mRNA expression (respectively) following muscle incubation in the presence or
the absence of 1400W. E) Insulin (INS) or basal (CTR) IRS-l associated PI 3-kinase
activity was measured in control (frozen) and incubated muscle.
Figure 4. NO donor increases GLUTI expression in L6 muscle cells. L6 muscle cells were
incubated with increasing doses of NOC-15 (a chemical NO donor) A) GLUTI and
GLUT4 were measured by RT-PCR (B) GLUTI protein expression. (C) Basal and (D)
insulin-stimulated 2-DG uptake were measured in L6 cells pretreated for 24 hours with
increasing doses ofNOC-15.

130
References
1. Mungrue, LN., et al. , From molecules to mammals: what's NOS got to do with it?
Acta Physiol Scand, 2003. 179(2): p. 123-35.
2. Wahl, S.M., et al. , Nitric oxide in experimental joint inflammation. Benefit or
detriment? Cells Tissues Organs, 2003. 174(1-2): p. 26-33.
3. Lowenstein, C.J. and E. Padalko, iNOS (NOS2) at a glance. J Cell Sci, 2004. 117(Pt
14): p. 2865-7.
4. Bedard, S. , B. Marcotte, and A. Marette, Cytokines modulate glucose transport in
skeletal muscle by inducing the expression of inducible nitric oxide synthase . . Biochem J,
1997. 325 ( Pt 2): p. 487-93.
5. Pilon, G. , P. Dallaire, and A. Marette, Inhibition of inducible nitric-oxide synthase
by activators of AMP-activated protein kinase: a new mechanism of action of insulin
sensitizing drugs. J Biol Chem, 2004.279(20): p. 20767-74.
6. Perreault, M. and A. Marette, Targeted disruption of inducible nitric oxide synthase
protects against obesity-linked insulin resistance in muscle. Nat Med, 2001.7(10): p. 1138-
43.
7. Noronha, B.T., et al. , Inducible nitric oxide synthase has divergent effects on
vascular and metabolic function in obesity. Diabetes, 2005. 54(4): p. 1082-9.
8. Sugita, H. , et al. , Inducible nitric-oxide synthase' and NO donor induce insulin
receptor substrate-1 degradation in skeletal muscle ceUs. J Biol Chem, 2005. 280(14): p.
14203-11.
9. Carvalho-Filho, M.A. , et al. , S-nitrosation of the insulin receptor, insulin receptor
substrate 1, and protein kinase B/Akt: a novel mechanism of insulin resistance. Diabetes,
2005.54(4): p. 959-67.
10. Torres, S.H., et al., Inflammation and nitric oxide production in skeletal muscle of
type 2 diabetic patients. J Endocrinol, 2004. 181(3): p. 419-27.
Il. Worm, D., et al. , Altered basal and insulin-stimulated phosphotyrosine phosphatase
(PTPase) activity in skeletal muscle from NIDDM patients compared with control subjects.
Diabetologia, 1996.39(10): p. 1208-14.

131
12. Henriksen, E.J. and J.O. Holloszy, Effect of diffusion distance on measurement of
rat skeletal muscle glucose transport in vitro. Acta Physiol Scand, 1991. 143(4): p. 381-6.
13. Kapur, S., et al., Expression ofnitric oxide synthase in skeletal muscle: a novel role
for nitric oxide as a modulator ofinsulin action. Diabetes, 1997.46(11): p. 1691-700.
14. Perreault, M., L. Dombrowski, and A. Marette, Mechanism ofimpaired nitric oxide
synthase activity in skeletal muscle of streptozotocin-induced diabetic rats. Diabetologia,
2000.43(4): p. 427-37.
15. Tremblay, P. , et al. , Dietary cod protein restores insulin-induced activation of
phosphatidylinositol 3-kinase/Akt and GLUT4 translocation to the T-tubules in skeletal
muscle ofhigh-fat-fed obese rats. Diabetes, 2003.52(1): p. 29-37.
16. Rao, A.M., et al. , Fluorometric assay of nitrite and nitrate in brain tissue after
traumatic brain in jury and cerebral ischemia. Brain Res, 1998. 793(1-2): p. 265-70.
17. Turinsky, J., Glucose and amino acid uptake by exercising muscles in vivo: effect of
insulin, jiber population, and denervation. Endocrinology, 1987.121(2): p. 528-35 .
18. Gosgnach, W. , et al., Shear stress induces iNOS expression in cultured smooth
muscle cells: role of oxidative stress. Am J Physiol Cell Physiol, 2000. 279(6): p. CI880-8.
19. Rubinstein, L, et al., Involvement of nitric oxide system in experimental muscle
crush in jury. J Clin Invest, 1998. 101(6): p. 1325-33.
20. Kuo, P.C., R.A. Schroeder, and J. Loscalzo, Nitric oxide and acetaminophen
mediated oxidative in jury: modulation of interleukin-l-induced nitric oxide synthesis in
cultured rat hepatocytes. J Pharmacol Exp Ther, 1997. 282(2): p. 1072-83.
21. Bellmann, K., et al., p38-dependent enhancement of cytokine-induced nitric-oxide
synthase gene expression by heat shock protein 70. J Biol Chem, 2000. 275(24): p. 18172-
9.
22. Paik, lY., et al. , Nitric oxide stimulates 18F-FDG uptake in human endothelial
cells through increased hexokinase activity and GLUTl expression. J Nucl Med, 2005.
46(2): p. 365-70.
23. Griscavage, J.M., A.J. Hobbs, and L.J. Ignarro, Negative modulation ofnitric oxide
synthase by nitric oxide and nitroso compounds. Adv Pharmacol, 1995.34: p. 215-34.
24. de Prutos, T., et al., NO from smooth muscle cells decreases NOS expression in
endothelial cells: role ofTNF-alpha. Am J Physiol, 1999.277(4 Pt 2): p. HI317-25.

132
25. Block, N.E., et al., Effect of denervation on the expression of Iwo glucose
transporter isoforms in rat hindlimb muscle. J Clin Invest, 1991. 88(5): p. 1546-52.
26. Worm, D., et al., Decreased skeletal muscle phosphotyrosine phosphatase (PTPase)
activity towards insulin receptors in insulin-resistant Zucker rats measured by delayed
Europium fluorescence. Diabetologia, 1996. 39(2): p. 142-8.

A.
C.
2
e-~o_
.~ E > ........... 0- en t E lU ...........
V) -o 0 z E s-
0
_iNOS
~eNOS
CTL INe
*
i cNOS
nNOS
GAPDH
OCTl
• INe
'*
61 iNOS
B.
D.
20
N
0 Z 10 ~ c
0
~~
eTL INe
iNOS
eNOS
nNOS
CTL INe
133
Figure 1

134
A.
al 1 2 3 4 5 6 7 8 9 1 0 11 1 2 +
B.
iNOS
GlUTl
GAPDH
CTL 1 2 3 4 5 6 7 8 9 1 0 11 1 2
:r: 1.2 0 1.0 Cl..
iNOS
-< O.~ \..? G.G ............ V")
o .• 0 z 0.2 0
CfR 12 3458 7 S 01011 12 :r: 1.5 0 Cl.. -< 1 ~O \..? ............
}:: :O~5 ~ ....J \..? 0 cm 1 2 3 45 e 7 $0 ·10 11 12
Figure 2

c.
A.
O buIJ 8 • in$ulin <lJ ~
7 tU ~
0.. ::J C
6 'f Q) V'l mS 0 U E ::J ......... rn ë5 4 >- E X 0 0. 3 Q)
"0 N 2
a 1400W
2 -,.---------.
o CTL INC INC
1400W
+ +
GLUTl
GAPDH
D.
o
B.
3
Q) ~
"0 2'2 Q) +J 0. 0 tU ::J ._
2 -S Q) +J
E V'l~ 0 ::J .~ U E V'l ::J ._ c: - +J
:.:: ~~ ::J x_ V'l 0 0 ~Q)~ .- '?
N
0
1400W
eTL INC INe 1400W
tGLUT4
GAPDH
"0 ~ tU -s E .~
V'l
.S -s
V'l
.S '+-0
-g.
135
*
+
E.
I ~ ." .. ~ 1
120
>- 100 ~
'S; SC) .~
U tU Q) 60 V'l tU c: 40 ~ M 20 a::
ins + + CTL INC
Figure 3

c.
A.
B.
CTL la 50 100 150 200
NOC-15 (J.lM)
GLUTl
GLUT4
GAPDH
1 ......... ---- .--, ·.--.,1 GLUT 1
CTL 1 50 100
NOC -15 (~lM)
D.
3·..,..... ......... ______ ,.,....",
2
0 : o CTL 5 25
NOC-15 (J.lM)
136
ins 5 25
ins + NOC-15 (J.lM)
Figure 4

Chapitre 4. L'inhibition de la synthase de NO inductible par les activateurs de la l kinase activée par l'AMP (AMPK): un nouveau mécanisme antiinflammatoire.
La kinase activée par l' AMP (AMPK), est une enzyme sensible aux niveaux d'énergie qui
est activée en réponse aux stress cellulaires et représente une étape critique pour la
régulation de plusieurs processus métaboliques. L'AMPK a récemment émergée comme
une nouvelle cible de choix pour le traitement de l' obésité et du diabète de type II puisque
son activation augmente l' oxydation des acides gras et améliore l 'homéostasie du glucose.
Nous avons démontré dans cet article que l' activation pharmacologique de l'AMPK par des
médicaments anti-diabétiques inhibait fortement la synthase de NO inductible (iNOS), un
élément pro-inflammatoire retrouvé dans le choc endotoxique et dans l' état d' inflammation
chronique relié à l'obésité. L'inhibition de iNOS par l'AMPK fut observée dans différents
types cellulaires (myocytes, macrophages, adipocytes) et serait le résultat d'une régulation
post-transcriptionnelle de la protéine iNOS. L' activation de L'AMPK in vivo bloque aussi
complètement l'induction de iNOS dans le muscle et dans le tissu adipeux de rats traités à
la LPS. Nous avons aussi observé que la réduction de l' expression de l'AMPK par des
petits ARN interférent renversait les effets inhibiteurs de l'AMPK sur l'expression de iNOS
et la production de NO dans les myocytes. Ces résultats indiquent que l' AMPK représente
une nouvelle avenue anti-inflammatoire, donc une cible thérapeutique pour des maladies
immunitaire inflammatoire.

138
Inhibition of inducible nitric oxide synthase by activators of AMP-activated protein
kinase: a new anti-inflammatory pathway
Geneviève Pilon, Patrice Dallaire and André Marette *
Department of Anatomy and Physiology, and Lipid Research Unit, Laval University
Hospital Research Center, Sainte-Foy, Québec, G 1 V 4G2, Canada.
RUNNING TITLE: activation of AMPK inhibits iNOS
* Address correspondence to: Dr. André Marette
Department ofPhysiology & Lipid Research Unit
Laval University Hospital Research Center
2705, Laurier Bld
Ste-Foy, (Québec), Canada, G1V 4G2
Tel: (4i8) 656-4141 ext. 7908
Fax: (418) 654-2176
E-mail:[email protected]

139
ABSTRACT
AMP-activated protein kinase (AMPK), an energy-sensing enzyme that is activated
in response to cellular stress, is a critical signaling molecule for the regulation of multiple
metabolic processes. AMPK has recently emerged as an attractive novel target for the
treatment of obesity and type 2 diabetes since its activation increases fatty acid oxidation
and improves glucose homeostasis. Here we show that pharmacological activation of
AMPK by anti-diabetic drugs markedly inhibits inducible nitric oxide (NO) synthase
(iN OS), a proinflammatory mediator in endotoxic shock and in chronic inflammatory states
including obesity-linked diabetes. AMPK-mediated iNOS inhibition was observed in
several cell types (myocytes, adipocytes, macrophages) and primarily resulted from post
transcriptional regulation of the iNOS proteine AMPK activation in vivo also blunted iNOS
induction in muscle and adipose tissues of endotoxin-challenged rats. Reduction of AMPK
expression by small interfering RNA reversed the inhibitory effects of AMPK activators on
iNOS expression and NO production in myocytes. These results indicate that AMPK is a
novel anti -inflammatory signaling pathway and thus represents a promising therapeutic
target for immune-inflammatory disorders.

140
AMP-activated protein kinase (AMPK) is emerglng as an important energy
sensing/signaling system in mammalian tissues. It is a member of a metabolite-sensing
protein kinase family that acts as a fuel gauge monitoring cellular energy levels 1. When
AMPK "senses" decreased energy storage, it acts to switch off ATP-consuming pathways
and switch on alternative pathways for ATP regeneration. AMPK is a heterotrimer
consisting of a catalytic a-subunit and two regulatory subunits, ~ and y. In response to
cellular energy depletion, as reflected by an increase in the AMP / A TP ratio, AMPK is
phosphorylated and activated by a still uncharacterized upstream AMPK kin~se2 . It can also
be activated allosterically by increases in the AMP/ ATP and creatine/creatine-P ratios. The
metabolic function of AMPK has perhaps been best documented ~n exercising skeletal
muscle, where its activation seems to contribute to increased glucose transport and fatty
acid oxidation3,4. AMPK can be activated chemically with 5-aminoimidazole-4-
carboxamide-riboside (AICAR), which is taken up by cells and phosphorylated by
adenosine kinase to form 5-aminoimidazole-4-carboxamide ribonucleoside (ZMP), a
nucleotide that mimics the effect of AMp5• More recent studies have also identified AMPK
as the mediator of the metabolic effects of the adipose-derived peptidic hormones leptin
and adiponectin in skeletal muscle and liver6-8.
Chronic treatment of animal models of type 2 diabetes with the AMPK activator
AICAR improves glucose homeostasis and insulin sensitivity9-14. · These beneficial effects
are thought to be mainly explained by the weIl known actions of AMPK on glucose
metabolism and lipid oxidation in muscle and liver. Accordingly, the anti-diabetic drugs
metformin and rosiglitazone also activate AMPK and this is believed to contribute to their
insulin-sensitizing actions in diabetic subjects1S-18. However, the potential effects of AMPK
activation on pro-inflammatory mediators of insulin resistance have not yet been
investigated. Indeed, an inflammatory component lS also present in obesity-linked
diabetes 19,20, as reflected by increased systemic and tissue concentrations of the
proinflammatory cytokines TNF-a and interleukin-6 in obese hum an subjects21 ,22 and
several animal models of obesiti3-2S . Furthermore, we have recently reported that inducible
nitric oxide synthase (iNOS), a cytokine-inducible proinflammatory mediator in several

141
pathological conditions, is overexpressed in muscle and fat of genetic and dietary models of
obesity and type 2 diabetes25. Targeted disruption of the iNOS gene was found to proteet
high-fat fed obese mice from developing insulin resistance and to significantly improve
glucose tolerance25. A role for iNOS in mediating insulin resistance is also supported by the
finding that pharmacological inhibition of iNOS reverses impaired insulin action in
cultured myocytes exposed to cytokines and the endotoxin LPS26. Since an inflammatory
component that includes iNOS is involved in the pathogenesis of muscle insulin resistance
in obesity-linked type 2 diabetes, we asked whether AMPK activation might improve
insulin action in muscle cells through inhibition of iNOS, and if so, whether AMPK is a
key iNOS inhibitory pathway in other cell types where increased iNOS expression is
pathogenic.
RESULTS
AMPK activators inhibit iNOS and reverse insulin resistance in skeletal muscle cells
We first tested the effect of the AMPK activator AICAR on iNOS induction in L6
myocytes chronically exposed to cytokines and LPS, a model of chronic inflammation
associated with insulin resistance26,27. As shown in Figure 1 A, treatment of muscle cells
with AICAR stimulated AMPK activity as reflected by increased phosphorylation of
AMPK on Thr172, a site known to activate the enzyme28, as weIl as by its ability to
enhance phosphorylation of acetyl-CoA carboxylase (ACC), a downstream target of AMPK
and a good correlate of its activation29• The anti-diabetic drug metformin and the PPAR-y
agonists troglitazone and 15dPGJ2 also activated AMPK in these cells (Fig. lA). Chronic
exposure of L6 myocytes to cytokines (TNF-a and IFN-y) and LPS markedly induced NO
production, as detected by the accumulation of the stable bioproduct nitrite in the culture
medium (Fig. lB). This effect was explained by a robust induction of iNOS mRNA and
prote in levels (Fig. 1 C-D). Neither eNOS nor nNOS are expressed in control or
cytokine/LPS-treated L6 myocytes, and NO production was completely inhibited by co
incubation with the specifie iNOS inhibitor 1400W3o (data not shown).
Addition of AICAR caused a dose-dependent reduction of cytokine/LPS-induced
NO production, reaching a maximal 80% inhibition at 2.5 mM (Figure lB). Treatment with

142
either metformin or PPARy agonists also decreased (by 50-90%) the effect of cytokine/LPS
on NO production. AlI AMPK activators inhibited NO production through reduction of
cellular iNOS protein levels (Fig. 1 C). AMPK activators failed to change the cellular
expression of desmin, the main intermediate filament protein in myocytes, here used as an
internaI control (data not shown). Reduction of iNOS protein expression was not linked to
suppression of iNOS transcription since iNOS mRNA levels were not affected by AICAR
or the anti-diabetic drugs metformin and troglitazone (Fig. ID). However, high
concentrations of 15dPGJ2 did reduce iNOS mRNA levels (see 25 JlM 15dPGJ2 in Fig.
ID). In additional studies, we also tested whether AICAR could inhibit iNOS-mediated NO
production either through reduction in substrate (L-arginine) uptake or by direct inhibition
of iNOS enzymatic activity. L-arginine uptake by muscle cells was not found to be affected
by AICAR (2.5 mM) (1297 ± 209 vs 1369 ± 90 pmol/min/mg for control and AICAR
treated cells, respectively). Furthermore, AICAR did not affect iNOS subunit dimerization
and failed to reduce iNOS catalytic activity in cytokine/LPS-treated myocytes, as assessed
by the formation of L-eH]citrulline form L-eH]-arginine (see Figures A and B,
supplementary materials). It has been reported that AICAR increases adenosine release by
ischemic heart tissue31 and this could contribute to iNOS inhibition. However, adenosine
per se failed to affect iNOS activity in L6 myocytes and the inhibitory effect of AICAR on
NO production was not decreased by blocking adenosine receptors with 8-(p-sulfophenyl)
theophylline (Figures C and D, supplementary materials) ruling out any role for adenosirie
release in AMPK-dependent iNOS inhibition.
To test whether AMPK-induced iNOS inhibition reverses the insulin resistance
promoting effects of cytokine/LPS, we next measured the effects of AMPK activators on
insulin action in L6 myocytes. We chose to assess insulin-induced activation of PI 3-kinase,
an essential mediator of insulin's metabolic actions, rather than glucose uptake since the
latter is increased by AMPK through insulin-independent pathways. As shown in Figures
2A and 2B, cytokines and LPS treatment caused insulin resistance as revealed by a 60%
reduction in the ability of insulin to stimulate IRS-l-associâted PI 3-kinase activity as
compared to control cells. However, addition of either AICAR, metformin or troglitazone
to cytokine/LPS-treated cells fully restored insulin action on PI 3-kinase. AMPK activating
---------------~ - - - ----

143
agents failed to increase PI 3-kinase in the absence of insulin (Figure 2A). The restoration
of insulin action by AMPK activating agents was similar to that obtained with the specific
iNOS inhibitor 1400W (Figure 2B).
AMPK activators inhibit iNOS in adipocytes and macrophages
We next tested whether AMPK activation inhibits iNOS in other cell types. We
have reported that adipose tissue is a major site of iNOS expression in endotoxemia and
that chronic treatment of adipocytes with cytokines and endotoxins reproduces this
inflammatory response in vitr032. As shown in Fig. 3A, addition of AICAR or anti-diabetic
drugs to cultured 3T3-Ll adipocytes activated AMPK, as reflected by enhanced ACC
phosphorylation. Activation of AMPK reduced cytokine/LPS-mediated NO production
(Fig. 3B) and cellular iNOS protein content (Fig. 3C) but not iNOS mRNA levels (Fig.
3D). Monocytes and macrophages are major sites of iNOS expression, particularly in
infectious or inflammatory diseases. Sustained production of NO endows macrophages
with cytostatic or cytotoxic activity against several pathogens and tumor cells but it can
also exert deleterious pro-inflammatory effects on host cells33. Activation of AMPK by
AICAR and metforrnin inhibited iNOS induction upon exposure of macrophages to
interferon-y (Fig. 3E and F). The PP ARy ligand troglitazone also increased AMPK and
caused a dose-dependent inhibition of NO production in these cells (Fig. 3E and F). The
inhibitory effects of AMPK activators was also linked to reduction in iNOS prote in
induction through a post-transcriptional mechanism (Fig. 3G and H). However, at higher
concentrations (> 10 J.lM) of troglitazone, a reduction in iNOS mRNA levels was also
observed.
Activation of AMPK by in vivo injection of AICAR and metformin inhibits iNOS in
muscle and adipose tissues of LPS-treated rats
To test whether AMPK also blunts iNOS induction in vivo, we assessed the ability
of AICAR and metforrnin to inhibit iNOS in rskeletal muscle and adipose tissue of rats
injected with the bacterial endotoxin LPS, a model of septic shock. Pre-treatment of LPS
challenged rats with AICAR or metforrnin increased ACC phosphorylation (Fig. 4A) and
significantly reduced NO production in the plasma as weIl as in muscle (gastrocnemius and

144
tibialis) and adipose tissues (Fig 4B). AICAR and metformin treatments prevented
induction of iNOS protein expression in muscle and fat tissues of septic rats (Fig. 4C). In
marked contrast, neither eNOS nor nNOS~ enzymes were affected by LPS or AMPK
activators in muscle and fat (nNOS~ was not detectable in fat) , highlighting the isoform
specific effect of AMPK on iNOS protein. As observed for celllines, the inhibitory effects
of AMPK activators on iNOS-mediated NO production was explained by post
transcriptional mechanisms si~ce LPS-induced iNOS mRNA expression was not affected
by AICAR or metformin treatments. (Fig. 4D). However, one notable exception is
metformin which causes a small but reproducible reduction in iNOS mRNA only in adipose
tissue.
RNA interference-mediated depletion of AMPK reverses the inhibitory effects of AMPK
activators on iNOS protein and NO production
To establish whether AICAR and anti-diabetic drugs inhibit iNOS through AMPK
activation, we next used RNA interference technology to «knockdown» endogenous
AMPK. Immunoblotting experiments confirmed that transfection of small interfering RNA
(siRNA) specific for the al and a2 catalytic subunits reduced AMPK expression to 53 ± 5
% of that of cells mock transfected with unre.1ated siRNA (Fig. 5A). Moreover, combined
siRNA treatment resulted in a comparable inhibition of AMPK and ACC phosphorylation
by AMPK activators (Fig. 5A). In AMPK-depleted myocytes, the ability of AI CAR,
metformin and troglitazone to inhibit cytokine/LPS-induced NO production and iNOS
prote in expression was significantly reduced as compared to cells treated with unrelated
siRNA (Fig. 5B). These results confirmed that these agents blunt iNOS expression and NO
production at least in part through activation of AMPK.
DISCUSSION
Elucidation of the multiple roles of NO in health and disease states has uncovered
many areas where manipulation of NO production would be of therapeutic bene fit. Thus,
whereas focal iNOS induction and NO production is an important anti-infectious and anti-

145
tumor mechanism of innate immunity34, high amounts of iNOS-derived NO can also lead to
self-tissue destruction in autoimmune diseases, allograft rejection, sepsis, rheumatoid
arthritis, chronic heart failure and other disorders accompanied by excessive activation of
the immune system (see3S-38). iNOS induction is also implicated in chronic metabolic
disorders such as atherosclerosis39,40 and obesity-linked diabetes2S,41 in which an
inflammatory condition is also believed to play a pathogenic role. Such wide implication
has produced an intense interest in understanding the modulation of iNOS expression and
activity, with the goal of finding novel mechanisms of control of this high output NO
pathway. For these reasons, the design of selective iNOS inhibitors and the identification of
iNOS inhibitory pathways have received much attention in recent years. In the present
study, we show that pharmacological activation of AMPK by AICAR and two classes of
anti-diabetic drugs inhibits iNOS induction in cells and tissues exposed to inflammatory
mediators. Gene silencing experiments confirmed that AMPK -activating agents blunt
iNOS-mediated NO production at least in part via activation of AMPK. Indeed, depletion
of AMPK in L6 myocytes by using the combination of al and a2-AMPK siRNA species
led to a significant loss in the ability of AICAR, metformin and troglitazone to reduce
cytokine/LPS-induced iNOS protein expression and NO production.
Metformin and the PPARy agonist rosiglitazone activate AMPK and this is believed
to contribute to their insulin-sensitizing actions in diabetic subjects9,lS-18. These anti
diabetic effects have been mainly attributed to the lipid and glucose-related metabolic
effects of AMPK activation. However, our data strongly suggest that inhibition of iNOS
induction and reduced NO formation a novel mechanism by which AMPK improves insulin
action in muscle. Indeed, we show here that AMPK activation -by several drugs reduces
iNOS induction in L6 myocytes chronically exposed to cytokines and LPS, an in vitro
model of insulin resistance for glucose transport26. Accordingly, we also found that iNOS
mediated impairment in insulin-induced PI 3-kinase activation can be reversed by AMPK
activators. Furthermore, the inhibitory effect of AMPK activation on iNOS was confirmed
in vivo in a model of septic shock associated with insulin resistance27. Further studies will
be needed to test whether long-term AMPK activation can also prevent or reduce iNOS

,.------ ------ ----~------ ~ - ~ - ---
146
induction in lower grade but chronic inflammatory disorders associated with insulin
resÎstance such as atherosclerosis and obesity-linked diabetes.
AMPK activation also suppressed iNOS protein expression and NO production in
cytokine/LPS-treated adipocytes and in adipose tissue of LPS-challenged rats. Whereas the
precise role of iNOS-derived NO in adipose cells is still not fully understood, it is thought
to be involved in the modulation of lipid metabolism in endotoxemia32,42 and may be a
paracrine mediator of skeletal muscle insulin resistance in obesity-linked diabetes25,43 .
Activation of AMPK in macrophages, an important site of iNOS expression, also caused
inhibition of IFNy-induced NO production. The latter finding is of important clinical
relevance since macrophage/monocyte iNOS induction has been implicated in the
pathogenesis or promotion of several inflammatory/immune disorders such as septic and
hemorrhagic shock44,45, multiple sclerosis46, myocardial infarction47,48, inflammatory bowel
disease49-51 , and pancreatic ~-cell dysfunction52,53.
It will be important to dissect out the molecular mechanisms by which AMPK
activators cause iNOS inhibition. It is well established that inflammatory cytokines and
LPS trigger iNOS transcription through a complex network of intracellular pathways
including NF-KR , JAKISTAT and MAP kinase54,55. Moreover, the PPARy igands
rosiglitazone and l5dPGJ2 were previously shown to reduce iNOS expression in several
cell types52,53,56 through transcriptional repression56. We were therefore expecting that
PPARy-dependent activation of AMPK would inhibit iNOS by reduction of its
transcription. Surprisingly, we found that all AMPK activating agents, including the
pp ARy agonists troglitazone and 15dPGJ2, decreased iNOS protein but barely affected
mRNA expression. Only when higher concentrations of troglitazone and 15dPGJ2 were
used did we observe a reduction in iNOS transcript levels. It should be noted that recent
studies have shown that inhibition of iNOS transcription by rosiglitazone may result from
activation of both pp ARô and pp ARy57. Thus high concentrations of rosiglitazone inhibit
iNOS in PPARy-l- macrophages at least in part by activating PPARy. Troglitazone is 100-
fold less potent than rosiglitazone58 for pp ARy but its affinity for pp ARô has not been
established. Therefore, it cannot be excluded that its inhibitory action on iNOS may be

147
mediated in part through PPAR8. Studies in cells lacking both PPAR8 and PPARy will be
needed to answer this important question. "
The inhibitory effect of AMPK activators on NO production were highly correlated with
the reduction in iNOS protein content, suggesting that AMPK modulates iNOS protein
turnover. In this regard, recent reports suggest that AMPK switches off protein synthesis
either through suppression of the mTOR-p70S6 kinase pathway59-61 , or by direct activation
of eukaryotic elongation factor 2 (eEF2) kinase, resulting in the phosphorylation and
inactivation of eEF262. It is also possible that AMPK reduces iNOS protein content by
promoting its ubiquitination, a process required for targeting iNOS through the proteasome
proteolysis pathway63. However, ubiquitination has also been reported to be involved in the
proteolytic degradation of nNOS64 whereas the inhibitory effects of AMPK on NO
production appears to be" specific for the iNOS isoform. Indeed, neither nNOS nor eNOS
proteins were affected by AICAR or metformin treatments in muscle and adipose tissue of
LPS-challenged rats, and AMPK activators blunted LPS-induced NO production but did
not reduce basal NO formation by constitutive NOS enzymes. This argues for inhibition of
prote in synthesis rather than ubiquitin-mediated proteasomal degradation as the principal
mechanism by which AMPK regulates iNOS prote in turnover.
The identification of AMPK as a selective iNOS-inhibitory pathway may provide a
molecular basis for the recent observations that exercise training, which increases AMPK
expression lO, improved exercise capacity in association with reduced skeletal muscle iNOS
expression in patients with chronic heart failure38. Our data further suggest that AMPK
represents a promising therapeutic target for immune-inflammatory disorders in which
iNOS induction is pathogenic but not critical for health (i.e. non-infectious diseases). These
include metabolic disorders such as atherosclerosis and obesity-linked diabetes. " However,
even for those inflammatory conditions, caution must be exerted since iNOS can also play
more subtle yet important physïological roles with potential adverse consequences
attendant on its inhibition in diseased states. For example, iNOS plays a protective role in
the development of transplant arteriosclerosis 65,66 and is also critical for optimal wound
healing67 which calls into question the use of iNOS inhibitory agents to treat diseases such

148
as atherosclerosisand diabetes. Bearing these cautions in mind, it is crucial to elucidate the
molecular mechanisms by which AMPK inhibits iNOS, since this may help design novel
strategies for an effective and tissue-specific inhibition of iNOS in inflammatory settings
while limiting adverse physiological consequences.
METHODS
Materials
AICAR was purchased from Toronto research chemicals (Toronto, ON, Canada).
Troglitazone and 15 deoxy PGJ2 were from Cayman Chemical company (Ann Arbor, MI) .
Interferon-y and TNF-a were from Research Diagnostics Inc. , (Flanders, NJ), and RD
systems (Minneapolis, MN), respectively. 1400W was from Biomol Research Laboratories
Inc. (Plymouth, PA) and Oligofectamine from Invitrogen (Burlington, Ontario, Canada). 2',
5'-ADP sepharose (4B) beads were from 'Amersham Pharmacia (Baie d'Urfe, Quebec,
Canada) and the Renaissance Enhanced chemiluminescence kit from NEN Life Science
Products (Boston, MA). AlI other chemicals were from Sigma (St.Louis~ MO). Monoclonal
antibodies against iNOS and eNOS and a polyclonal antibody against nNOS were obtained
from Transduction Laboratories (Mississauga, Ontario, Canada). Polyclonal antibodies
against AMPK, phospho-AMPK (Thr 172) and phospho-Acetyl-CoA Carboxylase (Ser 79)
were purchased from New England Biolabs (Beverly, MA). IRS-1 antibody (C-20) was
obtained from Santa Cruz Biotech (Santa Cruz, CA).
CelI culture
L6 myoblasts and 3T3-L1 fibroblasts were grown a-MEM (10% FBS) or DMEM (20 %
calf serum) containing 1 % antibiotic/antimycotic (100 000 units/ml penicilIin, 100 mg/ml
streptomycin, 0.25 mg/ml amphotericin) and differentiated into myotubes and adipocytes as
previously described26,32. Bone-marrow derived macrophages were a kind gift of Dr. M.
Olivier (CHUL Research Center, Laval University, Quebec, Canada). Generation and
immortalization of the mac~ophages from mouse femurs was performed as previously
described68. Induction of iNOS by cytokines and/or LPS in L6 myotubes, 3T3-L1

149
adipocytes and macrophages, and the effects of AMPK activating agents was carried out as
described in the figure legends. The accumulation of nitrite in the incubation medium was
used as an index of NO production. Nitrite was determined spectrophotometrically as
previously described26.
RNA interference
siRNAs were synthesized by Dharmacon Research (Lafayette, CO) and annealed according
to the manufacturer' s instructions. The sequence of the 19 nt (plus 2AA overhanging at S')
siRNAs were as follows: a1-AMPK: (GCAUAUGCUGCAGGUAGAU); a2-AMPK
(CGUCAUUGAUGAUGAGGCU), unrelated siRNA: (AUUGUAUGCGAUCGCAGAC).
L6 myotubes in 12-well plates were transfected 24 hours prior to cytokines/LPS treatment
with 0.27 JlM of a1- and a2-AMPK siRNAs by using Oligofectamine reagent. Mock
controls were transfected with unrelated siRN A.
AnimaIs
This study was approved by the Animal Care and Handling Committee of Laval University.
Male Sprague-Dawley rats (200-250 g) purchased from Charles River (Montréal, Qc,
Canada) were used in these studies. Rats were pretreated with a single i.p. injection of
AICAR (1 g/kg BW), metformin (100 mg/kg BW) or saline (vehicle) 30 min prior to LPS
(20 mg/kg BW ) or saline treatment. AnimaIs were sacrificed 4 hours later and hindlimb
muscles and white adipose tissues were removed and stored 'at -80°C until further
processlng.
Measurement of plasma and tiss~e nitrite and nitrate
Nitrite and nitrate levels in ,plasma, skeletal muscles and adipose tissue was measured by
fluorometry69. Briefly, blood was collected in tubes containing EDTA and centrifuged for
10 minutes at 3200 x g to obtain plasma. Tissues were grounded in liquid nitrogen using a
morter. The tissue powder was resuspended in 5 volumes of Tris-EDTA buffer (20 mM
Tris pH 7.5 , 10 mM EDTA) containing a protease inhibitor cocktail. Tissue lysates were
centrifuged at 2800 x g for 20 min and prote in concentration of the supematant was
measured by the BCA prote in assay. Plasma and tissue lysates were then centrifuged at

150
5000 x g (4°C) overnight in a Ultrafree®-Microcentrifuge 10 000 NMWL filter unit.
Nitrate was reduced to nitrite using nitrate reductase and the NADPH regenerating system
as described previously69. The fluorescence was measured at Àex 360 nm and !vern 450 nm.
Determination of NOS protein expression and Western blot analysis
Muscle and adipose tissue lysates (1 mg of protein) were used to purify NOS enzymes
using 2',5'-ADP sepharose resine as previously described25 . Samples were subjected to
sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-P AG~) and
immunoblotting was performed as previously described27. Immunoreactive bands were
detected by the enhanced chemiluminescence method.
RNA analysis
Total cellular RNA was isolated uSlng guanidium thiocyanate-phenol-chloroform
extraction and iNOS mRNA was measured by semi-quantitative reverse transcriptase
polymerase chain reaction as previously described26,32.
PI 3-Kinase Activity
Celllysates (500 Ilg) were immunoprecipitated with 2 Ilg of anti-IRS-1 ·coupled to protein
A-Sepharose overnight at 4°C. PI 3-kinase activity was determined as previously
described 70.
Data Analysis
AlI data are presented as means ± SE. The effects of the treatments were compared by an
ANOV A analysis followed by Pisher's post hoc test. Differences were considered to be
statistically significant at p<0.05.

151
ACKNOWLEDGMENTS
This work was supported by grants from the Canadian Institutes for Health Research
(CIHR) to A.M. A.M. is a CIHR Scientist. G. P. and P. D. were supported by CIHR and
Canadian Diabetes Association doctoral awards, respectively. We thank Bruno Marcotte
and Sandra F avret for expert technical assistance. We are grateful to Dr. Daniel Konrad for
his helpful advices on the use of siRNA and to Drs Yves Deshaies and Claude H. Côté for
critical reading of the manuscript.

152
REFERENCES
1. Hardie, D.G., Scott, l.W. , Pan, D.A. & Hudson, E.R. Management of cellular
energy by the AMP-activated prote in kinase system. FEBS Lett 546, 113-20 (2003).
2. Hong, S.P., Leiper, F.C. , Woods, A. , Carling, D. & Carlson, M. Activation of yeast
Snfl and mammalian AMP-activated protein kinase by upstream kinases. Proc Nat! Acad
Sci US A 100, 8839-43 (2003).
3. Winder, W.W. Energy-sensing and signaling by AMP-activated protein kinase in
skeletal muscle. J Appl Physiol91 , 1017-28 (2001).
4. Sakamoto, K. &"Goodyear, L.l. Invited review: intracellular signaling in contracting
skeletal muscle. J Appl Physiol93, 369-83 (2002).
5. Corton, l.M. , Gillespie, l.G. , Hawley, S.A. & Hardie, D.G. 5-aminoimidazole-4-
carboxamide ribonucleoside. A specifie method for activating AMP-activated protein
kinase in intact cells? Eur J Biochem 229, 558-65 (1995).
6. Minokoshi, Y. et al. Leptin stimulates fatty-acid oxidation by activating AMP-
activated prote in kinase. Nature 415, 339-43 (2002).
7. Tomas, E. et al. Enhanced muscle fat oxidation and glucose transport by ACRP30
globular domain: acetyl-CoA carboxylase inhibition and AMP-activated prote in kinase
activation. Proc Natl Acad Sci USA 99, 16309-13 (2002).
8. Yamauchi, T. et al. Adiponectin stimulates glucose utilization and fatty-acid
oxidation by activating AMP-activated protein kinase. Nat Med 8, 1288-95 (2002).
9. Musi, N. & Goodyear, L.l. Targeting the AMP-activated prote in kinase for the
treatment of type 2 diabetes. Curr Drug Targets Immune Endocr Metabol Disord 2, 119-27
(2002).
10. Winder, W.W. AMP-activated protein kinase: possible target for treatment of type 2
diabetes. Diabetes Technol Ther 2, 441-8 (2000).
Il. Holmes, B.F. , Kurth-Kraczek, E.l. & Winder, W.W. Chronic activation of 5'-AMP
activated prote in kinase increases GLUT-4, hexokinase, and glycogen in muscle. J Appl
Physiol87, 1990-5 (1999).

153
12. Halseth, A.E. , Ensor, N.J. , White, T.A. , Ross, S.A. & Gulve, E.A. Acute and
chronic treatment of ob/ob and db/db mice with AICAR decreases blood glucose
concentrations. Bioehem Biophys Res Commun 294, 798-805 (2002).
13. Fiedler, M. et al. 5-aminoimidazole-4-carboxy-amide-1-beta-D-ribofuranoside
treatment ameliorates hyperglycaemia and hyperinsulinaemia but not dyslipidaemia in
KKAy-CETP mice. Diabetologia 44, 2180-6 (2001).
14. Buhl, E.S. et al. Chronic treatment with 5-aminoimidazole-4-carboxamide-1-beta
D-ribofuranoside increases insulin-stimulated glucose uptake and GLUT4 translocation in
rat skeletal muscles in a 'fiber type-specific manner. Diabetes 50, 12-7 (2001).
15. Musi, N. et al. Metformin increases AMP-activated protein kinase activity ln
skeletal muscle of subjects with type 2 diabetes. Diabetes 51, 2074-81 (2002).
16. Hawley, S.A. , Gadalla, A.E. , Olsen, G.S. & Hardie, D.G. The antidiabetic drug
metformin activates the AMP-activated protein kinase cascade via an adenine nucleotide
independent mechanism. Diabetes 51, 2420-5 (2002).
17. Fryer, L.G., Parbu-Patel, A. & Carling, D. The Anti-diabetic drugs rosiglitazone and
metformin stimulate AMP-activated protein kinase through distinct signaling pathways. J
Biol Chem 277, 25226-32 (2002).
18. Zhou, G. et al. Role of AMP-activated protein kinase in mechanism of metformin
action. J Clin Invest 108, 1167-74 (2001).
19~ Pickup, J.C. & Crook, M.A. Is type II diabetes mellitus a disease of the innate
immune system? Diabetologia 41, 1241-8 (1998).
20. Marette, A. Mediators of cytokine-induced insulin resistance in obesity and other
inflammatory settings. Curr Opin Clin Nutr Metab Care 5, 377-83 (2002).
21. Hotamisligil, G.S. , Amer, P. , Caro, J.F. , Atkinson, R.L. & Spiegelman, B.M.
Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and
insulin resistance. J Clin Invest 95, 2409-15 (1995).
22. Yudkin, J.S. , Stehouwer, C.D. , Emeis, J.J. & Coppack, S.W. C-reactive protein in
healthy subjects: associations with obesity, insulin resistance, and endothelial dysfunction:
a potential role for cytokines originating from adipose tissue? Arterioseler Thromb Vase
Biol 19, 972-8 (1999).

154
23. Hotamisligil, G.S. , Shargill, N.S. & Spiegelman, B.M. Adipose expression of tumor
necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science 259, 87-91
(1993).
24. Hotamisligil, G.S. & Spiegelman, B.M. Tumor necrOSlS factor alpha: a key
component of the obesity-diabetes link. Diabetes 43, 1271-8 (1994).
25. PerreauIt, M. & Marette, A. ~argeted disruption of inducible nitric oxide synthase
protects against obesity-linked insulin resistance in muscle. Nat Med 7, 1138-43 (2001).
26. Bedard, S. , Marcotte, B. & Marette, A. Cytokines modulate glucose transport in
skeletal muscle by inducirig the expression of inducible nitric oxide synthase. Biochem J
325 ( Pt 2), 487-93 (1997).
27. Kapur, S., Bedard, S., Marcotte, B., Cote, C.H. & Marette, A. Expression of nitric
oxide synthase in skeletal muscle: a novel role for nitric oxide as a modulator of insulin
action. Diabetes 46, 1691-700 (1997).
28. Hawley, S.A. et al. Characterization of the AMP-activated protein kinase kinase
from rat liver and identification of threonine 172 as the major site at which it
phosphorylates AMP-activated prote in kinase. J Biol Chem 271, 27879-87 (1996).
29. Winder, W.W. & Hardie, D.G. AMP-activated protein kinase, a metabolic master
switch: possible roles in type 2 diabetes. Am J Physiol277, El-l 0 (1999).
30. Garvey, E.P. et al. 1400W is a slow, tight binding, and highly selective inhibitor of
inducible nitric-oxide synthase in vitro and in vivo. J Biol Chem 272, 4959-63 (1997).
31. Gruber, H.E. et al. lncreased adenosine concentration in blood from ischemic
myocardium by AlCA riboside. Effects on flow, granulocytes, and injury. Circulation 80,
1400-11 (1989).
32. Kapur, S. , Marcotte, B. & Marette, A. Mechanism of adipose tissue iNOS induction
in endotoxemia. Am J Physiol276, E635-41 (1999).
33. MacMicking, J. , Xie, Q.W. & Nathan, C. Nitric oxide and macrophage function.
Annu Rev Immunol15, 323-50 (1997).
34. Bogdan, C. , Rollinghoff, M. & Diefenbach, A. The role of nitric oxide in innate
immunity. lmmunol Rev 173, 17-26 (2000).
35. Nathan, C. lnducible nitric oxide synthase: what difference does it make? J Clin
lnvest 100, 2417-23 (1997).'

155
36. Parkinson, J.F. , Mitrovic, B. & Merrill, J.E. The role of nitric oxide in multiple
sclerosis. J Mol Med 75, 174-86 (1997).
37. Stichtenoth, D.O. & Frolich, J.C. Nitric oxide and inflammatory joint diseases. Br J
Rheumatol 37, 246-57 (1998).
38. Gielen, S. et al. Anti-inflammatory effects of exercise training in the skeletal muscle
of patients with chronic heart failure. J Am Coll Cardiol 42, 861-8 (2003).
39. Crornheeke, K.M. et al. Inducible nitric oxide synthase colocalizes with signs of
lipid oxidationlperoxidation in hum an atherosclerotic plaques. Cardiovasc Res 43, 744-54
(1999).
40. . Behr-Roussel, D. et al. Effect of chronic treatment with the inducible nitric oxide
synthase inhibitor N-iminoethyl-L-lysine or with L-arginine on progression of coronary and
aortic atherosclerosis in hypercholesterolemic rabbits. Circulation 102, 1033-8 (2000).
41. Shimabukuro, M. , Ohneda, M., Lee, Y. & Unger, R.H. Role of nitric oxide in
obesity-induced beta cell disease. J Clin Invest 100, 290-5 (1997).
42. Kapur, S. , Picard, F. , Perreault, M., Deshaies, Y. & Marette, A. Nitric oxide: a new
player in the modulation of energy metabolism. Int J Obes Relat Metab Disord 24 Suppl 4,
S36-40 (2000).
43. Pilon, G. , Penfomis, P. & Marette, A. Nitric oxide production by adipocytes: a role
in the pathogenesis of insulin resistance? Horm Metab Res 32, 480-4 (2000).
44. Angele, M.K. et al. Hypoxemia in the absence of blood loss upregulates iNOS
expression and activity in macrophages. Am J Physiol 276, C285-90 (1999).
45. Le Roy, D. et al. Nitric oxide production in experimental gram-negative infection:
studies with cytokine receptor-deficient mice. Shock 10, 37-42 (1998).
46. . Smith, K.J. & Lassmann, H. The role of nitric oxide in multiple sclerosis. Lancet
Neuroll , 232-41 (2002).
47. Bachmaier, K. et al. iNOS expressIon and nitrotyrosine formation in the
myocardium in response to inflammation is controlled by the interferon regulatory
transcription factor 1. Circulation 96, 585-91 (1997).
48. Wildhirt, S.M., Dudek, R.R., Suzuki, H. & Bing, R.J. Involvement of inducible
nitric oxide synthase in the inflammatory process of myocardial infarction. Int J Cardiol
50, 253-61 (1995).

156
49. Bauer, A.J. , Schwarz, N.T. , Moore, B.A. ,' Turler, A. & Kalff, J.C. Ileus in critical
illness: mechanisms and management. Curr Opin Crit Care 8, 152-7 (2002).
50. Hamalainen, M., Lahti, A. & Moilanen, E. Calcineurin inhibitors, cyclosporin A and
tacrolimus inhibit expression of inducible nitric oxide synthase in colon epithelial and
macrophage celllines. Eur J Pharmacol448, 239-44 (2002).
51. Southey, A. et al. Pathophysiological role of nitric oxide in rat experimental colitis.
Int J Immunopharmacol19 , 669-76 (1997).
52. Maggi, L.B. , Jr. et al. Anti-inflammatory actions of 15-deoxy~delta 12,14-
prostaglandin J2 and troglitazone: evidence for heat shock-dependent and -independent
inhibition of cytokine-induced inducible nitric oxide synthase expression. Diabetes 49,
346-55 (2000).
53. Kwon, G. , Xu, G. , Marshall, C.A. & McDaniel, M.L. Tumor necrosis factor alpha
induced pancreatic beta-cell insulin resistance is mediated by nitric oxide and prevented by
15-deoxy-DeltaI2,14-prostaglandin J2 and aminoguanidine. A role for peroxisome
proliferator-activated receptor gamma activation and inos expression. J Biol Chem 274,
18702-8 (1999).
54. Blanchette, J. , Jaramillo, M. & Olivier, M. Signalling events involved in interferon-
gamma-inducible macrophage nitric oxide generation. Immunology 108, 513-22 (2003).
55. Adams, V. et al. Induction of iNOS expression in skeletal muscle by IL-l beta and
NFkappaB activation: an in vitro and in vivo study. Cardiovasc Res 54, 95-104 (2002).
56. Li, M. , Pascual, G. & Glass, C.K. Peroxisome proliferator-activated receptor
gamma-dependent repression of the inducible nitric oxide synthase gene. Mol Cel! Biol 20,
4699-707 (2000).
57. WeI ch, J.S. , Ricote, M., Akiyama, T.E., Gonzalez, F.J. & Glass, C.K. PPARgamma
and PP ARdelta negatively regulate specific subsets of lipopolysaccharide and IFN -gamma
target genes in macrophages. Proc Nat! Acad Sci USA 100, 6712-7 (2003).
58. Young, P.W. et al. Identification of high-affinity binding sites for the insulin
sensitizer rosiglitazone (BRL-49653) in rodent and human adipocytes using a .
radioiodinated ligand for peroxisomal proliferator-activated receptor gamma. J Pharmacol
Exp Ther 284, 751-9 (1998).

157
59. Krause, U., Bertrand, L. & Hue, L. Control of p70 ribosomal prote in S6 kinase and
acetyl-CoA carboxylase by AMP-activated protein kinase and protein phosphatases in
isolated hepatocytes. Eur J Biochem 269, 3751-9 (2002).
60. Dubbelhuis, P.F. & Meijer, A.J. Hepatic amino acid-dependent signaling is under
the control of AMP-dependent protein kinase~ FEES Lett 521, 39-42 (2002).
61. Boister, D.R. , Crozier, S.J. , Kimball, S.R. & Jefferson, L.S. AMP-activated protein
kinase suppresses protein synthesis in rat skeletal muscle through down-regulated
mammalian target ofrapamycin (mTOR) signaling. J Biol Chem 277, 23977-80 (2002).
62. Horman, S. et al. Activation of AMP-activated prote in kinase leads to the
phosphorylation of elongation factor 2 and an inhibition of protein synthesis. Curr Biol 12,
1419-23 (2002).
63. Kolodziejski, P.J. , Musial, A. , Koo, J.S. & Eissa, N.T. Ubiquitination of inducible
nitric oxide synthase is required for its degradation. Proc Natl Acad Sci US A 99, 12315-20
(2002).
64. Bender, A.T., Demady, D.R. & Osawa, Y. Ubiquitination of neuronal nitric-oxide
synthase in vitro and in vivo. J Biol Chem 275, 17407-11 (2000).
65. Qian, Z. et al. lnducible nitric oxide synthase inhibition of weibel-palade body
release in cardiac transplant rejection. Circulation 104, 2369-75 (2001).
66. Koglin, J., Glysing-Jensen, T., Mudgett, J.S. & Russell , M.E. Exacerbated
transplant arteriosclerosis in inducible nitric oxide-deficient mice. Circulation 97, 2059-65
(1998).
67. Yamasaki, K. et al. ReversaI of impaired wound repair in iNOS-deficient mice by
topical adenoviral-mediated iNOS gene transfer. J Clin lnvest 101, 967-71 (1998).
68. Forget, G. et al. Role of host phosphotyrosine phosphatase SHP-1 ln the
development of murine leishmaniasis. Eur J lmmunol 31, 3185-96 (2001).
69. Rao, A.M., Dogan, A. , Hatcher, J.F. & Dempsey, R.J. Fluorometric assay of nitrite
and nitrate in brain tissue after traumatic brain injury and cerebral ischemia. Brain Res 793 ,
265-70 (1998).
70. Tremblay, F. & Marette, A. Amino acid and insulin signaling via the mTORlp70 S6
kinase pathway. A negative feedback mechanism leading to insulin resistance in skeletal
muscle cells. J Biol Chem 276, 38052-60. (2001).

158
LEGENDS TO FIGURES
Figure 1: Effects of AMPK activators on NO production and iNOS expression in L6
myocytes. Cells were treated for 24 hours with or without cytokines (TNF -u lOng/ml and
IFN-y, 200 units/ml) and LPS (10 ~g/ml) (Cyto/LPS) in the presence of AICAR (2.5 mM),
metformin (2 mM), troglitazone (10 ~M), or 15 deoxy PGl2 (10 and 25 ~M). A)
Representative immunoblots of ACC (ser-79) and AMPK (Thr 172) phosphorylation
following treatment with AMPK activators. B) NO production was measured by nitrite
accumulation in the incubation medium. Results are expressed as mean ± SE for 3-6
individual experiments. C) Representative immunoblots of iNOS protein expression. iNOS
migrated as a,,-, 130 kDa band. D) Representative RT-PCR for iNOS and GAPDH (internaI
control) mRNAs. Immunoblots and RT-PCR and representatives of 3-6 individual
experiments. * P < 0.05 as compared with cyto/LPS values.
Figure 2: Effects of cytokinelLPS and AMPK activators on IRS-I-associated PI 3-
kinase activity in L6 myocytes. PI 3-kinase was measured in anti-IRS-1
immunoprecipitates from lysatès of L6 myocytes treated for 24 hours with or without
cytokines (TNF-u 10 ng/ml and IFN-y, 200 units/ml) and LPS (10 ~g/ml) (Cyto/LPS) in
the presence of AICAR (2.5 mM), metformin (2 mM), troglitazone (10. J.lM), or the specifie
. iNOS inhibitor 1400W (0.1 mM). Quantification of 32p incorporated into PI 3-phosphate
(PI 3P) was expressed relative to insulin-stimulated values in control (CTR) cells.
Representative autoradiographs are shown in A. Results in B are expressed as mean ± SE
from 3-6 individual experiments. * P < 0.05 as compared with control values.
Figure 3: Effects of AMPK activators on NO production and iNOS expression in 3T3-
LI adipocytes and macrophages. 3T3-L1 adipocytes (panels A to D) were treated for 24
hours with or without cytokines (TNF-a 10 ng/ml and IFN-y, 200 units/ml) and LPS (10
J.lg/ml) (Cyto/LPS) in the presence of AICAR (2.5 mM), metformin (2 mM) or troglitazone
(10 J.lM). Macrophages (panels E to If) were treated for 10 hours with IFN-y (200 units/ml)
and the same AMPK activators. (A and E) Representative immunoblots of ACC ser-79

159
phosphorylation following treatment with AMPK activators. (B and F) Measurement of
nitrite production in the incubation medium. Data are from 2-4 independent experiments
and are expressed relative to nitrite production in cyto/LPS · cells in the absence of AMPK
activators. (C and G) Representative immunoblots of iNOS protein expression. (D and H)
Representative RT-PCR of iNOS and GAPDH mRNAs. Immunoblots and RT-PCR are
representative of 2-4 independent experiments * P < 0.05 as compared with cyto/LPS
values.
Figure 4: Effect of AICAR and metformin on iNOS in skeletal muscle and adipose
tissue in vivo. Rats were pre-treated with a single i.p. injection of AICAR (1 g/kg BW),
metformin (100 mg/kg BW) or saline (vehicle) 30 min prior to injection of LPS (20 mg/kg
BW, i.p.). Tissues and plasma were sampled 4 hours after LPS injection. A) immunoblots
of ACC (Ser 79) phosphorylation in muscle and! fat tissues, B) Measurement of nitrite and
nitrate in plasma, gastrocnemius, tibialis anterior and adipose tissue. Results are expressed
as mean ± SE from 4 animaIs. C) Representative immunoblots of eNOS, nNOSJ,l and iNOS
prote in expression in muscle and fat tissues. D) Representative RT-PCR of iNOS and
GAPDH mRNAs in muscle and adipose tissue. lmmunoblots and RT-PCR data are
representative of 4 independent experiments with different rats. + P < 0.05 as compared
with saline values.
Figure 5: siRNA-mediated reduction in AMPK expression reverses the inhibitory
effects of AMPK activators on iNOS expression and activity. L6 myocytes were
transfected with 0.27 J-lM of al-and a2-AMPK siRNAs by using Oligofectamine reagent.
Mock controls were transfected with the unrelated siRNA. Twenty-four hours after
transfection, cells were treated w~th cytokines, LPS and AMPK activators exactly as
described in Figure 1. A) Representative immunoblots of AMPK prote in expression,
AMPK (Thr 172) phosphorylation and ACC (Ser 79) phosphorylation in siRNA (a1/a2)
and mock transfected cells. B) Cellular iNOS protein expression and nitrite accumulation in
the medium of siRNA (a1/a2) and mock transfected cells. Nitrite data are mean ± SE of 3-
7 individual experiments. lmmunoblots in A and B are representative of at least 3
experiments. * P < 0.05 as compared with mock controls.
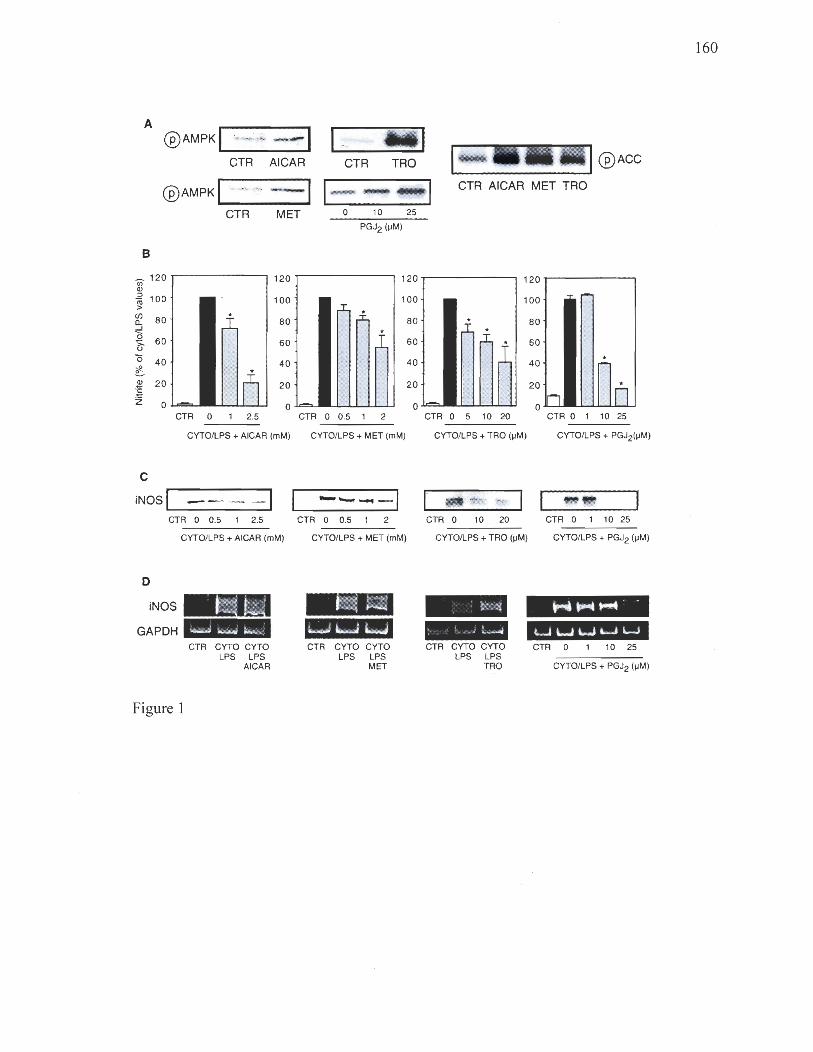
A @AMPK l ,,-,- --" 1
CTR AICAR CTR TRO @ACC
CTR AICAR MET TRO
CTR MET o 10 25
PGJ2 (IlM)
B
__ 120 120 (/)
120~----------~ 120 -r-------, Cl) :::l 100 ëti 100 >
Cf) a.. 80 80 ....1 Ô 60 >. 60 (.)
'0 40 40 ~ e..-
~ 20 20
Z 0 0 CTR 0 2.5
CYTO/LPS + AICAR (mM)
C
iN OS L-t ___ .", ""-" " _" " ___ , CTR 0 0.5 1 2.5
CYTO/LPS + AICAR (mM)
o
iNOS
GAPDH
Figure 1
CTR CYTO CYTO LPS LPS
AI CAR
100 1<'
/ )\ ,:[ * li
80
60 l '"""
40 1;:
: 20
Lc=::a..... {
o CTR 0 0.5 1 2
CYTO/LPS + MET (mM)
CTR 0 0 .5 2
CYTO/LPS + MET (mM)
CTA CYTO CYTO LPS LPS
MET
100
CTR 0 5 10 20
CYTO/LPS + TRO (IJM)
CTR 0 10 20
CYTOILPS + TRO (IJM)
CTR CYTO CYTO LPS LPS
TRO
80
60
40
20
a CTR 0 1 10 25
CYTO/LPS + PGJ2ÜJM)
•• CTR 0 1 10 25
CYTO/LPS + PGJ2 (!lM)
CTR 0 10 25
CYTO/LPS + PGJ2 (IJM)
160

A
PI(3)P ___ 1
1
r 1
Insulin
CYTO/LPS
Agent
B > 140 ...., "S: += (,)
120 cu (1.) <n mû) cO> 100 "- ::l ..:.:-lm M> -0 Il.!:: 80 "tJ§ (1)U ....,-('(Jo 60 ·ü~ o~ <n <n 40 CU
1 ,... 1
U) 20 Cl:
0
CTR
Figure 2
+ +
+ + +
+ +
+ +
AICAR MET TRO 1400W
CYTO/LPS
161
MET
TRO
1400W

A 3T3~L 1 ADIPOCYTES
CTA - AICAR MET TAO
CYTO/LPS
8
(il 120 Cl.> :::J ~ 100 >
Cf) c.. 80 -l 0 >. 60 ()
'0 ~ 4 0
~ 20 ~ z
0 CTA * AICAA MET TAO
CYTO/LPS
c
CTR - AICAA MET TAO
CYTOiLPS
o
CTR - AICAA MET TRO
CYTO/LPS
Figure3
E
@ACC
F
120
~1 00 :;:, ~ 80 > :;.-z ~ 60 '0 ê 4 0
S 20 E Z
0
G
iNOS
H
iNOS
GAPDH
MACROPHAGES
CTR - AICAR MET CTR - 10 20 40 100 CYTO/LPS IFNI' + TRO (~M)
~----------~ 120~--------------~
CTA - AICAR MET
IFN~y
CTR - AICAA MET
IFN~y
100
80
60
40
20
o CTR 0 10 20 40 100
IFNy + TRO ÜJM)
CTR 0 10 20 40 100
IFNy + TRO (lJM)
CTR - AICAR MET CTR 0 10 20 40 100
IFNy + TRO (~M) IFNy
162
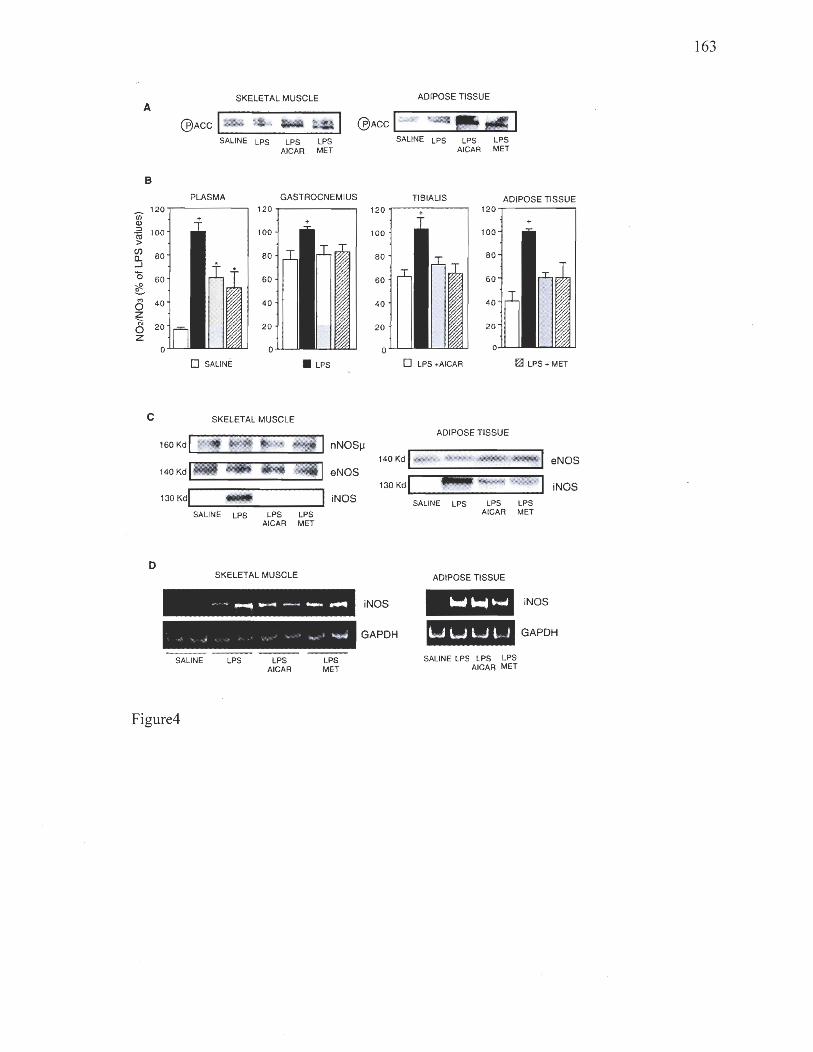
SKELET AL MUSCLE A
@ACC ' $$.$# rIe ,,; .. 1.1
B
(j) 120
Cl>
~ 100 >
~ 80 -l
60
8 40 Z ""N 20 o Z
o
c
140 Kd
SALINE LPS LPS LPS AICAR MET
PLASMA GASTROCNEMIUS 120..,.-------...,
100
80
60
40
20
o o SALlNÉ • LPS
SKELETAL MUSCLE
eNOS
130Kdl _ iNOS
SAUNE LPS LPS LPS
o
SALINE
Figure4
AICAR MET
SKELETAL MUSCLE
LPS LPS AICAR
LPS MET
163
ADIPOSE TISSUE
SALINE LPS LPS LPS AICAR MET
TISIALIS ADIPOSE TISSUE 1 20~-----"'" 120 -r-------,
+
100 100
80 BO
60 60
40 40
20 20
o o
o LPS +AICAR f2l LPS + MET
ADIPOSE TISSUE
eNOS
îNOS
SALINE LPS LPS LPS AICAR MET
ADIPOSE TISSUE
iNOS iNOS
GAPDH Wwuw GAPDH
SALINE LPS lPS LPS AICAR MET
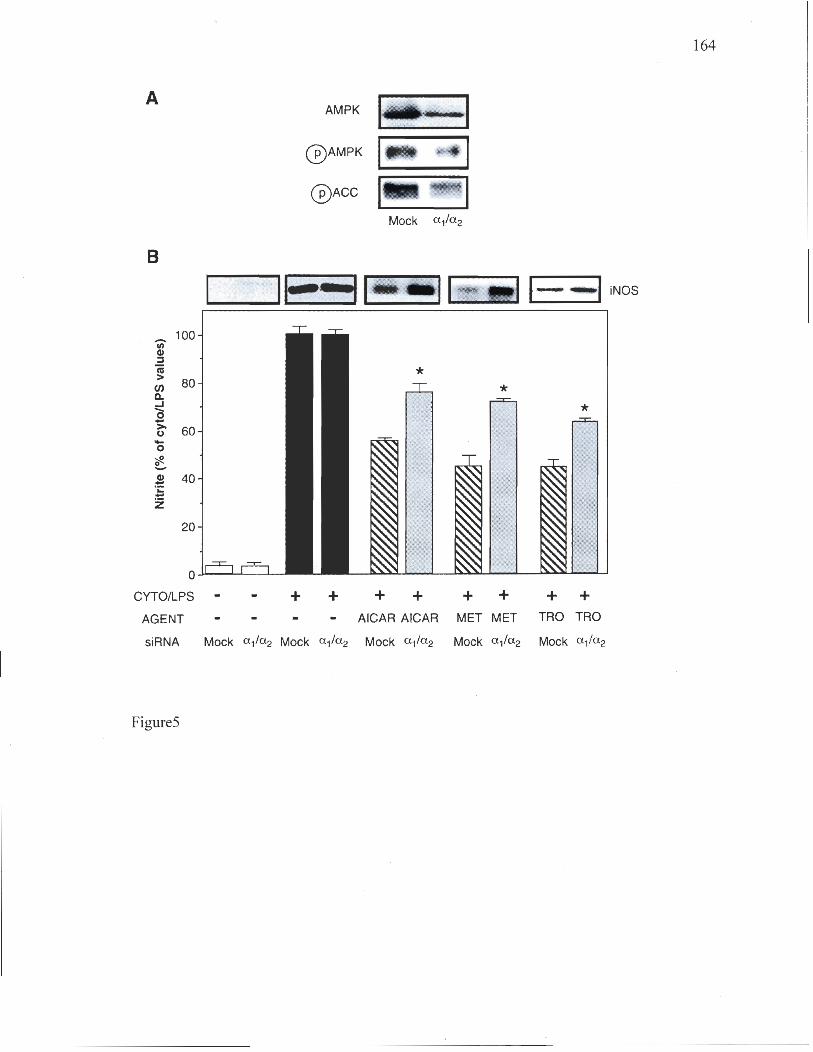
A
B
fi) 100
CI) ::l ëii > 80 (J) a. ...J ....... 0 '> 60 u ..... 0
~ CI) 40 -' '::: -Z
20
o ~-'--'---"'---CYTO/LPS
AGENT
siRNA
FigureS
164
AMPK
1--1 iNOS
+ + + + + + + + AICAR AICAR MET MET TRO TRO

Conclusion
Il est maintenant admis que l' obésité est caractérisée par un désordre inflammatoire associé
à la présence de cytokines telles que l' interleukine-6 (IL-6), le TNFu, et l' interferon-y.
L' implication de ces facteurs inflammatoires dans la résistance à l' insuline se précise de
plus en plus. La compréhension des mécanismes qui sous-tendent cette inflammation
chronique permettra de développer de nouvelles avenues afin de combattre ce dérangement
métabolique de plus en plus répandu.
Des études de notre laboratoire ont permis d' identifier l'enzyme iNOS comme cible
majeure à déchiffrer et à contrôler afin de combattre la résistance à l' insuline associée à
l' obésité. Les travaux présentés dans le présent ouvrage auront aidé à identifier le rôle de
iNOS dans la résistance à l'insuline selon les tissus, à préciser les mécanismes par lesquels
iNOS provoque ses effets délétères, à déterminer les implications physiologiques de
l' enzyme dans un modèle expérimental (soit l'incubation de muscles) et, enfin, à proposer
une nouvelle avenue pour limiter l'induction de iNOS soit l'activation de l'AMPK.
La production de monoxyde d'azote par les adipocytes : un rôle dans la pathogenèse de la résistance à l'insuline?
Notre première étude avait pour objectif de faire la revue comparative des effets du NO sur
le muscle et les tissus adipeux. Nous avions préalablement observé que l'induction d' iNOS
dans un modèle cellulaire de muscle (L6) causait la résistance à l'insuline au niveau du
transport du glucose. Les conséquences de l'induction de l'enzyme sur la sensibilité à
l' insuline dans les adipocytes restaient à déterminer. Nous avons donc utilisé un modèle
d' adipocytes blancs (3T3-Ll) et bruns (T37i) dans lesquels iNOS fut induit par un mélange
de cytokines (TNFu et IFNy) accompagné de LPS. Le rôle d'iNOS dans les effets observés
fut déterminé par l'utilisation d'un inhibiteur spécifique: le 1400W. Nous fûmes surpris de
constater que dans les adipocytes blancs et bruns en culture, contrairement à ce qui avait

166
préalablement été observé dans le modèle musculaire, que la résistance à l ' insuline induite
par la présence de cytokines et de LPS, n' impliquait pas iNOS puisque son inhibition par
le 1400W ne prévenait pas le défaut de transport du glucose stimulé par l ' insuline.
Pour tenter d' expliquer le rôle de iNOS dans la résistance à l ' insuline au niveau du muscle
il faut se questionner sur les avantages que procure ce défaut. Il faut d ' abord considérer
qu' iNOS fait partie de l' arsenal des défenses contre les infections. En pareils cas, on peut
penser que l ' organisme a évolué de façon à conserver l' énergie disponible en circulation
pour le cerveau et pour les cellules immuniatires qui, elles, n ' ont pas besoin d' insuline pour
utiliser le glucose. Il fut d ' ailleurs démontré qu'en situations pro-inflammatoires, il y a une
hausse de la gluconéogenèse, de la glycogénolyse et une augmentation de l 'utilisation du
glucose par les cellules immunitaires [478] [479]. On peut aussi observer dans ces
circonstances une augmentation des acides gras en circulation, ce qui contribue à
développer la résistance à l' insuline et diminue la capacité de l'hormone à inhiber la
production de glucose hépatique [480]. Comme le tissu musculaire consomme la majeure
partie du glucose en èirculation, il est concevable que l ' organisme développe cette
résistance à l' insuline plus fortement dans ce tissu pour utiliser l' énergie à meilleur escient
au moment de semblables situations pathologiques. De plus, il est maintenant établi que
l ' infiltration de macrophages au niveau du tissu adipeux peut contribuer à la résistance à
l ' insuline causée par l ' obésité. En effet, certains signaux tels que l' expression de monocyte
chemottractant protein-l (MCP-1) et de son récepteur C-C motif chemokin receptor-2
(CCR2), seraient impliqués dans l ' appel des macrophages aux tissus adipeux. Le groupe de
Martin Obin a d' ailleurs démontré qu'il y avait trente fois plus de mort cellulaire
d ' adipocytes chez les souris obèses db/db et chez les humains obèses et que cette nécrose
du tissu adipeux, occasionnait l ' appel des macrophages dans ce tissu. On peut donc émettre
l 'hypothèse que les adipocytes trop nombreux et trop gros sont moins efficacement irrigués
causant ainsi leur nécrose. L'induction de iNOS aux tissus adipeux pourrait alors présenter
un avantage puisque favorisant l'irrigation et l'angiogénèse du tissu, le NO contriburait à
une meilleure qualité du tissu et donc à une infiltration réduite de macrophages [481] [140].
Cela dit, nous avons tout de même observé - comme nous l'avons rapporté -une
résistance à l' insuline au tissu adipeux qui ne fait pas intervenir iNOS. Une étude

167
subséquente pourrait identifier la ou les causes de ce défaut. Il est probable que la présence
du TNFa en soit la cause. En eff~t, l' implication de cette cytokine fut mise en cause dans la
résistance à l' insuline dans les adipocytes bruns et blancs [482] [220].
Cette dichotomie dans la relation iNOS/insuline entre la cellule musculaire et celle du tissu
adipeux a déjà été observée dans notre laboratoire. Nous avons en effet démontré que
l'insuline avait la capacité d' inhiber l' induction d' iNOS dans les cellules de muscles L6 en
culture alors que le phénomène inverse était observé dans les adipocytes 3T3-L1 ([164] et
données non publiées de Penfornis et Marette). Il est alors concevable qu'au muscle
l' insuline tente de réprimer iNOS pour limiter la résistance à l' insuline induite par l' enzyme
alors qu'au tissu adipeux cette régulation n'est pas nécessaire puisque l'enzyme n'est pas
dans ce cas délétère. En fait, non seulement iNOS ne semble pas néfaste au point de vue de
la sensibilité à l'insuline au tissu adipeux mais on peut imaginer que sa présence pourrait
avoir des aspects bénéfiques. En effet, une étude de notre laboratoire a démontré que
l'induction d' iNOS inhibait la lipolyse, induite en présence de cytokines dans des adipocytes
en culture. En fait, la présence de cytokines et de LPS induisait la lipolyse mais la présence
d'un inhibiteur de iNOS augmentait cet effet suggérant un rôle de iNOS sur le processus
[483]. On peut donc en déduire que iNOS limitera la prod,uction d'acides gras libérés par le
tissus adipeux limitant ainsi la résistance à l'insuline causée par leur présence dans la
circulation. Il faut aussi dire qu'alors que l'activation de l'AMPK dans les cellules
musculaires a un effet positif sur le transport du glucose, cette même voie cause la
résistance à l'insuline dans les adipocytes [484] [485]. Les raisons de cette différence
marquée , demeurent nébuleuses pour le moment mais elles mettent tout de même en
lumière, une fois de plus, les différences complexes qui opposent le muscle au tissu
adipeux par rapport à la sensibilité à l' insuline.
Finalement, nous proposons dans cet article un modèle selon lequel les adipocytes qui
infiltrent le muscle expriment iNOS et y causent la résistance à l'insuline. En effet, des
études de notre laboratoire ont démontré qu' iNOS était clairement induit dans le tissu
adipeux de rats obèses sous un régime riche en gras [432]. Certains ont émis l'hypothèse
que lorsque le tissu adipeux prend de l'expansion, les adipocytes peuvent devenir

168
hypoxiques et sécréter des cytokines inflammatoires afin de favoriser l ' apport en sang
[486].
Le potentiel vasodilatateur et les propriétés d' angiogénèse du NO pourraient donc justifier
sa présence dans les adipocytes d' individus obèses mais, en contrepartie, créer la résistance
à l' insuline au muscle. Dans un même ordre d' idée, une étude, récente de notre laboratoire a
proposé un rôle indirect du NO produit par iNOS du tissu adipeux dans la résistance à
l'insuline du muscle. En effet, nous avons observé que la présence de NO causait
l' inhibition des pp ARy de cellules adipeuses diminuant ainsi l' adiponectine de haut poids
moléculaire en circulation [487]. Une diminution de cette forme particulière aura des
conséquences néfastes sur la sensibilité à l' insuline du muscle et particulièrement du foie.
Cette étude met une fois· de plus en lumière la complexité 'des effets du NO sur les
différents tissus. Des études plus approfondies seront nécessaires avant de développer des
outils thérapeutiques afin de mieux comprendre la relation entre les tissus individuellement
et cette molécule gazeuse.
Le peroxynitrite cause la résistance à l'insuline induite en présence d'iNOS par la nitration de IRS-l dans le muscle squelettique
Différentes études réalisées dan's notre laboratoire nous ont permIS de conclure à
l' implication d' iNOS dans la résistance à l'insuline dans le muscle squelettique. Le
mécanisme impliqué dans ce défaut induit par cette forte production de NO n 'avait
cependant pas encore été élucidé. Nous avons cherché à le faire. Nous avons trouvé que la
formation de peroxynitrite à partir du NO généré par iNOS est responsable de l ' inhibition
de l'action de l'insuline sur le transport du glucose. Dans cette seconde étude, nous avons
incidemment observé qu'une diminution de l'activité PI 3-kinase par la nitration de IRS-l
est la cible du ONOO-, un puissant oxydant dérivé du NO.
Il est admis qu'une surproduction de NO peut causer des dommages tissulaires par sa
réaction rapide avec le O2- en produisant du ONOO-. Cependant, quoique le ONOO- puisse
réagir avec des résidus tyrosines pour former des nitrotyrosines, la spécificité de cette
réaction au ONOO- est de plus en plus remise en question. Certains auteurs ont proposé que

169
le NO, le HN02 ainsi que le N02, en présence ou non de myeloperoxidase, pouvaient aussi
causer la nitration sur les résidus tyrosine [488] [489]. Afin de démontrer que le ONOO
pouvait bel et bien occasionner les effets observés par l' induction d' iNOS, nous avons
traité nos cellules directement avec l' oxydant. Observant les mêmes désordres
métaboliques selon un traitement aux cytokines et à la LPS ou encore au ONOO-, nous
avons conclu à l ' implication du ONOO- dans le processus.
Pour prouver hors de tout doute qu' il s ' agit bien du ONOO- généré en présence d' iNOS
qui cause la résistance à l' insuline, et que nous ne sommes pas en présence d'une
coïncidence voulant que le ONOO- et iNOS causent la résistance à l ' insuline par des
mécanismes similaires, il faudrait comprendre les mécanismes par lesquels le ONOO- est
formé en pareille circonstance. En effet, quoi qu' il soit bien entendu que le NO soit formé
par iNOS, la provenance du O2- n' a pas été strictement identifiée dans notre étude. On sait
cependant que les sources de O2- sont nombreuses dans une cellule: la NADPH oxydase, la
xanthine oxydase, la fumarate réductase, la succinate deshydrogénase ainsi que l' aspartate
oxidase [490] [491]. De plus, certaines de ces enzymes sont activées par le TNFa ou
l' IFNy, les deux cytokines employées dans notre étude [492] [493]. Une autre source de O2-
peut provenir de l'enzyme iNOS elle-même. En effet, en présence d'une faible
concentration de son substrat, la L-arginine, comme il fut rapporté lors de situations
inflammatoires, le O2- peut être généré par iNOS [494]. Dans cette optique, il est d ' ailleurs
important de mentionner que l'arginase II est aussi induite par certaines cytokines [495].
Ainsi, en situation inflammatoire, il est concevable que l'action des arginases contribuent à
la production de ONOO- pour combattre plus efficacement les pathogènes en diminuant les
niveaux d'arginine forçant ainsi iNOS à produire plus de O2-. Sans avoir identifié la source
de O2-, nous imaginons néanmoins qu'il s'agit d'un processus faisant intervenir l' action des
cytokines puisque des résultats préliminaires à l'aide d'un donneur de NO, le NOC-15, ne
nous a pas permis de recréer le défaut d'activation de la PI 3-kinase dans nos cellules de
muscle en culture.

170
Toujours dans l'optique de prouver l'implication du ONOO-, nous avons utilisé un
neutralisateur de l'oxydant provenant du thé: l' epigallocathéchin gallate (EGCG). Ces
expériences ont confirmé que la neutralisation du ONOO-. pouvait renverser la résistance à
l' insuline induite par le ONOO- et par les cytokines. Quoi qu' il fût rapporté que ce puissant
antioxydant puisse inhiber iNOS directement, nous n ' avons pas observé de réduction de
nitrite aux concentrations utilisées [496].
Une autre avenue intéressante, serait d' investiguer si, en plus de la nitration sur IRS-l , le
ONOO- ne causerait pas la résistance à l' insuline en activant certaines voies de
signalisation. En effet, des études ont démontré que le ONOO- pouvait activer la voie de
JNK et la voie de NF-KB, deux voies capables de causer la résistance à l' insuline [182]
[187]. Il fût aussi rapporté que la présence de ONOO- dans des cellules endothéliales
provoquaient l' activation de LKB 1 [497]. Quoi que cette kinase soit connue pour activer
l'AMPK ce qui provoque généralement des effets positifs sur la sensibilité à l' insuline, il
fût aussi observé que des souris invalidées pour le gène de LKB 1 spécifiquement aux
muscles étaient plus sensibles à l ' insuline [498]. Les souris n'exprimant pas LKBl au
muscle ont une plus grande activation de la voie AKT en réponse à Pinsuline puisque
l' expression de la protéine homologue à D. melanogaster tribbles (TRB3) est alors réduite.
En effet, TRB3 serait un facteur régulant négativement l' activation de l' AKT en réduisant
son expression. On peut donc concevoir qu'une suractivation de LKB 1 par la présence de
ONOO- puisse éventuellement causer la résistance à l'insuline via ce mécanisme.
Finalement, il faut être prudent avec l' interprétation de nitrotyrosine puisque dans certains
cas cette modification peut mimer la tyrosine phosphorylation [451]. En effet, la
nitrotyrosination est de plus en pl~lS perçue comme un mécanisme de signalisation
cellulaire permettant une perte ou parfois un gain de fonction [499]. Il est peu probable
cependant que ce fut le cas dans notre étude puisque la nitration sur les résidus tyrosine de
IRS-l est accompagnée d'une réduction de l' activité PI 3-kinase associée à IRS-l.
Malgré les conclusions de cette étude pointant le lien entre le ONOO- et la résistance à
l'insuline au muscle, il faut demeurer prudent avant de conclure que la neutralisation de cet
oxidant pourrait être la solution afin d'enrayer la résistance à l'insuline. En effet, la nature

171
évolue habituellement de façon intelligente de manière à protéger les organismes vivants.
Ainsi, il est concevable que la production de ONOO- ait sa raison d'être. En , effet, il fut
proposé que la présence de ONOO- causait l'inactivation rapide et irréversible des tyrosines
phosphatases PTP-1B, CD45 et LAR [466]. Comme les tyrosines phosphatases peuvent
bloquer les signaux envoyés par l'insuline, il est concevable que la résistance à l'insuline
puisse être amoindrie par la présence de ONOO- [3] [64]. Finalement, puisque certaines
tumeurs hormone-dépendantes sont associées à l'obésité, il est concevable que la présence
de ONOO- puisse avoir des effets protecteurs contre cet autre effet secondaire relié à une
surcharge pondérale en induisant l'apoptose de cellules tumorales.
L'incubation à long terme d'epitrochlearis cause l'induction d'iNOS un nouveau modèle de résistance à l'insuline
L' incubation de muscle in vitro peut s'avérer incontournable' lorsque l' on veut éliminer les
facteurs endogènes qui pourraient influencer notre interprétation de résultats. Cependant,
plusieurs auteurs ont mis en lumière certains déséquilibres au niveau du transport du
glucose basal et stimulé par l ' insuline dans de telles préparations [500] [501].
Comme nous l'avons mentionné à plusieurs reprises, nous avions observé que l' induction
d' iNOS pouvait être associée à des changements de transport du glucose au niveau du
muscle tant en situation basale qu'en présence d' insuline. Nous avons donc examiné si
l' induction d' iNOS pouvait expliquer les changements métaboliques observés lors de
l' incubation de muscles.
Nous avons observé que l' incubation d'epitrochlearis causait l' induction d' iNOS assez
rapidement lors de l' incubation. Plus précisément, nous n'observions pas d'iNOS dans les
muscles directement congelés après leur excision, mais dès que les muscles furent
préincubés 30 minutes pour être stabilisés, nous pouvions voir une très nette induction. Le
muscle était par la suite changé de milieu d' incubation toutes les deux heures. Nous fûmes
surpris de voir qu' après une heure d ' incubation, la présence d ' iNOS observée lors de
l' étape de stabilisation de 30 minutes n 'était plus apparente mais ne réapparaissait qu'après

172
deux heures pour atteindre son plus haut niveau après sept heures d' incubation. Comme
iNOS n'est pas présent normalement au muscle en absence de pathologie, il serait
surprenant que cette induction rapide en 30 minutes provienne effectivement du muscle. Il
est plutôt probable que la détection de l' enzyme soit plutôt due à la présence de
macrophages dans le muscle excisé. En effet, les macrophages répondent beaucoup plus
facilement à des stimuli inflammatoires. Cependant, au cours des lavages occasionnés par
les changements de milieu, on peut penser que la quantité de macrophages fut réduite.
Ainsi, lors de l' atteinte de la plus forte expression d' iNOS après sept heures, il y a fort à
parier que peu de macrophages étaient alors impliqués dans l' effet observé. Nonobstant la
provenance d' iNOS, la modification du transport du glucose basal et de la sensibilité à
l' insuline demeure et' elle doit être prise en considération lors d' études faisant intervenir
l' utilisation de muscles incubés.
Nous observons en effet que le transport du glucose basal est augmenté par la présence de
fortes concentrations de NO. L'association entre le NO et l' augmentation du transport du
glucose basal a déjà été rapportée dans divers modèles cellulaires [110] [109]. Cependant,
les mécanismes qui soutendent cette interaction n'ont jamais été précisément démontrés.
On sait par contre que la section non traduite de GLUT1 contient des éléments de réponse à
l'activator protein-l (AP-1 ) et au serum responsive element (SRE) [113]. Ces facteurs de
transcription pouvant être activés en présence d' oxydant, on peut imaginer que la forte
concentration de NO permettra leur activation et par conséquent l' induction de GLUT1.
Ces facteurs sont cependant reconnus pour être dépendants de l'activation de JNK. Si c'est
effectivement le cas dans la situation présente, il serait intéressant de savoir si l' activation
de cette voie contribue aussi, en partie, à l'état de résistance à l'insuline observée dans le
présent modèle à l' étude. Finalement, l' entrée massive de glucose occasionnée par
l' augmentation de l'expression de GLUT1 pourrait aussi avoir un rôle à jouer dans la
résistance à l'insuline. En effet, il fut proposé que les souris surexprimant GLUT1 au
muscle devenaient résistantes à l'insuline en raison de la modification de protéines
occasionnée par la voie de l'hexoxamine. En effet l'ajout de O-GlcNAC à certaines
protéines pouvait conduire à la résistance à l'insuline [502].

173
Dans le cas présent, le seul fait d' ajouter un inhibiteur spécifique d' iNOS rétablit la
sensibilité à l' insuline, ce qui laisse croire qu' il est à l' origine de tous les défauts
subséquents contribuant à une réduction de la sensibilité à l' insuline.
D'un autre côté, nous avons aussi observé que l' induction d' iNOS s' accompagnait d'une
réduction de l' activité des cNOS expliquée par une diminution de leur expression. Les
interrelations entre les trois enzymes NOS ont été abondamment rapportées, tantôt
démontrant que les enzymes répondaient aux mêmes stimuli, tantôt indiquant plutôt
qu' iNOS causait la réduction de ses deux consœurs [503] [357] [504]. La raison d'être de
ces différentes régulations s'explique difficilement puisqu'elle pourrait varier à mon avis
selon les tissus étudiés, la nature des cytokines présentes, la force d' induction de iNOS et la
nature des ROS présents dans le système. Il est donc probable que les niveaux
d'antioxydants et la présence de O2- aux différents tissus selon les différentes situations
pourraient expliquer la présence ou non de l' inhibition des cNOS par iNOS.
L'activation de l'AMPK : un nouvel outil pour réduire l'inflammation
Dans notre dernier article, nous avons démontré que iNOS est inhibé de façon post
transcriptionnelle par. les activateurs d'AMPK suivants : le AICAR, la metformine et les
agonistes de pp ARr : la troglitazone et la 15dPGJ2• Par la même occasion, nous avons
démontré que l' inhibition par iNOS de la PI 3-kinase était rétablie lorsque iNOS était
inhi bée en présence d' activateurs d' AMPK.
Il est déj à clairement établi que l' activation de l' AMPK présente des vertus non seulement
prometteuses pour la mise au point de futurs médicaments contre la résistance à l' insuline
mais 1; AMPK serait aussi responsable des propriétés antidiabétiques des médicaments les
plus utilisés présentement pour contrer la rési~tance à l' insuline, soit les TZDs et la
metformine.

174
Alors que la metformine active l' AMPK par un mécanisme dépendant de LKB-1 , la
relation entre les agonistes de PPARy et l'AMPK est plus compliquée à établir [505]. En
effet, il est . difficilement concevable que l' activation de récepteurs nucléaires régulant
normalement l' expression de gènes puisse causer une activation rapide de l' AMPK située
en grande partie dans ·le cytosol. Il serait sans doute fort utile de comprendre comment les
agonistes de pp ARy permettent l' activation de l ' AMPK afin de mettre au point d' autres
activateurs d ' AMPK.
Il serait surprenant, mais non impossible cependant, que l' activation des PPARy ne fasse
pas intervenir ces facteurs nucléaires. En effet, il fut proposé que la troglitazone occasionne
des changements au niveau du potentiel membranaire mitochondrial causant ainsi
l' activation de l'AMPK par les TZD [506]. Cependant, puisque tant les ligands
synthétiques TZD que le ligand naturel 15dPGJ2 activent l'AMPK, il serait surprenant que
ces ligands ne soient pas impliqués d'une façon ou d'une autre. Bien qu' il soit établi que les
pp ARy soient des récepteurs nucléaires, il est tout de même concevable que certains de ces
récepteurs puissent se retrouver dans le cytosol ou même à la membrane cellulaire. Ainsi
positionnés, ils pourraient permettre l' activation de voie de signalisation conduisant à
l' activation de l'AMPK plus directement.
Une autre piste de réponse envisageable pourrait se trouver ~u niveau des corépresseurs
situés sur les pp ARy non liés à un agoniste. En effet, Evans et ses collaborateurs ont
élaboré une hypothèse ouvrant à de plus grands horizons la conception de ces récepteurs
nucléaires. Dans le modèle proposé, la liaison d'un ligand au pp AR8 permet la libération
de corépresseurs inhibant certains gènes inflammatoires. Conséquemment, la présence de
pp AR8 non lié à un agoniste agit de façon pro-inflammatoire puisqu' ainsi ils agissent
comme agents neutralisants de corépresseurs qui, eux, sont anti-inflammatoires à l'état
libre. Sur la foi de cette hypothèse, il n'est pas impossible que la liaison de ligands des
pp ARy permette la libération de cofacteurs pouvant, eux, causer l' activation de l' AMPK.
Ainsi, l' interaction de ces facteurs libérés avec l' AMPK pourrait la rendre plus attrayante
pour l'une de ses kinases. Ou encore, ces facteurs pourraient interagir avec une kinase de
l'AMPK. Il est donc permis de croire que certains facteurs permettent le pont entre

175
l'AMPK et sa kinase rendant ainsi possible son activation. Ainsi donc, il ne faudrait pas
avoir une conception trop étroite de ces récepteurs PPAR et les déclarer d' emblée
incapables d' activer une kinase cytosolique.
Cette conception pourrait d' ailleurs expliquer pourquoi les souris hétérozygotes PPARy
ont, contre toute attente, une sensibilité à l' insuline améliorée [507]. En effet il est
surprenant de penser qu'une réduction de l' expression de PPARy cause une augmentation
de la sensibilité à l' insuline alors que l'activation de ces récepteurs cause le même effet.
Cela pourrait donc s' expliquer par le fait que la liaison des agonistes PPARy libère des
facteurs profitables pour la sensibilité à l'insuline et qu' ainsi une réduction du nombre de
pp AR Y augmente le nombre de ces facteuts à l' état libre, d' autant plus si ces facteurs
mènent à l'activation de l 'AMPK.
Cette étude nous a donc permis de conclure à l' implication de l'AMPK dans les effets
inhibiteurs sur iNOS du AICAR, de la metformin et de la troglitazone. Comme nous avons
démontré qu' iNOS était impliqué dans la résistance à l'insuline associée à l' obésité, si nous
sommes en mesure de trouver l' activateur idéal de l'AMPK, nous nous approcherons de la
drogue idéale pour contrer la résistance à l' insuline. Lors d'études futures , il pourrait être
intéressant d' envisager la relation inverse entre iNOS et l' AMPK. C'est-à-dire,
d' investiguer si l ' induction de iNOS peut causer l'inhibition de l'AMPK. Quoi que peu
d' indices nous laissent actuellement croire à cette hypothèse, il est possible d' établir
certaines pistes. Par exemple, les indices' fournies par les souris iNOS -/- de l' étude de
Mylène Perreault réalisée dans notre laboratoire. Il 'fut en effet noté que ces dernières
mangeaient considérablement plus que les souris sauvages sans cependant engraisser
davantage. Une relation d' inhibition entre iNOS et l'AMPK pourrait expliquer ces résultats.
En effet, si l'absence de iNOS dans l'hypothalamus permet une activation de base de
l'AMPK plus élevée dans cet organe, une prise alimentaire augmentée devrait en découler.
De plus, en périphérie, l' absence de iNOS pourrait, là aussi, lever une inhibition sur
l'AMPK causant une augmentation de la dépense énergétique par l'augmentation de la
mitochondriogénèse et l ' expression d'UCP3, expliquant pourquoi ces souris ne prennent
relativement peu de poids malgré une prise alimentaire grandement augmentée [432] [508].

176
Alors que les PPARy occasionnèrent une véritable révolution sur le plan pharmaceutique
dans le traitement du diabète de type2, les pp AR8 suscitent désormais de plus en plus
d' intérêt. Non seulement l'activation de ces récepteurs sensibilise à l'insuline, mais ils ne
causent pas l' augmentation, mais plutôt la diminution de la masse adipeuse, contrairement
à la sollicitation des PPARy [509].
Cependant, certains auteurs ont remis en doute l' importance de ces récepteurs dans la
sensibilité à l'insuline. En effet, le groupe de Terada et Han, n'a pas observé
d'augmentation du transport du glucose dans un modèle d' épitrochlearis incubé pendant six
heures en présence d ' un agoniste de PPARb. On peut cependant deviner que ce type de
modèle expérimental n 'est pas adéquat pour étudier la sensibilité à l' insuline pour les
raisons énumérées dans l ' étude du chapitre 3 de la présente thèse [510].
Malheureusement, pour l'instant, il n 'y a pas de médicament disponible pour activer les
PPAR8 chez l' humain, mais il s' agit sans aucun doute d'une cible prometteuse.
De notre côté, des études préliminaires nous ont permis de constater que l' activation des
PPAR8 à l' aide d'un agoniste spécifique; le GW0742 activait l'AMPK dans des cellules de
muscle L6 en culture et inhibait l' induction d' iNOS par un mécanisme post-transcriptionnel
pouvant impliquer l' inhibition par sa phosphorylation du facteur de traduction eEF2. En
effet, quoi que l'expression protéique de iNOS soit réduite, l'ARNm n'est pas modifiée.
D'un autre côté, il serait aussi intéressant d' étudier si cette régulation post
transcriptionnelle pourrait être expliquée par l'induction de micro ARN (mi RNA) par
l' activation de l'AMPK. En effet, les miRNA ont récemment été découverts et ont mis en
lumière un tout nouveau mode de régulation de la traduction [511]. Ces petits ARN
agissent en répresseurs post-transcriptionnels en s' appariant à des ARN messagers
particuliers réprimant ainsi leur traduction. Il serait donc intéressant de regarder si
l'activation de l'AMPK peut bloquer la traduction de la protéine iNOS par l' induction de
miRNA particuliers.
Pour conclure, même si pour le moment, plusieurs solutions pharmacologiques s' offrent à
nous, aucune n ' est sans conséquence ni totalement efficace pour contrer la résistance à

177
l' insuline. La prévention de l'obésité par l' éducation, l ' exercice et une alimentation
équilibrée demeure à mon sens l' avenue la plus efficace - sans être la plus simple, j ' en
cqnviens - pour lutter contre cette épidémie.

Bibliographie
1. Lazarus, N.R. , B. Davis, and K.J. O'Connor, An approach to a molecular understanding of exocytotic insulin release. J Physiol (Paris), 1976. 72(6): p. 787-94.
2. Wilden, P .A., et al. , The role of insulin receptor kinase domain autophosphorylation in receptor-mediated activities. Analysis with insulin and anti-receptor antibodies. J Biol Chem, 1992.267(19): p. 13719-27.
3. Elchebly, M., et al. , Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1 B gene. Science, 1999. 283(5407): p. 1544-8.
4. Ueki, K. , T. Kondo, and C.R. Kahn, Suppressor of cytokine signaling 1 (SOCS-l) and SOCS-3 cause insulin resistance through inhibition of tyrosine phosphorylation of insulin receptor substrate proteins by discrete mechanisms. Mol Cell Biol, 2004. 24(12): p. 5434-46.
5. Ramos, F.J. , et al., Grb10 mediates insulin-stimulated degradation of the insulin receptor: a mechanism of negative regulation. Am J Physiol Endocrinol Metab, 2006.290(6): p. E1262-6.
6. Dong, H., et al. , Increased hepatic levels of the insulin receptor inhibitor, PCl /NPP 1, induce insulin resistance and glucose intolerance. Diabetes, 2005. 54(2): p.367-72.
7. Sun, X.J. , et al. , Structure of the insulin receptor substrate IRS-1 defines a unique signal transduction protein. Nature, 1991. 352(6330): p. 73-7.
8. Nishiyama, M. and J.R. Wands, Cloning and increased expression of an insulin receptor substrate-1-like gene in human hepatoceUular carcinoma. Biochem Biophys Res Commun, 1992.183(1): p. 280-5.
9. Patti, M.E., et al. , 4PS/insulin receptor substrate (IRS)-2 is the alternative substrate of the insulin receptor in IRS-1-deficient mice. J Biol Chem, 1995. 270(42): p. 24670-3.
10. Lavan, B.E., et al., A novel 160-kDa phosphotyrosine protein in insulin-treated embryonic kidney ceUs is a new member of the insulin receptor substrate family. J Biol Chem, 1997.272(34): p. 21403-7.
Il. Fantin, V.R., et al., Characterization of insulin receptor substrate 4 in human embryonic kidney 293 ceUs. J Biol Chem, 1998. 273(17): p. 10726-32.
12. Cai, D. , et al., Two new substrates in insulin signaling, IRS5/DOK4 and IRS6/DOK5. J Biol Chem, 2003. 278(28): p. 25323-30.
13. Sesti, G. , et al. , Molecular mechanism of insulin resistance in type 2 diabetes meUitus: role of the insulin receptor variant forms. Diabetes Metab Res Rev, 2001. 17(5): p. 363-73.
14. Esposito, D.L., et al. , Tyr(612) and Tyr(632) in human insulin receptor substrate-1 are important for fuU activation of insulin-stimulated phosphatidylinositol 3-kinase activity and translocation ofGLUT4 in adipose ceUs. Endocrinology, 2001. 142(7): p.2833-40.

179
15. Velloso, L.A., et al., Glucose- and insulin-induced phosphorylation of the insulin receptor and its primary substrates IRS-1 and IRS-2 in rat pancreatic islets. FEBS Lett, 1995.377(3): p. 353-7.
16. Carvalho, C.R., et al. , Effect of aging on insulin receptor, insulin receptor substrate-1, and phosphatidylinositol 3-kinase in liver and muscle of rats. Endocrinology, 1996.137(1): p. 151-9.
17. Huang, C. , et al. , DifferentiaI contribution of insulin receptor substrates 1 versus 2 to insulin signaling and glucose uptake in 16 myotubes. J Biol Chem, 2005. 280(19): p. 19426-35.
18. Bouzakri, K., et al. , Reduced activation of phosphatidylinositol-3 kinase and increased serine 636 phosphorylation of insulin receptor substrate-1 in primary culture of skeletal muscle cells from patients with type 2 diabetes. Diabetes, 2003. 52(6): p. 1319-25.
19. Harrington, L.S. , et al. , The TSCl-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J Cell Biol, 2004. 166(2): p. 213-23.
20. Cai, D. , et al. , Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat Med, 2005. 11(2): p. 183-90.
21. Horike, N. , et al. , Adipose-specifie expression, phosphorylation of Ser794 in insulin receptor substrate-1, and activation in diabetic animaIs of salt-inducible kinase-2. J Biol Chem, 2003. 278(20): p. 18440-7.
22. Horike, N., et al., Roles of several domains identified in the primary structure of salt-inducible kinase (SIK). Endocr Res, 2002. 28(4): p. 291-4.
23. Yu, C., et al., Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J Biol Chem, 2002. 277(52): p. 50230-6.
24. Jacobs, R.L., et al. , Inhibition of hepatic phosphatidylcholine synthesis by 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside (AICAr) is independent of AMP-activated prote in kinase (AMPK) activation. J Biol Chem, 2006.
25. Kim, J.A., et al., Phosphorylation of Ser24 in the pleckstrin homology domain of insulin receptor substrate-1 by Mouse Pelle-like kinase/interleukin-1 receptorassociated kinase: cross-talk bellveen inflammatory signaling and insulin signaling that may contribute to insulin resistance. J Biol Chem, 2005. 280(24): p. 23173-83.
26. Hirosumi, J., et al., A central role for JNK in obesity and insulin resistance. Nature, 2002.420(6913): p. 333-6.
27. ' Hirashima, Y. , et al. , Insulin down-regulates insulin receptor substrate-2 expression through the phosphatidylinositol 3-kinase/Akt pathway. J Endocrinol, 2003. 179(2): p.253-66.
28. Shimomura, L, et al., Decreased IRS-2 and increased SREBP-1c lead to mixed insulin resistance and sensitivity in livers of lipodystrophic and ob/ob mice. Mol CeU, 2000.6(1): p. 77-86.
29. Clarke, J.F., et al., Inhibition of the translocation ofGLUT1 and GLUT4 in 3T3-L1 cells by the phosphatidylinositol 3-kinase inhibitor, wortmannin. Biochem J, 1994. 300 ( Pt 3): p. 631-5.
30. Kotani, K., et al., Requirement for phosphoinositide 3-kinase in insulin-stimulated GLUT4 translocation in 3T3-L1 adipocytes. Biochem Biophys Res Commun, 1995. 209(1): p. 343-8.

180
31. Rahn, T., et al. , Essential role of phosphatidylinositol 3-kinase in insulin-induced activation and phosphorylation of the cGMP-inhibited cAMP phosphodiesterase in rat adipocytes. Studies using the selective inhibitor wortmannin. FEBS Lett, 1994. 350(2-3): p. 314-8.
32. Cheatham, B., et al. , Phosphatidylinositol 3-kinase activation is required for insulin stimulation of pp70 S6 kinase, DNA synthesis, and glucose transporter translocation. Mol Cell Biol, 1994. 14(7): p. 4902-11.
33. Whitman, M., et al. , Type 1 phosphatidylinositol kinase makes a novel inositol phospholipid, phosphatidylinositol-3-phosphate. Nature, 1988.332(6165): p. 644-6.
34. Carpenter, C.L., et al. , Purification and characterization of phosphoinositide 3-kinase from rat liver. J Biol Chem, 1990. 265(32): p. 19704-11.
35. Escobedo, J.A. , et al. , cDNA cloning of a novel 85 kd prote in that has SH2 domains and regulates binding of PI3-kinase to the PDGF beta-receptor. Cell, 1991 . 65(1): p.75-82.
36. Skolnik, E.Y. , et al. , Cloning of PI3 kinase-associated p85 utilizing a novel method for expression/cloning of target proteins for receptor tyrosine kinases. Cell, 1991. 65(1): p. 83-90.
37. Ruiz-Larrea, F., et al. , Characterization of the bovine brain cytosolic phosphatidylinositol 3-kinase complexe Biochem J, 1993.290 (Pt 2): p. 609-16.
38. Backer, J.M. , et al., Phosphatidylinositol 3'-kinase is activated by association with IRS-l during insulin stimulation. Embo J, 1992.11(9): p. 3469-79.
39. Miralpeix, M., et al. , Insulin stimulates tyrosine phosphorylation of multiple high molecular weight substrates in Fao hepatoma cells. Biochemistry, 1992. 31(37): p. 9031-9.
40. Yu, J., et al., Regulation of the p85/pllO phosphatidylinositol 3 '-kinase: stabilization and inhibition of the pll Oalpha catalytic subunit by the p85 regulatory subunit. Mol Cell Biol, 1998.18(3): p. 1379-87.
41. Terauchi, Y. , et al. , Increased insulin sensitivity and hypoglycaemia in mice lac king the p85 alpha subunit ofphosphoinositide 3-kinase. Nat Genet, 1999.21(2): p. 230-5.
42. Mauvais-Jarvis, F., et al., Reduced expression of the murine p85alpha subunit of phosphoinositide 3-kinase improves insu/in signaling and ameliorates diabetes. J Clin Invest, 2002. 109(1): p. 141-9.
43. Ueki, K. , et al., Positive and negative roles of p85 alpha and p85 beta regulatory subunits of phosphoinositide 3-kinase in insulin signaling. J Biol Chem, 2003. 278( 48): p. 48453-66.
44. Ueki, K. , et al. , Positive and negative regulation of phosphoinositide 3-kinasedependent signaling pathways by three different gene products of the p85alpha regulatory subunit. Mol Cell Biol, 2000. 20(21): p. 8035-46.
45. Roche, S., et al., A function for phosphatidylinositol 3-kinase beta (p85alphapllObeta) in fibroblasts during mitogenesis: requirement for insulin- and lysophosphatidic acid-mediated signal transduction. Mol Cell Biol, 1998. 18(12): p. 7119-29.
46. Hooshmand-Rad, R., et al., The PI3-kinase isoforms pl 1 o (alpha) and pllO(beta) have differential roles in P DGF- and insulin-mediated signaling. J Cell Sci, 2000. 113 Pt 2: p. 207-14.

181
47. Asano, T., et al., pllObeta is up-regulated during differentiation of 3T3-Ll ceUs and contributes to the highly insulin-responsive glucose transport activity. J Biol Chem, 2000. 275(23): p. 17671-6.
48. Jiang, T., et al. , Membrane-permeant esters of phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem, 1998.273(18): p. 11017-24.
49. Hausdorff, S.F., et al., Identification of wortmannin-sensitive targets in 3T3-Ll adipocytes. DissociationoOf insulin-stimulated glucose uptake and glut4 translocation. J Biol Chem, 1999.274(35): p. 24677-84.
50. Somwar, R., et al. , DifferentiaI effects of phosphatidylinositol 3-kinase inhibition on intraceUular signaIs regulating GLUT4 translocation and glucose transport. J Biol Chem, 2001. 276(49): p. 46079-87.
51. Stephens, L., et al. , Characterization of a phosphatidylinositol-specific phosphoinositide 3-kinase from mammalian ceUs. CUIT Biol, 1994. 4(3): p. 203-14.
52. Virbasius, J.V. , A. Guilhenne, and M.P. Czech, Mouse pl 70 is a novel phosphatidylinositol 3-kinase containing ~ C2 domain. J Biol Chem, 1996.271(23): p. 13304~7.
53. Rozycka, M., et al., cDNA cloning of a third human C2-domain-containing class II phosphoinositide 3-kinase, PI3K-C2gamma, and chromosomal assignment of this gene (PIK3C2G) to 12p12. Genomics, 1998.54(3): p. 569-74.
54. Domin, J., et al., Cloning of a human phosphoinositide 3-kinase with a C2 domain that displays reduced sensitivity to the inhibitor wortmannin. Biochem J, 1997. 326 (Pt 1): p. 139-47.
55. Arcaro, A., et al., Human phosphoinositide 3-kinase C2beta, the role of calcium and the C2 domain in enzyme activity. J Biol Chem, 1998.273(49): p. 33082-90.
56. Chaussade, C. , et al., Expression of myotubularin by an adenoviral vector demonstrates its function as a phosphatidylinositol 3-phosphate [Ptdlns(3)P} phosphatase in muscle ceU lines: involvement of Ptdlns(3)P in insulin-stimulated glucose transport. Mol Endocrinol, 2003. 17(12): p. 2448-60.
57. Brown, R.A., et al., Insulin activates the alpha isoform of class II phosphoinositide 3-kinase. J Biol Chem, 1999.274(21): p. 14529-32.
58. Urso, B., et al., The alpha-isoform of class II phosphoinositide 3-kinase is more effectively activated by insulin receptors than IGF receptors, and activation requires receptor NPEYmotifs. FEBS Lett, 1999.460(3): p. 423-6.
59. Venable, C.L., et al., Overexpression of protein-tyrosine phosphatase-l B in adipocytes inhibits insulin-stimulated phosphoinositide 3-kinase activity without altering glucose transport or Akt/Protein kinase B activation. J Biol Chem, 2000. 275(24): p. 18318-26.
60. Mahadev, K., et al., Insulin-stimulated hydrogen peroxide reversibly inhibits prote in-tyrosine phosphatase 1 b in vivo and enhances the early insulin action cascade. J Biol Chem, 2001. 276(24): p. 21938-42.
61. Clement, S., et al., The lipid phosphatase SHIP2 controls insulin sensitivity. Nature, 2001.409(6816): p. 92-7.
62. Tang, X., et al., PTEN, but not SHIP2, suppresses insulin signaling through the phosphatidylinositol 3-kinase/Akt pathway in 3T3-Ll adipocytes. J Biol Chem, 2005. 280(23):p. 22523-9.
63. Sleeman, M.W., et al., Absence of the lipid phosphatase SHIP2 confers resistance to dietary obesity. Nat Med, 2005. 11(2): p. 199-205.

182
64. Dubois, M.J., et al., The SHP-l protein tyrosine phosphatase negatively modulates glucose homeostasis. Nat Med, 2006. 12(5): p. 549-56.
65. Dhand, R., et al., PI3-kinase is a dual specificity enzyme: autoregulation by an intrinsiç prote in-serine kinase activity. Embo J, 1994. 13(3): p. 522-33.
66. Foukas, L.C., et al., Regulation of phosphoinositide 3-kinase by its intrinsic serine kinase activity in vivo. Mol Cell Biol, 2004. 24(3): p. 966-75.
67. Tanti, J.F. , et al., Insulin receptor substrate 1 is phosphorylated by the serine kinase activity ofphosphatidylinositol 3-kinase. Biochem J, 1994.304 (Pt 1): p. 17-21.
68. Czupalla, C., et al., Identification and characterization of the autophosphorylation sites of phosphoinositide 3-kinase isoforms beta and gamma. J Biol Chem, 2003. 278(13): p. 11536-45.
69. Stokoe, D. , et al. , Dual role of phosphatidylinositol-3,4,5-trisphosphate in the activation ofprotein kinase B. Science, 1997.277(5325): p. 567-70.
70. Whiteman, E.L., H. Cho, and M.J. Bimbaum, Role of Akt/protein kinase B in metabolism. Trends Endocrinol Metab, 2002. 13(10): p. 444-51.
71. Alessi, D.R., et al. , Molecular basis for the substrate specificity of protein kinase B; comparison with MAPKAP kinase-l and p 70 S6 kinase. FEBS Lett, 1996.399(3): p. 333-8.
72. Rane, M.J. , et al. , p38 Kinase-dependent MAPKAPK-2 activation functions as 3-phosphoinositide-dependent kinase-2 for Akt in human ne utroph ils. J Biol Chem, 2001.276(5): p. 3517-23.
73. Delcommenne, M., et al., Phosphoinositide-3-0H kinase-dependent regulation of glycoge.n synthase kinase 3 and prote in kinase BIAKT by the integrin-linked kinase. Proc Natl Acad Sci USA, 1998.95(19): p. 11211-6.
74. Feng, J., et al., Identification of a PKBIAkt hydrophobie motif Ser-473 kinase as DNA-dependent protein kinase. J Biol Chem, 2004.279(39): p. 41189-96.
75. Sarbassov, D.D. , et al. , Phosphorylation and regulation of AktlPKB by the rictormTOR complex. Science, 2005.307(5712): p. 1098-101.
76. Chen, R., et al., Regulation of AktlPKB activation by tyrosine phosphorylation. J Biol Chem, 2001. 276(34): p. 31858-62.
77. Li, X., et al., Autophosphorylation of Akt at threonine 72 and serine 246. A potential mechanism of regulation of Akt kinase activity. J Biol Chem, 2006. 281(19): p. 13837-43.
78. Rommel, C., et al. , Mediation of IGF-l-induced skeletal myotube hypertrophy by PI(3)KlAktlmTOR and PI(3)KlAktIGSK3 pathways. Nat Cell Biol, 2001. 3(11): p. 1009-13. '
79. Nakae, J. , et al. , The forkhead transcription factor Foxol (Fkhr) confers insulin sensitivity onto glucose-6-phosphatase expression. J Clin lnvest, 2001. 108(9): p. 1359-67.
80. Kohn, A.D., et al., Expression of a constitutively active Akt SerlThr kinase in 3T3-Ll adipocytes stimulates glucose uptake and glucose transporter 4 translocation. J Biol Chem, 1996.271(49): p. 31372-8.
81. Wang, Q., et al., GLUT4 translocation by insulin in intact muscle ceUs: detection by afast and quantitative assay. FEBS Lett, 1998.427(2): p. 193-7.
82. Sano, H., et aL, ' Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. J Biol Chem, 2003. 278(17): p. 14599-602.
----------------------- - ---- - - --- - - - ---- - -----

183
83. Zeigerer, A., M.K. McBrayer, and T.E. McGraw, Insulin stimulation of GL UT4 exocytosis, but not its inhibition of endocytosis, is dependent on RabGAP AS160. Mol Biol Cell, 2004. 15(10): p. 4406-15.
84. Aledo, J.C., F. Darakhshan, and H.S. Hundal, Rab4, but not the transferrin receptor, is colocalized with GL UT 4 in an insulin-sensitive intraceUular compartment in rat skeletal muscle. Biochem Biophys Res Commun, 1995.215(1): p.321-8.
85. Sherman, L.A., et al., Differentiai effects of insulin and exercise on Rab4 distribution in rat skeletal muscle. Endoerinology, 1996. 137(1): p. 266-73.
86. Kramer, H.F., et al., Distinct signais regulate'AS160 phosphàrylation in response to insulin, A ICAR, and contraction in mouse skeletal muscle. Diabetes, 2006. 55(7): p. 2067-76.
87. Berwick, D.C., et al. , Protein kinase B phosphorylation of PIKfyve regulates the trafficking ofGLUT4 vesicles. J Cell Sei, 2004. 117(Pt 25): p. 5985-93.
88. Min, J., et al., Synip: a novel insulin-regulated syntaxin 4-binding protein mediating GLUT4 translocation in adipocytes. Mol Cell, 1999.3(6): p. 751-60.
89. Bandyopadhyay, G., et al., Activation ofprotein kinase C (alpha, beta, and zeta) by insulin in 3T3/Ll ceUs. Transfection studies suggest a role for -PKC-zeta in glucose transport. J Biol Chem, 1997.272(4): p. 2551-8.
90. Bandyopadhyay, G., et al., Effects of transiently expressed atypical (zeta, lambda), conventional (alpha, beta) and novel (delta, epsilon) protein kinase C isoforms on insulin-stimulated translocation of epitope-tagged GLUT4 glucose transporters in rat adipocytes: specific interchangeable effects of protein kinases C-zeta and Clambda. Biochem J, 1999. 337 ( Pt 3): p. 461-70.
91. Farese, R.V., Function and dysfunction of aPKC isoforms for glucose transport in insulin-sensitive and insulin-resistant states. Am J Physiol Endocrinol Metab, 2002. 283( 1 ): p . El-Il.
92. Bandyopadhyay, G., et al., Protein kinase C-Iambda knockout in embryonic stem ceUs and adipocytes impairs insulin-stimulated glucose transport. Mol Endocrinol, 2004. 18(2): p. 373-83.
93. Kotani, K., et al., Requirement of atypical protein kinase clambda for insulin stimulation of glucose uptake but not for Akt activation in 3T3-Ll adipocytes. Mol Cell Biol, 1998. 18(12): p. 6971-82.
94. Zhou, Q.L., et al., Analysis of insulin signaUing by RNAi-based gene silencing. Biochem Soc Trans, 2004. 32(Pt 5): p. 817-21.
95. Vollenweider, P., B. Menard, and P. Nicod, Insulin resistance, defective insulin receptor substrate 2-associated phosphatidylinositol-3' kinase activation, and impaired atypical protein kinase C (zeta/lambda) activation in myotubes from obese patients with impaired glucose tolerance. Diabetes, 2002.51(4): p. 1052-9.
96. Beeson, M., et al., Activation of protein kinase C-zeta by insulin and phosphatidylinositol-3,4,5-(P04)3 is defective in muscle in type 2 diabetes and impaired glucose tolerance: amelioration by rosiglitazone and exercise. Diabetes, 2003. 52(8): p. 1926-34.
97. Kim, Y.B., et al., Insulin-stimulated protein kinase C lambda/zeta activity is reduced in skeletal muscle of humans with obesity and type 2 diabetes: reversai with weight reduction. Diabetes, 2003. 52(8): p. 1935-42.

184
98. Joost, H.G. and B. Thorens, The extended GLUT-family of sugar/polyol transport facilitators: nomenclature, sequence characteristics, and potential function of its novel members (review). Mol Membr Biol, 2001.18(4): p. 247-56.
99. Miyamoto, K., et al., Role of liver-type glucose transporter (GLUT2) in transport across the basolateral membrane in rat jejunum. FEBS Lett, 1992. 314(3): p. 466-70.
100. Yasuda, K., et al. , Expression of GLUTl and GLUT2 glucose transporter isoforms in rat islets of Langerhans and their regulation by glucose. Diabetes, 1992.41(1): p. 76-81.
101. Bady, L, et al., Evidence from glut2-null mice that glucose is a critical physiological regulator offeeding. Diabetes, 2006.55(4): p. 988-95.
102. Thorens, B., A gene knockout approach in mice to identify glucose sensors controlling glucose homeostasis. Ptlugers Arch, 2003. 445(4): p. 482-90.
103. Yano, H., et al., Tissue distribution and species difference of the brain type glucose transporter (GLUT3). Biochem Biophys Res Commun, 1991.174(2): p. 470-7.
104. Wood, LS. and P. Trayhum, Glucose transporters (GLUT and SGLT): expanded families of sugar transport proteins. Br J Nutr, 2003.89(1): p. 3-9.
105. Wertheimer, E. , et al., The ubiquitous glucose transporter GLUT-l belongs to the glucose-regulated proteinfamily of stress-inducible proteins. Proc Natl Acad Sci U S A, 1991.88(6): p. 2525-9.
106. Rhoads, D.B., et al. , Evidence for expression of the facilitated glucose transporter in rat hepatocytes. Proc Natl Acad Sci USA, 1988.85(23): p. 9042-6.
107. Mitsumoto, Y., et al. , DifferentiaI expression of the GLUTl and GLUT4 glucose transporters during differentiation of L6 muscle cells. Biochem Biophys Res Commun, 1991. 175(2): p. 652-9.
108. Coderre, L. , et al., Alteration in the expression ofGLUT-l and GLUT-4 prote in and messenger RNA levels in denervated rat muscles. Endocrinology, 1992. 131( 4): p. 1821-5.
109. Paik, J.Y., et al., Nitric oxide stimulates 18F-FDG uptake in human endothelial cells through increased hexokinase activity and GLUTl expression. J Nucl Med, 2005.46(2): p. 365-70.
110. Bedard, S., B. Marcotte, and A. Marette, Cytokines modulate glucose transport in skeletal muscle by inducing the expression of inducible nitric oxide synthase. Biochem J, 1997.325 (Pt 2): p. 487-93.
111. Loike, J.D., et al., Hypoxia induces glucose transporter expression in endothelial cells. Am J Physiol, 1992.263(2 Pt 1): p. C326-33.
112. Sone, H., B.K. Deo, and A.K. Kumagai, Enhancement of glucose transport by vascular endothelial growth factor in retinal endothelial cells. Invest Ophthalmol Vis Sci, 2000. 41(7): p. 1876-84.
113. Kozlovsky, N., et al., Transcriptional activation of the Glutl gene in response to oxidative stress in L6 myotubes. J Biol Chem, 1997.272(52): p. 33367-72.
114. Battelino, T., et al., Tumor necrosis factor-alpha alters glucose metabolism in suckling rats. J Lab Clin Med, 1999. 133(6): p. 583-9.
115. Pfafflin, A., et al., Increased glucose uptake and metabolism in mesangial cells overexpressing glucose transporter 1 increases interleukin-6 and vascular endothelial growthfactor production: role of AP-l and HIF-lalpha. Cell Physiol Biochem, 2006. 18(4-5): p. 199-210.

185
116. Bames, K., et al. , Activation of GLUTl by metabolic and osmotic stress: potential involvement of AMP-activated protein kinase (A MPK). J Cell Sei, 2002. 115(Pt Il): p.2433-42.
117. Dohm, G.L. , et al. , Role of transverse tubules in insulin stimulated muscle glucose transport. J Cell Bioehem, 1993.52(1): p. 1-7.
118. Marette, A. , et al. , Abundance, localization, and insulin-induced translocation of glucose transporters in red and white muscle. Am J Physiol, 1992. 263(2 Pt 1): p. C443-52.
119. Klip, A. , et al. , Insulin-induced translocation of glucose transporters in rat hindlimb muscles. FEBS Lett, 1987.224(1): p. 224-30.
120. Roy, D. and A. Marette, Exercise induces the translocation ofGLUT4 to transverse tubules from an intracellular pool in rat skeletal muscle. Bioehem Biophys Res Commun, 1996.223(1): p. 147-52.
121. Douen, A.G., et al. , Exercise induces recruitment of the "insulin-responsive glucose transporter". Evidence for distinct intracellular insulin- and exercise-recruitable transporter pools in skeletal muscle. J Biol Chem, 1990.265(23): p. 13427-?0.
122. Foster, L.J. , et al. , Insulin accelerates inter-endosomal GLUT4 trafflc via phosphatidylinositol 3-kinase and protein kinase B. J Biol Chem, 2001. 276(47): p. 44212-21.
123. Tsakiridis, T. , M. Vranie, and A. Klip, Disassembly of the actin network inhibits insulin-dependent stimulation of glucose transport and prevents recruitment of glucose transporters to the plasma membrane. J Biol Chem, 1994. 269(47): p. 29934-42.
124. Tong, P. , et al., Insulin-induced cortical actin remodeling promotes GLUT4 insertion at muscle cell membrane ruffles. J Clin Invest, 2001.108(3): p. 371-81.
125. Omata, W., et al. , Actin filaments play a critical role in insulin-induced exocytotic recruitment but not in endocytosis ofGLUT4 in isolated rat adipocytes. Bioehem J, 2000.346 Pt 2: p. 321-8.
126. Guilherme, A. , et al. , Perinuclear localization and insulin responsiveness ofGLUT4 requires cytoskeletal integrity in 3T3-Ll adipocytes. J Biol Chem, 2000. 275(49): p. 38151-9.
127. Kanzaki, M., et al., Insulin stimulates actin comet tails on intracellular GLUT4-containing compartments in difJerentiated 3T3Ll adipocytes. J Biol Chem, 2001. 276(52): p. 49331-6.
128. Patki, V. , et al., Insulin action on GLUT4 trafflc visualized in single 3T3-11 adipocytes by using ultra-fast microscopy. Mol Biol Cell, 2001.12(1): p. 12'9-41. ,
129. Ishiki, M., et al. , Insulin regulates the membrane arrivaI, fusion, and C-terminal unmasking of glucose transporter-4 via distinct phosphoinositides. J Biol Chem, 2005.280(31): p. 28792-802. .
130. Maffueei, T. , et al. , Insulin induces phosphatidylinositol-3-phosphate formation through TC10 activation. Embo J, 2003. 22(16): p. 4178-89.
131. ~weeney, 9., et al. , Intracellular delivery of phosphatidylinositol (3,4,5)trisphosphate causes incorporation of glucose transporter 4 into the plasma membrane of muscle and fat cells without increasing glucose uptake. J Biol Chem, 2004. 279(31): p. 32233-42~
132. Somwar, R., et al. , A dominant-negative p38 MAPK mutant and novel selective inhibitors of p38 MAPK reduce insulin-stimulated glucose uptake in 3T3-Ll

186
adipocytes without affecting GLUT4 translocation. J Biol Chem, 2002. 277(52): p. 50386-95.
133. Konrad, D., et al., The antihyperglycemic drug alpha-lipoic acid stimulates glucose uptake via both GLUT4 translocation and GLUT4 activation: potential role ofp38 mitogen-activated protein kinase in GLUT4 activation. Diabetes, 2001. 50(6): p. 1464-71.
134. ~tonescu, C.N., et al. , Reduction of insulin-stimulated glucose uptake in L6 myotubes by the protein kinase inhibitor SB203580 is independent of p38MAPK activity. Endocrinology, 2005. 146(9): p. 3773-81.
135. Shepherd, P.R. and B.B. Kahn, Glucose transporters and insulin action-implications for insulin resistance and diabetes mellitus. N Engl J Med, 1999. 341(4): p. 248-57.
136. Kim, J.K. , et al., Prevention of fat-induced ïnsulin resistance by salicylate. J Clin Invest, 2001. 108(3): p. 437-46.
137. Abel, E.D., et al., Adipose-selective targeting of the GLUT4 gene impairs insulin action in muscle and liver. Nature, 2001. 409(6821): .p. 729-33.
138. Yalow, R.S. and S.A. Berson, Dynamics of insulin secretion in early diabetes in humans. Adv Metab Disord, 1970.1: p. Suppl 1 :95+.
139. Soukas, A., et al., Leptin-specific patterns of gene expression in white adipose tissue. Genes Dev, 2000. 14(8): p. 963-80.
140. Weisberg, S.P., et al., CCR2 modulates inflammatory and metabolic effects of highfat feeding. J Clin Invest, 2006. 116(1): p. 115-24.
141. Xu, H., et al., Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest, 2003. 112( 12): p. 1821-30.
142. Hotamisligil, G.S., N.S. Shargill, and B.M. Spiegelman, Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science, 1993.259(5091): p. 87-91.
143. Sethi, J.K. and G.S. Hotamisligil, The role of TNF alpha in adipocyte metabolism. Sernin Cell Dev Biol, 1999. 10(1): p. 19-29.
144. Hotamisligil, G.S., et al., Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J Clin Invest, 1995. 95(5): p. 2409-15.
145. Kem, P.A., et al., The expression oftumor necrosisfactor in human adipose tissue. Regulation by obesity, weight loss, and relationship to lipoprotein lipase. J Clin Invest, 1995. 95(5): p. 2111-9.
146. Saghizadeh, M., et al., The expression ofTNF alpha by human muscle. Relationship to insulin resistance. lClin Invest, 1996. 97(4): p. 1111-6.
147. Xu, H., et aL,. Altered tumor necrosis factor-alpha (l'NF-alpha) processing in adipocytes and increased expression of transmembrane TNF-alpha in obesity. Diabetes, 2002.51(6): p. 1876-83.
148. Uysal, K.T., et al., Protection from obesity-induced insulin resistance in mice lacking TNF-alphafunction. Nature, 1997.389(6651): p. 610-4.
149. Rui, L., et al., Insulin/IGF-l and TNF-alpha stimulate phosphorylation of IRS-l at inhibitory Ser307 via distinct pathways. J Clin Invest, 2001.107(2): p. 181-9.
150. Ozes, O.N., et al., A phosphatidylinositol 3-kinase/Akt/mTOR pathway mediates and PTEN antagonizes tumor necrosis factor inhibition of insulin signaling through insulin receptor substrate-l. Proc Nat! Acad Sci USA, 2001.98(8): p. 4640-5.

187
151. N akamo ri , Y., et al. , Myosin motor Myolc and its receptor NEMO/IKK-gamma promote TNF-alpha-induced serine307 phosphorylation of IRS-l. J Cell Biol, 2006. 173(5): p. 665-71.
152. Hotamisligil, G.S., et al., IRS-l-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science, 1996. 271(5249): p. 665-8.
153. Stephens, J.M., J. Lee, and P.F. Pilch, Tumor necrosis factor-alpha-induced insulin resistance in 3T3-Ll adipocytes is accompanied by a loss of insulin receptor substrate-l and GLUT4 expression without a loss of insulin receptor-mediated signal transduction. J Biol Chem, 1997.272(2): p. 971-6.
154. Steinberg, G.R. , et al. , Tumor necrosis factor alpha-induced skeletal muscle insulin resistance involves suppression of AMP-kinase signaling. Cell Metab, 2006. 4(6): p. 465-74.
155. Grunfeld, C. and K.R. Feingold, The metabolic effects oftumor necrosisfactor and other cytokines. Biotherapy, 1991. 3(2): p. 143-58.
156. Ryden, M., et al., Mapping of early signaling events in tumor necrosis factor-alpha -mediated lipolysis in humanfat ceUs. J Biol Chem, 2002. 277(2): p. 1085-91.
157. Peraldi, P., et al., Tumor necrosis factor (FNF)-alpha inhibits insulin signaling through stimulation of the p55 TNF receptor and activation of sphingomyelinase. J Biol Chem, 1996.271(22): p. 13018-22.
158. Stratford, S., et al., Regulation of insulin action by ceramide: dual mechanisms linking ceramide accumulation to the inhibition of A kt/prote in kinase B. J Biol Chem, 2004. 279(35): p. 36608-15. .
159. Summers, S.A., et al., Regulation ofinsulin-stimulated glucose transporter GLUT4 translocation and Akt kinase activity by ceramide. Mol Cell Biol, 1998. 18(9): p. 5457-64.
160. Teruel, T. , R. Hemandez, and M. Lorenzo, Ceramide mediates insulin resistance by tumor necrosis factor-alpha in brown adipocytes by maintaining Akt in an inactive dephosphorylated state. Diabetes, 2001. 50(11): p. 2563-71.
161. Adams, J.M., 2nd, et al., Ceramide content is increased in skeletal muscle from obese insulin-resistant humans. Diabetes, 2004.53(1): p. 25-31.
162. Kabayama, K., et al., TNFalpha-induced insulin resistance in adipocytes as a membrane microdomain disorder: involvement of ganglioside GM3. Glycobiology, 2005. 15(1): p. 21-9.
163. Yamashita, T., et al. , Enhanced insulin sensitivity in mice lacking ganglioside GM3. Proc Natl Acad Sci USA, 2003~ 100(6): p. 3445-9.
164. Bedard, S., B. Marcotte, ,and A. Marette, Insuliy! inhibits inducible nitric oxide synthase in skeletal muscle ceUs. Diabetologia, 1998.41(12): p. 1523-7.
165. Pilon, G., P. Dallaire, and A. Marette, Inhibition of inducible nitric-oxide synthase by activators of AMP-activated protein kinase: a new mechanism of action of insulin-sensitizing drugs. J Biol Chem, 2004. 279(20): p. 20767-74.
166. Maeda, N., et al., Diet-induced insulin resistance in mice lacking adiponectin/ACRP30. Nat Med, 2002.8(7): p. 731-7.
167. Ofei, F., et al., Effects of an engineered human anti-TNF-alpha antibody (CDP571) on insulin sensitivity and glycemic control in patients with NIDDM Diabetes, 1996. 45(7): p. 881-5.

188
168. Mohamed-Ali, V. , et al., Subcutaneous adipose tissue releases interleukin-6, but not tumor necrosis factor-alpha, in vivo. J Clin Endocrinol Metab, 1997. 82(12): p. 4196-200.
169. Bastard, J.P. , et al. , Adipose tissue IL-6 content correlates 11 ith resistance to insulin activation of glucose uptake both in vivo and in vitro. J Clin Endocrinol Metab, 2002. 87(5): p. 2084-9.
170. Rotter, V. , 1. Nagaev, and U. Smith, Interleukin-6 (IL-6) induces insulin resistance in 3T3-Ll adip 0 cytes and is, like IL-8 and tumor necrosis factor-alpha, overexpressed in human fat cel/s from insulin-resistant subjects.· J Biol Chem, 2003. 278( 46): p. 45777-84.
171. Senn, J.1. , et al. , Suppressor of cytokine signaling-3 (SOCS-3), a potential mediator of interleukin-6-dependent insulin resistance in hepatocytes. J Biol Chem, 2003. 278(16): p. 13740-6.
172. Mattacks, C.A. and C.M. Pond, Interactions of noradrenalin and tumour necrosis factor alpha, interleukin 4 and interleukin 6 in the control of lipolysis from adipocytes around lymph nodes. Cytokine, 1999. 11(5): p. 334-46.
173. . Tsigos, C. , et al. , Dose-dependent effects of recombinant human interleukin-6 on glucose regulation. J Clin Endocrinol Metab, 1997.82(12): p. 4167-70.
174. Wallenius, V. , et al. , Interleukin-6-deficient mice develop mature-onset obesity. Nat Med, 2002.8(1): p. 75-9.
175. Stenlof, K. , et al. , Interleukin-6 levels in the central nervaus system are negatively correlated with fat mass in overweight/obese subjects. J Clin Endocrinol Metab, 2003.88(9): p. 4379-83.
176. Crook, M.A. , P. Tutt, and J.C. Pickup, Elevated serum sialic acid concentration in NIDDM and its relationship to blood pressure. and retinopathy. Diabetes Care, 1993.16(1): p. 57-60.
177. Pickup, J.C. , et al. , NIDDM as a disease of the innate immune system: association of acute-phase reactants and interleukin-6 with metabolic syndrome X Diabetologia, 1997.40(11): p. 1286-92.
178. Daviaud, D. , et al., TNFalpha up-regulates apelin expression in human and mouse adipose tissue. Faseb J, 2006.20(9): p. 1528-30.
179. Curat, C.A., et al. , From blood monocytes ta adipose tissue-resident macrophages: induction of diapedesis by human mature adipocytes. Diabetes, 2004. 53(5): p. 1285-92.
180. Cousin, B., et al. , A raIe for preadipocytes as macrophage-like cel/s. Faseb J, 1999. 13(2):p.305-12. .
181. Strissel, K.J. , et al. , Adipocyte de a th, adipose tissue remodeling, and obesity complications. Diabetes, 2007. 56(12): p. 2910-8.
182. Aguirre, V. , et al., The c-Jun NH(2)-terminal kinase promotes insulin resistance during association with insulin · receptor substrate-l and phosphorylation of Ser(307). J Biol Chem, 2000. 275(12): p. 9047-54.
183. Nakatani, Y., et al. , Modulation of the JNK pathway in liver affects insulin resistance status. J Biol Chem, 2004. 279(44): p. 45803-9.
184. Jaeschke, A., M.P. Czech, and R.J. Davis, An essential raIe of the JIP 1 scaffold prote in for »(K activation in adipose tissue. Genes Dev, 2004. 18(16): p. 1976-80.

189
185. Solinas, G. , et al., JNKl in hematopoieticaUy derived ceUs contributes to dietinduced inflammation and insulin resistance without ajJecting obesity. Cell Metab, 2007.6(5): p. 386-97.
186. Nguyen, M.T., et al., A Subpopulation of Macrophages Infiltrates Hypertrophic Adipose Tissue and Is Activated by Free Fatty Acids via Toll-like Receptors 2 and 4 and JNK-dependent Pathways. J Biol Chem, 2007. 282(48): p. 35279-92.
187. Yuan, M. , et al. , ReversaI of obesity- and diet-induced insulin resistance with salicylates or targeted disruption oflkkbeta. Science, 2001. 293(5535): p. 1673-7.
188. Arkan, M.C. , et al. , IKK-beta links inflammation to obesity-induced insulin resistance. Nat Med, 2005.11(2): p. 191-8.
189. Yin, M.J. , Y. Yamamoto, and R.B. Gaynor, The anti-inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase-beta. Nature, 1998. 396(6706): p. 77-80.
190. Gao, Z. , et al. , Serine phosphorylation of insulin receptor substrate 1 by inhibitor kappa B kinase complex. J Biol Chem, 2002. 277(50): p. 48115-21.
191. Kaneto, H. , et al. , Oxidative stress, ER stress, and the JNK pathway in type 2 diabetes. J Mol Med, 2005. 83(6): p. 429-39.
192. Ozawa, K. , et al., The endoplasmic reticulum cha perone improves insulin resistance in type 2 diabetes. Diabetes, 2005.54(3): p. 657-63.
193. Pyrko, P. , et al. , HIV-l protease inhibitors neljinavir and atazanavir induce malignant glioma death by triggering endoplasmic reticulum stress. Cancer Res, 2007. 67(22): p. 10920-8.
194. Hsieh, Y.H., et al., DifferentiaI endoplasmic reticulum stress signaling pathways mediated by iNOS. Biochem Biophys Res Commun, 2007. 359(3): p. 643-8.
195. Yasukawa, H. , A. Sasaki, and A. Yoshimura, Negative regulation of cytokine signaling pathways. Annu Rev Immunol, 2000. 18: p. 143-64.
196. Emanuelli, B., et al., SOCS-3 is an insulin-induced negative regulator of insulin ,signaling. J Biol Chem, 2000. 275(21): p. 15985-91.
197. Kawazoe, Y., et al., Signal transducer and activator of transcription (STAT)induced STAT inhibitor 1 (SSI-l)/suppressor of cytokine signaling 1 (SOCSl) inhibits insulin signal transduction pathway through modulating insulin receptor substrate 1 (IRS-l) phosphorylation. J Exp Med, 2001. 193(2): p. 263-9.'
198. Mooney, R.A., et al. , Suppressors ofcytokine signaling-l and -6 associate with and inhibit the insulin receptor. A potential mechanism for cytokine-mediated insulin resistance. J Biol Chem, 2001. 276(28): p. 25889-93.
199. Rui, L. , et al., SOCS-l and SOCS-3 block insulin signaling by ubiquitin-mediated degradation of IRSl and IRS2. J Biol Chem, 2002. 277(44): p. 42394-8.
200. Hotta, K., et al., Plasma concentrations of a novel, adipose-specific prote in, adiponectin, in type 2 diabetic patients. Arterioscler Thromb Vasc Biol, 2000. 20(6): p. 1595-9.
201. Spranger, J., et al., Adiponectin and protection against type 2 diabetes meUitus. Lancet, 2003. 361(9353): p. 226-8.
202. Rajala, M.W. and P.E. Scherer, Minireview: The adipocyte--at the crossroads of energy homeostasis, inflammation, and atherosclerosis. Endocrinology, 2003. 144(9): p. 3765-73.
203. Scherer, P .E., et al., A novel serum protein similar to Cl q, produced exclusively in adipocytes. J Biol Chem, 1995.270(45): p. 26746-9.

190
204. Pajvani, D.B. and P .E. Scherer, Adiponectin: systemic contributor to insulin sensitivity. CUIT Diab Rep, 2003. 3(3): p. 207-13.
205. Yamauchi~ T., et al., Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med, 2002. 8(11): p. 1288-95.
206. Patel, L., et al., Resistin is expressed in human macrophages and directly regulated by PPAR gamma activators. Biochem Biophys Res Commun, 2003. 300(2): p. 472-6.
207. Steppan, C.M., et al. , The hormone resistin links obesity to diabetes. Nature, 2001. 409(6818): p. 307-12.
208. Banerjee, R.R., et al. , Regulation of fasted blood glucose by resistin. Science, 2004. 303(5661): p. 1195-8.
209. Busso, N., et al., Leptin signaling deficiency impairs humoral and cellular immune responses and attenuates experimental arthritis. J Immunol, 2002. 168(2): p. 875-82.
210. Krebs, M., et al., Free fatty acids inhibit the glucose-stimulated increase of intramuscular glucose-6-phosphate concentration in humans. J Clin Endocrinol Metab, 2001. 86(5): p. 2153-60.
211. Randle, P .1., et al., The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet, 1963.1: p. 785-9.
212. Jucker, B.M., et al., 13C and 31P NMR studies on the effects ofincreasedplasma free fatty acids on intramuscular glucose metabolism in the awake rat. J Biol Chem, 1997. 272(16):p. 10464-73.
213. Petersen, K.F., et al., Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science, 2003. 300(5622): p. 1140-2.
214. Dresner, A., et al., Effects of free fatty acids on glucose transport and IRS-lassociated phosphatidylinositol 3-kinase activity. J Clin Invest, 1999. 103(2): p. 253-9.
215. Itani, S.I., et al., Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IkappaB-alpha. Diabetes, 2002. 51(7): p. 2005-11.
216. Griffin, M.E., et al., Free fatty acid-induced insulin resistance is associated with activation of protein kinase C theta and alterations in the insulin signaling cascade. Diabetes, 1999.48(6): p. 1270-4.
217. Dey, D., et al., Involvement of novel PKC isoforms in FFA induced defects in insulin signaling. Mol Cell Endocrinol, 2006. 246(1-2): p. 60-4.
218. Anderwald, C., et al., Effects of insulin treatment in type 2 diabetic patients on intracellular lipid content in liver and skeletal muscle. Diabetes, 2002. 51(10): p. 3025-32.
219. Vettor, R., et al., Changes in FAT/CD36, UCP2, UCP3 and GLUT4 gene expression during lipid infusion in rat skeletal and heart muscle. Int J Obes Relat Metab Disord, 2002. 26(6): p. 838-47.
220. Nguyen, M.T., et al., JNK and tumor necrosis factor-alpha mediate free fatty acidinduced insulin resistance in 3T3-Ll adipocytes. J Biol Chem, 2005. 280(42): p. 35361-71.

191
221. Lam, T.K., et al., Free fatty acid-induced hepatic insulin resistance: a potential role for protein kinase C-delta. Am J Physiol Endocrinol Metab, 2002. 283(4): p. E682-91.
222. Jiang, R. and M. Carlson, Glucose regulates protein interactions within the yeast SNFl protein kinase complexe Genes Dev, 1996.10(24): p. 3105-15.
223. Beg, Z.H., D.W. All mann , and D.M. Gibson, Modulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity with cAMP and wth prote in fractions ofrat liver cytosol. Biochem Biophys Res Commun, 1973.54(4): p. 1362-9.
224. Carlson, C.A. and K.H. Kim, Regulation of hepatic acetyl coenzyme A carboxylase by phosphorylation and dephosphorylation. J Biol Chem, 1973.248(1): p. 378-80.
225. Carling, D. , V.A. Zammit, and D.G. Hardie, A common bicyclic protein kinase cascade inactivates the regulatory enzymes of fatty acid and cholesterol biosynthesis. FEBS Lett, 1987.223(2): p. 217-22.
226. Munday, M.R. , D. Carling, and D.G. Hardie, Negative interactions between phosphorylation of acetyl-CoA carboxylase by the ·cyclic AMP-dependent and AMPactivated protein kinases. FEBS Lett, 1988.235(1-2): p. 144-8.
227. Gao, G., et al., Non-catalytic beta- and gamma-subunit isoforms of the 5'-AMPactivated protein kinase. J Biol Chem, 1996. 271(15): p. 8675-~ 1.
228. Woods, A., et al., The alphal and alpha2 isoforms of the AMP-activated protein kinase have similar activities in rat liver but exhibit differences in substrate specificity in vitro. FEBS Lett, 1996.397(2-3): p. 347-51.
229. Michell, B.J., et al., Isoform-specific purification and substrate specificity of the 5'AMP-activated prote in kinase. J Biol Chem, 1996.271(45): p. 28445-50.
230. Hurley, R.L., et al., Regulation of AMP-activated protein kinase by multisite phosphorylation in response to agents that elevate cellular cAMP. J Biol Chem, 2006. 281(48):p. 36662-72.
231. Salt, I., et al., AMP-activated protein kinase: greater AMP dependence, and preferential nuclear localization, of complexes containing the alpha2 isoform. Biochem J, 1998.334 (Pt 1): p. 177-87.
232. Ai, H., et al., Effect of fiber type and nutritional state on AICAR- and contractionstimulated glucose transport in rat muscle. Am J Physiol Endocrinol Metab, 2002. 282(6): p. EI291-300.
233. Kawaguchi, T., et al., Mechanismfor fatty acid "sparing" effect on glucose-induced transcription: regulation of carbohydrate-responsive element-binding protein by AMP-activated prote in kinase. J Biol Chem, 2002. 277(6): p. 3829-35.
234. Foretz, M., et al., Short-term overexpression of a constitutively active form of AMPactivated protein kinase in the /iver leads to mild hypoglycemia and fatty liver. Diabetes, 2005.54(5): p. 1331-9.
235. Zhou, G., et al., Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest, 2001. 108(8): p. 1167-74.
236. Jorgensen, S.B., et al., The alpha2-5'AMP-activated protein kinase is · a site 2 glycogen synthase kinase in skeletal muscle and is responsive to glucose loading. Diabetes, 2004.53(12): p. 3074-81.
237. Viollet, B., et al., The AMP-activated protein kinase alpha2 catalytic subunit controls whole-body insulin sensitivity. J Clin Invest, 2003. 111 (1): p. 91-8.

192
238. Warden, S.M., et al., Post-translational modifications of the beta-l subunit of AMPactivated protein kinase affect enzyme activity and cel/ular localization. Biochem J, 2001. 354(Pt 2): p. 275-83.
239. Jiang, R. and M. Carlson, The Snfl protein kinase and its activating subunit, Snf4, interact with distinct domains of the Sipl/Sip2/GaI83 component in the kinase complex. Mol Cell Biol, 1997.17(4): p. 2099-106.
240. Sakoda, H., et al., Glycogen debranching enzyme association with beta-subunit regulates AMP-activated protein kinase activity. Am J Physiol Endocrinol Metab, 2005.289(3): p. E474-81.
241. Scott, J.W., et al., CBS domains form energy-sensing modules whose binding of adenosine ligands is disrupted by disease mutations. J Clin Invest, 2004. 113(2): p. 274-84.
242. Durante, P .E., et al., Effects of endurance training on activity and expression of AMP-activated protein kinase isoforms in rat muscles. Am J Physiol Endocrinol Metab, 2002.283(1): p. EI78-86.
243. Milan, D., et al., A mutation in PRKAG3 associated with excess glycogen content in pig skeletal muscle. Science, 2000. 288(5469): p. 1248-51.
244. Wilson, W.A., S.A. Hawley, and D.G. Hardie, Glucose repression/derepression in budding yeast: SNFl protein kinase is activated by phosphorylation under derepressing conditions, and this correlates with a high AMP:ATP ratio. CUIT Biol, 1996.6(11): p. 1426-34.
245. Hamilton, S.R., et al., AMP-activated protein kinase kinase: detection with recombinant AMPK alphal subunit. Biochem Biophys Res Commun, 2002. 293(3): p.892-8.
246. ' Hardie, D.G., Minireview: the AMP-activated protein kinase cascade: the key sen.sor of cel/ular energy status. Endocrinology, 2003.144(12): p. 5179-83.
247. Hawley, S.A., et al., 5'-AMP activates the AMP-activated protein kinase cascade, and Ca2+/calmodulin activates the calmodulin-dependent protein kinase 1 cascade, via three independent mechanisms. J Biol Chem, 1995.270(45): p. 27186-91.
248. Hawley, S.A., et al., Complexes between the LKBl tumor suppressor, STRAD alpha/beta and M025 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J Biol, 2003.2(4): p. 28.
249. Woods, A., et al., Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cel/s. Cell Metab, 2005. 2(1): p. 21-33.
250. Hurley, R.L., et al. , The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases.' J Biol Chem, 2005. 280(32): p. 29060-6.
251. Momcilovic, M., S.P. Hong, and M. Carlson, Mammalian TAKl activates Snfl protein kinase in yeast and phosphorylates AMP-activated prote in kinase in vitro. J Biol Chem, 2006. 281(35): p. 25336-43.
252. Ceddia, R.B. and G. Sweeney, Creatine supplementation increases glucose oxidation and AMP K phosphorylation and reduces lactate production in L6 rat skeletal muscle cel/s. J Physiol, 2004. 555(Pt 2): p. 409-21.
253. Rafaeloff-Phail, R., et al., Biochemical regulation of mammalian AMP-activated protein kinase activity by NAD and NADH J Biol Chem, 2004. 279(51): p. 52934-9.

193
254. Winder, W.W. and D.G. Hardie, Inactivation of acetyl-CoA carboxylase and activation of AMP-activated protein kinase in muscle during exercise. Am J Physiol, 1996. 270(2 Pt 1): p. E299-304.
255. Hutber, C.A. , D.G. Hardie, and W.W. Winder, Electrical stimulation inactivates muscle acetyl-CoA carboxylase and increases AMP-activated protein kinase. Am J Physiol, 1997. 272(2 Pt 1): p. E262-6.
256. Vavvas, D., et al. , Contr.action-induced changes in acetyl-CoA carboxylase and 5'AMP-activated kinase in skeletal muscle. J Biol Chem, 1997.272(20): p. 13255-61.
257. Kelly, M. , et al. , AMPK activity is diminished in tissues of IL-6 knockout mice: the effect of exercise. Bioehem Biophys Res Commun, 2004. 320(2): p. 449-54.
258. Carling, D., The AMP-activated protein kinase cascade--a unifying system for energy control. Trends Bioehem Sei, 2004. 29(1): p. 18-24.
259. Hardie, D.G. , AMP-activated protein kinase: a key system mediating metabolic responses to exercise. Med Sei Sports Exere, 2004.36(1): p. 28-34.
260. Kola, B., et al. , Cannabinoids and ghrelin have both central and peripheral metabolic and cardiac effects via AMP-activated prote in kinase. J Biol Chem, 2005. 280(26): p. 25196-20l.
261. Minokoshi, Y. , et al. , AMP-kinase regulates food intake by responding to hormonal and nutrient signaIs in the hypothalamus. Nature, 2004. 428(6982): p. 569-74.
262. Kim, M.S., et al. , Anti-obesity effects of alpha-lipoic acid mediated by suppression ofhypothalamic AMP-activated protein kinase. Nat Med, 2004.10(7): p. 727-33.
263. Lee, W.J. , et al., Alpha-lipoic acid increases insulin sensitivity by activating AMPK in skeletal muscle. Bioehem Biophys Res Commun, 2005. 332(3): p. 885-91.
264. Bimbaum, M.J. , Activating AMP-activated protein kinase without AMP. Mol Cell, 2005. 19(3): p. 289-90.
265. Namkoong, C., et al. , Enhanced hypothalamic AMP-activated protein kinase activity contributes to hyperphagia in diabetic rats. Diabetes, 2005. 54(1): p. 63-8.
266. Palanivel, R. and G. Sweeney, Regulation offatty acid uptake and metabolism in L6 skeletal muscle cells by resistin. FEBS Lett, 2005. 579(22): p. 5049-54.
267. Daval, M., et al., Anti-lipolytic action of AMP-activated protein kinase in rodent adipocytes. J Biol Chem, 2005.280(26): p. 25250-7.
268. Yin, W. , J. Mu, and M.J. Bimbaum, Role of AMP-activated protein kinase in cyclic AMP-dependent lipolysis In 3T3-Ll adipocytes. J Biol Chem, 2003. 278(44): p. 43074-80.
269. Luo, B., et al., Chronic hexosamine flux stimulates fatty acid oxidation byactivating AMP-activated prote in kinase in adipocytes. J Biol Chem, 2007.
210. Kimball, S.R., B.A. Siegfried, and L.S. Jefferson, Glucagon represses signaling through the mammalian target of rapamycin in rat liver by activating AMPactivated proteinkinase. J Biol Chem, 2004. 279(52): p. 54103-9.
271. GambIe, J. and G.D. Lopasehuk, Insulin inhibition of 5' adenosine monophosphateactivated protein kinase in the heart results in activation of acetyl coenzyme A carboxylase and inhibition of fatty acid oxidation. Metabolism, 1997. 46(11): p. 1270-4.
272. Makinde, A.O., J. GambIe, and G.D. Lopasehuk, Upregulation of 5'-AMP-activated protein kinase is responsible for the increase in myocardial fatty acid oxidation rates following birth in the newborn rabbit. Circ Res, 1997. 80(4): p. 482-9.

194
273. Soltys, C.L., S. Kovacic, and J.R. Dyck, Activation of cardiac AMP-activated protein kinase by LKBl expression or chemical hypoxia is blunted by increased Akt activity. Am J Physiol Heart Circ Physiol, 2006. 290(6): p. H2472-9.
274. Corton, J.M. , et al., 5-aminoimidazole-4-carboxamide ribonucleoside. A specific methodfor activating AMP-activated protein kinase in intact ceUs? Eur J Biochem, 1995.229(2): p. 558-65.
275. Witters, L.A., The blooming of the French lilac. r Clin Invest, 2001. 108(8): p. 1105-7.
276. Fryer, L.G. , A. Parbu-Patel, and D. Carling, The Anti-diabetic drugs rosiglitazone and metformin stimulate AMP-activated protein kinase through distinct signaling pathways. J Biol Chem, 2002. 277(28): p. 25226-32.
277. El-Mir, M.Y. , et al. , Dimethylbiguanide inhibits ceU respiration via an indirect effect targeted on the respiratory chain complex l J Biol Chem, 2000. 275(1): p. 223-8.
278. Brunmair, B. , et al. , Thiazolidinediones, like metformin, inhibit respiratory comp/ex 1: a common mechanism contributing to their antidiabetic actions? Diabetes, 2004. 53(4): p. 1052~9.
279. Kramer, D.K., et al. , Direct activation of glucose transport in primary human myotubes after activation of peroxisome prolijerator-activated receptor delta. Diabetes, 2005.54(4): p. 1157-63.
280. Horman, S. , et al. , Activation of AMP-activated protein kinase leads to the phosphorylation of elongation factor 2 and an inhibition of prote in synthesis. CUIT
Biol, 2002.12(16): p. 1419-23. 281. Boister, D.R., et al. , AMP-activated protein kinase suppresses protein synthesis in
rat skeletal muscle through down-regulated mammalian target of rapamycin (mTOR) signaling. J Biol Chem, 2002. 277(27): p. 23977-80.
282. Cheng, S.W., et al., Thr2446 is a novel mammalian target of rapamycin (mTOR) phosphorylation site regulated by nutrient status. J Biol Chem, 2004. 279(16): p. 15719-22.
283. Horman, S., et al., Myocardial ischemia and increased heart work 1r!odulate the phosphorylation state of eukaryotic elongation factor-2. J Biol Chem, 2003. 278( 43): p. 4 i 970-6.
284. Wang, W., et al., AMP-activated kinase regulates cytoplasmic HuR. Mol Cell Biol, 2002.22(10): p. 3425-36.
285. Wang, W. , et al., AMP-activated protein kinase-regulated phosphorylation and acetylation of importin ' alphal: involvement in the nuclear import of RNA -binding protein HuR. J Biol Chem, 2004. 279(46): p. 48376-88.
286. Di Marco, S., et al., NF-kappa B-mediated MyoD decay during muscle wasting requires nitric oxide synthase mRNA stabilization, HuR prote in, and nitric oxide release. Mol Cell Biol, 2005. 25(15): p. 6533-45.
287. Wojtaszewski, J.F., et al., Glycogen-dependent effects of 5-aminoimidazole-4-carboxamide (AICA)-riboside on AMP-activated protein kinase and glycogen synthase activities in rat skeletal muscle. Diabetes, 2002. 51(2): p. 284-92.
288. Mu, J. , et al. , A role for AMP-activated protein kinase in contraction- and hypoxiaregulated glucose transport in skeletal muscle. Mol Cell, 2001.7(5): p. 1085-94.

195
289. Fujii, N., et al., AMP-activated Protein Kinase (alpha}2 Activity Is Not Essentialfor Contraction- and Hyperosmolarity-induced Glucose Transport in Skeletal Muscle. J Biol Chem, 2005.280(47): p. 39033-41.
290. Russell, R.R., 3rd, et al., Translocation of myocardial GLUT-4 and increased glucose uptake through activation of AMPK by AICAR. Am J Physiol, 1999.277(2 Pt 2): p. H643-9.
291. Holmes, B.F., E.J. Kurth-Kraczek, and W.W. Winder, Chrônic activation of 5'AMP-activated protein kinase increases GLUT-4J hexokinaseJ and glycogen in muscle. J Appl Physiol, 1999.87(5): p. 1990-5.
292. Holmes, B.F., et al. , Regulation of muscle GLUT4 enhancer factor and myocyte enhancer factor 2 by AMP-activated protein kinase. Am J Physiol Endocrinol Metab, 2005.289(6): p. E1071-6.
293. Somwar, R., et al., Activation of p38 mitogen-activated protein kinase alpha and beta by insulin and contraction in rat skeletal muscle: potential role in the stimulation of glucose transport. Diabetes, 2000. 49(11): p. 1794-800.
294. Sweeney, G., et al., An inhibitor of p38 mitogen-activated protein kinase prevents insulin-stimulated glucose transport but not glucose transporter translocation in 3T3-Ll adipocytes and L6 myotubes. J Biol Chem, 1999.274(15): p. 10071-8.
295. Lemieux, K., et al., The AMP-activated protein kinase activator AICAR does not induce GLUT4 translocation to transverse tubules but stimulates glucose uptake and p38 mitogen-activated protein kinases alpha and beta in skeletal muscle. Faseb J, 2003.17(12): p. 1658-65.
296. Xi, X., J. Han, and J.Z. Zhang, Stimulation of glucose transport by AMP-activated protein kinase via activation of p38 "mitogen-activated protein kinase. J Biol Chem, 2001.276(44): p. 41029-34.
297. Li, J., et al., AMP-activated protein kinase activates p38 mitogen-activated prote in kinase by increasing recruitment ofp38 MAPK to TABl in the ischemic heart. Circ Res, 2005.97(9): p. 872-9.
298. Abbud, W., et al., Stimulation of AMP-activated protein kinase (AMPK) is associated with enhancement of Glutl-mediated glucose transport. Arch Biochem Biophys, 2000. 380(2): p. 347-52.
299. Minokoshi, Y., et al., Leptin stimulates jatty-acid oxidation by activating AMPactivated protein kinase. Nature, 2002.415(6869): p. 339-43.
300. Nath, N., et al., 5-aminoimidazole-4-carboxamide ribonucleoside: a novel immunomodulator with therapeutic ejJicacy in experimental autoimmune encephalomyelitis. J lmmunol, 2005. 175(1): p. 566-74.
301. Giri, S., et al., 5-aminoimidazole-4-carboxamide-l-beta-4-ribofuranoside inhibits proinflammatory response in glial ceUs: a possible role of AMP-activated protein kinase. J Neurosci, 2004. 24(2): p. 479-87.
302. Jhun, B.S., et al., Inhibition of AMP-activated protefn kinase suppresses IL-2 expression through down-regulation of NF-AT and AP-l activation in Jurkat T ceUs. Biochem Biophys Res Commun, 2006. 351(4): p. 986-92.
303. Hattori, Y., et al., Metformin inhibits cytokine-induced nuclear factor kappaB activation via AMP-activated protein kinase activation in vascular endothelial ceUs. Hypertension, 2006.47(6): p. 1183-8.

196
304. Ropelle, E.R., et al., A central role for neuronal adenosine 5'-monophosphateactivated prote in kinase in cancer-induced anorexia. Endocrinology, 2007. 148(11): p.5220-9.
305. Hallows, K.R., et al., Up-regulation of AMP-activated kinase by dysfunctional cystic fibrosis transmembrane conductance regulator in cystic fibrosis airway epithelial cells mitigates excessive inflammation. J Biol Chem, 2006. 281(7): p. 4231-41.
306. Cheng, P.Y., et al., The involvement of AMP-activated protein kinases in the antiinjlammatory efJect of nicotine in vivo and in vitro. Biochem Pharmacol, 2007. 74(12): p. 1758-65.
307. Bergeron, R., et al., Chronic activation of AMP kinase results in NRF-1 activation and mitochondrial biogenesis. Am J Physiol Endocrinol Metab, 2001. 281(6): p. E1340-6.
308. Koo, S.H., et al., The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature, 2005. 437(7062): p. 1109-11.
309. Leclerc, 1. and G.A. Rutter, AMP-activated protein kinase: a new beta-cell glucose sensor?: Regulation by amino acids and calcium ions. Diabetes, 2004. 53 Suppl 3: p. S67-74.
310. Chen, Z.P., et al., AMP-activated pro(ein kinase phosphorylation of endothelial NO synthase. FEBS Lett, 1999.443(3): p. 285-9.
311. Mount, P.F., et al., Acute renal ischemia rapidly activates the energy sensor AMP K but does not increase phosphorylation of eNOS-Ser 1177. Am J Physiol Renal Physiol, 2005. 289(5): p. F1103-15.
312. Thors, B., H. Halldorsson, and G. Thorgeirsson, Thrombin and histamine stimulate endothelial 'nitric-oxide synthase phosphorylation at Ser 1177 via an AMP K mediated pathway independent of P I3K-Akt. FEBS Lett, 2004. 573(1-3): p. 175-80.
313. Murad, F., Discovery of some of the biological efJects of nitric oxide and its role in cell signaling. Biosci Rep, 2004. 24(4-5): p. 452-74.
314. Bian, K. and F. Murad, Nitric oxide (NO)--biogeneration, regulation, and relevance to human diseases. Front Biosci, 2003. 8: p. d264-78.
315. Griffith, t.M., et al., The nature of endothelium-derived vascular relaxant factor. Nature, 1984.308(5960): p. 645-7.
316. Bredt, D.S. and S.H. Snyder, Isolation of nitric oxide , synthetase, a calmodulinrequiring enzyme. Proc Natl Acad Sci USA, 1990. 87(2): p. 682-5.
317. Hevel, J.M., K.A. White, and M.A. MarIetta, Purification of the inducible murine macrophage nitric oxide synthase. Identification as a jlavoprotein. J Biol Chem, 1991.266(34): p. 22789-91.
318. Forstermann, U., et al., Isoforms of nitric oxide synthase. Characterization and purification from difJerent cell types. Biochem Pharmacol, 1991. 42(10): p. 1849-57.
319. Ignarro, L.J., Nitric oxide. A novel signal transduction mechanismfor transcellular communication. Hypertension, 1990. 16(5): p. 477-83.
320. Moncada, S. and E.A. Higgs, Endogenous nitricoxide: physiology, pathology and ' clinical relevance. Eur J Clin Invest, 1991.21(4): p. 361-74.
321. McMillan, K. and B.S. Masters, Prokaryotic expression of the heme- and jlavinbinding domains of rat neuronal nitric oxide synthase as distinct polypeptides:

197
identification of the heme-binding proximal thiolate ligand as cysteine-415. Biochemistry, 1995.34(11): p. 3686-93.
322. Richards, M.K. and M.A. MarIetta, Characterization of neuronal nitric oxide synthase and a C415H mutant, purifiedfrom a baculovirus overexpression system. Biochemistry, 1994. 33(49): p. 14723-32.
323. Ghosh, D.K., H.M. Abu-Soud, and D.J. Stuehr, Reconstitution of the second step in NO synthesis using the isolated oxygenase and reductase domains of macrophage NO synthase. Biochemistry, 1995.34(36): p. 11316-20.
324. Klatt, P., et al. , Structural analysis of porcine brain nitric oxide synthase reveals a role for tetrG:hydrobiopterin and L-arginine in the formation of an SDS-resistant dimer. Embo J, 1995.14(15): p. 3687-95.
325. Alderton, W.K., C.E. Cooper, and R.G. Knowles, Nitric oxide synthases: structure, function and inhibition. Biochem J, 2001. 357(Pt 3): p. 593-615.
326. MarIetta, M.A., Nitric oxide synthase structure and mechanism. J Biol Chem, 1993. 268(17): p. 12231-4.
327. Garcia-Cardena, G., et al., Dissecting the interaction between nitric oxide synthase (NOS) and caveolin. Functional significance of the nos caveolin binding domain in vivo. J Biol Chem, 1997.272(41): p. 25437-40.
328. Montagnani, M., et al., Insulin-stimulated activation of eNOS is independent of Ca2+ but requires phosphorylation by Akt at Ser(1179). J Biol Chem, 2001. 276(32): p. 30392-8.
329. Zou, M.H., et al., Modulation by peroxynitrite of A kt- and AMP-activated kinasedependent Ser 1179 phosphorylation of endothelial nitric oxide synthase. J Biol Chem, 2002. 277(36): p. 32552-7.
330. Matsubara, M., et al., Regulation of endothelial nitric oxide synthase by protein kinase C. J Biochem (Tokyo), 2003.133(6): p. 773-81.
331. Lai, P.F., et al., Downregulation of eNOS mRNA expression by TNFalpha: identification and functional characterization of RNA -prote in interactions in the 3'UTR. Cardiovasc Res, 2003.59(1): p. 160-8.
332. Cardaropoli, S., et al., Infectious and inflammatory stimuli decrease endothelial nitric oxide synthase activity in vitro. J Hypertens, 2003. 21(11): p. 2103-10.
333. Saura, M., et al., Smad2 mediates transforming growth factor-beta induction of endothelial nitric oxide synthase expression. Circ Res, 2002.91(9): p. 806-13.
334. Abu-Soud, H.M., et al., Electron transfer, oxygen binding, and nitric oxide feedback inhibition in endothelial nitric-oxide synthase. J Biol Chem, ' 2000. 275(23): p. 17349-57.
335. Huang, P.L., et al., Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature, 1995. 377(6546): p. 239-42.
336. Xiao, Z., et al., Shear stress induction of the endothelial nitric oxide synthase gene is calcium-dependent but not calcium-activated. J Cell Physiol, 1997. 171(2): p. 205-11.
337. Buckley, B.J., Z. Mirza, and A.R. Whorton, Regulation ofCa(2+)-dependent nitric oxide synthase in bovine aortic endothelial cells. Am J Physiol, 1995. 269(3 Pt 1): p. C757-65.
338. Murad, F., What are the molecular mechanisms for the antiproliferative effects of nitric oxide and cGMP in vascular smooth muscle? Circulation, 1997. 95(5): p. 1101-3.

198
339. Cook, S., et al., Partial gene deletion of endothelial nitric oxide synthase predisposes to exaggerated high-fat diet-induced insulin resistance and arterial hypertension. Diabetes, 2004.53(8): p. 2067-72.
340. Chen, W., et al., Combined effects of endotheliaJ nitric oxide synthase gene polymorphism (G894T) and insulin resistance status on blood pressure and familial risk of hypertension in young adults: the Bogalusa Heart Study. Am 1 Hypertens, 2001.14(10): p. 1046-52.
341. Duplain, H., et al., Insulin resistance, hyperlipidemia, and hypertension in mice lacking endothelial nitric oxide synthase. Circulation, 2001. 104(3): p. 342-5.
342. Maniatis, N.A., et al. , Novel mechanism of endothelial nitric oxide synthase activation mediated by caveolae internalization in endothelial ceUs. Circ Res, 2006. 99(8): p. 870-7.
343. Connelly, L. , M. Madhani, and A.l. Hobbs, Resistance to endotoxic shock in endothelial nitric-oxide synthase (eNOS) knock-out mice: a pro-injlammatory role for eNOS-derived no in vivo. 1 Biol Chern, 2005. 280(11): p. 10040-6.
344. Peng, T., et al., Endothelial nitric-oxide synthase enhances lipopolysaccharidestimulated tumor necrosis factor-alpha expression via cAMP-mediated p38 MAPK pathway in cardiomyocytes. 1 Biol Chern, 2003.278(10): p. 8099-105.
345. Vo, P.A., et al., autoregulatory role of endothelium-derived nitric oxide (NO) on Lipopolysaccharide-induced vascular inducible NO synthase expression and function. 1 Biol Chern, 2005.280(8): p. 7236-43.
346. Kang, W.S., et al., Rapid up-regulation of endothelial nitric-oxide synthase in a mouse model of Escherichia coli lipopolysaccharide-induced bladder inflammation. 1 Pharrnacol Exp Ther, 2004. 310(2): p. 452-8.
347. Miyagoe-Suzuki, Y. and S.I. Takeda, Association ofneuronal nitric oxide synthase (nNOS) with alphal-syntrophin at the sarcolemma. Microsc Res Tech, 2001.55(3): p.164-70.
348. Wang, Y., D.C. Newton, and P.A. Marsden, Neuronal NOS: gene structure, mRNA diversity, andfunctional relevance. Crit Rev Neurobiol, 1999. 13(1): p. 21-43.
349. Brenrnan, 1.E., et al., Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and alphal-syntrophin mediated by PDZ domains. Cell, 1996.84(5): p. 757-67.
350. Silvagno, F., H. Xia, and D.S. Bredt, Neuronal nitric-oxide synthase-mu, an alternatively spliced isoform expressed in differentiated skeletal muscle. 1 Biol Chern, 1996.271(19): p. 112,04-8.
351. Beckman, 1.S., et al. , Apparent hydroxyl radical production by peroxynitrite: implications for endothelial in jury from nitric oxide and superoxide. Proc Nad Acad Sci USA, 1990.87(4): p. 1620-4.
352. Yan, X.B., et al., Brain ischemia induces serine phosphorylation of neuronal nitric oxide synthase by Ca(2+ )/calmodulin-dependent protein kinase II in rat hippocampus. Acta Pharrnacol Sin, 2004. 25(5): p. 617-22.
353. Lopez, F., et al., Neuronal nitric oxide synthase: a substrate for SHP-l involved in sst2 somatostatin receptor growth inhibitory signaling. Faseb 1, 2001. 15(12): p. 2300-2.
354. Korneirna, K. and Y. Watanabe, Dephosphorylation ofnNOS at Ser(847) by protein phosphatase 2A. FEBS Lett, 2001.497(1): p. 65-6.

199
355. Comini, L., et al., Effects of endotoxic shock on neuronal NOS and calcium transients in rat cardiac myocytes. Pharmacol Res, 2005.51(5): p. 409-17.
356. Ogando, D., et al., Steroid hormones augment nitric oxide synthase activity and expression in rat uterus. Reprod Fertil Dev, 2003. 15(5): p. 269-74.
357. Mathewson, A.M. and R.M. Wadsworth, Induction of iNOS restricts functional activity of both eNOS and nNOS in pig cerebral artery. Nitric Oxide, 2004. 11(4): p.331-9.
358. Sattler, R. , et al., Specific coupling of NMDA receptor activation to nitric oxide neurotoxicity by PSD-95 prote in. Science, 1999.284(5421): p. 1845-8.
359. Jaffrey, S.R., et al., Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat Cell Biol, 2001. 3(2): p. 193-7.
360. Brenman, J.E. , et al. , Regulation of neuronal nitric oxide synthase through alternative transcripts. Dev Neurosci, 1997.19(3): p. 224-31.
361. Tidball, J.G. and M. Wehling-Henricks, Expression of a NOS transgene in dystrophin-odeficient muscle reduces muscle membrane damage without increasing the expression of membrane-associated cytoskeletal proteins. Mol Genet Metab, 2004.82(4): p. 312-20.
362. Huang, P.L. and E.H. Lo, Genetic analysis of NOS isoforms using nNOS and eNOS knockout animais. Prog Brain Res, 1998. 118: p. 13-25.
363. Kovacs, G., et al., Neuronal nitric oxide synthase: its role and regulation in macula densa ceUs. J Am Soc Nephrol, 2003.14(10): p. 2475-83.
364. Lajoix, A.D., et al. , A neuronal isoform of nitric oxide synthase expressed in pancreatic beta-ceUs controls insulin secretion. Diabetes, 2001. 50(6): p. 1311-23.
365. Kanai, A.J., et al., Identification of a neuronal nitric oxide synthase in isolated cardiac mitochondria using electrocheinical detection. Proc Nad Acad Sci USA, 2001.98(24): p. 14126-31.
366. Frandsen, U., M. Lopez-Figueroa, and Y. Hellsten, Localization of nitric oxide synthase in human skeletal muscle. Biochem Biophys Res Commun, 1996. 227(1): p.88-93.
367. Bringold, U., P. Ghafourifar, and C. Richter, Peroxynitrite formed by mitochondrial NO synthase promotes mitochondrial Ca2+ release. Free Radic Biol Med, 2000. 29(3-4): p. 343-8.
368. Boveris, A., et al., Pharmacological regulation of mitochondrial nitric oxide synthase. Methods Enzymol, 2002. 359: p. 328-39.
369. Weitzberg, E. and J.O. Lundberg, Nonenzymatic nitric oxide production in humans. Nitric Oxide, 1998. 2(1): p. 1-7.
370. Cho, H.J., et al., Calmodulin is a subunit of nitric oxide synthase from macrophages. J Exp Med, 1992. 176(2): p. 599-604.
371. Kapur, S., et al., Expression of nitric oxide synthase in skeletal muscle: a novel role for nitric oxide as a modulator of insulin action. Diabetes, 1997. 46(11): p. 1691-700.
372. Rao, K.M., Molecular mechanisms regulating iNOS expression in various ceU types. J Toxicol Environ Health B Crit Rev, 2000.3(1): p. 27-58.
373. Nathan, C., Nitric oxide as a secretory product ofmammalian ceUs. Faseb J, 1992. 6(12): p. 3051-64.

200
374. Chan, E.D., J. Chan, and N.W. Schluger, What is the role ofnitric oxide in murine and human host defense against tuberculosis?Current knowledge. Am J Respir Cell Mol Biol, 2001.25(5): p. 606-12.
375. Bolli, R., B. Dawn, and Y.T. Xuan, Emerging role of the JAK-STAT pathway as a mechanism of protection against ischemia/reperfusion in jury. J Mol Cell Cardiol, 2001.33(11): p. 1893-6.
376. Kleinert, H., et al., Cytokine induction of NO synthase II in human DLD-l cel/s: roles of the JAK-STAT, AP-l and NF-kappaB-signaling pathways. Br J Pharrnacol, 1998.125(1): p. 193-201.
377. Janssen-Heininger, Y.M., 1. Macara, and B.T. Mossman, Cooperativity between oxidants and tumor necrosis factor in the activation of nuclear factor (NF)-kappaB: requirement of Ras/mitogen-activated protein kinases in the activation of NFkappaB by oxidants. Am J Respir Cell Mol Biol, 1999.20(5): p. 942-52.
378. Schroder, N.W., et al., Involvement of lipopolysaccharide binding protein, CD14, and Tol/-like receptors in the initiation of innate immune responses by Treponema glycolipids. J Immunol, 2000. 165(5): p. 2683-93.
379. Vitseva, 0.1., et al., Inducible Tol/-like receptor and NF-kappaB regulatory pathway expression in human adipose tissue. Obesity (Silver Spring), 2008. 16(5): p.932-7.
380. Froy, O., et al., Differentiai effect of insulin treatment on decreased levels of betadefensins and Tol/-like receptors in diabetic rats. Mol Immunol, 2007. 44(5): p. 796-802.
381. Geller, D.A., et al., A central rolefor IL-l beta in the in vitro and in vivo regulation of hepatic inducible nitric oxide synthase. IL-l beta induces hepatic nitric oxide synthesis. J Immunol, 1995. 155(10): p. 4890-8.
382. Hecker, M., M. Cattaruzza, and A.H. Wagner, Regulation of inducible nitric oxide synthase gene expression in vascular smooth muscle cel/s. Gen Phannacol, 1999. 32(1): p. 9-16.
383. Chu, S.C., et al., Analysis of the cytokine-stimulated human inducible nitric oxide synthase (iNOS) gene: characterization of differences between human and mouse iNOS promoters. Biochem Biophys Res Commun, 1998.248(3): p. 871-8.
384. Saura, M., et al., Interaction of interferon regulatory factor-l and nuclear factor kappaBduring activation of inducible nitric oxide synthase transcription. J Mol Biol, 1999.289(3): p. 459-71.
385. Korhonen, R., et al., Post-transcriptional regulation ofhuman inducible nitric-oxide synthase expression by the Jun N-terminal kinase. Mol Phannacol, 2007. 71(5): p. 1427-34.
386. Linker, K., et al., Involvement of KSRP in the post-transcriptional regulation of human iNOS expression-complex interplay of KSRP with TTP and HuR. Nucleic Acids Res, 2005. 33(15): p. 4813-27.
387. Ratovitski, E.A., et al., Kalirin inhibition of inducible nitric-oxide synthase. J Biol Chem, 1999.274(2): p. 993-9.
388. Ratovitski, E.A., et al., An inducible nitric-oxide synthase (NOS)-associated prote in inhibits NOS dimerization and activity. J Biol Chem, 1999.274(42): p. 30250-7.
389. Mori, M. and T. Gotoh, Regulation ofnitric oxide production by arginine metabolic enzymes. Biochem Biophys Res Commun, 2000. 275(3): p. 715-9.

201
390. Salh, B. , et al., Activation of phosphatidylinositol 3-kinase, protein kinase B, and p 70 S6 kinases in lipopolysaccharide-stimulated Raw 264.7 ceUs: differential effects of rapamycin, Ly294002, and wortmannin on nitric oxide production. 1 Immunol, 1998. 161(12): p. 6947-54.
391. Parikh, S. , G. Dorsey, and P.l. Rosenthal, Host polymorphisms and the incidence of malaria in Ugandan ch ildren. Am 1 Trop Med Hyg, 2004.71(6): p. 750-3.
392. Eissa, N.T., et al. , Alternative splicing of human inducible nitric-oxide synthase mRNA. tissue-specific regulation and induction by cytokines. 1 Biol Chem, 1996. 271(43): p. 27184-7.
393. Eissa, N.T. , et al. , Cloning and characterization of human inducible nitric oxide synthase splice variants: a domain, encoded by exons 8 and 9, is critical for dimerization. Proc Natl Acad Sci USA, 1998.95(13): p. 7625-30. '
394. Raso, G.M., et al. , Leptin potentiates IFN-gamma-induced expression of nitric oxide synthase and cyclo-oxygenase-2 in murine macrophage J774A.l. Br 1 Pharmacol, 2002. 137(6): p. 799-804.
395. Chang, P.C., et al. , Advanced glycosylation end products induce inducible nitric oxide synthase (iNOS) expression via a p38 MAPK-dependent pathway. Kidney Int, 2004.65(5): p.1664-75.
396. Ikeda, U. , et al., C-Reactive protein augments inducible nitric oxide synthase expression in cytokine-stimulated cardiac myocytes. Cardiovasc Res, 2002.56(1): p. 86-92.
397. Min, K.l., et al. , Gangliosides activate microglia viaprotein kinase C and NADPH oxidase. Glia, 2004.48(3): p. 197-206.
398. Woo, C.W., et al. , Homocysteine stimulates inducible nitric oxide synthase expression in macrophages: antagonizing effect of ginkgolides and bilobalide. Mol Cell Biochem, 2003. 243(1-2): p. 37-47.
399. Shimabukuro, M., et al., Role of nitric oxide in obesity-induced beta ceU disease. 1 Clin Invest, 1997.100(2): p. 290-5.
400. Won, 1.S., et al., The role of neutral sphingomyelinase produced ceramide in lipopolysaccharide-mediated expression of inducible nitric oxide synthase. J Neurochem, 2004.88(3): p. 583-93.
401. Pacheco, M.E., et al., High glucose enhances inducible nitric oxide synthase expression. Role of prote in kinase C-betaIl Eur J Pharmacol, 2006. 538(1-3): p. 115-23.
402. Guan, Z., et al., IGF-I and insulin amplify IL-l beta-induced nitric oxide and prostaglandin biosynthesis. Am 1 Physiol, 1998.274(4 Pt 2): p. F673-9.
403. Cerchiaro, G.A., et al., Inducible nitric oxide synthase in rat neutrophils: role of insulin. Biochem Pharmacol, 2001.62(3): p. 357-62.
404. Noh, H. , et al., High glucose increases inducible NO production in cultured rat mesangial ceUs. Possible role in fibronectin production. Nephron, 2002. 90(1): p. 78-85.
405. Sharma, K., et al., Enhanced expression of inducible nitric oxide synthase in murine macrophages and glomerular mesangial ceUs by elevated glucose levels: possible mediation via protein kinase C. Biochem Biophys Res Commun, 1995. 207(1): p. 80-8.

202
406. Powell, L.A., et al., High glucose decreases intracellular glutathione concentrations and upregulates inducible nitric oxide synthase gene expression in intestinal epithelial ceUs. 1 Mol Endoerinol, 2004. 33(3): p. 797-803.
407. Nakai, K., et al., EfJects ofhigh glucose on NO synthesis in human keratinocyte ceU line (HaCaT). 1 Dermatol Sei, 2003. 31(3): p. 211-8.
408. Nishio, E. and Y. Watanabe, Glucose-induced down-regulation of NO production and inducible NOS expression in cultured rat aortic vascular smooth muscle ceUs: role ofprotein kinase C. Bioehem Biophys Res Commun, 1996.229(3): p. 857-63.
409. Umansky, V. , et al. , Co-stimulatory effect ofnitric oxide on endothelial NF-kappaB implies a physiological self-amplifying mechanism. Eur 1 Immunol, 1998.28(8): p. 2276-82.
410. Connelly, L., et al., Biphasic regulation of NF-kappa B activity underlies the proand anti-injlammatory actions ofnitric oxide. 1 Imml:ffiol, 2001.166(6): p. 3873-81.
411. Giri, S., et al., The 15-deoxy-delta12, 14-prostaglandin J2 inhibits the injlammatory response in primary rat astrocytes via down-regulating n1-ultiple steps in phosphatidylinositol 3-kinase-Akt-NF-kappaB-p300 pathway independent of peroxisome proliferator-activated receptor gamma. 1 Immunol, 2004. 173(8): p. 5196-208.
412. Weber, S.M., A.L. Searim, and lA. Corbert, PPARgamma is not requiredfor the inhibitory actions of PGJ2 on cytokine signaling in pancreatic beta-cèlls. Am 1 Physiol Endoerinol Metab, 2004. 286(3): p. E329-36.
413. Melntyre, T.M., et al., Identification of an intracellular receptor for lysophosphatidic acid (LPA): LPA is "a transcellular PPARgamma agonist. Proe Nad Aead Sei USA, 2003.100(1): p. 131-6. .
414. Kadowaki, S., et al., Down-regulation of inducible nitric oxide synthase by lysophosphatidic acid in human respiratory epithelial ce Ils. Mol Cell Bioehem, 2004.262(1-2): p. 51-9.
415. Crosby, M.B., et al., Peroxisome proliferation-activated receptor (PPAR)gamma is not necessary for synthetic PPARgamma agonist inhibition ofinducible nitric-oxide synthase and nitric oxide. 1 Pharmaeol Exp Ther, 2005. 312(1): p. 69-76.
416. Hevener, A.L., et al., Macrophage PPAR gamma is required for normal skeletal muscle and hepatic insulin sensitivity and full antidiabetic effects of thiazolidinediones. 1 Clin Invest, 2007.117(6): p. 1658-69.
417. Odegaard, 1.1., et al., Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature, 2007. 447(7148): p. 1116-20.
418. Crosby, M.B., et al., A novel PPAR r~sponse element in the murine iNOS promoter. Mol Immunol, 2005.42(11): p. 1303-10.
419. "Weleh, 1.S., et al., PPARgamma and PPARdelta negatively regulate specific subsets of lipopolysaccharide and IFN-gamma target genes in macrophages. Proe Nad Aead Sei USA, 2003.100(11): p. 6712-7.
420. Cuzzoerea, S., et al., Rosiglitazone, a ligand of the peroxisome proliferatoractivated receptor-gamma, reduces acute injlammation. Eur 1 Pharmaeol, 2004. 483(1): p. 79-93.
421. Mendez, M. and M.C. LaPointe, PPARgamma inhibition of cyclooxygenase-2, PGE2 synthase, and inducible nitric oxide synthase in cardiac myocytes. Hypertension, 2003.42(4): p. 844-50.

203
422. Kim, 1., Y.S. Oh, and S.H. Shinn, Troglitazon.e reverses the inhibition of nitric oxide production by high glucose in cultured bovine retinal pericytes. Exp Eye Res, 2005.81(1): p. 65-70.
423. Cemuda-Morollon, E., et al., PPAR agonists amplify iNOS expression while inhibiting NF-kappaB: implications for mesangial cell activation by cytokines. 1 Am Soc Nephrol, 2002. 13(9): p. 2223-31.
424. Schulze, P.C., et al., Chronic heartfailure and skeletal muscle catabolism: effects of exercise training. Int 1 Cardiol, 2002.85(1): p. 141-9.
425. Gielen, S., et al., Anti-inflammatory effects of exercise training in the skeletal muscle of patients with chronic heart failure. 1 Am Coll Cardiol, 2003. 42(5): p. 861-8.
426. Yang, A.L. and H.I. Chen, Chronic exercise reduces adhesion molecules/iNOS expression and partially reverses vascular responsiveness in hypercholesterolemic rabbit aortae. Atherosclerosis, 2003. 169( 1): p. 11-7.
427. Pauli, 1.R., et al., Acute physical exercise reverses S-nitrosation of the insulin receptor, insulin receptor substrate 1, and protein Kinase B/A kt in dietary induced obese Wistar rats. 1 Physiol, 2007.
428. Morley, 1.E., et al., Measurement of nitric oxide synthase and its mRNA in genetically obese (ob/ob) mice. Life Sci, 1995.57(14): p. 1327-31.
429. Sugita, H., et al., Inducible nitric-oxide synthase and NO donor induce insulin receptor substrate-1 degradation in skeletal muscle cells. 1 Biol Chem, 2005. 280(14): p. 14203-11.
430. Fujimoto, M., et al., A role for iNOS in fasting hyperglycemia and impaired insu lin signaling in the liver of obese diabetic mice. Diabetes, 2005.54(5): p. 1340-8.
431. Zhou, Y.T., et al., Lipotoxic heart disease in obese rats: implications for human obesity. Proc Nad Acad Sci USA, 2000.97(4): p. 1784-9.
432. Perreault, M. and A. Marette, Targeted disruption of inducible nitric oxide synthase protects against obesity-linked insulin resistance in muscle. Nat Med, 2001. 7(10): p. 1138-43.
433. Engeli, S., et al., Regulation of the nitric oxide system in human adipose tissue. 1 . Lipid Res, 2004.45(9): p. 1640-8.
434. Choi, 1.W., et al., Increases in nitric oxide concentrations correlate strongly with body fat in obese humans. Clin Chem, 2001.47(6): p. 1106-9.
435. Lin, L.Y., et al., Nitric oxide production is paradoxically decreased after weight reduction surgery in morbid obesity patients. Atherosclerosis, 2006.
436. Noronha, B.T., et al., Inducible nitric oxide synthase has divergent effects on vascular and metabolic function in obesity. Diabetes, 2005. 54(4): p. 1082-9.
437. Torres, S.H., et al., Inflal1,1mation and nitric oxide production in skeletal muscle of type 2 diabetic patients. 1 Endocrinol, 2004. 181(3): p. 419-27.
438. Morris, B.l., et al., Influence of an inducible nitric oxide synthase promoter variant on clinical variables in patients with coronary artery disease. Clin Sci (Lond), 2001.100(5): p. 551-6.
439. Stamler, 1.S., et al., S-nitrosylation of proteins with nitric oxide: synthesis and characterization ofbiologically active compounds. Proc Nad Acad Sci USA, 1992. 89(1): p. 444-8.
440. Lander, H.M., et al., Nitric oxide-stimulated guanine nucleotide exchange on p21ras. 1 Biol Chem, 1995.270(13): p. 7017-20.

204
441. Park, H.S., et al., Nitric oxide negatively regulates c-Jun N-terminal kinase/stressactivated protein kinase by means of S-nitrosylation. Proc Nad Acad Sci USA, 2000.97(26): p. 14382-7 . .
442. Marshall, H.E. and 1.S. Starnler, Inhibition of NF-kappa B by S-nitrosylation. Biochernistry, 2001. 40(6): p. 1688-93.
443. Carvalho-Filho, M.A. , et al., S-nitrosation of the insulin receptor, insulin receptor substrate 1, and protein kinase B/A kt: a novel mechanism of insulin resistance. Diabetes, 2005. 54(4): p. 959-67.
444. Yasukawa, T. , et al. , S~nitrosylation-dependent inactivation of A kt/prote in kinase B in insulin resistance. J Biol Chern, 2005. 280(9): p. 7511-8.
445. Wadharn, C., et al. , High glucose attenuates protein S-nitrosylation in endothelial ceUs: role of oxidative stress. Diabetes, 2007. 56(11): p. 2715-21.
446. Radi, R. , et al. , Peroxynitrite oxidation of suljhydryls. The cytotoxic potential of superoxide and nitric oxide. J Biol Chem, 1991. 266(7): p. 4244-50.
447. Ischiropoulos, H., et al. , Peroxynitrite-mediated tyrosine nitration catalyzed by superoxide dismutase. Arch Biochern Biophys, 1992. 298(2): p. 431-7.
448. Stuehr, D.J., et al., N omega-hydroxy-L-arginine is an intermediate in the biosynthesis ofnitric oxide from L-arginine. J Biol Chem, 1991.266(10): p. 6259-63.
449. Heinzel, B., et al., Ca2+/calmodulin-dependent formation of hydrogen peroxide by brain nitric oxide synthase. Biochern J, 1992.281 (Pt 3): p. 627-30.
450. Albina, J.E. , et al. , Temporal expression of different pathways of 1-arginine metabolism in healing wounds. J Irnrnunol, 1990. 144(10): p. 3877-80.
451. MacMillan-Crow, L.A., J.P. Crow, and J.A. Thompson, Peroxynitrite-mediated inactivation of manganese superoxide dismutase involves nitration and oxidation of critical tyrosine residues. Biochemistry, 1998.37(6): p. 1613-22.
452. Baud, L., et al., Reactive oxygen production by cultured rat glomerular mesangial ceUs during phagocytosis is associated with stimulation of lipoxygenase activity. J Exp Med, 1983. 158(6): p. 1836-52.
453. Galbusera, C., et al., Superoxide radical production by aUopurinol and xanthine oxidase. Biochem Pharrnacol, 2006.71(12): p. 1747-52.
454. Natake, M. and M. Ueda, Changes in food proteins reacted with nitrite at gastric pH Nutr Cancer, 1986.8(1): p. 41-5.
455. Eiserich, J.P., et al., Formation of nitrating and chlorinating species by reaction of nitrite with hypochlorous acid. A novel mechanism for nitric oxide-mediated prote in modification. J Biol Chem, 1996.271(32): p. 19199-208.
456. Bian, K. , et al., Down-regulation of inducible nitric-oxide synthase (NOS-2) during parasite-induced gut inflammation: a path to identify a selective NOS-2 inhibitor. Mol Pharrnacol, 2001. 59(4): p. 939-47.
457. Lee, J.K. , et al., Experimental abdominal aortic aneurysms in mice lacking expression of inducible nitric oxide synthase. Arterioscler Thromb Vasc Biol, 2001. 21(9): p. 1393-401.
458. van der Vliet, A., et al. , Nitrotyrosine as biomarker for reactive nitrogen species. Methods Enzyrnol, 1996.269: p. 175-84.
459. Berlett, B.S., et al., Peroxynitrite-mediated nitration of tyrosine residues in Escherichia coli glutamine synthetase mimics adenylylation: relevance to signal transduction. Proc Nad Acad Sci USA, 1996.93(5): p. 1776-80.

205
460. Greenacre, S.A. and H. Ischiropoulos, · Tyrosine nitration: localisation, quantification, consequences for protein function and signal ,transduction. Free Radic Res, 2001. 34(6): p. 541-81.
461. Souza, l.M., et al., Proteolytic degradation of tyrosine nitrated proteins. Arch Biochem Biophys, 2000. 380(2): p. 360-6.
462. Marcondes, S., LV. Turko, and F. Murad, Nitration of succinyl-CoA:3-oxoacid CoA-transferase in rats after endotoxin administration. Proc Natl Acad Sci USA, 2001.98(13): p. 7146-51.
463. van Nassauw, L., L. ·Tao, and F. Harrisson, Localization of nitric oxide-related substances in the quai! ovary during foUiculogenesis. Histochem l , 1999. 31(7): p. 443-54.
464. Irie, Y., et al., Histone Hl.2 is a substrate for denitrase, an activity that reduces nitrotyrosine immunoreactivity in proteins. Proc Natl Acad Sci USA, 2003. 100(10): p. 5634-9.
465. MacMi11an-Crow, L.A., et al., Tyrosine nitration of c-SRC tyrosine kinase in human pancreatic ductal adenocarcinoma. Arch Biochem Biophys, 2000. 377(2): p. 350-6.
466. Takakura, K., et al., Rapid and irreversible inactivation of protein tyrosine phosphatases PTP 1 B, CD45, and LAR by peroxynitrite. Arch Biochem Biophys, 1999.369(2): p. 197-207.
467. Newman, D.K., et al., Nitration of PECAM-l ITIM tyrosines abrogates phosphorylation and SHP-2 binding. Biochem Biophys Res Commun, 2002. ·296(5): p. 1171-9.
468. Zou, M., C. Martin, and V. U11rich, Tyrosine nitration as a mechanism of selective inactivation of prostacyclin synthase by peroxynitrite. Biol Chem, 1997. 378(7): p. 707-13.
469. Daiber, A., et al., Nitration and inactivation of cytochrome P450BM-3 by peroxynitrite. Stopped-flow measurements prove ferryl intermediates. Eur 1 Biochem, 2000.267(23): p. 6729-39.
470. Blanchard-Fi11ion, B., et al., Nitration and inactivation of tyrosine hydroxylase by peroxynitrite. 1 Biol Chem, 2001. 276(49): p. 46017-23.
471. Cerie11o, A., et al., Detection of nitrotyrosine in the diabetic plasma: evidence of oxidative stress. Diabetologia, 2001. 44(7): p. 834-8.
472. Cerie11o, A., et al., Role of hyperglycemia in nitrotyrosine postprandial generation. Diabetes Care, 2002. 25(8): p. 1439-43.
473. Zou, M.H., C. Shi, and R.A. Cohen, High glucose via peroxynitrite causes tyrosine nitration and inactivation of prostacyclin synthase that is associated with thromboxane/prostaglandin H(2) receptor-mediated apoptosis and adhesion molecule expression in cultured human aortic endothelial ceUs. Diabetes, 2002. 51(1): p. 198-203.
474. Marfe11a, R., et al., Acute hyperglycemia induces an oxidative stress in healthy subjects. 1 Clin Invest, 2001.108(4): p. 635-6.
475. Chi, Q., T. Wang, ànd K. Huang, EjJect ofinsulin nitration by peroxynitrite on its biological activity. Biochem Biophys Res Commun, 2005. 330(3): p. 791-6.
476. Morris, l.W., D.A. Mercola, and E.R. Arqui Il a, Preparation and properties of 3-nitrotyrosine insulins. Biochemistry, 1970.9(20): p. 3930-7.

206
477. Shibuya, A., et al., Nitration of PPARgamma inhibits ligand-dependent translocation into the nucleus in a macrophage-like ceUline, RA W 264. FEBS Lett, 2002. 525(1-3): p. 43-7.
478. McGuinness, a.p., The impact of infection on gluconeogenesis in the conscious doge Shock, 1994. 2(5): p. 336-43.
479. Spitzer, 1.1., et al., Alterations in the metabolic control of carbohydrates in sepsis. Prog Clin Biol Res, 1989.308: p. 545-61.
480. Sindelar, D.K., et al., The role of fatty acids in mediating the effects of peripheral insulin on hepatic glucose production in the conscious doge Diabetes, 1997. 46(2): p. 187-96.
481. Kanda, H., et al., MCP-l contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. 1 Clin Invest, 2006. 116(6): p. 1494-505.
482. Femandez-Veledo", S., et al. , Ceramide mediates TNF-alpha-induced insulin resistance on GLUT4 gene expression in brown adipocytes. Arch Physiol Biochem 2006. 112(1): p. 13-22.
483. Penfomis, P. and A. Marette, lnducible nitric oxide synthase modulates lipolysis in adipocytes. 1 Lipid Res, 2005.46(1): p. 135-42.
484. Salt, I.P., 1.M. Connell, and G.W. Gould, 5-aminoimidazole-4-earboxamide ribonueleoside (AICAR) inhibits insulin-stimulated glucose transport in 3T3-Ll adipocytes. Diabetes, 2000. 49(10): p. 1649-56.
485. Gaidhu, M.P., S. Fediuc, and R.B. Ceddia, 5-Aminoimidazole-4-carboxamide-lbeta-D-ribofuranoside-induced AMP-aetivated protein kinase phosphorylation inhibits basal and insulin-stimulated glucose uptake, lipid synthesis, and fatty aeid oxidation in isolated rat adipoeytes. 1 Biol Chem, 2006. 281(36): p. 25956-64.
486. Pang, C., et al., Macrophage Infiltration into Adipose Tissue May Promote Angiogenesis for Adipose Tissue Remodeling in Obesity. Am 1 Physiol Endocrinol Metab, 2008.
487. Dallaire, P., et al., Obese mice lacking iNOS are sensitized to the metabolic actions of PP AR{ gamma} agonism. Diabetes, 2008.
488. Halliwell, B., What nitrates tyrosine? Is nitrotyrosine specifie as a biomarker of peroxynitriteformation in vivo? FEBS Lett, 1997.411(2-3): p. 157-60.
489. Pfeiffer, S., K. Schmidt, and B. Mayer, Dityrosine formation outcompetes tyrosine nitration at low steady-state concentrations of peroxynitrite. Implications for tyrosine modification by nitrie oxide/superoxide in vivo. 1 Biol Chem, 2000. 275(9): p.6346-52.
490. Messner, K.R. and 1.A. Imlay, Mechanism of superoxide and hydrogen peroxide formation by fumarate reductase, suecinate de hydroge nase, and aspartate oxidase. 1 Biol Chem, 2002. 277(45): p. 42563-71.
491. Inoguchi, T., et al., High glucose level and free fatty acid stimulate reaetive oxygen species productiC!n through protein kinase C--dependent activation of NAD(P)H oxidase in cultured vaseular eeUs. Diabetes, 2000.49(11): p. 1939-45.
492. Rinaldo, 1.E., et al., Nitric oxide inactivates xanthine dehydrogenase and xanthine oxidase in interferon-gamma-stimulated macrophages. Am 1 Respir Cell Mol Biol, 1994. 11(5): p. 625-30.

207
493. Kuwano, Y. , et al. , Interferon-gamma activates transcription of NADPH oxidase 1 gene and upregulates production of superoxide anion by human large intestinal epithelial ceUs. Am] Physiol Cell Physiol, 2006. 290(2): p. C433-43.
494. Louis, C.A., et al. , Distinct arginase isoforms expressed in primary and transformed macrophages: regulation by oxygen tension. Am ] Physiol, 1998. 274(3 Pt 2): p. R775-82.
495. Munder~ M., et al. , Thl/Th2-regulated expression of arginase isoforms in murine macrophages and dendritic ceUs. ] Immunol, 1999. 163(7): p. 3771-7.
496. Song, X.Z. , Z.G. Bi, and A.E. Xu, Green tea polyphenol epigaUocatechin-3-gallate inhibits the expression of nitric oxide synthase and generation of nitric oxide induced by ultraviolet B in HaCaT ceUs. Chin Med] (Engl), 2006. 119(4): p. 282-7.
497. Xie, Z. , et al. , Activation of protein kinase C zeta by peroxynitrite regulates LKB1-dependent AMP-activated prote in kinase in cultured endothelial ceUs. ] Biol Chem, 2006. 281(10):p. 6366-75.
498. Koh, H.]. , et al. , Skeletal muscle-selective knockout of LKBl increases insulin sensitivity, improves glucose homeostasis, and decreases TRB3. Mol Cell Biol 2006.26(22): p. 8217-27.
499. Radi, R. , Nitric oxide, oxidants, and protein tyrosine nitration. Proc Natl Acad Sci USA, 2004.101(12): p. 4003-8.
500. Bonen, A., M.G. Clark, and E.J. Henriksen, Experimental approaches in muscle metabolism: hindlimb perfusion and isolated muscle incubations. Am ] Physiol, 1994.266(1 Pt 1): p. EI-16.
501. Gras, F., et al. , Differences in troglitazone action on glucose metabolism in freshly isolated vs long-term incubated rat skeletal muscle. Br ] Pharmacol, 2003. 138(6): p. 1140-6.
502. Buse, M.G., et al., Enhanced O-GlcNAc protein modification is associated with insulin resistance in GLUT1-overexpressing muscles. Am ] Physiol Endocrinol Metab, 2002. 283(2): p. E241-50.
503. Rubinstein, 1., et al. , Involvement of nitric oxide system in experimenial muscle crush in jury. ] Clin Invest, 1998.101(6): p. 1325-33.
504. Schwartz, D., et al. , Inhibition of constitutive nitric oxide synthase (NOS) by nitric oxide generated by inducible NOS after lipopolysaccharide administration provokes renal dysfunction in rats. ] Clin Invest, 1997. 100(2): p. 439-48.
505. Shaw, R.J., et al., The kinase LKBl mediates glucose homeostasis in liver and therapeutic effects ofmetformin. Science, 2005. 310(5754): p. 1642-6.
506. Konrad, D., et al., Troglitazone causes acute mitochondrial membrane depolarisation and an AMPK-mediated increase in glucose phosphorylation in muscle ceUs. Diabetologia, 2005. 48(5): p. 954-66.
507. Yamauchi, T. , et al., The mechanisms by which both heterozygous peroxisome proliferator-activated receptor gamma (PPARgamma) deficiency and PPARgamma agonist improve insulin resistance. ] Biol Chem, 2001. 276(44): p. 41245-54.
508. Putman, C.T., et al. , AMPK 'activation increases uncoupling protein-3 expression and mitochondrial enzyme activities in rat muscle without fibre type transitions. ]
, Physiol, 2003. 551(Pt 1): p. 169-78. 509. Tanaka, T., et al., Activation of peroxisome proliferator-activated receptor delta
induces fatty acid beta-oxidation in skeletal muscle and attenuates metabolic syndrome. Proc Natl Acad Sci USA, 2003.100(26): p. 15924-9.

208
510. Terada, S., et al. , PPARdelta activator GW-501516 has no acute effect on glucose transport in skeletal muscle. Am J Physiol Endocrinol Metab, 2006. 290( 4): p. E607-11.
511. Lau, N.e., et al., An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science, 2001. 294(5543): p. 858-62.



















