
Rac1b Increases with Progressive Tau Pathology within Cholinergic Nucleus Basalis Neurons in...
15
Cell Injury, Repair, Aging, and Apoptosis Rac1b Increases with Progressive Tau Pathology within Cholinergic Nucleus Basalis Neurons in Alzheimer’s Disease Sylvia E. Perez,* Damianka P. Getova, † Bin He,* Scott E. Counts,* Changiz Geula, ‡ Laurent Desire, § Severine Coutadeur, § Helene Peillon, § Stephen D. Ginsberg, ¶ and Elliott J. Mufson* From the Department of Neurological Sciences,* Rush University Medical Center, Chicago, Illinois; the Department of Pharmacology and Clinical Pharmacology, † Medical University, Plovdiv, Bulgaria; the Cognitive Neurology and Alzheimer’s Disease Center, ‡ Northwestern University, Chicago Illinois; ExonHit SA, § Paris, France; and the Center for Dementia Research and Departments of Psychiatry and Physiology and Neuroscience, ¶ Nathan Kline Institute/New York University Langone Medical Center, New York, New York Cholinergic basal forebrain (CBF) nucleus basalis (NB) neurons display neurofibrillary tangles (NFTs) during Alzheimer’s disease (AD) progression, yet the mechanisms underlying this selective vulnerability are currently unclear. Rac1, a member of the Rho family of GTPases, may interact with the proapoptotic pan-neurotrophin receptor p75 NTR to induce neuro- nal cytoskeletal abnormalities in AD NB neurons. Herein, we examined the expression of Rac1b, a con- stitutively active splice variant of Rac1, in NB cholin- ergic neurons during AD progression. CBF tissues harvested from people who died with a clinical diag- nosis of no cognitive impairment (NCI), mild cogni- tive impairment, or AD were immunolabeled for both p75 NTR and Rac1b. Rac1b appeared as cytoplasmic diffuse granules, loosely aggregated filaments, or compact spheres in p75 NTR -positive NB neurons. Al- though Rac1b colocalized with tau cytoskeletal mark- ers, the percentage of p75 NTR -immunoreactive neu- rons expressing Rac1b was significantly increased only in AD compared with both mild cognitive im- pairment and NCI. Furthermore, single-cell gene ex- pression profiling with custom-designed microar- rays showed down-regulation of caveolin 2, GNB4, and lipase A in AD Rac1b-positive/p75 NTR -labeled NB neurons compared with Rac1b-negative/p75 NTR - positive perikarya in NCI. These proteins are in- volved in Rac1 pathway/cell cycle progression and lipid metabolism. These data suggest that Rac1b ex- pression acts as a modulator or transducer of vari- ous signaling pathways that lead to NFT formation and membrane dysfunction in a subgroup of CBF NB neurons in AD. (Am J Pathol 2012, 180:526 –540; DOI: 10.1016/j.ajpath.2011.10.027) Rac1, a member of the Rho family of GTPases, is a ubiquitous intracellular transducer protein that interacts with specific effectors (eg, p-21-activated kinase) to me- diate a diverse array of cellular functions, including cyto- skeleton remodeling, microtubule stability, gene tran- scription, and superoxide production. 1 Rac1 and related Rho GTPases regulate the formation, reorganization, and maturation of neuronal dendrites, 2,3 indicating a key role for these proteins in modulating cellular processes un- derlying normative brain functions. Conversely, Rac1 dysfunction has been implicated in mental retardation syndromes 3,4 and neurodegenerative diseases, includ- ing Parkinson’s disease 5 and Alzheimer’s disease (AD). 6 AD, the most common type of dementia in the elderly, is clinically characterized by a progressive cognitive de- cline. The hallmark neuropathologic features of the AD brain are insoluble extracellular deposits of amyloid (A), or amyloid plaques, and neurofibrillary tangles (NFTs), consisting of abnormal intracellular aggregates of the cytoskeletal protein, tau. 7–9 In addition to these Supported by NIH grants (P01AG14449, AG10668, and AG109466 to E.J.M.) and by a Fulbright Fellowship (D.P.G.). Accepted for publication October 11, 2011. Disclosures: L.D., S.C., and H.P. are employed by and hold stock options in ExonHit SA, which developed the expression profiling technol- ogy used in this work. E.M. has stock options in Ceregene, Inc. None of the other authors disclosed any relevant financial relationships. Supplemental material for this article can be found at http://ajp. amjpathol.org or at doi: 10.1016/j.ajpath.2011.10.027. Address reprint requests to Elliott J. Mufson, Ph.D., or Sylvia E. Perez, Ph.D., 1735 W. Harrison St., Ste. 300, Chicago, IL 60612. E-mail: emufson@ rush.edu or [email protected]. The American Journal of Pathology, Vol. 180, No. 2, February 2012 Copyright © 2012 American Society for Investigative Pathology. Published by Elsevier Inc. All rights reserved. DOI: 10.1016/j.ajpath.2011.10.027 526
Transcript of Rac1b Increases with Progressive Tau Pathology within Cholinergic Nucleus Basalis Neurons in...

The American Journal of Pathology, Vol. 180, No. 2, February 2012
Copyright © 2012 American Society for Investigative Pathology.
Published by Elsevier Inc. All rights reserved.
DOI: 10.1016/j.ajpath.2011.10.027
Cell Injury, Repair, Aging, and Apoptosis
Rac1b Increases with Progressive Tau Pathologywithin Cholinergic Nucleus Basalis Neurons in
Alzheimer’s DiseaseSylvia E. Perez,* Damianka P. Getova,† Bin He,*Scott E. Counts,* Changiz Geula,‡
Laurent Desire,§ Severine Coutadeur,§
Helene Peillon,§ Stephen D. Ginsberg,¶ andElliott J. Mufson*From the Department of Neurological Sciences,* Rush University
Medical Center, Chicago, Illinois; the Department of
Pharmacology and Clinical Pharmacology,† Medical University,
Plovdiv, Bulgaria; the Cognitive Neurology and Alzheimer’s
Disease Center,‡ Northwestern University, Chicago Illinois;
ExonHit SA,§ Paris, France; and the Center for Dementia
Research and Departments of Psychiatry and Physiology and
Neuroscience,¶ Nathan Kline Institute/New York University
Langone Medical Center, New York, New York
Cholinergic basal forebrain (CBF) nucleus basalis(NB) neurons display neurofibrillary tangles (NFTs)during Alzheimer’s disease (AD) progression, yet themechanisms underlying this selective vulnerabilityare currently unclear. Rac1, a member of the Rhofamily of GTPases, may interact with the proapoptoticpan-neurotrophin receptor p75NTR to induce neuro-nal cytoskeletal abnormalities in AD NB neurons.Herein, we examined the expression of Rac1b, a con-stitutively active splice variant of Rac1, in NB cholin-ergic neurons during AD progression. CBF tissuesharvested from people who died with a clinical diag-nosis of no cognitive impairment (NCI), mild cogni-tive impairment, or AD were immunolabeled for bothp75NTR and Rac1b. Rac1b appeared as cytoplasmicdiffuse granules, loosely aggregated filaments, orcompact spheres in p75NTR-positive NB neurons. Al-though Rac1b colocalized with tau cytoskeletal mark-ers, the percentage of p75NTR-immunoreactive neu-rons expressing Rac1b was significantly increasedonly in AD compared with both mild cognitive im-pairment and NCI. Furthermore, single-cell gene ex-pression profiling with custom-designed microar-rays showed down-regulation of caveolin 2, GNB4,and lipase A in AD Rac1b-positive/p75NTR-labeled
NB neurons compared with Rac1b-negative/p75NTR-526
positive perikarya in NCI. These proteins are in-volved in Rac1 pathway/cell cycle progression andlipid metabolism. These data suggest that Rac1b ex-pression acts as a modulator or transducer of vari-ous signaling pathways that lead to NFT formationand membrane dysfunction in a subgroup of CBFNB neurons in AD. (Am J Pathol 2012, 180:526–540; DOI:
10.1016/j.ajpath.2011.10.027)
Rac1, a member of the Rho family of GTPases, is aubiquitous intracellular transducer protein that interactswith specific effectors (eg, p-21-activated kinase) to me-diate a diverse array of cellular functions, including cyto-skeleton remodeling, microtubule stability, gene tran-scription, and superoxide production.1 Rac1 and relatedRho GTPases regulate the formation, reorganization, andmaturation of neuronal dendrites,2,3 indicating a key rolefor these proteins in modulating cellular processes un-derlying normative brain functions. Conversely, Rac1dysfunction has been implicated in mental retardationsyndromes3,4 and neurodegenerative diseases, includ-ing Parkinson’s disease5 and Alzheimer’s disease (AD).6
AD, the most common type of dementia in the elderly,is clinically characterized by a progressive cognitive de-cline. The hallmark neuropathologic features of the ADbrain are insoluble extracellular deposits of � amyloid(A�), or amyloid plaques, and neurofibrillary tangles(NFTs), consisting of abnormal intracellular aggregatesof the cytoskeletal protein, tau.7–9 In addition to these
Supported by NIH grants (P01AG14449, AG10668, and AG109466 toE.J.M.) and by a Fulbright Fellowship (D.P.G.).
Accepted for publication October 11, 2011.
Disclosures: L.D., S.C., and H.P. are employed by and hold stockoptions in ExonHit SA, which developed the expression profiling technol-ogy used in this work. E.M. has stock options in Ceregene, Inc. None ofthe other authors disclosed any relevant financial relationships.
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.10.027.
Address reprint requests to Elliott J. Mufson, Ph.D., or Sylvia E. Perez,Ph.D., 1735 W. Harrison St., Ste. 300, Chicago, IL 60612. E-mail: emufson@
rush.edu or [email protected].
Rac1b Up-Regulation in AD Neurons 527AJP February 2012, Vol. 180, No. 2
pathologic hallmark lesions, there is a reduction in cho-linergic basal forebrain (CBF) projection neurons locatedwithin the septal diagonal band complex and the nucleusbasalis of Meynert (NB)10–13 and a loss of cholineacetyltransferase (ChAT; the synthetic enzyme for ace-tylcholine) in CBF hippocampal and cortical targetfields in late AD.14 –18 These cholinergic deficits corre-late with cognitive decline and disease severity inAD.19 –21 NFTs are found in CBF neurons in the brainsof people who died with a clinical diagnosis of AD22–24
and mild cognitive impairment (MCI),24,25 a putativeprodromal AD stage. However, whether Rac1 expres-sion is associated with the onset of NFT pathologywithin CBF neurons during disease progression re-mains an open question.
Interestingly, CBF neurons depend on nerve growthfactor (NGF) for their survival, and in vitro studies haveshown that the proapoptotic pan-neurotrophin receptor,p75NTR, activates Rac1 in a NGF-dependent manner,which may initiate cytoskeletal abnormalities or apoptoticsignaling in AD.26 Moreover, data derived from in vivo andin vitro investigations show that A� interacts with Rac1 toinduce cytoskeletal changes related to NFT forma-tion27,28 and that RhoA proteins abnormally accumulatein NFT-bearing neurons in AD.6 Taken together, theseobservations suggest that dysregulation of the RhoGTPase Rac1 is associated with neuronal cytoskeletalalterations in AD.
A decade ago, a new isoform of Rac termed Rac1bwas identified in breast and colon cancers.29,30 Rac1b isa self-activated splicing variant of Rac1 that displays a19-amino acid (aa) insert from exon 3b that creates anovel effector-binding site absence in Rac1.29,31 Thepresence of these additional 19 aa altered the structureand activity of Rac1b,32,33 rendering it a constitutivelyactive variant of Rac1.34 Thus, Rac1b overactivation mayplay a prominent role in Rac1-related cellular neurode-generation. Recently, a transcriptomic profiling technol-ogy developed by Exonhit35 was used to compare alter-native RNA splicing events present in frontal cortex fromAD (Braak stage VI) and age-matched healthy controls,which identified splicing alterations occurring in Rac1 inAD brain.36 Moreover, in a preliminary report we foundRac1b immunoreactivity within NFT-bearing p75NTR-pos-itive NB neurons in end-stage AD.37 However, the poten-tial role of Rac1b in the development of NFT pathologywithin NB neurons during the progression of AD has notbeen explored. In the present study, we examined thespatiotemporal nature of Rac1b expression with respectto tau epitopes underlying NFT formation within p75NTR-positive NB neurons in postmortem tissue harvested frompeople who died with a clinical diagnosis of no cognitiveimpairment (NCI), MCI, or AD. Furthermore, we com-pared gene expression profiles of Rac1b-immunopositive(Rac1b�) and Rac1b-immunonegative (Rac1b�) NBneurons by laser capture microdissection coupled withcustom-designed microarray analysis to identify targetsof potential Rac1b-mediated mechanisms of neurode-
generation in AD.Materials and Methods
Alternative RNA Splicing for Rac1 in AD Brainand Other Tissues
Expression profiling technology developed by ExonHit(Paris, France)35 was used to compare alternative RNAsplicing events present in Rac1 within frozen brain tis-sues accrued from frontal cortex (Brodmann area 9) fromBraak stage VI AD and nondemented healthy controlsboth 70 years old. RT-PCR technology determined Rac1bexpression levels in different tissues, including brain.29
The primers used were F1, 5=-ATGCAGGCCATCAAGT-GTGTGG-3=; R1, 5=-TGGCATTGAGTGCGAAGGC-3=;and F2, 5=-AAAGACAAGCCGATTGCCG-3=. A discreteband representing a 492-bp amplicon that correspondsto the expected size of the Rac1b amplicon was detectedin the brain samples. The amplicon was cloned into TopoTA cloning vector (Invitrogen, Paris, France) and se-quenced with SP6 and T7 primers to confirm the se-quence identity of the Rac1b amplification product.
Generation and Specificity of a Rac1b Antibody
Generation of the Rac1b Antibody
An anti-Rac1b serum (SG-4088) was produced bySigma-Genosys (Sigma-Aldrich, St. Louis, MO) accord-ing to their standard immunization protocol by couplingthe peptide VGETYGKDITSRGKDKPIAC from AD frontalcortex to keyhole limpet hemocyanin. Serum was thenaffinity-purified on a Sepharose column with the use ofthe immunogen as bait.
Specificity of the Rac1b Antibody
Ten nanograms of either recombinant Rac1 or Rac1b[glutathione S-transferase (GST)-tagged; 50 kDa each;Cytoskeleton, Denver, CO] was separated by electropho-resis in a 12% SDS-PAGE system and transferred toHybond-C membranes (Amersham Biosciences, Paris,France). After transfer, membranes were blocked with5% nonfat milk in 0.1% PBS/0.1% Tween-20 and incu-bated overnight with the primary rabbit anti-Rac1b anti-body SG-4088 at a 1:5000 dilution, or with a mousemonoclonal anti-Rac1 antibody ARC03 (1:5000; Cyto-skeleton, Denver, CO). Immunologic complexes were de-veloped with an anti-rabbit or an anti-mouse peroxidaseantibody (1:5000; Jackson Laboratories, Paris, France)followed by enhanced chemiluminescence (AmershamBiosciences, Paris, France).
Cell Culture and Immunoblot Analysis
Myc (alias c-myc)-tagged Rac1b was cloned into theNIT tet off-expression vector,38 kindly provided by Dr.Fred H. Gage (The Salk Institute, La Jolla, CA). NIH3T3cells (LGC PromoChem, Molsheim, France) were grownin high-glucose Dulbecco’s modified Eagle’s mediumplus Glutamax supplemented with 10% newborn serum
and antibiotics. NIH3T3 cells were transiently transfected
528 Perez et alAJP February 2012, Vol. 180, No. 2
with NIT-Rac1b in the presence or absence of variousdoses of doxycycline (Sigma-Aldrich). Forty-eight hoursafter transfection, cells were lysed and 7.5 �g of proteinwas separated on a 12% SDS-PAGE gel and transferredto Hybond-C membranes (Amersham Biosciences). Aftertransfer, membranes were blocked with 5% nonfat milk in0.1% PBS/0.1% Tween-20 and incubated overnight withthe anti-Rac1b antibody at 1:5000, allowing for the de-tection of both endogenous and Myc-tagged Rac1b. Themembrane was then stripped at 55°C for 20 minutes andreprobed with an anti-Myc monoclonal antibody at 1:750(Tebu, Le Perray en Yvelines, France). Immunologic com-plexes were shown with anti-rabbit or an anti-mouse IgG(1:5000; Jackson Laboratories) followed by chemilumi-nescence (Amersham Biosciences).
Cell Culture and Immunocytochemistry
Human embryonic kidney (HEK) 293 cells were main-tained in modified Eagle’s medium plus Earle’s salt sup-plemented with 10% fetal bovine serum, 2 mmol/L L-glu-tamine (Sigma-Aldrich), nonessential amino acids, andantibiotics. 2 � 105 HEK293 cells/well were seeded in6-well plates for 24 hours and transiently transfected withthe Myc-tagged Rac1b expression vector or empty vec-tor with the use of Lipofectamine Plus (Invitrogen). Forty-eight hours after transfection, cells were fixed with 4%paraformaldehyde in PBS, permeabilized with 0.1% Tri-ton X-100, saturated in 5% nonfat milk/1% bovine serumalbumin, and processed for Rac1b fluorescence immu-nohistochemistry (SG-4088, 1:500).
Brain Tissue Accrual
The Human Investigations Committee of Rush UniversityMedical Center and Northwestern University School ofMedicine approved tissue usage for this study. Postmor-tem tissue containing the CBF neurons of the NB washarvested from 28 participants in the Religious OrdersStudy,39–42 a longitudinal clinical pathologic study of ag-ing and AD in older Catholic nuns, priests, and brothersconducted by Rush University Medical Center (Chicago,IL). Subjects were clinically characterized within a year ofdeath as having NCI (n � 12), MCI (n � 8, 6 with nonam-nestic MCI and 2 with amnestic MCI), and mild AD (n �7). All MCI and AD cases were evaluated clinically andpathologically as described previously.43–49 Cortical tis-sue was also harvested from four NCI, MCI, and ADcases to test the specificity of the Rac1b antibody.
In addition, brain tissue from 8 cases neuropathologi-cally characterized as severe (end-stage) AD, based onBraak stage, were accessed from the Rush Alzheimer’sDisease Brain Bank. A summary of the clinical and neu-ropathologic demographics of the cases examined in thisstudy is shown in Table 1. To test for disease specificityof Rac1b immunolabeling within CBF neurons of the NB,we examined tissue harvested from two other tauopa-thies: Lewy Body disease (LBD; n � 4) and frontotempo-ral dementia (FTD; n � 3) (see Table 2) accrued fromNorthwestern University School of Medicine (Chicago,
IL). Consensus criteria incorporated into the proceduresof the Uniform Data Set of the NIA Alzheimer’s DiseaseCenters program50,51 were used for the clinical diagnosisof FTD. Pathologic diagnosis of frontotemporal lobar de-generation (FTLD) and specification of its variants wererendered according to the published consensus criteriaset forth by the Consortium for FTLD.52 All tissue sampleswere fixed at autopsy in 4% paraformaldehyde, cryopro-tected, and cut at 40 �m on a freezing sliding microtomefor immunohistochemical and gene expression analysesas reported previously.43,44,53
Antibody Characterization
Antibodies used to detect cholinergic neurons of the NBincluded a mouse monoclonal antibody generatedagainst p75NTR (epitope aa 1 to 160) derived from A875melanoma cells (clone NGFR5; LabVision, Fremont, CA),which is an excellent marker of CBF neurons in the humanhealthy aged and AD brain54,55 and a goat polyclonal an-tiserum to ChAT (Millipore, Billerica, MA), which specificallymarks cholinergic neurons in human brain.56–58 These an-tibodies were used in colocalization experiments withRac1b and tau-specific epitope markers for NFTs. Antibodyspecificity of each cholinergic marker was reported by theirmanufacturers.
The tau antibodies used in this study included Alz50, amouse monoclonal antibody (gift from Dr. P. Davies, Al-bert Einstein School of Medicine, Bronx, NY), whichshows an early-stage conformational tau epitope (aa 5 to15, 312 to 322) and marks neurons undergoing earlydegenerative events associated with NFT formation,59–61
AT180 mouse monoclonal antibody (ThermoFisher, Pitts-burgh, PA), which detects tau phosphorylation at the thre-onine 231 site, an early event in the disassembly of tau,62,63
and the AT8 mouse monoclonal antibody (ThermoFisher),which marks tau phosphorylation at both serine 202 andthreonine 205 sites.64 The specificity of the tau antibodieshas been reported by numerous investigators59–64 andby their manufacturers.
Immunoblot and Immunohistochemistry
Cortical samples from NCI (n � 4) and severe AD (n � 4)were processed for Western blot analysis as reportedpreviously.65 Briefly, tissue was sonicated in ice-cold ho-mogenization buffer (20 mmol/L Tris, 1 mmol/L EGTA, 1mmol/L EDTA, 10% sucrose, pH 7.4) containing proteaseinhibitors, and protein concentration of S1 fraction wasdetermined by the Bradford method (Bio-Rad, Hercules,CA). Sample proteins (48 �g/sample) were separated bySDS-PAGE (7.5% acrylamide) and transferred electro-phoretically to polyvinylidene fluoride membranes (Immo-bilon P; Millipore). Membrane was incubated overnightwith rabbit anti-Rac1b antiserum (our SG-4088; 1:1000)and developed with a horseradish peroxidase-conju-gated goat anti-rabbit IgG (1:5000; Bio-Rad). Immunore-active proteins were visualized by enhanced chemilumi-nescence (Amersham Biosciences) on a Kodak ImageStation 440CF (Eastman Kodak, Cincinnati, Ohio).
Floating sections containing the NB region of the basal
forebrain extending from the decussation of the anterior
Rac1b Up-Regulation in AD Neurons 529AJP February 2012, Vol. 180, No. 2
commissure until its emergence from the temporal lobe,caudally, were dual immunolabeled for p75NTR (1:15,000)and the polyclonal rabbit Rac1b-specific antibody (1:1000) with the use of tissue sections from NCI, MCI,mild/moderate AD, severe AD, LBD, or FTD cases. Sec-tions were washed several times in Tris-buffered saline(TBS) before incubation with 0.1 mol/L sodium periodate(to inhibit endogenous peroxidase activity) in a TBS so-lution for 20 minutes. After several rinses in a solutioncontaining 0.25% Triton X-100 in TBS, the tissue wasplaced in a blocking solution containing TBS with 0.25%Triton X-100 and 3% goat serum for 1 hour. Sections weresubsequently incubated in rabbit anti-Rac1b antibody for24 hours in a medium containing TBS 0.25% Triton X-100and 1% goat normal serum. After washes with 1% goatnormal serum in TBS, sections were incubated with bio-tinylated goat anti-rabbit (Vector Laboratories, Burlin-game, CA) for 1 hour. After several washes in TBS thetissue was incubated for 60 minutes with an avidin-biotincomplex (1:500; “Elite Kit”; Vector Laboratories). Tissuewas rinsed in 0.2 mol/L sodium acetate, 1.0 mol/L imida-zole buffer (pH 7.4), and developed in an acetate-imida-zole buffer containing 2.5% nickel sulfate, 0.05% 3,3=-diaminobenzidine tetrahydrochochloride (Sigma-Aldrich)and 0.0015% H2O2. The reaction was terminated with anacetate-imidazole buffer solution. Subsequently, and af-ter several washes in TBS, sections were incubated withanti-p75NTR antibody for 24 hours in a medium containingTBS 0.25% Triton X-100 and 1% goat normal serum, andthen incubated with biotinylated goat anti-mouse (VectorLaboratories) and visualized with the chromogen 3,3=-diaminobenzidine tetrahydrochochloride (brown) withoutnickel intensification following the above protocol. Thisdual staining resulted in an easily identifiable two-coloredprofile: dark blue/black for Rac1b and brown for p75NTR-positive profiles.54 Bright-field images were acquired withNikon microphot microscope (Nikon, Melville, NY). Addi-tional sections were either double or triple immunofluo-rescence labeled with the use of antibodies directedagainst Rac1b (1:100; SG-4088), ChAT (1:100), the earlyconformational tau antibody Alz50 (1:4000), and the tauphosphorylation antibodies AT180 (1: 300) and AT8 (1:100). Fluorescence signals were shown with Cy3-, Cy2-,and Cy5-conjugated anti-mouse, rabbit, and goat IgGs(Jackson Immunoresearch, West Grove, PA), dependingon the primary antibody. Double and triple immunofluo-rescence images were acquired and analyzed with aZeiss Axioplan 2 (Zeiss, Thorndale, NY) and an Olympusconfocal microscope equipped with FluoroView 4.3 soft-ware (Olympus, Center Valley, PA), respectively. Omis-sion of primary antibodies resulted in a lack of immuno-reactivity in tissue sections. To test the specificity ofRac1b antiserum the antibody was preabsorbed againstpurified Rac1b protein (0.1 mg/mL), followed by immu-nostaining of NB tissue with the above protocol. Further-more, to test for the possible cross-reactivity between thetau antibodies and Rac1b, AT8 (1:1000) and Alz50 (1:10,000) were preabsorbed with Rac1b protein (0.1 mg/mL) overnight, followed by our standard immunostainingprotocols. Preabsorption with the Rac1b protein did not
prevent AT8 or Alz50 immunoreactivity.Cell Distribution and Quantitation
The distribution of NB neurons single- and double-stained for Rac1b and p75NTR receptor were charted withNeurolucida 8 (MicroBrightField Inc., Williston, VT) withthe aid of Nikon Optiphot-2 microscope. Counts of singlep75NTR and double Rac1b/p75NTR-immunoreactive (ir)cholinergic NB neurons were performed manually atmagnification of �40, and fiduciary landmarks were usedto prevent repeat counting of neurons. The number ofsections counted per case ranged from 6 to 11. Stereo-logic counting procedures were not used because thetotal number of neurons per region was not a requirementand the tissue was not prepared for unbiased counting.The results are presented as the percentage of Rac1b/p75NTR-immunopositive total neurons rather than abso-lute numbers of cell counts.
Single-Cell Gene Expression Analysis
Immunolabeled p75NTR-positive NB neurons (n � 140)from NCI cases (n � 6) and immunolabeled p75NTR anddouble-labeled Rac1b/p75NTR NB neurons (n � 260) fromsevere AD cases (n � 7) were accessed by laser capturemicrodissection (Arcturus XT; Applied Biosystems). Be-cause dual Rac1b�/p75NTR CBF labeling was negligible inthe MCI cases, gene expression comparisons were notperformed. The amplification of mRNA from individual neu-rons was performed with terminal continuation RNA ampli-fication methodology as previously described.39–42,53 Ter-minal continuation RNA amplification is a linear amplificationprocess that preserves the original quantitative relation-ships among the transcripts.39,53,66,67 Hybridization probeswere synthesized by in vitro transcription with the use of 33Pincorporation in 40 mmol/L Tris (pH 7.5); 6 mmol/L MgCl2;10 mmol/L NaCl; 2 mmol/L spermidine; 2.5 mmol/L DTT;125 �mol/L ATP, GTP, and CTP; 2.5 �mol/L cold UTP; 20U of RNase inhibitor; 2 kU of T7 RNA polymerase (Epi-center, Madison, WI); and 120 �Ci of 33P-UTP (Perkin-Elmer, Boston, MA).68 The labeling reaction was per-formed at 37°C for 4 hours. Radiolabeled terminalcontinuation RNA probes were hybridized to custom-de-signed microarrays without further purification.
Custom-Designed Microarray Platforms
Array platforms consist of 1 �g of linearized cDNA puri-fied from plasmid preparations adhered to high-densitynitrocellulose (Hybond XL; GE Healthcare, Piscataway,NJ) with the use of an arrayer robot (VersArray; Bio-Rad).39,68,69 Each cDNA and/or EST is verified by se-quence analysis and restriction digestion. Approximately576 cDNAs/ESTs are used on the array platform (seeSupplemental Table S1 at http://ajp.amjpathol.org). Ar-rays were hybridized for 24 hours, washed with serialdilutions of saline sodium citrate buffer, and then placedin a phosphor screen for 24 hours before development ona phosphor imager with ImageQuant 5.2 software (GEHealthcare).
Hybridization signal intensity of the empty vector (pBS;
double spotted on the array) identified background ac-
530 Perez et alAJP February 2012, Vol. 180, No. 2
tivity.39,53 Expression of terminal continuation-amplifiedRNA bound to each linearized cDNA was expressed as aratio of the total hybridization signal intensity of the array (ie,global normalization),39,69–72 thereby minimizing variationsbecause of differences in the specific activity of the probeand the absolute quantity of the probe present.73–75
Statistical Analysis
Demographic, clinical, and neuropathologic characteris-tics were compared across clinical diagnostic groupswith one-way analysis of variance, Kruskal-Wallis or Fish-er’s exact test follow by Dunn’s post hoc test for multiplecomparisons (Sigma Stat 3.5; Systat Software, Inc., SanJose, CA). Cell count data were analyzed with Kruskal-Wallis analysis of variance on ranks followed by Dunn’smethod for multiple comparisons, and correlations wereperformed with Spearman rank (Sigma Stat 3.5; SystatSoftware, Inc.). Single-cell gene array data were ana-lyzed with GeneLinker 4.6 bioinformatics software withthe use of a false discovery rate controlling proce-dure68,76 to reduce type I error (IOS, Kingston, ON).Levels of individual mRNAs were compared among NCIp75NTR, AD p75NTR, and AD Rac1b/p75NTR with the useof one-way analysis of variance with post hoc Holm-Sidaktesting for multiple comparisons (Sigma Stat 3.5, Systat
Table 1. Count Analysis: Clinical, Demographic, and Neuropatho
Clinical
NCI (n � 12) MCI (n � 8)
No. of females (%) 7 (84) 3 (35)Age at death (years)
Mean � SD 85.9 � 4.7 86.7 � 5.3Range 78.3–92.7 79.4–93.9
Postmortem intervals (hours)Mean � SD 5.5 � 2.1 6.6 � 3.4Range 3.0–9.7 2–11.5
Brain weight (g)Mean � SD 1159.9 � 86.5 1233.7 � 112.8Range 1000–1300 1060–1350
No. with ApoE e4 allele (%) 1 (8.3) 4 (50)Age during education
(years)Mean � SD 18 � 3.0 17.8 � 2.9Range 10–21 12–20
MMSE scoreMean � SD 28.1 � 1.4 26.7 � 2.1Range 26–28 22–27
Braak stage0 0 0I–II 3 1III–IV 8 7V–VI 1 0
National Institute on AgingReagan
No AD 0 0Low 6 4Intermediate 6 4High 0 0
Six of the eight patients with MCI were non-amnestic.*Fisher’s exact test.†One-way analysis of variance.‡
Kruskal-Wallis test.AD, Alzheimer’s disease; MCI, mild cognitive impairment; NA, not available;Software, Inc.). Descriptive statistical level of significancewas set at 0.05 (two-tailed).
Results
Subject Demographics
The portion of the study that evaluated Rac1b during theprogression of AD included 36 subjects divided into fourclinical diagnostic groups: 12 NCI, 8 MCI, 7 mild/moder-ate AD, and 8 severe AD (Table 1). No difference wasfound for age, sex, postmortem interval, brain weight,years of education, or Braak scores across groups. Theapolipoprotein E4 allele was most frequent in mild ADcompared with NCI cases (P � 0.05). Mini Mental StateExamination scores were significantly lower in mild ADthan in NCI (P � 0.05). Mini Mental State Examinationscores were not available for the severe AD cases. NIAReagan likelihood of AD scores was significantly higherin severe AD than in NCI and MCI (Table 1). We alsoevaluated three male and one female LBD cases with amean age of 79.2 years, and an average postmorteminterval of 7 hours as well as one male familial FTDLD-fused in sarcoma protein (FUS) and 1 male and 1 femalecase of FTLD-TAR DNA-binding protein 43 (TDP) with a
haracteristics by Diagnosis Category
sis
ild AD� 7)
Severe AD(n � 8) P values Pair-wise comparison
71) 6 (75) NS* �
7 � 6.0 82.5 � 7.4 NS† �3–94.5 67–92
8 � 2.8 7.6 � 4.0 NS† �5–8.1 3.3–16
4 � 138.7 1118.1 � 147.9 NS† �0–1460 940–132557.51) NA 0.038* NCI � AD
7 � 3.6 NA NS† �6–26
2 � 6.4 NA 0.010‡ NCI � AD0–28
0 0 NS† �1 03 63 2
0 0 0.001‡ NCI, MCI � severe AD1 04 02 8
logic C
diagno
M(n
5 (
876.
4.1.
1226.1064 (
19.1
20.1
NCI, no cognitive impairment; NS, not significant.

oral lobrotein in
Rac1b Up-Regulation in AD Neurons 531AJP February 2012, Vol. 180, No. 2
mean age of 59 years and an average postmortem inter-val of 12 hours (Table 2).
Generation and Characterization of Rac1bAntibody
Expression profiling showed a dysregulation of the splic-ing of exon 3b of the Rac1 gene (Figure 1A), leading tooverexpression of the Rac1b isoform in AD comparedwith age-matched control cortical brain tissue. Specifi-cally, the Rac1b isoform displayed 19 aa insert from exon3b, which is not present in Rac1 (Figure 1A). RT-PCRanalyses showed Rac1b transcripts in specific normal
Table 2. Demographic and Clinicopathologic Characteristics for
Age (years) Sex
LBD/pathologic diagnosisLBD 82 MLBD 82 MLBD 81 MLBD 72 FMean � SD 79.2 � 4.8
FTD/pathologic diagnosisFTDLD-FUS (familial) 64 MFTLD-TDP 67 MFTLD-TDP 46 FMean � SD 59 � 11.3
F, female; M, male; FTD, frontotemporal dementia; FTLD, frontotempdisease; NB, nucleus basalis; PMI, postmortem interval; TDP, ubiquitin p
nta
n int.
7 MB
231
canc
era t
e ca
ncer
nta
n int.
7 MB
231
canc
era t
e ca
ncer
Hea
rtLi
ver
Bra
inPl
acen
Sple
enSm
all
Col
onM
CF-
7M
DA
MR
enal
Pros
taH
2O
Hea
rtLi
ver
Bra
inPl
acen
Sple
enSm
all
Col
onM
CF-
7M
DA
MR
enal
Pros
taH
2O
A
B
Figure 1. A: Schematic representations of Rac1 and RAc1b mRNAs showingan extra exon (3b) in Rac1b and the locations of promoters used in RT-PCR.B: Agarose gels showing the levels of PCR products for Rac1 (F1/R1), Rac1b(F2/R1), and actin from heart, liver, brain (normal human frontal cortex),placenta, spleen, small intestine, colon, MCF-7 (human breast adenocarci-
noma cell line), MDA MB 231 (breast cancer), renal cancer, and prostatecancer.body tissues (eg, heart, liver, placenta, and spleen) andcancer tissues [including MCF-7, a human breast ade-nocarcinoma cell line, and MDA MB 231 (breast, renal,and prostate cancer cells], whereas Rac1 transcriptswere also detected in normal human frontal cortex, smallintestine, and colon (Figure 1B). Interestingly, this alter-native splicing of Rac1 was virtually undetectable withinprefrontal cortex from human healthy controls (Figure1B). Immunoblot analysis showed that the Rac1b antise-rum (SG-4088) did not cross-react with Rac1, whereasthe commercial anti-Rac1 antiserum (ARC03) recognizedRac1b protein (Figure 2A). Rac1b cDNA was cloned intoNIT tet off vector and transfected into NIH3T3 cells to testthe specificity of the antibody (SG-4088) generatedagainst the Rac1b isoform. Immunoblotting showed thatthe Rac1b antibody detected the expression of theRac1b vector in NIH3T3 cells cultured in the presence oflower levels or the absence of doxycycline, as expected(Figure 2B). Notably, this antibody recognized endoge-nous Rac1b expression in NIH3T3 cells (Figure 2B). Like-wise, HEK293 cells (Figure 2C) were immunopositive forRac1b after 48 hours of transfection with a vector con-taining Rac1b cDNA (Figure 2D), whereas HEK293 cellstransfected with empty vector were immunonegative (Fig-ure 2, E and F). Moreover, immunoblotting showed thatthe Rac1b antiserum indicated the presence of Rac1bprotein in the cortex of patients with AD, whereas noimmunoreactivity was seen in the cortex of normal humancontrols (Figure 3A). These results indicated that the an-tiserum generated against Rac1b does not cross-reactwith Rac1 protein and thus specifically recognizes theRac1b isoform in immunoblotting and immunocytochem-ical procedures.
General Characteristics of Rac1bImmunostaining in the NB
The immunohistochemical specificity of the Rac1b anti-serum in the NB was determined by pre-absorption withthe Rac1b protein, resulting in a lack of immunoreactiveprofiles within the NB (Figure 3B) compared with neuronsdually immunostained for Rac1b and p75NTR without pre-
d FTD
PMI (hours) Brain weight (g) Rac1b/p75 NB
4 1120 �8 1240 �6 1360 �
10 1220 �7 � 2.5 1235 � 98.4
13 1210 �9 1330 �
14 1150 �12 � 91.6
ar degeneration; FUS, FTD fused in sarcoma protein; LBD, Lewy bodyclusions.
LBD an
absorption (Figure 3C).

and the absence of immunostaining for Rac1b (F), respectively. Originalmagnification, �10.
532 Perez et alAJP February 2012, Vol. 180, No. 2
To examine the characteristics and distribution ofRac1b immunoreactivity in the NB during the progressionof AD, tissue from each clinical group was double immu-nostained for Rac1b and p75NTR. Although most of theRac1b-ir neurons also displayed p75NTR, not all p75NTR
co-expressed Rac1b (Figure 3C). Rac1b�/p75NTR-posi-tive NB neurons were more frequent in mild and severeAD cases (Figure 4, A–D), displaying a homogenousdistribution throughout the entire extent of the NB thatincluded its anteromedial (Ch4am), anterolateral (Ch4al),intermediate (Ch4i), and posterior (Ch4p)11,54,56 portions(Figure 5, A–F; see also Supplemental Figures S1–S4 athttp://ajp.amjpathol.org). Rac1b within both single- anddual-stained NB neurons appeared either as cytoplasmicgranules (Figure 5H), filaments (Figure 5, I and J), or as adense compact accumulation of filaments displacing thenucleus to the periphery of the cell (Figure 5K). In addi-tion, spheroids of twisted Rac1b-positive filaments werealso seen in the region of the NB (Figure 5L). Thesedifferent Rac1b-immunostained profiles were mainly ob-served in mild AD and severe AD and resembled thecytoskeletal abnormalities described within the NB withthe use of the pretangle marker AT8 during NFT forma-tion.23 Rac1b-positive cells with an NFT-like profile aswell as neuropil threads and dystrophic neurites (data notshown) were also seen in the cortex of mild and severeAD cases (Figure 5G) but not in NCI or MCI cortex (datanot shown). Pre-absorption with the Rac1b protein did notblock immunoreactivity for AT8 or Alz50-ir within the NB(Figure 5, M and N), suggesting that these tau antibodiesdid not cross-react with Rac1b. In the other tauopathiesexamined, only two LBD (Figure 5O) and two FTD (Figure5P) cases showed dual immunostaining for p75NTR and
50 kDa
NCI AD
Cortex
Anti-Rac1b50 kDa
B
A
C
Figure 3. A: Representative immunoblot of cortical samples from a NCI anda severe AD case probed with our generated antibody against Rac1b (SG-4088). B: Bright-field image showing the lack of Rac1b immunolabeling inNB neurons after pre-adsorption of the anti-Rac1b antibody (SG-4088) withRac1b protein. C: Bright-field image of double-immunolabeled Rac1b(black)/p75NTR (brown) and single-labeled p75NTR (brown, black arrow)NB neurons in an AD case. Inset shows higher magnification of a Rac1b/p75NTR neuron shown in panel C (open arrow). Scale bars: 30 �m (B andC); 20 �m (inset).
Rac1Anti-Rac1b Anti-Rac1
Rac1b Rac1 Rac1b
50 kDa
Anti-Rac1bc-myc-Rac1b
Endogenous-Rac1b
Anti cm cAnti-cmyc
C D
E F
A
B
Figure 2. A: Representative immunoblots of recombinant Rac1b and Rac1(50 kDa) proteins probed with our generated antibody against Rac1b(SG-4088; left) and with a commercial antibody against Rac1 (ARC03;right). Note that Rac1b antiserum (SG-4088) specifically recognizedRac1b protein and did not cross-react with Rac1, whereas the commercialRac1 antibody cross-reacted with Rac1b protein. B: Immunoblot fromNIH3T3 cell cultures transfected with Myc-tagged Rac1b into the NIT tetoff expression vector in the absence (-) or presence of different doses ofdoxycycline (dox) probed with anti-Rac1b and anti-Myc antibodies. Thebands from NIH3T3 cells cultured with the lowest concentration (0.2 pg)or absence of dox showed the highest Rac1b-ir band, which was con-firmed by reprobing the blots with the antibody against Myc. Rac1bantiserum also recognized endogenous Rac1b expression in NIH3T3 cellsthat was not shown with the Myc antibody. C and E: Contrast-phaseimages of HEK293 cells with Myc-tagged Rac1b vector (C) or empty vector(E), showing the overall cell culture appearance. D and F: Immunofluo-rescence images of C and E panels, showing Rac1b positive cells (red; D)
Rac1b in NB neurons (Table 2).
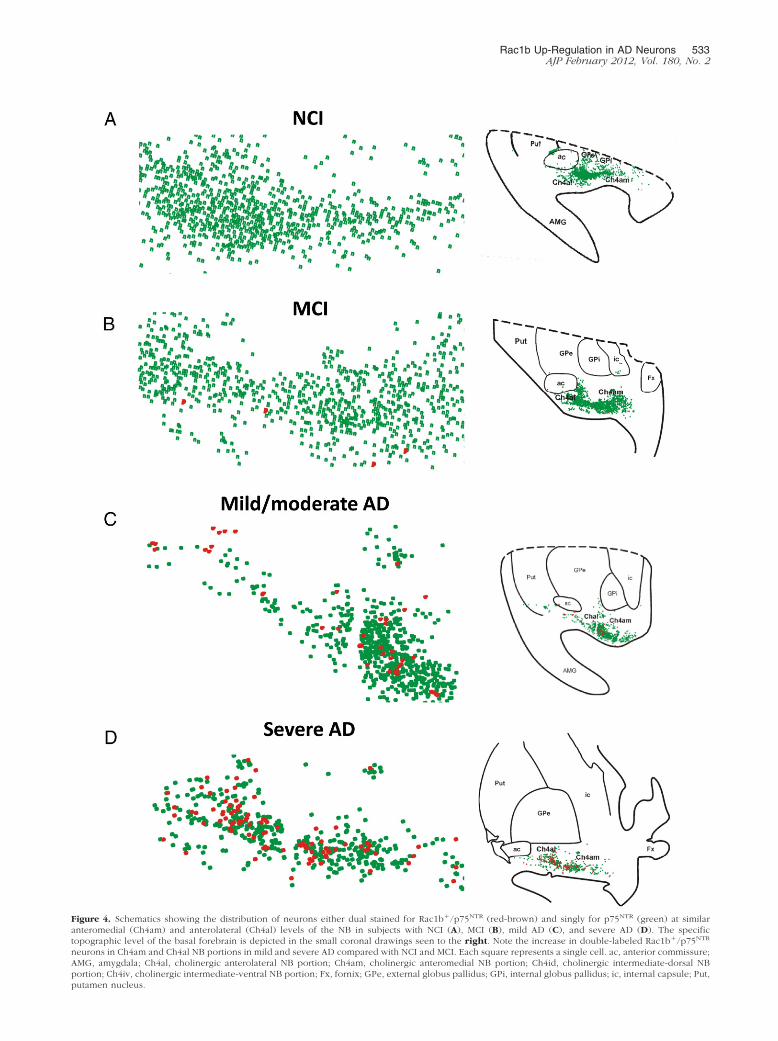
Rac1b Up-Regulation in AD Neurons 533AJP February 2012, Vol. 180, No. 2
Figure 4. Schematics showing the distribution of neurons either dual stained for Rac1b�/p75NTR (red-brown) and singly for p75NTR (green) at similaranteromedial (Ch4am) and anterolateral (Ch4al) levels of the NB in subjects with NCI (A), MCI (B), mild AD (C), and severe AD (D). The specifictopographic level of the basal forebrain is depicted in the small coronal drawings seen to the right. Note the increase in double-labeled Rac1b�/p75NTR
neurons in Ch4am and Ch4al NB portions in mild and severe AD compared with NCI and MCI. Each square represents a single cell. ac, anterior commissure;AMG, amygdala; Ch4al, cholinergic anterolateral NB portion; Ch4am, cholinergic anteromedial NB portion; Ch4id, cholinergic intermediate-dorsal NB
portion; Ch4iv, cholinergic intermediate-ventral NB portion; Fx, fornix; GPe, external globus pallidus; GPi, internal globus pallidus; ic, internal capsule; Put,putamen nucleus.
534 Perez et alAJP February 2012, Vol. 180, No. 2
Dual Labeling for Rac1b and TauImmunoreactivity within NB Neurons
To evaluate whether Rac1b profiles co-expressed differ-ent tau isoforms in NB neurons during the course of AD,immunofluorescence staining of Rac1b and selected tau
CA B D
FE
JH I
N
CO P
L M
epitopes was performed with the use of tissue from each
of the groups examined in this study (Figure 6, A–I).Virtually all Rac1b neurons colocalized with AT8, AT180,and Alz50 immunoreactivity in the NB during AD progres-sion (Figure 6, A–I). However, only an occasional singleRac1b-ir neuron was observed in AD (Figure 6, C and I).Rac1b-ir neurons expressing Alz50 and AT8 were seen
G
Figure 5. A. Bright-field image of double-immu-nolabeled Rac1b (black)/p75NTR (brown) and sin-gle-labeled p75NTR (brown) neurons in the antero-medial portion (Ch4am) of NB from a severe ADcase; inset shows double Rac1b/p75NTR and sin-gle p75NTR neurons from panel A (arrow). Thespecific level of Ch4am is depicted in the schematicdrawing shown in panel B. C: Low-power imageof double-immunolabeled Rac1b (black)/p75NTR
(brown) and single-labeled p75NTR (brown) neu-rons in the anterolateral portion (Ch4al) of the NBfrom a severe AD case; inset shows a doubleRac1b/p75NTR neuron from panel C (arrow). Thespecific level Ch4al is depicted in the schematicdrawing shown in panel D. E: Image showingdouble-immunolabeled Rac1b (black)/p75NTR
(brown) and single-labeled p75NTR (brown) neu-rons in the dorsal intermediate NB portion (Ch4id)of severe AD. The specific level of the dorsal-intermediate NB subfield is depicted in the sche-matic drawing in panel F. G: Rac1b-ir cells with anNFT-like prolife in the temporal cortex of a severeAD case; inset shows a high-power image of acortical Rac1b-ir neuron from panel G (arrow).H–K: High-power images showing subtypes ofneuronal cytoplasmic Rac1b immunoreactivity(black) in p75NTR-positive (brown) NB neuronswith a granular (H), filamentous (I and J), or com-pact spherical (K) appearance in severe AD. L:Twisted Rac1b-positive filaments in the NB in se-vere AD. M and N: AT8-ir and Alz50-positive neu-rons and processes in the NB after pre-adsorbingthe AT8 and Alz50 antibodies with Rac1b protein,respectively, in severe AD, indicating the specific-ity of the tau antibody staining. O: Bright-field im-age of double labeling for Rac1b (black) andp75NTR, showing their colocalization in a neuron(arrow) as well as single p75NTR neurons (brown)in NB in a LBD case; inset shows a higher mag-nification image of a Rac1b�/p75NTR neuron frompanel O (arrow). P: Bright-field image showingdouble-labeled Rac1b and p75NTR (arrow) andsingle-labeled p75NTR (brown) NB neurons in aFTD case; inset shows a detail of Rac1b�/p75NTR-positive NB neuron shown in panel P (arrow). ac,anterior commissure; AMG, amygdala; Ch4al, cho-linergic anterolateral NB portion; Ch4id, cholin-ergic intermediate-dorsal NB portion; Ch4iv, cho-linergic intermediate-ventral NB portion; Fx,fornix; GPe, external globus pallidus; GPi, internalglobus pallidus; ic, internal capsule; Put, putamennucleus. Scale bars: 100 �m (A and C); 40 �m(inset in A); 120 �m (E); 60 �m (G); 10 �m (H–L,insets in O and P); 30 �m (M–O and inset in C);20 �m (P and inset in G).
K
more often in MCI and AD (Figure 6, B, C, E, and F) than

AD (I) c
Rac1b Up-Regulation in AD Neurons 535AJP February 2012, Vol. 180, No. 2
in NCI cases. In addition, fluorescence triple labelingshowed that a few ChAT-ir neurons displayed Alz50 butnot Rac1b in NCI (Figure 6G) or MCI (Figure 6H) cases.In contrast, ChAT-ir neurons containing Alz50 also werereactive for Rac1b in AD (Figure 6I).
Rac1b�/p75NTR-ir NB Neuron NumberIncreases in AD
Neuronal counts were performed to determine thenumber of single p75NTR versus Rac1b�/p75NTR-posi-tive NB neurons across NCI, MCI, mild/moderate AD,and severe AD cases (Figure 7). Analysis of varianceshowed that the percentage of p75NTR-positive neu-rons expressing Rac1b protein was significantly in-creased in mild and severe AD than in MCI and NCI(Figure 7; P � 0.05). However, no significant differ-ences in the percentage of p75NTR-positive neuronscontaining Rac1b were detected between mild and
NCI MCA B
G H
D E
Figure 6. A–C: Immunofluorescence merged images showing dual-labeledNCI (A), MCI (B), and severe AD (C). Note the increase in single (open arrocase. D–F: Laser confocal merged images of Alz50 (green), AT8 (red), and RNCI case (D) and triple-labeled Rac1b, Alz50, and AT8 (pink/white, arrows)of ChAT (red), Alz50 (green), and Rac1b (blue), showing few double-labeledMCI (H; white arrow), whereas single Rac1b-ir neurons (blue) and triple stain the extent of double-labeled Alz50 and ChAT neuropil threads (yellow) in30 �m (F).
severe AD or between the MCI and NCI groups (Figure
7; P � 0.05). No correlations were observed betweenthe percentage of Rac1b�/p75NTR-positive neurons inthe NB and neuropathologic or clinical variables exam-ined across groups.
AD
C
I
F
ed) and Alz50 (green) NB-positive neurons (orange/yellow) in subjects withd double (white arrows) Rac1b and Alz50-positive neurons in a severe ADlue), showing double-labeled Rac1b and Alz50 NB neuron (pale green) in arons in MCI (E) and severe AD (F) cases. G–I: Laser confocal merged imagesnd ChAT (yellow) process and neurons in NB in NCI (G; white arrow) andRac1b, Alz50, and AT8 neuron (white) were seen in AD (I). Note the increaseompared with MCI (H). Scale bars: 40 �m (A, C, D, and E); 50 �m (B, G–I);
Figure 7. Bar graph showing the mean values of the percentage of Rac1b�/p75NTR-ir Ch4 NB neurons in NCI, MCI, and mild and severe AD. Statisticalanalysis indicated a significant increase in the percentage of double Rac1b�/
NTR
I
Rac1b (rws) anac1b (bNB neuAlz50 a
ined for
p75 -ir in NB neurons in mild and severe AD compared with the NCI andMCI groups (*P � 0.05).

536 Perez et alAJP February 2012, Vol. 180, No. 2
Single-Cell Gene Expression in p75NTR andRac1b�/p75NTR-Positive NB Neurons
Custom-designed microarrays were used to comparegene expression profiles of p75NTR single or Rac1b�/75NTR double-labeled NB neurons from AD cases in com-parison to p75NTR-labeled neurons from NCI. Althoughrelatively few genes displayed alterations across the in-dividual cell types, significant differences were found inthe expression levels for mRNAs encoding the G protein� polypeptide 4 (GNB4), fetal Alzheimer antigen (FALZ),lipase A (LIPA), and caveolin 2 (CAV2) genes (Figure 8;see also Supplemental Table S1 at http://ajp.amjpathol.org). Specifically, FALZ levels were significantly reducedin AD p75NTR compared with NCI p75NTR-positive NBneurons but not within AD Rac1b�/p75NTR NB-labeledneurons (P � 0.05; Figure 8A). By contrast, transcriptlevels of GNB4, LIPA, and CAV2 were significantly down-regulated in AD Rac1b�/p75NTR-positive NB neuronscompared with NCI p75NTR-ir NB neurons (P � 0.05;Figure 8, B–D). Moreover, CAV2 mRNA was reduced inAD Rac1b�/p75NTR-positive NB neurons compared withADp75NTR-positive NB neurons (Figure 8D; P � 0.05).
Discussion
Herein, we provide biochemical, genomic, and morpho-logic evidence that Rac1b, a constitutively active Rac1splice variant, accumulates within cholinergic NB neu-rons during the progression of AD. Rac1b protein expres-sion within a select group of p75NTR-positive NB neuronsis significantly increased in moderate and severe ADcompared with MCI and NCI cases. These Rac1b/p75NTR-positive neurons displayed a phenotype compa-rable to the tau cytoskeletal abnormalities described dur-ing the progression of NFTs within the NB in AD.23 Single-
cell gene array analysis of Rac1b/p75NTR dual-labeledNB neurons showed a decrease in GNB4, LIPA, andCAV2 transcripts in AD compared with singly labeledp75NTR-containing neurons in NCI, suggesting thatRac1b activity regulates lipid metabolism and cell-cyclepathways (see below). Hence, these signaling cascadesmay be linked to Rac1b-mediated cytoskeletal and mem-brane dysfunction, resulting in NB NFT pathology andultimately neuronal death in advanced AD.
The present study showed that Rac1b was selectivelyexpressed in a subpopulation of p75NTR-positive cholin-ergic neurons within the NB in AD. In this regard, thepercentage of the double-labeled Rac1b�/p75NTR-ir NBneurons increased in mild and severe AD compared withMCI and NCI. Although the functional consequences ofthe expression of Rac1b within NB neurons remain un-known, studies suggest that it may interact with the CBFneuron survival factor NGF and its receptors. The differ-entiation, survival, and function of CBF neurons dependon the binding of NGF to its cognate receptor TrkA77,78
and the pan-neurotrophin receptor p75NTR.79–81 Thesereceptors are produced in CBF neurons and transportedto their cortical projection sites.81,82 In general, the TrkAreceptor is associated with cell survival, whereas p75NTR
shows a dichotomous role that induces cell survival orapoptosis, depending on the cellular milieu. Clinicalpathologic studies show that the ratio of TrkA to p75NTR
receptors is decreased during the progression of AD,suggesting that the NGF system shifts from cell survivalto cell death during disease onset.83 Interestingly, re-ports indicate that p75NTR-mediated apoptosis requiresthe activation of Rac1.26,84 Therefore, the increase innumber of p75NTR-positive NB neurons expressingRac1b could be indicative of its involvement in p75NTR-mediated apoptotic signaling pathways within these cho-linergic neurons in AD. Although the mechanism wherebyRac1b expression might promote neurodegeneration is
Figure 8. Linear graphs showing the mean val-ues of the relative density of fetal Alzheimerantigen (FALZ) (A), GNB4 (B), LIPA (C), andCAV2 (D) transcripts in NCI and severe AD ob-tained from single p75NTR and AD doubleRac1b�/p75NTR-positive NB neurons with theuse of single-cell gene array technology. Signif-icant down-regulation in FALZ mRNA inp75NTR-ir Ch4 neurons was detected betweenNCI and AD (A). In addition, down-regulation ofGNB4 (B), LIPA (C), and CAV2 (D) transcripts inAD Rac1b�/p75NTR neurons compared with NCIp75NTR-positive NB neurons. Down-regulationof CAV2 (D) was also found between p75NTR-irand Rac1b�/p75NTR-positive neurons in severeAD (*P � 0.05).
unclear, it has been shown that Rac1b stimulates the

Rac1b Up-Regulation in AD Neurons 537AJP February 2012, Vol. 180, No. 2
expression of cyclin D1, a protein that promotes cellcycle (G1/S) progression.85 Indeed, NB neurons displaya greater expression of cell cycle-related proteins suchas cyclin D1 and proliferating cell nuclear antigen in MCIand AD compared with NCI.86,87 The presence of theseproteins in NB as well as in cortical neurons has beeninterpreted as a lethal reentrance into the cell cycle thatpromotes an aberrant DNA replication that precedes cel-lular death.86 Significantly, cell cycle kinases such ascdc2 have been shown to contribute to tau hyperphos-phorylation,88 suggesting a role for Rac1b in reactivatingcell cycle progression that contributes to neurofibrillarydegeneration in postmitotic neurons in AD. On the onehand, the expression of Rac1b in p75NTR NB-positiveneurons may stimulate anti-apoptotic signaling that, inturn, activates proteins associated with cell survival (suchas Akt) similar to those described for Rac1.34,85 WhetherRac1b interacts with p75NTR to promote cell death orsurvival during the course of AD is an area requiringcontinued investigation. On the other hand, the conceptthat intraneuronal p75NTR is a necessary precondition forthe induction of Rac1b is not supported by our observa-tions that cortical neurons also display Rac1b but arep75NTR immunonegative in AD.
Several lines of evidence suggest that dysregulation ofRac1 GTPase activity plays a key role in cytoskeletalperturbations that lead to the formation of NFTs inAD,6,27,28 particularly within CBF neurons.26 In the pres-ent study, we observed that Rac1b colocalizes with mark-ers of NFT formation within the NB cholinergic neuronsmainly in the later stages of the disease. In addition, themorphologic appearance of Rac1b in NB neurons wassimilar to the cytoskeletal changes described with theAT8 antibody, suggesting that Rac1b participates in theinduction of cytoskeletal alterations related to NFT forma-tion in AD. Our double/triple labeling experimentsshowed that most of the Rac1b-positive neurons in theNB colocalized with either phosphorylation-dependentand/or conformational tau markers in AD and to a lesserextent in MCI. We also found Rac1b-ir cholinergic neu-rons with the NB in LBD and FTD cases, suggesting thatthe expression of Rac1b in NB cholinergic neurons is acommon feature of tauopathies. Perhaps GTPase proteineffectors engage mechanisms related to tau aggrega-tion/NFT formation and other cytoskeletal-related inclu-sions such as the FTD fused in sarcoma protein.
Another contributing factor to the putative effects ofRac1b on NFT formation may be its interactions with A�,which is thought to be a prime agent in the pathogenesisof AD,89 including NFT formation.90–92 In this regard, wehave found that A� stimulates Rac1b recruitment in neu-rons (unpublished observations, Desire 2008). Moreover,in the present study we reported that Rac1b-positiveneurons, in areas of the cortex, contain A� pathology inmild and severe AD cases. In vivo and in vitro studieshave shown that A� modifies Rac1 activity, causingchanges in filamentous actin cytoskeletal dynamics.27,28
Moreover, abnormal accumulation of filamentous actinhas been associated with tau-induced neurodegenera-tion in mouse models of tauopathy.93 Although, virtually
no extracellular or intracellular A� is present in the regioncontaining the NB,94 we cannot exclude the possibilitythat cortical A� might induce a retrograde signal, whichactivates either Rac1 or Rac1b, resulting in cytoskeletalalterations underlying NFT formation within CBF neurons.
The present single-cell gene expression profiling ex-periments showed specific Rac1b-dependent reductionsin genes relevant to cell cycle re-entry (GNB4) and lipidmetabolism (CAV2 and LIPA) in AD p75NTR-positive NBneurons compared with NCI p75NTR-ir NB neurons. Thisgene signature provides a fairly unique expression profilethat has not been observed by our group in other types ofneurons in the NB or in hippocampal tangle-bearing neu-rons in AD.41,66,75 Because we also found Rac1b-positiveneurons in the AD cortex, it will be interesting to deter-mine whether these perikarya display a similar gene pro-file to that found in the NB cholinergic neurons. GNB4 isa transducer of membrane receptor signaling related tocell cycle. CAV2 is found in membrane lipid rafts andplays a role in membrane fluidity, protein trafficking, neu-rotransmission, and receptor trafficking.95 Dysfunction inlipid rafts occurs in normal aging and AD95,96 and mayplay a key role in the synthesis of A� and its oligomer-ization in AD.97 Although the relationship between CAV2and Rac1b remains unknown, we speculate that CAV2plays a role in the regulation of Rac1b in AD. Futurestudies will be necessary to determine the role and rela-tionship between caveolin proteins and Rac in AD. Like-wise, there was a reduction in LIPA transcript levels in ADRac1b�/p75NTR compared with NCI p75NTR-positive NBneurons. This enzyme, which is found in lysosomal mem-brane compartments, breaks down lipids such as cho-lesteryl esters and triglycerides, and mutations in LIPAresult in cholesteryl ester storage diseases, such as Wol-man disease. Taken together, these data suggest thatRac1b activity involves molecular interactions with path-ways that regulate lipid metabolism/plasma membranemaintenance, which potentially contribute to the patho-physiology seen in AD.98
In summary, our study shows biochemical, genomic,and morphologic evidence that Rac1b, a constitutivelyactive Rac1 spliced variant, increases in a subpopulationof p75NTR-positive NB neurons that colocalize with patho-logic tau epitopes during the course of AD. LikewiseRac1b�/p75NTR-positive neurons display a significantdown-regulation of GNB4, LIPA, and CAV2 mRNAs insevere AD. Overall, the present data suggest that in-creased Rac1b expression acts as a modulator ortransducer of various signaling pathways, includingcell-cycle re-entry and lipid metabolism that leads toNFT formation and membrane dysfunction in a sub-group of p75NTR cholinergic NB neurons in AD. Per-haps, Rac1b inhibitors may be useful to study thefunction of this splice variant in AD.33
Acknowledgments
We are indebted to the altruism and support of the hun-dreds of nuns, priests, and brothers participating in theReligious Orders Study, without whom this study could
not be possible. We thank the members of the Rush ADC,
538 Perez et alAJP February 2012, Vol. 180, No. 2
Irina Elarova, Dr. Melissa J. Alldred, Muhammad Na-deem, Sadiyat Shoaga, Lily Yu, and Michelle Doman fortechnical assistance, and Dr. Yaping Chu for aid in laserconfocal microscopy.
References
1. Bosco EE, Mulloy JC, Zheng Y: Rac1 GTPase: a “Rac” of all trades.Cell Mol Life Sci 2009, 66:370–374
2. Samuel F, Hynds DL: RHO GTPase signaling for axon extension: isprenylation important?. Mol Neurobiol 2010, 42:133–142
3. Newey SE, Velamoor V, Govek EE, Van Aelst L: Rho GTPases, den-dritic structure, and mental retardation. J Neurobiol 2005, 64:58–74
4. Ramakers GJ: Rho proteins and the cellular mechanisms of mentalretardation. Am J Med Genet 2000, 94:367–371
5. Chan D, Citro A, Cordy JM, Shen GC, Wolozin B: Rac1 proteinrescues neurite retraction caused by G2019S leucine-rich repeatkinase 2 (LRRK2). J Biol Chem 2011, 286:16140–16149
6. Huesa G, Baltrons MA, Gómez-Ramos P, Morán A, García A, HidalgoJ, Francés S, Santpere G, Ferrer I, Galea E: Altered distribution ofRhoA in Alzheimer’s disease and AbetaPP overexpressing mice. JAlzheimers Dis 2010, 19:37–56
7. Eriksen J, Janus C: Plaques, tangles, and memory loss in mousemodels of neurodegeneration. Behav Genet 2007, 37:79–100
8. Götz J, Streffer JR, David D, Schild A, Hoerndli F, Pennanen L,Kurosinski P, Chen F: Transgenic animal models of Alzheimer’s dis-ease and related disorders: histopathology, behavior and therapy.Mol Psychiatry 2004, 9:664–683
9. Spires TL, Hyman BT: Transgenic models of Alzheimer’s disease:learning from animals. NeuroRx 2005, 2:423–437
10. Coyle JT, Price DL, DeLong MR: Alzheimer’s disease: a disorder ofcortical cholinergic innervation. Science 1983, 219:1184–1190
11. Mesulam MM, Mufson EJ, Levey AI, Wainer BH: Cholinergic innerva-tion of cortex by the basal forebrain: cytochemistry and corticalconnections of the septal area, diagonal band nuclei, nucleus basalis(substantia innominata), and hypothalamus in the rhesus monkey.J Comp Neurol 1983, 214:170–197
12. Whitehouse PJ, Price DL, Clark AW, Coyle JT, DeLong MR: Alzheimerdisease: evidence for selective loss of cholinergic neurons in thenucleus basalis. Ann Neurol 1981, 10:122–126
13. Whitehouse PJ, Struble RG, Hedreen JC, Clark AW, Price DL: Alzhei-mer’s disease and related dementias: selective involvement of spe-cific neuronal systems. CRC Crit Rev Clin Neurobiol 1985, 1:319–339
14. Davies P: Neurotransmitter-related enzymes in senile dementia of theAlzheimer’s type. Brain Res 1979, 171:319–327
15. Davies P, Maloney AJ: Selective loss of central cholinergic neurons inAlzheimer’s disease (letter to the editor). Lancet 1976, 2:1403
16. Mufson EJ, Bothwell M, Kordower JH: Loss of nerve growth factorreceptor-containing neurons in Alzheimer’s disease: a quantitativeanalysis across subregions of the basal forebrain. Exp Neurol 1989,105:221–232
17. Richter JA, Perry EK, Tomlinson B: Acetylcholine and choline levels inpost-mortem human brain tissue: preliminary observations in Alzhei-mer’s disease. Life Sci 1980, 26:1683–1689
18. DeKosky ST, Ikonomovic MD, Styren SD, Beckett L, Wisniewski S,Bennett DA, Cochran EJ, Kordower JH, Mufson EJ: Upregulation ofcholine acetyltransferase activity in hippocampus and frontal cortexof elderly subjects with mild cognitive impairment. Ann Neurol 2002,51:145–155
19. Perry EK, Tomlinson BE, Blessed G, Bergmann K, Gibson PH, PerryRH: Correlation of cholinergic abnormalities with senile plaques andmental test scores in senile dementia. Br Med J 1978, 2:1457–1459
20. Wilcock GK, Esiri MM, Bowen DM, Smith CC: Alzheimer’s disease.Correlation of cortical choline acetyltransferase activity with the se-verity of dementia and histological abnormalities. J Neurol Sci 1982,57:407–417
21. DeKosky ST, Harbaugh RE, Schmitt FA, Bakay RA, Chui HC, Knop-man DS, Reeder TM, Shetter AG, Senter HJ, Markesbery WR: Corticalbiopsy in Alzheimer’s disease: diagnostic accuracy and neurochem-ical, neuropathological, and cognitive correlations. Intraventricular
Bethanecol Study Group. Ann Neurol 1992, 32:625–63222. Rasool CG, Svendsen CN, Selkoe DJ: Neurofibrillary degeneration ofcholinergic and noncholinergic neurons of the basal forebrain inAlzheimer’s disease. Ann Neurol 1986, 20:482–488
23. Sassin I, Schultz C, Thal DR, Rüb U, Arai K, Braak E, Braak H:Evolution of Alzheimer’s disease-related cytoskeletal changes in thebasal nucleus of Meynert. Acta Neuropathol 2000, 100:259–269
24. Vana LC, Kanaan NM, Ugwu IC, Wuu J, Mufson EJ, Binder, LI:Progression of tau pathology in cholinergic basal forebrain neurons inMCI and AD. Am J Pathol 2011, 179:2533–2550
25. Mesulam M, Shaw P, Mash D, Weintraub S: Cholinergic nucleusbasalis tauopathy emerges early in the aging-MCI-AD continuum.Ann Neurol 2004, 55:815–828
26. Harrington AW, Kim JY, Yoon SO: Activation of Rac GTPase by p75 isnecessary for c-jun N-terminal kinase-mediated apoptosis. J Neuro-sci 2002, 22:156–166
27. Mendoza-Naranjo A, Gonzalez-Billault C, Maccioni RB: Abeta1-42stimulates actin polymerization in hippocampal neurons throughRac1 and Cdc42 Rho GTPases. J Cell Sci 2007, 120:279–288
28. Petratos S, Li QX, George AJ, Hou X, Kerr ML, Unabia SE, Hatzini-siriou I, Maksel D, Aguilar MI, Small DH: The beta-amyloid protein ofAlzheimer’s disease increases neuronal CRMP-2 phosphorylation bya Rho-GTP mechanism. Brain 2008, 131:90–108
29. Jordan P, Brazåo R, Boavida MG, Gespach C, Chastre E: Cloning ofa novel human Rac1b splice variant with increased expression incolorectal tumors. Oncogene 1999, 18:6835–6839
30. Schnelzer A, Prechtel D, Knaus U, Dehne K, Gerhard M, Graeff H,Harbeck N, Schmitt M, Lengyel E: Rac1 in human breast cancer:overexpression, mutation analysis, and characterization of a newisoform. Rac1b. Oncogene 2000, 19:3013–3020
31. Fiegen D, Haeusler LC, Blumenstein L, Herbrand U, Dvorsky R, VetterIR, Ahmadian MR: Alternative splicing of Rac1 generates Rac1b, aself-activating GTPase. J Biol Chem 2004, 279:4743–4749
32. Haeusler LC, Hemsath L, Fiegen D, Blumenstein L, Herbrand U,Stege P, Dvorsky R, Ahmadian MR: Purification and biochemicalproperties of Rac1, 2, 3 and the splice variant Rac1b. MethodsEnzymol 2006, 406:1–11
33. Beausoleil E, Chauvignac C, Taverne T, Lacombe S, Pognante L,Leblond B, Pallares D, Oliveira CD, Bachelot F, Carton R, Peillon H,Coutadeur S, Picard V, Lambeng N, Désiré L, Schweighoffer F:Structure-activity relationship of isoform selective inhibitors ofRac1/1b GTPase nucleotide binding. Bioorg Med Chem Lett 2009,19:5594–5598
34. Singh A, Karnoub AE, Palmby TR, Lengyel E, Sondek J, Der CJ:Rac1b, a tumor associated, constitutively active Rac1 splice variant,promotes cellular transformation. Oncogene 2004, 23:9369–9380
35. Schweighoffer F, Ait-Ikhlef A, Resink AL, Brinkman B, Melle-Mi-lovanovic D, Laurent-Puig P, Kearsey J, Bracco L: Qualitative geneprofiling: a novel tool in genomics and in pharmacogenomics thatdeciphers messenger RNA isoforms diversity. Pharmacogenomics2000, 1:187–197
36. Lambeng N, Coutadeur S, Peillon H, Loiseau N, Bachelot F, DeOliveira C, Carton R, Leblond B, Beausoleil E, Chauvignac C, TaverneT, Schweighoffer F, Desire L: Development of a screening platform forthe identification of new Rac1/Rac1b inhibitors active in the preven-tion of amyloid-beta production. International Conference of Alzhei-mer’s Disease 2008, Abstract 256
37. Mufson EJ, Perez SE, Nadeem M, Coutadeur S, Peillon H, Desire L:Rac1b GTPase splicing variant colocalizes with select p75NTR cho-linergic basal forebrain neurons during neurofibrillary tangle forma-tion in mild cognitive impairment and Alzheimer’s disease. Society forNeuroscience 2008, Abstract 4401
38. Sakurada K, Ohshima-Sakurada M, Palmer TD, Gage FH: Nurr1, anorphan nuclear receptor, is a transcriptional activator of endogenoustyrosine hydroxylase in neural progenitor cells derived from the adultbrain. Development 1999, 126:4017–4026
39. Ginsberg SD, Che S: Combined histochemical staining. RNA ampli-fication, regional, and single cell cDNA analysis within the hippocam-pus. Lab Invest 2004, 84:952–962
40. Counts SE, Chen EY, Che S, Ikonomovic MD, Wuu J, Ginsberg SD,Dekosky ST, Mufson EJ: Galanin fiber hypertrophy within the cholin-ergic nucleus basalis during the progression of Alzheimer’s disease.Dement Geriatr Cogn Disord 2006, 21:205–214
41. Ginsberg SD, Che S, Wuu J, Counts SE, Mufson EJ: Down regulation
of trk but not p75NTR gene expression in single cholinergic basal
Rac1b Up-Regulation in AD Neurons 539AJP February 2012, Vol. 180, No. 2
forebrain neurons mark the progression of Alzheimer’s disease.J Neurochem 2006, 97:475–487
42. Counts SE, He B, Che S, Ikonomovic MD, DeKosky ST, Ginsberg SD,Mufson EJ: Alpha7 nicotinic receptor up-regulation in cholinergicbasal forebrain neurons in Alzheimer disease. Arch Neurol 2007,64:1771–1776
43. Gilmor ML, Erickson JD, Varoqui H, Hersh LB, Bennett DA, CochranEJ, Mufson EJ, Levey AI: Preservation of nucleus basalis neuronscontaining choline acetyltransferase and the vesicular acetylcholinetransporter in the elderly with mild cognitive impairment and earlyAlzheimer’s disease. J Comp Neurol 1999, 411:693–704
44. Mufson EJ, Chen EY, Cochran EJ, Beckett LA, Bennett DA, KordowerJH: Entorhinal cortex beta-amyloid load in individuals with mild cog-nitive impairment. Exp Neurol 1999, 158:469–490
45. Bennett DA, Wilson RS, Schneider JA, Evans DA, Beckett LA, Aggar-wal NT, Barnes LL, Fox JH, Bach J: Natural history of mild cognitiveimpairment in older persons. Neurology 2002, 59:198–205
46. Mitchell TW, Mufson EJ, Schneider JA, Cochran EJ, Nissanov J, HanLY, Bienias JL, Lee VM, Trojanowski JQ, Bennett DA, Arnold SE:Parahippocampal tau pathology in healthy aging, mild cognitive im-pairment, and early Alzheimer’s disease. Ann Neurol 2002, 51:182–189
47. McKhann G, Drachman D, Folstein M, Katzman R, Price D, StadlanEM: Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health andHuman Services Task Force on Alzheimer’s Disease. Neurology1984, 34:939–944
48. Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM,Vogel FS, Hughes JP, van Belle G, Berg L: The Consortium to Estab-lish a Registry for Alzheimer’s Disease (CERAD). Part II: standardiza-tion of the neuropathologic assessment of Alzheimer’s disease. Neu-rology 1991, 41:479–486
49. Braak H, Braak E: Neuropathological stageing of Alzheimer-relatedchanges. Acta Neuropathol 1991, 82:239–259
50. Morris JC, Weintraub S, Chui HC, Cummings J, DeCarli C, Ferris S,Foster NL, Galasko D, Graff-Radford N, Peskind ER, Beekly D, RamosEM, Kukull WA: The Uniform Data Set (UDS): clinical and cognitivevariables and descriptive data from Alzheimer Disease Centers. Alz-heimer Dis Assoc Disord 2006, 20:210–216
51. Weintraub S, Salmon D, Mercaldo N, Ferris S, Graff-Radford NR, ChuiH, Cummings J, DeCarli C, Foster NL, Galasko D, Peskind E, DietrichW, Beekly DL, Kukull WA, Morris JC: The Alzheimer’s Disease Cen-ters’ Uniform Data Set (UDS): the neuropsychologic test battery.Alzheimer Dis Assoc Disord 2009, 23:91–101
52. Cairns NJ, Bigio EH, Mackenzie IR, Neumann M, Lee VM, HatanpaaKJ, White CL III, Schneider JA, Grinberg LT, Halliday G, DuyckaertsC, Lowe JS, Holm IE, Tolnay M, Okamoto K, Yokoo H, Murayama S,Woulfe J, Munoz DG, Dickson DW, Ince PG, Trojanowski JQ, MannDM: Neuropathologic diagnostic and nosologic criteria for frontotem-poral lobar degeneration: consensus of the Consortium for Fronto-temporal Lobar Degeneration. Acta Neuropathol 2007, 114:5–22
53. Che S, Ginsberg SD: Amplification of RNA transcripts using terminalcontinuation. Lab Invest 2004, 84:131–137
54. Mufson EJ, Bothwell M, Hersh LB, Kordower JH: Nerve growth factorreceptor immunoreactive profiles in the normal, aged human basalforebrain: colocalization with cholinergic neurons. J Comp Neurol1989, 285:196–217
55. Mufson EJ, Ma SY, Dills J, Cochran EJ, Leurgans S, Wuu J, BennettDA, Jaffar S, Gilmor ML, Levey AI, Kordower JH: Loss of basalforebrain P75(NTR) immunoreactivity in subjects with mild cognitiveimpairment and Alzheimer’s disease. J Comp Neurol 2002, 443:136–153
56. Mesulam MM, Mufson EJ, Wainer BH, Levey AI: Central cholinergicpathways in the rat: an overview based on an alternative nomencla-ture (Ch1-Ch6). Neuroscience 1983, 10:1185–1201
57. Kordower JH, Gash DM, Bothwell M, Hersh L, Mufson EJ: Nervegrowth factor receptor and cholineacetyltransferase remain colocal-ized in the nucleus basalis (Ch4) of Alzheimer’s patients. NeurobiolAging 1989, 10:67–74
58. Mesulam MM, Geula C, Bothwell MA, Hersh LB: Human reticularformation: cholinergic neurons of the pedunculopontine and lat-erodorsal tegmentalnuclei and some cytochemical comparisons to
forebrain cholinergic neurons. J Com Neurol 1989, 283:611–63359. Carmel G, Mager EM, Binder LI, Kuret J: The structural basis ofmonoclonal antibody Alz50’s selectivity for Alzheimer’s disease pa-thology. J Biol Chem 1996, 271:32789–32795
60. Jicha GA, Berenfeld B, Davies P: Sequence requirements for forma-tion of conformational variants of tau similar to those found in Alzhei-mer’s disease. J Neurosci Res 1999, 55:713–723
61. Jicha GA, Bowser R, Kazam IG, Davies P: Alz-50 and MC-1, a newmonoclonal antibody raised to paired helical filaments, recognizeconformational epitopes on recombinant tau. J Neurosci Res 1997,48:128–132
62. Mercken M, Vandermeeren M, Lübke U, Six J, Boons J, Van deVoorde A, Martin JJ, Gheuens J: Monoclonal antibodies with selectivespecificity for Alzheimer Tau are directed against phosphatase-sen-sitive epitopes. Acta Neuropathol 1992, 84:265–272
63. Goedert M, Jakes R, Crowther RA, Cohen P, Vanmechelen E, Van-dermeeren M, Cras P: Epitope mapping of monoclonal antibodies tothe paired helical filaments of Alzheimer’s disease: identification ofphosphorylation sites in tau protein. Biochem J 1994, 301:871–877
64. Goedert M, Jakes R, Vanmechelen E: Monoclonal antibody AT8recognises tau protein phosphorylated at both serine 202 and thre-onine 205. Neurosci Lett 1995, 189:167–169
65. Perez SE, He B, Muhammad N, Oh K-J, Fahnestock M, Ikonomovic M,Mufson EJ: Cholinotrophic basal forebrain system alterations in3xTg-AD transgenic mice. Neurobiol Dis 2010, 41:338–352
66. Ginsberg SD, Hemby SE, Lee VM, Eberwine JH, Trojanowski JQ:Expression profile of transcripts in Alzheimer’s disease tangle-bear-ing CA1 neurons. Ann Neurol 2000, 48:77–87
67. Alldred MJ, Che S, Ginsberg SD: Terminal continuation (TC) RNAamplification without second strand synthesis. J Neurosci Methods2009, 177381–177385
68. Ginsberg SD: RNA amplification strategies for small sample popula-tions. Methods 2005, 37:229–237
69. Ginsberg SD, Elarova I, Ruben M, Tan F, Counts SE, Eberwine JH,Trojanowski JQ, Hemby SE, Mufson EJ, Che S: Single-cell geneexpression analysis: implications for neurodegenerative and neuro-psychiatric disorders. Neurochem Res 2004, 29:1053–1064
70. Eberwine J, Kacharmina JE, Andrews C, Miyashiro K, McIntosh T,Becker K, Barrett T, Hinkle D, Dent G, Marciano P: mRna expressionanalysis of tissue sections and single cells. J Neurosci 2001, 21:8310–8314
71. Hemby SE, Ginsberg SD, Brunk B, Arnold SE, Trojanowski JQ, Eber-wine JH: Gene expression profile for schizophrenia: discrete neurontranscription patterns in the entorhinal cortex. Arch Gen Psychiatry2002, 59:631–640
72. Hemby SE, Trojanowski JQ, Ginsberg SD: Neuron-specific age-re-lated decreases in dopamine receptor subtype mRNAs. J CompNeurol 2003, 456:176–183
73. Ginsberg SD, Alldred MJ, Counts SE, Cataldo AM, Neve RL, Jiang Y,Wuu J, Chao MV, Mufson EJ, Nixon RA, Che S: Microarray analysis ofhippocampal CA1 neurons implicates early endosomal dysfunctionduring Alzheimer’s disease progression. Biol Psychiatry 2010, 68:885–893
74. Ginsberg SD, Che S, Hashim A, Zavadil J, Cancro R, Lee SH, PetkovaE, Sershen HW, Volavka J: Differential regulation of catechol-O-meth-yltransferase expression in a mouse model of aggression. BrainStruct Funct 2011, 216:347–356
75. Ginsberg SD, Mufson EJ, Alldred MJ, Counts SE, Wuu J, Nixon RA,Che S: Upregulation of select rab GTPases in cholinergic basalforebrain neurons in mild cognitive impairment and Alzheimer’s dis-ease. J Chem Neuroanat 2011, 42:102–110
76. Reiner A, Yekutieli D, Benjamini Y: Identifying differentially expressedgenes using false discovery rate controlling procedures. Bioinformat-ics 2003, 19:368–375
77. Huang EJ, Reichardt LF: Neurotrophins: roles in neuronal develop-ment and function. Annu Rev Neurosci 2001, 24:677–736
78. Kaplan D, Zirrgiebel U, Atwal J: Center stage for NGF in peripheral(but not central) sensory neuron outgrowth. Neuron 2000, 25:253–254
79. Barrett GL, Bartlett PF: The p75 nerve growth factor receptor medi-ates survival or death depending on the stage of sensory neurondevelopment. Proc Natl Acad Sci U S A 1994, 91:6501–6505
80. Frade JM, Barde YA: Nerve growth factor: two receptors, multiple
functions. Bioessays 1998, 20:137–145
540 Perez et alAJP February 2012, Vol. 180, No. 2
81. Lad SP, Neet KE, Mufson EJ: Nerve growth factor: structure, functionand therapeutic implications for Alzheimer’s disease. Curr Drug Tar-get CNS Neurol Disord 2003, 2:315–334
82. Sobreviela T, Clary DO, Reichardt LF, Brandabur MM, Kordower JH,Mufson EJ: TrkA-immunoreactive profiles in the central nervoussystem: colocalization with neurons containing p75 nerve growthfactor receptor, choline acetyltransferase, and serotonin. J CompNeurol 1994, 350:587–611
83. Mufson EJ, Counts SE, Perez SE, Ginsberg SD: Cholinergic systemduring the progression of Alzheimer’s disease: therapeutic implica-tions. Expert Rev Neurother 2008, 8:1703–1718
84. Yoon SO, Casaccia-Bonnefil P, Carter B, Chao MV: Competitivesignaling between TrkA and p75 nerve growth factor receptors de-termines cell survival. J Neurosci 1998, 18:3273–3281
85. Matos P, Jordan P: Expression of Rac1b stimulates NF-kappaB-mediated cell survival and G1/S progression. Exp Cell Res 2005,305:292–299
86. Yang Y, Geldmacher DS, Herrup K: DNA replication precedes neu-ronal cell death in Alzheimer’s disease. J Neurosci 2001, 21:2661–2668
87. Yang Y, Mufson EJ, Herrup K: Neuronal cell death is preceded by cellcycle events at all stages of Alzheimer’s disease. J Neurosci 2003,23:2557–2563
88. Vincent I, Jicha G, Rosado M, Dickson DW: Aberrant expression ofmitotic cdc2/cyclin B1 kinase in degenerating neurons of Alzheimer’sdisease brain. J Neurosci 1997, 17:3588–3598
89. Selkoe DJ: Alzheimer’s disease: genes, proteins, and therapy.Physiol Rev 2001, 81:741–766
90. Götz J, Chen F, van Dorpe J, Nitsch RM: Formation of neurofibrillarytangles in P301l tau transgenic mice induced by Abeta 42 fibrils.
Science 2001, 293:1491–149591. Oddo S, Caccamo A, Kitazawa M, Tseng BP, LaFerla FM: Amyloiddeposition precedes tangle formation in a triple transgenic model ofAlzheimer’s disease. Neurobiol Aging 2003, 24:1063–1070
92. Bolmont T, Clavaguera F, Meyer-Luehmann M, Herzig MC, Radde R,Staufenbiel M, Lewis J, Hutton M, Tolnay M, Jucker M: Induction oftau pathology by intracerebral infusion of amyloid-beta-containingbrain extract and by amyloid-beta deposition in APP x Tau transgenicmice. Am J Pathol 2007, 171:2012–2020
93. Fulga TA, Elson-Schwab I, Khurana V, Steinhilb ML, Spires TL, Hy-man BT, Feany MB: Abnormal bundling and accumulation of F-actinmediates tau-induced neuronal degeneration in vivo. Nat Cell Biol2007, 9:139–148
94. Jacobs RW, Duong T, Scheibel AB: Immunohistochemical analysis ofthe basal forebrain in Alzheimer’s disease. Mol Chem Neuropathol1992, 17:1–20
95. Ohno-Iwashita Y, Shimada Y, Hayashi M, Inomata M: Plasma mem-brane microdomains in aging and disease. Geriatr Gerontol Int 2010,10:S41–S52
96. Williamson R, Sutherland C: Neuronal membranes are key to thepathogenesis of Alzheimer’s disease: the role of both raft and non-raftmembrane domains. Curr Alzheimer Res 2011, 8:213–221
97. Rushworth JV, Hooper NM: Lipid rafts: linking Alzheimer’s amyloid-�production, aggregation, and toxicity at neuronal membranes. Int JAlzheimers Dis 2010, 2011:603052
98. Oh D, Han S, Seo J, Lee JR, Choi J, Groffen J, Kim K, Cho YS, ChoiHS, Shin H, Woo J, Won H, Park SK, Kim SY, Jo J, Whitcomb DJ, ChoK, Kim H, Bae YC, Heisterkamp N, Choi SY, Kim E: Regulation ofsynaptic Rac1 activity, long-term potentiation maintenance, andlearning and memory by BCR and ABR Rac GTPase-activating pro-
teins. J Neurosci 2010, 30:14134–14144



















