
Regulation of Monocyte Chemoattractant Protein-1 Expression by Tumor Necrosis Factor-α and...
10
Regulation of monocyte chemoattractant protein (MCP)-1 transcription by interferon-gamma (IFN-g) in human astrocytoma cells: postinduction refractory state of the gene, governed by its upstream elements Z.-H. LUCY ZHOU, 1 YULONG HAN, TAO WEI, SUMER ARAS, 2 PRIYA CHATURVEDI, 3 SARAH TYLER, 4 M. R. SANDHYA RANI, AND RICHARD M. RANSOHOFF* ,5 Department of Neurosciences, The Lerner Research Institute, and *Department of Neurology and The Mellen Center for Multiple Sclerosis Treatment and Research, Cleveland Clinic Foundation, Cleveland, Ohio 44195, USA ABSTRACT Monocyte chemoattractant protein (MCP)-1 is expressed by astrocytes in diverse inflam- matory states and is a key regulator of monocyte recruitment to the central nervous system (CNS). In the current study, we addressed mechanisms by which transcription of the human MCP-1 gene (hMCP-1) was terminated, after induction by interferon (IFN)-g. Our results demonstrated that IFN-g-induced transcription of hMCP-1 was followed by a refractory state, during which hMCP-1 was resistant to restimulation by either IFN-g or heterologous activators such as TNF-a. This refractory state affected the hMCP-1 gene selectively, as other IFN-g-inducible genes remained responsive to restimulation. The IFN-g-induced hMCP-1 refractory state was governed at the transcriptional level and was sensitive to protein synthesis inhibitors, suggesting a requirement for newly expressed components. A mini- mal 213 base pair hMCP-1 regulatory element directed both IFN-g-mediated transcription and the subsequent refractory state. We previously demonstrated that IFN-g treatment resulted in coordinate protein occu- pancy in vivo of two hMCP-1 promoter elements, a gamma-activated site (GAS) and a GC-rich element. During the refractory state, IFN-g treatment failed to induce protection of either the hMCP-1 GAS element or the GC box. These results furnish insight into the expression of hMCP-1 during CNS inflammation and provide the first delineation of an IFN-g-induced tran- scriptional refractory state.—Zhou, Z.-H. L., Han, Y., Wei, T., Aras, S., Chaturvedi, P., Tyler, S., Rani, M. R. S., Ransohoff, R. M. Regulation of monocyte chemoattractant protein (MCP)-1 transcription by inter- feron-gamma (IFN-g) in human astrocytoma cells: postinduction refractory state of the gene, governed by its upstream elements. FASEB J. 15, 383–392 (2001) Key Words: chemokine z gene expression z interferons z STAT factors z Sp1 transcription factor Monocyte chemoattractant protein 1 (MCP-1), a member of the chemokine superfamily, is implicated in immune regulation, inflammatory responses, the pathogenesis of atherosclerosis, wound healing, tissue remodeling, and modulation of tumor behavior (1–7). In human monocytes, MCP-1 induces chemotaxis, cal- cium flux, and the respiratory burst and up-regulates adhesion molecule expression and cytokine production (2). MCP-1 is secreted by many cell types in response to lipopolysaccharide, or cytokines including interleukin (IL) -1, IL-4, interferon g (IFN-g), and tumor necrosis factor (TNF)-a (8, 9). Four closely related murine and human MCPs have been identified, all of which exhibit monocyte che- moattractant activity in vitro. The specific functions of murine MCP-1 (mMCP-1) in vivo were investigated by construction and analysis of mMCP-1-deficient mice. Given the presence of four similar MCPs, it is surprising that these studies indicated that deletion of MCP-1 abrogated or altered diverse acute and chronic im- mune and inflammatory processes (3–7). It is uncertain whether expression patterns or structural characteris- tics account for the nonredundant functions exerted by mMCP-1. At the genetic level, mMCP-1 was first characterized as JE, an immediate-early gene that was activated in quiescent NIH 3T3 fibroblasts by exposure to platelet derived growth factor (PDGF) (12). Human MCP-1 (hMCP-1) encodes a protein that is 55% homologous to its mouse counterpart (1). Despite the relative lack of coding sequence identity, mMCP-1 and hMCP-1 are both highly expressed in diverse pathological states, 1 Current address: Department of Neurology, Stanford Uni- versity, 3801 Miranda Ave., Palo Alto, CA, USA. 2 Current address: Science Faculty, Ankara University, An- kara, Turkey. 3 Current address: Department of Oncology, SmithKline Beecham Pharmaceuticals, King of Prussia, PA 19406, USA. 4 Current address: Princeton University, Princeton, NJ, USA. 5 Correspondence: Department of Neurosciences, Lerner Re- search Institute, NC30, Cleveland Clinic Foundation, 9500 Eu- clid Ave., Cleveland, OH 44195, USA. E-mail: [email protected] 383 0892-6638/01/0015-0383 © FASEB
Transcript of Regulation of Monocyte Chemoattractant Protein-1 Expression by Tumor Necrosis Factor-α and...

Regulation of monocyte chemoattractant protein(MCP)-1 transcription by interferon-gamma (IFN-g)in human astrocytoma cells: postinduction refractorystate of the gene, governed by its upstream elements
Z.-H. LUCY ZHOU,1 YULONG HAN, TAO WEI, SUMER ARAS,2 PRIYA CHATURVEDI,3
SARAH TYLER,4 M. R. SANDHYA RANI, AND RICHARD M. RANSOHOFF*,5
Department of Neurosciences, The Lerner Research Institute, and *Department of Neurology andThe Mellen Center for Multiple Sclerosis Treatment and Research, Cleveland Clinic Foundation,Cleveland, Ohio 44195, USA
ABSTRACT Monocyte chemoattractant protein(MCP)-1 is expressed by astrocytes in diverse inflam-matory states and is a key regulator of monocyterecruitment to the central nervous system (CNS). In thecurrent study, we addressed mechanisms by whichtranscription of the human MCP-1 gene (hMCP-1) wasterminated, after induction by interferon (IFN)-g. Ourresults demonstrated that IFN-g-induced transcriptionof hMCP-1 was followed by a refractory state, duringwhich hMCP-1 was resistant to restimulation by eitherIFN-g or heterologous activators such as TNF-a. Thisrefractory state affected the hMCP-1 gene selectively,as other IFN-g-inducible genes remained responsive torestimulation. The IFN-g-induced hMCP-1 refractorystate was governed at the transcriptional level and wassensitive to protein synthesis inhibitors, suggesting arequirement for newly expressed components. A mini-mal 213 base pair hMCP-1 regulatory element directedboth IFN-g-mediated transcription and the subsequentrefractory state. We previously demonstrated thatIFN-g treatment resulted in coordinate protein occu-pancy in vivo of two hMCP-1 promoter elements, agamma-activated site (GAS) and a GC-rich element.During the refractory state, IFN-g treatment failed toinduce protection of either the hMCP-1 GAS elementor the GC box. These results furnish insight into theexpression of hMCP-1 during CNS inflammation andprovide the first delineation of an IFN-g-induced tran-scriptional refractory state.—Zhou, Z.-H. L., Han, Y.,Wei, T., Aras, S., Chaturvedi, P., Tyler, S., Rani,M. R. S., Ransohoff, R. M. Regulation of monocytechemoattractant protein (MCP)-1 transcription by inter-feron-gamma (IFN-g) in human astrocytoma cells:postinduction refractory state of the gene, governed byits upstream elements. FASEB J. 15, 383–392 (2001)
Key Words: chemokine z gene expression z interferons z STATfactors z Sp1 transcription factor
Monocyte chemoattractant protein 1 (MCP-1), amember of the chemokine superfamily, is implicated in
immune regulation, inflammatory responses, thepathogenesis of atherosclerosis, wound healing, tissueremodeling, and modulation of tumor behavior (1–7).In human monocytes, MCP-1 induces chemotaxis, cal-cium flux, and the respiratory burst and up-regulatesadhesion molecule expression and cytokine production(2). MCP-1 is secreted by many cell types in response tolipopolysaccharide, or cytokines including interleukin(IL) -1, IL-4, interferon g (IFN-g), and tumor necrosisfactor (TNF)-a (8, 9).
Four closely related murine and human MCPs havebeen identified, all of which exhibit monocyte che-moattractant activity in vitro. The specific functions ofmurine MCP-1 (mMCP-1) in vivo were investigated byconstruction and analysis of mMCP-1-deficient mice.Given the presence of four similar MCPs, it is surprisingthat these studies indicated that deletion of MCP-1abrogated or altered diverse acute and chronic im-mune and inflammatory processes (3–7). It is uncertainwhether expression patterns or structural characteris-tics account for the nonredundant functions exerted bymMCP-1.
At the genetic level, mMCP-1 was first characterizedas JE, an immediate-early gene that was activated inquiescent NIH 3T3 fibroblasts by exposure to plateletderived growth factor (PDGF) (12). Human MCP-1(hMCP-1) encodes a protein that is 55% homologousto its mouse counterpart (1). Despite the relative lackof coding sequence identity, mMCP-1 and hMCP-1 areboth highly expressed in diverse pathological states,
1 Current address: Department of Neurology, Stanford Uni-versity, 3801 Miranda Ave., Palo Alto, CA, USA.
2 Current address: Science Faculty, Ankara University, An-kara, Turkey.
3 Current address: Department of Oncology, SmithKlineBeecham Pharmaceuticals, King of Prussia, PA 19406, USA.
4 Current address: Princeton University, Princeton, NJ,USA.
5 Correspondence: Department of Neurosciences, Lerner Re-search Institute, NC30, Cleveland Clinic Foundation, 9500 Eu-clid Ave., Cleveland, OH 44195, USA. E-mail: [email protected]
3830892-6638/01/0015-0383 © FASEB

suggesting that the two proteins may represent func-tional orthologs. Genomic elements regulating expres-sion of the mMCP-1 gene have been characterized: fourdistal elements including two nuclear factor kappa-B(NFkB) binding sites and a seven base pair (bp)element in the 39-untranslated region of the gene wereresponsible for induction by various stimuli, includingPDGF and TNF-a (13, 14).
Both mMCP-1 and hMCP-1 are produced by astro-cytes of the central nervous system (CNS) in responseto diverse insults, including immune-mediated, post-traumatic, and ischemic inflammatory states, as well asduring HIV encephalopathy (15–21). Therefore, eluci-dating the regulation of MCP-1 in astrocytes is criticalto understanding the role of this key mediator of CNSinflammation.
We previously described the mechanism wherebyIFN-g treatment induced transcription of the hMCP-1gene in astrocytic cells (22). For these experiments, weused CRT astrocytoma cells, which closely mimic thecytokine response characteristics of human astrocytesin primary culture (23, 24). Initial studies establishedrequirement of a cis-acting gamma-activated site (GAS)and the trans-acting factor signal transducer and activa-tor of transcription (STAT)-1a for IFN-g-inducedMCP-1 transcription (22). Analysis of the proximalpromoter region revealed a GC-rich consensus bindingsite for the ubiquitous transcription factor, Sp1, cen-tered ;90 bp downstream of the GAS site. In vivogenomic footprinting (IVGF) revealed that the hMCP-1GC box was not protected until 15–30 min after IFN-gtreatment, temporally coincident both with induciblemethylation-resistance of the GAS site and transcrip-tional activation of hMCP-1 (22). Further functionalanalyses of the hMCP-1 promoter by transient transfec-tion of a series of site-directed mutants indicated acooperative interaction between the GAS element andthe GC box. Electrophoresis mobility shift assays(EMSA) showed that Sp1 was constitutively abundant innuclear extracts of CRT cells and that levels of Sp1 wereunaltered at any time point after IFN-g treatment (22).These observations led us to propose the hypothesisthat hMCP-1 induction by varied stimuli would producechanges in access to the regulatory regions of thepromoter, allowing binding of inducible transcriptionfactors (STATs, NFkB family members) to their cog-nate sites and recruitment of ubiquitous and constitu-tive Sp1 to the GC-rich element (22).
Elegant studies of the mMCP-1 gene by Boss andcolleagues confirmed and extended this hypothesis(25–6). Using a variety of approaches in Sp1-deficientmammalian and insect cells, these workers demon-strated that a proximal regulatory region containingthe Sp1 binding site was essential for mMCP-1 transcrip-tion in response to either TNF-a or PDGF, even thoughthe mechanisms of induction by these two agentsclearly differed (25, 26). The absence of functional Sp1in mammalian cells precluded in vivo assembly oftranscription factor NFkB on a distal regulatory ele-ment of the mMCP-1 promoter in TNF-a-treated cells,
despite the demonstration of abundant and functionalNFkB in vitro in cell extracts (26). Finally, blockade ofPDGF-induced MCP-1 transcription by trans-retinoicacid was associated with defective occupancy in vivo ofthe Sp1 binding site and other proximal regulatoryelements. Similar concepts had been developed byCollins et al. in studies of the vascular cell adhesionmolecule promoter (27).
During CNS inflammation, MCP-1 expression is strik-ingly transitory; this characteristic implies the presenceof a negative regulatory mechanism (15). Gene induc-tion by IFNs, which are implicated in the pathogenesisof immune-mediated inflammatory states, is known tobe transient in many cases (28). The present studyextends our characterization of IFN-g regulation ofhMCP-1 expression in astrocytoma cells. Unexpectedly,we found that the hMCP-1 gene was resistant to re-stimulation after IFN-g-induced transcription had ter-minated. This resistance to restimulation (termed arefractory state in this report) was long-lasting andoperated at the level of gene transcription. The initialIFN-g-induced transcription and postinduction refrac-tory state of hMCP-1 were both mediated through a 213bp element upstream of the structural gene, which wascharacterized in our previous report (22).
During the IFN-g-mediated refractory state, hMCP-1transcription could not be induced either by reexpo-sure to IFN-g or by heterologous stimuli such as TNF-a.The refractory state was selective for hMCP-1, as otherchemokine genes including IP-10 remained responsiveto transcriptional activators, including IFN-g. Inhibi-tion of protein synthesis during the initial exposure toIFN-g fully reversed the refractory state, suggestingdependence on newly synthesized components. Analy-sis of promoter occupancy in vivo during the refractorystate revealed impaired IFN-g-inducible protection ofthe GAS site and the GC-rich element, suggestingdecreased availability of the hMCP-1 promoter to fac-tors required for gene transcription.
This is the first report of an IFN-g-inducible tran-scriptional refractory state, and suggests mechanisms bywhich MCP-1 expression in vivo may be temporallyrestricted. Further, these data provide additional sup-port for the hypothesis that cytokine-regulated cis-elements and the GC box cooperate to govern tran-scription of both human and murine MCP-1.
MATERIALS AND METHODS
Cell culture
The CRT astrocytoma cell line, as described previously (24)was derived from a grade IV human astrocytoma. Experi-ments reported here were done between passages 10 and 40.CRT cells were routinely maintained in RPMI 1640, supple-mented with 2 mM L-glutamine and 10% fetal bovine serum.
Reagents
Purified human recombinant IFN-g (1.93107 units/mg pro-tein) was purchased from Genentech Inc. (South San Fran-
384 Vol. 15 February 2001 ZHOU ET AL.The FASEB Journal

cisco, Calif.). TNF-a was purchased from Becton-Dickinson(San Jose, Calif.). Polybrene (hexadimethrine bromide), cy-cloheximide, and dimethyl sulfoxide were purchased fromSigma Chemical Company (St. Louis, Mo.).
RNA isolation, Northern blot, and RNase protectionanalysis
Initial studies established optimal time points for determina-tion of steady-state levels of hMCP-1 mRNA by Northern andnuclease protection analyses. Although the fold induction ofMCP-1 message varied among experiments, the time courseof accumulation and decay was reproducible. Total cellularRNA was isolated from 90% confluent CRT cells using TRIzol(GIBCO BRL, Grand Island, N.Y.) according to the manufac-turer’s instructions; 30 mg of total RNA was denatured withformaldehyde, electrophoresed, and transferred to Gene-Screen nylon membrane (Biotechnology Systems, NEN Re-search Products, Boston, Mass.). Hybridization was carriedout at 42°C for 16 h in a solution containing denaturedhuman MCP-1 cDNA probe (13106 cpm/ml). A 740 bphuman MCP-1 cDNA hybridization probe was generated byPstI digestion of pGEM-hJE34 (a generous gift from Dr. B.J.Rollins, Dana-Farber Cancer Institute). The gel-isolated insertwas radiolabeled by random priming as described previously(22). Autoradiograms show results typical of those obtainedfrom three separate experiments. Subsequent analyses ofhMCP-1 mRNA accumulation were performed by RNaseprotection, using riboprobes, as described (22). These exper-iments were repeated twice and representative results areshown.
Nuclear run-on analysis
Initial studies established optimal time points for analyzinghMCP-1 gene transcription by nuclear run-on assays. For eachdata point, 5 3 106 cells at 70–80% confluency were washed,scraped in ice-cold phosphate-buffered saline (PBS), andpelleted. Nuclei were isolated and nascent transcripts werelabeled with [32P]-UTP at 25°C for 45 min, as describedpreviously (24). As hybridization substrates, plasmid DNAswere denatured and spotted onto nitrocellulose membranes,which were excised with a hole punch. Hybridization withindividual substrates was carried out in minimal volumes at42°C for 3 days, with 107 cpm/ml of radiolabeled RNA probegenerated from nuclei representing various experimentalconditions. After high-stringency washes at 65°C, individualnitrocellulose membrane circles were mounted on filterpaper supports to generate autoradiograms. For some exper-iments, hybridization signals were quantitated in a Phosphor-Imager. Transcriptional activation was calculated as theMCP-1/b-actin densitometric ratio. These experiments wererepeated twice and representative results are shown.
Promoter-reporter construction
Construction of expression plasmids containing a series ofdeletion and substitution mutants of the MCP-1 promoter,directing either CAT or luciferase reporters, has been de-scribed in detail (22, 29). GL-IP10 was generated by insertionof a 972 bp IP-10 genomic fragment into the promoterlesspGL3-basic vector (30).
Transient transfection
Polybrene with 50 mg of supercoiled test plasmid DNA wasused to transfect CRT astrocytoma cells, as described (22). As
an internal control for transfection efficiency, 1 mg of asimian virus 40 promoter-b-galactosidase reporter plasmid,pCH110 (Pharmacia, Piscataway, N.J.), was cotransfected witheach test plasmid. After cells were transfected, heat-shocked,and rested overnight, they were pooled and split into 100 mmdishes to control for differential transfection efficiency.
Initial studies established optimal time points for analyzinghMCP-1 promoter-reporter expression in transient transfec-tion assays. Using this information as a guide, cells weresubjected to various protocols of cytokine stimulation, wash-ing, and restimulation; cells were then incubated overnight incomplete cytokine-free medium to allow CAT or luciferaseprotein to accumulate, before lysis and enzyme assay.
CAT and luciferase assays were performed using standard-ized protocols (22). b-Galactosidase activity was measured incell lysates with the b-galactosidase enzyme assay system kit(Promega, Madison, Wis.). Luciferase or CAT activities ofcytokine-exposed or control cells were normalized to b-galac-tosidase activity. Results presented in this study were obtainedfrom three to four separate experiments. For statistical anal-ysis, the t test was used to evaluate paired samples, withsignificance set at P,0.05.
IVGF
Initial studies established optimal time points for analyzingIFN-g-induced occupancy of the hMCP-1 promoter by IVGF.In vivo methylation of cellular DNA and DNA preparationwere performed as described (14, 22, 30). Ligation-mediatedpolymerase chain reaction was carried out according to theprocedure of Mueller et al. (31), as adapted for mMCP-1 byPing et al. and with modifications for hMCP-1, as we previ-ously described (22, 30). Both strands of the 213 bp promoterproximal region of the hMCP-1 gene were analyzed. Thisanalysis was repeated twice; representative results for thenoncoding strand (the site of asymmetric Sp1 binding to theGC-rich element) are shown. The annealing temperatures forthe coding strand primers were 59°C, 66°C, and 69°C. Codingstrand primers were:
59-TGTGGTTCAAGGAGAAGAAGAGGG-3959-GCTATGAGCAGCAGGCAC-AGAAGG-3959-CAGGCACAGAAGGGCGGCAGAGAC-39.The annealing temperatures for noncoding strand primers
were 59°C, 66°C, and 69°C. Primers for the noncoding strandwere:
59-CCCTCTTAGTTCACATCTGTGGTCAG-3959-CCCATCCTCCCCATTTGCTCATT-3959-TCCCCATTTGCTCATTTGGTCTCAGCAG-39.
RESULTS
After induction with IFN-g, the hMCP-1 gene isrefractory to restimulation
As shown in Fig. 1, hMCP-1 mRNA accumulated rapidlyafter IFN-g treatment reached a maximum by 8 h anddecayed markedly by 24 h in the continued presence ofIFN-g.
To determine (Fig. 2) whether this down-regulationwas associated with postinduction repression ofhMCP-1 mRNA expression, we incubated CRT cellswith IFN-g for 24 h, terminated IFN-g stimulation byextensive washing, incubated in IFN-g-free media forvarying times, and restimulated with IFN-g. Unexpect-edly, hMCP-1 was resistant to re-induction by IFN-g
385IFN-g-INDUCED REFRACTORY STATE OF THE HMCP-1 GENE

after down-regulation (Fig. 2, lanes 4–6 vs. lane 2). Thisrefractory state was equally robust after 4, 8, or 24 h ofIFN-g-free incubation (Fig. 2, lanes 4–6). Thus, IFN-g-mediated induction of MCP-1 was succeeded by arefractory state during which the gene was resistant tore-induction with IFN-g. In parallel studies, the IP-10gene remained responsive to reinduction with IFN-g(Fig. 2, lanes 4–6), indicating that IFN-g receptordown-regulation or transcription factor exhaustion wasnot responsible for the inability of restimulation withIFN-g to induce hMCP-1 mRNA accumulation.
After induction with IFN-g, the hMCP-1 gene isresistant to restimulation with TNF-a
TNF-a is a potent and well-characterized stimulus forMCP-1 transcription. We asked whether the hMCP-1gene was resistant to induction with TNF-a during the
IFN-g-induced refractory state. After treatment for 4 hwith either IFN-g or TNF-a (Fig. 3, lanes 2, 5), CRTcells accumulated abundant hMCP-1 mRNA. Whencells were pretreated with IFN-g for 4 h, washed exten-sively, incubated in cytokine-free medium for 4 h, andreexposed either to IFN-g or TNF-a, MCP-1 mRNA wasnot induced above baseline levels (Fig. 3, compare lane1 with lanes 3 and 4). This result indicated that theIFN-g-mediated postinduction refractory state ren-dered the hMCP-1 gene resistant to induction byTNF-a, an efficient heterologous stimulus.
The refractory state of the MCP-1 gene occurs at thelevel of transcription
To determine whether the IFN-g-induced refractorystate of the hMCP-1 gene was governed at the transcrip-tional level, nuclear run-on experiments were con-ducted. In IFN-g-treated CRT cells, hMCP-1 transcrip-
Figure 2. Refractory state of hMCP-1 but not IP-10 afterinduction with IFN-g. CRT cells were treated with IFN-g (100U/ml) for 4 h (lane 2) or 24 h (lane 3) before Northernanalysis, demonstrating down-regulation of hMCP-1 but notIP-10 mRNA after 24 h exposure to IFN-g. The refractory stateof hMCP-1 is shown in lanes 4–6: after 24 h treatment withIFN-g, cells were washed five times with PBS and incubated incytokine-free medium for 4 h (lane 4), 8 h (lane 5), or 24 h(lane 6). Cells were then restimulated with IFN-g for 4 h(lanes 4–6). hMCP-1, IP-10, and b-actin mRNAs were ana-lyzed by Northern blotting, and quantitated on a Phospho-rImager; bar histograms indicate chemokine/b-actin ratios.Representative results of three independent experiments areshown.
Figure 1. Time course of hMCP-1 mRNA accumulation in-duced by IFN-g. CRT cells were treated for the indicatedtimes with IFN-g (100 U/ml); mRNA levels were determinedby Northern blot analysis and results quantitated on a Phos-phorImager (Molecular Dynamics, Sunnyvale, Calif.), withhMCP-1/b-actin ratios shown in the bar histogram. Theresults shown are representative of three separate experi-ments.
386 Vol. 15 February 2001 ZHOU ET AL.The FASEB Journal

tion was induced at 2 h and declined markedly 1 h later(Fig. 4: compare 2 h and 3 h). Restimulation of cellswith IFN-g failed to recover transcription of thehMCP-1 gene (Fig. 4: 3 h1rest1restim).
The transcriptional refractory state of the MCP-1gene was selective, as the IP-10 gene remained fullyresponsive to IFN-g restimulation (Fig. 4). The tran-scription factor STAT-1a is required for the IFN-g-inducible expression of both hMCP-1 and IP-10 in CRTcells (22, 30, 32). Therefore, these data indicated thatpost-IFN-g receptor events, including activation of theSTAT-1a transcription factor, were maintained duringthe refractory state and that their failure did notaccount for the unresponsive state of the hMCP-1 gene.
The IFN-g-induced refractory state of the hMCP-1gene is directed by a 213 bp element upstream of thestructural gene
We previously characterized a 213 bp element of thehMCP-1 gene that directed the transcriptional responseto IFN-g in CRT cells (22). To test whether the IFN-g-induced refractory state was governed by upstreamelements of the hMCP-1 gene, we modified transienttransfection assays, using hMCP-1 promoter-reporterconstructs documented in previous studies. Thepremise of these experiments was that repetitive cyclesof cytokine stimulation should produce additive accu-mulation of CAT or luciferase activity if the promoterremained responsive to restimulation. Therefore, ex-periments were performed to determine whether re-peated cycles of stimulation would result in additiveaccumulation of hMCP-1 promoter-reporter CAT orluciferase activity.
We tested this assay, using a previously characterizedIP-10 promoter-reporter (GL-IP-10) (30). Luciferaseactivity was strongly induced by IFN-g (100 U/ml)treatment (Fig. 5A, 6 h). After extensive washing,incubation for 3 h in cytokine-free medium, and IFN-g
(100 U/ml) restimulation, GL-IP-10 reporter activityaccumulated in a nearly additive fashion (Fig. 5A,compare 6 h with 6 h1Restim), indicating that thereporter protein remained stable in IFN-g-treated CRTcells during the time frame of the experiment. Thisresult indicated that a transcriptional refractory statecould be detected as a failure of reporter activity toaccumulate in additive fashion after repetitive cycles ofcytokine stimulation.
Serial deletion constructs containing various lengthof 59-flanking sequences of the hMCP-1 promoter wereinserted upstream of CAT reporter gene, reserved ascontrols (Fig. 5B, lanes 1 for each construct), orinduced (Fig. 5B, lanes 2) with IFN-g (100 U/ml; 2 h).Significantly increased CAT activity was induced byIFN-g (P,0.001; t test) from constructs containing 3.5kb, 394 bp, 292 bp, or 213 bp of the hMCP-1 promoter,whereas a 141 bp hMCP-1 promoter-reporter constructfailed to respond to stimulation (Fig. 5B, compare lanes1 and 2). These results reproduced data from ourprevious report (22) indicating that the 213 bp con-struct contains a minimal IFN-g-inducible hMCP-1 pro-moter.
Figure 4. The IFN-g-induced refractory state occurs at thelevel of transcription and affects hMCP-1 but not IP-10.Transcription rates of the hMCP-1, IP-10, and b-actin geneswere analyzed by evaluation of nascent transcripts, afterisolation of nuclei from cells exposed to IFN-g (500 U/ml) forvarying times. For restimulation, cells were exposed for 3 h toIFN-g, washed, incubated in cytokine-free medium for 1 h,and exposed IFN-g for 2 h. Densitometric signals werequantitated on a PhosphorImager and chemokine gene tran-scription was normalized to b-actin. Representative data fromone of two experiments are shown.
Figure 3. During the IFN-g-mediated refractory state,hMCP-1 is resistant to in-duction by TNF-a. Duringthe first treatment, as indi-cated, CRT cells were incu-bated in cytokine-free me-dium (lanes 1, 2, 5) orinduced with IFN-g (100U/ml; lanes 3 and 4) for4 h. Cells were then exten-sively washed and incu-bated for 4 h during thesecond treatment, as indi-cated, with medium (lane1), IFN-g (lanes 2, 3), or
TNF-a (lanes 4, 5) before preparation of total cellular RNAand analysis of MCP-1 and g-actin accumulation by nucleaseprotection assay. Representative results from two separateexperiments are shown. After exposure to IFN-g during thefirst treatment (lane 4), CRT cells were resistant to inductionof hMCP-1 by TNF-a.
387IFN-g-INDUCED REFRACTORY STATE OF THE HMCP-1 GENE
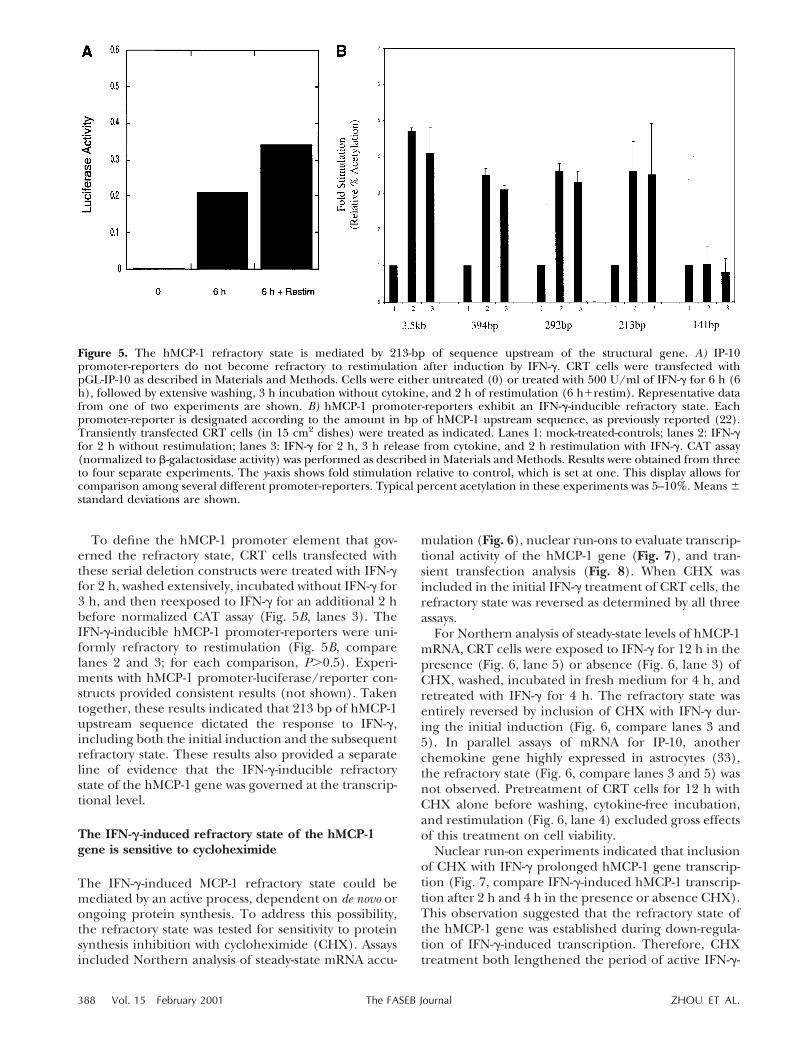
To define the hMCP-1 promoter element that gov-erned the refractory state, CRT cells transfected withthese serial deletion constructs were treated with IFN-gfor 2 h, washed extensively, incubated without IFN-g for3 h, and then reexposed to IFN-g for an additional 2 hbefore normalized CAT assay (Fig. 5B, lanes 3). TheIFN-g-inducible hMCP-1 promoter-reporters were uni-formly refractory to restimulation (Fig. 5B, comparelanes 2 and 3; for each comparison, P.0.5). Experi-ments with hMCP-1 promoter-luciferase/reporter con-structs provided consistent results (not shown). Takentogether, these results indicated that 213 bp of hMCP-1upstream sequence dictated the response to IFN-g,including both the initial induction and the subsequentrefractory state. These results also provided a separateline of evidence that the IFN-g-inducible refractorystate of the hMCP-1 gene was governed at the transcrip-tional level.
The IFN-g-induced refractory state of the hMCP-1gene is sensitive to cycloheximide
The IFN-g-induced MCP-1 refractory state could bemediated by an active process, dependent on de novo orongoing protein synthesis. To address this possibility,the refractory state was tested for sensitivity to proteinsynthesis inhibition with cycloheximide (CHX). Assaysincluded Northern analysis of steady-state mRNA accu-
mulation (Fig. 6), nuclear run-ons to evaluate transcrip-tional activity of the hMCP-1 gene (Fig. 7), and tran-sient transfection analysis (Fig. 8). When CHX wasincluded in the initial IFN-g treatment of CRT cells, therefractory state was reversed as determined by all threeassays.
For Northern analysis of steady-state levels of hMCP-1mRNA, CRT cells were exposed to IFN-g for 12 h in thepresence (Fig. 6, lane 5) or absence (Fig. 6, lane 3) ofCHX, washed, incubated in fresh medium for 4 h, andretreated with IFN-g for 4 h. The refractory state wasentirely reversed by inclusion of CHX with IFN-g dur-ing the initial induction (Fig. 6, compare lanes 3 and5). In parallel assays of mRNA for IP-10, anotherchemokine gene highly expressed in astrocytes (33),the refractory state (Fig. 6, compare lanes 3 and 5) wasnot observed. Pretreatment of CRT cells for 12 h withCHX alone before washing, cytokine-free incubation,and restimulation (Fig. 6, lane 4) excluded gross effectsof this treatment on cell viability.
Nuclear run-on experiments indicated that inclusionof CHX with IFN-g prolonged hMCP-1 gene transcrip-tion (Fig. 7, compare IFN-g-induced hMCP-1 transcrip-tion after 2 h and 4 h in the presence or absence CHX).This observation suggested that the refractory state ofthe hMCP-1 gene was established during down-regula-tion of IFN-g-induced transcription. Therefore, CHXtreatment both lengthened the period of active IFN-g-
Figure 5. The hMCP-1 refractory state is mediated by 213-bp of sequence upstream of the structural gene. A) IP-10promoter-reporters do not become refractory to restimulation after induction by IFN-g. CRT cells were transfected withpGL-IP-10 as described in Materials and Methods. Cells were either untreated (0) or treated with 500 U/ml of IFN-g for 6 h (6h), followed by extensive washing, 3 h incubation without cytokine, and 2 h of restimulation (6 h1restim). Representative datafrom one of two experiments are shown. B) hMCP-1 promoter-reporters exhibit an IFN-g-inducible refractory state. Eachpromoter-reporter is designated according to the amount in bp of hMCP-1 upstream sequence, as previously reported (22).Transiently transfected CRT cells (in 15 cm2 dishes) were treated as indicated. Lanes 1: mock-treated-controls; lanes 2: IFN-gfor 2 h without restimulation; lanes 3: IFN-g for 2 h, 3 h release from cytokine, and 2 h restimulation with IFN-g. CAT assay(normalized to b-galactosidase activity) was performed as described in Materials and Methods. Results were obtained from threeto four separate experiments. The y-axis shows fold stimulation relative to control, which is set at one. This display allows forcomparison among several different promoter-reporters. Typical percent acetylation in these experiments was 5–10%. Means 6standard deviations are shown.
388 Vol. 15 February 2001 ZHOU ET AL.The FASEB Journal

induced hMCP-1 gene transcription (Fig. 7) and abro-gated the refractory state (Fig. 6). These results arecompatible with those previously reported by Larnerand colleagues, who described IFN-b-mediated regula-tion of two genes. These workers reported that induc-tion of these two genes by IFN-b was followed by adramatic transcriptional down-regulation, which wasabrogated by CHX, suggesting an active mechanism ofsuppression (34). Exposure to CHX did not extend, butmoderately impaired, IFN-g-induced transcription ofthe IP-10 gene (Fig. 7), further indicating selectiveregulation of hMCP-1 by IFN-g.
Transient transfection experiments (Fig. 5B) demon-strated that the refractory state of the hMCP-1 gene wasgoverned by upstream elements of the gene. This assaywas also used to address the dependence of the refractorystate on ongoing protein synthesis. As previously ob-served, repetitive cycles of stimulation with IFN-g failed toinduce CAT reporter activity above levels obtained afterindividual cycles of stimulation (Fig. 8, compare ‘stim’
Figure 6. CHX treatment during IFN-g stimulation abolishesthe subsequent refractory state of the hMCP-1 gene. CRT cellswere reserved as controls (lane 1) or treated for 4 h (lane 2)with IFN-g (100 U/ml) to demonstrate induction of hMCP-1or IP-10 mRNA, as assayed by Northern blotting. To analyzethe refractory state (lanes 3 and 5), CRT cells were treatedwith IFN-g (100 U/ml) for 12 h, extensively washed andincubated in cytokine-free medium for 1 h, and retreated for4 h with IFN-g before analysis of chemokine and b-actinmRNAs. With the initial 12 h incubation, CHX (50 mg/ml)was either included (lane 5) or omitted (lane 3). Lane 4: as aviability control for exposure to CHX, CRT cells were incu-bated with CHX alone (50 mg/ml) for 12 h before washing,rest for 1 h, treatment for 4 h with IFN-g (100 U/ml), andNorthern analysis.
Figure 7. CHX treatment during IFN-g stimulation prolongstranscription of the hMCP-1, but not the IP-10, gene. CRTcells were treated with IFN-g (500 U/ml) for 2 or 4 h, asindicated, with or without CHX (50 mg/ml). Autoradiogramsdemonstrating transcription of hMCP-1, IP-10, and b-actinwere generated on a PhosphorImager. pBS (pBluescript;Promega) vector: negative control for nonspecific hybridiza-tion. Transcription of hMCP-1 but not IP-10 was markedlyprolonged at both 2 h and 4 h by inclusion of CHX withIFN-g. CHX alone did not stimulate transcription of hMCP-1(not shown).
Figure 8. CHX treatment during IFN-g stimulation abrogatesresistance of a minimal hMCP-1 promoter-reporter to IFN-grestimulation. CRT cells were transfected with the 213 bphMCP-1 promoter-reporter, as described in Materials andMethods; cells were then incubated in the presence orabsence of IFN-g with or without CHX, as indicated, beforeincubation overnight in medium, lysis, and normalized assayof CAT reporter expression. Control: mock-treated; stim:treated with IFN-g for 2 h; stim-re/stim: 2 h of IFN-g,extensive washing; 3 h of cytokine-free incubation and 2 h ofrestimulation with IFN-g; stim-re/stim-CHX: 2 h of CHX withIFN-g, extensive washing; 3 h of cytokine-free incubation and2 h of restimulation with IFN-g alone; CHX: cycloheximidealone for 2 h; stim1CHX: treatment with CHX and IFN-g for2 h. The y-axis shows fold stimulation relative to control,which is set at one.
389IFN-g-INDUCED REFRACTORY STATE OF THE HMCP-1 GENE

with stim-re/stim). However, when CHX was includedwith IFN-g during the initial induction (stim-re/stim-CHX), accumulation of CAT activity increased markedlyafter a second cycle of stimulation with IFN-g (Fig. 8,compare stim-re/stim with stim-re/stim-CHX). CHXalone did not induce expression of the promoter-reporter(Fig. 8, compare control with CHX).
Addition of CHX to IFN-g did not augment inductionof the promoter-reporter (Fig. 8, compare stim withstim-CHX). This result was compatible with observationsshown in Figs. 6 and 7 in which CHX prolonged transcrip-tion of the hMCP-1 gene, but did not markedly increasemRNA accumulation. Together, observations describedin Figs. 6–8 indicated that the IFN-g-induced transcrip-tional refractory state of the hMCP-1 gene was reversedentirely by inhibiting protein synthesis with CHX.
In vivo state of the IFN-g-regulated MCP-1 promoter
Our previous analysis showed that IFN-g-mediated oc-cupancy of the upstream GAS and GC-rich elements ofthe hMCP-1 gene occurred coincident with hMCP-1gene transcription (22). Functional assays of site-di-rected mutants of the GAS site and GC box demon-strated the critical importance of both elements forefficient induction of hMCP-1 gene transcription byIFN-g (22). To determine the in vivo occupancy ofthese elements during the IFN-g-induced transcrip-tional refractory state, genomic footprinting analyses ofthe hMCP-1 promoter were conducted (Fig. 9). CRTcells were treated for 15 min with IFN-g and immedi-ately assayed (during gene induction) or washed andincubated in IFN-g-free medium for 3 h before retreat-ment with IFN-g (during the refractory state).
After 15 min of IFN-g treatment (Fig. 9A, lane 3),strong protection of the core residues of the GAS site—G2209 and G2210— was observed (arrows), along withmethylation hypersensitivity of flanking residue G2201
and weak protection of G2200. Residues outside theregulatory element (G2154 and nearby G residues) wereunaffected. After 5 h, the pattern of methylation resis-tance of these GAS residues returned to that observed inmock-treated cells (Fig. 9A, compare lanes 2 and 5).During the refractory state, IFN-g-induced occupancy atthe GAS element of the hMCP-1 promoter was markedlyaltered, and resembled the pattern observed in mock-treated cells or after the termination of transcription after5 h of IFN-g (Fig. 9A). In particular, protection at G2209,G2210, and G2200 was not induced by IFN-g treatmentduring the refractory state (Fig. 9A, lane 4). ResidueG2201 exhibited methylation hypersensitivity during therefractory state, suggesting incomplete assembly of tran-scription factors on the refractory hMCP-1 promoter.Concurrently, after 15 min of IFN-g treatment (Fig. 9B,lane 3) the GC box of the noncoding strand became DMSresistant, most evident at G2123 and nearby residue G2140
(arrowhead). Located between the two regulatory ele-ments, residue G2154 was unaffected by cytokine treat-ment at any time (Fig. 9A, B). During the refractoryperiod, IFN-g treatment failed to induce altered methyl-
ation sensitivity in the extended GC box (Fig. 9B, com-pare lanes 3 and 4). These results suggested that theIFN-g-induced refractory state of the hMCP-1 gene inCRT astrocytoma cells was associated with impaired accessof critical transcription factors to regulatory elements ofthe gene.
DISCUSSION
In the present study, we provide evidence that IFN-g-induced transcription of the hMCP-1 gene in CRT
Figure 9. Altered hMCP-1 promoter occupancy during theIFN-g-induced refractory state. A) Occupancy of the hMCP-1GAS element. The pattern of methylation resistance andhypersensitivity in the region of the GAS site of the MCP-1promoter (noncoding strand) is shown. Lane assignment andsymbol representation are indicated in the legend of panel B.Two GAS element methylation-resistant residues are indi-cated (arrows). B) Occupancy of the hMCP-1 GC box. CRTcells were mock-treated (lane 2) or exposed to IFN-g (500U/ml) for 15 min (lane 3); treated with IFN-g for 15 min,followed by extensive washing, 3 h of cytokine-free incuba-tion, and 15 min of restimulation (lane 4); or treated withIFN-g for 5 h (lane 5). Genomic DNA was prepared andanalyzed by IVGF as described in Materials and Methods.Lane 1 shows analysis of DNA methylated in vitro. The regionof the GC box on the noncoding strand is shown in theautoradiogram, with numbering by reference to the transcrip-tional start site. Arrow indicates an IFN-g-inducible methyla-tion-resistant guanine residue.
390 Vol. 15 February 2001 ZHOU ET AL.The FASEB Journal

astrocytoma cells is succeeded by a refractory state ofthe gene, a novel phenomenon for IFN-g-induciblegenes. Further, this postinduction repression is medi-ated through upstream elements of the hMCP-1 geneand depends on an active process. Our conclusions arebased on the following observations: 1) treatment ofCRT cells with IFN-g resulted in a rapid, transientaccumulation of hMCP-1 mRNA, governed at the levelof transcription; 2) after induction by IFN-g, thehMCP-1 gene (but not other IFN-g-responsive genes)became resistant to restimulation with either IFN-g orTNF-a; 3) the refractory state was abrogated by expo-sure to CHX during the initial induction with IFN-g.
The phenomenon of a postinduction transcriptionalrefractory state has been well established, and severalmechanisms, including inducible repressors, have beenpostulated (34, 35). Maniatis et al. examined virus-mediated induction and postinduction repression ofthe IFN-b gene. It was convincingly demonstrated thata virus-inducible cellular component termed PRDI-BFIacted as a postinduction repressor by binding directlyto positive regulatory elements of the IFN-b promoter(36–38). Larner and Darnell showed that the transcrip-tional response of two IFN-b-inducible genes was fol-lowed by a potent refractory state, which required denovo or ongoing protein synthesis (34). Our studies alsosuggested that the refractory state of the hMCP-1 genewas dependent on newly expressed proteins. In partic-ular, CHX completely reversed IFN-g-induced resis-tance to restimulation (Fig. 6) and prolonged theduration of hMCP-1 transcription (Fig. 7). These re-sults are thoroughly compatible with those reported byLarner and colleagues in their studies of postinductionrepression of IFN-b-inducible genes (34). Consistentwith this interpretation, CHX treatment also abolishedthe IFN-g-induced resistance of hMCP-1 promoter-reporters to restimulation (Fig. 8).
In vivo genomic footprinting (IVGF) proved useful toanalyze the refractory state of the hMCP-1 promoter.This analysis was performed after cells were treated for15 min with IFN-g (Fig. 9, lanes 3) or treated, rested,and restimulated with IFN-g for 15 min (Fig. 9, lanes 4)during the refractory state. The results indicated sub-stantial differences in hMCP-1 promoter occupancy incells permissive for transcription (Fig. 9, lanes 3) ascompared with cells expressing the refractory state (Fig.9, lanes 4), suggesting that impaired access of thepromoter to STAT-1a homodimers (at the GAS site)and Sp1 (at the GC box) rendered the promotertranscriptionally inactive (22).
Recent work by Boss and colleagues characterizedthe transcriptional response of the mMCP-1 gene tovaried stimuli, including PDGF and TNF-a (25–26).Our current results demonstrate strikingly similar reg-ulation of hMCP-1 and mMCP-1. In both cases, induc-ible and constitutive transcription factors are readilydetected in cell extracts by EMSA yet fail to associate invivo with MCP-1 promoters unless conditions are per-missive for gene transcription. Further, the presentreport supports the critical involvement of the GC box
for expression of the hMCP-1 gene, an attribute docu-mented by Boss et al. for mMCP-1.
We considered the possible involvement of previ-ously described IFN-g-inducible inhibitory trans-actingfactors such as interferon regulatory factor 2 (IRF-2) orICSBP. Sequence analysis of the 213 bp hMCP-1 regu-latory region revealed an IRF binding motif near thetranscription start site, but no binding activity wasdetected by EMSA when using a probe derived fromthis region (not shown).
As shown in our previous studies, STAT-1a wasessential for IFN-g induction of the hMCP-1 gene.Therefore, the recently described suppressors of cyto-kine signaling (SOCS) proteins (39–41) were potentialmediators of the specific down-regulation and refrac-tory state of hMCP-1. However, intact expression ofSTAT-dependent genes such as IP-10 during the refrac-tory state excluded this possibility, as SOCS act up-stream of tyrosine phosphorylation, nuclear transloca-tion, and DNA binding by STATs (39). For similarreasons, the inducible (42) Janus kinase inhibitorswould also appear to be unlikely candidates to explainour findings in these studies.
In summary, this report documents a selective refrac-tory state of hMCP-1, an IFN-g-inducible gene. Thedown-regulation and refractory state of the hMCP-1gene indicate the presence of a gene-specific induciblenegative feedback mechanism that operates at thetranscriptional level. Elucidating this refractory statewill provide useful information about how hMCP-1, akey regulatory factor for CNS inflammation, is gov-erned in physiology and disease.
This study was supported by NIH grants 2RO1-NS32151,2PO1-CA62220 and National Multiple Sclerosis Society grantRG2362 to R.M.R. and by the Williams Family MultipleSclerosis Research Fund.
REFERENCES
1. Rollins, B., Stier, P., Ernst, T., and Wong, G. (1989) The humanhomolog of the JE gene encodes a monocyte secretory protein.Mol. Cell. Biol. 9, 4687–4689
2. Rollins, B. J. (1996) Monocyte chemoattractant protein 1: apotential regulator of monocyte recruitment in inflammatorydisease. Mol. Med. Today 2, 198–204
3. Gu, L., Rutledge, B., Fiorillo, J., Ernst, C., Grewal, I., Flavell, R.,Gladue, R., and Rollins, B. (1997) In vivo properties of mono-cyte chemoattractant protein-1. J. Leukoc. Biol. 62, 577–580
4. Lu, B., Rutledge, B. J., Gu, L., Fiorillo, J., Lukacs, N. W., Kunkel,S. L., North, R., Gerard, C., and Rollins, B. J. (1998) Abnormal-ities in monocyte recruitment and cytokine expression in mono-cyte chemoattractant protein 1-deficient mice. J. Exp. Med. 187,601–608
5. Gu, L., Okada, Y., Clinton, S. K., Gerard, C., Sukhova, G. K.,Libby, P., and Rollins, B. J. (1998) Absence of monocytechemoattractant protein-1 reduces atherosclerosis in low densitylipoprotein receptor-deficient mice. Mol. Cell. 2, 275–281
6. Gosling, J., Slaymaker, S., Gu, L., Tseng, S., Zlot, C. H., Young,S. G., Rollins, B. J., and Charo, I. F. (1999) MCP-1 deficiencyreduces susceptibility to atherosclerosis in mice that overexpresshuman apolipoprotein B. J. Clin. Invest. 103, 773–778
7. Gu, L., Tseng, S., Horner, R. M., Tam, C., Loda, M., and Rollins,B. J. (2000) Control of TH2 polarization by the chemokine
391IFN-g-INDUCED REFRACTORY STATE OF THE HMCP-1 GENE

monocyte chemoattractant protein-1. Nature (London) 404, 407–411
8. Rollins, B. J. (1997) Chemokines. Blood 90, 909–9289. Rollins, B., ed. (1999) Chemokines and Cancer, Humana Press,
Totowa, N.J.10. Ransohoff, R. M. (1997) Chemokines in neurological disease
models: correlation between chemokine expression patternsand inflammatory pathology. J. Leukoc. Biol. 62, 645–652
11. Ransohoff, R. M., and Tani, M. (1998) Do chemokines mediateleukocyte recruitment in post-traumatic CNS inflammation?Trends Neurosci. 21, 154–159
12. Cochran, B. J., Reffel, A. C., and Stiles, C. D. (1983) Molecularcloning of gene sequences regulated by platelet-derived growthfactor. Cell 33, 939–947
13. Freter, R. R., Irminger, J.-C., Porter, J. A., Jones, S. D., and Stiles,C. D. (1992) A novel 7-nucleotide motif located in 39 untrans-lated sequences of the immediate-early gene set mediates plate-let-derived growth factor induction of the JE gene. Mol. Cell.Biol. 12, 5288–5300
14. Ping, D., Jones, P. L., and Boss, J. M. (1996) TNF regulates thein vivo occupancy of both distal and proximal regulatory regionsof the MCP-1/JE gene. Immunity 4, 455–469
15. Ransohoff, R. M., Hamilton, T. A., Tani, M., Stoler, M. H., Shick,H. E., Major, J. A., Estes, M. L., Thomas, D. M., and Tuohy, V. K.(1993) Astrocyte expression of mRNA encoding cytokines IP-10and JE/MCP-1 in experimental autoimmune encephalomyelitis.FASEB J. 7, 592–602
16. Gourmala, N. G., Buttini, M., Limonta, S., Sauter, A., andBoddeke, H. W. (1997) Differential and time-dependent expres-sion of monocyte chemoattractant protein-1 mRNA by astro-cytes and macrophages in rat brain: effects of ischemia andperipheral lipopolysaccharide administration. J. Neuroimmunol.74, 35–44
17. Conant, K., Garzino-Demo, A., Nath, A., McArthur, J., Halliday,W., Power, C., Gallo, R., and Major, E. (1998) Induction ofmonocyte chemoattractant protein-1 in HIV-1 Tat stimulatedastrocytes and elevation in AIDS dementia. Proc. Natl. Acad. Sci.USA 95, 3117–3121
18. McManus, C., Berman, J. W., Brett, F. M., Staunton, H., Farrell,M., and Brosnan, C. F. (1998) MCP-1, MCP-2 and MCP-3expression in multiple sclerosis lesions: an immunohistochem-ical and in situ hybridization study. J. Neuroimmunol. 86, 20–29
19. Van Der Voorn, P., Tekstra, J., Beelen, R. H., Tensen, C. P., VanDer Valk, P., and De Groot, C. J. (1999) Expression of MCP-1 byreactive astrocytes in demyelinating multiple sclerosis lesions.Am. J. Pathol. 154, 45–51
20. Simpson, J. E., Newcombe, J., Cuzner, M. L., and Woodroofe,M. N. (1998) Expression of monocyte chemoattractant pro-tein-1 and other beta-chemokines by resident glia and inflam-matory cells in multiple sclerosis lesions. J. Neuroimmunol. 84,238–249
21. Glabinski, A. R., Balasingam, V., Tani, M., Kunkel, S. L., Strieter,R. M., Yong, V. W., and Ransohoff, R. M. (1996) Chemokinemonocyte chemoattractant protein-1 is expressed by astrocytesafter mechanical injury to the brain. J. Immunol. 156, 4363–4368
22. Zhou, Z. H., Chaturvedi, P., Han, Y.-L., Aras, S., Li, Y.-S.,Kolattukudy, P. E., Ping, D., Boss, J. M., and Ransohoff, R. M.(1998) Interferon-g induction of the human monocyte che-moattractant (hMCP)-1 gene in astrocytoma cells: functionalinteraction between a gamma activated site (GAS) and GC-richelement. J. Immunol. 160, 3908–3917
23. Barna, B. P., Chou, S. M., Jacobs, B., Yen-Lieberman, B., andRansohoff, R. M. (1989) Interferon-beta impairs induction ofHLA-DR antigen expression in cultured adult human astrocytes.J. Neuroimmunol. 23, 45–53
24. Ransohoff, R. M., Devajyothi, C., Estes, M., Babcock, G., Rudick,R., Frohman, E., and Barna, B. (1991) Interferon-b specificallyinhibits interferon-g-induced class II major histocompatibilitycomplex gene transcription in a human astrocytoma cell line.J. Neuroimmunol. 33, 103–112
25. Ping, D., Boekhoudt, G., and Boss, J. M. (1999) trans-Retinoicacid blocks platelet-derived growth factor-BB-induced expres-sion of the murine monocyte chemoattractant-1 gene by block-
ing the assembly of a promoter proximal Sp1 binding site.J. Biol. Chem. 274, 31909–31916
26. Ping, D., Boekhoudt, G., Zhang, F., Morris, A., Philipsen, S.,Warren, S. T., and Boss, J. M. (2000) Sp1 binding is critical forpromoter assembly and activation of the MCP-1 gene by tumornecrosis factor. J. Biol. Chem. 275, 1708–1714
27. Neish, A. S., Khachigian, L. M., Park, A., Baichwal, V. R., andCollins, T. (1995) Sp1 is a component of the cytokine-inducibleenhancer in the promoter of vascular cell adhesion molecule-1.J. Biol. Chem. 270, 28903–28909
28. Friedman, R. L., Manly, S. P., McMahon, M., Kerr, I. M., andStark, G. R. (1984) Transcriptional and post-transcriptionalregulation of interferon-induced gene expression in humancells. Cell 38, 745–755
29. Li, Y. S., Shyy, Y. J., Wright, J. G., Valente, A. J., Cornhill, J. F.,and Kolattukudy, P. E. (1993) The expression of monocytechemotactic protein (MCP-1) in human vascular endotheliumin vitro and in vivo. Mol. Cell. Biochem. 126, 61–68
30. Majumder, S., Zhou, L. Z., Chaturvedi, P., Babcock, G., Aras, S.,and Ransohoff, R. M. (1998) p48/STAT-1alpha-containing com-plexes play a predominant role in induction of IFN-gamma-inducible protein, 10 kDa (IP-10) by IFN-gamma alone or insynergy with TNF-alpha. J. Immunol. 161, 4736–4744
31. Mueller, P. R., Garrity, P. A., and Wold, B. (1994) Ligation-mediated PCR for genomic sequencing and foot printing. InCurrent Protocols in Molecular Biology (Ausubel, F. M., Brent, R.,Kingston, R. E., Moore, D. D., Seidman, J. G., Smith, J. A., andStruhl, K., eds) Vol. 2, John Wiley and Sons, Boston
32. Majumder, S., Zhou, Z.-H. L., and Ransohoff, R. (1996) Tran-scriptional regulation of chemokine gene expression in astro-cytes. J. Neurosci. Res. 45, 758–769
33. Tani, M., Glabinski, A. R., Tuohy, V. K., Stoler, M. H., Estes,M. L., and Ransohoff, R. M. (1996) In situ hybridization analysisof glial fibrillary acidic protein mRNA reveals evidence ofbiphasic astrocyte activation during acute experimental autoim-mune encephalomyelitis. Am. J. Pathol. 148, 889–896
34. Larner, A. C., Chaudhuri, A., and Darnell, J. E., Jr. (1986)Transcriptional induction by interferon. New protein(s) deter-mine the extent and length of the induction. J. Biol. Chem. 261,453–459
35. Pine, R., Decker, T., Kessler, D. S., Levy, D. E., and Darnell, J. E.,Jr. (1990) Purification and cloning of interferon-stimulatedgene factor 2 (ISGF2): ISGF2 (IRF-1) can bind to the promotersof both b interferon- and interferon-stimulated genes but is nota primary transcriptional activator of either. Mol. Cell. Biol. 10,2448–2457
36. Goodbourne, S., and Maniatis, T. (1988) Overlapping positiveand negative regulatory domains of the human beta-interferongene. Proc. Natl. Acad. Sci. USA 85, 1447–1451
37. Keller, A. D., and Maniatis, T. (1991) Identification and char-acterization of a novel repressor of beta-interferon gene expres-sion. Genes Dev. 5, 868–879
38. Maniatis, T., Whittemore, L. A., and Du, W. (1992) Positive andnegative control of human interferon-beta gene expression. InTranscriptional regulation, Part 2 (McKnight, S. L., ed) pp. 1193–1220, Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
39. Song, M. M., and Shuai, K. (1998) The suppressor of cytokinesignaling (SOCS) 1 and SOCS3 but not SOCS2 proteins inhibitinterferon-mediated antiviral and antiproliferative activities.J. Biol. Chem. 273, 35056–35062
40. Starr, R., and Hilton, D. J. (1998) SOCS: suppressors of cytokinesignalling. Int J. Biochem. Cell Biol. 30, 1081–1085
41. Nicholson, S. E., and Hilton, D. J. (1998) The SOCS proteins: anew family of negative regulators of signal transduction. J. Leu-koc. Biol. 63, 665–668
42. Sakamoto, H., Yasukawa, H., Masuhara, M., Tanimura, S.,Sasaki, A., Yuge, K., Ohtsubo, M., Ohtsuka, A., Fujita, T., Ohta,T., Furukawa, Y., Iwase, S., Yamada, H., and Yoshimura, A.(1998) A Janus kinase inhibitor. JAB, is an interferon-gamma-inducible gene and confers resistance to interferons. Blood 92,1668–1676
Received for publication May 26, 2000.Revised for publication July 14, 2000.
392 Vol. 15 February 2001 ZHOU ET AL.The FASEB Journal




















