
Dynamic regulation of the proinflammatory cytokine, interleukin-1β: Molecular biology for...
33
ELSEVIER life Sciences, Vol. 65, No. 5. pp. 44941. 1999 Copyright 8 1999 Elsevier Science Inc. Printed in the USA.All rights reserved 0024-3205~~see front matter PI1 SOO24-3205(99)00095-8 MINIREVIEW DYNAMIC REGULATION OF THE PROINFLAMMATORY CYTOKINE, INTERLEUKINJP: MOLECULAR BIOLOGY FOR NON-MOLECULAR BIOLOGISTS Linda R. Watkins,* Michael K. Hansen, Kien T. Nguyen, Jacqueline E. Lee” and Steven F. Maier Department of Psychology & aDepartment of Molecular, Cellular & Developmental Biology, University of Colorado at Boulder Boulder, CO 80309 U.S.A. (Received in final form April 5, 1999) Interleukin-lp (IL-lp) is a key mediator and modulator of a wide array of physiological responses important for survival. It is created by a variety of cell types, including immune cells, glia, and neurons. It is a very potent biological molecule, acting both at the periphery as well as within the central nervous system. The production and release of ILlp is tightly regulated by far more complex processes than previously thought. An appreciation of this complexity is necessary for proper interpretation of apparent contradictions in the literature where different aspects of ILlp expression are measured. Given that many researchers are not molecular biologists by training, yet need an appreciation of the controls that regulate the function of key proteins such as IL-l@, this review is aimed at both: (a) clarifying the multiple levels at which IL-lp production is modulated and (b) using IL-lp regulation to explain the dynamics of gene regulation to non-molecular biologists. Three major topics will be discussed. First, regulation of IL10 production will be examined at every level from extracellular signals that trigger gene activation through release of active protein into the extracellular fluid. Second, regulation of ILlp bioavailability and bioactivity will be discussed. This section examines the fact that even after ILlp is released, it may or may not be able to exert a biological action due to multiple modulatory factors. Last is the introduction of the idea that IL-lp regulation is, at times, beyond the direct control of host; that is, when IL1 p production becomes dysregulated by pathogens. Key Words: gene regulation, interleukin- 1p, proinflammatory cytokine *Corresponding author: Dr. L.R. Watkins, address as above. phone: 303-492-7034; FAX: 303-492-2967; email: [email protected]
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of Dynamic regulation of the proinflammatory cytokine, interleukin-1β: Molecular biology for...

ELSEVIER
life Sciences, Vol. 65, No. 5. pp. 44941. 1999 Copyright 8 1999 Elsevier Science Inc. Printed in the USA. All rights reserved
0024-3205~~see front matter
PI1 SOO24-3205(99)00095-8
MINIREVIEW
DYNAMIC REGULATION OF THE PROINFLAMMATORY CYTOKINE,
INTERLEUKINJP: MOLECULAR BIOLOGY FOR NON-MOLECULAR BIOLOGISTS
Linda R. Watkins,* Michael K. Hansen, Kien T. Nguyen, Jacqueline E. Lee” and Steven F. Maier
Department of Psychology & aDepartment of Molecular, Cellular & Developmental Biology, University of Colorado at Boulder
Boulder, CO 80309 U.S.A.
(Received in final form April 5, 1999)
Interleukin-lp (IL-lp) is a key mediator and modulator of a wide array of physiological responses important for survival. It is created by a variety of cell types, including immune cells, glia, and neurons. It is a very potent biological molecule, acting both at the periphery as well as within the central nervous system. The production and release of ILlp is tightly regulated by far more complex processes than previously thought. An appreciation of this complexity is necessary for proper interpretation of apparent contradictions in the literature where different aspects of ILlp expression are measured. Given that many researchers are not molecular biologists by training, yet need an appreciation of the controls that regulate the function of key proteins such as IL-l@, this review is aimed at both: (a) clarifying the multiple levels at which IL-lp production is modulated and (b) using IL-lp regulation to explain the dynamics of gene regulation to non-molecular biologists. Three major topics will be discussed. First, regulation of IL10 production will be examined at every level from extracellular signals that trigger gene activation through release of active protein into the extracellular fluid. Second, regulation of ILlp bioavailability and bioactivity will be discussed. This section examines the fact that even after ILlp is released, it may or may not be able to exert a biological action due to multiple modulatory factors. Last is the introduction of the idea that IL-lp regulation is, at times, beyond the direct control of host; that is, when IL1 p production becomes dysregulated by pathogens.
Key Words: gene regulation, interleukin- 1 p, proinflammatory cytokine
*Corresponding author: Dr. L.R. Watkins, address as above. phone: 303-492-7034; FAX: 303-492-2967; email: [email protected]

450 Dynamics of lnterleukin-la Gene Regulation Vol. 65, No. 5. 1999
Interleukin-lp (IL-lp) is a protein released by a wide variety of cell types distributed throughout the body. Although best characterized as a chemical signal between immune cells during infection and injury, there is growing recognition that it is intimately involved in a broad array of physiological responses, both in the periphery as well as within the central nervous system (l-3). For example, IL- 1 p released from activated peripheral immune cells is a critical mediator of immune-to-brain communication (3,4). In this role, IL-lp can activate sensory nerves (5,6), thereby signaling the brain to create “sickness responses” including fever, decreases in food and water intake, increased sleep, exaggerated pain, and so forth (4,5,7). Intriguingly, activated peripheral immune cells induce IL- 1 p gene activation and increases in IL- 1 Q protein within the CNS itself (8- 10). This CNS-created IL- 1 p then initiates classical sickness responses (1). Within the CNS, IL-lp is created and released by various types of glia, and possibly by neurons as well (11,12). Here, IL- 1 p serves a wide variety of roles including regulation of core body temperature, sleep, pain responsivity, learning and memory, and hypothalamo-pituitary hormones (1,3,13,14). In addition, IL-lb within the brain and spinal cord serves as an intercellular signal during infection and injury (15).
Thus, both at the periphery and within the CNS, IL- 1 p dynamically regulates a wide variety of key systems important for survival. In addition, IL- 1 p is a very potent biological molecule, producing large effects when administered into the brain in the femptogram to picogram range (16,17). Appropriately, production and release of IL-1 p are tightly regulated. As more of the molecular biology of IL- 1 l3 gene regulation is understood, it is becoming clear that its regulation is more complex than initially thought. An appreciation of this complexity is necessary for proper interpretation of apparent contradictions in the literature in which different aspects of IL-ll3 synthesis and release are measured (e.g., messenger RNA levels, protein levels, immunoreactivity, bioactivity) and treated as if they assess the same process. The literature on the effects of “stress” on IL-l p is a good example, and is hopelessly chaotic without an understanding of how IL- 1 p is actually regulated (see ( 18) for discussion). It is also clear that the terminology and concepts used in the molecular biology literature are largely mysterious and inaccessible to many non-molecular biologists. The purpose of this review is two-fold: (a) to examine the multiple levels at which IL- 1 p production is modulated in order to provide a clearer understanding of how this critical signaling molecule responds to the changing internal environment of the organism, and (b) to use IL-lp to explain the dynamics of gene regulation to non-molecular biologists. Because this review requires the use of molecular biology terms that may be unfamiliar to many readers, key terms will be italicized in the text where they are defined.
In the sections that follow, regulation of IL-1 p will be divided into 3 major topics. The first section focuses on the regulation of IL-lp production (Figure 1) by examining: (A.) extracellular signals, second messenger systems, and nuclear factors that signal the DNA to create the new messenger RNA @RNA) used to create IL-1 p protein; (B.) factors within the nucleus that regulate the processing of newly created IL-ll3 mRNA, (C.) control over transport of the mRNA from the nucleus to specific cytoplasmic sites where IL- 1 l3 protein will be created; (D.) factors that regulate the rate at which the mRNA is destroyed; (E.) factors that regulate the synthesis of immature IL-19 protein (pro-IL-lp) from its mRNA, and (F.) regulation of the conversion of this inactive pro-IL- 1 l3 (the precursor form of IL-lp) into the active mature form of IL-lp. As will be described, conversion of pro-IL-ll3 to the mature form is linked to release of IL-lp from the cell. This sets the stage for the second section of this review, namely regulation of IL- 1 p bioavailability/bioactivity. IL- 1 p bioavuikzbiZify/bioactivify refers to the fact that even after IL-lp is released, it may or may not be able to exert a biological action. Indeed, there are five ways to modulate the biological effects of mature IL-l@ (A.) modulation of the availability of the membrane-bound receptors for IL- 1 p that create cell signaling; (B.) altered expression of so-called “decoy” receptors that bind up extracellular IL- 1 l3, thus preventing it from exerting a physiological action; (C.) production of an extracellular IL-lp antagonist which blocks binding of IL-l 8 to its receptor; (D.) production of an intracellular version of this same IL-lp

Vol. 65, No. 5, 1999 Dynamics of lnterkukin- 1 b Gene Regulation 451
antagonist which disrupts intracellular signaling initiated in response to extracellular binding of IL- 1 g; or (E.) altered expression of proteins that regulate the function of IL-1 receptors. The last section introduces the idea that IL-lb regulation is not always in the control of the organism. Although detection of pathogens (bacteria, viruses, parasites, etc.) by immune cells certainly stimulates the creation of IL-l p, pathogens have also evolved a variety of tactics to circumvent their own destruction by the host. Part of the pathogen’s dance with death suppresses the host’s production and bioavailability of IL-1 p.
As a last point, it should be noted that this review is not intended to provide an exhaustive review of all literature related to IL-lp regulation. The reader should be aware that there are apparent discrepancies in the literature as to the effects of various transmitters and drugs on IL- 1 p regulation. When reading such literature, one should be cognizant that such differing outcomes can result from multiple causes. As two simple examples, consider (a) in vitro vs. in vivo experiments, and (b) experiments using different cell types. Regarding in vitro vs. in vivo differences, they are complex. On the one hand, one could place high value on in vitro data since the conditions to which the cells are exposed appear to be completely under the control of the experimenter. On the other hand, such situations are highly artificial as they cannot take into account the natural milieu of dynamically changing hormones, transmitters, surrounding cells, etc. to which in vivo cells are exposed. Furthermore, for IL-lp regulation at least, in vitro conditions can create spurious results simply because the cells are %apped” in culture along with whatever endproducts accumulate there. Just as an example, a given drug could directly stimulate the release of both ILlB and some other endproduct that, in turn, suppresses IL-lg production. As this “‘other endproduct” accumulates in the cell culture, the in vitro result measured may be an overall suppression of IL-1B production, despite the fact that the direct effect of the drug itself is stimulatory. Regarding experiments using different cell types, the differences in apparent outcome to any given experimental manipulation could again arise from multiple sources. For example, responses of cells within the central nervous system may differ from those in the peripheral nervous system, and different types of immune cells may respond differentially to the same experimental manipulation. In part, such differences in response may result from the very different milieu to which the cells are exposed. In addition, true differences can exist in how various cells respond, based on different types of receptors and internal cell signaling systems expressed by various cell types, and so forth. But even with such complexities in mind, this review should help familiarize the reader with the numerous molecules capable of altering IL- 1 p gene expression and the multiple levels of regulation that exist.
Gene Regulation: Overview
The regulation of gene expression is a very complex, highly controlled process that ultimately allows different cell types in a multicellular organism to differ dramatically in both structure and function. For example, if a mammalian neuron and macrophage are compared, the differences generated during cell differentiation are so extreme that it is difficult to imagine that the two cells contain the same genmaic DNA; that is, the same genetic information that encodes all the RNA and protein molecules required to construct its cells. In addition to the process of cell differentiation, gene expression can also be dynamically modulated after development is complete. Indeed, most of the specialized cells in a multicellular organism alter their patterns of gene expression in response to extracellular stimuli, such as hormones, neurotransmitters, and cytokines.
Regulation of gene expression begins within the nucleus at the level of DNA and usually concludes with the creation of active protein (Figure 1). This involves a cascade of steps, each of which will be examined in detail in separate sections that follow. As a brief overview, DNA is an enormously long,

452 Dynamics of Int&ukin-lp Gene Regulation Vol. 65, No. 5, 1999
Basic Steps In Gene Regulation
Nucleus : u Cytoplasm
Inactive mRNA
DNA + Pre-mRNA + mRNA ---_)mRNA A.
n
C. Transcriptional &A mRNA -4
D. mRNA Degradation Control
Control Processing Control
Transport Control
E. Translational Control
Protein Precursor
1
F. Post-Translational Control
Active Protein
FIG. 1.
Schematic of gene regulation. The 6 points of regulation (A.-F.) refer to sections discussed in detail in the text. Transcriptional control (A.) is exerted at the level of the DNA such that creation of pre-mRNA is regulated. Pre-mRNA must be modified before it can be used for creation of protein. RNA processing control (B.) regulates this maturation process in the formation of mRNA. The mRNA then moves from the nucleus into the cytoplasm where protein will be created. mRNA transport control (C.) defines where the mRNA is moved to by carrier proteins, dependent upon whether the protein product is to be packaged into vesicles or not. Once the mature mRNA has moved to the site of translation, it has a finite lifespan. mlXNA degradation control (D.) regulates how rapidly mRNA is destroyed, an event which disrupts further protein synthesis. While the mRNA is still intact, it can be used to create protein via translation. Translational control (E.) regulates whether protein will be formed, and how efficiently this occurs. Many proteins, after being synthesized, must be modified prior to use. Post-translational control (F.) regulates this maturation of the protein to form the final active product.
unbranched, linear polymer formed from covalently linked deoxyribonucleic acids. The genetic information of a cell is contained in the linear order of the nucleotides in its DNA. A gene is composed of a specific region of DNA that contains all of the information needed to make a particular protein. In general, each gene is composed of 3 main parts (Figure 2): (a) the region of DNA that gets copied into RNA (trunscripriunal region), (b) the region in the nucleotide sequence in DNA where RNA synthesis begins (the prom&r region), and (c) sites that upregulate or downregulate gene expression (DNA regulatory sites). These DNA regulatory sites occur within 2 regions of the gene. First, they occur within the promoter region itself. Second, they occur at distant sites that are located before (“upstream” to) the promoter region and are therefore called the upsrream DNA regulatory region (Figure 2). The process whereby RNA is synthesized from the DNA is called transcription and occurs by using one strand of DNA as the template for enzymatically creating (“transcribing”) a complementary RNA strand. This newly formed molecule, called pre-&WA, is then modified to become messenger RNA (mRNA), by a process known as splicing in which certain portions of the nucleotide sequence are removed from the pre-mRNA. After transport of the mRNA from the nucleus to the cytoplasm, the sequence of nucleotides in the mRNA directs the linear linking of amino acids to form protein in a process known as translation. Finally, after synthesis, many proteins require modifications before they are ready to function as active proteins.
Thus, as shown in Figure 1, gene expression can be regulated at multiple steps in the pathway from DNA to RNA to protein. These include: controlling when and how often a given gene is transcribed (transcriptional control); controlling how the pre-mRNA is processed to create mature mRNA (RNA

Vol. 65. No. 5, 1999 Dynsmics of Interleukln- I fi Gene Regulation 453
The Interleukin-lb Gene
p negative Glucocorticoid Response Element p Repressor Site i 7 NFKB i ; / ]
p W-1
/ j ] FNFPA
: i f f j ; p NF-IL6 : ::
I j j /j
i ; ; /j r spi-1 ; ii ; I;
: y NF-IL6 ; i ; ! / /I
3 ; ; :
; / ] j/ ; i
i 1 i ?
5’ ; I : ii
; ii i i
i : i j I b 3’ : : ii : !
I Up8tram DNA Regulatory Region I Prcmot6l Rag&n Exon 1. Intro0 1, Exon 2. - . ..._....” . . . . . bltron 8, Exon 7
; ji j / ! i 1 CAMP Response Element
/ I- Nqq;;‘NFrB i!y if7
Transcriptional Region
Start Site
LPS Response Enhancer
CAAT Box TATA Box
FIG. 2.
Schematic of the IL1 p gene. Ibis diagram is derived primarily from genomic DNA studies of human and mouse IL10 genes. This diagram is provided for two purposes. First, for readers relatively unfamiliar with molecular biology, this diagram serves to provide a concrete view of the physical organization of a gene. Second, for readers expert in molecular biology, this diagram summarizes what is currently understood about the organization and regulation of the ILIP gene. For this second group of readers, relevant information is provided here rather than in the text to enhance readability of the text for a general audience. Referring now to the figure, the IL-lg gene, like other genes, is composed of 3 regions: upstream DNA regulatory regions at the 5’ end. the promoter region, and the transcriptional region at the 3’ end which contains the code for making pre-mRNA. The upstream regulatory region spans from hundreds to thousands of nucleotides away from the transcriptional region. It includes binding sites for various transcription factors, a tonic repressor protein, a negative glucocorticoid response element, a CAMP response element, and an LPS response enhancer. The known binding sites and their relative sizes are illustrated by black bars. The vertical dotted lines extending from the black bars to the gene indicate the relative position of these binding sites on the gene. At the 3’ end, the coding region contains 7 exons separated by 6 introns. Transcription begins at the promoter start site adjoining exon 1 and linearly progresses along all exons and introns. For transcription to begin, a series of nuclear regulatory proteins (transcription factors) and the enzyme for transcription of mRNA (RNA polymerase) must bind in a specific order to specific positions within the promoter region (79,274). Together, these form the initiation complex (see Figure 3C). Transcription factors that bind specific regions of the promoter regulate the ease with which transcription occurs. These transcription factors are induced in response to the extracellular presence of hormones, cytokines, transmitters, and so forth (79,274-276). Numerous binding sites for transcription factors can exist within the promoter region, as it spreads out from the start site to about 100 nucleotides upstream (in the 5’ direction). Like the promoters of most genes, the promoter for the ILIP gene contains a octomer of thymines (T) and adenines (A) called a TATA box. The TATA box is where the first transcription factor (TATA-binding protein) binds, and it is important for correct positioning of the growing initiation complex (79,274). Many promoters, including the promoter for the ILIP gene, also contain a CAAT box, named after its nucleotide sequence containing cytosines (C), A, and T. The CAAT box has a major impact on the efficiency of the promoter for initiating transcription (79.274). GC boxes (hexamers of guanine [G] and C) in the promoter are binding sites for the transcription factor SP-1 and, again, influence the frequency of transcription initiation (79,274). Lastly, the promoter of the IL-lp gene also contains binding regions for tissue-specific factors that control whether the gene can be expressed in various cell types. All of the factors that bind to the promoter region Iunction by modulating the efficiency of the initiation complex (274). This reflects the fact that initiation, rather than completion, of transcription is tightly regulated. Adding to the complexity of transcriptional regulation, however, is the fact that the gene itself is not the only site of regulation. Transcription factors may modulate the action of other transcription factors, may act only in the presence of one or more other proteins, or may modulate RNA polymerase directly. Thus, the mapping in this figure of where various modulators bind to the IL 1 p gene yields important but incomplete information as to the dynamics of transcriptional regulation (78,79,274,276).

454 D ynamic.~ of lmrhkin- 16 Gene Regulation Vol. 65, No. 5. 1999
prmeming control); controlling the transport of the completed mFWAs from the nucleus to specific sites in the cytoplasm @RNA transport control); regulating the half-life of individual mRNAs in the cytoplasm (mRNA degradation control); selecting which mRNAs in the cytoplasm are translated into protein (translational control); and lastly, selectively activating, inactivating, or compartmentalizing protein molecules after they have been made (post-translational control). It seems that every step in gene expression that can be controlled, in principle, is likely to be regulated under some circumstances for some genes. The IL,-1 p gene is likely to be one of these genes.
A. Transcriptional Control
The basics. As noted above, transcription is the process whereby RNA is synthesized on a DNA template. This process occurs using an enzyme called RNA polymeruse that binds to a start (initiation) site within the promoter region in the DNA and begins RNA synthesis here (Figures 2 & 3C). RNA synthesis continues until the enzyme encounters a stop (termination) signal in the DNA, whereupon both the polymerase and its completed RNA chain (pre-mRNA) are released (Figure 3D). This pre- mRNA is retained in the nucleus for further processing (see Section LB, below). By convention, the end of the pre-mRNA that is created first is called the 5’ end, what is created last is called the 3 ’ end (Figure 3 C,D). DNA has a corresponding 5’ and 3’ orientation (Figure 2).
Transcriptional controls (step A in Figure 1) are generally considered to be the most important control point in gene regulation because initiation, rather than completion, of transcription is tightly regulated. Thus, one can think of the creation of each mRNA molecule as an all-or-none process. Gene transcription is regulated by transcription factors (gene regulatory proteins; Figure 3C), which recognize and bind short stretches of DNA of defined sequence (DNA regulatory sites) and thereby determine which of the thousands of genes in a cell will be transcribed. In addition, although the promoter contains the nucleotide sequence in the DNA to which RNA polymerase binds to begin transcription (start site of Figure 2), RNA polymerases cannot initiate transcription on their own. They require a set of regulatory proteins that must be assembled at the promoter before transcription can begin (initiation complex of Figure 3C). This assembly process provides multiple steps at which the rate of transcription initiation can be increased or decreased in response to regulatory signals, and many transcription factors operate by influencing these steps (19,20). Furthermore, many transcription factors can act hundreds to thousands of nucleotides away from the promoter they influence (upstream DNA regutafory region of Figure 2). The result is that a single promoter can be controlled by an almost unlimited number of regulatory sequences scattered along the DNA. Finally, different genes may share gene regulatory proteins. These properties allow for the simultaneous modulation of many genes and can explain why one extracellular signal can result in the production of several different proteins; for example, to create a concerted cellular response to challenge.
ILl~trunscription The genomic DNA for ILlp has been isolated in human, mouse, and pig (21-24). The significance of knowing the genomic structure is that much of the knowledge of IL-1 l3 transcriptional regulation thus comes from studies done in these species, since these are the only species for which the DNA regulatory sites and promoter regulatory sites are known. For example, the promoter region as well as various DNA regulatory sites have been characterized in the IL- 1 p gene (Figure 2). The relative positions and sixes of the various known DNA regulatory sites within both the promoter region and upstream regulatory region of the IL-ll3 gene are illustrated as black boxes in Figure 2 simply to provide the reader with a feel for the physical layout of this regulated gene (see caption of Figure 2 for detail beyond that provided in the text, if desired).
In addition, lL-lp complementu~ DNA (cDNA), which are DNA molecules artificially synthesized in the laboratory as a complementary copy of the mature mRNA, has been cloned in many species, including human, mouse, rat, rabbit, monkey, sheep, horse, cattle, cat, dog, deer, pig, chicken, and carp (25-32). The major advantage of cDNA clones is that they lack the introns (non-coding sequences; see
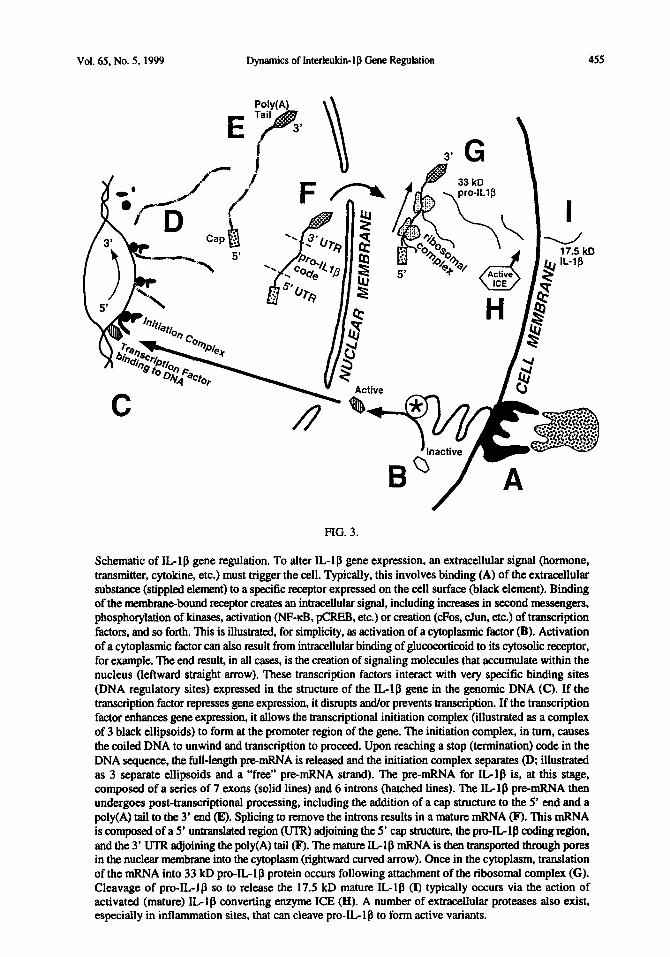
Vol. 65, No. 5. 1999 Dynamics of Interleukin- 1 g Gene Regulation
Schematic of IL 1 g gene regulation. To alter IL- 1 p gene expression, an extracellular signal (hormone, transmitter, cytokine, etc.) must trigger the cell. Typically, this involves binding (A) of the extracellular substance (stippled element) to a specific receptor expressed on the cell surface (black element). Binding of the membranebound receptor creates an intracellular signal, including increases in second messengers, phosphotylatlon of kinases. activation @IF&, pCRE%, etc.) or creation (cFos, cJun, etc.) of transcription factors, and so forth. This is illustrated, for simplicity, as activation of a cytoplasmlc factor (B). Activation of a cytoplasmic factor can also result from intracellular binding of glucocorticoid to its cytosolic receptor, for example. The end result, in all cases, is the creation of signaling molecules that accumulate within the nucleus (leftward straight arrow). These transcription factors interact with very specific binding sites (DNA regulatory sites) expressed in the structure of the ILIP gene in the genomic DNA (C). If the transcription factor represses gene expression, it disrupts and/or prevents transcription. If the transcription factor enhances gene expression, it allows the transcriptional initiation complex (illustrated as a complex of 3 black ellipsoids) to form at the promoter region of the gene. The initiation complex, in turn, causes the coiled DNA to unwind and transcription to proceed. Upon reaching a stop (termination) code in the DNA sequence, the h&length premRNA is released and the initiation complex separates @; illustrated as 3 separate ellipsoids and a “free” pre-mRNA strand). The pre-mRNA for IL-lp is, at this stage, composed of a series of 7 exons (solid lines) and 6 introns (hatched lines). The Llg premRNA then undergoes post-transcriptional processing, including the addition of a cap structure to the 5’ end and a poly(A) tail to the 3’ end (E). Splicing to remove the introns results in a mature mRNA (F). This mRNA is composed of a 5’ untranslated region (UTR) adjoining the 5’ cap structure, the pro-IL 1 g coding region, and the 3’ UTR adjoining the poly(A) tail 0. The mature IL1 p mRNA is then transported through pores in the nuclear membrane into the cytoplasm (rightward curved arrow). Once in the cytoplasm, translation of the mRNA into 33 lcD pro-IL-lg protein occurs following attachment of the ribosomal complex (G). Cleavage of pro-IL-lg so to release the 17.5 kD mature IL-lg (I) typically occurs via the action of activated (mature) ILlg converting enzyme ICE (II). A number of extracellular proteases also exist, especially in inflammation sites, that can cleave pro-IL-lp to form active variants.

456 Dynamics of Interleukin-le Gene Regulation Vol. 65, No. 5. 1999
Section LB, below) that are present in genomic DNA. Thus, they can be used to determine the amino acid sequence of a protein by DNA sequence analysis and this has allowed extensive studies on the biological properties of IL-Ip. Furthermore, they can be used for determining changes in mRNA expression using molecular biology techniques such as Northern blot analysis, in situ hybridization, RNase protection assays, or reverse transcription-polymerase chain reaction (RT-PCR) (for reviews of these methods, see (33,34)).
Comtifufive transcription of ZL-1 /I? It is controversial whether the IL- 1 p gene tonically creates pre- mRNA (that is, whether constitutive transcription occurs) or whether this gene is silent until stimulated by extracellular signals. It had been thought that constitutive transcription of the IL- 1 p gene normally occurred only in a few specific cell types under basal conditions (35). However, these conclusions were principally based on studies done in tissue culture using techniques with limited sensitivity. Thus, one must give careful consideration to such studies when examining in vivo basal levels of IL-lp transcription. For example, with the advent of molecular biology techniques such as RT-PCR, very small quantities of mRNA can now be detected, which may allow for a more accurate determination of basal levels of IL-lp transcription. Furthermore. several studies using tissue culture reported no constitutive transcription of the IL- 1 p gene, yet IL- 1 p transcription could be readily induced by the addition of bacterial cell products such as lipopolysaccharide (LPS; also known as endotoxin) (36-38). This may be a key observation inasmuch as strong evidence now suggests that normal animals show spontaneous bacterial translocation, a process whereby whole bacteria, LPS, or other bacterial cell products cross the gut barrier. LPS is a normal constituent of portal venous blood in man (39), LPS fed to normal rabbits is detected in liver macrophages (Kupffer cells) (40), and LPS is found in serum from normal rats (Nguyen et al., unpublished observations). Such observations bring into question what “basal levels” of IL-lp gene transcription really mean in an intact organism where tonic stimulation by bacterial products may be the norm. Finally, constitutive levels of IL-l p mRNA have been detected in many organs and tissues in the periphery, as well as many brain regions such as the hypothalamus, hippocampus, brain stem, cerebral cortex, and pituitary (9,4 1,42). Collectively, these data suggest constitutive transcription of the IL-l p gene in vivo.
Transcription factors. As noted above, gene transcription is controlled by gene regulatory proteins called transcription factors. These proteins recognize DNA in much the same way as any ligand recognizes its receptor; that is, they first form a tertiary structure that is compatible with the surface with which they interact and establish numerous atomic interactions (19). These high-affinity interactions in the nucleus between transcription factors and DNA sequences play a key role in governing gene transcription.
A variety of classical transcription factors can alter IL-lp gene transcription (Table 2). One of the best known and most widely studied transcription factors that activates IL- 1 p gene transcription is nuclear factor (NF)-kappaB (NF-d), which will be used as a model to explain how a transcription factor can regulate gene transcription. In most cells, NF-KB is sequestered in the cytoplasm because of its association with a specific inhibitor protein called IKB (43), although constitutive nuclear expression of NF-KB is observed in some mature B and T lymphocyte and macrophage cell lines (44-46). Upon cell activation and phosphorylation of IIcB, NFKB is released from this complex and translocates to the nucleus, where it interacts with its DNA recognition sites to mediate gene transcription (Figure 2 illustrates the locations of 2 known NF-KB binding sites in the upstream regulatory region of the IL-lp gene) (47). One of the key features of NF-KB transcriptional control is that it is used in many different gene systems. Nuclear expression of NF-KB is induced by multiple stimuli, including granulocyte- macrophage colony stimulating factor (GM-CSF), macrophage-CSF (M-CSF), IL- 1 p, bradykinin, tumor necrosis factor-alpha (TNF-a), protein kinase C (PKC) activators, increases in cyclic AMP (CAMP), Sendai virus induced myeloid extracts, LPS, calcium ionophores, and reactive oxygen intermediates (48-58). Furthermore, NF-KB transcriptional control is very fast. For example, nuclear expression of NF-KB is detectable within 2 minutes and maximal lo-15 minutes after TNF-cc treatment

Vol. 65, No. 5, 1999 Dynamics of Interleukin-I p Gene Regulation 457
TABLE 1A A: Effect of extracellular signals on IL-1 p mKNA synthesis, IL-l p mRNA steady-state levels, and IL- 1 l3 mRNA stability (half-life). B: Effect of extracellular signals on synthesis of pro-IL 1 p protein and on release of mature IL-ll3 protein. Numbers in brackets identify representative references cited. Abbreviations: Acetylch.; acetylcholine; ATP: extracellular adenosine triphosphate; P-Adren.: P-adrenergic agonists; Brady.: bradykinin; CSa: unglycosylated complement factor 5a; CCK: cholecystokinin; CRH: corticotropin releasing hormone; DA: dopamine; EGF: epidermal growth factor; Glucocort.: gluococortioids, including dexamethasone; GM-CSF: granulocyte-macrophage colony stimulating factor; His/IL-la: synergy observed by combined application of histamine and interleukin-la; ICAM-I: intercellular adhesion molecule-l; IFNP: interferon beta; IFNy: interferon-gamma; IFN/G/I: synergy observed by combined application of interferon-gamma, granulocyte-macrophage colony stimulating factor, and interleukin-3; IL- 1 CL: interleukin- 1 alpha; IL- 1 l3: interleukin- 1 beta; ILlRIW synergy observed by combined application of interleukin-1 and tumor necrosis factor; IL-2: interleukin-2; IL-3: interleukin-3; IL-4: interleukin-4; IL-6: interleukind; IL-lo: interleukin-10; IL-17: interleukin-17; IL-18 interleukin-18; Leukotr.: leukotrienes; LPS: lipopolysaccharide, the cell walls of gram negative bacteria; M-CSF: macrophage colony stimulating factor; Nitric Ox.: nitric oxide; Noradren.: noradrenalin; PGE2: prostaglandin E2; Subs. P: substance P; TGFa: transforming growth factor alpha; TGFPl: transforming growth factor beta- 1; TNF-a: tumor necrosis factor-alpha; TNF-J3: tumor necrosis factor-beta.
mRNA Svnthesis mRNA Levels Increase Decrease Increase No Effect
mRNA Stabilitv Decrease Increase
Acetylch. No Effect Decrease
ATP jj-Adren. (114) Brady. (48) C5a (85,153) CC CR::
(85)
DA (111)
-. . EGF (108) (108.277) (277) Glucocor. (87,W GE-E:‘, (54.80)
(103) (54)
(88.144,145) (35J4.80)
(86) HMLla (86) ICAM-I (63) mfl ~’
(278) (35,116) (192)
rJ_ IL:;
(35) (109) (54)
u-4 (35,146) (54)
g-6 IL:10
( ) (::)
(117) (143,146)
IL-17 IL-18 Lepti Leukotr. LPS M-CSF Nitric Ox.
(;y;)
(112)
19.103) (153.154) (54)
(85) (35.54)
Noradren. PGE2 (84) Subs. P
(84)
ItX$Z (277) TGFP I (81)
(277)
TNF-a (82.91) (106) (81)
TNF-f3 U!ZI (128,129)

458 Dynamics of Interbkin-la Gene Regutation Vol. 65. No. 5, 1999
TABLE 1B
mo IL-lb Synthesis MaturelL_lB Release Increase No Effect Decrease Increase No Effect Decrease
Acetylch. (193) ATP (167,178,179)
B- Adreu Brady. .
I1 14.1881
C5a (85,150) C CCRHK
(1831
(111) DA (193) EGF (108) Gbc;; (103) (184)
(279) ne (193)
HislILla ICAM- 1 (63)
E# (280)
(116,278) (116) FNIGII
3 (38.1161
(104) (38) ILI/TNF (104) _^
(185) L-6 IL-10 IL-17 IL-18
(to’)
Leptin Leukotr. (177) L i&SF $ 28 1.282
Nitric Ox. Noradren. PGE2 Subs. P TGFa TGFB 1 TNF-a
(1;;)
(176) (181,182)
(81) (104,279)
(59). NF-KB can also be detected in nuclear extracts within 15 minutes of either LPS or IL-1 treatment (60). The rapidity of these responses is likely due to the fact that, unlike many other transcription factors, the immediate induction of NF-KB by stimuli is independent of new NF-KB protein synthesis
(61).
It is important to note that NFKB plays only one part in an ensemble of regulatory factors in controlling IL- 1 p gene transcription. Additional transcription factors that increase IL- 1 p transcription by interacting with the IL-lfi gene itself and/or with components of the initiation complex that transcribes the gene include (Table 2): (a) AP-1 (activator protein-l; a heterodimer of c-3un and c-Fos) induced by cross-linking cell-surface adhesion molecules (ICAM-l), IL-lp, CAMP, and PKC (35,53,62X&); (b) a tyrosine-phosphorylated protein induced by both LPS and IL-l, which is similar to Signal Transducers and Activators of Transcription [STAT] proteins used by other cytokines (65); (c) a heterodimer of nuclear factor-interleukin-6 (NF-IL-6) and CAMP response element binding (CREB) protein induced by LPS (66); (d) a heterodimer of NF-IL-6 and a non-CREB protein induced by LPS (66); (e) NF-IL-6 induced by lipoarabinomannan (Mycobacterium tuberculosis cell wall component), LPS and TN&x (67); (f) CREB proteins and other CAMP responsive CREB/activating transcription factor (ATF) family members induced by LPS, phorbol ester, and TNF-a (68); and (g) NF-PA and immediate-early 1 (IEl) induced by cytomegalovirus (69,70). The relative sizes and locations of known binding sites in the IL-lp gene for such transcription factors are illustrated in Figure 2.

TA
BL
E
2 E
ffec
t of
cyt
opla
smic
si
gnal
s an
d tr
ansc
ript
ion
fact
ors
on I
L-1
0 m
RN
A s
ynth
esis
, IL
-l p
mR
NA
ste
ady-
stat
e le
vels
, IL
-lp
mR
NA
sta
bilit
y, p
ro-I
L-1
p p
rote
in s
ynth
esis
, and
rel
ease
of
mat
ure
IL-1
l3 pr
otei
n. N
umbe
rs i
n br
acke
ts i
dent
ify
repr
esen
tativ
e re
fere
nces
cite
d.
Abb
revi
atio
ns:
AP-
1: A
ctiv
atio
n pr
otei
n-l
whi
ch i
s a
hete
rodi
mer
of
c-F
os a
nd c
-Jun
tra
nscr
iptio
n fa
ctor
s;
CA
MP
: cy
clic
ad
enos
ine
mon
opho
spha
te;
CR
EB
: C
AM
P r
espo
nse
elem
ent
bind
ing
prot
ein;
G
luco
cort
.: gl
uoco
cort
ioid
s,
incl
udin
g de
xam
etha
sone
; N
FlM
/E
Il : n
ucle
ar f
acto
r be
ta-A
and
ear
ly-i
mm
edia
te
1 cy
tom
egal
ovir
al
prot
ein,
tran
scri
ptio
n fa
ctor
s; N
FIL
6: n
ucle
ar f
acto
r in
terl
euki
nd,
a tr
ansc
ript
ion
fact
or;
NFI
L6/
CR
EB
: nu
clea
r fa
ctor
int
erle
ukin
-6 a
ctin
g to
geth
er
with
CA
MP
resp
onse
ele
men
t bi
ndin
g pr
otei
n, t
rans
crip
tion
fact
ors;
NF-
KB
: nuc
lear
fac
tor
kapp
a B
, tr
ansc
ript
ion
fact
or;
Hea
t f
Shoc
k 1:
fact
or 1
of
the
heat
sho
ck p
rote
in f
amily
of
tran
scri
ptio
n fa
ctor
s; P
KC
: pr
otei
n ki
nase
C;
Rep
ress
or:
toni
c re
pres
sor
s,
tran
scri
ptio
n fa
ctor
; ST
AT
-lik
e: a
tran
scri
ptio
n fa
ctor
sim
ilar
to S
igna
l T
rans
duce
rs
and
Act
ivat
ors
of T
rans
crip
tion
(ST
AT
) pr
otei
ns c
omm
on t
o cy
toki
nes.
1
pRN
A
Svnt
hesi
s __
m
RN
A
Lev
els
mR
NA
St
abili
ty
p’~,
~E-l
@
Svnt
hesi
s M
atur
e IL
-Ifl
R
elea
se
? In
re
ase
Dec
reas
e In
crea
se
Dec
reas
e In
crea
se
Dec
reas
e A
P- 1
e
Dec
reas
e In
crea
se
c(35
,53,
63)
Dec
reas
e -
Cal
cium
w
:’
(182
) $
Cal
mod
ulin
C
AM
P
(648
%
(90)
(I
II)
[No
Eff
ect
(84,
90)]
(1
03)
(180
) (1
86-1
88)
F
CR
EB
(6
8,84
,9
1)
%
~luc
ocor
t. 18
7,88
1 (1
031
NFp
AIE
11
(88.
144.
145)
IZ;j7
0)
f
NFI
L6
Nm
6I
CR
F.B
16
6)
NF-
KB
(4
9.52
.53.
56)
Hea
t Sh
ock
1 (7
3)
PKC
(6
4.92
) (1
101
(128
.142
) R
epre
ssor
f1
89-1
911
(7 1
.72)
ST
AT
-lik
e (6
5)
ms
(253
) (1
23)
(134
.253
) (1
6X25
2.25
6)

460 Dynamics of Interleukin-lp Gene Regulation Vol. 65. No. 5, 1999
In terms of negative transcriptional signals, both tonic and phasic factors are involved. Evidence exists for tonic suppression of IL-18 transcription by a short-lived, and as yet unidentified, transcriptional repressor protein (Figure 2), since IL-ll3 transcription is rapidly induced by cycloheximide, a compound that disrupts protein synthesis (71) (cf (62)). Tonic transcriptional repressor proteins are also known to physically prevent the binding of the transcription factor NF-KB to the IL-ll3 gene (72). Transcription factors that phasically suppress IL-ll3 gene activation include those activated by extracellular substances and/or cellular stressors. IL- 1 p gene transcription is suppressed by heat shock factor 1 (73) and by a 150 kD complex formed by activation of intracellular glucocorticoid receptors. This glucocorticoid-receptor complex translocates to the nucleus where it binds to a negative glucocorticoid response element (nGRE) in the IL- 1 p gene (Figure 2) (74). There is evidence that this binding of activated glucocorticoid receptors to nGREs results in a physical disruption of tbe ability of AP- 1, CREB, NF-IL-6, and NFKB to bind, thus disrupting gene activation (75-77).
Ex?rucellulur and inrrucelluhrfuctors. A variety of extracellular and intracellular factors can alter IL-lj3 transcription. Extracellular signals refer to the multiple hormones, cytokines, and transmitters that bind to a cell and trigger IL-ll3 transcription (Table 1A). Intracellular signals refer to the various second messengers used in many signal transduction pathways (Table 2); however, they do not enter the nucleus. Thus, for most of these factors, the mechanism of altering gene transcription ultimately must occur through activation of transcription factors and their subsequent interactions with DNA in the nucleus. In addition, although many studies have examined IL-lp mRNA, not all of these studies reflect changes in the rate of IL-ll3 transcription. In fact, most studies simply report levels of IL-l l3 mRNA which, as should be apparent in Figure 1 and in the sections that follow, could in principle reflect changes in the rate of transcription or alterations in the degradation rate of preexisting mRNA (see Section I.D, below). We will begin by describing those studies in which extracellular and intracellular signals have been shown to alter the rate of IL- 1 p transcription.
Ex?rucellufur signals. A variety of extracellular signals increase the rate of IL-l p gene transcription (Table IA), including cross-linking of ICAM- adhesion molecules (78,79), GM-CSF (54,80), M-CSF (54), transforming growth factor p 1 (TGFPl) (81) IL-la (82), IL-ll3 (83), TNF-a (82), IL-2 (35), prostaglandin E, (PGE,) (84), bradykinin (48), and complement factor 5a (C5a) (85). In addition, interferon gamma (IFNy) enhances IL-lp mRNA transcription in LPS-stimulated cells (35) as well as in both resident and thioglycollate-elicited peritoneal macrophages (7 1). Synergy in inducing IL- 1 p transcription is also seen between IFNy, GM-CSF and IL-3 (35) and between histamine and IL-la
(86).
Extracellular substances that suppress IL-1 p gene activation include anti-inflammatory cytokines and steroids (Table 1A). That is, IL-4, IL-6 and IL-10 can each suppress LPS-induced IL-lp expression (35). IL-3 also decreases IL-lp transcription rate (35). Finally, as previously mentioned, glucocorticoids and dexamethasone also inhibit IL-lp transcription, effects blocked by RU486 (87,88), a receptor antagonist that blocks glucocorticoid actions.
Intrucellulur signals. A variety of cytoplasmic signals have been implicated in IL-ll3 gene transcription (Table 2). CAMP, for example, generally increases IL-ll3 gene transcription (89), although it decreases IL-lp transcription in astrocytes (90). PGE, and TNF-a increase IL-l l3 transcription, in part, via increases in CAMP and CREB (84,91). In addition, CAMP increases IL-lp gene activation via activation of PKC (64). PKC is a serine-tbreonine kinase activated by phorbol ester to produce IL-lp mRNA and protein. PKC is also involved in LPS effects since LPS-induced IL-lp mRNA is downregulated by a PKC inhibitor (92).
A variety of protein kinases, in addition to PKC, lead to IL-lfi gene activation (Table 2). One way that such protein kinases (extracellular signal-regulated kinase [ERK], ~38, c-Jun NH2 terminal kinase [JNK], etc) are activated is by formation of reactive oxygen intermediates. These rapidly form upon

Vol. 65, No. 5, 1999 Dynamics of Interleukin-lp Gene Regulation 461
stimulation by IL-la, IL-lfi, TNF-a, ultraviolet light, or growth factors (93). The activated protein kinases, in turn, phosphorylate, and thus activate, transcription factors implicated in IL-1B transcription (see above) (94,95). Arachidonic acid (but not its cyclooxygenase or lipooxygenase met&o&es) also activates protein kinases such as JNK, which, in turn, activates c-Jun, one of several transcription factors regulating IL-1 mRNA (96). Dexamethasone, amongst its other actions, inhibits activation of JNK (97).
Intriguingly, IL-l itself may be an intracellular signal at cytoplasmic and nuclear sites, as well as a classic extracellular signal acting by actions exerted via membrane-bound receptors. Internalization of IL-l-bound IL-l receptors provides the potential for prolonged intracellular signaling. Internalized IL- 1 a and IL- 1 B remain bound to the IL- 1 receptor without degradation for up to 8 hours in T cell lines and libroblasts (98-100). These internalized receptors, bound by IL- 1, translocate either to the Golgi apparatus (101) or to the nucleus (98-100) allowing the potential to alter protein production or gene transcription, respectively. For activation of transcription, a 50 amino acid segment of the cytoplasmic domain of the IL-l receptor is thought to be essential (102).
IL-l/lmRNA steady stare levels. As previously mentioned, many studies in the literature simply report IL-1p mRNA levels; however, changes in steady state levels can result from altered transcription or changes in the half-life of preexisting n-RNA. A variety of extracellular and intracellular signals modulate IL-lp mRNA levels (Tables 1A and 2). Increases occur in response to LPS (9,103), IL-la (104), IL-ll3 (9,104,105), TNF-a (106), TNF-p (106), IL-17 (107), epidermal growth factor (log), IL- 2 (109), PKC (1 lo), CAMP (11 l), corticotrophin releasing hormone (CRH) (11 I), leptin (112), and IL-l 8 (113). b-adrenergic agonists increase IL-l l3 mRNA levels, yet decrease IL- 1 p release; a- adrenergic agonists have no effect (114,115). Synergy occurs between TNF and either IL- 1 a or IL- 10 (104). IFNy has no effect on IL-lp mRNA levels yet enhances LPS-induced release of mature IL-lb (116). In contrast, in response to silica, IFNy reduces both levels of IL-l l3 mRNA and release of mature IL-I p (116). Lastly, both dexamethasone (103) and the anti-inflammatory cytokine IL-4 (117) suppress IL- 1 l3 mRNA expression.
B. Pre-mFWA Processing Control
The basics. After the initiation of transcription, the pre-mRNA, which is commonly referred to as “immature mRNA” as it cannot yet be used to create protein, undergoes a series of covalent modifications that occur while the pre-mRNA is still in the nucleus of the cell (Figures IB & 3E,F). First, the 5’ end (which is the end synthesized first during the process of transcription) is cupped by the addition of a methylated guanine (G) nucleotide (the 5’ cup structure). In addition, the DNA code of virtually all known eukaryotic mRNAs contains a poly-udenylurion signal sequence (that is, a series of adenylate (A) and uridylate (U) nucleotides of the sequence AAUAAA) (78,118). During transcription, this poly-adenylation signal sequence is detected and bound by a specific catalytic complex within the nucleus, causing cleavage of the mRNA beyond it (that is, in the 3’ direction from the polyadenylation signal sequence), followed by the addition of a 3’ tail of 50-250 adenylate (A) nucleotides (called the 3’poly(A) tail ) (78,119). Each of these modifications impacts the function of the mRNA at several steps along the pathway to protein. Initially, the 5’ cap structure protects the growing pre-mRNA transcript from degradation; pre-mRNA is inherently unstable and may be destroyed before use. In addition, the cap structure is important for regulating the splicing process described below (120,121) and will be seen in sections that follow to also be involved in: (a) transport of the mRNA from the nucleus to the cytoplasm, (b) regulating how long the mRNA survives in the cytoplasm prior to its destruction, and (c) regulating the translation process. The poly(A) tail has several functions as described below: (a) it aids in the export of mature mRNA from the nucleus to the cytoplasm, (b) it has a major influence on the life-span of its mRNA, and (c) it influences the efficiency of translation (for reviews, see (119,121-123)) (see Section I.E., below).

462 Dynamics of Interleukin-la Gem Regulation Vol. 65, No. 5, 1999
In addition to these covalent modifications, eukaryotic pre-mRNA requires the deletion of specific nucleotide sequences that are not useful for creating protein, and would disrupt protein synthesis if they were allowed to remain. Eukaryotic genes contain two types of sequences, introns and exons, both of which are initially transcribed into pre-mRNA (Figure 3E). Although a few functions have been assigned to introns, most introns are simply regarded as unnecessary sequences that need to be deleted. The exo11s, on the other hand, contain both the essential codes for creating protein and for controlling this protein-synthetic process, called translation. Exons at the beginning (5’) and end (3’) of the mRNA (Figure 3F) do not contain code for making protein during translation. Rather, as will be described in subsequent sections, these 5’ and 3’ untranslated regions (5’ UTR and 3’ UTR) are important for regulation of: (a) efficiency of translation, (b) stability of the mRNA, and (c) routing of the mRNA from the nucleus to the cytoplasmic site where translation will occur.
Alter the cap structure and poly(A) tail are added to the pre-mRNA (see above), the introns are very precisely deleted by a process called splicing. Splicing occurs in the nucleus within a complex called a spliceosome that contains proteins and small nuclear ribonucleoprotein particles (78). Introns are spliced out of the pre-mRNA strand by recognition of specific exon-intron boundaries as well as a conserved segment within the intron called a branch site (78). This splicing process yields the mature form of mRNA composed of sequentially joined exons (Figure 3F). Once processed, the pre-mRNA is called mRNA and can be used to create protein.
How splicing occurs can be modulated, allowing variation in the number of exon sequences retained in the final mature mRNA used for translation of protein. This is called alternative splicing and it: (a) can affect how easily the resultant mRNA can be translated into protein, and (b) generates different forms of protein, created from the same gene, which differ in length and/or sequence. Alternative splicing allows the final protein product to change based on changing conditions within the cell (for review, see (124,125)).
IL-l/3 pre-mRNA processing. IL-lp pre-mRNA, for human and mouse at least, consists of a continuous sequence of 7 exons separated by 6 introns (Figures 2 & 3 D,E) (23,118). The first (5’) exon and most of the last (3’) exon contain the UTRs. Exons 2-6 and part of Exon 7 contain the code for creating IL- 1 p protein (126). The immature mRNA for IL- 1 l3 is very unstable. This unstable nature of IL- 1 p pre-mRNA is thought to be due, at least in part, to the presence of repressor proteins which bind to the IL-lp pre-mRNA and suppress its processing into the more stable mature form (127). Indeed, some cell types (such as libroblasts) constitutively transcribe IL-ll3 mRNA so inherently unstable that no accumulation of this mRNA even occurs under basal conditions (128,129).
Alternative splicing of the IL-ll3 mRNA has also been reported, at least in horses (130). Here, alternative splicing has been observed in response to LPS, causing deletion of an entire exon from the mature IL-l p mRNA (130). This results in the deletion of 162 nucleotides corresponding to exon 5 of the human and mouse IL-lp genes (130). The factors controlling the maturation and alternative splicing steps have yet to be defined.
C. mRNA Transport Control
The basics & transport of IL-Ip ARNA. Translation of mRNA into protein occurs at various sites within the cytoplasm. As noted previously, the 5’ cap and poly(A) tail play important roles in the transport of all mRNAs from the nucleus to the cytoplasm. These structures are recognized by specialized transport proteins that ferry the mature mRNA from the nucleus to the cytoplasmic site appropriate for the type of protein to be made (Figures lC, 3F,G) (120). In general, mRNAs that encode for proteins destined to be secreted are directed to the endoplasmic reticulum by a signal sequence at the protein’s amino (N-) terminus. The protein coding region of such mRNAs creates a 15-20 amino acid N-terminal sequence (the signal sequence) which allows insertion of the protein into

Vol. 65, No. 5, 1999 Dynamics of tnterleukin-I p Gene Regulation 463
the endoplasmic reticulum for packaging (131). This is the route taken for the mRNA of the interleukin-1 receptor antagonist (IL-lra) (132), for example. On the other hand, mRNAs that lack a typical signal sequence (e.g.. the mRNA for IL-lp) are directed to specific intracellular locations where the entire protein is synthesized by free ribosomes in the cytoplasm (for review see (120,133,134)). For these mRNAs, the signal is thought to be in the 3’ UTR (135). This is the route taken by mRNA for IL-l p, as well as for IL-la (132) and is likely important in the processing of pro- IL-1B protein to the mature form (see Section 1.F).
D. mRNA Degradation Control
The basics. There are several factors that determine how long the mature mRNA survives (Figure 1D) prior to enzymatic destruction (e.g., its half-life). As previously noted, the 5’ cap and poly(A) tail initially protect the pre-mRNA from degradation in the nucleus; however, both of these structures also protect the mature n-RNA from degradation in the cytoplasm. Indeed, it is clear that destruction of the cap structure always precedes destruction of the protein coding region of the mature mRNA ( 120,12 1). In addition, as the mRNA ages, a progressive shortening of the poly(A) tail occurs via sequential removal of each successively exposed terminal nucleotide. When the poly(A) tail is shortened below about 10 nucleotides, it can no longer bind its stabilizing, protective coat of poly(A) tail binding proteins (136). In turn, this earmarks the mRNA for destruction, triggering first the destruction of the cap structure, followed by destruction of the mRNA itself ( 119,12 1 - 123).
In addition to stability conferred by the cap structure and poly(A) tail, the half-life of mRNA is also markedly affected by the existence of specific sequences encoded within its 3’ UTR. A common instability sequence found in the 3’ UTR of many unstable mRNAs is called the AURE, for Adenylate- Uridylate Rich Element, and it consists of 50-150 nucleotide sequences containing reiterations of the pentanucleotide AUUUA and other U-rich regions (122). The AUUUA segments facilitate degradation of the mRNA “body” while the U-rich sequences promote degradation of the poly(A) tail (122). AUREs are contained in mRNAs that require tight control of the duration of protein synthesis; that is, where a short burst of protein signal is desired. Such mRNAs include transcription factors like CFOS and cJun, adhesion molecules, and proinflammatory cytokines including IL-l p ( 119,121- 123). Indeed, AUREs are so characteristic of these proteins that their presence has been used as a screen for isolating new DNA sequences for such substances (137). With no AURE present, mRNA half-lives are generally on the order of 12 or more hours (119,122). The AURE, by targeting its mRNA for rapid destruction, creates the 5 minute half-lives of transcription factor mRNAs and the 3 to 4 hour half-life of IL-ll3 mRNA (119,122,138).
Finally, many mRNAs do not have fixed half-lives. Indeed, in mammalian cells, the abundance of mRNA can fluctuate many-fold following a change in mRNA half-life, without any alteration in transcription rate (122). mRNA stability can change in response to cytokines, environmental factors, cell growth rates, etc. (see below). This happens, in part, by regulation of AUREs once the mature mRNA has been transported to the cytoplasm (122,139). The AURE contains binding sites for various substances that can modulate the impact of the AURE on mRNA stability (122,139). Substances that have been reported to modulate AURE function include PKC, intracellular calcium ions, cytokines, regulatory AURE-binding proteins, growth factors, LPS, reactive oxygen species, a variety of hormones, and so forth (122,123,140).
IL-1pmRNA stabilization. As noted above, the mRNA for IL- 1 p contains an AURE, whose effects on mRNA stability can be modulated. Indeed, a number of factors have been identified that can increase IL- 1 p mRNA half-lives in this manner (Tables 1A & 2). For example, this 3’ UTR of IL- 1 p mRNA contains an LPS response element. Binding of an LPS-induced protein to this region removes a tonic destabilizing signal, thereby increasing mRNA stability (141). GM-CSF, M-CSF, IFNy, and TNF each increase IL- 1 l3 mRNA half-life (35,54,80,128,129). For TNF, at least, this effect is mediated by PKC, since PKC stimulators increase IL-lp mRNA stability and PKC inhibitors both prevent

464 Dynamics of Interleukin-I p Gene Regulation Vol. 65. No. 5, 1999
accumulation of IL- 1 p mRNA by TNF and accelerate decay of IL-1 p mRNA in cells pretreated with TNF (128,142).
On the other hand, glucocorticoids, histamine, calmodulin antagonists, and calcium antagonists each decrease IL-lp mRNA half-life (8688143-145). The anti-inflammatory cytokine IL-4 also decreases IL- 1 p mRNA half-life, with this effect produced via induction of a protein that directly destabilizes the mRNA (143,146).
E. Translational Control
The basics. The basic mechanics of translation are the same, regardless of whether proteins are synthesized on ribosomes associated with the endoplasmic reticulum or on free ribosomes in the cytoplasm (Figures 1E & 3G). The cap structure and 5’ UTR of the mRNA are critical regions for controlling the start of protein synthesis (133,147,148). Initiation, rather than completion, of translation is the tightly controlled step of the process, just as it is for transcription (134,147). A number of cytoplasmic factors (including a variety of eukaryoric inifiutionfacfors [eZFs], ribosomes, etc.) must act in concert for translation to occur. These progressively complex together as each successive factor binds to the cap structure. Formation of this complex first attracts a small ribosomal unit to the mRNA and stimulates it to bind. Upon binding, the 5’ UTR is “scanned” to locate the optimal translation start site, denoted in part by a specific code in the mRNA (that is, AUG). When this is found, a large ribosomal unit (which expresses the enzymatic activity necessary to link amino acids together) is attracted and bound to the complex. Translation then begins, wherein the amino acids carried by transfer RNAs are sequentially incorporated as the ribosomal complex moves down the mRNA (Figure 3G). Upon reaching any of three possible mRNA termination sequences (that is, UAA, UAG, or UGA), the ribosomal complex dissociates and the newly formed protein is released. Thus, every protein molecule formed represents one complete iteration of the process just described.
Although it may seem that protein production should naturally occur as long as the mRNA is present, this is not necessarily so. Indeed, transcription can be an isolated event from translation. For example, IL- 1 p mRNA has been observed to be associated with ribosomal complexes yet fail to create protein (149). A variety of stimuli (LPS, IL-l, unglycosylated C5a, TNF, TGFPl, CAMP, etc.) can increase IL-18 transcription without translation (35,81,89,103,150). Indeed, both positive and negative regulation of translation occur at multiple sites of modulation along the translation pathway. Six of the best documented will be briefly described below.
First, there is regulation of whether the mRNA can even enter into translation. mRNA, when not being translated, is sequestered in a functionally inactive state within structures called messenger ribonucleoprotein (mRNP) particles (15 1). Modulation of the percentage of mRNA sequestered in mRNP particles can markedly regulate translation. Indeed, one effect of dexamethasone is to shift mRNA from association with ribosomal units to sequestration in such particles (15 1).
Second, there are specific sequences (recognition sites) within the cap structure that regulate translation. Recall that the cap structure is the site where the initial interaction with translation machinery occurs. This cap structure expresses binding sites for modulatory proteins that, upon binding, physically obscure the cap and disrupt initiation of translation (148).
Third, the 5’ UTR can contain specific sequences that contort the linear structure of the mRNA, making initiation of translation difficult (148). The disruptive effects of such sequences are modulated by proteins binding at specific regulatory sites within the 5’ UTR, and binding of the modulatory proteins can increase or decrease translation by altering the conformation of the 5’ UTR (148). Binding of such proteins can also physically obscure the 5’ UTR, thereby disrupting the ability of the translation machinery to “scan” the 5’ UTR for the translation start site (148).

Vol. 65. No. 5, 1999 Dynamics of Interleukin- lp Gene Regulation 465
Fourth, the structure of various components of the translation machinery can be modulated. For example, several factors involved in translation initiation can be activated or deactivated based on their phosphorylation state. Factors such as PKC and growth factors activate translation by phosphorylating eukaryotic initiation factors 4F (eIF4F) and eIF4E (135,151). In contrast, heat shock proteins dephosphorylate eIF4F and eIF4E and thereby suppress translation (15 1). eIF4F is also targeted by polioviruses during infection of the host. Poliovirus halts translation of host mRNAs by proteolytic cleavage of eIF4F. This allows poliovirus to take over host translational machinery for its own use, since translation of viral mRNAs occurs independently of eIF4F (151). As a countermeasure, the host responds to viral infection by suppressing translation via phosphorylation inactivation of eIF2 (15 1).
Fifth, the poly(A) tail and its associated poly(A) tail binding prorein synergistically interact with the cap structure and 5’ UTR to enhance translation (134). Once aging of the mRNA shortens the poly(A) tail below about 10 nucleotides, it can no longer interact with 5’ UTR or cap structure, thereby disrupting translation. The poly(A) tail binding protein stimulates translation via enhancement of binding of the large ribosomal subunit to the translation machinery at the mRNA start site (135).
Lastly, the presence of an AURE in the 3’ UTR modulates translation, as well as affecting mRNA half- life (see above) (134,135,151). AUREs modulate translation by: (a) altering the interaction of the poly(A) tail with the cap and 3’ UTR structures, and/or (b) directly affecting these 3’ translational regulators (137). AUREs modulate translation stability both by tonically decreasing the efficiency with which protein is formed and by expression of binding sites for proteins that can enhance or suppress this AURE effect (134,137,152). For example, AUREs found in cytokine mRNAs are thought to tonically suppress translation via binding of a constitutive repressor protein. LPS enhances translation via de-repression; that is, by creating a protein that binds the AURE in a manner which dislodges the tonic repressor (137,153,154). Dexamethasone binds to a specific site on the AURE. Its binding prevents LPS-induced translation by preventing LPS from dislodging the repressor protein (137). On the other hand, IL- 1 p may enhance its own synthesis by binding to and stabilizing its own mRNA, through actions at the AURE (155).
Pro-IL-l/3?ranslation. Creation of the 31 kD inactive pro-IL-lp protein is controlled by both the general and cytokine-specific forms of translational control reviewed above (Tables 1B & 2; Figure 3G). For example, the 3’ UTR of IL-lb mRNA contains an LPS response element which both facilitates protein production and increases the half-life of the IL- 1 p mRNA (141). Translation of IL- 1 p is also increased by epidermal growth factor (log), CRH (111) and cross-linking of ICAM- 1 adhesion molecules (63). In contrast, dexamethasone inhibits IL- 1 p translation, likely via increases in CAMP (103). In addition, changes in pro-IL-ll3 protein levels can occur either via alterations in the half-life (approx. 3 hours) of the pro-IL-lp protein (138) or by changes in the activity of the IL-l converting enzyme which cleaves pro-IL-lp into active IL-ll3 (see below).
Quantitative assessment of 3 1 kD pro-IL- 1 p levels is currently hindered by technical difficulties. Virtually all anti-IL-l p antisera available to date bind with high affinity to the 17.5 kD mature IL- 1 p protein but are insensitive to pro-IL-lp (156-158). This remarkable difference in detectability is due to the N-terminal amino acid sequence of the pro-IL- 1 p, which causes a conformation change in the C-terminal portion of pro-IL- 1 p ( 156). Because anti-IL- 1 l3 antisera are directed against the C-terminal portion of the protein, this shift in the 3-dimensional structure causes pro-IL-ll3 levels to be underestimated by 1 O-fold or more ( 156,158).
F. Post-Translational Control
The basics and pro-IL-lpprocessing. Like many proteins, pro-IL-ll3 requires post-translational modifications before it is ready to function as an active protein. Pro-IL-1P must be proteolytically cleaved and extracellularly released to become active, although not necessarily in that order (Figure

466 Dynamics of Interleukin-1 p Gene Regulation Vol. 65, No. 5, 1999
3 H.1). Some cells (e.g., fibroblasts, keratinocytes) do not normally synthesize enzymes to cleave pro- IL- 1 p into its active form, but rather secrete inactive pro-IL- 1 p into the extracellular fluid ( 159). Here, it is cleaved by proteolytic enzymes including chymase, elastase, cathepsin G, and collagenase, the major proteases released at inflammation sites by a variety of immune cells and tissue damage ( 160,16 1). Each of these enzymes cleaves the pro-IL- 1 p into a form similar in size and specific activity as the 17.5 kD mature IL1 p protein (160). Regardless of these considerations, the cleavage of pro-IL- 1 p to its mature form and its release from the cell are prerequisites for biological function.
The IL-I converting enzyme. Most cells that produce pro-IL- 1 p also produce IL-1 converting enzyme (ICE). ICE is perhaps best known for its role in programmed cell death. The ICE gene is the mammalian homologue of the ~uenohabditis glegans death (Ced) gene family of usteine--artate protea (caspases) (132,162). Based on this lineage, ICE is also referred to as Ced-3 or caspase-1. Unlike IL-lra and IL- la, the ICE gene is not found in close association with the gene for IL- 1 p; rather, it is found on chromosome 11 in mouse and human (163,164).
ICE, like IL-l& is first synthesized as an inactive precursor (pro-ICE) that is found free in the cytoplasm, rather than packaged in vesicles (156). Unlike IL-lfi, the mature active form of ICE is also found free within the cytoplasm (Figure 3H) (165). ICE activity must therefore be regulated, since both ICE and pro-IL- 1 p co-exist without mature IL- lfl being formed (166). Regulation of ICE activity is as yet poorly understood. To date, calcium ionophores (13 1) and extracellular ATP (167) are known to dramatically enhance processing and release of IL-lp, most likely via modulation of ICE. ICE mRNA is decreased by stress exposure (168) and increased by both adrenalectomy (168) and LPS (169,170).
ICE specifically cleaves the Aspartate (amino acid position 116)-Alanine (amino acid position 117) bond in pro-IL-l p to generate mature IL- 1 p (132). This cleavage of pro-IL- 1 p is generally thought to be an obligatory step for IL-lp release, since treatment with most ICE inhibitors fails to cause detectable levels of extracellular pro-IL- 1 p (132). This association between cleavage and release is not absolute, however. Cleavage and release can be dissociated by some types of ICE inhibitors, including the peptidyl aldehyde L-709,049 (17 1) and YVAD-CHO (172). These cause pro-IL- 1 p to be secreted instead of the mature form (132,171). As noted previously, the presence of many extracellular enzymes that can cleave pro-IL-lp to active forms suggests that such ICE inhibitors actually would not functionally block IL- 1 p activity.
Mature IL-1/3release. Secretion of the mature form of IL-lp occurs through an ill-defined process. As previously mentioned, pro-IL- l/3 cleavage and release often co-occur (165). Here, it is thought that pro-ICE present in the cytoplasm is proteolytically activated, assembled, and inserted into the cell membrane. This multimeric ICE pore complex would then cleave pro-IL- 1 p and release the mature form of IL-lp into the extracellular environment (165). In addition, mature IL-lp is thought to be released by a novel secretory pathway (173). Indeed, it was shown that IL-lp is contained, in part, within intracellular vesicles that protect it from protease digestion and active secretion of IL-lp requires its translocation across the cellular membrane (173). Regardless of these possibilities, the release of IL- 1 p clearly occurs via pathways different from the classical endoplasmic reticulum-Golgi- secretory vesicle pathway common for most secreted proteins. In agreement, processing and membrane translocation of pro-IL-lp can also be facilitated by myristyl acylation, a common form of post- translational modification for facilitating release of cellular proteins not contained in classical secretory vesicles (174). For example, Lipid A (the biologically active component of LPS) stimulates myristyl acylation of pro-IL-lp (174).
Studies on IL-l@ release into the extracellular fluid do not typically assess mechanism of action. Changes in protein product release could result from effects anywhere along the molecular biological cascade from transcription to post-translational processing and release (Figure 31; Tables 1B & 2).

Vol. 65, No. 5. 1999 Dynamics of Interleukin-lp Gene Regulation 467
Thus, for the literature reviewed below, increased IL-1B protein levels could reflect increases in protein translation and release, increases in processing of pre-existing pools of pro-IL-l B (see below), or increases in the half-life of IL-ll3 protein (138). Enhanced release of mature IL-l B has been reported following administration of IL-la (38), IL-lp (38), LPS (36,37), nitric oxide (175), PGE, (176), leukotriene inhibitors (177), P2Z purinergic receptor agonists such as extracellular ATP (167,178,179), CAMP (180), substance P (181,182), calcium ionophores (182), IPNy (116), cholecystokinin (CCK) (183), IL-17 (107) and IL-18 (113) (Tables 1B & 2). In contrast, release of IL-l p has been reported to be inhibited by dexamethasone ( 184), IL-4 ( 185), CAMP ( 186- 188), PKC inhibitors (189-l 91), P-adrenergic agonists ( 114,188), IPNy ( 116,192) intracellular calcium buffers (182), and IL-18 (113).
In addition to classical views of IL-lp regulation, recent evidence suggests that release of mature IL- 1 l3 does not require ongoing protein synthesis. That is, evidence is beginning to accrue in support of pre-existing releasable pools. Low levels of stimulation of human monocytes cause intracellular accumulation of pro-IL- 1 p, which can be released by LPS as mature IL- 1 l3 despite pharmacological disruption of new protein synthesis (171). The existence of pre-formed pools of IL-1B within the central nervous system is supported by basal IL-l 0 release from hypothalamic explants. This basal release can be inhibited by acetylcholine and histamine, and increased by noradrenalin, dopamine, prostaglandin, bacterial cell walls, and nitric oxide (175.176.193.194). Constitutive, as well as inducible, IL-18 protein levels have also been observed in multiple brain and spinal cord regions (510,195).
Overview
IL-l p is one member of a family of molecules, which includes three ligands (IL-l a, IL-l p, and IL- 1 receptor antagonist [IL-lra]), two membrane-bound receptors, an IL-1 receptor-associated kinase (IRAK), ICE, the IL-l receptor accessory protein (IL-lRAcP), and various soluble receptors (for review, see (155)). In principle, the upregulation or downregulation of any one component of the IL- 1 family could influence the level of activation of the entire IL- 1 system. Regulation of IL- 1 p and ICE has been reviewed in earlier sections. In this section, we will review the best documented pathways by which changes in the levels of various IL-l family members can affect the bioavailability and/or bioactivity of mature IL-l 8 protein.
A. IL-1 Type I Receptors
Extracellular IL-lj3 can bind to either IL.-I rype I or type II receptors (IL-1RI or IL-MI, respectively). These 2 receptor subtypes are derived from different genes on chromosome 2 (196) and serve different physiological functions. It is generally agreed that cell signaling occurs via binding of IL- 1 RI, rather than to IL-1RII (see Section ILC, below) (155,197,198). When IL-19 binds to membrane-bound IL- lRI, it initially does so with low affinity. High affinity binding, with resultant intracellular signaling, is achieved upon binding of the IL-lp/IL-1RI complex to another membrane-associated molecule called IL-l receptor accessory protein (IL-1RAcP; see Section II.B, below) (155).
IL-W exists in both membrane-bound and soluble forms (199,200). The soluble form of the IL-1RI receptor is derived by post-translational processing involving proteolytic cleavage and release of its extracellular portion (155,199). While this soluble receptor can bind ILIp, under normal basal conditions it is unlikely to markedly alter the availability of extracellular IL-lp to membrane-bound IL-1RI. This is both because: (a) soluble IL-1RI does not preferentially bind IL-l 8, as indicated by its binding profile of IL-lra > IL-la > IL-ll3 (155); and (b) soluble IL-1RI is not associated with IL- lRAcP, and therefore binds IL-l B only with low affinity (155).

468 Dynamics of Interleukin-lp Gene Regulation Vol. 65, No. 5, 1999
The fact that both soluble and membrane-bound IL-N derive from the same mRNA may complicate interpretation of alterations in gene expression (Table 3). This is because, at first glance, increases in mRNA for IL-1RI could produce increases in cell signaling (via increases in membrane-bound IL- lRI), potential decreases in cell signaling (via increases in soluble IL-IRI), or both. However, as noted above, soluble IL- 1RI actually preferentially binds IL- lra rather than IL- 18. Indeed, increases in soluble IL-lR1 may interfere with antagonist actions of extracellular IL-lra (201). Thus, overall, changes in IL-lR1 mRNA could increase the bioactivity of IL-ll3 both by increasing availability of membrane bound IL-lR1 for cell signaling and by increasing availability of soluble IL- IRI for sequestration of the endogenous IL-l antagonist.
To date, increases in IL-N n-RNA in peripheral tissues have been reported (Table 3) in response to dexamethasone (202-205), GM-CSF (204), IFN-a (206), IL-la (207,208), IL-4 (197,209). IL-6 (205,210), IL-13 (202,211), LPS (205,210,212), PKC (208), and TNF-a (210). IFNy can either increase (204,206,211) or decrease (204) IL-WI mRNA. IL-lp also increases IL-1RI mRNA in most cell types studied (204,208,209) but decreases IL-N mRNA in bovine peripheral blood mononuclear cells (204). In brain, mRNA for IL-lR1 is increased by i.p. LPS (212,213), intracerebroventricular (i.c.v.) LPS (214), and chronic i.c.v. IL-lp (105,215). In anterior pituitary cell lines, IL-lp and TNF-a also upregulate IL-N mRNA (216). Decreases in IL-N mRNA occur in glial cultures in response to either P-adrenergic agonists or CAMP (217).
Similar to IL-lp (see above), transcription of IL-N mRNA can be dissociated from translation (155). Thus direct measures of expressed IL-lR1 protein may be especially relevant for assessing the physiological impact of altered gene expression (155). Increases in membrane-bound (cell signaling) IL-1RI protein have been reported after PKC, PG&, dexamethasone, epidermal growth factor, IL-2, and IL-4 (155). In contrast, TGFP and IL-l can downregulate surface expression of IL-N (155).
B. IL-1 Type II (Decoy) Receptors
IL-1RII are called “decoy” receptors since IL-1 binding fails to induce cell signaling (218). Both membrane-bound and soluble forms of IL-lRI1 exist (199,200,219), with soluble IL-lRI1 being created either by alternative splicing of the mRNA (199) or by post-translational cleavage and release of the extracellular portion of the membrane-bound receptor (155,199). Membrane-bound and soluble IL-1RII bind both pro-IL-18 (released by tibroblasts, keratinocytes, etc.) and mature IL-lI3 (220). Binding of pro-IL-lp to either form of IL-1RII prevents its processing to a mature form, while binding of mature IL- 1 p to IL- 1RII blocks its ability to bind to the physiologically functional IL- 1 RI expressed nearby (218,220). Sequestration of IL-l 8 by IL-1RII likely disrupts the physiological impact of extracellular IL-18. This is due both to (a) the marked preference that IL-lRI1 has for IL-18, as reflected in its binding profile of IL- 1 p > IL- la > IL-l ra; and (b) the fact that both mature IL- 1 p and pro-IL-l/3 bind with higher affinity to IL-lRI1 than to IL-N (155,220). Indeed, binding of soluble IL-1RII to IL-lp is essentially irreversible (155). These points, in addition to the fact that extracellular concentrations of soluble IL- 1 RI1 greatly exceed those of soluble IL- 1 RI ( 155). support the idea that IL-IRII, rather than soluble IL-lR1, has a major role in regulating IL-lp actions.
Clearly, any agent that can modulate the production of IL-1 RI1 can regulate IL- 18 bioactivity (Table 3). At the level of mRNA, increases in IL-1RII mRNA in peripheral tissues occur after dexamethasone (202,203), GM-CSF (204), IL-la (207,208), IL-4 (202,204,211), IL-13 (202,211). LPS (212,221). and PKC (208). For glucocorticoids, at least, increased soluble IL-1RII mRNA results both from increases in transcription rate and from increases in its half-life (203,222). Furthermore, IL-1RII mRNA can be either increased (197,208) or decreased (204,211) by IL-18 and IFNy. In brain, mRNA for IL-lRI1 is increased by i.p. LPS (212). In anterior pituitary cell lines, mRNA for IL-WI is increased by either IL- 1 l3 or TNF-a (2 16).

Vol. 65, No. 5, 1999 Dynamics of Intedeukin-1 p Gene Regulation 469
TABLE 3 Effect of various agents on the bioavailability/bioactivity of released IL-l p. Bioavailability/bioactivity can be influenced by changes in the expression of the IL-l receptor type I (IL-M), the IL-l receptor type II (IL-MI) which has also been referred to as decoy receptors, extracellular (soluble) IL- 1 receptor antagonist (sIL- lra), and by intracellular IL- 1 receptor antagonist (icIL-lra). Numbers in brackets identify representative references cited. Abbreviations: CAMP: cyclic adenosine monophosphate; UEBP: CAAT/enhancer binding protein, a transcription factor; CRH: corticotropin releasing hormone; Glucocort.: glucocortioids, including dexamethasone; IFNa: interferon-alpha; IFNP: interferon-beta; IFNy: interferon-gamma; IL- la: interleukin-1 alpha; IL-l@: interleukin-1 beta; IL-4: interleukin-4; IL- 6: interleukind; IL-l& interleukin- 10; IL- 13: interleukin- 13; LPS: lipopolysaccharide, the cell walls of gram negative bacteria; NF-KB: nuclear factor kappa B, transcription factor; ROIs: reactive oxygen intermediates; TGFPl: transforming growth factor beta-l; TNF-a: tumor necrosis factor-alpha.
IL-lR1 IL-1RII (De v) sIL-lra Levels icIL-lra Levels Increase DeCreZW Increase D&Se Incresse lrio Effect Decrease Increase No Effed DeWease
Calcium (283) ChW
CRHB QEP
(;;‘)
(111) Glucocon. (202-205) (202.203,222) (284) (226.240) G-CSF (285.2861 GM-CSF (204) (204) (287) Histamine (86) IE@ (206) IFNO (280) IFNy (197) (204,21 I) (226,278) (226) ILL IL:,;
(20$;06,21 I) (204) (20 . 081 f207.208) (86. 86) (204,208,209) (204) (205) (2W
IL-3 ;;;;
E-4 (-02-04.2 1 I ) (GO5.110)
~204.211.218~ (234.278) (226.235) U-6 (233) IL10 (211) u 13 LPS
(20’.2111 (202.2 I 1.222) (562.278) f 321 (226.23%
(205.210,212) (212,221) (262,263) (237) NF-kS (233) &tx Oxide (288) PKC (208) (290) (237) ROls
yj2s9)
JGFDI I 361 (236‘1 (129) TNF-a (210) (283) Vitues (132.252.261)
(is:, (129) C-5 )
Increases in membrane-bound IL- lRI1 protein levels occur in response to IL-4 (202), IL- 13 (202,223) and dexamethasone (202,203). Increased release of soluble IL-1RII has also been reported in response to reactive oxygen intermediates (224), IL-4 (202,218,222), IL-13 (202,218,222,223), and dexamethasone (which may also act, in part, by its induced increase of IL-4 and IL-13) (202,203,221,222,225) (Table 3). While LPS alone did not induce release of soluble IL-1RII receptors, it dramatically synergized with dexamethasone to induce release of these decoy receptors (210). In human keratinocytes, increased mRNA for IL-lRI1 is observed after IFNy (197) and this is correlated with release of soluble IL-lRII(197). In human monocytes, where decreased mRNA for IL,-1RII is observed after IFNy (211), a correlated decrease in release of soluble IL-1RII occurs (211).
C. Soluble (Extracellular) IL-ha
There are three forms of IL-Zru: soluble IL-lra (sIL-lra) which is released extracellularly, and two forms of intracellular IL-lra (icIL-lra, types I and II) (226-228). All three forms arise from a single IL-lra gene. This gene is strikingly similar to the IL-lp gene in terms of intron-exon organization (126) and chromosomal location (229,230). In fact, the marked homologies between the genes for IL-

410 Dynamics of InterleAn-lfl Gene Regulation Vol. 65, No. 5. 1999
lra and IL- 1 p suggest that IL- lra arose by duplication of the IL- 1 B gene with loss of expression of the IL-lp exons 2-4 (231).
Extracellularly released IL-lra competitively inhibits binding of IL- 1 p to its receptor. The affinity of IL-lra for IL-1RI (see above) is approximately equal to that of IL-l 8. However, due to the marked “spare receptor” effect for the IL-l system, whereby very few receptors need be bound for a physiological response to occur, ILlra is a weak inhibitor of physiological responses to IL-18 (155). Thus it would be expected that only marked increases in extracellular IL-lra would cause significant changes in IL-Q bioactivity. As noted above, IL-lra has only weak affinity for the L-1 decoy receptor, in keeping with the role of the decoy receptor in suppressing IT.,- 1 p actions (232).
Like all other aspects of IL-lp regulation, IL,-lra formation can be modulated (Table 3). Increases in sIL-lra mRNA and/or protein occur in response to IL-la (86), IL-lp (233), IL-6 (233), antiinflammatory cytokines (e.g., IL-4, IL-13) (234,235). IFNy (226) and CRH (111). The effect of CRH appears to be exerted via elevated CAMP (11 l), whereas IL- 18 and IL-6 actions are mediated via activation of Nl-KB and CAAT/enhancer binding protein (C/EBP) transcription factors (233). sIL- lra mRNA half-life is enhanced by TGFPl (236). In contrast, sIL-lra levels are reported to not be affected by histamine (86).
D. Intracellular IL-b
As noted above, two forms of intracellular IL- lra (icIL- lra, types I and II) result from activation of the IL-lra gene (226-228). Alternative splicing, controlled, in part, by tissue specific prmzders (i.e., sites on the DNA that “allow” only specific types of tissues to produce icIL-lra) (237), is used to create the two icIL-lra forms of the IL-lra protein (226,227,230,233,238). icIL-lra is constitutively expressed in epithelial cells and can also be induced in other cell types (e.g., macrophages) after stimulation by LPS or phorbol ester (237).
The function of icIL-lra has been unknown until very recently. icIL-lra inhibits the bioactivity of IL- 18, not by blocking IL-lp binding to extracellular receptors, but rather by blocking intracellular actions induced by IL-10 (239). When IL-l p binds to IL-lRI, cell signaling occurs, leading to the formation of second messengers and transcription factors, which in turn increase the transcription of a variety of genes. icILlra does not alter any of these early steps in cell signaling by extracellular IL- 18. Rather, icIL-lra disrupts the response to bound IL-l p by decreasing the stability of mRNAs induced by the cascade of events initiated by extracellular IL-lg (239). The recent recognition of multiple forms of icIL-lra suggests that multiple physiological functions may be under their control (228).
Like sIL-lra, icIL-lra formation is regulated (Table 3). icIL-lra mRNA and protein are increased in response to either IL-4 or IL-13 (226,234,235), corticosteroids (226,240). and IFNy (226). icIL-lra mRNA and icIL-lra protein levels are not affected by either TNFa or TGFPl (129). On the other hand, icIL-lra mRNA level and half-life have also been reported to be enhanced by TGFBl (236).
E. IL-1 Receptor Accessory- and IL-1 Receptor-Related Proteins
As noted above, IL-18 exerts its cell signaling effects via binding to membrane-bound ILlRI. In many tissues, including brain, IL-1RI is found in association with IL-1 Receptor Accessory Protein (ZL- IRAcP) (241-243). IL-1RAcP forms a complex with IL-1RI and either IL-la or B-18, but not IL-lra (24 1). IL- 1RAcP serves to increase binding affinity between the IL- 1RI and IL- 1 (24 1). Indeed, cell lines that express IL-1RI without IL-1RAcP do not respond to IL-l (244,245). Binding of IL-1 induces the formation of a complex containing both IL-lR1 and IL-1RAcP; however, ILlRAcP does not directly bind IL-l (155). IL-1RAcP is required for both internalization of the activated IL-1 receptor complex as well as for intracellular signaling to occur (245). Intracellular signaling ensues following

Vol. 65, No. 5, 1999 Dynamics of Interleukin-lfl Gene Regulation 471
activation of IL-l receptor associated kinase (IRAK), a serine-threonine protein kinase attached to the receptor complex via its linkage to IL- 1 RAcP (246,247).
Very little is known to date regarding the regulation of IL-1RAcP. Both membrane-bound and soluble forms of ILlRAcP have been demonstrated, with the soluble form created by alternative processing (155,241). However, the physiological significance of the soluble form is unclear since no IL-1 binding capacity has yet been reported for either membrane-bound or soluble IL-lRAcP, nor is soluble IL-1RAcP known to interact with soluble IL-N (241). Chronic i.c.v. IL-lfi increases soluble, but not membrane-bound, IL-1RAcP mRNA in brain (214,215). In contrast, LPS does not alter IL-1RAcP mRNA in brain, despite increasing IL-N mRNA (242). In liver, LPS can either increase (9) or decrease IL-lR4cP mRNA (243). In response to systemic IL-ll3, no change in either liver or brain IL 1 RAcP mRNA was observed (9). In contrast, systemic IL- 1 a increases IL- 1 RAcP mRNA in lung and spleen, has no effect in brain, and decreases expression in the liver (24 1).
In contrast to IL- 1 RAcP, IL1 receptor-related proteins (IL-l Rrp) are thought to be receptors despite the fact that this group of proteins fails to bind any known IL-l ligands (248,249). Indeed, IL-1Rrp may actually be a receptor for IL-l 8 rather than IL-l(250). Fit-l proteins are also thought to be receptors distantly related to IL-lR, but bind IL-1 only with very low afftnity (251). To date, no information is available regarding potential regulation of these IL-l related receptor groups, but simply that they exist further substantiates the growing diversity in possibilities for IL-l regulation.
. * . v Pmost Da
One last mechanism by which &lp can be dramatically regulated is through infection of the host by bacteria, viruses, or parasites. Classically, one immediately thinks of increases in IL- 1 p production by the host’s immune cells in response to pathogens. While tbis is indeed true, and in no need for review here, what this classic view fails to appreciate is the ability of the pathogen to dance around the host’s defenses. Since this is a lesser known and highly relevant aspect of cytokine regulation, this topic will be included here. Indeed, the effect of pathogens on host immunologic defenses include, but are by no means limited to, disruption of IL-lp synthesis and bioavailability. A broad disruption of immunologic responses by pathogens is to be expected since their virulence and survival depends on their ability to elude and counter host defenses.
Bacteria and viruses. Toward this end, bacteria and viruses have evolved cytokine response modifier genes whose products inhibit the synthesis and/or release of cytokines including IL-lp (252). These pathogen defense mechanisms are aimed at a variety of steps in the host production of cytokines. In terms of disrupting IL-lp synthesis, there are both specific and global mechanisms. For example, Hepatitis B virus selectively blocks LPS-induced production of both IL-lp mRNA and protein (253). Respiratory syncytial virus has adopted a different approach and disrupts IL- 1 p synthesis via a virus- induced release of the anti-inflammatory cytokine, IL-10 (254). On the other hand, viruses can also cause generalized disruption of host mRNA synthesis, an effect which would disrupt IL- 1 p synthesis as well as the synthesis of most other proteins. Many viruses globally destabilize host cell mRNAs within 3 hours of infection. The viruses spare ribosomal RNAs and transfer RNAs (which are both used in various aspects of the translation process), presumably so that they can be used to create virally-directed proteins (123). Viruses create this generalized disruption of host mRNAs by creating viron host shutoff phosphoprotein which, by unknown mechanisms, causes only host mRNAs to rapidly degrade (123). Poliovirus, as noted previously (see Section I.E. above) specifically inactivates an initiation factor (eIF4F) required for host, but not viral, translation to occur (15 1).
Beyond synthesis, pathogens can also disrupt IL- 1 B release by actions at the level of ICE regulation. One of the many responses that host cells have to infection is the activation of ICE. Detection of viral double-stranded RNA by the host potently induces IFNy which, in turn, is a potent inducer of ICE

412 Dynamics of Interleukin-lp Gene Regulation Vol. 65. No. 5, 1999
(255). To counter this host defense, baculo-, cowpox, vaccinia, and myxoma viruses produce a variety of potent inhibitors of ICE activation or action (162,256-258).
Pathogens can disrupt IL-18 function following release, as well. Pox viruses and vaccinia viruses, amongst other pathogens, release a viral version of IL- 1 “decoy” receptors (132,259,260), in addition to “decoy” receptors for TNF, IL-6, and IFNy (252,261). Bacteria can also disrupt IL-lp function by causing the release of both IL-1 decoy receptors and soluble IL-lra (221,262,263). Lastly, retroviruses inhibit IL- 1 mediated responses by blocking cellular events beyond the level of IL-l p binding to its receptor; that is, it disrupts IL-18 mediated cell signaling (264).
Parasites. Like bacteria and viruses, parasites can disrupt IL- 1 p production and function via diverse mechanisms. Since IL-lp has anti-parasitic activity, blockade of this cytokine enhances parasite survival (265). Many parasites have evolved to avoid induction of IL-lfl during infection, as well as to suppress IL-ll3 production by other subsequent infective agents such as bacteria (266-269). Various parasites also inhibit IL-ll3 production indirectly by tonically inducing the host to release high levels of the anti-inflammatory cytokines IL-4 and IL-10 (Table IA) (270,271). In addition, Schistosomiasis worms (270) and Brugiu makzyi worms (271) suppress IFNy production, an effect which, as noted above, would suppress induction of ICE (255).
The means by which parasites disrupt IL- 1 p function are diverse as well. For example, the larval form of the cestode Tueniu raeniaeformis creates a protease inhibitor that disrupts IL-l p activity, such as IL- 1 p induced increases in immunocyte replication (272). Schistosoma munsoni, on the other hand, disrupts IL- lp activity by inducing the host’s release of a-melanocyte stimulating hormone (a-MSH) (267), an endogenous, functional interleukin-1 antagonist (273).
As can be appreciated from the discussion above, IL-lp is not regulated at only a single step in its synthesis pathway. Rather, it can be regulated at every step that has been examined: at the level of pre- transcriptional processes (second messengers, creation of transcription factors, etc), transcription, splicing, mRNA stability, translation of mRNA into an inactive pro-IL-lp protein, post-translational processing of pro-IL-l p into its active form, regulation of the enzyme required for post-translational processing, and release of the mature protein. The bioavailability and bioactivity of IL- 1 p is regulated as well, both by regulation of the IL-1 receptors responsible for cell signaling, by release of “decoy” receptors that bind IL- 10 without creating receptor-mediated effects, by regulating receptor accessory proteins that modulate IL- 1 signaling, and by creation of IL- lra which modulates IL- 1 p actions both extracellularly and intracellularly.
What becomes clear from the literature is that there are many substances that influence IL-1B production and function. Some of these substances exert apparently opposing effects at different levels of gene regulation (increasing mRNA but decreasing protein release, for example); some of these substances increase IL 1 p production by themselves yet decrease IL- 1 p production in the presence of other modulators; some exert no effect on their own yet modulate IL-If3 production induced by immune stimulators such as endotoxin; some do not regulate IL- 1 p directly but rather cause cells to release IL-l decoy receptors or alter soluble or intracellular IL-lra.
This means that to understand regulation of IL-l l3 in particular, and gene regulation in general, it is necessary to come to grips with the reality that gene regulation is very different than many of the synthesis pathways with which non-molecular biologists are comfortable. The fact of most immediate impact is that there is no single rate-limiting step. Neither mRNA level nor pro-protein level nor any other single molecular measure necessarily reflects the final outcome. A study of how exposure to

Vol. 65, No. 5, 1999 Dynamics of Intexleukin-1 p Gene Regulation 413
stressors or pathogens or hormones affects the various levels of gene expression yields important insights into the similarities and differences in the mechanisms involved. However, to truly understand the biological impact of altered gene expression requires a level of analysis not yet approached by the field. Perhaps the most critical question should be whether gene regulation in the cell under study has any biologically measurable result on Q@ cells.
For example, consider the question of whether stress increases IL- 1 p in brain. It is quite possible that stress could increase IL-l g mRNA as measured by in situ hybridization or RT-PCR, but that IL-lp signaline remains unaffected by the stressor. Pro-IL-l g might not be created by translation, pro-IL- 1 l3 might not be processed into mature IL 1 p, decoy receptors might be upregulated to absorb any mature IL- 1 p produced, IL- 1 RAcP might be decreased and thereby IL- 1 g signaling in the target cell blocked, etc. Conversely, IL-ll3 signaling could be increased by stress (or anything else) even if mRNA remains unchanged. ICE activity could be increased thus cleaving pre-existing pro-IL- 1 p into mature IL- 1 p, decoy receptors could be downregulated, IL-l type I receptor and IL-1RAcP upregulated, etc.
Thus, what appear to be simple questions to the neuroscientist or other non-molecular biologist are vastly complex, and careful work will be required to produce answers. An appreciation of the multiple stages of regulation of IL- 1 p likely hints at the importance of tightly controlled IL- 1 g protein levels. Furthermore, it provides some understanding of why the research literature is confused and seemingly contradictory. As noted above, a perusal of the literature concerning the relationship between stress and IL-l p yields findings in every conceivable direction (for review, see (18)). However, the studies in the literature use varying stressors and measure different aspects of IL-1p function and regulation. Apparent chaos is, therefore, to be expected. It is hoped that an understanding of the underlying molecular biology will help to produce clarity and order out this chaos.
This work was supported by NIH grants MH55283, MH45045, MHO03 14, MH01558, NS35 118, and a Juvenile Diabetes Foundation International Career Development Award (J.E.L.)
1. 2. 3. 4. 5.
6.
7.
8. 9.
10.
11. 12.
13. 14.
Reference
N.J. ROTHWLL and G. LUHESHI, Adv. Pharmacol. 25 l-20 (1994). C.A. DINARELLG, FASEB J 2 108-l 15 (1988). SF. MAIER and L.R. WATKINS, Psych. Review LQS 83-107 (1998). L.R. WATKINS, S.F. MAIER and L.E. GGEHLER, Life Sci. 2 101 l-1026 (1995). S.F. MAIER, L.E. GGEHLER, M. FLESHNER and L.R. WATKINS, De role of the vaggs nerve in vtohne to bmn
. . _ _ wmanwcatlon Vol. 840, S.M. McCann, J.M. Lipton, E.M. Stemberg, G.P. Chrousos, P.W. Gold and C.C. Smith (Eds), 289-301, NYAS, NY (1998). J.L. HINDE, S.E. HAMMACK, R.P.A. GAYKEMA, L.E. GGEHLER, S.F. MAIER and L.R. WATKINS, Proc. Sot. Neurosci. 24 (1998). S. KENT, R.-M. BLUTHE, K.W. KELLEY and R. DAN’IZER, Trends in Pharmacol. Sci. u 24-28 (1992). S. LAYE, P. PARNET, E. GGUJON and R. DANTZER, Mol. Brain Res. 22 157-62 (1994). M.K. HANSEN, P. TAISHI, Z. CHEN and J.M. KRUEGER, J. Neurosci. 1l2247-2253 (1998). K.T. NGUYEN, T. DEAK, S.M. OWENS, T. KOHNO, M. FLESHNER, L.R. WATKINS and S.F. MAIER, J. Neurosci. & 2239-2246 (1998). C.D. BREDER, C.A. DINARELLG and C.B. SAPER, Science 24e 321-324 (1988). J.A. DeLEO and R.W. COLBURN, Proinflammatorv cvto- cells. the r role m m *i ’
a, L.R. Watkins and S.F. Maier (Eds), in press, Birkhauser, Amsterdam (1998). J.M. KRUEGER and J.A. MAJDE, Crit. Rev. Immunol. 14 355-379 (1994). M.J. KLUGER, Physiol. Rev. 2193-127 (1991).

414 Dynamics of Interleukin-lp Gene Regulation Vol. 65. No. 5. 1999
15. 16.
17.
18.
19. 20. 21.
22.
23.
24.
25.
26. 27.
28.
29.
30.
31.
32.
33.
34. 35. 36.
37.
38. 39.
40. 41.
42.
43.
44. 45. 46.
47. 48.
NJ. ROTHWELL, G. LUHESHI and S. TOULMOND, Pharmacol Ther fi 85-95 (1996). N.S. MORROW, G. QUINONEZ, H. WEINER, Y. TACHE and T. GARRICK. Amer. J. Physiol. m G196-202 (1995). T. NAKAGAWA. M. MINAMI, S. KATSUMATA, Y. IENAGA and M. SATOH. Brit. J. Pharmacol. m 2661-2666 (1995). L.R. WATKINS, K.T. NGUYEN, J. LEE and SF. MAIER, Q cvtokines. R. Danker, E.E. Woolmann and R. Yirmiya @is), in press, Plenum, London (1998). P.F. JOHNSON and S.L. M&NIGHT, Amm. Rev. Biochem. s 799-839 (1989). L. ZAWEL and D. REINBERG, Prog. Nucleic Acid Res. Mol. Biol. 44 67-108 (1993). G. BENSI, G. RAUGEI, E. PALLA, V. CARINCI, D. TORNESE BUONAMASSA and M. MELLI, Genea95-lOl(l987). B.D. CLARK, K.L. COLLINS, M.S. GANDY. A.C. WEBB and P.E. AURON. Nucleic Acids Res 14 7897-7914 (1986). J.L. TELFORD. G. MACCHIA, A. MASSONE, V. CARINCI, E. PALLA and M. MELLI, Nucleic Acids Res 14 9955-9963 (1986). K. VANDENBROECK, P. FITEN, E. BEUKEN, E. MARTENS, A. JANSSEN. J. VAN DAMME, G. OPDENAKKER and A. BILLIAU, Eur. J. Biochem. 2u 45-52 (1993). P.E. AURON, A.C. WEBB, L.J. ROSENWASSER, S.F. MUCCI, A. RICH, S.M. WOLFF and CA. DINARELLO. Proc. Natl. Awl. Sci. SL 7907-7911 (1984). C. FISKERSTRAND and D. SARGAN, Nucleic Acids Res. &j 7165 (1990). Y. FURUTANI. M. NOTAKE. M. YAMAYOSHI, J. YAMAGISHI, H. NOMURA, M. OHUE, R. FURUTA, T. KUKUI, M. YAMADA and S. NAKAMURA, Nucleic Acids Res. u 5869-5882 (1985). M.J. HEUTHER, G. LIN, D.M. SMITH, M.P. MURTAUGH andT.W. MOLITOR, Genem 285-289 (1993). H. KATO, T. OHASHI, N. NAKAMURA, Y. NISHIMURA, T. WATARI, R. GOITSUKA. H. TSUJIMOTO and A. HASEGAWA, Vet. Immunol. Immunopathol. 48 221-231 (1995). P.T. LOMEDICO, U. GUBLER, C.P. HELLMANN, M. DUKOVICH, J.G. GIRL Y.C. PAN, K. COLLIER, R. SEMIONOW, A.O. CHUA and S.B. MIZEL, Nature u 458-462 (1984). C.R. MALISZEWSKI, P.E. BAKER, M.A. SCHOENBORN, B.S. DAVIS, D. COSMAN, S. GILLIS and D.P. CERRE’ITI, Mol. Immunol. a 429-437 (1988). F. VILLINGER, S.S. BRAR, A. MAYNE, N. CHIKKALA and A.A. ANSARI, J. Immunol. m 3946-3954 (1995). M.J. DALLMAN, R.A. MONTGOMERY, C.P. LARSEN, A. WANDERS and A.F. WELLS, Immunol. Rev. 119 163-179 (1991). P.C. EMSON, Trends Neurosci. 16 9-16 (1993). M.J. FBNTON, Int J Immunopharmacol14 401-411 (1992). P. LIBBY, J.M. ORDOVAS, K.R. AUGER, A.H. ROBBINS, L.K. BIRINYI and C.A. DINARELLO, Am. J. Pathol. m 179-185 (1986). P. LIBBY, J.M. ORDOVAS, L.K. BIRINYI, K.R. AUGER and C.A. DINARELLO. J. Clin. Invest. 18 1432-1438 (1986). S.J. WARNER, K.R. AUGER and P. LIBBY, J. Immunol. m 1911-1917 (1987). A.I. JACOB, P.K. GOLDBERG, N. BLOOM, G.A. DEGENSHEIN and P.J. KOZINN, Gastroenterology 22 1268- 1270 (1977). D. ROWLEY, J.G. HOWARD and C.R. JENKIN, Landet 1366-367 (1956). P. TAISHI, S. BREDOW, N. GUHA-THAKURTA, F. OBAL, JR. and J.M. KRUEGER, J. Neuroimmunol. a 69-74 (1997). S. LAYE, R.M. BLUTHE, S. KENT, C. COMBE, C. MEDINA, P. PARNET, K. KELLEY and R. DAN’IZER, Am. J. Physiol. mR1327-R1331 (1995). A.A. BEG, T.S. VINCO, P.V. NANERMEI and A.S. BALDWIN, JR., Mol. Cell Biol. 12 3301-3310 (1993). P.A. BAEUERLE, Biochem. Biophys. Acta m 63-80 (1991). M. GRILLI, J.J. CHIU and M.J. LENARDO, Int. Rev. Cytol. m l-62 (1993). G.E. GRIFFIN, K. LEUNG, T.M. FOLKS, S. KUNKEL and G.J. NABEL, Res. Virol. m 233-238 (1991). M.J. LENARDO and D. BALTIMORE, Cell 58 227-229 (1989). Z.K. PAN, B.L. ZURAW, C.C. LUNG, E.R. PROSSNITZ, D.D. BROWNING and R.D. YE, J Clin Invest !j& 2042-2049 (1996).

Vol. 65, No. 5, 1999 Dynamics of Interleukin- 1 p Gene Regulation 415
49.
50.
51.
52.
53.
54.
55.
56.
57. 58.
59.
60.
61. 62.
63.
64. 6.5.
66.
67. 68.
69.
70.
71.
72. 73.
74. 75. 76.
77.
78. 79. 80. 81. 82.
J.P. COGSWELL, M.M. GODIEVSKI. G.V. WISELY, W.C. CLAY, L.M. LEESNIIZER, J.P. WAYS and G. J.G, J Immunol m712-723 (1994). B. STRULOVICI. S. DANIELISSAKANI, E. O’IO, N.J. JR., H. CHAN and A.P. TSOU, Biochemistry 28 3569-3576 (1989). R. NEWTON, I.M. ADCOCK and P.J. BARNES, Biochem Biophys Res Commun ~ 518-523 (1996). J. HISCOTT, J. MAROIS, J. GAROUFALIS. M. D’ADDARIO, A. ROULSTON, I. KWAN, N. PEPIN, J. LACOS’IE, H. NGUYEN and G. BENSI, Mol Cell Biol 12 623 l-6240 ( 1993). M. ABE, Y. TANAKA, K. SAITO, F. SHIRAKAWA, Y. KOYAMA, S. GOT0 and S. ETO, J Rheumatol24 420-429 (1997). W. OSTER, M.A. BRACH, H.J. GRUSS, R. MERTELSMANN and F. HERRMANN, Blood z 1260. 1265 (1992). B. MEIER, H.H. RADEKE, S. SELLE, M. YOUNES, H. SIES, K. RESCH and G.G. HABERMEHL, Biochem J 262 539-545 (1989). T. HENKEL, T. MACHLEIDT, I. ALKALAY, M. KRONKE, Y. BEN-NERIAH and P.A. BAEUERLE, Nature= 182-185 (1993). R. SCHRECK. P. RIEBER and P.A. BAEUERLE, EMBO J 1Q 2247-2258 (1991). S. LEGRAND-POELS, S. MANIGLIA, J.R. BOELAERT and J. PIElTE, Biochem Ph armacol s 339- 346 (1997). H.P. HOHMANN, R. KOLBECK, R. REMY and A.P. VAN LOON, Mol. Cell Biol. fi 23 15-23 18 (1991). F. SHIRAKAWA, M. SHEDID, J. SUTILES, B.A. POLLOK and S.B. MIZEL. Mol. Cell Biol. 2 959- 964 (1989). R. SEN, Baltimore, D., Cell Q 921-928 (1986). M. TURNER, D. CHANTRY, G. BUCHAN, K. BARRETT and M. FELDMANN, J Immunol m 3556-3561 (1989). Y.KOYAMA,Y.TANAKA, K.SAlTO,M.ABE,K.NAKATSUKA,I.MORIMOTO,P.E. AURON and S. ETG. J Immunol u 5097-5103 (1996). E. SERKKOLA and M. HURME, Eur J Biochem 212 243-249 (1993). J. TSUKADA, W.R. WAlERMAN, Y. KOYAMA, A.C. WEBB and P.E. AURON, Mol Cell Biol 16 2183-2194 (1996). J. TSUKADA, K. SAITQ, W.R. WATERMAN, A.C. WEBB and P.E. AURON, Mol Cell Biol 14 7285-7297 (1994). Y. ZHANG and W.N. ROM, Mol Cell Biol u 3831-3837 (1993). G. CHANDRA.J.P.COGSWELL,L.R.MILLER,M.M.GODLEVSKI, S.W. STINNEIT, S.L. NOEL, S.H. KADWELL, T.A. KOST and J.G. GRAY, J Immunol m 45354543 (1995). G.W. HUNNINGHAKE, B.G. MONKS, L.J. GEIST, M.M. MONICK, M.A. MONROY. M.F. STINSKI, A.C. WEBB, J.M. DAYER. P.E. AU’ION and M.J. FENTON, Mol Cell Biol 12 3439-3448 (1992). J.W. CRUMP, L.J. GEIST, P.E. AURON, A.C. WEBB, M.F. STINSKI and G.W. HUNNINGHAKE, Am J Respir Cell Mol Biol6 674-677 (1992). M.A. COLLART, D. BELIN, J.D. VASSALLI, S. DE KOSSODO and P. VASSALLI, J Exp Med 164 2113-2118 (1986). T.V. LEBEDEVA and A.K. SINGH, Biochim Biophys Acta jw 32-38 (1997). C.M. CAHILL, W.R. WATERMAN, Y. XIE, P.E. AURON and S.K. CALDERWOOD, J Biol Chem m 2487424879 (1996). G. ZHANG, L. ZHANG and G.W. DUFF, DNA Cell Biol L6 145-152 (1997). A. RAY and K.E. PREFONTAINE, Proc. Natl. Acad. Sci. u 752-6 (1994). LM. ADCOCK, C.R. BROWN, C.M. GELDER, H. SHIRASAKI, M.J. PETERS and P.J. BARNES, Am J Physiola C331C338 (1995). R.I. SCHEINMAN, A. GUALBERT0,C.M. JEWELL, J.A. CIDLOWSKI andA.S.J. BALDWIN, Mol Cell Biol fi 943-953 (1995). M. KRAUSE, Methods in Cell Biology 48483-512 (1995). B. LEWIN, Genes Oxford Univ. Press, Oxford (1995). M.C. FERNANDEZ, J. WALTERS and P. MARUCHA. J Leukoc Biol 2 598603 (1996). D. CHANTRY, M. TURNER, E. ABNEY and M. FELDMANN. J 1mmuno1~4295-4300 (1989). J.L. SLACK, J. NEMUNAlTlS, D.F.D. ANDREW& J.W. SINGER and D.F. ANDREWS. Blood a 2319-2327 (1990).

476 Dynamics of Interteukin-lfl Gene Regulation Vol. 65, No. 5, 1999
83.
84. 85.
86. 87. 88.
89. 90. 91.
92. 93. 94. 95.
96. 97. 98.
99.
100. 101.
102.
103. 104.
105.
106.
107.
108. 109.
110. 111.
112. 113.
114. 115.
116. 117.
118.
M.J. FENTON, M.W. VERMEULEN, B.D. CLARK, AC. WEBB and P.E. AURON, J Immunol m 2267-2273 (1988). Y. OHMORI, G. STRASSMAN and T.A. HAMILTON, J Immunol m 3333-3339 (1990). T. GEIGER, C. RORDORF, N. GALAKATOS, B. SELIGMANN, R. HENN, J. LAZDINS and K. VOSBECK, 143 l(l992). E. VANNIER and C.A. DINARELLO, J Clin Invest a 281-287 (1993). N. BUSSO, M. COLLART, J.D. VASSALLI and D. BELIN, Exp Cell Res 123 425-430 (1987). S.W. LEE, A.P. TSOU, H. CHAN, J. THOMAS, K. PETRIE, E.M. EUGUI and A.C. ALLISON, Proc Nat1 Acad Sci U S A&j 1204-1208 (1988). S.S. SUNG and J.A. WALTERS, J Clin Invest 88 19151923 (1991). S.A. WILLIS and P.D. NISEN, J Immunol m 1399- 1406 (1995). J.J. LORENZ, P.J. FURDON, J.D. TAYLOR, M.W. VERGHESE, G. CHANDRA, T.A. KOST, S.A. HANELINE, L.A. RONER and J.G. GRAY, J Immunol m 836-844 (1995). E.M. SERKKOLA, Diss Abstr Int s 230 (1995). C. ROSETE and M. KARIN, Science 224 1194-7 (1996). A. KOJ, Biochim. Biophys. Acta 12u. 84-94 (1996). J. RAINGEAUD, S. GUPTA, J.S. ROGERS, M. DICKENS, J. HAN and R.J. ULEVI’ICH. J. Biol. Chem. 2zp 7420-6 (1995). M.T. RIZZO and C. CARLO-STALLA, Blood &3792-800 (1996). J.L. SWAN’TEK, M.H. COBB and T.D. GEPPERT, Mol Cell Biol. u 6274-82 (1997). B.M. CURTIS, M.B. WIDMER, P. DEROOS and E.E. QWARNSTROM, J. Immunol. 144 1295-303 (1990). S.B. MIZEL, P.L. KILIAN, J.C. LEWIS, K.A. PAGANELLI and R.A. CHIZZONITE, J. Immunol. m 2906-12 (1987). E.E. QWARNSTROM, R.C. PAGE, S. GILLIS and S.K. DOWER, J. Biol. Chem. m 8261-9 (1988). G. PRAAST, R. HOFMEISTER, K. GRUBE, W. HANS, T. KUHLWEIN, P.H. KRAMMER and W. FALK, I. Inflamm. &j 125-38 (1995-96). A. HEGUY, C. BALDARI, K. BUSH, R. NAGELE, R.C. NEWTON, R.J. ROBB, R. HORUK, J.L. TELFORD and M. MELLI, Cell Growth Differ. 2 3 1 l-5 (1991). P.J. KNUDSEN, C.A. DINARELLO and T.B. STROM, J Immunol 122 3 189-3 194 (1986). J.A. ELIAS, M.M. REYNOLDS, R.M. KOTLOFF and J.A. KERN, Proc Natl Acad Sci U S A 86 6171-6175 (1989). S.E. ILYIN, G. SONTI, D. GAYLE and C.R. PLATA-SALAMAN, J. Mol. Neurosci. 2 169-181 (1996). K. KAUSHANSKY, V.C. BROUDY, J.M. HARLAN and J.W. ADAMSON, J Immunol u 3410- 3415 (1988). D.V. JOVANOVIC, J.A. DIBATTISTA, J. MARTEL-PELLETIER, F.C. JOLICOEUR, Y. HE, M. ZHANG, F. MINEAU and J.P. PELLETIER, J. Immunol. 164 3513-21 (1998). K.S. SCHLUNS, J.E. COOK and P.T. LE, J Immunol m 2704-2712 (1997). M. ADACHI, H. INOUE, S. ARINAGA, J. LI, H. UEO, M. MORI and T. AKIYOSHI, Cancer Immunoo. Immunother. 44 329-34 (1997). M.F.J. SMITH, F. KUEPPERS and J.C. LEE, Lymphokine Cytokine Res. 1p 397403 (1991). P.M. PAEZ, J. SAUER, CC. PEREZ, S. FINKIELMAN, G.K. STALLA, F. HOLSBOER and E. ARZT, Endocrinology m 5504-5510 (1995). L.P. KAPCALA, J.R. HE, Y. GAO, C. LIU and E.B. DE SOUZA, Proc. Sot. Neurosci. 2;! 145 (1997). A.J.PURAN,G.FANTUZZI,Y. GU,M.S.S. SUandC.A. DINARELLO, J. Clin. Invest.lQl711-721 (1998). E. HETIER, J. AYALA, A. BOUSSEAU and A. PROCHIAN’IZ, Exp Brain Res 86 407-413 (1991). K. YABUUCHI, E. MARUT.A, J. YAMAMOTO, A. NISHIYORI, S. TAKAMI, M. MINAMI and M. SATOH, Eur. J. Pharmacol. m 133-140 (1997). M. SIHVOLA and M. HURME, Stand J Immunol24.689-698 (1989). s. BRANTSCHEN, J.F. GAUCHAT, A.L. DE WECK and B.M. STADIER, Eurj Immunol 1p 2017- 2023 (1989). B.D. CLARK, K.L. COLLINS, M.S. GANDY, AC. WEBB and P.E. AURON, Nucleic Acids Res ti 868 (1987).
119. A.B. SACHS, Cell 14 413-421(1993). 120. J.D. LEWIS and E. IZAURRALDE, Eur. J. Biochem. 242 461-9 (1997).

Vol. 65. No. 5, 1999 Dynamics of laterleukin-lp Gene Regulation 417
121.
122. 123. 124. 125. 126.
127. 128. 129. 130.
131. 132.
133. 134. 135. 136. 137. 138. 139. 140. 141. 142. 143. 144. 145. 146.
147. 148.
149. 150. 151. 152. 153. 154.
155. 156. 157.
158. 159.
160.
161.
162.
163.
164.
M. WICKENS, P. ANDERSON and R.J. JACKSON, Current Opinion in Genetics & Development z 220-232 (1997). J. ROSS, Microbial. Rev. 2 423-450 (1995). J. ROSS, Trends in Genetics j,2 (1996). D. HODGES and S.I. BERNSTEIN, Adv. Genet. fi 207-281 (1994). P.A. NORTON, J. Cell. Sci. _lQZ 1-7 (1994). S.P. EISENBERG, M.T. BREWER, E. VERDERBER, P. HEIMDAL, B.J. BRANDHUBER and R.C. THOMPSON, Proc Nat1 Acad Sci U S A 88 5232-5236 (1991). N. JARROUS and R. KAEMPFER, J Biol Chem B 23141-23149 (1994). M. GOROSPE, S. KUMAR and C. BAGLIONI, J Biol Chem a68 6214-6220 (1993). K. KANG, C. HAMMERBERT and K.D. COOPER, Exp Dermatol 2 218-226 (1996). H. KATO, H.Y. YOUN, T. OHASHI, T. WATARI, R. GOITSUKA, H. TSUJIMOTO and A. HASEGAWA, Gene m 11-16 (1996). J. SUTTLES, J.G. GIRI and S.B. MIZEL, 1 Immunol j& 175-182 (1990). D.K. MILLER, J.R. CALAYCAY, K.T. CHAPMAN, A.D. HOWARD, M.J. KOSTURA, S.M. MOLINEAUX and N.A. THRONBERRY, Ann. N.Y. Acad. Sci m 133-148 (1993). R. GREEN and H.F. NOLLER, Ann. Rev. Biochem. 66 679-716 (1997). V. PAIN, Eur. J. Biochem. m747-71 (1996). R.J. JACKSON, Cell B 9-14 (1993). I. STANSFIELD, K.M. JONES and M.F. TUlTE, TIBS 24489-4491(1995). V. KRUYS and G. HUEZ, Biochimie 24 862-866 (1994). D.J. HERZYK, J.N. ALLEN, C.B. MARSH and M.D. WEWERS, J Immunol. m3052-3058 (1992). L.E. RAJAGOPALAN and J.S. MALTER, Prog. Nucleic Acid Res. Mol. Biol. 56 257-286 (1997). C.Y.A. CHEN and A.B. SHYU, TIBS 211465-470 (1995). J.A. KERN, L.J. WARNOCK and J.D. MC CAFFERTY, J Immunol 158 1187-l 193 (1997). K. YAMATO, Z. EL-HAIJAOUI and H.P. KOEFFLER, J Cell Physiol m 610-616 (1989). Y. OHMORI and T.A. HAMILTON, J Immunol m 538-545 (1992). Y. AMANO, S.W. LEE and A.C. ALLISON, Mol Pharmacol Q 176-182 (1993). D.W. KIMBERLIN, S.A. WILLIS, M.G.H. JR. andP.D. NISEN, J. Clin. Immunol. fi 199-204 (1995). R.P. DONNELLY, M.J. FENTON, J.D. KAUFMAN and T.L. GERRARD, J Immunol 146 343 l-3436 (1991). M.W. HEN’IZE, Current Opinion in Cell Biology 2 393-398 (1995). M. JANSEN, C.H. DE MOOR, J.S. SUSSENBACH and L.J. VAN DEN BRANDE, Pediatric Research a 681-686 (1995). R.L. KASPAR and L. GEHRKE, J. Immunol. m 277-286 (1994). C.A. DINARELLO, 228-232 (1992). J.W.B. HERSHEY, Annu. Rev. Biochem. 4p717-755 (1991). V. KRUYS, 0. MARINX, G. SHAW, J. DESCHAMPS and G. HUEZ, Science m 852-855 (1989). C.A. DINARELLO, Proc Sot Exp Biol Med 2ap 228-232 (1992). D. CAPUT, B. BEU’ILER, K. HARTOG, R. THAYER, S. BROWN-SHIMER and A. CERAMI, Proc Nat1 Acad Sci USAU 1670-1674 (1986). C.A. DINARELLO, Blood u 2095-2147 (1996). M.D. WEWERS, H.A. POPE and D.A. MILLER, J. Immunol. Meth. 145 269-78 (1993). D.J. HERZYK, A.E. BERGER, J.N. ALLEN and M.D. WEWERS, J. Immunol. Meth. 148 243-254 (1992). C.A. DINARELLO, J. Immunolog. Meth. m 255-9 (1992). K. ZEPTER, A. HAFFNER, L.F. SOOHOO, D. DE LUCA, H.P. TANG, P. FSHER, J. CHAVINSON and C.A. ELMETS, J. Immunol. 15e 6203-6208 (1997). D.J. HAZUDA, J. STRICIUER, F. KUEPPERS, P.L. SIMON and P.R. YOUNG, J Biol Chem m 63 18-6322 (1990). H. MIZUTANI, N. SCHECHTER, G. LAZARUS, R.A. BLACK and T.S. KUPPER, J Exp Med 124 821-825 (1991). N.M. ROBERTSON, J. ZANGRILLI, T. FERNANDES-ALNEMRI, P.D. FRIESEN, G. LITWACK and E.S. ALNEMRI, Cancer Res 52 43-47 (1997). D.P. CERRETII, L.T. HOLLINGSWORTH, C.J. KOZLGSKY, M.B. VALENTINE, D.N. SHAPIRO, S.W. MORRIS and N. NELSON, Genomics 2p 468-73 (1994). F.J. CASANO, A.M. ROLANDO, J.S. MUDGETI and S.M. MOLINEAUX, Genomics 2p 474-481 (1994).

478 Dynamics of Interleukin-10 Gene Regulation Vol. 65. No. 5, 1999
165.
166.
167.
168.
169.
170.
171. 172. 173. 174.
175.
176.
177.
178. 179.
180. 181.
182.
183.
184.
185.
186.
187.
188.
189.
190. 191. 192. 193.
194.
195.
196. 197.
1.1. SINGER, S. SCOlT, J. CHIN, E.K. BAYNE. G. LIMJUCO, J. WEIDNER, D.K. MILLER, K. CHAPMAN and M.J. KOSTURA, J Exp Med m 1447-1459 (1995). R. LALIBERTE, D. PERREGAUX, L. SVENSSON, C.J. PAZOLES and C.A. GABEL, J Immunol u 2168-2179 (1994). D. FERRARI, P. CHIOZZI. S. FALZONI, M. DAL SUSINO, L. MELCHIORRI, O.R. BARICORDI and F. DI VIRGILIO, J Immunol m 1451-1458 (1997). S. LAYE, E. GOUJON. C. COMBE, R. VAN HOY. K.W. KEI.IY, P. PARNET and R. DAN’lZER, J Neuroimmunol68 6 l-66 (1996). K.M. KEANE. D.A. GIEGEL, W.J. LIPINSKI, M.G. CALLAHAN and B.D. SHIVERS, Cytokine z 105-110 (1995). S. TINGSBORG, M. ZEITERSTROM, K. ALHEIM, H. HASANVAN, M. SCHULTZBERG and T. BARTFAI, Brain Res. 212 153-158 (1996). J. CHIN and M.J. KOSTURA, J. Immunology m 5574-5585 (1993). D.G. PERREGAUX and CA. GABEL, J. Imm~nol. 164 2469-2477 (1998). A. RUBARTELLI, F. COZZOLINO, M. TALIO and R. SITIA. EMBO J 2 1503-1510 (1990). S.L. BURSTEN, R.M. LOCKSLEY, J.L. RYAN and D.H. LOVE’IT, J Clin Invest 82 1479-1488 (1988). C. MANCUSO, G. TRINGALI, A. GROSSMAN, P. PREZIOSI and P. NAVARRA. Endo. m 1031-7 (1998). P. NAVARRA, G. POZZOLI. C. BECHERUCCI, P. PREZIOSI, A.B. GROSSMAN and L. PARENTE, Neuroendo. 2 257-261 (1993). S.P. SIRKO, R. SCHINDLER, M.J. DOYLE, S.M. WEISMAN and CA. DINARELLO, Eur J Immunol21243-250 (1991). D.G. PERREGAUX, L. SVENSSON and CA. GABEL, J Immunol m 57-64 (1996). R.J. GRIFFITHS, E.J. STAM, J.T. DOWNS and I.G. OTIERNESS, J. Immunol m 2821-2828 (1995). H. OKAMOTO, C. OH and K. NAKANO, Immunology 2p 186-190 (1990). J. VIAC, A. GUENICHE, J.D. DO UlREMEPUICH, U. REICHBRT, A. CLAUDY and D. SCHMm, Arch Dennatol Res 288 85-90 ( 1996). F.C. MARTIN, A.C. CHARLES, M.J. SANDERSON and J.E. MERRILL, Brain Res m 13-18 (1992). M.E. CUNNINGHAM, T.A. SHAW-STIFFEL, L.H. BERNSTEIN, T.J. TINGHlTELLA, R.E. CLAUS, D.A. BROGAN and M.A. MCMILLEN, Amer. J. Gastroentoerol. ep 621-626 (1995). J.A. KERN, R.J. LAMB, J.C. REED, R.P. DANIELE and P.C. NOWELL, J Clin Invest fi 237-244 (1988). P. MIOSSEC, J. BRIOLAY, J. DECHANET, J. WIJDENES, H. MARTINEZ-VALDEZ and J. BANCHEREAU, Arthritis Rheum 25 874-883 (1992). J. VIHERLUO’IO, T. PALKAMA. 0. SILVENNOINEN and M. HURME, Scan J Immunol 3 121- 125 (1991). K. IWAZ, E. KOUASSI, S. LAFONT and J.P. REVILLARD. Int J Immunophannacol12 631-637 (1990). T. YOSHIMURA, C. KURITA, T. NAGAO, E. USAMI, T. NAKAO, S. WATANABE, J. KOBAYASHI, F. YAMAZAKI, H. TANAKA, N. INAGAKI and H. NAGAI, Pharmacology % 144- 152 (1997). L. SHAPIRA, S. TAKASHIBA, C. CHAMPAGNE, S. AMAR and T.E. VAN DYKE, J. Immunol. kQ 1818-24 (1994). 0. BAKOUCHE. J.L. MOREAU and L.B. LACHMAN, J Immunol148 84-91 (1992). T. KRAKAUER, Cell Immunol122 224-228 (1996). C.S. CHUJOR, L. KLEIN and C. LAM, Eur J Immunol26 1253-1259 (1996). G. TRINGALI, M.A., C. MANCUSO, G. GUERRIERO, P. PREZIOSI and P. NAVARRA, Pharmacol. Res. s 269-73 (1997). G. TRINGALI, C. MANCUSO, A. MIRTELLA, G. POZZOLI, L. PARENTE, P. PREZIOSI and P. NAVARRA. Neurosci. Lett. 219 143-6 (1996). E.D. MILLIGAN, K.T. NGUYEN, J.L. HINDE, K. MEHMERT. M. BOHL, SF. MAIER and L.R. WATKINS, Proc. Sot. Neurosci. 24 (1998). J.E. SIMS, S.L. PAINTER and I.R. GOW, Cytokine 1483-490 (1995). R.W. GROVES, J. GIRI, J. SIMS, S.K. DOWER and T.S. KUPPER, J. Immunol. m 4065-4072 (1995).

Vol. 65, No. 5, 1999 Dynamics of Iaterleukia-lp Gene Regulation 479
198.
199.
200. 201.
202.
203.
204. 205.
206.
207.
208.
209.
210. 211. 212. 213.
214.
215. 216. 217. 218.
219.
220. 221. 222.
223.
224.
225.
226. 227. 228.
229.
230.
J.E. SIMS, M.A. GAYLE, J.L. SLACK, M.R. ALDERS0N.T.A. BIRD, J.G. GIRL F. COLOTI’A, F. RE, A. MANTOVANI, K. SHANEBECK, ET AL, Proc. Natl. Acad. Sci. U S A ~ 6155-6159 (1993). C. LIU, R.P. HART, X.J. LIU, W. CLEVENGER. R.A. MAKI and E.B. DE SOUZA. J. Biol. Chem. m 2096520972 (1996). R. FERNANDEZ-BOTRAN, P.M. CHILTON and Y. MA, Adv. Immunol. 62 269-336 (1996). M. SVENSON, M.B. HANSEN, P. HEEGAARD, K. ABELL and K. BEND’IZEN. Cytoldne 2 427-35 (1993). F. COLGTTA, S. SACCANI, J.G. GIRL S.K. DOWER, J.E. SIMS, M. INTRONA and A. MANTOVANI, J. Immunol. m 2534-2541 (1996). F. RE, M. MUZIO. M. DEROSSI, N. POLENTARU’ITI, J.G. GIRL A. MANTOVANI and F. COLOTTA, J Exp Med n 739-743 (1994). P.W. YU, L.A. SCHULER and C.J. CZUPRYNSKI, Clin. Diagn. Lab Immunol. 4 769-773 (1997). A. ITO, T. TAKII, N. GOTO, Y. KIT0 and K. ONOZAKI, J. Interferon Cytokine Res. 12 413-417 (1997). T. TAKII, N. NIKI, D. YANG, H. KIMURA, A. ITO, H. HAYASHI and K. ONOZAKI. J. Interferon. Cytokine Res. fi 10651073 (1995). C.J. VAN DER LAKEN, O.C. BOERMAN, W.J. OYEN, P. LAVERMAN, M.T. VAN DE VEN, F.H. CORSTENS and J.W. VAN DER MEER, J. Infect. Dis. 12z 1398-1401 (1998). J. BRISTULF, S. GA’ITI, D. MALINOWSKY, L. BJORK, A.K. SUNDGREN and T. BARTFAI, Eur Cytokine Netw 2 3 19-330 (1994). D. KUNZ, G. WALKER, W. EBERHARDT, U.K. MESSMER, A. HUWILER and J. PFEILSCHIFIER, J. Clin. Invest. UM 2800-2809 (1997). A. ITO, T. TAKII, T. SOJI and K. ONOZAKI, J. Interferon. Cytokine Res. 1z 55-61 (1997). H.L. DICKENSHEETS and R.P. DONNELLY, J. Immunol. 1Ip 6226-6233 (1997). M.M. GABELLEC, R. GRIFFAIS, G. FILLION and F. HAOUR, J. Neuroimmunol. 66 65-70 (1996). N. REINISCH, M. WOLKERSDORFER, C.M. KAHLER, K. YE, C.A. DINARELLG and C.J. WIEDERMANN, Neurosci. Lett. j&j 165-167 (1994). S.E. ILYIN, D. GAYLE, NC. FLYNN and C.R. PLATA-SALAMAN, Brain Res. Bull. U 507-515 (1998). D. GAYLE, S.E. ILYIN and C.R. PLATA-SALAMAN, Brain Res. Bull. 44 311-317 (1997). J. BRISTULF and T. BARTFAI, Neurosci. Lett. 182 53-56 (1995). Y. TOMOZAWA, T. INOUE and M. SATGH, Neurosci. Lett. les. 57-60 (1995). F. COLOTTA, F. RE. M. MUZIO, R. BERTINI. N. POLENTARU’ITI, M. SIRONI, J.G. GIRL S.K. DOWER, J.E. SIMS and A. MANTOVANI, Science 26L 472-475 (1993). J.G. GIRL J. WELLS, S.K. DOWER, C.E. MCCALL, R.N. GUZMAN, J. SLACK, T.A. BIRD, K. SHANEBECK, K.H. GRABSTEIN, J.E. SIMS and M.R. ALDERSON, J. Immunol. m 5802-5809 (1994). J.A. SYMONS, P.R. YOUNG and G.W. DUFF, Proc Nat1 Acad Sci U S A % 1714-1718 (1995). E.A. BROWN, H.A. DARE, C.B. MARSH and M.D. WEWERS, Cytokine&828-836 (1996). F. COLOTTA, F. RE, M. MUZIO, N. POLENTARUTI’I, A. MINTY, D. CAPUT, P. FBRRARA and A. MANTOVANI, J Biol Chema 12403-12406 (1994). F. COLO’ITA, F. RE. M. MUZIO, N. POLENTARUTI’I, A. MINTY, D. CAPUT, P. FERRARA and A. MANTOVANI. J. Biol. Chem. 26e 12403-12406 (1994). P. SAMBO, E.J. FADLON, M. SIRONI, C. MATTEUCCI, M. INTRONA, A. MANIGVANI and F. COLOTTA, Blood 811682-1686 (1996). F. RAMIERZ, D.J. FOWELL, M. PUKLAVEC, S. SIMMONDS and D. MASON, J. Immunol. m 2406-12 (1996). S.J. LEVINE, T. WU and J.H. SHELHAMER, J Immunol m 5949-5957 (1997). A. MANTOVANI and F. COLOTTA, J Exp Med m 623-628 (1995). L. WEISSBACH, K. ‘IRAN, S.A. COLQUHOUN, M.F. CHAMPLIAUD and C.A. ‘IOWLE, Biochem. Biophys. Res. Commun. 244 91-95 (1998). A. STEINKASSERER. N.K. SPURR, S. COX, P. JEGGO and R.B. SIM, Genomics 11654-657 (1992). A. LENNARD, P. GORMAN, M. CARRIER, S. GRIFFITHS, H. SCOTNEY, D. SHEER and R. SOLARI, Cytokine 4 83-89 (1992).
231. A.L. HUGHES, J. Mol. Evol. a6-12 (1994). 232. C.H. EVANS and P.D. ROBBINS, Receptor 49-15 (1994).

480 Dynamics of lnterleukin-lp Gene Regulation Vol. 65. No. 5, 1999
233. 234.
235.
236.
237.
238. 239.
240.
241.
242.
243. 244. 245. 246. 247.
248.
249.
250.
251.
252. 253.
254.
255.
256.
257.
258.
259. 260.
261. 262.
263.
264.
C. GABAY, M.F. SMITH, D. EIDLEN and W.P. AREND, J Clin Invest s 2930-2940 (1997). F. RE, M. MENGOZZI. M. MUZIO, C.A. DINARELLO, A. MANTOVANI and F. COLG’ITA, Eur J Immunol 22 570-573 (1993). M. MUZIO, F. RE, M. SIRONI, N. POLENTARUTTI, A. MINTY, D. CAPUT, P. FERRARA, A. MANTOVANI and F. COLOTTA, Blood fi 1738-1743 (1994). M. MUZIO. M. SIRONI, N. POLENTARUTTI, A, MANTOVANI and F. COLGITA, Em J Immunol a 3194-3198 (1994). J.K. JENKINS, R.F. DRONG, M.E. SHUCK, M.J. BIENKOWSKI, J.L. SLIGHTOM. W.P. AREND and M.F.J. SMITH, J Immunol m 748-755 (1997). A. CORRADI, A.T. FRANZI and A. RUBARTELLI, Exp Cell Res m 355-362 (1995). J.M. WATSON, A.K. LGFQUIST, CA. RINEHART, J.C. OLSEN, S.S. MAKAROV, D.G. KAUFMAN and J.S. HASKILL, J Immunol 155 4467-4475 (1995). S.J. LEVINE, T. BENFIELD and J.H. SHELHAMER, Amer. J. Respir. Cell Mol. Biol. fi 245-51 (1996). S.A. GREENFEDER, P. NUNES, K. KWEE, M. LAEOW, R.A. CHIZZONITE and G. JU, J. Biol. Chem. 22p 13757-13765 (1995). M.M. GABELLEC, M. JAFARIAN-TEHRANI, R. GRIFFAIS and F. HAOUR, Neuroimmunol. 1304- 309 (1996). C. LIU, D. CHALMERS, R. MAKI and E.B. DE SOUZA, J. Neuroimmunol. @41-48 (1996). H. WESCHE, D. NEUMANN, K. RESCH and M.U. MARTIN, FEBS Lett. m 104-108 (1996). C. KORHERR, R. HOFMEISTER, H. WESCHE and W. FALK, Eur. J. Immunol. 21262-267 (1997). J. HUANG, X. GAO, S. LI and Z. CAO, Proc. Natl. Acad. Sci. N 12829-12832 (1997). F. VOLPE, J. CLATWORTHY, A. KAPTEIN, B. MASCHERA, A.M. GRIFFIN and K. RAY, FEBS L&t. $#_@41-44 (1997). P. PARNET, K.E. GARKA, T.P. BONNERT, S.K. DOWER and J.E. SIMS, J. Biol. Chem. m3967- 3970 (1996). T.W. LOVENBERG, P.D. CROWE, C. LlU, D.T. CHALMERS, X.J. LIU, C. LIAW, W. CLEVENGER, T. OLTERSDGRF, E.B. DE SOUZA and R.A. MAKI, J. Neuroimmunol. ZQ 113- 122 (1996). K. TORIGOE, S. USHIO, T. OKURA, S. KOBAYASHI, M. TANIAI, T. KUNIKATA, T. MURAKAMI, 0. SANOU, H. KOJIMA, M. FUJII, T. OHTA, M. IKEDA, H. IKEGAMI and M. KURIMOTO, J. Biol. Chem. 222 25737-25742 (1997). A. REIKERSMRFER, H. HOLZ, H.G. STUNNENBERG and M. BUSSLINGER, J. Biol. Chem. m 17645-17648 (1996). D.J. PICKUP, Infect Agents Dis 2 116-127 (1994). J. OQUENDO, S. DUBANCHET, F. CAPEL, H. MABIT and M.A. PETIT, Eur. Cytokine Network ~793-800 (1996). Q. NING, D. BROWN, J. PARODO, C.J. ROBERGE, P.E. POUBELIE, A.D. BEAULIEU. D. I-IEf-lZ and J. GOSSELIN, J. Immunol~4884-4891 (1996). T. TAMURA, S. UEDA, M. YOSHIDA, M. MATSUZAKI, H. MOHRI and T. OKUBO, Biochem Biophys Res Commun 229.21-26 (1996). A.D. HOWARD, O.C. PALYHA, P.R. GRIFFIN, E.P. PETERSON, A.B. LENNY, G.J. DING, D.J. PICKUP, N.A. THORNBERRY, J.A. SCHMIDT and M.J. TOCCI, J Immunol m 2321-2332 (1995). S. KETrLE, A. ALCAMI, A. KHANNA, R. EHRET, C. JASSOY and G.L. SMITH, J Gen Virol28 677-685 (1997). F. PETIT, S. BERTAGNOLI, J. GELFI, F. FASSY, C. BOUCRAUT-BARALON and A. MILON, J Virol Zp 5860-5866 (1996). A. ALCAMI and G.L. SMITH, Cella 153-167 (1992). M.K. SPRIGGS, D.E. HRUBY, CR. MALISZEWSKI, D.J. PICKUP, J.E. SIMS, M.L. BULLER and J. VAN SLYKE, Cell u 145-152 (1992). G.L. SM1THandY.S. CHAN,JGenVirolz511-518(1991). E.V. GRANOWITZ, A.A. SANTOS, D.D. POUTSIAKA, J.G. CANNON, D.W. WILMORE, S.M. WOLFF and C.A. DINARELLG, Lancet m 1423-1424 (1991). M.A. CASSATELLA, L. MEDA, S. GASPERINI, F. CALZETTI and S. BONORA, J. Exp. Med. 129. 1695-1699 (1994). R.A. GOTI’LIEB, W.J. LENNARZ, R.D. KNOWLES, G.J. CIANCIOLO, C.A. DINARELLO, L.B. LACHMAN and ES. KLEINERMAN, J. Immunol. m4321-4328 (1989).

Vol. 65, No. 5, 1999 Dynamics of Interleukin-1 p Gene Regulation 481
265.
266.
267.
268.
269. 270.
271.
272.
273. 274. 275. 276. 277.
278.
279. 280. 281.
282. 283.
284.
28.5.
286. 287. 288.
289.
290.
S. PIED, A. WAS, F. BERLUI’-PICARD, L. RENIA, F. MIL’IGEN, M. GENTILINI, J. DOLY and D. MAZIER, J. Immunol. 148 197-201 (1992). S. FRANKENBURG, V. LEIBOVICI, N. MANSBACH, S.J. TURCO and G. ROSEN, J. Immunol. 145 4284-4289 (1990). 0. DUVAUX-MIRET, G.B. STEFANO, E.M. SMITH, C. DISSOUS and A. CAPRON, Proc. Natl. Acad. Sci. & 778-781 (1992). N.E. REINER, W. NG, C.B. WILSON, W.R. MCMASTER and S.K. BURCHE’IT, J. Clin. Invest. @ 1914-1924 (1990). G.D. CRAWFORD, D.J. WYLER and C.A. DINARELLO, J. Infect. Dis. 152315-322 (1985). C.L. KING, A. MEDHAT, I. MALHOTRA, M. NAFEH, A. HELMY, J. KHAUDARY, S. IBRAHIM, M. EL-SHERBINY, S. ZAKY, R.J. STUPI, K. BRUSTOSKI, M. SHEHATA and M.T. SHATA, J. Immunol. m4715-4721 (1996). R.A. LAWRENCE, J.E. ALLEN, J. OSBORNE and R.M. MAIZELS, J. Immunol. m 1216-1224 (1994). R.W. LEID, C.M. SUQUET, H.G. BOUWER and D.J. HINRICHS, J. Immunol. H 2700-2702 (1986). J.M. LIPTON and A. CATANIA, Imrnunol Today J.& 140-145 (1997). J.S. FASSLER and G.N. GUSSIN, Methods Enzymol. m 3-29 (1996). C.S. HILL and R. TREISMAN, Cell m 199-211 (1995). K.J. HERTEL, K.W. LYNCH and T. MANIATIS, Cm-r. Opin. Cell Biol. 2 350-357 (1997). P.T. LB. S. LAZORICK, L.P. WHICHARD, B.F. HAYNES and K.H. SINGER, J Exp Med 124 1147- 1157 (1991). J.N. KLINE, P.A. FISHER, M.M. MONICK and G.W. HUNNINGHAKE, Amer. J. Physiol. m L92- 98 (1995). M. MALYAK, M.F.J. SMITH, A.A. ABEL and W.P. AREND, J Clin Imjunol 14 20-30 (1994). J. COCLET-NININ, J.M. DAYER and D. BURGER, Eur. Cytokine Network & 345-349 (1997). D. PERREGAUX, J. BARBERIA, A.J. LANZE’ITI, K.F. GEOGHEGAN, T.J. CARTY and C.A. GABEL, J Immunol m 1294- 1303 (1992). D. PERREGAUX and C.A. GABEL, J Biol Chemm 15195-15203 (1994). S. ORLANDO, C. MATI-EUCCI, E.J. FADLON, W.A. BUURMAN, M.T. BARDELLA, F. COLOTTA, M. INTRONA and A. MANTOVANI, J. Immunol. m 3861-3868 (1997). J. SAUER, M. CASTREN, U. HOPFNER, F. HOLSBOER, G.H. STALLA and E. ARZT, J. Clin. Endocrinol. Metabl. fi 73-79 (1996). T. POLLMACHER, C. KORTH, W. SCHREIBER, D. HERMANN and J. MULLINGTON, Cytokine 8 799-803 (1996). J.K. JENKINS and W.P. AREND, Cytokine 2 407-415 (1993). S.R. MC COLL, R. PAQUIN, C. MENARD and A.D. BEAULIEU, J. Exp. Med. 126 593-598 (1992). J.P. PELLETIER, F. MINEAU, P. RANGER, G. TARDIF and J. MARTEL-PELLETIER, Osteoarthritis Cartilage 3 77-84 (1996). F. COLO’ITA, S. ORLANDO, E.J. FADLON, S. SOZZANI, C. MATTEUCCI and A. MANTOVANI, J. Exp. Med. LB1 2181-2186 (1995). B. GALVE-DEROCHEMONTEIX, L.P. NICOD, R. CHICHEPORTICHE, S. LACRAZ, C. BAUMBERGER and J.M. DAYER, Amer. J. Respir. Cell. Mol. Biol. & 160-168 (1993).




















