
Are purified or expanded cord blood-derived CD133+ cells better at improving cardiac function

Pravastatin Improves Function in Hibernating Myocardiumby Mobilizing CD133� and cKit� Bone Marrow ProgenitorCells and Promoting Myocytes to Reenter the Growth Phase
of the Cardiac Cell CycleGen Suzuki, Vijay Iyer, Thomas Cimato, John M. Canty, Jr
Abstract—3-Hydroxy-3-methyl glutaryl coenzyme A reductase inhibitors have been reported to increase circulating bonemarrow progenitor cells and variably improve global function in heart failure. The potential role of improved perfusionversus direct effects of statins on cardiac myocytes has not been established. We chronically instrumented swine witha left anterior descending artery (LAD) stenosis to produce chronic hibernating myocardium with regional contractiledysfunction in the absence of heart failure. Hemodynamics, function, perfusion, and histopathology were assessed inpigs treated for 5 weeks with pravastatin (n�12) versus untreated controls (n�10). Regional LAD wall thickening wasdepressed under baseline conditions (LAD 3.7�0.3 versus 6.6�0.3 in remote regions, P�0.01). It remained unchangedin untreated animals but increased from 3.8�0.6 to 5.2�0.5 mm after pravastatin (P�0.01). There was no increase inmyocardial perfusion at rest or during vasodilation. Pravastatin mobilized circulating CD133�/cKit� bone marrowprogenitor cells and increased myocardial tissue levels (LAD CD133� cells from 140�33 to 884�167 cells/106
myocyte nuclei and cKit� cells from 223�49 to 953�123 cells/106 myocyte nuclei). Pravastatin increased myocytesin mitosis (phospho–histone-H3; 9�5 to 43�7 nuclei/106 myocyte nuclei, P�0.05) and the growth phase of the cellcycle (Ki67; 410�82 to 1261�235 nuclei/106 myocyte nuclei, P�0.05) in diseased but not normal hearts. As a result,pravastatin increased LAD myocyte nuclear density from 830�41 to 1027�55 nuclei/mm2 (P�0.05). These dataindicate that, in the absence of impaired endothelial function and heart failure, dysfunctional hibernating myocardiumimproves after pravastatin. This effect is independent of myocardial perfusion and related to mobilization ofCD133�/cKit� bone marrow progenitor cells which stimulate myocyte proliferation resulting in quantitative increasesin myocyte nuclear density. (Circ Res. 2009;104:255-264.)
Key Words: statins � hibernating myocardium � cardiac repair � bone marrow progenitor cells
Although 3-hydroxy-3-methyl glutaryl coenzyme A(HMG-CoA) reductase inhibitors were originally devel-
oped as lipid-lowering drugs and were demonstrated to retardthe progression of atherosclerosis and improve endothelialfunction, pleotrophic actions of statins may be of equalimportance in reducing death and disability from cardiovas-cular disease. Statins have been shown to have antiinflamma-tory, antihypertrophic, and proangiogenic actions that mayfavorably impact ventricular function independently of theiractions on stabilizing and/or regressing atherosclerotic le-sions.1,2 In addition, statins have been demonstrated to mo-bilize endothelial progenitor cells (EPCs) into the circulationin humans and animals.3,4 Preclinical studies have demon-strated that EPCs can arise from bone marrow and incorpo-rate into the vascular wall.5 Some studies have also demon-strated the ability of bone marrow progenitor cell (BMPC)populations to differentiate into cardiac myocytes.6,7 Several
small randomized clinical studies have demonstrated objec-tive increases in myocardial function after statins8,9 but thismay not be a class effect.10 Although angiogenesis could playa role in some of these actions, improvements in myocardialfunction have also been demonstrated in nonischemiccardiomyopathy.
We performed the present study to identify the effects ofpravastatin on myocardial perfusion and ischemic myocardialdysfunction that are independent of cholesterol lowering. Weused a well-characterized porcine model of collateral-dependent myocardium resulting in hibernating myocardiumfrom a chronic LAD stenosis. Because regional rather thanglobal left ventricular (LV) dysfunction is present, the con-founding role of statins on inflammation, cytokines, andneurohormonal activation in heart failure are absent.11 Inaddition, because hypercholesterolemia and atherosclerosisare absent, the effects of pravastatin on angiogenesis were
Original received October 1, 2008; revision received November 7, 2008; accepted December 4, 2008.From the Veterans Affairs Western New York Healthcare System, Departments of Medicine and Physiology and Biophysics, and Center for Research
in Cardiovascular Medicine, University at Buffalo, The State University of New York.Correspondence to John M. Canty, Jr, Division of Cardiology, University at Buffalo, Biomedical Research Building, Room 345, 3435 Main St, Buffalo,
NY 14214. E-mail [email protected]© 2009 American Heart Association, Inc.
Circulation Research is available at http://circres.ahajournals.org DOI: 10.1161/CIRCRESAHA.108.188730
255 by guest on September 18, 2015http://circres.ahajournals.org/Downloaded from by guest on September 18, 2015http://circres.ahajournals.org/Downloaded from by guest on September 18, 2015http://circres.ahajournals.org/Downloaded from by guest on September 18, 2015http://circres.ahajournals.org/Downloaded from by guest on September 18, 2015http://circres.ahajournals.org/Downloaded from by guest on September 18, 2015http://circres.ahajournals.org/Downloaded from by guest on September 18, 2015http://circres.ahajournals.org/Downloaded from by guest on September 18, 2015http://circres.ahajournals.org/Downloaded from by guest on September 18, 2015http://circres.ahajournals.org/Downloaded from by guest on September 18, 2015http://circres.ahajournals.org/Downloaded from by guest on September 18, 2015http://circres.ahajournals.org/Downloaded from by guest on September 18, 2015http://circres.ahajournals.org/Downloaded from

studied independently of inhibiting plaque progression andimproving endothelium-dependent vasodilation. Our resultsdemonstrate that pravastatin mobilizes BMPCs in hibernatinghearts and promotes myocytes to reenter the growth andmitotic phase of the cell cycle. As a result, myocyte nucleardensity is increased and myocardial function improves with-out a change in myocardial perfusion.
Materials and MethodsProcedures and protocols conformed to institutional guidelines forthe care and use of animals in research. Detailed experimental andhistological protocols are described in the online data supplement,available at http://circres.ahajournals.org.
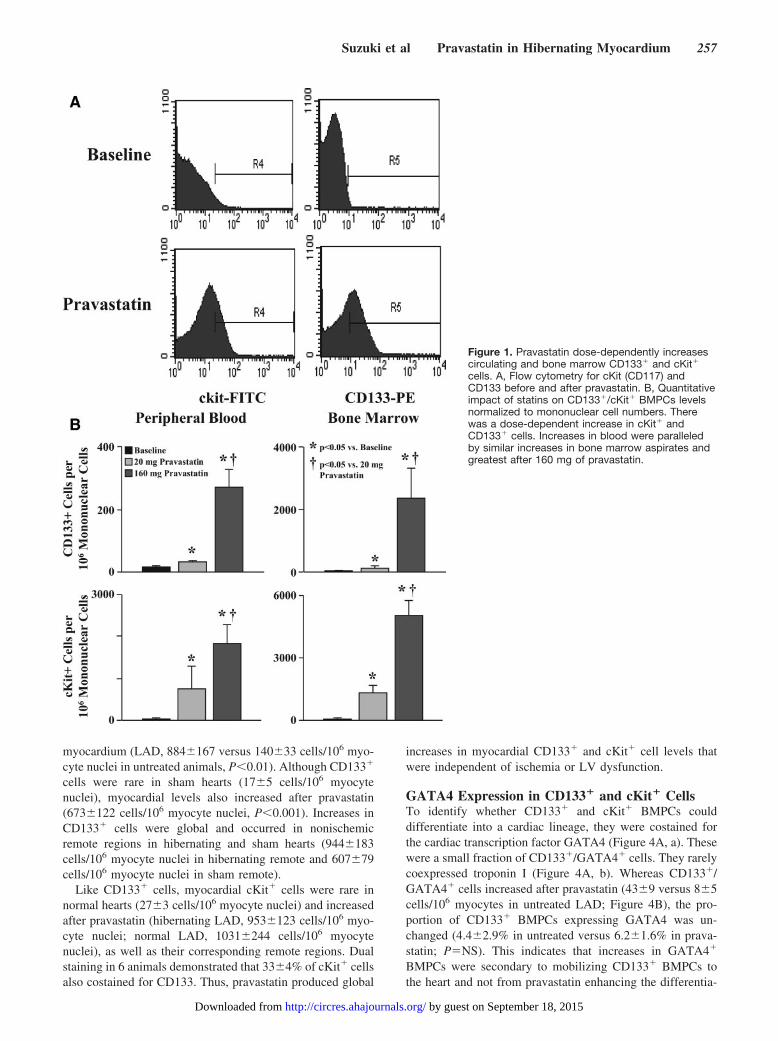
Mobilization of BMPCs With PravastatinWe initially assessed the dose dependency of pravastatin to mobilizeBMPCs in normal swine using flow-cytometry (FACS, n�11).Baseline measurements were compared to those 2 weeks after dailytreatment with low-dose (20 mg/d) or high-dose (160 mg/d) prava-statin. We assessed changes in peripheral blood (30 mL) as well asbone marrow (30 mL). Data from FACS analysis are expressed asCD133� or cKit� cells per million mononuclear cells.
Effects of Pravastatin on Flow and Function inHibernating MyocardiumPigs were chronically instrumented with a 1.5-mm Delrin stenosis onthe proximal LAD as described previously (online data supple-ment).12 We performed baseline physiological studies 4 months afterinstrumentation (n�22) when hibernating myocardium with reduc-tions in LAD wall thickening and resting LAD perfusion werepresent. Under propofol sedation, a Millar catheter was inserted intothe left ventricle for microsphere injection. Regional wall thickeningwas assessed with transthoracic echocardiography. Microsphere flowwas assessed at rest and following adenosine vasodilation. Animalswere subsequently treated with pravastatin (160 mg/d, n�12) for 5weeks and compared to untreated controls (n�10). At the end of thestudy, physiological studies were repeated and the heart was excisedfor 5-triphenyltetrazolium chloride (TTC), flow, and histologicalanalysis.
Myocyte Nuclear Density and MorphometrySamples from hibernating LAD and normal remote regions werefixed and paraffin-embedded. Trichrome-stained sections were usedto quantify connective tissue.13 Periodic acid Schiff–stained sectionswere used to quantify myocyte diameter, nuclear length, and regionalmyocyte nuclear density. Histological results in hibernating animalswere compared to normal sham animals (pravastatin, n�10; un-treated, n�5).
Quantitative Analysis of Myocyte Proliferation andCD133�/cKit� CellsTo identify myocyte proliferation, anti-Ki67 and anti–phospho–histone-H3 staining were performed.14 CD133�/cKit� BMPCs inmyocardial tissue were quantified using confocal immunofluores-cence (online data supplement). Preliminary studies demonstratedthat tissue CD133�/cKit� BMPCs were CD45 negative. Toevaluate whether CD133�/cKit� BMPCs differentiated into acardiac lineage we costained BMPCs with GATA-4 and cardiactroponin I antibodies.14
StatisticsData are expressed as means�SE. Differences between groups wereassessed by 2-way ANOVA and the post hoc Holm–Sidak test.Temporal physiological changes between initial and final studieswere assessed by paired t tests. P�0.05 was considered significant.
ResultsPigs were in good health at the time of study, and TTCstaining showed no significant infarction. Initial physiologi-cal studies were performed 130�3 days after instrumentationand repeated 5 weeks after pravastatin (166�3 days).
Pravastatin Dose-Dependently Mobilizes CD133�
and cKit� BMPCsFigure 1 summarizes flow cytometric data before andafter low- and high-dose pravastatin for 2 weeks. Therewere no differences in circulating mononuclear cells(n�11, 11.6�106�1.8�106 cells/mL at baseline versus16.6�106�4.7�106 cells/mL after pravastatin; P�NS). Incontrast, pravastatin dose-dependently increased bothCD133� and cKit� cells in blood and bone marrowaspirates. Circulating and bone marrow CD133� and cKit�
cells were much higher after a 160-mg dose of pravastatin.For example, circulating CD133� cells averaged 18�3cells/106 mononuclear cells at baseline, 32�5 cells/106
mononuclear cells after 20 mg pravastatin (P�0.05 versusbaseline), and 272�54 cells/106 mononuclear cells after160 mg of pravastatin (P�0.05 versus baseline and 20 mgof pravastatin).
Effects of Pravastatin on Function and Flow inHibernating MyocardiumHemodynamics (supplemental Table I) and echocardiograph-ic measurements of function (supplemental Table II) aresummarized in the online data supplement. Hemodynamicswere similar between treatment groups with the exception ofLV end-diastolic pressure, which tended to be lower inanimals receiving pravastatin. Pravastatin increased LADsystolic wall thickening (percentage WT, 28�2.8% versus48�5.1% after pravastatin, P�0.05; supplemental Table II)with systolic excursion (ESWT-EDWT) summarized in Fig-ure 2A. There were no differences in remote regions. Serialanalysis of function within the same animal confirmed asimilar improvement, with no change in untreated controls.Likewise, global function was normal and did not changeafter pravastatin (ejection fraction, 64�5% versus 69�2%after pravastatin; P�NS). Although pravastatin improvedregional function in hibernating myocardium, there was noeffect on resting flow (Figure 2B). When analyzed in terms ofrelative full-thickness perfusion, relative reductions in restingflow (LAD/remote, 0.79�0.07 in pravastatin versus0.70�0.05 in untreated) were similar, with no serial changesbetween studies in the same animal. Likewise, relativeadenosine flow (LAD/remote 0.27�0.04 in pravastatin ver-sus 0.23�0.02 in untreated) was not significantly different,nor were there serial changes over time. Thus, althoughpravastatin increased LAD wall thickening, there was nofunctionally significant angiogenesis.
Increase in Myocardial CD133� and cKit� CellsAfter PravastatinWe analyzed myocardial tissue to determine whether theincrease in circulating CD133�/cKit� BMPCs produced cor-responding increases in myocardial tissue levels (Figure 3Aand 3B). Pravastatin increased CD133� cells in hibernating
256 Circulation Research January 30, 2009
by guest on September 18, 2015http://circres.ahajournals.org/Downloaded from
myocardium (LAD, 884�167 versus 140�33 cells/106 myo-cyte nuclei in untreated animals, P�0.01). Although CD133�
cells were rare in sham hearts (17�5 cells/106 myocytenuclei), myocardial levels also increased after pravastatin(673�122 cells/106 myocyte nuclei, P�0.001). Increases inCD133� cells were global and occurred in nonischemicremote regions in hibernating and sham hearts (944�183cells/106 myocyte nuclei in hibernating remote and 607�79cells/106 myocyte nuclei in sham remote).
Like CD133� cells, myocardial cKit� cells were rare innormal hearts (27�3 cells/106 myocyte nuclei) and increasedafter pravastatin (hibernating LAD, 953�123 cells/106 myo-cyte nuclei; normal LAD, 1031�244 cells/106 myocytenuclei), as well as their corresponding remote regions. Dualstaining in 6 animals demonstrated that 33�4% of cKit� cellsalso costained for CD133. Thus, pravastatin produced global
increases in myocardial CD133� and cKit� cell levels thatwere independent of ischemia or LV dysfunction.
GATA4 Expression in CD133� and cKit� CellsTo identify whether CD133� and cKit� BMPCs coulddifferentiate into a cardiac lineage, they were costained forthe cardiac transcription factor GATA4 (Figure 4A, a). Thesewere a small fraction of CD133�/GATA4� cells. They rarelycoexpressed troponin I (Figure 4A, b). Whereas CD133�/GATA4� cells increased after pravastatin (43�9 versus 8�5cells/106 myocytes in untreated LAD; Figure 4B), the pro-portion of CD133� BMPCs expressing GATA4 was un-changed (4.4�2.9% in untreated versus 6.2�1.6% in prava-statin; P�NS). This indicates that increases in GATA4�
BMPCs were secondary to mobilizing CD133� BMPCs tothe heart and not from pravastatin enhancing the differentia-
Figure 1. Pravastatin dose-dependently increasescirculating and bone marrow CD133� and cKit�
cells. A, Flow cytometry for cKit (CD117) andCD133 before and after pravastatin. B, Quantitativeimpact of statins on CD133�/cKit� BMPCs levelsnormalized to mononuclear cell numbers. Therewas a dose-dependent increase in cKit� andCD133� cells. Increases in blood were paralleledby similar increases in bone marrow aspirates andgreatest after 160 mg of pravastatin.
Suzuki et al Pravastatin in Hibernating Myocardium 257
by guest on September 18, 2015http://circres.ahajournals.org/Downloaded from

tion of CD133� BMPCs to myocytes. Similar results werefound for cKit� cells.
Increased Myocytes in the Growth Phase of theCell Cycle After PravastatinWe quantified the frequency of myocyte nuclei expressingKi67, a marker of the growth phase of the cell cycle andphospho–histone-H3, a marker of mitosis (Figure 5). Un-treated hibernating animals had low values of Ki67 positivity(LAD, 410�82 nuclei/106 myocyte nuclei; remote, 285�88nuclei/106 myocyte nuclei). After pravastatin, Ki67 positivityincreased (LAD, 1261�235 nuclei/106 myocyte nuclei; re-mote, 969�132 nuclei/106 myocyte nuclei; P�0.05 versusuntreated). Surprisingly, although CD133� and cKit� cellsincreased in sham hearts, there was no increase in Ki67positivity after pravastatin (LAD, 274�82 nuclei/106 myo-cyte nuclei, versus remote, 284�69 nuclei/106 myocytenuclei; P�NS versus untreated). Likewise, myocyte nuclearphospho–histone-H3 positivity was selectively increased inpravastatin-treated hibernating animals versus sham-treatedanimals (LAD, 43�7 versus 9�5 nuclei/106 myocyte nuclei;
remote, 34�9 versus 6�3 nuclei/106 myocyte nuclei;P�0.05, respectively). Thus, pravastatin only increased myo-cytes in the growth phase of the cell cycle in diseased hearts.
Effects of Pravastatin on Myocyte Nuclear Densityin Hibernating MyocardiumTo determine whether increases in cell cycle markers led tomyocyte regeneration, we assessed the effects of pravasta-tin on myocyte nuclear density (Figure 6). Whereas LADconnective tissue was mildly increased in hibernatinganimals receiving pravastatin (LAD, 8.3�1.9%, versusremote, 4.1�0.7%; P�0.05), it was similar to untreatedanimals (LAD, 6.9�0.7%, versus remote, 4.5�0.2%,P�0.05; pravastatin versus untreated, P�NS, respec-tively). In untreated animals, LAD myocyte nuclear den-sity was reduced (LAD, 830�41 nuclei/mm2, versus re-mote, 1027�55 nuclei/mm2; P�0.05), as we havedemonstrated previously.13 After pravastatin, LAD myo-cyte nuclear density increased from 830�41 to 1054�19nuclei/mm2 (P�0.01), but values remained significantlylower than normal myocardium (P�0.05). Measurements
Figure 2. Pravastatin improves regional function inhibernating myocardium. A, In hibernatingmyocardium, systolic LAD wall thickening (ESWT-EDWT) was depressed and significantly increasedafter pravastatin (160 mg/d) in comparison tountreated animals. There was no significant changein remote zone wall thickening. Similar changeswere seen for percentage wall thickening. B, Therewas no effect of pravastatin on myocardial perfu-sion in hibernating myocardium.
258 Circulation Research January 30, 2009
by guest on September 18, 2015http://circres.ahajournals.org/Downloaded from

of myocyte diameter (Figure 7) demonstrated that in-creases in myocyte nuclear density after pravastatin wereaccompanied by a reduction in myocyte size (mean diam-eter, 13.2�0.6 �m, versus 15.7�0.4 �m; P�0.05). The
diameter of Ki67-positive myocytes was even smaller(11.9�0.2 �m, P�0.05). Thus, the increase in myocytenuclear density coupled with a reduction in myocyte size isconsistent with myocyte proliferation after pravastatin inhibernating myocardium.
Figure 3. Pravastatin increases myocardial CD133� and cKit�
BMPCs in hibernating and sham animals. A, Both cKit� (a) andCD133� (b) cells (stained green) localized in interstitial regionsand were frequently clustered. Myocytes were stained with tro-ponin I (red) and nuclei with TO-PRO-3 (blue). B, There was aglobal increase in myocardial CD133� and cKit� cells in LADand remote regions after 5 week of pravastatin. Levels weresimilar in hearts from hibernating and sham animals.
Figure 4. Coexpression of GATA4 in CD133� and cKit�
BMPCs. A, Example of a CD133� cell costaining for GATA4 (a)and troponin I (b). Similar changes were seen in cKit� cells. B,Pravastatin increased myocardial CD133� and cKit� BMPCscoexpressing GATA4 in normal and hibernating hearts. Becausethe percentage of CD133�/cKit� BMPCs coexpressing GATA4remained fairly constant (4.4�2.9% vs 6.2�1.6% after pravasta-tin), the increase reflected a greater total number of CD133�/cKit� cells in the tissue (Figure 3B).
Suzuki et al Pravastatin in Hibernating Myocardium 259
by guest on September 18, 2015http://circres.ahajournals.org/Downloaded from

DiscussionThere are several important new findings from our study.First, pravastatin dose-dependently increases CD133� andcKit� cells in bone marrow and blood. This leads to corre-sponding increases in myocardial tissue from normal anddiseased hearts. Second, in hibernating myocardium, prava-statin improves regional wall thickening with no demonstra-ble effect on myocardial perfusion, indicating that functionalimprovement is independent of angiogenesis. Third, immu-nohistochemical analysis of myocardial tissue demonstratesthat improved function is accompanied by an increase in
myocytes in the proliferative phase of the cell cycle. Thisleads to corresponding increases in myocyte nuclear densityand reductions in myocyte diameter in hibernating myocar-dium. Although statins mobilized CD133� and cKit� BMPCsto normal hearts, evidence of cardiac myocyte proliferationwas absent. Collectively, the observations are consistent withthe notion that chronic mobilization of CD133�/cKit�
BMPCs with pravastatin leads to significant myocyte regen-eration in diseased hearts.
Mobilization of BMPCs and Localization inthe HeartThere is now substantial support for the notion that BMPCscan facilitate myocyte regeneration under selected conditions.Orlic et al demonstrated the ability of cKit� bone marrowcells injected into murine infarcts to regenerate myocardium.15 In asubsequent study, myocyte regeneration was effected bymobilizing BMPCs with stem cell factor and granulocytecolony-stimulating factor.16 Although controversial, the abil-ity of BMPCs to repair the heart in humans is supported bymyocyte chimerism in transplant recipients.17,18 This, as wellas other preclinical work, stimulated rapid translation of thesefindings to early clinical trials using a variety of largelyunfractionated intracoronary and intramyocardial adult bonemarrow preparations in patients with myocardial infarction.Although the therapy appears safe and functional effects aregenerally favorable, the magnitude of improvement ismodest (reviewed in a recent metaanalysis19). Importantly,BMPC mobilization strategies using granulocyte colony-stimulating factor in humans have not demonstrated sig-nificant functional improvement.20 Thus, myocardial hom-ing, as well as mobilization, is required for a physiologicaleffect.
Our results demonstrate that pravastatin affords an alter-native approach to effect bone marrow–mediated cardiacrepair. Because statins have been demonstrated to mobilizeEPCs, previous basic and clinical studies have focused onevaluating their effect on angiogenesis and vasculogenesisrather than myocyte regeneration. A variety of HMG-CoAreductase inhibitors can mobilize bone marrow– derivedCD34� and CD133� EPCs. These can differentiate intoendothelial cells and have been demonstrated to improveperfusion in the severely ischemic murine hind limbmodel.21,22 Dimmeler et al demonstrated that simvastatin canalso increase bone marrow cKit� cells.22 Previous studieshave established that EPC mobilization involves activation ofthe phosphatidylinositol 3-kinase/Akt pathway by statins.22,23
Our results extend these studies in swine by showing aparallel dose-dependent increase in cKit� and CD133� cellsin bone marrow and blood. This supports the notion that theprimary action of statins is to increase the proliferation ofCD133� and cKit� cells in bone marrow. It is also possiblethat CD133�/cKit� BMPCs arise from other tissue reservoirswhich will require further study. The corresponding globalincrease in myocardial CD133� and cKit� cell levels innormal as well as hibernating hearts is probably a function oftheir circulating concentration. Although the number ofCD133�/GATA-4� and cKit�/GATA-4� cells increased,pravastatin did not enhance differentiation into a cardiac
Figure 5. Pravastatin increases Ki67 and phospho–histone-H3–positive myocytes in hibernating hearts. A, Pravastatin increasedmyocyte Ki67 (green) (a and b) and phospho–histone-H3 (pHH3)(c), indicating an increased number of myocytes in the growthand mitotic phase of the cell cycle. Ki67- and pHH3-positivenuclei were confirmed as myocytes by costaining with troponin I(red). B, Quantitative data demonstrated significant increases inKi67 and pHH3 in the LAD and remote regions in treated pigswith hibernating myocardium. In contrast, these were not alteredin sham animals despite increased myocardial CD133� andcKit� BMPCs after pravastatin.
260 Circulation Research January 30, 2009
by guest on September 18, 2015http://circres.ahajournals.org/Downloaded from

lineage because the percentage of GATA-4� cells wasunchanged.
Lack of Effect of Pravastatin on Perfusion inHibernating MyocardiumMobilization of EPCs can salvage tissue in rodent hind limbischemia models via increases capillary density, suggestingsmall vessel proliferation. The ability of statins to increasemyocardial perfusion is less clear. Indeed, this questionprovided the initial rationale for our study, where pravastatinwas anticipated to improve perfusion to collateral-dependenthibernating myocardium. Surprisingly, there was no differ-ence in resting or vasodilated flow in LAD or remotemyocardium after pravastatin (online data supplement). Like-wise, relative perfusion differences between LAD and remoteregions were unchanged. Although we did not assess ana-tomic variables such as capillary density, the failure ofcoronary flow to increase in ischemic myocardium supportsthe notion that statins do not have any meaningful effect on
coronary collateral resistance vessels. Dissociations betweenflow and capillary density can occur because the latter vesselscontribute negligibly to coronary vascular resistance.24 Nev-ertheless, because there were small increases in end-diastolicwall thickness after pravastatin (�15%), we cannot excludethe possibility that angiogenesis matched to changes inregional mass occurred.
A potential explanation for the failure of pravastatin toincrease perfusion may relate to a biphasic dose-dependenteffect on angiogenesis. Weis et al found that low-dosecerivastatin or atorvastatin potentiated endothelial cell prolif-eration, whereas higher doses inhibited it.25 In vivo, Boodh-wani et al demonstrated that high-dose atorvastatin (1.5mg/kg per day) impairs collateral development in normocho-lesterolemic swine.26 Our findings are similar using a lesspotent HMG-CoA reductase inhibitor. In disease states whereendothelium-dependent vasodilation of coronary resistancevessels is impaired, such as atherosclerosis, statins may haveimportant beneficial effects in improving endothelium-dependent vasodilation of resistance arteries, including col-lateral resistance vessels.24 These functional effects ratherthan de novo arteriogenesis or vasculogenesis likely contrib-ute to the well-known improvement in symptomatic myocar-dial ischemia after administering statins to patients.27
Effect of Statins on Myocardial FunctionPrevious animal studies have demonstrated beneficial effectsof statins on global myocardial function in a variety ofmodels of heart disease. In general, all have used high dosesof statins that greatly exceed the pharmacological range usedin humans (eg, 20 mg/kg simvastatin per day). These are alsoeven higher than doses demonstrated to impair angiogenesisin vivo and in vitro. In rodent models of infarction, statinsimproved global LV function by preventing deleterious LVremodeling without reducing infarct size.28–31 Statins havealso been demonstrated to prevent the progression of hyper-trophy following angiotensin infusion and aortic banding32
and to prevent heart failure in genetic models of cardiomy-opathy.31 Studies evaluating the effect of statins on heartfailure in large animals are limited. In dogs with heart failure,
Figure 6. Pravastatin increases myocytenuclear density in hibernating myocardium.There was a regional reduction in LAD myo-cyte nuclear density in untreated hibernatinganimals that was greatest in the subendo-cardium. After pravastatin, LAD myocytenuclear density increased but remained sig-nificantly lower than values found in normalsham animals. This demonstrates a cumula-tive increase in myocytes after 5 weeks ofpravastatin.
Figure 7. Redistribution of LAD myocyte diameters in hibernat-ing myocardium. After pravastatin, LAD myocyte diameters weresignificantly smaller (mean, 13.2�0.6 �m) than untreated ani-mals (15.7�0.4 �m, P�0.05). Ki67-positive myocytes were evensmaller (11.9�0.2, P�0.01). The shift in size supports the notionthat proliferating myocytes arose from a stem cell population.
Suzuki et al Pravastatin in Hibernating Myocardium 261
by guest on September 18, 2015http://circres.ahajournals.org/Downloaded from

statins attenuate the rate of decline in LV function inpacing-induced cardiomyopathy33 and cardiomyopathy attrib-utable to coronary microembolization.34 In general, mecha-nistic insight has largely focused on the ability of statins toimprove NO signaling and reduce circulating markers ofinflammation. None of the previous studies has elucidated aquantitative contribution of statins to myocyte regeneration.
Our results demonstrate that pravastatin increases regionalfunction in hibernating myocardium in the absence of infarc-tion or heart failure. Although functional improvement wasnot related to improved perfusion, it was accompanied by anincrease in regional end-diastolic wall thickness and anincrease in myocyte nuclear density. Resolution of myocytecellular hypertrophy in LAD myocardium could also contrib-ute improved wall thickening. Thus, the mechanism ofimprovement appears to be related to myocyte regenerationthat restores regional myocyte loss from apoptosis during thedevelopment of hibernating myocardium.13 Although clinicaltrials in hibernating myocardium are lacking, Gheorghiade etal reported a dramatic serial functional improvement in apatient with hibernating myocardium from a chronic LADocclusion after simvastatin.35
Statins Increase Myocytes in the Growth Phase ofthe Cell Cycle in Diseased HeartsWe found that statins increased myocytes in the proliferativephase of the cell cycle in a fashion similar to that which wehave reported after intracoronary administration of AdvFGF-5.14 Although this was accompanied by a global increase inmyocardial tissue CD133� and cKit� BMPCs, increases inmyocyte Ki67 and phospho–histone-H3 staining were re-stricted to hibernating hearts. The possibility that increasedcirculating CD133�/cKit� BMPCs leads to increased myo-cyte regeneration is unlikely because pravastatin increasedCD133�/cKit� BMPCs but did not cause myocyte prolifer-ation in normal hearts. Thus, additional myocardial factorsare probably required to stimulate myocyte proliferation.Because Ki67 increased in the remote myocardial region ofhibernating hearts, as well as the dysfunctional LAD region,the stimulus is global. Circulating factors related to neuro-hormonal activation such as elevated catecholamines, angio-tensin, endothelin, and cytokines are plausible but seemunlikely in this model because global function is normal andhemodynamic evidence of neurohormonal activation is ab-sent. A more likely possibility is that a factor releasedfollowing transient myocardial ischemia recirculates to nor-mal as well as ischemic myocardium. Alternatively, it ispossible that myocardial stretch related to increased preloadstimulates the expression of preload-induced myocardialgrowth factors and genes that facilitate myocyte proliferationin conjunction with the increase in tissue CD133� and cKit�
cells.36 Additional studies will be required to address thesepossibilities directly. Finally, although statins could have in-creased myocyte number by inhibiting apoptosis, we havepreviously demonstrated that apoptosis has returned to baseline4 months after instrumentation in this model, which is the timeframe over which the present studies were conducted.14
The actual origin of cycling myocytes cannot be unambig-uously determined from our study. One possibility is that the
CD133� and cKit� cells differentiate directly into cardiacmyocytes.37 This is supported by the observation that aportion of them express GATA4 and troponin I. Anotherpossibility is that CD133� and cKit� cells stimulate residentcardiac stem cells (CSCs) to proliferate with Ki67� myocytesarising from CSCs, as has been suggested after administrationof mesenchymal stem cells.38 It is also plausible that CD133�
and cKit� cells fuse with cardiac myocytes and facilitate theirreentry into the growth phase of the cell cycle.39 Finally,paracrine factors released from CD133� and cKit� cellscould stimulate CSC proliferation or facilitate myocytes toreenter the cell cycle.40 Additional studies using models usinggenetic fate mapping and stem cell tracking will be requiredto address these possibilities.36,41 Although the precise sig-naling is unknown, the increase in myocyte nuclear density,smaller myocyte size, and improvement in regional functiondemonstrated in vivo all suggest an effect of pravastatin onmyocyte regeneration.
Clinical ImplicationsOur results support the hypothesis that the improvement inejection fraction in patients with heart failure previouslydemonstrated with simvastatin8 and atorvastatin9 may resultfrom stimulation of endogenous cardiac repair mechanisms.Nevertheless, this may not be a class effect, as evidenced bythe failure of the extremely potent drug rosuvastatin toimprove ejection fraction.10 In concert with the latter study,the recently completed large placebo controlled clinical trialsof rosuvastatin in patients who have predominantly class IIand III heart failure with depressed ejection fraction (CORONA42
and GISSI-HF43) have failed to show any impact of this drugon cardiovascular mortality. Results of rosuvastatin on serialfunction in these studies have not yet been reported. Individ-ual statins likely vary in their ability to affect myocardialfunction through differential potency (or dose range), as wellas their ability to affect relevant pleotrophic pathways (eg,upregulation of Akt or inhibiting myocardial proliferativepathways such as Rho, Rac, and Ras). Finally, age-relatedimpairment in cardiac stem cell repair mechanisms may beimportant,11 and the clinical trials have focused on elderlypatients with heart failure (mean ages of 68 to 73 years)versus younger patients in small positive studies using myo-cardial function as an end point.
In summary, our study demonstrates functional improve-ment in hibernating myocardium after pravastatin that isindependent of angiogenesis or atherosclerotic plaque regres-sion that occurs in the absence of a proinflammatory state orneurohormonal activation. Beneficial effects may be absentwhen infarction rather than hibernating myocardium predom-inates because residual perfusion to scar is low. Nevertheless,because patients with ischemic cardiomyopathy usually havepatchy fibrosis averaging �20% of LV mass, our observa-tions may be very relevant to understanding statin-mediatedfunctional improvement in nonischemic cardiomyopathies, aswell as settings where dysfunction from reversible ischemiaand LV remodeling play major roles. Finally, the ability ofstatins to mobilize circulating BMPCs may be an importantvariable to consider in understanding the independent effects
262 Circulation Research January 30, 2009
by guest on September 18, 2015http://circres.ahajournals.org/Downloaded from

of exogenous cell-based therapies administered in clinicaltrials examining cardiac repair.
AcknowledgmentsWe thank Anne Coe, Deana Gretka, Elaine Granica, and AmyJohnson for technical assistance.
Sources of FundingSupported by the Veterans Affairs Administration; American HeartAssociation; Buswell Fellowship; National Heart, Lung, and BloodInstitute; Albert and Elizabeth Rekate Fund; and Empire State StemCell Board.
DisclosuresNone.
References1. Lipinski MJ, Abbate A, Fuster V, Vetrovec GW. Drug insight: statins for
nonischemic heart failure–evidence and potential mechanisms. Nat ClinPract Cardiovasc Med. 2007;4:196–205.
2. Ramasubbu K, Estep J, White DL, Deswal A, Mann DL. Experimentaland clinical basis for the use of statins in patients with ischemic andnonischemic cardiomyopathy. J Am Coll Cardiol. 2008;51:415–426.
3. Vasa M, Fichtlscherer S, Adler K, Aicher A, Martin H, Zeiher AM,Dimmeler S. Increase in circulating endothelial progenitor cells by statintherapy in patients with stable coronary artery disease. Circulation. 2001;103:2885–2890.
4. Deschaseaux F, Selmani Z, Falcoz PE, Mersin N, Meneveau N, PenfornisA, Kleinclauss C, Chocron S, Etievent JP, Tiberghien P, Kantelip JP,Davani S. Two types of circulating endothelial progenitor cells in patientsreceiving long term therapy by HMG-CoA reductase inhibitors. EurJ Pharmacol. 2007;562:111–118.
5. Asahara T, Masuda H, Takahashi T, Kalka C, Pastore C, Silver M,Kearne M, Magner M, Isner JM. Bone marrow origin of endothelialprogenitor cells responsible for postnatal vasculogenesis in physiologicaland pathological neovascularization. Circ Res. 1999;85:221–228.
6. Badorff C, Brandes RP, Popp R, Rupp S, Urbich C, Aicher A, Fleming I,Busse R, Zeiher AM, Dimmeler S. Transdifferentiation of blood-derivedhuman adult endothelial progenitor cells into functionally active cardio-myocytes. Circulation. 2003;107:1024–1032.
7. Iwasaki H, Kawamoto A, Ishikawa M, Oyamada A, Nakamori S,Nishimura H, Sadamoto K, Horii M, Matsumoto T, Murasawa S, ShibataT, Suehiro S, Asahara T. Dose-dependent contribution of CD34-positivecell transplantation to concurrent vasculogenesis and cardiomyogenesisfor functional regenerative recovery after myocardial infarction.Circulation. 2006;113:1311–1325.
8. Node K, Fujita M, Kitakaze M, Hori M, Liao JK. Short-term statintherapy improves cardiac function and symptoms in patients with idio-pathic dilated cardiomyopathy. Circulation. 2003;108:839–843.
9. Sola S, Mir MQ, Lerakis S, Tandon N, Khan BV. Atorvastatin improvesleft ventricular systolic function and serum markers of inflammation innonischemic heart failure. J Am Coll Cardiol. 2006;47:332–337.
10. Krum H, Ashton E, Reid C, Kalff V, Rogers J, Amarena J, Singh B,Tonkin A. Double-blind, randomized, placebo-controlled study ofhigh-dose HMG CoA reductase inhibitor therapy on ventricularremodeling, pro-inflammatory cytokines and neurohormonal parametersin patients with chronic systolic heart failure. J Card Fail. 2007;13:1–7.
11. Dimmeler S, Leri A. Aging and disease as modifiers of efficacy of celltherapy. Circ Res. 2008;102:1319–1330.
12. Fallavollita JA, Perry BJ, Canty JM Jr. 18F-2-deoxyglucose depositionand regional flow in pigs with chronically dysfunctional myocardium:evidence for transmural variations in chronic hibernating myocardium.Circulation. 1997;95:1900–1909.
13. Lim H, Fallavollita JA, Hard R, Kerr CW, Canty JM Jr. Profoundapoptosis-mediated regional myocyte loss and compensatory hypertrophyin pigs with hibernating myocardium. Circulation. 1999;100:2380–2386.
14. Suzuki G, Lee TC, Fallavollita JA, Canty JM Jr. Adenoviral gene transferof FGF-5 to hibernating myocardium improves function and stimulatesmyocytes to hypertrophy and reenter the cell cycle. Circ Res. 2005;96:767–775.
15. Orlic D, Kajstura J, Chimenti S, Jakoniuk I, Anderson SM, Li B, PickelJ, McKay R, Nadal-Ginard B, Bodine DM, Leri A, Anversa P. Bone
marrow cells regenerate infarcted myocardium. Nature. 2001;410:701–705.
16. Orlic D, Kajstura J, Chimenti S, Limana F, Jakoniuk I, Quaini F, Nadal-Ginard B, Bodine DM, Leri A, Anversa P. Mobilized bone marrow cellsrepair the infarcted heart, improving function and survival. Proc NatlAcad Sci U S A. 2001;98:10344–10349.
17. Quaini F, Urbanek K, Beltrami AP, Finato N, Beltrami CA, Nadal-GinardB, Kajstura J, Leri A, Anversa P. Chimerism of the transplanted heart.N Engl J Med. 2002;346:5–15.
18. Minami E, Laflamme MA, Saffitz JE, Murry CE. Extracardiac progenitorcells repopulate most major cell types in the transplanted human heart.Circulation. 2005;112:2951–2958.
19. Abdel-Latif A, Bolli R, Tleyjeh IM, Montori VM, Perin EC, HornungCA, Zuba-Surma EK, Al-Mallah M, Dawn B. Adult bone marrow-derived cells for cardiac repair: a systematic review and meta-analysis.Arch Intern Med. 2007;167:989–997.
20. Zohlnhofer D, Dibra A, Koppara T, de Waha A, Ripa RS, Kastrup J,Valgimigli M, Schomig A, Kastrati A. Stem cell mobilization by granu-locyte colony-stimulating factor for myocardial recovery after acute myo-cardial infarction: a meta-analysis. J Am Coll Cardiol. 2008;51:1429–1437.
21. Llevadot J, Murasawa S, Kureishi Y, Uchida S, Masuda H, Kawamoto A,Walsh K, Isner JM, Asahara T. HMG-CoA reductase inhibitor mobilizesbone marrow–derived endothelial progenitor cells. J Clin Invest. 2001;108:399–405.
22. Dimmeler S, Aicher A, Vasa M, Mildner-Rihm C, Adler K, Tiemann M,Rutten H, Fichtlscherer S, Martin H, Zeiher AM. HMG-CoA reductaseinhibitors (statins) increase endothelial progenitor cells via the PI3-kinase/Akt pathway. J Clin Invest. 2001;108:391–397.
23. Kureishi Y, Luo Z, Shiojima I, Bialik A, Fulton D, Lefer DJ, Sessa WC,Walsh K. The HMG-CoA reductase inhibitor simvastatin activates theprotein kinase Akt and promotes angiogenesis in normocholesterolemicanimals. Nat Med. 2000;6:1004–1010.
24. Canty JM Jr. Coronary blood flow and myocardial ischemia. In: Libby P,Bonow RO, Mann DL, Zipes DP, eds. Braunwald’s Heart Disease. 8thed. Philadelphia, Pa: Elsevier; 2007:1167–1194.
25. Weis M, Heeschen C, Glassford AJ, Cooke JP. Statins have biphasiceffects on angiogenesis. Circulation. 2002;105:739–745.
26. Boodhwani M, Mieno S, Voisine P, Feng J, Sodha N, Li J, Sellke FW.High-dose atorvastatin is associated with impaired myocardial angio-genesis in response to vascular endothelial growth factor in hypercholes-terolemic swine. J Thorac Cardiovasc Surg. 2006;132:1299–1306.
27. Stone PH, Lloyd-Jones DM, Kinlay S, Frei B, Carlson W, Rubenstein J,Andrews TC, Johnstone M, Sopko G, Cole H, Orav J, Selwyn AP,Creager MA. Effect of intensive lipid lowering, with or without anti-oxidant vitamins, compared with moderate lipid lowering on myocardialischemia in patients with stable coronary artery disease: the VascularBasis for the Treatment of Myocardial Ischemia Study. Circulation.2005;111:1747–1755.
28. Bauersachs J, Galuppo P, Fraccarollo D, Christ M, Ertl G. Improvementof left ventricular remodeling and function by hydroxymethylglutarylcoenzyme a reductase inhibition with cerivastatin in rats with heart failureafter myocardial infarction. Circulation. 2001;104:982–985.
29. Hayashidani S, Tsutsui H, Shiomi T, Suematsu N, Kinugawa S, Ide T,Wen J, Takeshita A. Fluvastatin, a 3-hydroxy-3-methylglutaryl coenzymea reductase inhibitor, attenuates left ventricular remodeling and failureafter experimental myocardial infarction. Circulation. 2002;105:868–873.
30. Landmesser U, Engberding N, Bahlmann FH, Schaefer A, Wiencke A,Heineke A, Spiekermann S, Hilfiker-Kleiner D, Templin C, Kotlarz D,Mueller M, Fuchs M, Hornig B, Haller H, Drexler H. Statin-inducedimprovement of endothelial progenitor cell mobilization, myocardial neo-vascularization, left ventricular function, and survival after experimentalmyocardial infarction requires endothelial nitric oxide synthase.Circulation. 2004;110:1933–1939.
31. Abraham SS, Osorio JC, Homma S, Wang J, Thaker HM, Liao JK, MitalS. Simvastatin preserves cardiac function in genetically determined car-diomyopathy. J Cardiovasc Pharmacol. 2004;43:454–461.
32. Takemoto M, Node K, Nakagami H, Liao Y, Grimm M, Takemoto Y,Kitakaze M, Liao JK. Statins as antioxidant therapy for preventingcardiac myocyte hypertrophy. J Clin Invest. 2001;108:1429–1437.
33. Trochu JN, Mital S, Zhang X, Xu X, Ochoa M, Liao JK, Recchia FA,Hintze TH. Preservation of NO production by statins in the treatment ofheart failure. Cardiovasc Res. 2003;60:250–258.
Suzuki et al Pravastatin in Hibernating Myocardium 263
by guest on September 18, 2015http://circres.ahajournals.org/Downloaded from

34. Zaca V, Rastogi S, Imai M, Wang M, Sharov VG, Jiang A, Goldstein S,Sabbah HN. Chronic monotherapy with rosuvastatin prevents progressiveleft ventricular dysfunction and remodeling in dogs with heart failure.J Am Coll Cardiol. 2007;50:551–557.
35. Gheorghiade M, Klein L, Stone NJ, Bonow RO, Kim RJ. Improvement inthe function of hibernating myocardium in a patient with heart failure dueto coronary artery disease receiving high-dose simvastatin. Ital Heart J.2004;5:160–162.
36. Muller P, Kazakov A, Semenov A, Bohm M, Laufs U. Pressure-inducedcardiac overload induces upregulation of endothelial and myocardialprogenitor cells. Cardiovasc Res. 2008;77:151–159.
37. Rota M, Kajstura J, Hosoda T, Bearzi C, Vitale S, Esposito G, IaffaldanoG, Padin-Iruegas ME, Gonzalez A, Rizzi R, Small N, Muraski J, AlvarezR, Chen X, Urbanek K, Bolli R, Houser SR, Leri A, Sussman MA,Anversa P. Bone marrow cells adopt the cardiomyogenic fate in vivo.Proc Natl Acad Sci U S A. 2007;104:17783–17788.
38. Mazhari R, Hare JM. Mechanisms of action of mesenchymal stem cells incardiac repair: potential influences on the cardiac stem cell niche. NatClin Pract Cardiovasc Med. 2007;4(suppl 1):S21–S26.
39. Matsuura K, Wada H, Nagai T, Iijima Y, Minamino T, Sano M, AkazawaH, Molkentin JD, Kasanuki H, Komuro I. Cardiomyocytes fuse with
surrounding noncardiomyocytes and reenter the cell cycle. J Cell Biol.2004;167:351–363.
40. Urbich C, Aicher A, Heeschen C, Dernbach E, Hofmann WK, ZeiherAM, Dimmeler S. Soluble factors released by endothelial progenitor cellspromote migration of endothelial cells and cardiac resident progenitorcells. J Mol Cell Cardiol. 2005;39:733–742.
41. Fazel S, Cimini M, Chen L, Li S, Angoulvant D, Fedak P, Verma S,Weisel RD, Keating A, Li RK. Cardioprotective c-kit� cells are from thebone marrow and regulate the myocardial balance of angiogenic cyto-kines. J Clin Invest. 2006;116:1865–1877.
42. Kjekshus J, Apetrei E, Barrios V, Bohm M, Cleland JG, Cornel JH,Dunselman P, Fonseca C, Goudev A, Grande P, Gullestad L, HjalmarsonA, Hradec J, Janosi A, Kamensky G, Komajda M, Korewicki J, Kuusi T,Mach F, Mareev V, McMurray JJ, Ranjith N, Schaufelberger M,Vanhaecke J, van Veldhuisen DJ, Waagstein F, Wedel H, Wikstrand J.Rosuvastatin in older patients with systolic heart failure. N Engl J Med.2007;357:2248–2261.
43. Gissi-Hf Investigators. Effect of rosuvastatin in patients with chronicheart failure (the GISSI-HF trial): a randomised, double-blind, placebo-controlled trial. Lancet. 2008;372:1231–1239.
264 Circulation Research January 30, 2009
by guest on September 18, 2015http://circres.ahajournals.org/Downloaded from

Supplemental Material
Online Supplement
Pravastatin Improves Function in Hibernating Myocardium by Mobilizing
CD133+ and cKit+ Bone Marrow Progenitor Cells and Promoting Myocytes to Reenter the
Growth Phase of the Cardiac Cell Cycle
Gen Suzuki
Vijay Iyer
Thomas Cimato
and
John M. Canty, Jr.
From the VA WNY Health Care System and the Departments of Medicine and Physiology&Biophysics and the Center for Research in
Cardiovascular Medicine at the University at Buffalo.
Supported by the VA, AHA, Buswell Fellowship NHLBI, Albert and Elizabeth Rekate Fund and the Empire State Stem Cell Board.
Correspondence:
John M. Canty, Jr. Division of Cardiology University at Buffalo Biomedical Research Building, Room 345 3435 Main Street Buffalo, NY 14214 Phone: 716-829-2663 [email protected]
[7]Chronic Ischemic Heart Disease, [130] Animal Model of Human Disease,
1

Supplemental Material
Materials and Methods
Procedures and protocols conformed to institutional guidelines for the care and use of animals
in research.
Flow Cytometry and Mobilization of Bone Marrow Progenitor Cells with Pravastatin
We initially assessed the dose-dependency of pravastatin to mobilize bone marrow progenitor
cells in normal swine (n=11). Baseline measurements were compared to those after 2-weeks of
treatment with low dose (20 mg/day) or high dose (160 mg/day) pravastatin. Mononuclear cells were
isolated from peripheral blood (30 ml) and bone marrow (30ml) using the Becton Dickinson CPT cell
separation system. White cell and monocyte counts were performed with a hemocytometer.
Approximately 10-20x106 mononuclear cells were analyzed by FACS after staining for CD133 (PE
conjugated, Miltenyi Biotech) and cKit (FITC conjugated, eBioscience). Isotype controls were used
as negative controls. Single stains were performed to determine quality control and multichannel
compensation. Data are expressed as progenitor cells (CD133+ or cKit+) per million mononuclear
cells. All counts were corrected for the absolute mononuclear cell count.
Effects of Pravastatin (160 mg/day) on Flow and Function in Hibernating Myocardium
Hibernating myocardium was produced as previously described1. Briefly, pigs were sedated
(Telazol; tiletamine 50mg/ml and zolazepam 50 mg/ml)/xylazine (100 mg/ml, 0.022 mg/kg i.m.),
intubated and ventilated with a 0.5–2% isoflurane-oxygen mixture. Through a limited pericardiotomy,
the proximal LAD was instrumented with a Delrin occluder (1.5 mm i.d.). Antibiotics (cefazolin, 25
mg/kg and gentamicin, 3 mg/kg i.m.) were given 1-hour before surgery and repeated after closing the
chest. Following analgesia was performed by using intercostal nerve block (0.5% Marcaine),
intramuscular doses of butorphanol (2.2 mg/kg q6h) and flenixin (1-2 mg/kg q.d.).
2

Supplemental Material
Serial Physiological Studies - Studies began 4-months after instrumentation (n=22) at which time
reductions in flow, function and coronary flow reserve indicative of hibernating myocardium were
present2. Sedation was initiated with Telazol/xylazine and maintained with propofol (5-10 mg/kg/hr
i.v.). Under sterile conditions, we inserted a 6-Fr introducer into the left brachial artery. A Millar
Micro-Tip pressure catheter was inserted into the left ventricle (LV) apex for microsphere injection.
The introducer side port was used to monitor aortic pressure and perform blood withdrawal for
microspheres. Animals were heparinized (100 U/kg), and hemodynamics allowed to equilibrate for at
least 30-minutes. Regional wall-thickening was assessed with transthoracic echocardiography from a
right parasternal approach3. All pigs showed anterior dysfunction but dyskinesis was not present
under any condition. Systolic wall-thickening (ΔWT=ESWT-EDWT) was measured in LAD and
remote regions. Ventricular dimensions and LV mass were calculated using ASE criteria. This was
followed by LV microsphere injection to assess resting perfusion. Finally, pharmacological
vasodilation was produced using adenosine (0.9mg/kg/min i.v.) with phenylephrine infused and
titrated to maintain mean blood pressure ~100 mmHg and microsphere flow repeated. At the end of
the study, catheters were removed and pigs were brought back to the animal facility. Animals were
treated with pravastatin (160 mg/day for five weeks, n=12) and compared to untreated controls (n=10).
We also studied the effects of pravastatin on normal control animals without a stenosis or LV
dysfunction (pravastatin treated n=10, untreated n=5).
Animals were brought back for repeat physiological studies five weeks later after which they
were euthanized under anesthesia. The LV was immediately excised, weighed and sectioned into 1-
cm rings parallel to the AV groove from apex to base. Thin rings between each major ring were
incubated in TTC to quantify infarction. Central areas of the ischemic LAD and remote region were
taken for flow and histology as previously demonstrated4 and outlined below.
3

Supplemental Material
Microsphere Perfusion - Resting and vasodilated myocardial perfusion were assessed using
fluorescent microspheres (15μm) as previously described1, 3. We injected ~3x106 microspheres into
the LV over 10 seconds while an arterial reference sample was withdrawn at 6 ml/min (90-seconds).
Midventricular rings were divided into twelve circumferential wedges with each cut into three
transmural layers5. Dyes were extracted using standard techniques and fluorescence quantified at
selected excitation wavelengths6. In each animal the circumferential flow distribution during
adenosine was analyzed to identify the hibernating risk region5. From this we eliminated border
regions and derived weighted averages from central hypoperfused or normally-perfused remote
regions. Relative perfusion was determined by dividing the flow in each individual piece by the
corresponding average full-thickness value from normal myocardium.
Myocardial Histopathology Myocyte Nuclear Density and Morphometry - Samples adjacent to LAD (hibernating) and posterior
descending arteries (normal) were fixed (10% formalin) and paraffin-embedded. Point-counting of
trichrome-stained sections was used to quantify connective tissue1. PAS stained sections were used to
quantify myocyte diameter and nuclear length (100 longitudinal myocytes per region) in
subendocardial and subepicardial thirds of the left ventricle5. Myocyte diameter from the pravastatin
group was compared to age-matched animals with untreated hibernating myocardium and sham
controls. We also assessed regional myocyte nuclear density as previously described7.
Quantitative Immunohistochemistry of Progenitor Cells and Myocyte Proliferation - To identify
myocyte proliferation, frozen tissue sections (5μm) were incubated with anti-Ki67 (clone MIB-1,
Dako, 1:200) to detect myocytes in the growth phase of the cell cycle. An anti-phospho-Histone H3
antibody (upstate, 1:1000) was used to detect cells in the mitotic phase of the cell cycle7. Co-staining
with anti-cTnI (Santa Cruz, 1:200) was used to confirm these changes were in cardiac myocytes.
4

Supplemental Material
Hematopoietic progenitor cells were quantified using the cell surface marker CD133 (immature HSC
epitope, Miltenyi Biotec, 1:100)8, cKit (CD117, Santa-Cruz, 1:200) and CD45 (BD biosciences,
1:200). Samples were post-treated with FITC and TRITC conjugated secondary antibody (Zymed).
Nuclei were stained with TO-PRO-3 (Invitrogen, 1μM). To quantify the percentage of CD133+/cKit+
cells expressing cardiac lineage markers, we co-stained sections with GATA-4 (Santa Cruz, 1:100)
and cTnI (Santa Cruz, 1:100). All of these antibodies have previously been used in swine5, 8. Images
were acquired with a confocal microscope (Bio-Rad MRC 1024). CD133+/cKit+ cells as well as
myocytes in the proliferative phase of the cell cycle were expressed in relation to myocyte nuclear
density measurements. We assessed expressed the frequency of Ki67 positive myocyte nuclei per
million myocyte nuclei as we have previously escibed.5 An average of 745±74 fields (~116 mm2)
were examined per slide for each stain.
Statistics
Data are expressed as mean±standard error. Our primary analysis focused upon differences in
hemodynamics, flow, function and quantitative immunohistochemical parameters between pravastatin
and untreated animals using a two-way ANOVA with post-hoc difference assessed using the Holm-
Sidak test (Sigma Stat 3.0). We also assessed serial physiological changes in animals studied at five
weeks as compared to baseline measurements using paired t-tests with each animal used as its own
control. A similar approach was used to assess differences in perfusion between vasodilation and rest
as well as between LAD and remote regions of the same heart. Differences of p<0.05 were considered
significant.
5

Supplemental Material
Results
Serial hemodynamics in each of the treatment groups are presented in Online Table 1. Online
Table 2 summarizes serial measurements in myocardial function assessed using echocardiography.
Baseline findings at the initial study confirmed dysfunctional hibernating myocardium. Animals in
each treatment group had similar resting hemodynamics and systolic function as summarized in the
Tables and the following presents the average baseline data for the entire group. Regional systolic
LAD wall thickening (ESWT-EDWT) was reduced in comparison to remote normally perfused
regions (3.7±0.3 mm vs. 6.6±0.3 mm, p<0.001). There were reductions in resting full-thickness
perfusion (LAD 0.72±0.06 vs. 0.94±0.05 ml/min/g in remote, p<0.01) with the greatest reduction in
the subendocardium (0.66±0.06 vs. 1.05±0.06 ml/min/g in remote, p<0.001). Full-thickness flow
during adenosine was severely attenuated (LAD 1.22±0.22 vs. 4.47±0.25 ml/min/g in remote,
p<0.001) and subendocardial flow did not increase above the resting value (adenosine LAD flow
0.68±0.15 ml/min/g, p-ns. vs. rest).
6

Supplemental Material
References
1. Fallavollita JA, Perry BJ, Canty JM, Jr. 18F-2-deoxyglucose deposition and regional flow in
pigs with chronically dysfunctional myocardium: Evidence for transmural variations in
chronic hibernating myocardium. Circulation. 1997;95:1900-1909.
2. Fallavollita JA, Logue M, Canty JM, Jr. Stability of hibernating myocardium in pigs with a
chronic left anterior descending coronary artery stenosis: Absence of progressive fibrosis in the
setting of stable reductions in flow, function and coronary flow reserve. J Am Coll Cardiol.
2001;37:1989-1995.
3. Malm BJ, Suzuki G, Canty JM, Jr., Fallavollita JA. Variability of contractile reserve in
hibernating myocardium: Dependence on the method of stimulation. Cardiovasc Res.
2002;56:422-433.
4. Luisi AJ, Jr., Fallavollita JA, Suzuki G, Canty JM, Jr. Spatial inhomogeneity of sympathetic
nerve function in hibernating myocardium. Circulation. 2002;106:779-781.
5. Suzuki G, Lee TC, Fallavollita JA, Canty JM, Jr. Adenoviral gene transfer of FGF-5 to
hibernating myocardium improves function and stimulates myocytes to hypertrophy and
reenter the cell cycle. Circ Res. 2005;96:767-775.
6. Glenny RW, Bernard S, Brinkley M. Validation of fluorescent-labeled microspheres for
measurement of regional organ perfusion. J Appl Physiol. 1993;74:2585-2597.
7. Lim H, Fallavollita JA, Hard R, Kerr CW, Canty JM, Jr. Profound apoptosis-mediated regional
myocyte loss and compensatory hypertrophy in pigs with hibernating myocardium. Circulation.
1999;100:2380-2386.
7

Supplemental Material
8
8. Vacanti V, Kong E, Suzuki G, Sato K, Canty JM, Jr., Lee TC. Phenotypic changes of adult
porcine mesenchymal stem cells induced by prolonged passaging in culture. J Cell Physiol.
2005;205:194-201.

Supplemental Material
Online Table I. Effects of Pravastatin on Hemodynamics.
Values are Mean ± SEM; *p<0.05 vs. Untreated, †p<0.05 vs. Initial; LVEDP – Left Ventricular End Diastolic Pressure
Systolic Pressure (mm Hg)
Mean Aortic Pressure (mm Hg)
Heart Rate
(bpm)
LVEDP (mm Hg)
LV dP/dt Max
(mm Hg/sec) REST Initial Untreated 120±3 94±4 94±8 24±2 2055±197 Pravastatin 114±4 83±3 106±4 18±2 2013±103 Final Untreated 122±7 96±7 96±7 24±3 2024±111 Pravastatin 126±5† 93±4† 102±5 16±2* 2538±125*† ADENOSINE Initial Untreated 125±5 95±4 95±4 36±3 2149±278 Pravastatin 127±4 90±4 102±5 30±5 1945±134 Final Untreated 129±7 92±7 92±4 32±2 2022±199 Pravastatin 128±6 88±4 96±6 24±2* 2281±183
LV dP/dt – first derivative of LV pressure

Supplemental Material
Online Table II. Effects of Pravastatin on Echocardiographic Measurements of Regional Function. LAD Remote
EDWT (mm)
ΔWT (mm)
WT (%)
EDWT (mm)
ΔWT (mm)
WT (%)
Initial Untreated 10.4±0.4 3.5±0.2 34.4±2.0 7.5±0.4 6.0±0.4 84.0±8.6 Pravastatin 9.8±0.6 3.8±0.6 40.4±4.0 8.5±0.7 7.1±0.5 87.5±9.4 Final Untreated 10.8±0.6 2.9±0.4 28.6±2.8 9.3±0.3† 7.0±0.5 75.5±4.6 Pravastatin 11.1±0.7† 5.2±0.5*† 48.3±5.1*† 9.4±0.9 8.1±0.9 89.3±10.4 Values are mean ± SEM; *p<0.05 vs. Untreated; †p<0.05 vs. Initial; LAD – Left Anterior Descending Artery; LV – Left Ventricular; EDWT – End-Diastolic Wall Thickening; WT – Wall Thickening

Gen Suzuki, Vijay Iyer, Thomas Cimato and John M. Canty, JrPhase of the Cardiac Cell Cycle
Bone Marrow Progenitor Cells and Promoting Myocytes to Reenter the Growth+cKit and+Pravastatin Improves Function in Hibernating Myocardium by Mobilizing CD133
Print ISSN: 0009-7330. Online ISSN: 1524-4571 Copyright © 2008 American Heart Association, Inc. All rights reserved.is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Circulation Research
doi: 10.1161/CIRCRESAHA.108.1887302009;104:255-264; originally published online December 18, 2008;Circ Res.
http://circres.ahajournals.org/content/104/2/255World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://circres.ahajournals.org/content/suppl/2008/12/18/CIRCRESAHA.108.188730.DC1.htmlData Supplement (unedited) at:
http://circres.ahajournals.org//subscriptions/
is online at: Circulation Research Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer about this process is available in the
located, click Request Permissions in the middle column of the Web page under Services. Further informationEditorial Office. Once the online version of the published article for which permission is being requested is
can be obtained via RightsLink, a service of the Copyright Clearance Center, not theCirculation Researchin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on September 18, 2015http://circres.ahajournals.org/Downloaded from
Copyright © 2022 FDOKUMEN




















