
Preparation of bone carbonate for stable isotope analysis: the effects of treatment time and acid...
14
Preparation of bone carbonate for stable isotope analysis: the effects of treatment time and acid concentration Sandra J. Garvie-Lok a *, Tamara L. Varney b , M. Anne Katzenberg c a Department of Anthropology, University of Alberta, 13–15 HM Tory Building, Edmonton, Alberta T6G 2H4, Canada b Department of Anthropology, University of Manitoba, Winnipeg, Manitoba R3T 5V5, Canada c Department of Archaeology, University of Calgary, Calgary, Alberta T2N 1N4, Canada Received 27 October 2002; received in revised form 22 August 2003; accepted 27 October 2003 Abstract While apatite carbonate 13 C analysis is a useful tool in palaeodietary research, the vulnerability of carbonates to diagenesis necessitates careful sample preparation. The most common preparation method involves dilute acetic acid treatments to remove highly soluble mineral contaminants. However, there is variation in both treatment time and acid concentration. This study addresses the impact of such variation on carbonate 13 C and 18 O, sample loss in solution, and sample Fourier transform infrared spectroscopy (FTIR) characteristics. Samples of fresh and archaeological bone were treated with 1.0 M and 0.1 M acetic acid. Carbonate 13 C and 18 O, sample loss and FTIR characteristics were measured at treatment times of 4 to 24 h. The results suggest that 4 h of treatment suffice to remove highly soluble contaminants, and that longer treatment times can lead to sample recrystallization. Some samples treated with 1.0 M acid showed unacceptable loss or possible recrystallization, suggesting that 0.1 M acid treatment is preferable. However, the more dilute acid caused smaller shifts in 13 C and 18 O for all samples and treatment times. Thus, while 0.1 M acid appears to produce superior sample quality, it may not produce results directly comparable to those of 1.0 M acid treatments. This has important implications for the comparison of apatite carbonate stable isotope values reported in the literature. 2003 Elsevier Ltd. All rights reserved. Keywords: Archaeological bone; Bone carbonate; Bioapatite; Stable carbon isotopes; Stable oxygen isotopes; Sample preparation 1. Introduction The analysis of stable isotopes of carbon in preserved bone collagen for the purpose of reconstructing past diet and ecology has become routine in bioarchaeological studies since its introduction in the 1970s [40,41]. More recently the carbonate in bone mineral has been used as a source of biologically deposited carbon for stable isotope studies. Bone carbonate is present in samples that are very ancient [16] and in samples from tropical regions where preservation is often very poor [12]. When collagen is preserved, analysis of stable carbon isotopes from both collagen and carbonate is useful because the two tissues reflect different dietary sources. Specifically, collagen is formed largely from ingested protein while carbonate is deposited from dissolved bicarbonate in the circulation, which is drawn from all dietary components [1,14,38]. While the use of apatite carbonate 13 C analysis in palaeodietary reconstruction is becoming increasingly common, the vulnerability of bone mineral to diagenesis necessitates careful preparation and evaluation of car- bonate samples to ensure that the biogenic isotopic signals are being obtained. This paper presents the results of a test of varying preparation methods for the isolation of bone carbonate and discusses the chemical properties of samples prepared using these methods. 1.1. Background Sullivan and Krueger [36] first provided evidence that carbon isotopes from carbonate provide useful * Corresponding author. Tel.: +1-708-492-0137; fax: +1-708-492-5273. E-mail address: [email protected] (S.J. Garvie-Lok). Journal of Archaeological Science 31 (2004) 763–776 SCIENCE Journal of Archaeological http://www.elsevier.com/locate/jas SCIENCE Journal of Archaeological http://www.elsevier.com/locate/jas 0305-4403/04/$ - see front matter 2003 Elsevier Ltd. All rights reserved. doi:10.1016/j.jas.2003.10.014
Transcript of Preparation of bone carbonate for stable isotope analysis: the effects of treatment time and acid...

Preparation of bone carbonate for stable isotope analysis:the effects of treatment time and acid concentration
Sandra J. Garvie-Loka*, Tamara L. Varneyb, M. Anne Katzenbergc
aDepartment of Anthropology, University of Alberta, 13–15 HM Tory Building, Edmonton, Alberta T6G 2H4, CanadabDepartment of Anthropology, University of Manitoba, Winnipeg, Manitoba R3T 5V5, Canada
cDepartment of Archaeology, University of Calgary, Calgary, Alberta T2N 1N4, Canada
Received 27 October 2002; received in revised form 22 August 2003; accepted 27 October 2003
Abstract
While apatite carbonate �13C analysis is a useful tool in palaeodietary research, the vulnerability of carbonates to diagenesisnecessitates careful sample preparation. The most common preparation method involves dilute acetic acid treatments to removehighly soluble mineral contaminants. However, there is variation in both treatment time and acid concentration. This studyaddresses the impact of such variation on carbonate �13C and �18O, sample loss in solution, and sample Fourier transform infraredspectroscopy (FTIR) characteristics. Samples of fresh and archaeological bone were treated with 1.0 M and 0.1 M acetic acid.Carbonate �13C and �18O, sample loss and FTIR characteristics were measured at treatment times of 4 to 24 h. The results suggestthat 4 h of treatment suffice to remove highly soluble contaminants, and that longer treatment times can lead to samplerecrystallization. Some samples treated with 1.0 M acid showed unacceptable loss or possible recrystallization, suggesting that0.1 M acid treatment is preferable. However, the more dilute acid caused smaller shifts in �13C and �18O for all samples andtreatment times. Thus, while 0.1 M acid appears to produce superior sample quality, it may not produce results directly comparableto those of 1.0 M acid treatments. This has important implications for the comparison of apatite carbonate stable isotope valuesreported in the literature.� 2003 Elsevier Ltd. All rights reserved.
Keywords: Archaeological bone; Bone carbonate; Bioapatite; Stable carbon isotopes; Stable oxygen isotopes; Sample preparation
1. Introduction
The analysis of stable isotopes of carbon in preservedbone collagen for the purpose of reconstructing past dietand ecology has become routine in bioarchaeologicalstudies since its introduction in the 1970s [40,41]. Morerecently the carbonate in bone mineral has been used asa source of biologically deposited carbon for stableisotope studies. Bone carbonate is present in samplesthat are very ancient [16] and in samples from tropicalregions where preservation is often very poor [12]. Whencollagen is preserved, analysis of stable carbon isotopesfrom both collagen and carbonate is useful because thetwo tissues reflect different dietary sources. Specifically,
collagen is formed largely from ingested protein whilecarbonate is deposited from dissolved bicarbonate in thecirculation, which is drawn from all dietary components[1,14,38].
While the use of apatite carbonate �13C analysis inpalaeodietary reconstruction is becoming increasinglycommon, the vulnerability of bone mineral to diagenesisnecessitates careful preparation and evaluation of car-bonate samples to ensure that the biogenic isotopicsignals are being obtained. This paper presents theresults of a test of varying preparation methods for theisolation of bone carbonate and discusses the chemicalproperties of samples prepared using these methods.
1.1. Background
Sullivan and Krueger [36] first provided evidencethat carbon isotopes from carbonate provide useful
* Corresponding author. Tel.: +1-708-492-0137;fax: +1-708-492-5273.
E-mail address: [email protected] (S.J. Garvie-Lok).
Journal of Archaeological Science 31 (2004) 763–776
SCIENCE
Journal of
Archaeological
http://www.elsevier.com/locate/jas
SCIENCE
Journal of
Archaeological
http://www.elsevier.com/locate/jas
0305-4403/04/$ - see front matter � 2003 Elsevier Ltd. All rights reserved.doi:10.1016/j.jas.2003.10.014

information similar to that derived from collagen andthat it may still be possible to obtain dietary informationfrom bone when collagen preservation is poor. However,this was disputed by Schoeninger and DeNiro [30] whofound inconsistent results when comparing stable car-bon isotope ratios from the two tissues. The debate thatensued revolved partially around conflicting assump-tions about the meaning of collagen-carbonate �13Cspacing, with some (e.g., [29,32]) arguing that thisspacing should be consistent and others (e.g., [14]) that itshould vary. This debate was complicated by the varietyof sample preparation methods used [30,31,36,37]. Thecontroversy is reviewed more completely by Krueger [13]and Lee-Thorp and van der Merwe [19]. Further workhas clarified the relationship between collagen andcarbonate �13C in mammals [1,38] and suggested aconsistent method for sample preparation [13,16], form-ing a theoretical background for stable isotope researchusing archaeological tooth and bone carbonates.
1.2. Current preparation methods
Current preparation methods for archaeological boneand tooth enamel carbonate are generally based on thetechnique developed by Lee-Thorp [16,19]. Briefly, themethod involves treating samples with a soak in 1.0 Macetic acid solution for 24 to 36 h in order to remove themost soluble mineral fraction. The rationale behind thisapproach is to remove as much as possible of the labilecarbonate present in the sample.1 It is reasoned that thiscarbonate, which is the most easily dissolved, is also themost likely to be contaminated, since it includes themost easily contaminated fraction of the carbonatepresent in life as well as the more soluble exogenouscontaminants such as calcite which may have beendeposited in the sample during diagenesis [16]. However,it is important to recognize that the solubility of diage-netic contaminants of bone and teeth varies. Undersome conditions, recrystallization of bone or tooth min-eral during diagenesis can lead to the formation ofhighly soluble, high-carbonate recrystallized apatite inarchaeological samples. Such recrystallized material,which can be dissolved with acetic acid treatment, hasbeen documented in a number of studies [11,16,46].However, under different conditions archaeologicalsamples may become contaminated with recrystallizedapatite showing high crystallinity, low carbonate content
and FTIR profiles characteristic of fluorine-containingapatite [11,23,35,46]. Such material, being less solublethan the intact bioapatite, will not be removed by aceticacid treatment.
While the original acetic acid treatment technique asdeveloped by Lee-Thorp [16] is used extensively in thecurrent literature (e.g., [8–10,28,33,39,42,46]), otherstudies have implemented modifications. These includevariations in both acid solution concentration and acidtreatment time. For example, some researchers (e.g.,[4,5,45,47]) have used buffered 1.0 M acetic acid solutionwhile others (e.g., [2,18,22,23]) have used 0.1 M aceticacid solution. As well, some researchers (e.g., [2,5,18])have limited treatment time in the acetic acid to lessthan the 24 to 36 h limit suggested by Lee-Thorp andvan der Merwe [19]. The variation seen in currentpreparation methods stems from concerns about theeffects of overly harsh acid treatment on apatite samples,which may include treatment-induced recrystallizationand excessive sample loss.
The potential for bone or tooth mineral to recrystal-lize during sample preparation is clear. As reviewed byLeGeros [20], bone and tooth mineral can be recrystal-lized in vitro at a range of pH values and solution ionconcentrations. The crystal species that will formdepends on factors including temperature, solutionacidity and the concentration and type of ions presentin the solution. As recrystallization during samplepreparation may incorporate exogenous minerals intothe sample, defeating the purpose of the treatmentprocess, the risk that it will occur deserves consideration.
The observations of researchers working witharchaeological and modern bone and tooth mineralconfirm that recrystallization occurs under the con-ditions typical of sample preparation, and suggest thatthe risk of such recrystallization increases with longertreatment times and more concentrated acid solutions.Lee-Thorp [16,19] documented the gradual formation ofbrushite in modern bone treated with 1.0 M acetic acid.No brushite was present prior to treatment; after 48 h oftreatment, brushite was detectable, and after 7 days oftreatment, the majority of the sample was brushite.Recrystallization can also occur after long treatmentwith more dilute acetic acid. Koch et al. [11] reported thedevelopment of brushite in modern dentine samplestreated with 0.1 M acetic acid for 72 h. In a wider study,Nielsen-Marsh and Hedges [21] explored the effects ofacetic, proprionic and hydrochloric acid treatments ofvarying duration and concentration on bone samples.Some treatments caused demonstrable recrystallizationto brushite; for example, brushite formed in modernbone after 72 h of treatment with 1.0 M acetic acid.However, of those that did not result in brushiteformation, some treatments caused increases in samplecrystallinity that continued after net sample dissolution(as measured by loss of sample weight) was complete.
1 The carbonate of bone and tooth mineral includes structural andlabile carbonate. As reviewed by LeGeros [20], structural carbonate issubstituted into the crystal lattice at the OH� and PO4
3� positions;these substitutions are referred to respectively as Type A and Type Bstructural carbonate. Labile (adsorbed) carbonate is not associatedwith a well-defined lattice position. Instead, it is adsorbed on to thesurfaces of apatite crystals or incorporated into less mature, moredisorganized regions of apatite crystals [3,6,25,27]. It is more easilydissolved and altered than the more stable structural carbonate.
S.J. Garvie-Lok et al. / Journal of Archaeological Science 31 (2004) 763–776764

Nielsen-Marsh and Hedges observed [21, pp. 489–490]that although brushite was not detected in thesesamples, the continued increase in crystallinity in theabsence of net sample dissolution strongly suggestedrecrystallization. Considering their results for acetic acidtreatments alone, shorter treatment times, more dilutesolutions and lower solution acidity were all linked toless crystallinity increase. Taken together, the results ofthe three studies suggest that shorter treatment timesand buffered or more dilute acid preparations mayreduce the risk of sample recrystallization.
Excessive loss of sample during treatment is anotherproblem sometimes associated with the use of 1.0 Macetic acid. These authors have observed unacceptableloss of sample (well over 50% of sample weight) in somesamples treated with 1.0 M acetic acid. Other studies inthe literature report similar observations, and suggestcompensating by reducing solution strength or treat-ment time. Balasse et al. [2] report that 24 h treatmentswith 1.0 M acetic acid solution and 6 h treatments with0.2 M acetic acid solution both caused unacceptabledissolution in finely powdered enamel samples. How-ever, when solution concentration was reduced to 0.1 Mand treatment time reduced to 4 h, sample loss wasreduced to acceptable levels. Koch et al. [11] have alsodemonstrated that using 0.1 M or buffered 1.0 M aceticacid solutions to treat samples reduces sample dissol-ution. Finally, Lee-Thorp et al. [18] have suggestedreducing treatment times for very small and finelypowdered samples to as little as 15 min to prevent exces-sive loss. Excessive sample loss is not solely related to acidsolution strength or treatment time. It can be linked to theparticle size to which the sample is ground; for example,the extremely fine enamel powder produced by a dentaldrill is more vulnerable to dissolution in treatment thanis the larger powder produced by a Spex freezer mill [18].The degree of dissolution may also reflect the maturityof the tissue under study. As the crystallinity of imma-ture bone mineral is relatively low (e.g., [24,26]), samplesfrom juveniles may be more vulnerable to dissolutionduring treatment. These factors aside, however, moreconcentrated acid solutions and longer treatment timesboth increase the risk of unacceptable sample loss.
Thus, recent experiments have suggested that the useof shorter treatment times and more dilute or bufferedacetic acid solutions may lessen the risk of recrystalliz-ation and reduce sample loss in treatment. However, theresults are still somewhat contradictory. Some datasuggest good sample quality after 24 h or more in 1.0 Macetic acid [13,17,19]. Other results show sampledegeneration after treatment with 1.0 M acetic acid butgenerally good quality after treatment with 0.1 M aceticacid [2,11,34]. Yet others suggest significant alterationafter as little as an hour in 0.1 M acetic acid [21]. Animportant factor in these variable results appears to bevariation in sample vulnerability to the effects of acid
treatment due to differences in diagenetic history. Thecurrent preparation methods were initially developed totreat fossilized material [16], and successful studies onfossilized bones (e.g., [16,17]) show that well-preservedmineral may remain in bone for millions of years.However, work on more recently deposited, non-fossilized bones shows that variation in diagenetichistory affects the success of acetic acid treatment. Whensamples are contaminated only with highly solublediagenetic material such as calcite, treatment with aceticacid can potentially eliminate the diagenetic con-taminants and leave the remaining bone mineral in goodcondition. However, when recent archaeologicalmaterial is contaminated with carbonate-poor recrystal-lized apatite, which is of lower solubility than theremaining intact bone mineral, poor final results areobtained regardless of acid solution concentration [23].
A final point must be raised when discussing thedocumented effects of different acid preparations onbone and tooth apatite samples. While acetic acid solu-tion concentration has an impact on sample loss andrecrystallization, it also appears to have an impact onsample stable isotope values. Studies in which fresh ornearly fresh modern bones and teeth were treated withacid solutions [11,15,16,46] have documented changes insample carbonate �13C and �18O after acid treatment,with �13C values dropping and �18O values increasing.Koch et al. [11] found that this effect was more pro-nounced for 1.0 M acetic acid than for 0.1 M acetic acid.Since these stable isotope shifts in fresh samples cannotbe attributed to the removal of diagenetic material, theymust be due to a fundamental effect on the bone ortooth mineral itself. Koch et al. [11] propose that theymay be due to the differential removal of structuralcarbonate substitutions in A or B lattice sites by the acidtreatments, suggesting that the stable isotope values oftype A and B structural carbonate differ systematically.2
Whatever the cause, these effects are a concern whenconsidering a change in sample preparation methods, assamples treated with different acid preparations may nothave directly comparable stable isotope values.
1.3. Questions to be resolved
The variation in preparation methodology seen in theliterature and the inconclusive results of recent researchpose some questions which this study attempts toresolve. First, does 0.1 M acetic acid offer superiorresults to 1.0 M acetic acid when used to treat recentarchaeological bone? Second, how rapidly are the stableisotope values and other traits of powdered bone
2 Type A and B carbonate substitutions are defined in Note 1. Thecarbonate in these positions has been shown to behave differently inlife and during diagenesis [24,26,27,34]. Thus, it is reasonable tosuggest that they also show systematic differences in stable isotoperatio.
S.J. Garvie-Lok et al. / Journal of Archaeological Science 31 (2004) 763–776 765

samples altered during the first few hours of exposure to0.1 M and 1.0 M acetic acid solutions? The effects of1.0 M and 0.1 M acetic acid on bone and enamel mineralhave been compared by Koch et al. [11], Nielsen-Marshand Hedges [21] and Balasse et al. [2]. However, Kochet al. [11] and Balasse et al. [2] did not use similartreatment times when comparing the 1.0 M and 0.1 Mtreatment solutions, and Nielsen-Marsh and Hedges [21]and Balasse et al. [2] did not compare the stable isotopevalues of their samples. Thus, a comparison of theeffects of similarly timed 1.0 M and 0.1 M acetic acidtreatments on the quality and stable isotope values ofbone samples will provide novel information. This infor-mation will help to clarify the relative merits of shortexposures to 1.0 M acetic acid and longer exposures to0.1 M acid. It will also address the question of whetherthe stable isotope values of samples prepared usingdifferent acid treatments are comparable. This questionis important, as preparation methods for bone mineralare not uniform from one laboratory to another, and arechanging over time. If different methods produce sys-tematically different results, data from different studiesmay not be directly comparable.
2. Methods
Archaeological mammal bones of four species werechosen to represent a range of preservation conditionstypical of recent (<2 kya) archaeological bone (seeTable 1). Thus, the study material is more comparable tothe Holocene bone examined by Koch et al. [11] andBalasse et al. [2] than to the much older fossil materialexamined in work such as Lee-Thorp [16]. Archaeol-ogical sample preservation was assessed based on appar-ent condition (informally assessed from gross signs suchas friability and surface cracking), carbonate contentand crystallinity as measured by FTIR and organiccontent based on loss of mass in NaOCl (Table 1). Masslost in NaOCl does not represent the total amount oforganic material in the bone, as 10% to 15% of theorganic matter in fresh bone is found in apatite crystalaggregates where it is not vulnerable to attack by theNaOCl solution [44]. However, aggregate organic matteris relatively invulnerable to diagenesis; although theoverall organic content of bone decreases during dia-genesis, the amount of organic matter seen in crystalaggregates does not [7]. Thus, while the proportion of abone sample lost in NaOCl does not reflect its totalorganic content, it does reflect the survival of thatorganic fraction which is susceptible to loss duringdiagenesis. A fresh modern cow bone obtained from abutcher was also included in the sample set. The inclu-sion of a fresh bone sample served to document theeffects of acid treatment on bone which had not beendiagenetically altered, and to further examine the effects
of 1.0 M and 0.1 M acetic acid on fresh bone that werenoted by Koch et al. [11].
Samples were prepared for acid treatment as follows.Archaeological samples were scrubbed with distilledwater and any cancellous bone was removed to eliminatepossible trapped soil. They were then cleaned ultra-sonically in double-distilled water, and air-dried for48 h. The fresh bone was manually freed of soft tissue,then cleaned in the same way. All samples were thenreduced to a uniform powder by grinding for 3 min in aSpex liquid nitrogen freezer mill. For each sample,thirteen 250 mg aliquots were weighed into lidded 14 mlcentrifuge tubes. To remove most of the organic com-ponent, the subsamples were treated with 2% sodiumhypochlorite. Ten ml of solution was added to eachtube, shaken with the powder and allowed to stand for12 h, after which the tubes were centrifuged, the super-natant discarded, and 10 ml of fresh solution added.This process was repeated three times for a total of 48 hof treatment; all subsamples were then rinsed to neu-trality with repeated changes of double-distilled water.For each sample, one subsample was lyophilized forFTIR and stable isotope analysis. This subsampleallowed weight loss due to the water and sodiumhypochlorite treatment to be determined and the stableisotope values and FTIR profiles of the sample prior toacid treatment to be defined. The remainder of thesubsamples were passed on to the acid treatmentprocess.
Subsamples were divided into two acid treatmentgroups, one to be treated with 1.0 M acetic acid solutionand one to be treated with 0.1 M acetic acid solution.10 ml of solution was added to each tube; the volumeof solution used in proportion to sample weight(0.04 ml/mg) was based on the procedure of Koch et al.[11]. Individual subsamples were rinsed to neutrality at4 h intervals from 4 to 48 h. Each subsample received a5 min exposure to vacuum halfway through treatment toencourage complete exposure of crystal surfaces to theacid solution [13]. After rinsing, all samples were lyo-philized and weighed. Stable isotope and FTIR analysiswas performed on the 4, 12 and 24 h groups, with othergroups retained for later analysis if necessary.
Stable isotope analysis was performed at theIsotope Science Laboratory of the University of CalgaryDepartment of Physics and Astronomy. CO2 wasliberated from the samples by combination with 100%anhydrous phosphoric acid under vacuum; the reactionwas allowed to proceed at room temperature untilcomplete. CO2 gas was cryogenically purified andcollected for analysis on a VG SIRA mass spectrometer.Correction of �13C and �18O was done directly to aninternal standard (Lublin calcite) prepared and rununder the same conditions whose �13C and �18O valuesrelative to international standards had been previouslydetermined in the same laboratory (this assumed
S.J. Garvie-Lok et al. / Journal of Archaeological Science 31 (2004) 763–776766

Table 1Initial condition of samples
Sample Species Origin Context Age (y) Appearance Weight lost in NaOCl (%) FTIR CI FTIR C/P
Fresh cow Canada – 0 fresh 26.6 2.9 0.231 cow Greece midden 700–800 excellent (very hard, little colour change) 22.8 2.8 0.342 human Antigua plough zone 1400–1800 poor (chalky, badly leached) 13.2 2.9 0.333 bison Canadian Plains kill site 800–1000 poor (soft, surface cracked and badly discoloured) 18.1 3.8 0.134 caribou Canadian Arctic living floor ca. 700 good (firm, moderately discoloured) 28.7 2.8 0.27
S.J.
Garvie-L
oket
al./
Journalof
Archaeological
Science
31(2004)
763–776767

the �18O fractionation of the reaction between apatitecarbonate and H3PO4 to be identical to that for calcite).The precision of the stable isotope analysis was �0.1‰.
FTIR analysis was performed at the University ofCalgary Department of Chemistry. 1.5 mg of samplewas ground with 200 mg of analytical quality KBr in anagate mortar and pestle and sintered into a disk in ahydraulic press at 10,000 lb of pressure for 2 min.Spectra were collected from 4000 to 400 cm�1 using 16scans and a resolution of 8 cm�1 on a Mattson GenesisSeries FTIR analyzer. Spectra were baseline correctedand heights at 565, 590, 605, 1035 and 1415 cm�1 weremeasured. Peak heights were used to calculateFTIR indices of crystallinity and carbonate content.Crystallinity (FTIR CI) was defined as [565ht+605ht]/590ht, where Xht is height at X wavenumbers (e.g.,[21–23,43,46]). Carbonate content (FTIR C/P) wasdefined as 1415ht/1035ht, a ratio shown to have a goodlinear relationship with overall carbonate content asmeasured by other means [34,46]. Duplicate FTIR scanswere performed on some samples to determine therepeatability of the CI and C/P measurements. Repeatmeasurements of FTIR CI fell within 0.1 of each other,while repeat measurements of FTIR C/P fell within 0.03of each other. An illustration of the calculation of FTIRCI and C/P ratios from a scan is provided in Fig. 1.
3. Results
3.1. Initial sample quality
Measures of the initial quality of the samples prior toacid treatment are provided in Table 1. The samples’
loss of mass in NaOCl generally reflects their apparentcondition, with the fresh and well-preserved bones show-ing higher organic content than the more poorly pre-served bones. The exception is sample 4, which has ahigher organic content than even the fresh bone,although its texture suggests that significant diagenesishas occurred. This may indicate that most of thecollagen present in the sample has been degraded intoshort segments that were retained in the bone, makingthe sample more friable but not affecting its organiccontent. FTIR data for the fresh bone (CI=2.9,C/P=0.23) are similar to values reported by Wright andSchwarcz [46] for fresh bone. Spectra of samples 1, 2 and4 are similar to that of fresh bone apatite, yielding CIvalues similar to that of the fresh bone and C/P valuessomewhat higher (Table 1). The FTIR data for sample 3suggest poorer mineral preservation. With higher CI,lower C/P and an abrupt shoulder at 1096 cm�1, it issimilar to bones described in other studies whose apatitehas partially recrystallized to francolite, a low-carbonateapatite with fluoride substitutions [23,46].
3.2. Response to acid treatment
Dissolution in acid treatment was determined fromthe final weight of dry prepared bone mineral remainingafter the treatment was complete, taken as a percentageof the original weight present. This measure will bereferred to from this point as the mineral yield of thesamples. Mineral yields for all samples for 0 through48 h of treatment with 1.0 and 0.1 M acid solutions aredepicted in Fig. 2. Note that the mineral yield values at0 h are less than 100%; this original value reflects theloss of sample weight due to the water and sodiumhypochlorite treatment. Both 0.1 M and 1.0 M acidsolutions produce a sharp drop in mineral yield for allsamples within the first 4 h. After this point almost all ofthe samples show a rough plateau; the exception is the0.1 M treatment of sample 3, which does not establish aplateau until the 16 h point.
Crystallinity shows little change in general (Table 2).Acid treatment does not have a significant effect onsample CI in most cases. However, archaeologicalsamples 2 and 3 show slight CI increases (+0.2), and thefresh bone shows decreased CI when treated with 1.0 Macid. The meaning of this apparent drop in crystallinityis discussed further below.
The response of carbonate content to acid treatmentis more variable (Table 3). C/P of fresh bone shows aslight decrease (0.23 to 0.19) after treatment with 0.1 Macid, and an increase after treatment with 1.0 M acid.The behaviour of C/P in the archaeological bonesamples varies. Sample 2 shows a sharp initial decline inC/P, more marked for the 1.0 M acid, followed by aplateau. However, samples 1 and 3 show little change,and sample 4 shows an apparent increase in carbonate
Fig. 1. Calculation of FTIR CI and FTIR C/P. Calculation of FTIRCI and C/P: baselines are drawn as shown and the heights of thepeaks at 565, 590, 605, 1035 and 1415 wavenumbers are measured.FTIR indices are calculated as follows, where Xht is the height ofthe peak at X wavenumbers: FTIRCI=(565ht+605ht)/590ht; FTIRC/P=1415ht/1035ht.
S.J. Garvie-Lok et al. / Journal of Archaeological Science 31 (2004) 763–776768

content similar to that seen in the 1.0 M acid treatmentof fresh bone.
The initial carbonate �13C values of the samples afterNaOCl treatment, and �13C values for samples at 4, 12and 24 h of acetic acid treatment, are shown in Fig. 3
and Table 4. Information for �18O is presented in Fig. 4and Table 5. For the fresh bone, the acid treatmentsresult in lower �13C and higher �18O; both changes aremore marked in the 1.0 M acid treatment. The patternfor archaeological samples 1, 2 and 4 is similar. Sample
0.1 M
0%
20%
40%
60%
80%
100%
0 4 8 12 16 20 24 28 32 36 40 44 48
Treatment time (hours)
Min
eral
Yie
ld (%
orig
inal
wei
ght)
1.0 M
0%
20%
40%
60%
80%
100%
Treatment time (hours)
Fresh
Sample 1
Sample 2
Sample 3
Sample 4
0 4 8 12 16 20 24 28 32 36 40 44 48
Fig. 2. Sample loss by time and acid concentration.
Table 2Changes in FTIR CI after treatment with 1.0 M and 0.1 M aceticacid
Sample FTIR CI after acid treatment
0 h 4 h 12 h 24 h
Fresh 1.0 M 2.9 2.7 2.5 2.80.1 M 2.9 3.0 3.0 2.7
Sample 1 1.0 M 2.8 2.8 2.8 2.80.1 M 2.8 2.9 2.7 2.8
Sample 2 1.0 M 2.9 2.9 3.2 3.10.1 M 2.9 3.2 3.0 3.0
Sample 3 1.0 M 3.8 3.8 3.8 3.80.1 M 3.8 4.0 3.9 4.0
Sample 4 1.0 M 2.8 2.9 2.9 2.90.1 M 2.8 2.8 2.8 2.8
Table 3Changes in FTIR C/P after treatment with 1.0 M and 0.1 M aceticacid
Sample FTIR C/P after acid treatment
0 h 4 h 12 h 24 h
Fresh 1.0 M 0.23 0.41 0.45 0.370.1 M 0.23 0.22 0.19 0.19
Sample 1 1.0 M 0.34 0.30 0.33 0.330.1 M 0.34 0.33 0.34 0.26
Sample 2 1.0 M 0.33 0.19 0.19 0.180.1 M 0.33 0.24 0.22 0.21
Sample 3 1.0 M 0.13 0.10 0.09 0.100.1 M 0.13 0.12 0.10 0.11
Sample 4 1.0 M 0.27 0.59 0.50 0.270.1 M 0.27 0.37 0.27 0.26
Table 4Changes in carbonate �13C after treatment with 1.0 M and 0.1 Macetic acid
Sample Carbonate �13C after acid treatment (‰ PDB)
0 h 4 h 12 h 24 h
Fresh 1.0 M �13.6 �13.5 �15.2 �15.20.1 M �13.6 �14.5 �14.6 �14.5
Sample 1 1.0 M �9.5 �12.0 �11.3 �11.50.1 M �9.5 �11.1 �11.0 �10.6
Sample 2 1.0 M �8.5 �10.8 �10.8 �10.80.1 M �8.5 �9.6 �9.7 �9.8
Sample 3 1.0 M �7.7 �7.8 �8.9 �9.00.1 M �7.7 �8.7 �8.7 �9.0
Sample 4 1.0 M �10.7 �10.5 �11.0 �12.10.1 M �10.7 �10.4 �10.6 �10.7
Table 5Changes in carbonate �18O after treatment with 1.0 M and 0.1 Macetic acid
Sample Carbonate �18O after acid treatment (‰ PDB)
0 h 4 h 12 h 24 h
Fresh 1.0 M �14.9 �10.0 �10.1 �10.70.1 M �14.9 �12.0 �12.9 �14.2
Sample 1 1.0 M �7.8 �3.3 �3.4 �3.40.1 M �7.8 �5.1 �4.6 �3.6
Sample 2 1.0 M �7.8 �2.8 �2.4 �2.80.1 M �7.8 �4.2 �4.0 �4.2
Sample 3 1.0 M �15.3 �16.9 �12.1 �12.30.1 M �15.3 �14.7 �14.1 �13.1
Sample 4 1.0 M �17.9 �14.2 �15.2 �15.60.1 M �17.9 �13.5 �16.7 �16.2
S.J. Garvie-Lok et al. / Journal of Archaeological Science 31 (2004) 763–776 769

3 shows a small change in �13C for both treatments, and�18O changes similar to those seen in the other samples.For all samples except archaeological sample 3, thegreatest change in �13C and �18O is seen in the first 4 h oftreatment. After this point, stable isotope values tend toestablish a rough plateau; this is more uniform for �13Cthan for �18O.
During the course of the study, the systematic effectof 1.0 M and 0.1 M acetic acid on sample stable isotopevalues was noted, and the possibility was considered that
it was a function of the proportion of the sample thatwas dissolved, with more sample dissolution leading to agreater change in stable isotope value. This possibilitywas tested by performing 4 h 1.0 M and 0.1 M aceticacid treatments on a repeat set of samples using doublethe proportion of solution to sample. As expected, thedoubled solution volumes dissolved a substantiallygreater amount of mineral. However, there was nocorresponding increase in �13C or �18O shifts. For mostsamples, the shift in �13C and �18O was close to that
Fresh Bone
-16
-15
-14
-13
-12
-11
-10
-9
-8
-7
0 4 8 12 16 20 24
Treatment time (hours)
Car
bona
teδ
13C
(‰PD
B)
Archaeological Sample 1
-16
-15
-14
-13
-12
-11
-10
-9
-8
-7
0 4 8 12 16 20 24
Treatment time (hours)
Car
bona
te δδ
13C
(‰PD
B)
Archaeological Sample 2
-16
-15
-14
-13
-12
-11
-10
-9
-8
-7
0 4 8 12 16 20 24
Treatment time (hours)
Car
bona
teδ
13C
(‰PD
B)
Archaeological Sample 3
-16
-15
-14
-13
-12
-11
-10
-9
-8
-7
0 4 8 12 16 20 24
Treatment time (hours)
Car
bona
teδ
13C
(‰PD
B)
Archaeological Sample 4
-16
-15
-14
-13
-12
-11
-10
-9
-8
-7
0 4 8 12 16 20 24
Treatment time (hours)
Car
bona
teδ
13C
(‰PD
B)
Fig. 3. Sample �13C by time and acid concentration (C=1.0 M acetic acid; B=0.1 M acetic acid).
S.J. Garvie-Lok et al. / Journal of Archaeological Science 31 (2004) 763–776770
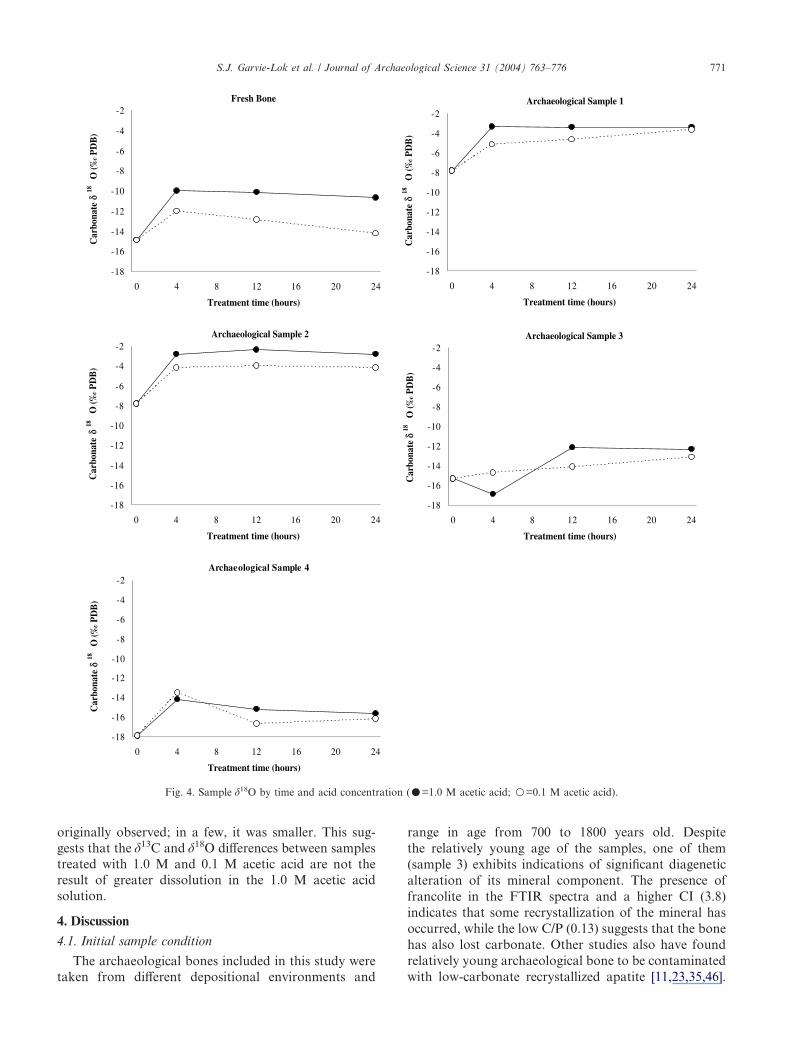
originally observed; in a few, it was smaller. This sug-gests that the �13C and �18O differences between samplestreated with 1.0 M and 0.1 M acetic acid are not theresult of greater dissolution in the 1.0 M acetic acidsolution.
4. Discussion
4.1. Initial sample condition
The archaeological bones included in this study weretaken from different depositional environments and
range in age from 700 to 1800 years old. Despitethe relatively young age of the samples, one of them(sample 3) exhibits indications of significant diageneticalteration of its mineral component. The presence offrancolite in the FTIR spectra and a higher CI (3.8)indicates that some recrystallization of the mineral hasoccurred, while the low C/P (0.13) suggests that the bonehas also lost carbonate. Other studies also have foundrelatively young archaeological bone to be contaminatedwith low-carbonate recrystallized apatite [11,23,35,46].
Fresh Bone
-18
-16
-14
-12
-10
-8
-6
-4
-2
0 4 8 12 16 20 24
Treatment time (hours)
Car
bona
teδ
18O
(‰ P
DB
)Archaeological Sample 1
-18
-16
-14
-12
-10
-8
-6
-4
-2
0 4 8 12 16 20 24
Treatment time (hours)
Car
bona
teδ
18O
(‰ P
DB
)
Archaeological Sample 2
-18
-16
-14
-12
-10
-8
-6
-4
-2
0 4 8 12 16 20 24
Treatment time (hours)
Car
bona
teδ
18O
(‰ P
DB
)
Archaeological Sample 3
-18
-16
-14
-12
-10
-8
-6
-4
-2
0 4 8 12 16 20 24
Treatment time (hours)
Car
bona
te δδ
18O
(‰ P
DB
)
Archaeological Sample 4
-18
-16
-14
-12
-10
-8
-6
-4
-2
0 4 8 12 16 20 24
Treatment time (hours)
Car
bona
teδ
18O
(‰ P
DB
)
Fig. 4. Sample �18O by time and acid concentration (C=1.0 M acetic acid; B=0.1 M acetic acid).
S.J. Garvie-Lok et al. / Journal of Archaeological Science 31 (2004) 763–776 771

Samples contaminated in this way, whether fossil orarchaeological, have not been found to respond well toacetic acid treatment [22,46].
4.2. Dissolution in acetic acid
Both fresh and archaeological samples show rapiddissolution in the acetic acid treatment. Sample loss isof a greater magnitude for both fresh bone and thearchaeological samples in the 1.0 M acid solution. Thefresh bone shows the greatest dissolution having lost68% and 35% of the original sample weight after 4 h in1.0 M acid and 0.1 M acid solutions respectively. Theimproved mineral yields in 0.1 M acid are consistentwith other data on sample dissolution and acid solutionconcentration [2,11,21]. Almost all of the samples showa rough plateau in mineral yield after the 4 h mark. Theone exception is the 0.1 M treatment of sample 3, whichdoes not appear to establish a plateau until the 16 hpoint. Minor fluctuations in the mineral yield plateauxlikely reflect experimental error due to differences in thevigour with which subsamples were shaken during mix-ing and rinsing, or in the amount of extremely finesample powder that was lost during rinsing. The con-stancy of mineral yield after a period of time suggeststhat the solutions involved reached saturation. Thiseffect is expected for proportions of acid solution tosample similar to the 0.04 ml/mg used in this study[21,22].
4.3. Impact of acetic acid treatment on CI
In general, the changes in FTIR CI seen after acidtreatment do not exceed the measurement uncertainty of�0.1. However, increases larger than this are seen inarchaeological sample 2 in both 1.0 M and 0.1 M acidtreatments, and in archaeological sample 4 in 1.0 Macid, while a CI decrease is seen in fresh bone treatedwith 1.0 M acid. The CI values of the treated samples donot stay constant from 4 to 24 h of treatment time;instead, they fluctuate somewhat. While this may reflectmeasurement error between the different subsamples,Nielsen-Marsh and Hedges [21] demonstrated that thecrystallinity of a sample in treatment may alter withoutnet dissolution being seen, and this opens the possibilitythat the fluctuations are genuine. Nielsen-Marsh andHedges [21] reported a far greater increase in the CI offresh bone after brief treatments with 1.0 M and 0.1 Macetic acid. The difference in the two sets of results maypartially be due to the fact that most samples here arearchaeological; the least crystalline portions of theirbone mineral may already have already dissolved duringdiagenesis, leaving the rest of the mineral more resistantto CI increase during treatment. However, the differentresults obtained for fresh bone are more difficult toexplain, especially the significant decrease in CI seen
after the 1.0 M acid treatment. Crystallinity decrease isnot predicted in the model developed by Nielsen-Marshand Hedges [21], and runs counter to changes generallyrecorded in archaeological bone. In comparison to theFTIR scan at 4 h of 0.1 M acid treatment, the scan at4 h of 1.0 M acid treatment appears to show weakeningof the peak at 605 cm�1 (see Fig. 5). The reason for thischange is unclear, although it presumably reflects attackon the bone by the more concentrated solution, whichwould agree with the large amount of mineral thatdissolved. The change has led to decrease in the CIvalue, as the signal of the peak at 605 cm�1 is weakenedin comparison to the signal at 590 cm�1.
Fig. 5. Modern bone after 4 h treatments with 1.0 M and 0.1 M aceticacid. FTIR spectra of modern bone after 4 h treatments with 0.1 Macid and 1.0 M acid. Note the more marked changes in the bonesubjected to 1.0 M acid.
S.J. Garvie-Lok et al. / Journal of Archaeological Science 31 (2004) 763–776772

4.4. Impact of acetic acid treatment on C/P
Based on prior observations of C/P and %CO2
decrease in modern bone and enamel samples treatedwith acid [11,15,16,46], fresh bone C/P was expected todecrease substantially after acid treatment. However,this does not occur. The C/P value of the fresh bonechanges only slightly after treatment with 0.1 M acid,decreasing from an initial value of 0.23 to 0.19 at 12 htime. A similar unexpectedly small decrease wasreported for fresh bone analysed by Wright andSchwarcz [46]. While the reason for such small decreasesin C/P is unclear, it may be related to natural variationin the C/P values of fresh bone.
In 1.0 M acid, the C/P value of the fresh boneincreases. This cannot reflect a true addition of carbon-ate as no source of additional carbonate was avail-able. Examination of FTIR spectra suggests that theincrease reflects a proportionate loss in intensity of the1035 cm�1 peak. The reason for this increase is unclear.Nielsen-Marsh and Hedges [23] report a small C/Pincrease (from 0.34 to 0.41) in modern bone treated with0.1 M acetic acid. They remark ([23], p. 1153) that theobservation seems to be anomalous, but do not com-ment further. The behaviour of C/P indices in freshbone, both treated and untreated, may be clarified infurther research.
The behaviour of C/P in the archaeological bonesamples varies. Sample 2 shows a sharp initial decline inC/P, followed by a plateau in C/P values for bothtreatments. The initial decrease in C/P is more markedfor 1.0 M acid. This pattern of change reflects thebehaviour of the mineral yield in the sample, and wasexpected based on prior descriptions of C/P and %CO2
in acid-treated archaeological bone [15,16,46]. However,it is not observed in the other archaeological samples.Samples 1 and 3 show little change, while sample4 shows increases in C/P similar to those of thefresh bone. The FTIR spectrum of sample 4 showsa loss of intensity in the 1035 cm�1 peak similar tothat seen in the 1.0 M acid treatment of the freshsample.
The variation in the C/P profiles of the archaeologi-cal bones may reflect differences in their preservationand diagenetic history. For example, it is tempting tosuggest that sample 3, with its high CI, low originalC/P and other signs of recrystallization, had beendegraded to the point where it was resistant to furtherchanges in C/P, while sample 2 had been contaminatedwith easily soluble excess carbonate which was dis-solved in treatment. Other research has also docu-mented variation in the response of archaeologicalbone C/P to acid treatment [23,46]. Samples thatshowed no change at all were interpreted as indicatingthe absence of diagenetic carbonate in the originalbone [23].
4.5. Impact of acetic acid treatment on stable isotopevalues
The effects of acetic acid treatment on isotopic valuesare consistent with previous work [11,15,16,46]. Forfresh bone, the acid treatments result in lower �13C. Thearchaeological samples also show lower �13C values,with the exception of the 0.1 M treatment of sample 4.For 1.0 M and 0.1 M treatments of fresh bone, thegreatest shift in �13C values occurs in the first 4 hfollowed by a plateau. The 0.1 M treatments of archaeo-logical samples 1, 2 and 3 show the same pattern.However, the 1.0 M treatments deviate from this pat-tern, only establishing a �13C plateau at the 12 h point.The �13C changes seen in sample 4 are different; �13Cshows no significant change over time in 0.1 M acid, andundergoes a gradual decrease in 1.0 M acid that showsno sign of stopping at the 24 h point. Thus, all archaeo-logical samples treated with 1.0 M acetic acid show �13Calteration up to the 12 or 24 h point, well after the 4 hpoint at which sample dissolution seems to be complete.The observation of �13C alteration in the absence ofsample dissolution is worrying, as it may indicateproblems with recrystallization.
The findings of this study echo those of several others[11,15,16,46] in documenting �13C change due to aceticacid treatment in fresh bone which cannot have con-tained diagenetic material. As found by Koch et al. [11],the shift in �13C value is greater for the 1.0 M treatmentthan the 0.1 M treatment. While these findings wereexpected, those seen for archaeological bone are moresurprising. The �13C values of archaeological samplesfrom various sites are altered consistently in the samedirection as that seen for fresh bone, and C/P values donot suggest that diagenetic carbonates were removedfrom some of these archaeological samples. Somestudies attribute �13C changes in archaeological bonemineral after acid treatment to the distinctive �13Cvalues of the mineral contaminants removed [16,17,46].However, given these observations, it seems more likelythat the �13C changes seen in the archaeological materialstem at least partially from the inherent effect of aceticacid treatment on bone apatite carbonate �13C, as hasbeen suggested for modern bone by a number ofresearchers (e.g., [11,13,19]).
The behaviour of �18O during acid treatment is con-sistent with that of �13C. As reported by other investiga-tors [11,46], the acid treatments produce higher �18Ovalues in fresh bone. This effect is seen in all the archaeo-logical samples save for the 4 h 1.0 M treatment ofsample 3, which shows a more negative value. This maybe indicative of a variable �18O response to acid treat-ment similar to that reported for archaeological bone byWright and Schwarcz [46]. However, as the 12 h and 24 h�18O values for this sample are less negative, thismeasurement may be anomalous. As with �13C, �18O
S.J. Garvie-Lok et al. / Journal of Archaeological Science 31 (2004) 763–776 773

changes are seen in the fresh bone, and seen in archaeo-logical bones in the absence of FTIR evidence for theremoval of carbonate from those samples. Again, thissuggests that the �18O change in these samples reflects aninherent effect of acetic treatment on bone carbonate�18O rather than the removal of diagenetic carbonates.The fresh bone and archaeological samples 1 and 2 showa pattern similar to that seen in the �13C data, in that�18O shows the largest shift between 0 and 4 h of treat-ment and larger stable isotope shifts are associated with1.0 M treatment. Archaeological samples 3 and 4 departfrom this pattern; as mentioned above, the 1.0 M acidtreatment of sample 3 causes an initial negative shift in�18O, and in the 4 h treatments of sample 4, 0.1 M acidcauses a greater isotopic shift than 1.0 M acid. By the24 h point, all samples display higher �18O values for1.0 M treatments than for 0.1 M treatments. In the caseof fresh bone, the �18O difference is substantial (3.5‰).In some cases, such as both treatments of sample 2 andthe 1.0 M treatment of sample 1, �18O values establish arough plateau after the 4 h point. However, the 0.1 Mtreatments of samples 1 and 3 show a steady increaserather than a plateau, and values for both treatmentsof sample 4 and the fresh bone fall back towards theoriginal value by the 24 h point.
While �13C and �18O changes in the treated samplesfollow similar patterns in general, changes in the twovalues are clearly not linked, since �13C and �18O valuescan change independently of each other. For example,the 0.1 M acid treatments of the fresh bone and ofarchaeological samples 1 and 3 produce a �13C plateauafter the 4 h mark, but do not show a plateau in �18O;instead, �18O values vary while �13C values remainconstant. While it is possible that this variation stemsfrom measurement error or other factors, it is alsopossible that it is indicative of ongoing recrystallizationof the samples in the absence of further dissolution asmeasured by weight loss. Nielsen-Marsh and Hedges[21] have demonstrated that such alteration can occur insamples left in acid solutions for long periods. Thus, thepotential that it has occurred in these samples should beconsidered.
5. Conclusions and implications for archaeologicalcarbonate research
The results of this study have implications both forthe preparation of bone mineral samples for carbonatestable isotope analysis and for interpreting carbonatestable isotope data.
5.1. Acid solution concentration
The data support the use of 0.1 M acetic acid ratherthan 1.0 M acetic acid for preparing fresh and recentarchaeological bone. While FTIR CI and C/P values
suggest that most samples suffer little or no recrystalliz-ation after 1.0 M acetic acid treatments of up to 24 h,shifts in �13C in 1.0 M acid after sample dissolution iscomplete suggest otherwise. As well, the anomalous CIand C/P values of the fresh bone and archaeologicalsample 4 after short 1.0 M acetic acid treatments suggestundesirable changes in the mineral component of thosesamples. 1.0 M acid treatments also cause more sampledissolution, a potential problem when dealing with smallsamples. Thus, it appears that treatment with 0.1 M acidis preferable, especially when dealing with fresh or veryrecent bone, as the fresh bone shows the most problemsin 1.0 M acetic acid.
5.2. Acid treatment time
The plateau in sample dissolution past the 4 h markfor all samples and treatments reflects saturation of theacid solution [23]. For most samples, �13C, C/P and CIalso follow a plateau from 4 to 24 h of exposure. Basedon these observations, it might seem that longer acidtreatments will have no significant effect on bonemineral samples. However, changes in �13C, C/P and CIseen in some treatments past the 4 h point may indicaterecrystallization, as may the fluctuating �18O values seenin general from 4 to 24 h of treatment. The potentialfor bone to recrystallize during acid treatment in theabsence of any net dissolution was demonstrated byNielsen-Marsh and Hedges [21]. These data supportthose findings. Based on them, we suggest that acidtreatments be applied for no longer than 4 h even whenthe more dilute 0.1 M acetic acid solution is used.
5.3. Differential isotopic effects of treatment with 1.0 Mand 0.1 M solutions
As in Koch et al. [11], a differential offset is seen in�13C and �18O values after exposure to 1.0 M and 0.1 Macetic acid solutions, with consistently greater isotopicshifts seen in the 1.0 M solution. This differential offsetis seen in fresh material as well as in archaeologicalmaterial, indicating that it must be characteristic ofacetic acid treatments of bone in general. It doesnot appear to result from increased sample dissolution in1.0 M acetic acid, since increasing the proportion ofsolution to sample (and thus increasing the amount ofsample dissolved) did not cause a corresponding increasein the isotopic shift. It also does not appear to resultfrom the preferential loss of labile carbonate from thebone, since archaeological samples have likely lost muchof their labile carbonate [13] and yet in this study, aconsistent �13C effect of acetic acid treatment is seen inarchaeological samples. Instead, the data suggest anexplanation similar to that proposed by Koch et al. [11].Those authors suggest that the differential effects of1.0 M and 0.1 M acetic acid treatments of biological
S.J. Garvie-Lok et al. / Journal of Archaeological Science 31 (2004) 763–776774

apatite reflect effects on the structural carbonate of thematerial, perhaps related to differences between type Aand type B carbonate substitutions. The consistent dif-ference between 1.0 M and 0.1 M acetic acid treatmentssuggests caution when comparing literature �13C and�18O data for archaeological bone and tooth carbonates.As reviewed above, various acid solutions, including1.0 M acetic acid, 0.1 M acetic acid and bufferedpreparations, have been used by different researchers toprepare bone and tooth carbonates for analysis. Thepresence of systematic differences in stable carbon andoxygen isotope ratios between material treated with the1.0 M and 0.1 M solutions implies systematic variabilityin published �13C and �18O values for this material. Thepossibility that such treatment-induced variability ispresent in the literature must be considered, and cautionshould be used when comparing published �13C and�18O values for material prepared using different acidtreatment protocols.
Acknowledgements
The authors gratefully acknowledge the financialsupport for this research provided by Social Sciencesand Humanities Research Council of Canada doctoralfellowships held by SGL and TLV. We wish to thank DrB. Kooyman of the University of Calgary Departmentof Archaeology, Dr R. Murphy of the National ParkAuthority of Antigua and Barbuda and the AthenianAgora Excavations (American School of ClassicalStudies at Athens) for providing bone samples. We alsothank S. Taylor and the staff of the Isotope ScienceLaboratory of the University of Calgary Department ofPhysics and Astronomy for sample �13C and �18O analy-sis. Finally, thanks go to Dr R. Yamdagni and the staffof the University of Calgary Department of ChemistryInstrumentation Facility for access to, and training on,their FTIR equipment.
References
[1] S.H. Ambrose, L. Norr, Experimental evidence for the relation-ship of the carbon isotope ratios of whole diet and dietary proteinto those of bone collagen and carbonate, in: J.B. Lambert, G.Grupe (Eds.), Prehistoric Human Bone—Archaeology at theMolecular Level, Springer-Verlag, New York, 1993, pp. 1–37.
[2] M. Balasse, S.H. Ambrose, A.B. Smith, T.D. Price, The seasonalmobility model for prehistoric herders in the south-western Capeof South Africa assessed by isotopic analysis of sheep toothenamel, Journal of Archaeological Science 29 (2002) 917–932.
[3] F. Betts, C. Blumenthal, A.S. Posner, Bone mineralization,Journal of Crystal Growth 53 (1981) 63–73.
[4] H. Bocherens, Preservation of isotopic signals (13C, 15N) inPleistocene mammals, in: S. Ambrose, M.A. Katzenberg (Eds.),Biogeochemical Approaches to Paleodietary Analysis, KluwerAcademic/Plenum Publishers, New York, 2000, pp. 65–88.
[5] H. Bocherens, M. Fizet, A. Mariotti, Diet, physiology andecology of fossil mammals as inferred from stable carbon andnitrogen isotope biogeochemistry: implications for Pleistocenebears, Palaeogeography, Palaeoclimatology, Palaeoecology 107(1994) 213–225.
[6] N.P. Camacho, S. Rinnerthaler, E.P. Paschalis, R. Mendelsohn,A.L. Boskey, P. Fratzl, Complementary information on boneultrastructure from scanning small angle x-ray scattering andFourier-transform infrared microspectroscopy, Bone 25 (1999)287–293.
[7] M.J. DeNiro, S. Weiner, Organic matter within crystallineaggregates of hydroxyapatite: A new substrate for stable isotopicand possibly other biogeochemical analyses of bone, Geochimicaet Cosmochimica Acta 52 (1988) 2415–2423.
[8] D.R. Grocke, Stable-isotope studies on the collagenic andhydroxylapatite components of fossils: Palaeoecologicalimplications, Lethaia 30 (1997) 65–78.
[9] A.H. Jahren, L.C. Todd, R.G. Amundson, Stable isotope dietaryanalysis of bison bone samples from the Hudson-Meng bonebed:effects of paleotopography, Journal of Archaeological Science 25(1998) 465–475.
[10] M.A. Katzenberg, A. Weber, Stable isotope ecology andpalaeodiet in the Lake Baikal region of Siberia, Journal ofArchaeological Science 26 (1999) 651–659.
[11] P.L. Koch, N. Tuross, M.L. Fogel, The effects of sample treat-ment and diagenesis on the isotopic integrity of carbonate inbiogenic hydroxylapatite, Journal of Archaeological Science 24(1997) 417–429.
[12] J. Krigbaum, Human paleodiet in tropical Southeast Asia: Iso-topic evidence from Niah Cave and Gua Cha. PhD Thesis, NewYork University, New York, 2001.
[13] H.W. Krueger, Exchange of carbon with biological apatite,Journal of Archaeological Science 18 (1991) 355–361.
[14] H.W. Krueger, C.H. Sullivan, Models for carbon isotope frac-tionation between diet and bone, American Chemical SocietySymposium Series 258 (1984) 205–222.
[15] L.S. Land, E.L. Lundelius Jr, S. Valastro Jr, Isotopic ecology ofdeer bones, Palaeogeography, Palaeoclimatology, Palaeoecology32 (1980) 143–151.
[16] J.A. Lee-Thorp, Stable carbon isotopes in deep time: Diet of fossilfauna and hominids, PhD Thesis, University of Cape Town, CapeTown, 1989.
[17] J.A. Lee-Thorp, Preservation of biogenic carbon isotopic signalsin Plio-Pleistocene bone and tooth mineral, in: S. Ambrose, M.A.Katzenberg (Eds.), Biogeochemical Approaches to PaleodietaryAnalysis, Kluwer Academic/Plenum Publishers, New York, 2000,pp. 89–115.
[18] J.A. Lee-Thorp, L. Manning, M. Sponheimer, Problems of andprospects for carbon isotope analysis of very small samples oftooth enamel, Bulletin de la Societe geologique de la France 168(1997) 767–773.
[19] J.A. Lee-Thorp, N.J. van der Merwe, Aspects of the chemistry ofmodern and fossil biological apatites, Journal of ArchaeologicalScience 18 (1991) 343–354.
[20] R.Z. LeGeros, Calcium Phosphates in Oral Biology andMedicine, Monographs in Oral Science 15, Karger, Basel, 1991.
[21] C.M. Nielsen-Marsh, R.E.M. Hedges, Dissolution experimentson modern and diagenetically altered bone and the effect on theinfrared splitting factor, Bulletin de la Societe geologique de laFrance 168 (1997) 485–490.
[22] C.M. Nielsen-Marsh, R.E.M. Hedges, Patterns of diagenesisin bone I: The effects of site environments, Journal ofArchaeological Science 27 (2000) 1139–1150.
[23] C.M. Nielsen-Marsh, R.E.M. Hedges, Patterns of diagenesis inbone II: Effects of acetic acid treatment and the removal ofdiagenetic CO2
3�, Journal of Archaeological Science 27 (2000)1151–1159.
S.J. Garvie-Lok et al. / Journal of Archaeological Science 31 (2004) 763–776 775

[24] E.P. Paschalis, E. DiCarlo, F. Betts, P. Sherman, R. Mendelsohn,A.L. Boskey, FTIR microspectroscopic analysis of humanosteonal bone, Calcified Tissue International 59 (1996) 480–487.
[25] C. Rey, B. Collins, T. Goehl, I.R. Dickson, M.J. Glimcher, Thecarbonate environment in bone mineral: A resolution-enhancedFourier transform infrared spectroscopy study, Calcified TissueInternational 45 (1989) 157–164.
[26] C. Rey, V. Renugopalakrishnan, B. Collins, M.J. Glimcher,Fourier transform infrared spectroscopic study of the carbonateions in bone mineral during aging, Calcified Tissue International49 (1991) 251–258.
[27] C. Rey, V. Renugopalakrishnan, M. Shimizu, B. Collins, M.J.Glimcher, A resolution-enhanced Fourier transform infraredspectroscopic study of the environment of the CO3
2� ion in themineral phase of enamel during its formation and maturation,Calcified Tissue International 49 (1991) 259–268.
[28] J-.F. Saliege, A. Person, F. Paris, Preservation of 13C/12C originalratio and 14C dating of the mineral fraction of human bones fromSaharan tombs, Niger, Journal of Archaeological Science 22(1995) 301–302.
[29] M.J. Schoeninger, Reconstructing prehistoric human diet, in:D.H. Price (Ed.), The Chemistry of Prehistoric Human Bone,Cambridge University Press, Cambridge, 1989, pp. 38–67.
[30] M.J. Schoeninger, M.J. DeNiro, Carbon isotope ratios of apatitefrom fossil bone cannot be used to reconstruct diets of animals,Nature 297 (1982) 577–578.
[31] M.J. Schoeninger, M.J. DeNiro, Reply to Sullivan and Krueger,Nature 301 (1983) 178.
[32] H.P. Schwarcz, J. Melbye, M.A. Katzenberg, M. Knyf, Stableisotopes in human skeletons of Southern Ontario: reconstructingpalaeodiet, Journal of Archaeological Science 12 (1985) 187–206.
[33] A. Sillen, J.A. Lee-Thorp, Trace element and isotopic aspects ofpredator-prey relationship in terrestrial foodwebs, Palaeogeogra-phy, Palaeoclimatology, Palaeoecology 107 (1994) 243–255.
[34] M. Sponheimer, J.A. Lee-Thorp, Alteration of enamel carbonateenvironments during fossilization, Journal of ArchaeologicalScience 26 (1999) 143–150.
[35] H. Le, Q. Stuart-Williams, H.P. Schwarcz, C.D. White, M.W.Spence, The isotopic composition and diagenesis of humanbone from Teotihuacan and Oaxaca, Mexico, Palaeogeography,Palaeoclimatology, Paleoecology 126 (1996) 1–14.
[36] C.H. Sullivan, H.W. Krueger, Carbon isotope analysis of separatechemical phases in modern and fossil bone, Nature 292 (1981)333–335.
[37] C.H. Sullivan, H.W. Krueger, Carbon isotope ratios of boneapatite and animal diet reconstruction, Nature 301 (1983) 177.
[38] L.L. Tieszen, T. Fagre, Effect of diet quality and composition onthe isotopic composition of respiratory CO2, bone collagen,bioapatite, and soft tissues, in: J.B. Lambert, G. Grupe (Eds.),Prehistoric Human Bone—Archaeology at the Molecular Level,Springer-Verlag, New York, 1993, pp. 121–155.
[39] D.H. Ubelaker, M.A. Katzenberg, L.G. Doyon, Status and dietin precontact highland Ecuador, American Journal of PhysicalAnthropology 97 (1995) 403–411.
[40] N.J. van der Merwe, J.C. Vogel, 13C content of human collagen asa measure of prehistoric diet in Woodland North America,Nature 276 (1978) 815–816.
[41] J.C. Vogel, N.J. van der Merwe, Isotopic evidence for early maizecultivation in New York state, American Antiquity 42 (1977)238–242.
[42] Y. Wang, T.E. Cerling, A model of fossil tooth and bonediagenesis: implications for paleodiet reconstruction from stableisotopes, Palaeogeography, Palaeoclimatology, Palaeoecology107 (1994) 281–289.
[43] S. Weiner, O. Bar-Yosef, States of preservation of bonesfrom prehistoric sites in the Near East: a survey, Journal ofArchaeological Science 17 (1990) 187–196.
[44] S. Weiner, P. Price, Disaggregation of bone into crystals, CalcifiedTissue International 39 (1986) 365–375.
[45] F.B. Wiedemann, H. Bocherens, A. Mariotti, A. von den Driesch,G. Grupe, Methodological and archaeological implications ofintra-tooth isotopic variations (�18O, �13C) in herbivores from AinGhazal (Jordan, Neolithic), Journal of Archaeological Science 26(1999) 697–704.
[46] L. Wright, H.P. Schwarcz, Infrared and isotopic evidence fordiagenesis of bone apatite at Dos Pilas, Guatemala: Palaeodietaryimplications, Journal of Archaeological Science 23 (1996)933–944.
[47] L. Wright, H.P. Schwarcz, Stable carbon and oxygen isotopes inhuman tooth enamel: identifying breastfeeding and weaning inprehistory, American Journal of Physical Anthropology 106(1998) 1–18.
S.J. Garvie-Lok et al. / Journal of Archaeological Science 31 (2004) 763–776776




















