
Regulation and Traffic of Ceramide 1-Phosphate Produced by Ceramide Kinase: COMPARATIVE ANALYSIS TO...
14
Regulation and Traffic of Ceramide 1-Phosphate Produced by Ceramide Kinase COMPARATIVE ANALYSIS TO GLUCOSYLCERAMIDE AND SPHINGOMYELIN * □ S Received for publication, August 24, 2007, and in revised form, December 7, 2007 Published, JBC Papers in Press, December 16, 2007, DOI 10.1074/jbc.M707107200 Alistair Boath 1 , Christine Graf, Emilie Lidome 1 , Thomas Ullrich, Peter Nussbaumer, and Fre ´de ´ ric Bornancin 2 From the Novartis Institutes for BioMedical Research, Vienna, Brunnerstrasse 59, A-1235 Wien, Austria Ceramide 1-phosphate (C1P) has been characterized as a sphingolipid that participates in cell signaling. Although C1P synthesis is thought to occur via phosphorylation of ceramide by ceramide kinase (CerK), the processes that regulate C1P forma- tion and fate remain largely unknown. In this study we analyzed bone marrow-derived macrophages (BMDM) from CerK-null mice (Cerk / ) and found significant levels of C1P, suggesting that previously unrecognized pathways may also lead to C1P formation. After these experiments we used an overexpression system, BMDM from Cerk / mice, and short-chain fluorescent ceramides to trace CerK-dependent formation of C1P. Because the ceramide analogs can also be converted to glucosylceramide (GlcCer) and sphingomyelin (SM), they allowed us to directly compare all three metabolites. We found that C1P produced by CerK is turned over rapidly when serum is removed or upon calcium chelation, whereas GlcCer and SM are stable under these conditions. We further demonstrated that ceramide must be transported to the Golgi complex to be phosphorylated by CerK. Inhibition of the ceramide transfer protein slowed down SM formation without decreasing C1P, suggesting an alternate route of ceramide transport. Other experiments indicated that, like GlcCer and SM, C1P traffics along the secretory pathway to reach the plasma membrane. Furthermore, in BMDM C1P was secreted more readily than was GlcCer or SM. Altogether, our results indicate that CerK is essential to C1P formation via phos- phorylation of Cer, providing the first insights into mechanisms underlying ceramide access to CerK and C1P trafficking as well as clarifying C1P as a signaling entity. Sphingolipids, a class of lipids found in all eukaryotic cell membranes, also function as signaling molecules. De novo syn- thesis of sphingolipids occurs through a well documented path- way beginning with condensation of serine and palmitoyl-CoA and eventually leading to ceramide (Cer), 3 which is a central intermediate (1, 2). Pathways that regulate Cer levels prompt intense scientific interest because of the role of Cer in many cellular mechanisms including apoptosis and differentiation as well as implications for disease (1–7). Cer metabolism to more complex sphingolipids is important to understand because it impacts Cer levels and can produce bioactive lipids (2–5). Cer is converted to glucosylceramide (GlcCer) by a glucosylceramide synthase and to sphingomyelin (SM) by sphingomyelin syn- thases (SMS) (1). Recently, a ceramide kinase (CerK) was iden- tified that can phosphorylate Cer to ceramide 1-phosphate (C1P) (8). Although C1P was first described about 20 years ago (9, 10), regulation of its synthesis and fate has not yet been stud- ied. Synthesis is believed to occur mainly through the phos- phorylation of Cer by CerK. C1P is classified as a signaling lipid based on various reports showing that C1P participates in phagocytosis (11), eicosanoid synthesis (12, 13), plays a role in mast cell degranulation (14), controls apoptosis in bone marrow-derived macrophages (15), mobilizes calcium in GH4C1 pituitary cells (16) and Jurkat T-cells (17), stimu- lates dopamine release in PC12 cells (18), and activates cell proliferation and survival (19, 20). In the present work we provide the first evidence that CerK is not the only source of C1P. After making this discovery using cells isolated from Cerk / 4 mice, we focused on refining our understanding of C1P by differentiating CerK-mediated phos- phorylation of Cer from other mechanisms that produce C1P. To do so we used short-chain fluorescent ceramides as preci- sion tools to study only CerK-dependent C1P formation. Importantly, these tools have enabled us to show for the first time that C1P is a short-lived metabolite compared with GlcCer and SM, strongly supporting the hypothesis that C1P is a sig- naling messenger whose turnover is tightly regulated. Further- more, because of the unique properties of the fluorescent cera- mide analogs used here, we have been able to probe other important aspects of C1P metabolism. We found, for example, that Cer is not phosphorylated at the endoplasmic reticulum (ER) but must be transported to the Golgi complex for phos- * The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertise- ment” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact. □ S The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1–3. 1 Supported by a fellowship from the European Commission Leonardo da Vinci program. 2 To whom correspondence should be addressed. Tel.: 43-80166-335; Fax: 43-86634 –582; E-mail: [email protected]. 3 The abbreviations used are: Cer, ceramide; CerK, Cer kinase; BAPTA, 1,2- bis(2-aminophenoxy)ethane-N,N,N,N-tetraacetic acid; BMDM, bone mar- row-derived macrophage(s); BODIPY, boron dipyrromethene difluoride; C1P, ceramide 1-phosphate; DMB, N-(5-(5,7-dimethyl BODIPY)); DMB-Cer, N-(5-(5,7-dimethyl-BODIPY)-L-pentanoyl)-D-erythrosphingosine; ER, endoplasmic reticulum; NBD-Cer, N-(7-(4-nitrobenzo-2-oxa-l,3-diazole))-6- aminocaproyl-D-erythrosphingosine; GlcCer, glucosylceramide; SM, sphin- gomyelin; SMS, SM synthase; TRB-Cer, N-((4-(4,4-difluoro-5- (2-thienyl)-4- bora-3a,4a-diaza-s-indacene-3-yl)phenoxy)acetyl)sphingosine; HPA-12, N-(hydroxy-1-hydroxymethyl-3-phenylpropyl) dodecanamide; CER, Cer transfer protein; DMEM, Dulbecco’s modified Eagle’s medium; FCS, fetal calf serum; HBSS, Hanks’ balanced salt solution; WT, wild type. 4 The generation of Cerk / will be described elsewhere. THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 283, NO. 13, pp. 8517–8526, March 28, 2008 © 2008 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in the U.S.A. MARCH 28, 2008 • VOLUME 283 • NUMBER 13 JOURNAL OF BIOLOGICAL CHEMISTRY 8517 by guest on April 21, 2016 http://www.jbc.org/ Downloaded from by guest on April 21, 2016 http://www.jbc.org/ Downloaded from by guest on April 21, 2016 http://www.jbc.org/ Downloaded from by guest on April 21, 2016 http://www.jbc.org/ Downloaded from by guest on April 21, 2016 http://www.jbc.org/ Downloaded from
Transcript of Regulation and Traffic of Ceramide 1-Phosphate Produced by Ceramide Kinase: COMPARATIVE ANALYSIS TO...

Regulation and Traffic of Ceramide 1-Phosphate Producedby Ceramide KinaseCOMPARATIVE ANALYSIS TO GLUCOSYLCERAMIDE AND SPHINGOMYELIN*□S
Received for publication, August 24, 2007, and in revised form, December 7, 2007 Published, JBC Papers in Press, December 16, 2007, DOI 10.1074/jbc.M707107200
Alistair Boath1, Christine Graf, Emilie Lidome1, Thomas Ullrich, Peter Nussbaumer, and Frederic Bornancin2
From the Novartis Institutes for BioMedical Research, Vienna, Brunnerstrasse 59, A-1235 Wien, Austria
Ceramide 1-phosphate (C1P) has been characterized as asphingolipid that participates in cell signaling. Although C1Psynthesis is thought to occur via phosphorylation of ceramide byceramide kinase (CerK), the processes that regulate C1P forma-tion and fate remain largely unknown. In this study we analyzedbone marrow-derived macrophages (BMDM) from CerK-nullmice (Cerk�/�) and found significant levels of C1P, suggestingthat previously unrecognized pathways may also lead to C1Pformation. After these experiments we used an overexpressionsystem, BMDMfromCerk�/�mice, and short-chain fluorescentceramides to trace CerK-dependent formation of C1P. Becausethe ceramide analogs can also be converted to glucosylceramide(GlcCer) and sphingomyelin (SM), they allowed us to directlycompare all three metabolites. We found that C1P produced byCerK is turned over rapidly when serum is removed or uponcalcium chelation, whereas GlcCer and SM are stable underthese conditions. We further demonstrated that ceramide mustbe transported to the Golgi complex to be phosphorylated byCerK. Inhibition of the ceramide transfer protein slowed downSM formation without decreasing C1P, suggesting an alternateroute of ceramide transport. Other experiments indicated that,like GlcCer and SM, C1P traffics along the secretory pathway toreach the plasma membrane. Furthermore, in BMDM C1P wassecreted more readily than was GlcCer or SM. Altogether, ourresults indicate thatCerK is essential toC1P formation via phos-phorylation of Cer, providing the first insights intomechanismsunderlying ceramide access to CerK and C1P trafficking as wellas clarifying C1P as a signaling entity.
Sphingolipids, a class of lipids found in all eukaryotic cellmembranes, also function as signaling molecules. De novo syn-thesis of sphingolipids occurs through awell documented path-way beginning with condensation of serine and palmitoyl-CoAand eventually leading to ceramide (Cer),3 which is a central
intermediate (1, 2). Pathways that regulate Cer levels promptintense scientific interest because of the role of Cer in manycellular mechanisms including apoptosis and differentiation aswell as implications for disease (1–7). Cer metabolism to morecomplex sphingolipids is important to understand because itimpacts Cer levels and can produce bioactive lipids (2–5). Cer isconverted to glucosylceramide (GlcCer) by a glucosylceramidesynthase and to sphingomyelin (SM) by sphingomyelin syn-thases (SMS) (1). Recently, a ceramide kinase (CerK) was iden-tified that can phosphorylate Cer to ceramide 1-phosphate(C1P) (8).Although C1P was first described about 20 years ago (9,
10), regulation of its synthesis and fate has not yet been stud-ied. Synthesis is believed to occur mainly through the phos-phorylation of Cer by CerK. C1P is classified as a signalinglipid based on various reports showing that C1P participatesin phagocytosis (11), eicosanoid synthesis (12, 13), plays arole in mast cell degranulation (14), controls apoptosis inbone marrow-derived macrophages (15), mobilizes calciumin GH4C1 pituitary cells (16) and Jurkat T-cells (17), stimu-lates dopamine release in PC12 cells (18), and activates cellproliferation and survival (19, 20).In the present workwe provide the first evidence that CerK is
not the only source of C1P. After making this discovery usingcells isolated from Cerk�/� 4 mice, we focused on refining ourunderstanding of C1P by differentiating CerK-mediated phos-phorylation of Cer from other mechanisms that produce C1P.To do so we used short-chain fluorescent ceramides as preci-sion tools to study only CerK-dependent C1P formation.Importantly, these tools have enabled us to show for the firsttime thatC1P is a short-livedmetabolite comparedwithGlcCerand SM, strongly supporting the hypothesis that C1P is a sig-naling messenger whose turnover is tightly regulated. Further-more, because of the unique properties of the fluorescent cera-mide analogs used here, we have been able to probe otherimportant aspects of C1P metabolism. We found, for example,that Cer is not phosphorylated at the endoplasmic reticulum(ER) but must be transported to the Golgi complex for phos-* The costs of publication of this article were defrayed in part by the payment
of page charges. This article must therefore be hereby marked “advertise-ment” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
□S The on-line version of this article (available at http://www.jbc.org) containssupplemental Figs. 1–3.
1 Supported by a fellowship from the European Commission Leonardo daVinci program.
2 To whom correspondence should be addressed. Tel.: 43-80166-335; Fax:43-86634 –582; E-mail: [email protected].
3 The abbreviations used are: Cer, ceramide; CerK, Cer kinase; BAPTA, 1,2-bis(2-aminophenoxy)ethane-N,N,N�,N�-tetraacetic acid; BMDM, bone mar-row-derived macrophage(s); BODIPY, boron dipyrromethene difluoride;C1P, ceramide 1-phosphate; DMB, N-(5-(5,7-dimethyl BODIPY)); DMB-Cer,
N-(5-(5,7-dimethyl-BODIPY)-L-pentanoyl)-D-erythrosphingosine; ER,endoplasmic reticulum; NBD-Cer, N-(7-(4-nitrobenzo-2-oxa-l,3-diazole))-6-aminocaproyl-D-erythrosphingosine; GlcCer, glucosylceramide; SM, sphin-gomyelin; SMS, SM synthase; TRB-Cer, N-((4-(4,4-difluoro-5- (2-thienyl)-4-bora-3a,4a-diaza-s-indacene-3-yl)phenoxy)acetyl)sphingosine; HPA-12,N-(hydroxy-1-hydroxymethyl-3-phenylpropyl) dodecanamide; CER, Certransfer protein; DMEM, Dulbecco’s modified Eagle’s medium; FCS, fetalcalf serum; HBSS, Hanks’ balanced salt solution; WT, wild type.
4 The generation of Cerk�/� will be described elsewhere.
THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 283, NO. 13, pp. 8517–8526, March 28, 2008© 2008 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in the U.S.A.
MARCH 28, 2008 • VOLUME 283 • NUMBER 13 JOURNAL OF BIOLOGICAL CHEMISTRY 8517
by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from
by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from
by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from
by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from
by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from

phorylation by CerK. Trafficking of Cer to CerK, however, doesnot appear to rely on the same pathway as that of SM synthesisbecause pharmacological inhibition of the Cer transfer protein(CERT) did not slow down C1P synthesis. Finally, we observedthat C1P readily traffics from the Golgi network to the plasmamembrane, as do GlcCer and SM (21).In short, using a dual gain-of-function/loss-of function strat-
egy (by overexpressing CerK and by working with Cerk�/�
cells) as well as sensitive and specific fluorescent ceramide ana-logs, we explain here for the first time how CerK-dependentC1P is formed, regulated, and trafficked compared with GlcCerand SM.
EXPERIMENTAL PROCEDURES
Materials—NBD-C6 ceramide, DMB-C5 ceramide, andTRB-ceramide were from Molecular Probes; monensin wasfrom Sigma; recombinant mouse macrophage colony stimula-tion factor was from R&D systems; and Dulbecco’s modifiedEagle’s medium (DMEM), Roswell Park Memorial Institute1640 (RPMI) medium, and fetal calf serum (FCS) were fromInvitrogen. N-(Hydroxy-1-hydroxymethyl-3-phenylpropyl)dodecanamide (HPA-12) and its inactive enantiomer mix werekindly donated by Dr. K. Hanada (National Institute of Infec-tious Diseases, Tokyo). PIK93 was synthesized at NovartisInstitutes for BioMedical Research. All other reagents werefrom Sigma unless otherwise stated.Preparation of DMB-labeled Sphingolipids—DMB-C14 sphin-
gosine ((2S,3R)-2-amino-14-(4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacen-3-yl)-tetradec-4-ene-1,3-diol) was pre-pared as reported previously (22). DMB-C14 sphinganine((2S,3R)-2-amino-14-(4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacen-3-yl)-tetradecane-1,3-diol) was synthesized byhydrogenation ofN-Boc-oxazolidine-protectedDMB-C14 sphin-gosine (22) usingWilkinson’s catalyst followed by deprotection asreported for DMB-C14 sphingosine. A ceramide analog, labeledon the sphingosine chain, DMBSphC14-C6 ceramide ((2S,3R)-14-(4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacen-3-yl)-2-hexanoylamino-tetradec-4-ene-1,3-diol) was synthesizedfrom C6 ceramide and 3-(undec-10-enyl)-4,4-difluoro-5,7-di-methyl-4-bora-3a,4a-diaza-s-indacen by cross-metathesis reac-tion as described in Nussbaumer et al. (23).Cell Culture and Treatments—COS-1 cells were cultured in
DMEM supplemented with 10% FCS at 37 °C, 5% CO2 in ahumidified atmosphere. ACOS-CerK cell line was generated aspreviously described (24). For bone marrow-derived macro-phage (BMDM) preparation, wild type and Cerk�/� Balb/c4mice were sacrificed via cervical dislocation. Femurs wereextracted and washed twice in phosphate-buffered saline. Endsof the bones were cut, and bone marrow was extruded withRPMI, 10% heat-inactivated FCS and antibiotics (culturemedium) using a 25-gauge syringe. Cells were filtered through a70-�m cell strainer, and the filter was washed with culturemedium. Cells were spun down at 150 � g for 10 min at roomtemperature. Cells were re-suspended in culture medium andseeded in 10-cm dishes incubated at 37 °C, 5% CO2 in a humid-ified atmosphere. After 24 h non-adherent cells were extractedand seeded in multi-well culture plates. Cells differentiated tomacrophages with 40 ng/ml recombinant mouse macrophage
colony stimulation factor for 7 days (media were changed after3 days). For treatment with transport inhibitors, cells weregrown to confluence inmulti-well culture plates before incuba-tion with inhibitors or cell culture medium as a control for 10min at 37 °C. Cells were then incubatedwith 5�MDMB-C5Cerat 4 °C for 30 min before being warmed to 37 °C.Fluorescence Microscopy—COS-1 cells were seeded into
4-chambered coverslips (LabTEKTM II; Nalge Nunc) at 5� 104cells per chamber. 24 h later cells were cooled down to 4 °C for15 min, then washed with cold 1% FCS-containing DMEMmedium and incubated for 30 min with 5 �M fluorescent lipid.Cells were washed again with cold 1% FCS-containing mediumand either imaged immediately or further incubated in thepresence of ceramide transport inhibitors for 15 min at 4 °Cbefore incubation at 25 °C for various time points and imaging.For fixation, cell monolayers were incubated for 30min at 25 °Cin Hanks’ balanced salt solution (HBSS) (without calcium andmagnesium) containing 3% paraformaldehyde. Monolayerswere then washed 5 times for 5 min in cold HBSS. 5 �M fluo-rescent lipid was applied to fixed cells in FCS-free cold DMEMand incubated on ice for 30 min. Monolayers were finallywashed 4 times for 30 min at 25 °C in HBSS containing 3.4mg/ml bovine serum albumin before imaging. Labeling of theendoplasmic reticulum with the ER-Tracker reagent (0.1�g/ml) was done on live cells as described by the manufacturer(Molecular Probes). Live-cell fluorescencemicroscopywas per-formed on an inverted microscope Axiovert 200M equippedwith a high resolution microscopy camera AxioCam MRc(Zeiss) and an objective Plan-Neofluar 32x/1.30 DIC using thefollowing filter set: excitation 470/emission 525 nm for greenfluorescence (NBD-Cer, DMB-Cer, and ER-Tracker) and 546/590 nm for red fluorescence (TRB-Cer).Lipid Extraction and Thin Layer Chromatography (TLC)—
Cells were rinsed with HBSS buffer supplemented with 10 mM
EDTA. Lipids were then extractedwith 200�l ofmethanol, 200�l of chloroform, and 150 �l of HBSS/EDTA. Samples werevortexed and spun in a tabletop centrifuge. The organic phasewas then taken and dried using a Thermo SPDSpeedVac. Driedsamples (corresponding to total lipid extracts from equivalentnumber of cells) were dissolved in 10 �l of methanol:chloro-form 1:1 and analyzed on Silica Gel 60 HPTLC plates (Merck)using butanol:acetic acid:water, 3:1:1, as themobile phase. TLCplates were dried and imaged using Fuji film LAS3000 intelli-gent dark box in SYBR Green fluorescent light. Quantitativeanalysis of TLCplateswas performed using the ImageJ softwareobtained from http://rsb.info.nih.gov/ij/download.html.Radiolabeling and Two-dimensional TLC Analysis—Cells
were seeded to a 6-well plate. On day 7 cells were washed andpreincubated for 6 h in phosphate-free medium (MEM Earl)supplemented with 10% FCS (dialyzed against Tris-bufferedsaline). 15 min before the end of the preincubation period,inhibitors were added. Medium was then replaced by freshmedium containing 200 �Ci/ml [33P]orthophosphate (GEhealthcare) as well as the indicated concentrations of inhibitoror an equivalent dilution of vehicle (Me2SO). After 2 h of incu-bation, the medium was removed, and cells were washed twicewith phosphate-buffered saline, scraped with 0.5 ml of cold
Ceramide 1-Phosphate Formation and Traffic
8518 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 283 • NUMBER 13 • MARCH 28, 2008
by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from

methanol, and transferred to Eppendorf vials. After the addi-tion of 0.5 ml of chloroform and 0.45 ml of 0.5 N HCl/2 M NaCl,samples were vortexed, and after a short centrifugation thechloroform phase was analyzed by two-dimensional TLC (Sil-ica 60 HPTLC plates, Merck) with chloroform methanol, 25%NH4OH, 60:35:8, for the first dimension and chloroform/meth-anol/acetone/acetic acid/water, 10:4:3:2:1, for the seconddimension. An acetone wash step was included before the sec-ond dimension.Determination of C1P Levels by Liquid Chromatography/
Electrospray Ionization/Tandem Mass Spectroscopy—Cells(1–2 � 106/sample) were lysed by freeze/thawing in 400 �l ofphosphate-buffered saline. Two hundred �l were transferredinto a glass tube, and 300 �l of 2 M KCl, 4% HCl as well as 1875�l ofmethanol/chloroform2:1was added. Sampleswere spikedwith an internal standard (C8-C1P, Avanti Polar Lipids). Aftervortexing and sonication, 675 �l of chloroformwas, added, andafter further vortexing, 500 �l of water was added. The samplewas finally centrifuged for 10 min at 2600 rpm at 4 °C. Theorganic phase was dried in an autosampler vial. Samples weredissolved in mobile phase and subjected to high performanceliquid chromatography (HPLC 1100; Agilent), performed asdescribed in Merrill et al. (25) with some modifications. Inshort, a SupelcoDiscovery C18HS column (2.1� 50mm, 3-�mparticle size) was used and eluted with a gradient (eluent A, 5
mM ammonium formiate � 1% for-mic acid inmethanol/water 70/30�2% tetrahydrofuran; eluent B, 5 mMammonium formiate � 1% formicacid in methanol � 2% tetrahydro-furan; 70–100% B in 1.8 min) at aflow of 400 �l/min at 60 °C. Electro-spray ionization with tandem massspectroscopy using an API 4000QTrap instrument (MDS Sciex) wasemployed to detect C1Pwith positiveionization. The optimal collisionenergy for C16-C1P was �51 V andfor C8-C1P was �39 V; the multiplereaction-monitoring transitions fol-lowed were m/z 618.6/264.1 and506.4/264.2, respectively. This proce-dure allows for detection of C1Pformed by CerK. Alternative proto-cols have been reported where KOHis used at the extraction step. How-ever, if the reaction is not adequatelyneutralized afterward, SM is readilyhydrolyzed into C1P during the sub-sequent dry-down step, leading tooverestimation of C1P levels (26).
RESULTS
Unique Role for CerK in the Pro-duction of C1P from Cer—Weselected BMDMfor use in this studybecause we found these primarycells have significant CerK activity.
Thus, we were surprised to find that C16-C1P, a major subspe-cies of C1P, was not abolished in BMDM from Cerk�/� mice(Fig. 1A). Labeling of BMDMusing [33P]orthophosphate clearlyshowed that BMDM from Cerk�/� mice cannot phosphorylateCer to produce C1P (Fig. 1B). These results indicate that othersources for C1P formation exist and demonstrate the criticalrole of CerK in the de novo phosphorylation of Cer.Current hypotheses for the formation of C1P focus on
phosphorylation of Cer by lipid kinases such as CerK. How-ever, the inability of a recently identified CerK-like proteinto phosphorylate ceramide (27) has renewed interest in find-ing alternate pathways of C1P formation; in particular, viaacylation of the lysosphingolipid sphingosine 1-phosphate.To investigate this potential pathway, we applied DMB(dimethyl-BODIPY)-labeled sphingosine to WT and Cerk�/�
BMDM; DMB-sphingosine is a fluorescent sphingosine ana-log, which has been validated as a substrate for intracellularsphingosine kinases (28). Although we detected DMB-sphingosine 1-phosphate, we found no evidence of DMB-C1P under these conditions (Fig. 1C). We also evaluatedDMB-dehydrosphingosine as a substrate because dehy-drosphingosine is an anabolite intermediate of the de novosphingolipid synthesis pathway that can also be phosphoryl-ated by sphingosine kinases (29). DMB-dehydrosphingosinewas indeed converted to its phosphorylated counterpart;
FIGURE 1. Pathways leading to C1P formation. A, determination of C16-C1P levels by liquid chromatogra-phy/mass spectroscopy in BMDM from WT and Cerk�/� animals. The C1P values represent the mean � S.D. ofvalues obtained from n � 7 animals. Statistical significance was evaluated using the Student t test. B, endog-enous C1P formation probed in Cerk�/� and WT BMDM upon a 6-h labeling with [33P]orthophosphate followedby lipid extraction and two-dimensional TLC analysis. The area corresponding to the Rf of endogenous C1P isshown. C, metabolism of fluorescent sphingosine (Sph) and dihydrosphingosine (DH-Sph) in WT and Cerk�/�
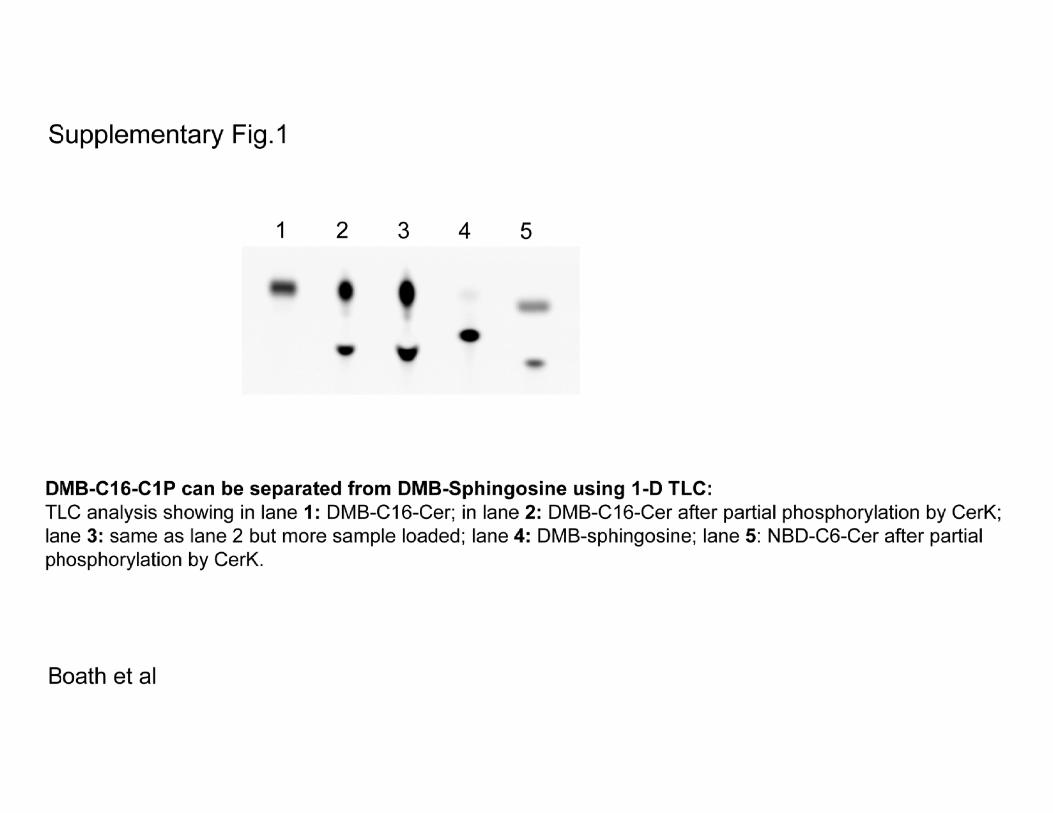
BMDM; 5 �M concentrations of each analog was added to subconfluent BMDM and allowed to incubate for 3 hbefore lipid harvest and TLC analysis. Standards 1 and 2 represent fluorescent sphingosine and fluorescentdihydrosphingosine, respectively. That C1P, if formed, was not running on the TLC together with sphingosineor sphinganine and might, as a result, have gone undetected was checked and is shown in supplemental Fig.1. D, possible pathways leading to C1P formation.
Ceramide 1-Phosphate Formation and Traffic
MARCH 28, 2008 • VOLUME 283 • NUMBER 13 JOURNAL OF BIOLOGICAL CHEMISTRY 8519
by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from

however, we detected no further acylation (Fig. 1C). Theseresults suggest that acylation of sphingosine 1-phosphate (orDH-sphingosine 1-phosphate) is not likely to be a physiolog-ical route for the production of C1P (DH-C1P) (Fig. 1D). Therest of our experiments in this present study focused exclu-sively on C1P that originates from CerK-catalyzed phospho-rylation of Cer.In previous studies we used a short chain [nitrobenzo-2-oxa-
1,3-diazole]-labeled ceramide (NBD-Cer) to assess CerK activ-ity in cell-based assays (27). In most cell lines NBD-Cer isreadily converted to GlcCer and SM, but C1P is barelydetectable unless CerK is overexpressed, as shown in Fig. 2A.In this study we establish that this assay is entirely selectivefor CerK because no phosphorylation of NDB-Cer occurs inCerk�/� BMDM (Fig. 2A).The sensitivity of this TLC-basedreadout is well in the range of the standard in vitro CerKactivity assay (compare Fig. 2, A and B). Moreover, fluores-cent ceramides and cell-based assays enabled us to simulta-neously analyze, along C1P, the two major Cer metabolites,GlcCer and SM. NBD-Cer is known to be a marker of thetrans-Golgi network (30), where the respective enzymes,including CerK (31), can be found. Competition for substrateis illustrated in Fig. 2A; in the presence of CerK, the levels ofNBD-GlcCer and NBD-SM diminish, as does the residualcellular pool of NBD-Cer. Because of the specificity, sensi-
tivity, and multiplicity of readouts,we have selected this experimentalparadigm in our present study.C1P Produced by CerK Is a Short-
lived Sphingolipid Metabolite—IfC1P can act as a signaling molecule,its turnover should be regulated.To analyze serum dependence inC1P formation, we serum-starvedBMDM for 2 h and then re-intro-duced serum in some BMDM, incu-bating cells with NBD-Cer for 2 h.We found that without serum, littleC1P was formed, but when serumwas re-introduced to the medium,C1P levels sharply increased (Fig.3A). The positive correlation withserum was stronger for C1P forma-tion (�328%) than for GlcCer(�103%) or for SM (�17%). There-fore, steady-state levels of C1P aremore dependent on serum than arethose of GlcCer and SM.When cellswere first incubated for 1 h withNBD-Cer in serum-containingmedium and then incubated in aserum-free medium, C1P levelsdecreased 70%, although levels ofothermetabolitesdidnot change (Fig.3B). These results indicate thatC1P ismore labile than GlcCer and SM andreinforce dependence of CerK/C1Pon factors present in serum.
CerK is activated by calcium ions (8), and a Ca2�-calmodulinbinding domain was recently characterized in the C-terminalpart of the protein (32). In vitro, CerK activity is dependent ondivalent ions, particularly calcium ions (33), and no CerK activ-ity is found in the presence of saturating amounts of chelatorssuch as EDTA (supplemental Fig. 2). To study the dependenceof CerK on calcium ions, we used an NBD-Cer cell-based assayand the intracellular calcium chelator BAPTA (1,2-bis(2-aminophenoxy)ethane-N,N,N�,N�-tetraacetic acid). BAPTAdecreased C1P formation almost 100% (Fig. 3C) while decreas-ing GlcCer and SM synthesis 70 and 30%, respectively (Fig. 3C).When BAPTA was added to a serum-containing medium for1 h after a prior incubationwithNBD-Cer, C1P levels decreasedmore than 90% compared with controls; levels of the other twometabolites, however, were largely maintained (Fig. 3D). Theseresults from a cell-based assay demonstrate the total depend-ence of CerK on Ca2� ions and the remarkably short half-life ofC1P compared with GlcCer and SM.Transport of Cer to the Golgi Complex Allows for Phosphoryl-
ation by CerK—NBD-Cer is able to reach the Golgi membranedirectly, even in fixed cells (30) (Fig. 4Aa). DMB-Cer (dimethylBODIPY-Cer), an alternate fluorescent ceramide, accumulatesat the ER if cells are fixed (Fig. 4Ab). In live cells, DMB-Certraffics from the ER to the Golgi complex, relying on a cytosolictransport protein namedCERT (34, 35) (Fig. 4A, c and e). Com-
FIGURE 2. NBD-C1P formation specifically accounts for CerK activity. CerK activity was determined inCOS-1cells (COS), COS-1 cells overexpressing CerK (COS-CerK), and BMDM from WT and Cerk�/� animalsusing either a cell-based assay with NBD-Cer (A) or an in vitro kinase assay with 32P ATP (B). A, left, 5 �M
NBD-Cer was added to each cell type seeded to equivalent densities in a 24-well plate format. Cells wereincubated for 2h at 37 °C before lipid extraction. The entire lipid extracts were loaded on a TLC plate andrun to resolve the different lipid species (details are described under “Experimental Procedures”). Right,TLC densitometry histogram of the amount of GlcCer (open circles), C1P (black circles), and SM (gray circles)formed �S.D. from triplicates of a representative experiment. Std, standard. B, in vitro kinase assay. Thisassay is an established, and a selective assay for CerK and was done as described by Rovina et al. (47).Activity levels are depicted as the mean � S.D. of eight determinations obtained on two separate occa-sions. CerK activity was determined in parallel for COS cells and BMDM and normalized to protein levels,therefore allowing for direct comparison.
Ceramide 1-Phosphate Formation and Traffic
8520 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 283 • NUMBER 13 • MARCH 28, 2008
by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from

pared with NBD-Cer, DMB-Cer is poorly converted to GlcCer(Figs. 4B and 2A), although the reason why is not entirely clear(36). In contrast, SM is readily synthesized using DMB-Cer assubstrate, which is delivered to SMS-1 by CERT (35). We thenfound that DMB-Cer can also serve as a substrate for recombi-nant CerK in COS-1 cells (Fig. 4B, left) andmade similar obser-vations about endogenous CerK in BMDM (Fig. 4B, right). Weused a second BODIPY-based fluorescent ceramide, TRB-Cer;this analog accumulates at the ER as DMB-Cer but is barelytransported to the Golgi complex (37) (Fig. 4Ad). We foundTRB-Cer to be very poorlymetabolized into any detectable spe-cies, particularly C1P (Fig. 4C). Altogether, these data establishthat Cer must be transported to the Golgi complex to be con-verted into C1P.Our observations suggest that CerK and SMS-1 might com-
pete for ceramide provisioned by CERT-mediated transport.To test this hypothesis we used HPA-12, a ceramide analogsynthesized by Hanada and co-workers (38) that is a directantagonist of theCERTprotein. InChinese hamster ovary cells,the (1R,3R) HPA-12 enantiomer inhibited the transport ofexogenous DMB-Cer and endogenous Cer for conversion toDMB-SM and SM, respectively (38).We observed that HPA-12also inhibits DMB-Cer transport in COS-cells (Fig. 4Ae).Therefore, we investigated the effect of (1R,3R) HPA-12 treat-ment onDMB-Cermetabolism inCOS-CerK cells and BMDM.In COS-CerK cells, pretreatment with 5 �M (1R,3R) HPA-12decreased the levels of SM synthesized from DMB-Cer 15 minafter incubation at 37 °C compared with untreated controls(Fig. 5A, top panel), findings consistent with data fromHanada
and co-workers (38). Unexpectedly, the levels of C1P did notdecrease with drug treatment but in fact increased (Fig. 5A, toppanel). In BMDM, 5 �M (1R,3R) HPA-12 also decreased SMlevels, whereas it increased DMB-C1P synthesis �100% com-pared with controls (Fig. 5A, lower panel). In parallel to theactive (1R,3R) HPA-12 compound, we tested a 5 �M racemicmixture of (1S,3R) (3S,1R) enantiomers as an inactive control(38). This inactive compound had no effect on the amount ofC1P and SM synthesized from DMB-C5 Cer in either COS-CerK cells or in BMDM compared with the untreated samples(Fig. 5A, right). These experiments show that pharmacologicalinhibition of CERT slows down SM formation but does notinhibit the production of C1P.CERT bears a pleckstrin homology domain, which is known
to interact with phosphatidylinositol 4-phosphate (35).Recently, phosphatidylinositol 4-kinase III� was shown to playa role in the transport of DMB-Cer to the Golgi complex inCOS-cells (37). Inhibition of phosphatidylinositol 4-kinase III�by the selective inhibitor PIK93 negatively impacted ceramidetransport and, consequently, SM synthesis (37). Therefore, wetreated COS-CerK cells and BMDM with PIK93. As expected,metabolism of DMB-Cer to SM was greatly reduced (Fig. 5B).However, there was no concomitant decrease in C1P levelscompared with control levels (Fig. 5B and not shown).
Using [33P]orthophosphate labeling, we also checked forendogenous C1P formation in response to treatment witheither HPA-12 or PIK93. Consistent with our observationsusingDMB-Cer, endogenousC1P formationwas not decreasedafter CERT inhibition (Fig. 5C). Endogenous C16-C1P levels
FIGURE 3. C1P is a short-lived metabolite. A, after 2 h of serum starvation, BMDM were incubated for 2 h with 5 �M NBD-Cer in DMEM in the absence orpresence of 10% FCS. Histogram representation for each metabolite of the amount made in 0% FCS versus the amount made in 10% FCS. Results are the meanof triplicate determinations �S.D. B, BMDM incubated with 5 �M NBD-Cer for 1 h in DMEM � 10% FCS with or without follow up with 1 h of incubation withDMEM � 0% FCS. Histogram representation for each metabolite of the amount made “with” the subsequent incubation versus the amount obtained “without.”Mean of triplicate determinations �S.D. C, BMDM-treated �20 �M BAPTA for 30 min followed by 1 h of incubation with 5 �M NBD-Cer. A histogram represen-tation for each metabolite shows the amount made upon BAPTA treatment versus the amount obtained with vehicle. Results are of triplicate determinations�S.D. D, BMDM incubated with 5 �M NBD-Cer for 1 h followed by an additional hour �20 �M BAPTA. Shown is a histogram representation for each metaboliteof the residual amount after BAPTA treatment versus the amount obtained with vehicle. Results are the mean of triplicate determinations �S.D. All results in thisfigure show one of three experiments with similar results.
Ceramide 1-Phosphate Formation and Traffic
MARCH 28, 2008 • VOLUME 283 • NUMBER 13 JOURNAL OF BIOLOGICAL CHEMISTRY 8521
by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from

were further verified by liquid chromatography followed bymass spectrometry and found to be unmodified after treatmentwith HPA-12 or PIK93 (not shown). Overall, these data showthat inhibition of CERT, be it direct (with HPA-12) or indirect(with PIK93), has no effect on C1P synthesis, suggesting thatCERT-mediated Cer transport is not critical for CerK to accessits substrate.C1P Traffics to the Plasma Membrane—After NBD-Cer is
applied to cells, it is readily metabolized into NBD-GlcCer andNBD-SM (39), and these two metabolites traffic along thesecretory pathway to finally reach the plasmamembrane.At theplasma membrane, they can be removed by a so-called back-
exchange process, whereby the lipidexits the external leaflet of theplasma membrane and moves intoan acceptor in the medium, such asproteins (e.g. bovine serum albu-min) and lipoproteins (40). Thisback-exchange is, therefore, an indi-cator of localization at the plasmamembrane. No prior knowledge ofthis process exists for C1P. In ourstudy, when we treated COS-CerKcells with 5 �M NBD-Cer and incu-bated them at 37 °C, we observedNBD-C1P in the medium in a man-ner consistent with NBD-GlcCerand NBD-SM (Fig. 6, A and B). Thisobservation indicates that NBD-C1P has been transported to theplasma membrane and back-ex-changed into themedium. To checkfor the dependence of this phenom-enon on the secretory pathway, weincubated cells with the ionophoremonensin, which blocks vesiculartrafficking between the medial andthe trans-Golgi network (41). Weobserved that, in the presence of10 �M monensin, all NBD-Cermetabolites including C1P accu-mulated in cells and decreasedtheir release into the medium (Fig.6A). NBD-C1P did not accumulateto the same extent as NBD-GlcCeror NBD-SM, which may indicateinhibition of CerK by monensin orresult from the short half-life ofC1P.Trafficking along the secretory
pathway is blocked at low tempera-tures (42). Thus, we analyzed therelease of the various NBD-labeledmetabolites during cell treatmentand incubation at 18 °C. WhenCOS-CerK cells were incubatedwith NBD-Cer for 3 h at 18 °C,metabolism to NBD-GlcCer, NBD-
SM, and NBD-C1P was comparable with that obtained in cellsincubated at 37 °C. However, at 18 °C there was no significantrelease of any NBD-Cer metabolites into the medium (Fig. 6B),thereby confirming the involvement of the secretory pathwayin the trafficking of C1P to the plasma membrane.Moreover, when WT BMDM were incubated with 5 �M
NBD-Cer, all detected metabolites were released into themedium (Fig. 6C, left). Remarkably, in these cells NBD-C1Pwasby far the most readily secreted NBD-Cer metabolite. Ratios ofC1P secreted to C1P synthesized reached 40% after 60 min ofincubation at 37 °C without any lag time. During the same timeperiod and after a slower onset, the ratios for NBD-SM and
FIGURE 4. Cer needs transport to the Golgi complex for conversion into C1P. A, COS-1 cells were fixed andstained with NBD-Cer (a) or DMB-Cer (b) as described under “Experimental Procedures.” One representative cellis shown. NBD-Cer stains the Golgi complex only, whereas DMB-Cer stains all of the perinuclear region. Uponincubation for 30 min at 25 °C in live cells, DMB-Cer can exit the ER and accumulates at the Golgi complex (c);there is no more perinuclear staining; under similar conditions TRB-Cer remains in the ER as seen by co-stainingwith the ER-Tracker reagent (d). Kinetics of transport of DMB-Cer at 25 °C (e). At t � 0 the labeled ceramide isfound at the plasma membrane, and at t � 10 min the dye has reached the cytoplasm and starts appearing atthe Golgi complex (red arrows). Enrichment at the Golgi complex increases at t � 20 min and is almost completeat t � 30 min. In the presence of 10 �M HPA-12, DMB-Cer accumulation at the Golgi complex is slowed down.The red arrows indicate the diffuse labeling by DMB-Cer and the persistent perinuclear staining in the presenceof this compound. B, metabolism of DMB-Cer in COS or COS-CerK cells (left) or BMDM from Cerk�/� or WT mice(right) after a 2-h incubation at 37 °C with 5 �M DMB-Cer; lipids were extracted and the whole extract wasanalyzed on TLC. GlcCer is poorly produced from DMB-Cer. However, if the DMB label is carried by the sphin-gosine chain, GlcCer can be made (cf. supplemental Fig. 3). C, TRB-Cer is not readily metabolized in either COSor COS-CerK cells. TLC analysis is after a 3-h incubation at 37 °C with 10 �M TRB-Cer.
Ceramide 1-Phosphate Formation and Traffic
8522 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 283 • NUMBER 13 • MARCH 28, 2008
by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from

NBD-GlcCer reached only 15 and 10%, respectively (Fig. 6C,right).
DISCUSSION
CerK, aMajor but Not Exclusive Pathway to C1P—Our studyconfirms that CerK is essential to the formation of C1P throughthe phosphorylation of Cer. Cells from Cerk�/� mice areunable to form C1P, as verified by three assays highly specificfor CerK activity: (i) pulse labeling with [33P]orthophosphate(Fig. 1B), (ii) NDB-Cer or DMB-Cer as substrates (Figs. 2A and4B), and (iii) a standard in vitro kinase assay (Fig. 2A). Our studyalso demonstrates for the first time that CerK is not the solesource of C1P, a discovery based on our measurements of sig-nificant residual levels of C1P in cells fromCerk�/� animals. Toinvestigate other possible mechanisms of C1P synthesis, weused the fluorescent lysolipids sphingosine and sphinganine asprecursors for their corresponding lysophospholipids and
asked whether the latter could be further acylated to build C1P.Such activity, however, was not observed (Fig. 1C). Therefore,an alternate pathway leading to C1P has yet to be discovered,and it may be interesting to explore whether a type D sphingo-myelinase could be implicated (Fig. 1D).Our work then focused on studying C1P produced by CerK.
We used fluorescent ceramide analogs for three important rea-sons: (i) they can specifically probe C1P made by CerK (seeabove); (ii) they can be metabolized by CerK, glucosylceramidesynthase, and SMS so that the fate of the three metabolites,C1P, GlcCer, and SM can be studied side by side, and (iii) theirtransport pathways could shed light on themechanisms of syn-thesis of ceramide metabolites.Probing CerK-dependent C1P Turnover and Transport with
NBD-Cer—The first analog we used is a short-chain ceramide(C6) with a fluorescent NBD label (NBD-Cer). NBD-Cer is rap-
FIGURE 5. Pharmacological inhibition of CERT favors C1P formation over SM formation. A, COS-CerK cells or WT BMDM were preincubated at 37 °C with5 �M HPA-12 or controls and then incubated with 5 �M DMB-C5 Cer for 30 min at 4 °C. Cells were warmed to 37 °C for the stated times. Left, TLC analysis ofDMB-C5 Cer metabolites. Middle, histogram representing amount of C1P (black) and SM (gray) in drug-treated samples as a % of untreated control amounts.Right, line graph representing amount of C1P made as a ratio to SM in HPA-12, HPA-12 inactive mix, and untreated samples. Results are the mean of fourmeasurements obtained in two separate experiments �S.D. B, effect of phosphatidylinositol 4-kinase III� inhibition on metabolism of DMB-C5 Cer in WTBMDM. Cells were preincubated with 250 nM PIK93 at 37 °C and then incubated with 5 �M DMB-C5 Cer for 30 min at 4 °C. Cells were warmed to 37 °C for thestated times. Left, TLC analysis of DMB-C5 Cer metabolites. Right, histogram representation of the levels of C1P (black) and SM (gray) in PIK93-treated samplesas a percentage of control values. Results are the mean of four measurements taken from two separate experiments �S.D. C, two-dimensional-TLC analysis ofBMDM endogenous phospholipids after treatment with various inhibitors and labeling with [33P]orthophosphate, as described under “Experimental Proce-dures.” Only a part of the TLC plates is shown. a, vehicle (Me2SO 0.05%); b, 5 �M HPA-12; c, 250 nM PIK93; d, treatment with a potent CerK inhibitor (data notshown); arrowheads indicate labeled endogenous C1P species.
Ceramide 1-Phosphate Formation and Traffic
MARCH 28, 2008 • VOLUME 283 • NUMBER 13 JOURNAL OF BIOLOGICAL CHEMISTRY 8523
by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from

idly taken up by cells and accumulates at the Golgi complexeven after cell fixation (30) (Fig. 4Aa), i.e.nophysiological trans-port is required. NBD-Cer is a good substrate for glucosylcera-mide synthase, SMS (39, 40), andCerK (Fig. 2A); furthermore, itis easily extracted from membranes so that the trafficking ofNBD-Cer metabolites can be probed using a so-called “backexchange” procedure (40). In NBD-Cer- and cell-based assays,we have demonstrated competition betweenCerK, glucosylcer-amide synthase, and SMS for Cer access at the Golgi complex;C1P formation in COS-CerK and BMDM decreased the levelsof the two other fluorescent ceramide metabolites in com-parison to COS-1 cells or Cerk�/� cells (Fig. 2A); this con-firms that CerK can localize to the Golgi complex as we haveshown previously (31). NBD-Cer was also used to identifythe short half-life of C1P and to monitor trafficking to theplasma membrane.C1P is believed to act as a signaling molecule based on the
pioneering study by Chalfant and co-workers (12) showing thatC1P mediates cytokine signaling in eicosanoid synthesis inA549 lung carcinoma cells. A key characteristic of a signalingintermediate is its transient appearance resulting from on andoff signals, which are tightly regulated.We have confirmed thischaracteristic based on our comparative analyses of C1P,GlcCer, and SM, which are formed when incubated with NBD-
Cer under regulated conditions such as reduced serum or cal-cium chelation. We found that NBD-C1P synthesis was moresensitive to serum compared with NBD-GlcCer and NBD-SMsynthesis, as observed either directly (Fig. 3A) or indirectly (Fig.3B). This suggests that, in the absence of serum, CerK may beless active or that C1Pmay be degradedmore significantly thanGlcCer and SM. To test these two possibilities, we used theintracellular calcium chelator BAPTA to inhibit CerK, becausecalcium ions are required for CerK activity in vitro (8, 33) (sup-plemental Fig. 2). During treatment with BAPTA, CerK andglucosylceramide synthase were strongly inhibited (reduced to0% of activity and 30%, respectively, Fig. 3C). We used this par-adigm to test the stability of NBD-Cer metabolites and foundthat NBD-C1P, but not NBD-GlcCer or NBD-SM, is rapidlyturned over (Fig. 3D). This finding is the first evidence that C1Pis a short-livedmetabolite and strongly supports the role of C1Pas a signaling molecule. We hypothesize that “on” signals mayresult from post-translational modifications of CerK (e.g. phos-phorylation) that increase CerK activity and C1P formation.“Off” signals may trigger deactivation of CerK and/or dephos-phorylation of C1P through the action of broad-specificity lipidphosphatases or a C1P phosphatase (43, 44) or through degra-dation of C1P by other means. All these possibilities warrant
FIGURE 6. C1P traffics to the plasma membrane and is readily secreted by BMDM. A, effect of 10 �M monensin on back-exchange of NBD-Cer metabolites.Left, TLC analysis of NBD-Cer metabolites in cells and media (�monensin). Right, histogram representation of levels of GlcCer (white), SM (gray), and C1P (black)back-exchanged as a percentage of the total made after 120 min incubation with 5 �M NBD-Cer. Comparison between control and monensin-treated samplesas a mean of triplicate determinations � S.D. This is one of three experiments with similar results. B, effect of temperature block on back-exchange of NBD-Cermetabolites. Cells were incubated for 3 h at 37 °C (left) or 18 °C (right) with 5 �M NBD-Cer. Lipids were extracted from cells and media and analyzed by TLC.C, left, TLC analysis of NBD-Cer metabolites in cells (top) and cell culture media (bottom) from BMDM incubated with 5 �M NBD-Cer for stated times at 37 °C.Right, line graph representing the amount of C1P, SM, and GlcCer secreted as a percentage of the total amount (secreted � cellular) of metabolite made in WTBMDM. Mean values obtained from four independent measurements � S.D. (S.D., when not visible, are smaller than the symbol).
Ceramide 1-Phosphate Formation and Traffic
8524 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 283 • NUMBER 13 • MARCH 28, 2008
by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from

further testing because our current understanding of the regu-lation of CerK/C1P is still limited.After synthesis, NBD-C1P is trafficked out of the Golgi com-
plex. Inhibition of intra-Golgi transport in COS-CerK cellsdecreased the amount of NBD-C1P that was available for back-exchange from the plasma membrane. This decrease was par-alleled by an increase of NBD-C1P inside the cell (Fig. 6A). At18 °C, when vesicular transport is blocked, no significant secre-tion of NBD-C1P was detected (Fig. 6B). These results demon-strate that C1P, like SM and GlcCer, is trafficked through theGolgi complex “en route to the plasma membrane” (40). InBMDM, the release of NBD-C1P into the medium was evenmore striking; NBD-C1P was secreted readily and more exten-sively than NBD-GlcCer or NBD-SM (Fig. 6C).Probing Cer Access to CerK with TRB-Cer, DMB-Cer, HPA-
12, and PIK93—We then used two BODIPY-labeled fluores-cent ceramide analogs, TRB-Cer and DMB-Cer, to investigatethemechanisms of ceramide access toCerK. TRB-Cer accumu-lates at the ER and cannot be efficiently transported to theGolgicomplex (37) (Fig. 4Ad). Using TRB-Cer, we have shown thatCer transport from the ER to the Golgi complex is required forC1P synthesis; we have also confirmed this requirement for SM
and GlcCer synthesis (Fig. 4C). These observations are consist-ent with published immunohistochemistry data showing thatCerK does not significantly localize to the ER (45). The secondfluorescent ceramide, DMB-Cer, has been validated by manystudies (36–38, 46) as a tool to follow the synthesis of SM. Thetransport of DMB-Cer from the ER to the Golgi complex (Fig.4A, b, c, and e) relies, at least in part, on a recently identifiedceramide transfer protein named CERT (35). When CERT isinhibited, the accumulation of SM is slowed down (38). GlcCeris poorly produced from DMB-Cer (36), and inhibiting CERThas amarginal impact onGlcCer synthesis (38).We report herethat DMB-Cer can readily serve as a substrate for CerK (Fig.4B), and therefore, we asked whether CERTwould be as criticalfor C1P formation as it is for SM formation. Surprisingly, inhi-bition of CERT-dependent DMB-Cer transport to the Golgicomplex either directly with HPA-12 (Fig. 4Ae) or indirectlywith PIK93, a phosphatidylinositol 4-kinase III� inhibitor,slowed down synthesis of DMB-SM but did not inhibit DMB-C1P formation (Fig. 5, A and B). In fact, DMB-C1P levelsincreased during HPA-12 treatment in COS-CerK cells and inBMDM (Fig. 5A). An inactive enantiomer mix of HPA-12 hadno effect on either DMB-SM or DMB-C1P formation (Fig. 5A,right). Similar observations were made when monitoringendogenous C1P formation (Fig. 5C), ruling out the possibilitythat the observed effects of these inhibitors of ceramide trans-port apply only to the use of artificial substrates. In summary,through a combination of direct and indirect pharmacologicalinhibition of CERT, it has been shown that the transport of Certo CerK is not critically dependent on CERT, contrary toSMS-1, which relies on CERT to use Cer as a substrate (35, 38).Moreover, the lack of effect of PIK93 on C1P formation alsosuggests that phosphatidylinositol 4-phosphate is not criticalfor the localization and function of CerK via its pleckstrinhomology domain (47).While our work was in progress, Lamour et al. (45) reported
that CERT is amajor provider of Cer for C1P synthesis, findingscontrary to our results. Although we cannot account for alldiscrepancies between the two studies, some important differ-ences may shed some light. First, the method of inhibition andthe duration of the studies differ. Lamour et al. (45) used RNAinterference to inhibit CERT, and therefore, readouts weretaken after prolonged incubation times. In our study, we usedshort-term pharmacological inhibition of CERT, the effects ofwhich can be analyzedwithinminutes. Short-term inhibition ofCERT may not impact C1P formation, whereas this may occurwhen CERT is stably down-regulated. The inhibitors we usedhave been validated for transient use only (37–38) so our studydid not address both short- and long-term responses. Nonethe-less, workingwith pharmacological tools to inhibit CERT eitherdirectly or indirectly and using a close but inactive analog areclear advantages of our study. Second, because the cell typesexamined in each study were not the same, we might expectthat the subcellular repartition of CerKwould differ. In a recentreport we found microtubules to be responsible for the rapidtranslocation of CerK between the Golgi complex and theplasma membrane (47). The differential localization of CerKmay, therefore, depend on the cell type and its activation status,as depicted in Fig. 7.
FIGURE 7. CerK localization and C1P formation. The picture depicts currentunderstanding of Cer metabolism with focus on CerK-catalyzed productionof C1P. Cer at the ER can be transported on COP II vesicles or using the CERTprotein to reach various locations and enzymes (48, 49) (anabolic pathway).CERT might play a role in providing Cer to CerK (45), but the present studysuggests an alternative route(s). The topology of SMS-1 indicates an intralu-minal active site, whereas the active site of glucosylceramide synthase (GCS)is on the cytosolic side (50 –52). CerK is myristoylated (31), supporting cyto-solic orientation. CerK bears a pleckstrin homology (PH) domain important forlocalization and activity (8, 31) and reported to bind phosphoinositides (47,53). Calmodulin (CaM) acts as a calcium sensor for CerK (32). CerK can trafficbetween the Golgi complex and the plasma membrane using microtubules(31, 47) (dashed line with two arrowheads). At the plasma membrane (PM),CerK may be poised to act downstream sphingomyelinase activity to convertthe produced Cer into C1P (catabolic pathway) (54).
Ceramide 1-Phosphate Formation and Traffic
MARCH 28, 2008 • VOLUME 283 • NUMBER 13 JOURNAL OF BIOLOGICAL CHEMISTRY 8525
by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from

In conclusion, our results identify some novel aspects of C1Pbiology. CerK-mediated phosphorylation of Cer is a major, butnot an exclusive pathway to the formation of C1P. A regulationprocess controls the steady-state levels of C1P generated byCerK, thereby satisfying an essential criterion for signalingfunction. Moreover, CerK can build C1P at the Golgi complexby relying in part on aCer transportmechanism that remains tobe identified. Finally, C1P readily traffics along the secretorypathway to reach the plasma membrane, a finding consistentwith reports that describe C1P in synaptic vesicles (9) and sug-gest its importance to processes such as phagocytosis (11).
Acknowledgments—We are grateful to Dr. Kentaro Hanada(National Institute of Infectious Diseases, Tokyo, Japan) who kindlyprovided us with the HPA-12 compounds. We thank Novartis Insti-tutes for BioMedical Research colleagues Ian Bruce, Cathy Leblanc,and Ivan Cornella-Taracido for providing PIK93, Nicole Urtz forguidance in the isolation of bone marrow, Philipp Rovina for adviceon the preparation ofmacrophages, andThomasBaumruker for help-ful comments on the manuscript.
REFERENCES1. Futerman, A. H., and Riezman, H. (2005) Trends Cell Biol. 15, 312–3182. Ogretmen, B., and Hannun Y. A. (2004) Nat. Rev. Cancer 4, 604–6163. Hait, N. C., Oskeritzian, C. A., Paugh, S. W., Milstien, S., and Spiegel, S.
(2006) Biochim. Biophys. Acta 1758, 2016–20264. Zeidan, Y. H., and Hannun, Y. A. (2007) Trends Mol. Med. 13, 327–3365. Lahiri, S., and Futerman, A. H. (2007) Cell. Mol. Life Sci. 64, 2270–22846. Schenck,M., Carpinteiro, A., Grassme, H., Lang, F., andGulbins, E. (2007)
Arch. Biochem. Biophys. 462, 171–1757. Baumruker, T., Bornancin, F., and Billich, A. (2005) Immunol. Lett. 96,
175–1858. Sugiura, M., Kono, K., Liu, H., Shimizugawa, T., Minekura, H., Spiegel, S.,
and Kohama, T. (2002) J. Biol. Chem. 277, 23294–233009. Bajjalieh, S. M., Martin, T. F., and Floor, E. (1989) J. Biol. Chem. 264,
14354–1436010. Kolesnick, R.N., andHemer,M. R. (1990) J. Biol. Chem. 265, 18803–1880811. Hinkovska-Galcheva, V. T., Boxer, L. A., Mansfield, P. J., Harsh, D., Black-
wood, A., and Shayman, J. A. (1998) J. Biol. Chem. 273, 33203–3320912. Pettus, B. J., Bielawska, A., Spiegel, S., Roddy, P., Hannun, Y. A., and Chal-
fant, C. E. (2003) J. Biol. Chem. 278, 38206–3821313. Pettus, B. J., Bielawska, A., Subramanian, P., Wijesinghe, D. S., Maceyka,
M., Leslie, C. C., Evans, J. H., Freiberg, J., Roddy, P., Hannun, Y. A., andChalfant, C. E. (2004) J. Biol. Chem. 279, 11320–11326
14. Mitsutake, S., Kim, T. J., Inagaki, Y., Kato, M., Yamashita, T., and Igarashi,Y. (2004) J. Biol. Chem. 279, 17570–17577
15. Gomez-Munoz, A., Kong, J. Y., Salh, B., and Steinbrecher, U. P. (2004) J.Lipid Res. 45, 99–105
16. Tornquist, K., Blom, T., Shariatmadari, R., and Pasternack, M. (2004) Bio-chem. J. 380, 661–668
17. Colina, C., Flores, A., Castillo, C., Garrido,M., del, R., Israel, A., DiPolo, R.,and Benaim, G. (2005) Biochem. Biophys. Res. Commun. 336, 54–60
18. Jeon, H. J., Lee, D. H., Kang, M. S., Lee, M. O., Jung, K. M., Jung, S. Y., andKim, D. K. (2005) J. Neurochem. 95, 811–820
19. Gomez-Munoz, A., Duffy, P. A., Martin, A., O’Brien, L., Byun, H. S., Bitt-man, R., and Brindley, D. N. (1995)Mol. Pharmacol. 47, 833–839
20. Gomez-Munoz, A., Kong, J. Y., Parhar, K., Wang, S. W., Gangoiti, P.,Gonzalez, M., Eivemark, S., Salh, B., Duronio, V., and Steinbrecher, U. P.(2005) FEBS Lett. 579, 3744–3750
21. Rosenwald, A. G., and Pagano, R. E. (1993) Adv. Lipid Res. 26, 101–118
22. Peters, C., Billich, A., Ghobrial, M., Hogenauer, K., Ullrich, T., and Nuss-baumer, P. (2007) J. Org. Chem. 72, 1842–1845
23. Nussbaumer, P., Ettmayer, P., Peters, C., Rosenbeiger, D., and Hogenauer,K. (2005) Chem. Commun. 40, 5086–5087
24. Graf, C., Rovina, P., Tauzin, L., Schanzer, A., and Bornancin, F. (2007)Biochem. Biophys. Res. Commun. 354, 309–314
25. Merrill, A. H., Jr., Sullards, M. C., Allegood J. C., Kelly, S., and Wang, E.(2005)Methods 36, 207–224
26. Nussbaumer, P. (2008)ChemMedChem, in press27. Bornancin, F., Mechtcheriakova, D., Stora, S., Graf, C., Wlachos, A.,
Devay, P., Urtz, N., Baumruker, T., and Billich, A. (2005)Biochim. Biophys.Acta 1687, 31–43
28. Ettmayer, P., Billich, A., Baumruker, T., Mechtcheriakova, D., Schmid, H.,and Nussbaumer, P. (2004) Bioorg. Med. Chem. Lett. 14, 1555–1558
29. Kohama, T., Olivera, A., Edsall, L., Nagiec,M.M., Dickson, R., and Spiegel,S. (1998) J. Biol. Chem. 273, 23722–23728
30. Pagano, R. E., Sepanski, M. A., and Martin, O. C. (1989) J. Cell Biol. 109,2067–2079
31. Carre, A., Graf, C., Stora, S., Mechtcheriakova, D., Csonga, R., Urtz, N.,Billich, A., Baumruker, T., and Bornancin, F. (2004)Biochem. Biophys. Res.Commun. 324, 1215–1219
32. Mitsutake, S., and Igarashi, Y. (2005) J. Biol. Chem. 280, 40436–4044133. Bajjalieh, S., and Batchelor, R. (1999)Methods Enzymol. 311, 207–21534. Fukasawa, M., Nishijima, M., and Hanada, K. (1999) J. Cell Biol. 144,
673–68535. Hanada, K., Kumagai, K., Yasuda, S.,Miura, Y., Kawano,M., Fukasawa,M.,
and Nishijima, M. (2003) Nature 426, 803–80936. Pagano, R. E., Martin, O. C., Kang, H. C., and Haugland, R. P. (1991) J. Cell
Biol. 113, 1267–127937. Toth, B., Balla, A.,Ma, H., Knight, Z. A., Shokat, K.M., and Balla, T. (2006)
J. Biol. Chem. 281, 36369–3637738. Yasuda, S., Kitagawa, H., Ueno, M., Ishitani, H., Fukasawa, M., Nishijima,
M., Kobayashi, S., andHanada, K. (2001) J. Biol. Chem. 276, 43994–4400239. Lipsky, N. G., and Pagano, R. E. (1983) Proc. Natl. Acad. Sci. U. S. A. 80,
2608–261240. Lipsky, N. G., and Pagano, R. E. (1985) J. Cell Biol. 100, 27–3441. Griffiths, G., Quinn, P., and Warren, G. (1983) J. Cell Biol. 96, 835–85042. Helms, J. B., Karrenbauer, A., Wirtz, K. W., Rothman, J. E., and Wieland,
F. T. (1990) J. Biol. Chem. 265, 20027–2003243. Shinghal, R., Scheller, R. H., and Bajjalieh, S. M. (1993) J. Neurochem. 61,
2279–228544. Boudker, O., and Futerman, A. H. (1993) J. Biol. Chem. 268, 22150–2215545. Lamour, N. F., Stahelin, R. V., Wijesinghe, D. S., Maceyka, M., Wang, E.,
Allegood, J. C., Merrill, A. H., Jr., Cho, W., and Chalfant, C. E. (2007) J.Lipid Res. 48, 1293–1304
46. Giussani, P., Maceyka, M., Le Stunff, H., Mikami, A., Lepine, S., Wang, E.,Kelly, S., Merrill, A. H., Jr., Milstien, S., and Spiegel, S. (2006) Mol. Cell.Biol. 26, 5055–5069
47. Rovina, P., Jaritz, M., Hoefinger, S., Graf, C., Devay, P., Billich, A., Baum-ruker, T., and Bornancin, F. (2006) Biochem. J. 400, 255–265
48. Hanada, K., Kumagai, K., Tomishige, N., and Kawano, M. (2007) Biochim.Biophys. Acta 1771, 644–653
49. Ardail, D., Popa, I., Bodennec, J., Louisot, P., Schmitt, D., and Portoukalian,J. (2003) Biochem. J. 371, 1013–1019
50. Futerman, A. H., Stieger, B., Hubbard, A. L., and Pagano, R. E. (1990)J. Biol. Chem. 265, 8650–8657
51. Jeckel, D., Karrenbauer, A., Burger, K. N., van Meer, G., and Wieland, F.(1992) J. Cell Biol. 117, 259–267
52. Huitema, K., van den Dikkenberg, J., Brouwers, J. F., and Holthuis, J. C.(2004) EMBO J. 23, 33–44
53. Kim, T. J., Mitsutake, S., and Igarashi, Y. (2006) Biochem. Biophys. Res.Commun. 342, 611–617
54. Dressler, K. A., and Kolesnick, R. N. (1990) J. Biol. Chem. 265,14917–14921
Ceramide 1-Phosphate Formation and Traffic
8526 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 283 • NUMBER 13 • MARCH 28, 2008
by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from



Frédéric BornancinAlistair Boath, Christine Graf, Emilie Lidome, Thomas Ullrich, Peter Nussbaumer and
SPHINGOMYELINCOMPARATIVE ANALYSIS TO GLUCOSYLCERAMIDE AND
Regulation and Traffic of Ceramide 1-Phosphate Produced by Ceramide Kinase:
doi: 10.1074/jbc.M707107200 originally published online December 16, 20072008, 283:8517-8526.J. Biol. Chem.
10.1074/jbc.M707107200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
Supplemental material:
http://www.jbc.org/content/suppl/2007/12/18/M707107200.DC1.html
http://www.jbc.org/content/283/13/8517.full.html#ref-list-1
This article cites 53 references, 28 of which can be accessed free at
by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from





![Arrangement of ceramide [EOS] in a stratum corneum lipid model matrix: new aspects revealed by neutron diffraction studies](https://static.fdokumen.com/doc/165x107/631f0e12198185cde200ea75/arrangement-of-ceramide-eos-in-a-stratum-corneum-lipid-model-matrix-new-aspects.jpg)














