
Autoregulation and Heterogeneity in Expression of Human Cripto-1

Vol. 20, No. 7, 2007 / 769
MPMI Vol. 20, No. 7, 2007, pp. 769–780. doi:10.1094 / MPMI -20-7-0769. © 2007 The American Phytopathological Society
Promoters of Orthologous Glycine max and Lotus japonicus Nodulation Autoregulation Genes Interchangeably Drive Phloem-Specific Expression in Transgenic Plants
Sureeporn Nontachaiyapoom,1,2 Paul T. Scott,1 Artem E. Men,1,3 Mark Kinkema,1 Peer M. Schenk,2 and Peter M. Gresshoff1,2 1Australian Research Council Centre of Excellence for Integrative Legume Research, University of Queensland, St. Lucia, QLD 4072, Australia; 2School of Integrative Biology, University of Queensland, St. Lucia, QLD 4072, Australia; 3Australian Genome Research Facility, University of Queensland, St. Lucia, QLD 4072, Australia
Submitted 21 December 2006. Accepted 22 February 2007.
The nodule autoregulation receptor kinase (GmNARK) of soybean (Glycine max) is essential for the systemic auto-regulation of nodulation. Based on quantitative reverse-transcriptase polymerase chain reaction, GmNARK is ex-pressed to varying levels throughout the plant; the tran-script was detected at high levels in mature leaves and roots but to a lesser extent in young leaves, shoot tips, and nodules. The transcript level was not significantly affected by Bradyrhizobium japonicum during the first week follow-ing inoculation. In addition, the activities of the promoters of GmNARK and Lotus japonicus HAR1, driving a β-glu-curonidase (GUSPlus) reporter gene, were examined in stably transformed L. japonicus and transgenic hairy roots of soybean. Histochemical GUS activity in L. japonicus plants carrying either a 1.7-kb GmNARKpr::GUS or 2.0-kb LjHAR1pr::GUS construct was clearly localized to living cells within vascular bundles, especially phloem cells in leaves, stems, roots, and nodules. Phloem-specific expres-sion also was detected in soybean hairy roots carrying these constructs. Our study suggests that regulatory ele-ments required for the transcription of these orthologous genes are conserved. Moreover, rapid amplification of 5′ cDNA ends (5′ rapid amplification of cDNA ends) revealed two major transcripts of GmNARK potentially originating from two TATA boxes. Further analysis of the GmNARK promoter has confirmed that these two TATA boxes are functional. Deletion analysis also located a region control-ling phloem-specific expression to a DNA sequence between 908 bp and 1.7 kb upstream of the translation start site of GmNARK.
Additional keywords: NRR, promoter deletion, supernodulation.
Even though nitrogen is the most abundant gas in the Earth’s atmosphere, most living systems cannot directly utilize it. Leguminous plants overcome this limitation through symbiotic association with nitrogen-fixing bacteria in soil, collectively known as rhizobia. Successful symbiotic association results in the formation of highly specialized organs called root nodules, which provide an environment suitable for bacterial colonization
and biological nitrogen fixation. However, nodule initiation and proliferation, in much the same manner as the proliferation of other meristems in plants, are controlled in order to attain equi-librium between cell proliferation and differentiation (Beveridge et al. 2007).
Nodule proliferation is controlled primarily by autoregula-tion of nodulation (AON) (Carroll et al. 1985a and b; Delves et al. 1986; Pierce and Bauer 1983). Genes responsible for this process have been isolated by positional cloning from soybean (GmNARK), Lotus japonicus (LjHAR1) and Medicago trunca-tula (MtSUNN), and by homology from pea (PsSYM29) (Krusell et al. 2002; Nishimura et al. 2002; Searle et al. 2003; Schnabel et al. 2005). Mutants defective in these genes display supernodulation or hypernodulation phenotypes in the pres-ence of rhizobia. Grafting between supernodulating and wild-type plants showed that long-distance signaling was involved in AON (Delves et al. 1986; Jiang and Gresshoff 2002; Krusell et al. 2002; Men et al. 2002; Penmetsa et al. 2003) and the leaf genotype controlled proliferation of nodule primordia (Delves et al. 1992; Francisco and Harper 1995).
The expression of genes controlling AON has been described in general terms. Nishimura and associates (2002) revealed by Northern hybridization that the LjHAR1 transcript was present in leaves, shoots, roots, flowers, and, at a much lower level, nodules; they did not detect LjHAR1 transcript in shoot apices. Concurrently, Krusell and associates (2002) demonstrated, again by Northern hybridization, that the PsSYM29 transcript was present in roots, leaves, and nodules of pea. Further, by Northern hybridization and quantitative reverse-transcriptase polymerase chain reaction (QRT-PCR), Krusell and associates (2002) reported that the LjHAR1 transcript was detected in roots, nodules, and leaves of L. japonicus. Neither rhizobia nor KNO3 altered LjHAR1 transcript level. Schnabel and associates (2005) revealed that MtSUNN was expressed in shoots, flowers, and roots, and that expression of MtSUNN mRNA was reduced in sunn-1 roots. Searle and associates (2003) investigated GmNARK expression in leaves and shoot apical meristems (SAMs) by QRT-PCR and reported that the GmNARK transcript level in leaves was 20 times higher than that in the SAMs. A reduction in GmNARK transcript level in leaves of one of the two nts mutants also was observed. The results of Schnabel and associates (2005) and Searle and associates (2003) suggest that different non-sense alleles differentially affect mRNA sta-bility. Arai and associates (2005) again studied the expression
Corresponding author: P. M. Gresshoff; Fax: +61 7 3365-3556; E-mail: [email protected]
770 / Molecular Plant-Microbe Interactions
of GmNARK using nonquantitative RT-PCR. They reported that GmNARK was transcribed constitutively in soybean cotyledons, leaves, nodules, and leaf buds, regardless of the presence of the rhizobial symbiont, stage of growth, or GmNARK genotype.
In the present study, we used QRT-PCR to examine the ex-pression of GmNARK in different tissues of soybean and analyze the possible effect of Bradyrhizobium inoculation on GmNARK transcript levels. Localization of GmNARK expression in a tis-sue-specific manner was confirmed and expanded upon by his-tochemical analysis of GmNARKpr::GUS reporter gene expres-sion in transgenic plants. In addition, the activity of the GmNARK promoter also was compared with the L. japonicus LjHAR1 promoter in both stably transformed L. japonicus and transgenic soybean hairy roots. The results demonstrate clearly that both promoters function in a similar fashion in either homologous or heterologous promoter–plant configurations. The phloem-specific expression supports a possible role for GmNARK/LjHAR1 in the regulation of a translocatable bio-logically active signal or signals. Additionally, 5′ rapid ampli-fication of cDNA ends (RACE) revealed two major transcripts of GmNARK. Further analysis of GmNARK promoter suggested the presence of two functional TATA boxes and a region appar-ently controlling phloem-specific expression of GmNARK.
RESULTS
GmNARK shows tissue-specific expression. Primers for QRT-PCR were designed to anneal to the 3′ un-
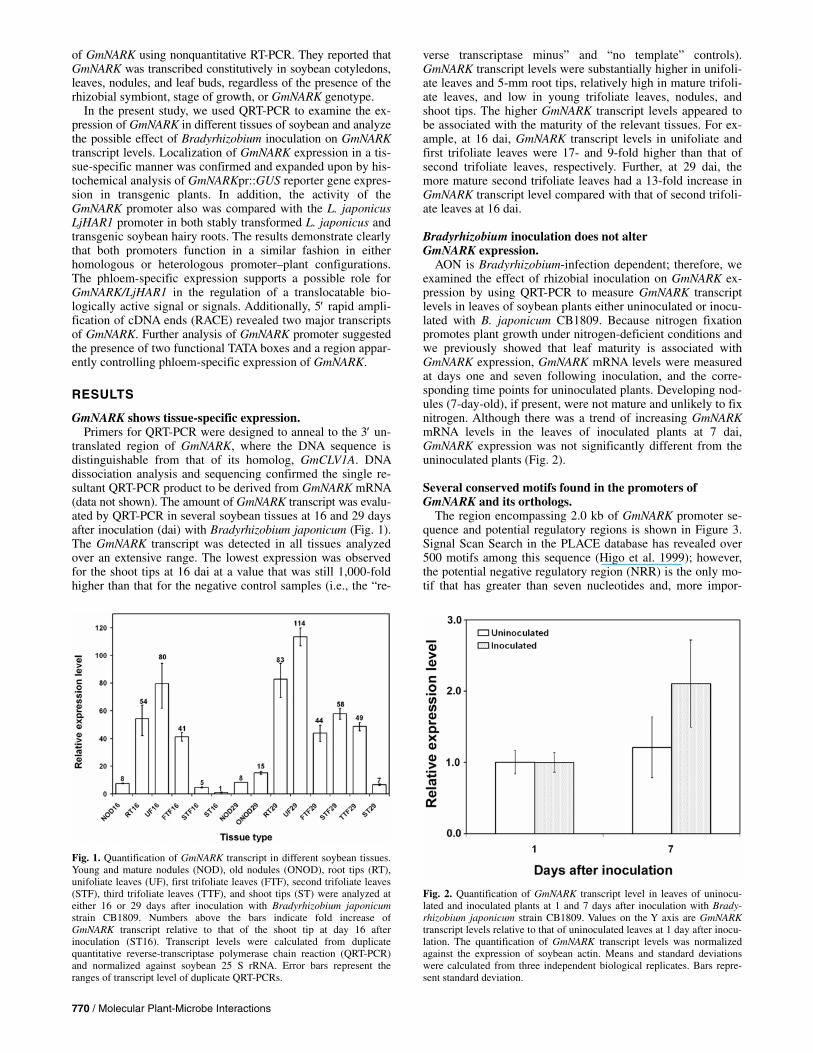
translated region of GmNARK, where the DNA sequence is distinguishable from that of its homolog, GmCLV1A. DNA dissociation analysis and sequencing confirmed the single re-sultant QRT-PCR product to be derived from GmNARK mRNA (data not shown). The amount of GmNARK transcript was evalu-ated by QRT-PCR in several soybean tissues at 16 and 29 days after inoculation (dai) with Bradyrhizobium japonicum (Fig. 1). The GmNARK transcript was detected in all tissues analyzed over an extensive range. The lowest expression was observed for the shoot tips at 16 dai at a value that was still 1,000-fold higher than that for the negative control samples (i.e., the “re-
verse transcriptase minus” and “no template” controls). GmNARK transcript levels were substantially higher in unifoli-ate leaves and 5-mm root tips, relatively high in mature trifoli-ate leaves, and low in young trifoliate leaves, nodules, and shoot tips. The higher GmNARK transcript levels appeared to be associated with the maturity of the relevant tissues. For ex-ample, at 16 dai, GmNARK transcript levels in unifoliate and first trifoliate leaves were 17- and 9-fold higher than that of second trifoliate leaves, respectively. Further, at 29 dai, the more mature second trifoliate leaves had a 13-fold increase in GmNARK transcript level compared with that of second trifoli-ate leaves at 16 dai.
Bradyrhizobium inoculation does not alter GmNARK expression.
AON is Bradyrhizobium-infection dependent; therefore, we examined the effect of rhizobial inoculation on GmNARK ex-pression by using QRT-PCR to measure GmNARK transcript levels in leaves of soybean plants either uninoculated or inocu-lated with B. japonicum CB1809. Because nitrogen fixation promotes plant growth under nitrogen-deficient conditions and we previously showed that leaf maturity is associated with GmNARK expression, GmNARK mRNA levels were measured at days one and seven following inoculation, and the corre-sponding time points for uninoculated plants. Developing nod-ules (7-day-old), if present, were not mature and unlikely to fix nitrogen. Although there was a trend of increasing GmNARK mRNA levels in the leaves of inoculated plants at 7 dai, GmNARK expression was not significantly different from the uninoculated plants (Fig. 2).
Several conserved motifs found in the promoters of GmNARK and its orthologs.
The region encompassing 2.0 kb of GmNARK promoter se-quence and potential regulatory regions is shown in Figure 3. Signal Scan Search in the PLACE database has revealed over 500 motifs among this sequence (Higo et al. 1999); however, the potential negative regulatory region (NRR) is the only mo-tif that has greater than seven nucleotides and, more impor-
Fig. 1. Quantification of GmNARK transcript in different soybean tissues.Young and mature nodules (NOD), old nodules (ONOD), root tips (RT),unifoliate leaves (UF), first trifoliate leaves (FTF), second trifoliate leaves(STF), third trifoliate leaves (TTF), and shoot tips (ST) were analyzed ateither 16 or 29 days after inoculation with Bradyrhizobium japonicumstrain CB1809. Numbers above the bars indicate fold increase ofGmNARK transcript relative to that of the shoot tip at day 16 afterinoculation (ST16). Transcript levels were calculated from duplicatequantitative reverse-transcriptase polymerase chain reaction (QRT-PCR) and normalized against soybean 25 S rRNA. Error bars represent theranges of transcript level of duplicate QRT-PCRs.
Fig. 2. Quantification of GmNARK transcript level in leaves of uninocu-lated and inoculated plants at 1 and 7 days after inoculation with Brady-rhizobium japonicum strain CB1809. Values on the Y axis are GmNARKtranscript levels relative to that of uninoculated leaves at 1 day after inocu-lation. The quantification of GmNARK transcript levels was normalized against the expression of soybean actin. Means and standard deviationswere calculated from three independent biological replicates. Bars repre-sent standard deviation.

Vol. 20, No. 7, 2007 / 771
tantly, also could be found in the promoters of LjHAR1 and MtSUNN. This NRR previously was identified in the promoter of the extensin gene from Brassica napus and has been re-ported to be responsible for phloem-specific expression of this gene (Elliott and Shirsat 1998). A comparison of the putative
GmNARK and LjHAR1 promoters showed that several highly conserved motifs are present in both of them. Three motifs with the highest homology are i) the 14-bp motif showing 100% identity, found at –107 and –104 bp (with respect to the translation start site [TSS]) in the GmNARK and LjHAR1 pro-
Fig. 3. Sequence of GmNARK promoter. Nucleotides are numbered relative to the translation start site (TSS, +1). Three highly conserved motifs, the 14-, 36-, and 37-bp motifs, identified by Multiple Em for Motif Elicitation are indicated as white text on pink background and the potential negative regulatory regionidentified by Signal Scan Search is indicated as white text on red background. Two putative TATA boxes, TATA1 and TATA2, are written on gray and green backgrounds, respectively. The transcription start sites determined by 5′ rapid amplification of cDNA ends are indicated as white text on either gray or green background according to the background color of their proximate TATA box. The termination points of the 2.0-kb GmNARK promoter, the 1.7-kb GmNARKpromoter, and five other truncations used in the promoter deletion analysis are indicated by bent arrows. Forward primer used in the polymerase chain reaction amplification of each truncation is underlined. The T2mT1::GUS construct had the same termination point as that of 123-bp GmNARKpr::GUS, but nucleotide substitution were made in its TATA1 element (TATATAA → TGTCTGC).

772 / Molecular Plant-Microbe Interactions
moters, respectively; ii) the 36-bp motif showing 89% sequence identity, found at –1,009 and –517 bp in the GmNARK and LjHAR1 promoters, respectively; and iii) the 37-bp motif showing 89% sequence identity, found at –1,679 and –1,504 bp in the GmNARK and LjHAR1 promoters, respectively. Inter-estingly, the second motif also was found at –494 bp in the MtSUNN promoter with sequence identity as high as 92% of that of GmNARK and 97% of that of LjHAR1 (Fig. 4). Poten-tial NRRs in the LjHAR1 and MtSUNN promoters were found in these 36-bp motifs.
GmNARK has two transcription initiation regions. In silico analysis of the GmNARK promoter sequence adja-
cent to the TSS has revealed several potential TATA boxes. Two of them, TATA1 and TATA2 (Fig. 3), conform to consen-sus the TATA box sequence of leghaemoglobin and nodulin genes (TATATAA) (Joshi 1987) and are located at 24 and 25 nucleotides upstream of the predicted transcription start sites (Neural network promoter prediction website). The transcrip-tion start sites of GmNARK were determined experimentally twice by 5′ RACE. Both experiments showed two discrete bands of amplification products on ethidium bromide-stained agarose gels, whereas amplification of the RT-minus controls (i.e., RNA samples that were treated similarly but without the addition of reverse-transcriptase enzyme) gave no detectable band (data not shown). Sequencing of RACE clones revealed multiple transcription start sites for GmNARK (Fig. 5). The first RACE experiment (RACE1) failed to identify the precise transcription start site at position –91 or –90 because dCTP was used in the tailing of cDNA. The second RACE experi-ment (RACE2) using dGTP determined the site to be at –91 (G). A purine, particularly adenine, was the preferred base at the transcription start site. Transcription start sites of GmNARK could be grouped into two regions: i) –91 to –83 and ii) –19 to –14. The transcription start sites at –92, –8, and –7 could not be repeatedly mapped and, therefore, were not in-cluded in these regions. More importantly, the regions at –91 to –83 and –19 to –14 were located 24 to 33 nucleotides down-stream of TATA2 and TATA1, respectively. The presence of two transcription initiation regions was not affected by Brady-rhizobium inoculation (RACE2) (Fig. 5) or the types of tissue (root or leaf) (Fig. 6).
The 1.7-kb GmNARK and 2.0-kb LjHAR1 promoters interchangeably drive phloem-specific expression in transgenic plants.
Southern blot analysis was carried out on 13 independent transgenic lines of L. japonicus carrying the 1.7-kb GmNARKpr::GUS construct. Eight transgenic lines were char-acterized as carrying one to three copies of T-DNA insert (data not shown). These lines showed no difference in the β-glu-curonidase (GUS) staining pattern, though variation of the GUS intensity was observed. Further analysis was carried out on these lines. L. japonicus plants regenerated from hypocotyl-derived calli (T0) as well as their seedlings (T1) were subjected to histochemical staining for GUS activity. A slight difference in the GUS distribution was observed; further studies carried out on T1 seedlings revealed that this difference resulted from the presence of sucrose in the culture medium accompanied by nonfixation of tissue (data not shown). Seedlings grown in
Fig. 5. Sites of transcription initiation determined by 5′ rapid amplification of cDNA ends (RACE) experiments. In all, 12 clones obtained by the first 5′RACE (RACE1) and 26 clones obtained by the second 5′ RACE (RACE2) were analyzed. Numbers in the table indicate 5′ RACE clones whose 5′ end corre-sponded to the particular nucleotide. The precise transcription start site at the position –90/–91 could not be identified by RACE1 because dCTPs were used for the homopolymeric tailing of the cDNA. Note that only partial sequence of the 5′ untranslated region of GmNARK is shown in this figure.
Fig. 4. Alignment of the 36-bp motifs identified by Multiple Em for Motif Elicitation in LjHAR1, MtSUNN, and GmNARK promoters. Potential negative regulatory regions (indicated as white text on black background) in MtSUNN and LjHAR1 promoters are located in these motifs. Numbers show the positions of these regions relative to their translation start sites. Stars indicate consensus nucleotides.
Fig. 6. Nonquantitative reverse-transcriptase polymerase chain reaction(RT-PCR) analysis of GmNARK transcripts in soybean root and unifoliate leaf. The cDNAs obtained from two independent experiments (R1 and R2) were used as templates. PCRs labeled with TATA2 used the forward primer that annealed to the 5′ untranslated region sequence of GmNARKupstream of the TATA1, PCRs labeled with TATA1+TATA2 used the for-ward primer that annealed to the sequence downstream of the TATA1, and PCRs labelled with 25S rRNA used 25S rRNA primers. RT-minus controls of the same cDNA samples were used previously in the quantitative RT-PCR experiments. They demonstrated the lack of a significant amount of genomic DNA in these cDNA samples (data not shown).

Vol. 20, No. 7, 2007 / 773
half-strength B5 medium solidified with agar (without sucrose) displayed a GUS staining pattern consistent with seedlings grown in vermiculite; thus, both growth conditions were used for L. japonicus seedlings.
The effect of different concentrations of potassium ferri-/fer-rocyanide (0.5, 1, and 5 mM) on the GUS staining pattern was examined. The results showed that these concentrations of po-tassium ferri-/ferrocyanide did not significantly affect the cell-specific localization of GUS. However, the 5 mM potassium ferri-/ferrocyanide treatment dramatically reduced the intensity of GUS staining (data not shown). Accordingly, 1 mM potas-sium ferri-/ferrocyanide and an increased concentration of 5-bromo-4-chloro-3-indolyl-beta-D-glucuronic acid, cyclohexyl-ammonium salt (X-Gluc) were later used in all GUS staining solutions.
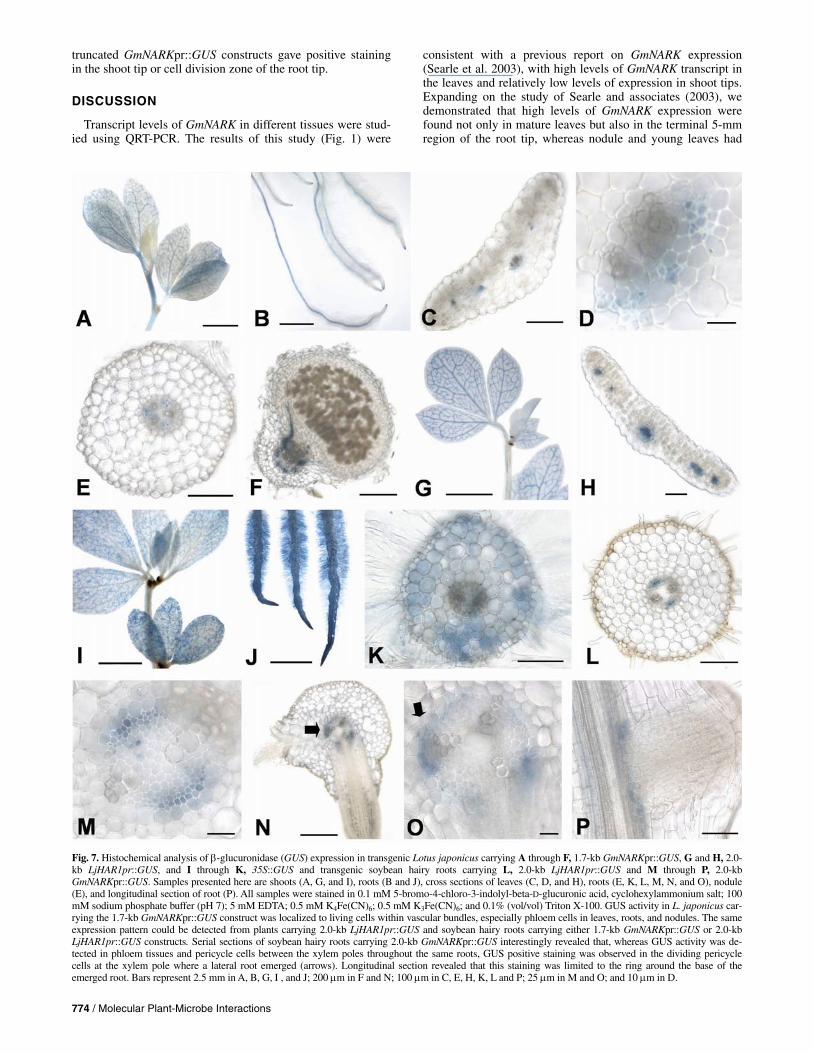
Stained L. japonicus seedlings carrying the 1.7-kb GmNARKpr::GUS construct revealed GUS staining in the vas-cular bundles of leaves, stems, and roots but not in shoot tips, very young leaves, and the zone of cell division in roots (Fig. 7A and B). Cross sections showed that this GUS activity was restricted to the phloem tissue in the leaves (Fig. 7C and D). In roots, GUS activity was detected in phloem tissue and several pericycle cells adjacent to the phloem between the xylem poles (Fig. 7E). GUS activity in nodules, similar to other parts of the plants, was limited to phloem tissues (Fig. 7F). Based on histo-chemical analysis, the inoculation of plants with Mesorhizo-bium loti did not have a significant effect on GUS activity. However, it was observed that 4-week-old plants that had been inoculated with M. loti were slightly more advanced in growth than uninoculated plants and, consequently, had more intense GUS activity in the most mature leaves (data not shown).
The same expression pattern was detected in transgenic L. japonicus plants carrying the 2.0-kb LjHAR1 promoter::GUS construct as for the L. japonicus plants carrying the 1.7-kb GmNARKpr::GUS construct (Fig. 7G). The GUS activity was specific to the vascular cylinder, particularly phloem tissues, of leaves (Fig. 7H) and all other organs analyzed (data not shown).
In contrast to GUS expression driven by the 1.7-kb GmNARKpr::GUS or the 2.0-kb LjHARpr::GUS constructs, GUS expression driven by the Cauliflower mosaic virus (CaMV) 35S promoter was detected in all tissues analyzed (Fig. 7I, J, and K). GUS activity was not restricted to the phloem and could be detected in the meristematic cells of the root tip. The promoterless GUS construct failed to give detect-able GUS activity in eight transgenic lines of L. japonicus (Fig. 8E, close-up image of a cotyledon), though two addi-tional lines carrying the same construct displayed GUS activity (data not shown). PCR confirmed that these transgenic plants had T-DNA inserts (data not shown). The positive GUS activ-ity most likely was due to the insertion of T-DNA adjacent to endogenous promoters (Buzas et al. 2005).
The introduced NcoI site at the ATG start of the 1.7-kb GmNARKpr::GUS construct did not affect the GUS expression pattern because both the 1.7-kb GmNARKpr::GUS and 2.0-kb GmNARKpr::GUS constructs displayed the same expression pattern. The transgenic hairy roots of soybean carrying the 1.7-kb GmNARKpr::GUS (not shown), 2.0-kb GmNARKpr::GUS (Fig. 7M to P), or 2.0-kb LjHARpr::GUS constructs (Fig. 7L) had the same GUS expression pattern as the roots of transgenic L. japonicus carrying the 1.7-kb GmNARKpr::GUS and 2.0-kb LjHARpr::GUS constructs. Analysis of the GUS expression pat-tern in transgenic soybean hairy roots revealed that, although GUS activity was detected in phloem and pericycle cells be-tween the xylem poles throughout the same roots (Fig. 7M, N, and O), positive GUS staining was observed in dividing peri-cycle cells at a xylem pole associated with lateral root emer-
gence (Fig. 7N and O, arrows). Longitudinal sections revealed that this positive staining was limited to the ring around the base of the root (Fig. 7P). It is important to note that, in addi-tion to the phloem and pericycle, GUS activity frequently was detected in root caps of L. japonicus and soybean roots carry-ing the 1.7-kb (or 2.0-kb) GmNARKpr::GUS and 2.0-kb LjHARpr::GUS constructs; however, this positive staining was line dependent and, thus, it was excluded as a typical expres-sion pattern driven by these promoters.
Deletion analysis of the GmNARK promoter identifies a region directing phloem-specific GUS expression.
Bioinformatics analysis indicated several cis elements that potentially could contribute to the activity of the 1.7-kb GmNARK promoter, including its phloem-specific expression. Therefore, additional GmNARKpr::GUS constructs containing 908, 465, 300, 123, or 49 bp of promoter sequence immediately upstream of the translation start site were used to define and characterize these elements. Because two functional TATA boxes were found in the GmNARK promoter, an additional con-struct containing 123 bp of the GmNARK promoter sequence, but with modifications to the TATA1 element (TATATAA to TGTCTGC; namely T2mT1::GUS), was used to test the ex-pression pattern driven by this truncated promoter in the ab-sence of a functional copy of TATA1. To provide nearly identi-cal growth conditions for all L. japonicus seedlings, seedlings were grown under controlled in vitro conditions. Histochemi-cal GUS assays were performed on 1- to 2-week-old seedlings. In all, 41 independent transgenic lines carrying 908, 465, 300, 123, and 49 bp, and T2mT1 GmNARKpr::GUS constructs (five to nine lines for each construct), were assayed for histochemi-cal GUS activity. Approximately 7% of the transgenic lines displayed an irregular GUS pattern (presumably due to posi-tional effects) and 27% of the transgenic lines gave no detect-able GUS staining; these lines were excluded from further analysis. The remaining 27 independent transgenic lines (66%) displayed the same GUS expression pattern, though variation of the GUS intensity was observed (discussed below).
Images of typical stained cotyledon, trifoliate leaf, and seed-ling root carrying the 1.7-kb GmNARKpr::GUS are shown in Figure 8 (A, F, and J respectively). Deletion of the region span-ning –1.7 kb to –908 bp of the GmNARK promoter resulted in significant changes in the GUS staining pattern. In leaves (Fig. 8B and G), GUS staining occasionally was maintained in vas-cular tissue of cotyledons and trifoliate leaves; however, the in-tensity of GUS staining frequently was reduced. Concurrently, GUS activity became prominent in nonvascular tissues. Micro-scopic observation revealed that epidermal cells, especially guard cells, were mainly responsible for this positive staining (data not shown). The leaves obtained from L. japonicus lines carrying the 1.7-kb GmNARKpr::GUS construct scarcely showed GUS activity in epidermal cells, even though several blue guard cells in one leaf could be detected. In roots (Fig. 8K and M), GUS staining was detectable in all cell types, in-cluding epidermal and cortical cells. In contrast to the reduc-tion of GUS staining in leaf vascular bundles, an increase in GUS intensity was observed in root vascular tissues. The leak-ing of intensified GUS enzyme most likely is responsible for the detection of GUS staining in nonliving cells of vascular bundles. Further deletion of the GmNARK promoter did not change the GUS expression pattern from that of the 908-bp GmNARKpr::GUS construct. Variation of the GUS staining intensity commonly was observed among different transgenic lines, including those carrying the same construct. Examples of stained cotyledons, leaves, and roots of different truncated lines are shown in Figure 8 (B to D and G to I). It is important to note that none of the L. japonicus transgenic lines carrying
774 / Molecular Plant-Microbe Interactions
truncated GmNARKpr::GUS constructs gave positive staining in the shoot tip or cell division zone of the root tip.
DISCUSSION
Transcript levels of GmNARK in different tissues were stud-ied using QRT-PCR. The results of this study (Fig. 1) were
consistent with a previous report on GmNARK expression (Searle et al. 2003), with high levels of GmNARK transcript in the leaves and relatively low levels of expression in shoot tips. Expanding on the study of Searle and associates (2003), we demonstrated that high levels of GmNARK expression were found not only in mature leaves but also in the terminal 5-mm region of the root tip, whereas nodule and young leaves had
Fig. 7. Histochemical analysis of β-glucuronidase (GUS) expression in transgenic Lotus japonicus carrying A through F, 1.7-kb GmNARKpr::GUS, G and H, 2.0-kb LjHAR1pr::GUS, and I through K, 35S::GUS and transgenic soybean hairy roots carrying L, 2.0-kb LjHAR1pr::GUS and M through P, 2.0-kb GmNARKpr::GUS. Samples presented here are shoots (A, G, and I), roots (B and J), cross sections of leaves (C, D, and H), roots (E, K, L, M, N, and O), nodule (E), and longitudinal section of root (P). All samples were stained in 0.1 mM 5-bromo-4-chloro-3-indolyl-beta-D-glucuronic acid, cyclohexylammonium salt; 100 mM sodium phosphate buffer (pH 7); 5 mM EDTA; 0.5 mM K4Fe(CN)6; 0.5 mM K3Fe(CN)6; and 0.1% (vol/vol) Triton X-100. GUS activity in L. japonicus car-rying the 1.7-kb GmNARKpr::GUS construct was localized to living cells within vascular bundles, especially phloem cells in leaves, roots, and nodules. The sameexpression pattern could be detected from plants carrying 2.0-kb LjHAR1pr::GUS and soybean hairy roots carrying either 1.7-kb GmNARKpr::GUS or 2.0-kb LjHAR1pr::GUS constructs. Serial sections of soybean hairy roots carrying 2.0-kb GmNARKpr::GUS interestingly revealed that, whereas GUS activity was de-tected in phloem tissues and pericycle cells between the xylem poles throughout the same roots, GUS positive staining was observed in the dividing pericyclecells at the xylem pole where a lateral root emerged (arrows). Longitudinal section revealed that this staining was limited to the ring around the base of theemerged root. Bars represent 2.5 mm in A, B, G, I , and J; 200 μm in F and N; 100 μm in C, E, H, K, L and P; 25 μm in M and O; and 10 μm in D.

Vol. 20, No. 7, 2007 / 775
relatively low levels of expression. Our data showed that leaf maturity correlated strongly with GmNARK transcript levels. In addition, Bradyrhizobium inoculation did not significantly alter the transcript level (Fig. 2). The results of QRT-PCR were in good agreement with the histochemistry of the GmNARK promoter-GUS reporter lines. Histochemical analysis had an advantage over QRT-PCR because it enabled the activity of GmNARK to be examined at tissue and cellular levels. For ex-ample, the absence of GUS activity in the zone of cell division in the root was distinguishable from the GUS-positive region by histochemical assay (Fig. 7B), whereas the 5-mm root tip was shown to have a relatively high level of GmNARK tran-script by QRT-PCR. Histochemical analysis also confirmed that rhizobial inoculation did not have a significant effect on GUS activity.
It is widely known that the shoot plays a major role in AON via long-distance signaling (Delves et al. 1986, 1992; Francisco
and Harper 1995; Men et al. 2002; Sheng and Harper 1997); therefore, the finding that the 1.7-kb GmNARK promoter drives phloem-specific expression in the shoot (Fig. 5A, C, and D) is not surprising. The results of this study support and substantiate the model for AON signaling pathways proposed by Caetano-Anollés and Gresshoff (1990), in which the perception of the root-derived signal or ligand activated in response to the root-derived signal by GmNARK is likely to happen in the phloem, resulting in a signal transduction cascade that generates the shoot-derived inhibitor (SDI). The SDI is then directly translo-cated to the root, where further nodule development is inhibited.
The high GmNARK transcript level in the root possibly can be explained by the minor role of the root in AON (Francisco and Harper 1995; Sheng and Harper 1997) or the involvement of GmNARK in autoregulation of mycorrhization (Meixner et al. 2005). Another possible explanation is that GmNARK has functions in other developmental processes in addition to
Fig. 8. Histochemical analysis of β-glucuronidase (GUS) expression in transgenic Lotus japonicus carrying A, F, J, and L, 1.7-kb GmNARKpr::GUS; B, G, K,and M, 908-bp GmNARKpr::GUS; C, 300-bp GmNARKpr::GUS; D and H, 123-bp GmNARKpr::GUS; I, 49-bp GmNARKpr::GUS; and E, promoterless GUS. Samples represented here are cotyledons (A through E), trifoliate leaves (F through I), roots (J and K), and cross sections of roots (L and M). All samples werefixed in 0.5% paraformaldehyde and stained in 0.2 mM 5-bromo-4-chloro-3-indolyl-beta-D-glucuronic acid, cyclohexylammonium salt; 100 mM sodium phos-phate buffer (pH 7); 5 mM EDTA; 1.0 mM K4Fe(CN)6; 1.0 mM K3Fe(CN)6; and 0.1% (vol/vol) Triton X-100. In plants carrying 1.7-kb GmNARKpr::GUS, GUSexpression was restricted to vascular tissues in leaf and root. This pattern was no longer observed in plants carrying 908-bp GmNARKpr::GUS and further trun-cated constructs. The promoterless GUS construct did not give detectable GUS activity. Variation of the GUS staining intensity was observed among transgeniclines carrying the same constructs (data not shown). Bars represent 500 μm in A through E, 1 mm in F through K, and 100 μm in L and M.

776 / Molecular Plant-Microbe Interactions
AON, such as negative control of cell division in pericycle or procambial cells (Beveridge et al. 2007; Buzas and Gresshoff 2007). It was observed that GUS activity could be detected in seedlings as young as 1 week old grown under axenic condi-tions. Moreover, GUS expression directed by the 2.0-kb GmNARK promoter (or 1.7-kb promoter) (data not shown) in soybean roots was seen in phloem tissues and pericycle cells between the xylem poles at almost all stages of root and lateral root development (Fig. 7E, M, N, and O). The fact that GUS expression was detected in pericycle cells at the xylem pole around the base of emerging roots (Fig. 7N, O, and P), a pat-tern similar to GUS expression in the Arabidopsis thaliana enhancer trapped line LRB10 during lateral root emergence (Malamy and Benfey 1997), implies the involvement of GmNARK either directly or indirectly in lateral root develop-ment. Nodulation and lateral root development have been re-ported to share similarities in organogenesis, gene expression, and responses to hormones or signaling molecules (Mathesius et al. 2000; Nakagawa and Kawaguchi 2006; Nutman 1948; Olah et al. 2005). However, this proposed role for GmNARK is likely to be obscured by other functionally redundant genes, because GmNARK mutants did not show any nonsymbiotic phenotype (Carroll et al. 1985a and b).
Results from the 5′ RACE experiments and truncated GmNARKpr::GUS lines (–123 bp, –49 bp, and T2mT1) con-firmed that the in silico-predicted TATA1 and TATA2 in the GmNARK promoter are indeed functional. In plants, the TATA box is typically located 32 ± 7 nucleotides upstream of the transcription start site (Joshi 1987). We could map transcrip-tion initiation repeatedly to regions that were 24 to 29 and 25 to 33 nucleotides downstream of TATA1 and TATA2, respec-tively. No other AT-rich regions could be located within 39 nu-cleotides upstream of these transcription initiation sites (Fig. 3). Interestingly, the longer transcripts mediated by TATA2 contain two AUG sites upstream of the proposed TSS (Searle et al. 2003), with one site in-frame with the AUG start codon. How-ever, these two AUG sequences are closely followed by in-frame stop codons and, therefore, are unlikely to serve as func-tional start codons. Several plant genes were reported to have transcription directed by two or multiple TATA boxes (Bassett et al. 2004; Doyle and InSeob 2001; Grace et al. 2004; Lee et al. 1994). Some of these genes were observed to use the TATA boxes differentially in a tissue-dependent (Bassett et al. 2004) or stimulus-dependent manner (Doyle and InSeob 2001; Lee et al. 1994). In the case of the Phaseolus vulgaris β-phaseolin promoter, two TATA boxes functioned in concert to ensure high levels of transcription (Grace et al. 2004). Because GmNARK is responsible for symbiotic phenotypes and has been shown to function mainly in the shoot, we compared the activity of the two TATA boxes when plants were inoculated with Bradyrhizobium japonicum with that when plants were uninoculated (Fig. 5) and in the shoot with that in root tissue (Fig. 6). The results demonstrated that both TATA boxes were used in shoot and root tissues and their activity was unaffected by Bradyrhizobium inoculation. Additionally, the 123-bp (hav-ing both TATA2 and TATA1), 49-bp (having only TATA1), and T2mT1 (having TATA2 and a mutated TATA1) GmNARKpr:: GUS constructs were capable of GUS transcription and they did not give rise to any difference in GUS staining pattern (Fig. 8, partly shown). Together, our results suggest that the two TATA boxes in the GmNARK promoter cooperatively direct transcription of GmNARK and only one of them is adequate for the basal transcription.
A comparison of 2.0 kb of DNA sequence of the GmNARK and LjHAR1 promoters using Signal Scan Search indicated the presence of over 70 putative motifs (data not shown). Fourteen of these motifs, including an NRR, have an equal to or greater
than 7-bp sequence and are less likely to be found by chance. In addition, longer highly conserved regions have been identi-fied by Multiple Em for Motif Elicitation; three motifs show-ing the highest homology (89 to 100%) are shown in Figure 3. The QRT-PCR results of GmNARK expression (Figs. 1 and 2) are in strong agreement with the expression of LjHAR1 previ-ously reported by Krusell and associates (2002) and Nishimura and associates (2002). More importantly, our study showed that the 1.7 kb of GmNARK promoter and the 2.0 kb of LjHAR1 promoter are interchangeable in driving GUS expres-sion in either stable transgenic L. japonicus or transgenic hairy roots of soybean (Fig. 7). The expression studies and bioinfor-matics analysis suggest that cis- and trans-acting elements re-quired for the transcriptional regulation of these orthologs are likely to be conserved.
Sequence analysis of 2.0 kb of the GmNARK promoter re-vealed only one putative cis-acting element (NRR) that has been reported to function in phloem-specific expression. Inter-estingly, this putative NRR motif also was found in the LjHAR1 promoter which, according to our study, also drives phloem-specific expression. Therefore, we were tempted to conclude that the putative NRR motif found at –308 bp in the GmNARK promoter was truly responsible for the phloem-specific expression. However, the promoter deletion analysis clearly demonstrated that this is not the case and the true phloem-specific element must reside within 1.7 kb and 908 bp upstream of the TSS in the GmNARK promoter. Two other can-didate regions are the 37- and 36-bp motifs, which show ho-mology to the corresponding regions of the LjHAR1 promoter. The 36-bp motif also was found in the MtSUNN promoter at a location similar to that of LjHAR1 (Fig. 4). Supposing that the promoter of MtSUNN drives phloem-specific expression, the 36-bp motif is more likely to contain the regulatory element driving this common expression pattern. Notably, whereas the potential NRRs were found in the 36-bp motifs of the LjHAR1 and MtSUNN promoters, only a variant form of NRR (one G was replaced by C and one A was replaced by T) (Fig. 4) was found in the 36-bp motif of GmNARK promoter. It is possible that the transcription factor responsible for the phloem-specific expression of GmNARK might recognize this variant form of NRR. Interestingly, an identical palindromic sequence separated by a 3-bp spacer (GAATACGATTC) also was observed in the 36-bp motif of these orthologous genes. Some protein-binding sites are bipartite and the bipartite motifs often are palin-dromic. The palindromic sequence found here is very similar to recognition sequence of AtHB-9 (GTAAT(G/C)ATTAC), a member of the homeodomain-leucine zipper family, whose members are characterized by expression in the vascular tissue (Baima et al. 2001; Sessa et al. 1998). Further analyses, includ-ing internal deletions, are needed to determine whether these potential motifs are truly responsible for the phloem-specific gene expression of GmNARK. Additionally, the lack of GUS activity in the shoot and root meristems of L. japonicus transgenic lines carrying either the 1.7-kb GmNARKpr::GUS construct or its derivatives suggests that the element or elements that positively mediate the expression in these regions reside upstream of the 1.7-kb GmNARK promoter; or, alternatively, regulatory elements that negatively regulate these regions may reside within the 49-bp GmNARK promoter.
MATERIALS AND METHODS
Plant growth and tissue collection. Soybean (Glycine max (L.) Merr. cv. Bragg) plants were
grown in pots containing vermiculite and kept under a regime of 16 and 8 h, 28 and 25°C, day and night, respectively. They were irrigated three times a week with B&D nutrient solution

Vol. 20, No. 7, 2007 / 777
(Broughton and Dilworth 1971) supplemented with 0.5 to 2 mM KNO3. Plants used for the tissue-specific GmNARK ex-pression analysis were inoculated with 4-day-old cultures of B. japonicum CB1809 grown in YMB (Vincent 1970) at 5 days post germination. Mature and young nodules (NOD), 5-mm root tips (RT), unifoliate leaves (UF), first trifoliate leaves (FTF), second trifoliate leaves (STF), and 3-mm shoot tips (ST) were collected at 16 dai. Additionally, old nodules (ONOD) (characterized by the larger sizes and well-developed lobes), NOD, RT, UF, FTF, STF, third trifoliate leaves (TTF), and ST were collected at 29 dai. Different tissue types were collected from approximately 40 plants and pooled together. Leaves harvested from 29-day-old plants also were used in RACE1 experiment as well as the nonquantitative RT-PCR analysis of GmNARK transcripts. Leaves and roots harvested at 30 dai from independently grown but similarly treated soybean plants also were used in the nonquantitative RT-PCR analysis of GmNARK transcripts. Leaves harvested from 18-day-old soybean plants either treated as described above or similarly but without inoculation were used in the second RACE experi-ment (RACE2). Soybean plants used for the analysis of Bra-dyrhizobium inoculation-dependent GmNARK expression were inoculated with B. japonicum CB1809 grown in YMB or an equivalent volume of YMB at 10 days post germination. Leaf tissue was harvested from four plants at 1 and 7 dai. All tissues were harvested into 50-ml Falcon tubes submerged in liquid nitrogen and stored at –80°C.
Seed of L. japonicus transgenic lines were scarified and sub-sequently sterilized in 3% H2O2, 70% ethanol for 10 min. The seed then were rinsed three times in sterile water and placed on germination medium (half-strength B5, 1.2% agar) in Phy-tatray II (Sigma-Aldrich, St. Louis; cat. no. P5929). They were kept under a day-and-night regime of 12 and 12 h, 18 and 21°C, respectively. Histochemical GUS assay was performed using 1- to 4-week-old seedlings. To study the expression of promoter::GUS fusion in nodules, L. japonicus plants were grown in vermiculite. Induction of nodulation in L. japonicus was performed by inoculating each plant with 107 cells of M. loti NZP2235.
RNA isolation and cDNA synthesis, Total RNA used for the QRT-PCR analysis of the tissue-
specific GmNARK expression and nonquantitative RT-PCR of GmNARK transcripts was extracted from 250 mg of each plant tissue sample using a modification of the phenol/sodium dode-cyl sulfate (SDS) method of Wilkins and Smart (1996). First, the plant tissue was ground to a fine powder in liquid nitrogen. Next, cells were lysed in 500 μl of the homogenization buffer (100 mM Tris HCl, pH 8.5; 100 mM LiCl; 5 mM EDTA, pH 8.0; 100 mM NaCl; 1% [wt/vol] SDS; and 2% β-mercapto-ethanol). Proteins and other soluble organic compounds were removed by an extraction three times with phenol/chloroform/ isoamyl alcohol (25:24:1) and once with chloroform/isoamyl alcohol (24:1). The separation of RNA from DNA was per-formed by the selective precipitation of RNA using LiCl as de-scribed by Wilkins and Smart (1996). The RNA then was treated with amplification-grade DNaseI (Invitrogen, Carlsbad, CA, U.S.A.) as described by the manufacturer. The cDNA was synthesized from approximately 3.0 μg of total RNA with Su-perscript II reverse transcriptase (Invitrogen) in the presence of 62.5 ng of random primers (Invitrogen) and 1.25 μl of 0.5 μM oligo(dT) mix (5′-GACCACGCGTATCGATGTCGAC(T16)V-3′) following the manufacturer’s instructions. For QRT-PCR of GmNARK, 25 μl of the cDNA was diluted with water to a final volume of 100 μl. The cDNA solution was diluted a further 1,500-fold for endogenous control QRT-PCR using 25S ribo-somal RNA primers. For nonquantitative RT-PCR analysis, the
cDNA solution was diluted sixfold with water. Total RNA used for the analysis of GmNARK rhizobium inoculation-dependent expression was prepared using a Nucleospin RNA Plant Kit (Macherey-Nagel, Düren, Germany) according to the manu-facturer’s instructions. Approximately 1 μg of total RNA was DNase-treated (MBI Fermentas, Burlington, Canada) at 37°C for 40 min and 0.5 μg of the DNase-treated RNA was reverse transcribed into first-strand cDNA using Superscript III reverse transcriptase (Invitrogen). The cDNA was diluted 1:10 prior to QRT-PCR analysis. For all experiments, RT-minus controls were included as a control for subsequent PCR reactions.
QRT-PCR. An independent study was carried out to evaluate the ex-
pression stability of five soybean reference genes: i) 25S rRNA, ii) actin (Soy57; GenBank accession number U60500), iii) ubiquitin, iv) β-tubulin (GenBank accession number D26092), and v) actin 1 (Sac1; GenBank accession number J01298) across a variety of tissue types. The results suggested that 25S rRNA was the most stable control gene (data not shown). Therefore, the 25S rRNA was chosen as reference gene for tissue-specific expression study. The analysis of Bradyrhizo-bium inoculation-dependent GmNARK expression used one type of tissue (i.e., leaf) and soybean actin was chosen as refer-ence gene. The sequences of GmNARK forward and reverse primers were 5′-TTATTTGCTCATCGATCCAGATTC-3′ and 5′-TCGGACAAACAACAGATGAAAGTTA-3′, the soybean actin forward and reverse primers were 5′-GCCTGCCATGTATGTT GCAA-3′ and 5’-CGTGTGGCTCACACCATCA-3′, and the 25S rRNA forward and reverse primers were 5′- TTAACAGC CTGCCCACCCTGG-3′ and 5′-ATCCATTTTGCCGACTTCC C-3′, respectively. Each PCR was carried out in a total volume of 25 μl containing 12.5 μl of 2× SYBR Green master mix (Applied Biosystems, Foster City, CA, U.S.A.), 5 μl of the cDNA template, and 5 μl of forward and reverse primer mix. The final concentrations of GmNARK, soybean actin, and 25S rRNA primers were 300, 300, and 200 nM, respectively. All reactions were performed in an ABI PRISM 7700 sequence detection system (Applied Biosystems) using the following cy-cle conditions: 1 cycle of initial denaturation (95°C for 10 min), 45 cycles of amplification and quantification (95°C for 15 s and 60°C for 1 min), and 1 cycle of a melting curve pro-gram (60 to 95°C with a continuous fluorescence measure-ment). All PCRs were carried out in duplicate. Specific ampli-fication was confirmed by sequencing the PCR products. For the analysis of tissue-specific GmNARK expression, the PCR efficiency of each pair of primers was predetermined based on template dilution series as described by Rasmussen (2001). For the analysis of rhizobium inoculation-dependent GmNARK expression, the efficiency of amplification was determined using LinRegPCR software (Ramakers et al. 2003) and subsequently used for the calculation of relative expression level.
Sequence analysis of GmNARK promoter. Approximately 2.1 kb of incomplete DNA sequence for the
GmNARK promoter was obtained from GenBank (accession number AP005667) and T. S. Laniya (personal communica-tion). This putative DNA sequence was used to design more sequencing primers to construct a single contiguous sequence for the GmNARK promoter. The bacterial artificial chromo-some (BAC) 75M10 from Soybean BAC Library PI437.654, previously identified to contain GmNARK (Searle et al. 2003), was obtained from Clemson University Genomics Institute. Plasmid DNA was prepared using a Plasmid Midi Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Each DNA sequencing reaction contained 1 μl of BDT v3.1 Reaction Mix (Applied Biosystems), 3 μl of 5× sequencing

778 / Molecular Plant-Microbe Interactions
buffer, 333 nM primer, and approximately 1 μg of BAC DNA in a final reaction volume of 12 μl, and was performed under the following cycle conditions: 1 cycle of 95°C for 5 min and 50 cycles of 96°C for 30 s, 50°C for 15 s, and 60°C for 4 min. The sequencing reactions then were subjected to capillary gel electrophoresis in an ABI 3730xl DNA Analyzer (Applied Bio-systems). All sequences were assembled into one contig using the Contig Assembly Program on the University Claude Bernard-Lyon 1 website. DNA sequences comprising approxi-mately 2.0 kb of the GmNARK promoter, LjHAR1 promoter (GenBank accession number AP005667), and MtSUNN pro-moter (GenBank accession number AY769943) were analyzed for the presence of potential cis-acting regulatory elements using Signal Scan Search (Prestridge 1991) available in the PLACE database (Higo et al. 1999). The search for highly conserved regions in the promoters of these orthologous genes was per-formed using Multiple Em for Motif Elicitation (Bailey and Elkan 1994).
5′ RACE. Total RNA used in RACE1 was prepared from 100 mg of
leaves using RNeasy plant mini kit (Qiagen). The integrity of total RNA was determined by agarose gel electrophoresis. The cDNA was synthesized from 3 μg of total RNA using GmNARK-specific primer-1 (GSP1) 5′-GAGGTGCTTGAGG GAAGTGA-3′ and Superscript III reverse transcriptase (Invi-trogen) following the manufacturer’s instructions. The reaction product was purified with a QIAquick PCR purification kit (Qiagen) and 10 μl of the purified cDNA sample was used in a tailing reaction. TdT tailing of cDNA, PCR of dC-tailed cDNA, and nested amplification were performed using the 5′ RACE System for Rapid Amplification of cDNA Ends (ver-sion 2.0; Invitrogen). The gene-specific primers for PCR of dC-tailed cDNA and the nested amplification were GSP2 5′-GTGAGGTTGTTCTGCGAGAC-3′ and GSP3 5′-GGCGGAA GGTGACCGAAGAG-3′, respectively. The PCR products then were cloned into pCR4-TOPO using a TOPO TA Cloning Kit (Invitrogen) and analyzed by DNA sequencing. The 5′ RACE experiment was repeated with the following modifications: i) two RNA samples were obtained from soybean leaves harvested from plants either inoculated with Bradyrhizobium or uninocu-lated, using the modified phenol/SDS extraction combined with LiCl precipitation as described in RNA isolation and QRT-PCR; ii) the dGTPs were used for homopolymeric tailing of the first-strand cDNA instead of dCTPs and, consequently, two newly designed anchor primers, NAP1 containing 3′ sequence complementary to the dG-tailed cDNA (5′-CGGAATTCGGC GTGGACAGG(C16)-3′) and NAP2 (5′-CGGAATTCGGCGT GGACAGG-3′), were used in the PCR of dC-tailed cDNA and the nested amplification, respectively; and iii) Pfu DNA poly-merase (MBI Fermentas) was used in both PCRs.
Nonquantitative RT-PCR analysis of GmNARK transcripts. A 25-μl PCR reaction contained 2 μl of 1:6 diluted cDNA,
1.25 units of Taq DNA polymerase (Invitrogen), 1× PCR buffer (20 mM Tris-HCl, 50 mM KCl, pH 8.4), 1.5 mM MgCl2, 0.25 mM dNTPs, and 400 nM forward and reverse primers. The PCR conditions consisted of a preliminary incu-bation at 95°C for 2 min; followed by 40 cycles of 94°C for 1 min, 55°C for 30 s, and 72°C for 30 s; with a final extension at 72°C for 10 min. Three sets of forward and reverse primers used were i) TestTATA2-FP 5′-GTAAAAATAAGGACGCAG GCAAA-3′ and NARK250DS-RP 5′-GAAGAGAGGAACAA ACGAGAC-3′, ii) TestTATA1-FP 5′-GACATGAGAAGCTGT GTGTGC-3′ and TestTATA1-RP 5′-GCGAGCTCCTTGGGA AGTA-3′, and iii) 25S rRNA forward and reverse primers. The TestTATA2-FP was designed to anneal specifically to the 5′
untranslated region sequence of GmNARK upstream of the TATA1. The TestTATA1-FP and TestTATA1-RP were carefully designed to avoid the amplification of GmCLV1A.
Gene reporter constructs. The 465-bp GmNARK promoter region was amplified from
the BAC clone 75M10 (Soybean BAC Library PI437.654, Clemson University Genomics Institute, Clemson, SC, U.S.A.) using a forward primer that annealed to the sequence 465 bp upstream of the GmNARK TSS (Fig. 3) and a reverse primer, PNARK-RP 5′-CTTCCCATGGCTCTCTGA-3′, that generated an NcoI site (underlined) at ATG start codon. Subsequently, the PCR product was cloned into pCR4-TOPO vector (Invitro-gen) and the correct promoter sequence was confirmed by se-quencing analysis. A 485-bp PstI-NcoI fragment containing this region of GmNARK promoter was then transcriptionally fused to the GUS gene containing the catalase intron from pCAMBIA1305.1 (GenBank accession number AF354045) and subcloned as a promoter::GUS fusion into pCAMBIA1300 (GenBank accession number AF234296) in an orientation such that the 5′ end of the promoter was located next to the T-DNA right border. The 1.7-kb, 908-bp, and 300-bp GmNARKpr::GUS constructs were accomplished as given above, except that different forward primers (Fig. 3) were used in PCR amplifications and PstI-NcoI fragments containing the different lengths of GmNARK promoters from pCR4-TOPO vectors were used directly to replace the PstI-NcoI fragments in the 465-bp GmNARKpr::GUS plasmids. The forward primers used in the construction of 123- and 49-bp GmNARKpr::GUS plasmids were as shown in Figure 3, except with a PstI site (CTGCAG) added to the 5′ end of these sequences. They were paired with a reverse primer located within the GUS gene 5′-CGGTGAACATCACTGGATCA-3′. PCR products were puri-fied, cut with PstI and MluI, and used to replace the PstI-MluI fragment in the 465-bp GmNARKpr::GUS plasmid. To deter-mine transcription directed by the 123-bp GmNARK promoter in the absence of a functional TATA1, mutations were intro-duced into the TATA1 element by PCR-based site-directed mutagenesis. In brief, two DNA fragments carrying mutations in the TATA1 sequence (TGTCTGC) were amplified from the 465-bp GmNARKpr::GUS plasmid using partially overlapping primers in two separate reactions. The correct sizes of the two PCR products were confirmed on a 2.5% agarose gel. The PCR products then were purified, and 10 ng of each PCR product was mixed together and used as template for the sec-ond PCR reaction. This PCR product was purified, cut with PstI and MluI, and used to replace the PstI-MluI fragment in the 465-bp GmNARKpr::GUS plasmid. The PCR-based site-directed mutagenesis also was used to generate the 2.0-kb GmNARKpr::GUS construct so that the 2.0-kb GmNARK pro-moter could be transcriptionally fused to the GUS gene with-out the introduction of NcoI site at ATG start codon. The 2.0-kb LjHAR1 promoter region was amplified from L. japonicus ecotype Miyakojima MG-20 genomic DNA using a 2.1-kb up-stream primer 5′-GTTTGTGGAGCGGAAAGGTA-3′ and a reverse primer with an introduced NcoI site (underlined) at ATG start codon 5′-TGCAACCATGGCAATTTTGTTTAATG CTTCTTCCTCAG-3′. Because there is a PstI site located 2.0 kb upstream of the LjHAR1 promoter, the PCR product was directly purified, cut with PstI and NcoI, and used to replace the promoter fragment in the 465-bp GmNARKpr::GUS vector. For the promoterless construct, the GmNARK promoter was removed from the 465-bp GmNARKpr::GUS plasmid by diges-tion with HindIII and NcoI. The linear plasmid DNA was puri-fied from an agarose gel and self-ligated. The pCAMBIA1305.1 carrying the GUS gene under the control of CaMV 35S pro-moter was used as a positive control vector.

Vol. 20, No. 7, 2007 / 779
Plant transformation. Plasmid DNA was introduced into either Agrobacterium tu-
mefaciens AGL1 or A. rhizogenes K599 by electroporation in 1-mm cuvettes at 2.20 kV in a micropulser (BioRad, Hercules, CA, U.S.A.). Transformed bacteria were selected on Luria-Bertani medium supplemented with rifampicin (50 μg/ml) and kanamycin (50 μg/ml).
Hypocotyl transformation of L. japonicus was performed as described by Stiller and associates (1997) with the modifica-tion that pre-cut hypocotyls were cocultivated with A. tumefa-ciens for 5 days and hygromycin B (15 μg/ml; Sigma-Aldrich) was used as an antibiotic for the selection of transgenic plants. L. japonicus T0 plants were transferred to pots containing ver-miculite and kept under a day-and-night regime of 18 and 6 h, 23 and 18°C, respectively. Southern blot analysis was per-formed using genomic DNA isolated from 13 T0 transgenic lines of L. japonicus carrying 1.7-kb GmNARKpr::GUS as de-scribed by Dellaporta and associates (1983) with the modifica-tion that DNA was further purified by phenol/chloroform/isoa-myl alcohol (25:24:1) and chloroform/isoamyl alcohol extrac-tions. Approximately 10 μg of DNA was digested with either HindIII or BglII. Digested products were separated by electro-phoresis on a 0.8% agarose gel and transferred onto a Hybond-N+ Nylon membrane (Amersham Pharmacia Biotech, Piscata-way, NJ, U.S.A.). Prehybridization was carried out at 65°C overnight in the hybridization buffer (0.25 M Na2HPO4; 7% SDS; 1 mM EDTA) in the presence of salmon DNA at 0.208 mg/ml. A 1.4-kb fragment of GUSPlus was amplified using the internal FP and RP primers located within the GUS gene and the 1.7-kb GmNARKpr::GUS plasmid as the template. It was subsequently 32P-labeled using the DecaLabel DNA labeling Kit (MBI Fermentas). Hybridization was carried out overnight at 65°C in 50 ml of the hybridization buffer containing 100 ng of the labeled probe. The membrane was washed twice in 1× SSC (0.15 M NaCl plus 0.015 M sodium citrate)and 0.1% SDS at 65°C for 15 min and subsequently exposed to Kodak BioMax MS-1 Film (Kodak, Rochester, NY, U.S.A.) for 2 days at –80°C.
Hairy root transformation of soybean was performed as de-scribed by Kereszt and associates (2007) with some modifica-tions. In brief, 6-day-old seedlings were injected with a few drops of a 2-day-old culture of A. rhizogenes K599 at the hy-pocotyl area using a syringe fitted with a 25G needle (Terumo, Tokyo). Soybean plants with hairy roots were transferred to new pots containing vermiculite and allowed to grow under a day-and-night regime of 16 and 8 h, 28 and 25°C, respectively.
Histochemical GUS assay. GUS activity was determined histochemically as described
by Larkin and associates (1996) with some modifications. Two different but similar recipes for staining solution were used: i) 0.1 mM X-Gluc, 100 mM sodium phosphate buffer (pH 7), 5 mM EDTA, 0.5 mM K4Fe(CN)6, 0.5 mM K3Fe(CN)6, and 0.1% (vol/vol) Triton X-100; and ii) the same recipe except that the concentration of X-Gluc, K4Fe(CN)6, and K3Fe(CN)6 were raised to 0.2, 1.0, and 1.0 mM, respectively. Briefly, tis-sues were directly stained in X-Gluc solution under vacuum infiltration for 15 min and then incubated at 37°C for a further 2 h (for soybean hairy roots) or 24 h (for L. japonicus seed-lings). Shoots were cleared in 70% ethanol before visualization. Tissues chosen for sectioning were fixed in 0.5% paraformal-dehyde in 100 mM sodium phosphate buffer (pH 7) on ice un-der vacuum for 45 min, rinsed three times in 100 mM sodium phosphate buffer (pH 7) at room temperature, stained in X-Gluc solution, embedded in 3% agarose, and sectioned to 100-μm thickness with a vibratome (Lancer Series 1000; Ted Pella, Irvine, CA, U.S.A.).
ACKNOWLEDGMENTS
We thank the ARC for provision of a Centre of Excellence grant and the Queensland State Government Smart State Innovation Scheme as well as the University of Queensland for support. We would like to thank R. Simp-son (School of Molecular and Microbial Sciences, University of Queens-land, St Lucia, QLD, Australia) for providing 25S rRNA primers for QRT-PCR and his assistance with the optimization of QRT-PCR. We also wish to thank T. S. Laniya for providing the GmNARK promoter sequence and C. Vickers (Department of Biology, University of Essex, Colchester, U.K.) for providing a plasmid used in the construction of GmNARK promoter and GUSPlus fusion. S. Nontachaiyapoom acknowledges the Ph.D. schol-arship from the Institute for the Promotion of Teaching Science and Tech-nology (IPST), Thailand.
LITERATURE CITED
Arai, M., Hayashi, M., Takahashi, M., Shimada, S., and Harada, K. 2005. Expression and sequence analysis of systemic regulation gene for sym-biosis, NTS1/GmNARK in supernodulating soybean cultivar, Sakukei 4. Breed. Sci. 55:147-152.
Bailey, T. L., and Elkan, C. 1994. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Pages 28-36 in: Proc. Second Int. Conf. Intelligent Syst. Mol. Biology. The Association for the Advancement of Artificial Intelligence Press, Menlo Park, CA, U.S.A..
Baima, S., Possenti, M., Matteucci, A., Wisman, E., Altamura, M. M., Ruberti, I., and Morelli, G. 2001. The Arabidopsis ATHB-8 HD-zip protein acts as a differentiation-promoting transcription factor of the vascular meristems. Plant Physiol. 126:643-655.
Bassett, C. L., Nickerson, M. L., Farrell, R. E., and Harrison, M. 2004. Multiple transcripts of a gene for a leucine-rich repeat receptor kinase form morning glory (Ipomoea nil) originate from different TATA boxes in a tissue-specific manner. Mol. Gen. Genet. 271:752-760.
Beveridge, C. A., Mathesius, U., Rose, R. J., and Gresshoff, P. M. 2007. Common regulatory themes in meristem development and whole-plant homeostasis. Curr. Opin. Plant Biol. 10:44-51.
Broughton, W. J., and Dilworth, M. J. 1971. Control of leghaemoglobin synthesis in snake beans. Biochem. J. 125:1075-1080.
Buzas, D. M., and Gresshoff, P. M. 2007. Short- and long-distance control of root development by LjHAR1 during the juvenile stage of Lotus japonicus. J. Plant Physiol. 164: 452-459.
Buzas, D. M., Lohar, D., Sato, S., Nakamura, Y., Tabata, S., Vickers, C. E., Stiller, J., and Gresshoff, P. M. 2005. Promoter trapping in Lotus ja-ponicus reveals novel root and nodule GUS expression domains. Plant Cell Physiol. 46:1202-1212.
Caetano-Anollés, G., and Gresshoff, P. M. 1990. Early induction of feed-back regulatory responses governing nodulation in soybean. Plant Sci. 71:69-81.
Carroll, B. J., McNeil, D. L., and Gresshoff, P. M. 1985a. Isolation and properties of soybean (Glycine max (L.) Merr.) mutants that nodulate in the presence of high nitrate concentrations. Proc. Natl. Acad. Sci. U.S.A. 82:4162-4166.
Carroll, B. J., McNeil, D. L., and Gresshoff, P. M. 1985b. A supernodula-tion and nitrate tolerant symbiotic (nts) soybean mutant. Plant Physiol. 78:34-40.
Dellaporta, S., Wood, J., and Hicks, J. B. 1983. A plant DNA miniprepara-tion: Version II. Plant Mol. Biol. Rep. 1:19-21.
Delves, A. C., Mathews, A., Day, D. A., Carter, A. S., Carroll, B. J., and Gresshoff, P. M. 1986. Regulation of the soybean-Rhizobium nodule symbiosis by shoot and root factors. Plant Physiol. 82:588-590.
Delves, A. C., Higgins, A., and Gresshoff, P. M. 1992. The shoot apex is not the source of the systemic signal controlling nodule numbers in soy-bean. Plant Cell Environ. 15:249-254.
Doyle, M. C., and InSeob, H. 2001. The roles of two TATA boxes and 3′-flanking region of soybean beta -tubulin gene (tubB1) in light-sensitive expression. Mol. Cells 12:197-203.
Elliott, K. A., and Shirsat, A. H. 1998. Promoter regions of the extA ex-tensin gene from Brassica napus control activation in response to wounding and tensile stress. Plant Mol. Biol. 37:675-687.
Francisco, P. B., and Harper J. E. 1995. Translocatable leaf signal auto-regulates soybean nodulation. Plant Sci. 107:167-176.
Grace, M. L., Chandrasekharan, M. B., Hall, T. C., and Crowe, A. J. 2004. Sequence and spacing of TATA box elements are critical for accurate initiation from the β-phaseolin promoter. J. Biol. Chem. 279:8102-8110.
Higo, K., Ugawa, Y., Iwamoto, M., and Korenaga, T. 1999. Plant cis-acting regulatory DNA elements (PLACE) database: 1999. Nucleic Acids Res. 27:297-300.

780 / Molecular Plant-Microbe Interactions
Jiang, Q., and Gresshoff, P. M. 2002. Shoot-control and genetic mapping of the har1-1 (hypernodulation and aberrant root formation) mutant of Lotus japonicus. Funct. Plant Biol. 29:1371-1376.
Joshi, C. P. 1987. An inspection of the domain between putative TATA box and translation start site in 79 plant genes. Nucleic Acids Res. 15:6643-6653.
Kereszt, A., Li, D., Indrasumunar, A., Nguyen, C. D. T., Nontachaiyapoom, S., Kinkema, M., and Gresshoff, P. M. 2007. Agrobacterium rhizogenes-mediated transformation of soybean to study root biology. Nat. Protoc. 2:948-952.
Krusell, L., Madsen, L. H., Sato, S., Aubert, G., Genua, A. Szczyglowski, K., Duc, G., Kaneko, T., Tabata, S., de Bruijn, F., Pajuelo, E., Sandal, N., and Stougaard, J. 2002. Shoot control of root development and nodulation is mediated by a receptor-like kinase. Nature 420:422-426.
Larkin, P.J., Gibson, J. M., Mathesius, U., Weinman, J. J., Gärtner, E., Hall, E., Tanner, G. J., Rolfe, B. G., and Djordjevic, M. A. 1996. Transgenic white clover. Studies with the auxin responsive promoter, GH3, in root gravitropism and secondary root development. Transgenic Res. 5:325-335.
Lee, W., Heinz, R., Robb, J., and Nazar, R. N. 1994. Differential utiliza-tion of alternate initiation sites in a plant defense gene responding to environmental stimuli. Eur. J. Biochem. 226:109-114.
Malamy, J. E., and Benfey, P. N. 1997. Organization and cell differentia-tion in lateral roots of Arabidopsis thaliana. Development 124:33-44.
Mathesius, U., Weinman, J. J., Rolfe, B. G., and Djordjevic, M. A. 2000. Rhizobia can induce nodules in white clover by “hijacking” mature cor-tical cells activated during lateral root development. Mol. Plant-Microbe Interact. 13:170-182.
Meixner, C., Ludwig-Muller, J., Miersch, O., Gresshoff, P. M., Staehelin, C., and Vierheilig, H. 2005. Lack of mycorrhizal autoregulation and phytohormonal changes in the supernodulating soybean mutant nts1007. Planta 222:709-715.
Men, A. E., Laniya., T. S., Searle, I. R., Iturbe-Ormaetxe, I., Gresshoff, I., Jiang, Q., Carroll, B. J., and Gresshoff, P. M. 2002. Fast neutron muta-genesis of soybean (Glycine soja L.) produces a supernodulating mutant containing a large deletion in linkage group H. Genome Lett. 3:147-155.
Nakagawa, T., and Kawaguchi, M. 2006. Shoot-applied MeJA suppresses root nodulation Lotus japonicus. Plant Cell Physiol. 47:176-180.
Nishimura, R., Hayashi, M., Wu, G., Kouchi, H., Imaizumi-Anraku, H., Murakami, Y., Kawasaki, S., Akao, S., Ohmori, M., Nagasawa, M., Harada, K., and Kawaguchi, M. 2002. HAR1 mediates systemic regula-tion of symbiotic organ development. Nature 420:426-429.
Nutman, P. S. 1948. Physiological studies on nodule formation I. The rela-tion between nodulation and lateral root formation in red clover. Ann. Bot. 12:81-96.
Olah, B., Brieré, C., Becard, G., Dénarié, J., and Gough, C. 2005. Nod fac-tors and a diffusible factor form arbuscular mycorrhizal fungi stimulate
lateral root formation in Medicago truncatula via the DMI1/DMI2 sig-nalling pathway. Plant J. 44:195-207.
Penmetsa, R.V., Frugoli, J., Smith, L., Long, S. R., and Cook, D. 2003. Genetic evidence for dual pathway control of nodule number in Medi-cago truncatula. Plant Physiol. 131:998-1008.
Pierce, M., and Bauer, W. D. 1983. A rapid regulatory response governing nodulation in soybean. Plant Physiol. 73:286-290.
Prestridge, D. S. 1991. Signal Scan: A computer program that scans DNA sequences for eukaryotic transcriptional elements. Comput. Appl. Bio-sci. 7:203-206.
Ramakers, C., Ruijter, J. M., Deprez, R. H. L., and Moorman, A. F. M. 2003. Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neurosci. Lett. 339:62-66.
Rasmussen, R. 2001. Quantification on the LightCycler. Pages 21-34 in: Rapid Cycle Real-Time PCR, Methods and Applications. S. Meuer, C. Wittwer, and K. Nakagawara. eds. Springer Press, Heidelberg, Germany.
Searle, I. R., Men, A. E., Laniya, T. S., Buzas, D. M., Iturbe-Ormaetxe, I., Carroll, B. J., and Gresshoff, P. M. 2003. Long-distance signalling in nodulation directed by a CLAVATA1-like receptor kinase. Science 299:109-112.
Sessa, G., Steindler, C., Morelli, G., and Ruberti, I. 1998. The Arabidopsis ATHB-8, -9 and -14 genes are members of a small gene family coding highly related HD-Zip proteins. Plant Mol. Biol. 38:609-622.
Schnabel, E., Journet, E., Carvalho-Niebel, F., Duc, G., and Frugoli, J. 2005. The Medicago truncatula SUNN gene encodes a CLV1-like leu-cine-rich repeat receptor kinase that regulates nodule number and root length. Plant Mol. Biol. 58:809-822.
Sheng, C., and Harper J. E. 1997. Shoot versus root signal involvement in nodulation and vegetative growth in wild-type and hypernodulating soybean genotypes. Plant Physiol. 113:825-831.
Stiller, J., Martirani, L., Tuppale, S., Chian, R, Chiurazzi, M., and Gresshoff, P. M. 1997. High frequency transformation and regeneration of transgenic plants in the model legume Lotus japonicus. J. Exp. Bot. 48:1357-1365.
Vincent, J. M. 1970. A Manual for the Practical Study of the Root-Nodule Bacteria. Blackwell Scientific Publications, Oxford.
Wilkins, T. A., and Smart, L. B. 1996. Isolation of RNA from plant tissue. Pages 21-42 in: A Laboratory Guide to RNA Isolation, Analysis and Synthesis. P. A. Kreig, ed. Wiley-Liss,New York.
AUTHOR-RECOMMENDED INTERNET RESOURCES
Neural network promoter prediction server: www.fruitfly.org/seq_tools/promoter.html
University Claude Bernard-Lyon 1 website: pbil.univ-lyon1.fr/cap3.php
Copyright © 2022 FDOKUMEN




















