Possible Lamotrigine-Induced Mania in a Child With Autism Spectrum Disorder and Epilepsy
20
Independent Effect of Paliperidone Extended Release on Social Functioning Beyond Its Effect on Positive and Negative Symptoms of Schizophrenia A Mediation Analysis To the Editors: S chizophrenia is a severe, chronic, debil- itating mental illness affecting up to 1% of the population worldwide. It is charac- terized by a range of symptoms, including positive and negative symptoms and neuro- cognitive deficits, and is often characterized by periods of relative stability and recur- rences of symptoms. Even during periods of relative stability, it is unusual for patients to fully recover from social functioning deficits. 1,2 The inability of patients to com- petently perform activities of daily living, attend to personal hygiene, maintain social relationships, and work or live indepen- dently results in significant financial and social costs to society, their families, and caregivers. 3 Finding treatments to improve social functioning is important, and in research, measurement of social function- ing improvements is challenging. Positive and negative symptom control can be asso- ciated with improvement in various aspects of social functioning and is a challenge to differentiate how much of the improvement is due to the nonspecific improvement in positive and negative symptoms and how much is an independent effect on social functioning. 3 Although both symptom control and social functioning can improve with treatment, not all treatment-related improvements in social functioning need to be attributed to symptom improvement. Paliperidone extended-release tablets (paliperidone ER; INVEGA [ALSA Cor- poration, Mountain View, Calif]) have re- cently been approved in the United States and the EU (prolonged release) as a new chemical entity and new antipsychotic drug. In the paliperidone ER program, the Positive and Negative Syndrome Scale (PANSS) was used as the primary outcome measure. Given the growing importance of measuring functional improvements, a clinician-reported measure of social func- tioning, the Personal and Social Perfor- mance (PSP) scale, 4 was included as a key secondary outcome measure because it measures a different concept from PANSS. Three 6-week, placebo-controlled studies of similar design established the efficacy, safety, and tolerability of paliper- idone ER and significantly showed that symptom severity and social functioning improve with paliperidone ER treatment. 5 This letter presents the results of a mediation analysis showing a statistically significant effect of paliperidone ER on social functioning independent from its effect on symptom improvement. The analysis is based upon pooled data from the three 6-week studies in patients with schizophrenia treated with fixed daily doses of paliperidone ER (3 mg, 6 mg, 9 mg, and 12 mg) or placebo. The intent-to-treat pop- ulation and the last-observation-carried- forward approach were used for efficacy analysis. 5 The primary efficacy end point was change from baseline in PANSS total score at the end of treatment. The key secondary end point was change from baseline to end point in PSP score. Positive and Negative Syndrome Scale assessments were performed at baseline, days 4, 8, and 15 and then every 7 days until day 43 or end of treatment. Personal and Social Perfor- mance scale assessments were performed at baseline and at day 43 or end of treatment. The Baron and Kenny mediation model 6 assesses the degree of treatment effect upon a response variable in the presence of another variable (ie, the mediating vari- able). The model allows the examining of the degree of mediation (either as partial or complete mediation). We show that there is only a partial mediation of PANSS upon PSP in our setting and thus are able to conclude that there is an independent effect of paliperidone ER on social functioning that is over and above its effects on symptoms. Change in PANSS is considered a partial mediator if (i) treatment (paliper- idone ER) significantly predicts a change in PSP; (ii) treatment significantly predicts a change in PANSS; (iii) independent of treatment, change in PANSS linearly sig- nificantly predicts the change in PSP; and (iv) treatment significantly predicts a change in PSP even when controlling for the significant effect of change in PANSS. Complete mediation occurs if in condition (iv), described earlier, the effect of treat- ment was no more significant. Our model extends the Baron and Kenny 6 mediation model (which use regression models) by using analysis of covariance models to handle conditions (i), (ii), (iii), and (iv), where the response variable was either change in PSP or change in PANSS. Terms for treatment, protocol, baseline TABLE 1. Establishment of the Partial Mediation of Change in PANSS Total Score (and Factor Scores) and the Independent Effect of Treatment on Change in PSP Dependent Variable (Model No.*) Independent Variable(s) Parameter Estimates (P) PSP (i) Treatment 7.61 †‡ PANSS (ii) Treatment 12.49 †‡ PSP (iv) Treatment 1.80 †§ PANSS 0.47 ‡|| PSP (i) Treatment 7.61 †‡ Positive (ii) Treatment 3.66 †‡ PSP (iv) Treatment 3.03 †‡ Positive 1.25 ‡|| PSP (i) Treatment 7.61 †‡ Negative (ii) Treatment 2.64 †‡ PSP (iv) Treatment 4.38 †‡ Negative 1.23 ‡|| PSP (i) Treatment 7.61 †‡ Disorganized (ii) Treatment 2.82 †‡ PSP (iv) Treatment 3.09 †‡ Disorganized 1.68 ‡|| PSP (i) Treatment 7.61 †‡ Uncontrolled (ii) Treatment 1.96 †‡ PSP (iv) Treatment 3.98 †‡ Uncontrolled 1.79 ‡|| PSP (i) Treatment 7.61 †‡ Anxiety (ii) Treatment 1.48 †‡ PSP (iv) Treatment 5.23 †‡ Anxiety 1.77 ‡|| *Results for model (iii) are not presented. Because all treatment-by-PANSS total or sub- scale factor interaction terms were not signif- icant, results from model (iii) are similar to the partial effects presented in model (iv). † These quantities represent the average treatment effect at end point (relative to baseline). ‡ Significance is P G 0.001. § Significance is P = 0.01. || These quantities represent the estimate of the common slopes. PANSS indicates change from baseline in PANSS total score; PSP, change from baseline in PSP scale score; Positive, change from baseline in positive symptoms score; Negative, change from baseline in negative symptoms score; Disorganized, change from baseline in disorganized thought score; Uncontrolled, change from baseline in uncontrolled hostility/ excitement score; Anxiety, change from base- line in anxiety/depression score. LETTERS TO THE EDITORS 496 www.psychopharmacology.com Journal of Clinical Psychopharmacology & Volume 29, Number 5, October 2009 9 Copyright @ 200 Lippincott Williams & Wilkins. Unauthorized reproduction of this article is prohibited.
Transcript of Possible Lamotrigine-Induced Mania in a Child With Autism Spectrum Disorder and Epilepsy

Independent Effect ofPaliperidone Extended
Release on SocialFunctioning Beyond ItsEffect on Positive andNegative Symptoms of
SchizophreniaA Mediation Analysis
To the Editors:
Schizophrenia is a severe, chronic, debil-itating mental illness affecting up to 1%
of the population worldwide. It is charac-terized by a range of symptoms, includingpositive and negative symptoms and neuro-cognitive deficits, and is often characterizedby periods of relative stability and recur-rences of symptoms. Even during periodsof relative stability, it is unusual for patientsto fully recover from social functioningdeficits.1,2 The inability of patients to com-petently perform activities of daily living,attend to personal hygiene, maintain socialrelationships, and work or live indepen-dently results in significant financial andsocial costs to society, their families, andcaregivers.3 Finding treatments to improvesocial functioning is important, and inresearch, measurement of social function-ing improvements is challenging. Positiveand negative symptom control can be asso-ciated with improvement in various aspectsof social functioning and is a challenge todifferentiate how much of the improvementis due to the nonspecific improvement inpositive and negative symptoms and howmuch is an independent effect on socialfunctioning.3 Although both symptomcontrol and social functioning can improvewith treatment, not all treatment-relatedimprovements in social functioning need tobe attributed to symptom improvement.
Paliperidone extended-release tablets(paliperidone ER; INVEGA [ALSA Cor-poration, Mountain View, Calif ]) have re-cently been approved in the United Statesand the EU (prolonged release) as a newchemical entity and new antipsychoticdrug. In the paliperidone ER program, thePositive and Negative Syndrome Scale(PANSS) was used as the primary outcomemeasure. Given the growing importanceof measuring functional improvements, aclinician-reported measure of social func-tioning, the Personal and Social Perfor-mance (PSP) scale,4 was included as akey secondary outcome measure becauseit measures a different concept from
PANSS. Three 6-week, placebo-controlledstudies of similar design established theefficacy, safety, and tolerability of paliper-idone ER and significantly showed thatsymptom severity and social functioningimprove with paliperidone ER treatment.5
This letter presents the results of amediation analysis showing a statisticallysignificant effect of paliperidone ER onsocial functioning independent from itseffect on symptom improvement. Theanalysis is based upon pooled data fromthe three 6-week studies in patients withschizophrenia treated with fixed daily dosesof paliperidone ER (3 mg, 6 mg, 9 mg, and12 mg) or placebo. The intent-to-treat pop-ulation and the last-observation-carried-forward approach were used for efficacyanalysis.5 The primary efficacy end pointwas change from baseline in PANSS totalscore at the end of treatment. The keysecondary end point was change frombaseline to end point in PSP score. Positiveand Negative Syndrome Scale assessmentswere performed at baseline, days 4, 8, and15 and then every 7 days until day 43 or endof treatment. Personal and Social Perfor-mance scale assessments were performed atbaseline and at day 43 or end of treatment.The Baron and Kenny mediation model6
assesses the degree of treatment effectupon a response variable in the presence ofanother variable (ie, the mediating vari-able). The model allows the examining ofthe degree of mediation (either as partial orcomplete mediation). We show that there isonly a partial mediation of PANSS uponPSP in our setting and thus are able toconclude that there is an independent effectof paliperidone ER on social functioningthat is over and above its effects onsymptoms. Change in PANSS is considereda partial mediator if (i) treatment (paliper-idone ER) significantly predicts a change inPSP; (ii) treatment significantly predictsa change in PANSS; (iii) independent oftreatment, change in PANSS linearly sig-nificantly predicts the change in PSP; and(iv) treatment significantly predicts achange in PSP even when controlling forthe significant effect of change in PANSS.Complete mediation occurs if in condition(iv), described earlier, the effect of treat-ment was no more significant. Our modelextends the Baron and Kenny6 mediationmodel (which use regression models) byusing analysis of covariance models tohandle conditions (i), (ii), (iii), and (iv),where the response variable was eitherchange in PSP or change in PANSS.Terms for treatment, protocol, baseline
TABLE 1. Establishment of the PartialMediation of Change in PANSS TotalScore (and Factor Scores) and theIndependent Effect of Treatment onChange in PSP
DependentVariable(Model No.*)
IndependentVariable(s)
ParameterEstimates
(P)
PSP (i) Treatment 7.61†‡
PANSS (ii) Treatment 12.49†‡
PSP (iv) Treatment 1.80†§
PANSS 0.47‡||
PSP (i) Treatment 7.61†‡
Positive (ii) Treatment 3.66†‡
PSP (iv) Treatment 3.03†‡
Positive 1.25‡||
PSP (i) Treatment 7.61†‡
Negative (ii) Treatment 2.64†‡
PSP (iv) Treatment 4.38†‡
Negative 1.23‡||
PSP (i) Treatment 7.61†‡
Disorganized (ii) Treatment 2.82†‡
PSP (iv) Treatment 3.09†‡
Disorganized 1.68‡||
PSP (i) Treatment 7.61†‡
Uncontrolled (ii) Treatment 1.96†‡
PSP (iv) Treatment 3.98†‡
Uncontrolled 1.79‡||
PSP (i) Treatment 7.61†‡
Anxiety (ii) Treatment 1.48†‡
PSP (iv) Treatment 5.23†‡
Anxiety 1.77‡||
*Results for model (iii) are not presented.Because all treatment-by-PANSS total or sub-scale factor interaction terms were not signif-icant, results from model (iii) are similar to thepartial effects presented in model (iv).
†These quantities represent the averagetreatment effect at end point (relative tobaseline).
‡Significance is P G 0.001.§Significance is P = 0.01.||These quantities represent the estimate of
the common slopes.
PANSS indicates change from baseline inPANSS total score; PSP, change from baselinein PSP scale score; Positive, change frombaseline in positive symptoms score; Negative,change from baseline in negative symptomsscore; Disorganized, change from baseline indisorganized thought score; Uncontrolled,change from baseline in uncontrolled hostility/excitement score; Anxiety, change from base-line in anxiety/depression score.
LETTERS TO THE EDITORS
496 www.psychopharmacology.com Journal of Clinical Psychopharmacology & Volume 29, Number 5, October 2009
9Copyright @ 200 Lippincott Williams & Wilkins. Unauthorized reproduction of this article is prohibited.

score, and center within protocol (fixedeffect) were included to mirror the originalanalysis undertaken by Meltzer et al.5 Theeffect of the interaction of change inPANSS by treatment was also examinedto validate the assumption of a commonlinear effect of change in PANSS uponchange in PSP across the paliperidone ERtreatment groups and placebo. Appropriatecontrasts were included in the model to testthe average effect of treatment across the 4paliperidone dose groups and the effect ofthe individual doses.
The pooled intent-to-treat population(n = 1192) was 63% white, 62% male,with a mean age (TSD) of 38.4 T 11.0years, mean baseline PANSS total score(TSD) of 93.6 T 11.7, and mean baselinePSP score (TSD) of 47.7 T 14.1. Table 1provides the results of the analysis ofcovariance models used to establish partialmediation. All relevant parameters are sig-nificantly different from zero (P G 0.02).There is a statistically significant positiveaverage effect of paliperidone ER treat-ment on the change from baseline to endpoint in PSP score (P = 0.0103) afteraccounting for (a) the significant effect ofpaliperidone ER on the change from base-line in PANSS and (b) the linear predictiverelationship of change in PANSS upon thechange in PSP. Thus, the significant posi-tive effect of paliperidone ER on socialfunctioning (PSP) is independent fromthe effect on symptoms (PANSS). Theinteraction term, treatment by change inPANSS, was not significant (P = 0.4342)when included in model (iv), but thecommon change in PANSS was signifi-cant (P G 0.001), thus validating model(iii). Hence, model (iv) did not include theinteraction term (Table 1). Furthermore,using the different contrasts versus place-bo to assess the mediating effect of eachindividual dose resulted in similar conclu-sions. In addition to the global symptomchanges assessed by the PANSS, themediating role of each of the 5 symptomdomains based on the Marder criteria (pos-itive symptoms, negative symptoms, dis-organized thoughts, uncontrolled hostility/excitement, and anxiety/depression) on therelationship between paliperidone and PSPwas also assessed. Results similar to thoseobtained for the total PANSS were ob-tained (Table 1). The treatment by changein PANSS subscale factor interactions wasall not significant.
DISCUSSIONCurrent atypical antipsychotics pro-
vide effective treatment for both positiveand negative symptoms of schizophrenia.However, social functioning deficits oftenremain only partially treated. These con-
tinuing social functioning impairmentsresult in significant costs to society in lostproductivity, health care costs, and the needto provide supervised housing and ongoingassistance to families and caregivers.3 Thus,diminished social functioning in schizo-phrenia is probably responsible for moreburden in patients, families, and caresystems than residual symptoms. Findinga psychotropic treatment that improvessocial functioning is critically important.The clinical program of paliperidone ERwas designed to incorporate the PSP as ameasure of social functioning. All paliper-idone ER doses were shown to be superiorto placebo across both the primary (totalPANSS and the 5 subscale factors accord-ing to the Marder criteria) and key second-ary (PSP) variables.5 In this letter, we reportfurther analyses that demonstrated that theameliorative effect of paliperidone ER up-on social functioning (PSP) was above andbeyond its effect upon symptom severity(total PANSS and its 5 subscale factors) inpatients with schizophrenia.
ACKNOWLEDGMENTSAllan Sampson contributed in his
capacity as a statistical consultant toJohnson & Johnson Pharmaceutical Re-search & Development. The authors thankInes Adriaenssen of Johnson & JohnsonPharmaceutical Services for her encour-agement in the development of the manu-script. The authors also thank MichelleTickner, PhD, of Medicus International forher editorial assistance.
This study was supported and fundedby Johnson & Johnson PharmaceuticalResearch & Development. Editorial sup-port for the preparation of this manuscriptwas supported and funded by Johnson &Johnson Pharmaceutical Services, LLC.
AUTHOR DISCLOSUREINFORMATION
D. Hough, I.F. Nuamah, P. Lim, D.D.Gagnon, and M. Rothman are fulltimeemployees of Johnson & Johnson. A.Sampson is employed by the Universityof Pittsburgh, acted as a statistical con-sultant for Johnson & Johnson, and hasno financial conflicts to disclose.
David Hough, MD
Isaac F. Nuamah, PhD
Pilar Lim, PhDJohnson & Johnson Pharmaceutical
Research & DevelopmentTitusville, NJ
Allan Sampson, PhDDepartment of StatisticsUniversity of Pittsburgh
Pittsburgh, PA
Dennis D. Gagnon, MA
Margaret Rothman, PhDJohnson & Johnson Pharmaceutical Services
Raritan, NJ
REFERENCES
1. Berndt ER. Changes in the costs of treatingmental health disorders. Pharmacoeconomics.2004;22(suppl 2):37Y50.
2. Addington J, Addington D. Neurocognitiveand social functioning in schizophrenia: a2.5 year follow-up study. Schizophr Res.2000;44:47Y56.
3. Priebe S. Social outcomes in schizophrenia.Br J Psychiatry. 2007;191(suppl 50):s15Ys20.
4. Morosini PL, Magliano L, Brambilla L,et al. Development, reliability andacceptability of a new version of theDSM-IV Social and Occupational FunctionAssessment Scale (SOFAS) to assess routinesocial function. Acta Psychiatr Scand.2000;101:323Y329.
5. Meltzer HY, Bobo WV, Nuamah I, et al.Efficacy and tolerability of oral paliperidoneextended-release tablets in the treatment ofacute schizophrenia: pooled data from three,6-week, placebo-controlled studies. J ClinPsychiatry. 2008;69:817Y829.
6. BaronRM,KennyDA. Themoderator-mediatordistinction in social psychological research:conceptual, strategic and statisticalconsiderations. J Pers Soc Psychol.1986;51:1173Y1182.
Atypical NeurolepticMalignant Syndrome
With QuetiapineA Case Report and Review
of the LiteratureTo the Editors:
There has been a growing number ofcase reports describing possible neu-
roleptic malignant syndrome (NMS) as-sociated with atypical antipsychoticmedications.1 Many of these reports,however, reveal a symptom constellationthat is not consistent with classic featuresof NMS. This has led to the conceptual-ization of a variant of NMS occurringsecondary to atypical antipsychotics andhas been termed atypical NMS.2 Hyper-thermia, a core feature of standard NMS,is absent in many cases of atypical NMSsecondary to atypical antipsychotics.1 De-spite growing support for atypical NMSas a legitimate entity, the concept andcriteria of atypical NMS remain contro-versial.3 We present a case consistent withatypical NMS secondary to quetiapine.Although quetiapine has been implicated
Journal of Clinical Psychopharmacology & Volume 29, Number 5, October 2009 Letters to the Editors
* 2009 Lippincott Williams & Wilkins www.psychopharmacology.com 497
9Copyright @ 200 Lippincott Williams & Wilkins. Unauthorized reproduction of this article is prohibited.

as an etiological factor in NMS,4Y15 pre-vious reports have not established whetherthe reaction to quetiapine fits with aclassic or instead atypical NMS presenta-tion. In this report, we review existing casereports of quetiapine-induced NMS in anattempt to clarify whether their presenta-tions were more consistent with classic oratypical NMS.
CASE REPORTA 52-year-old man with chronic
major depressive disorder was admitted toour psychiatric ward with a severe majordepressive episode and suicidal ideation.For the 2 years before admission, he hadbeen receiving a daily combination of ris-peridone at 2 mg, valproic acid at 750 mg,clonazepam at 2.5 mg, zopiclone at 7.5 mg,lamotrigine at 200 mg, and citalopram at80 mg. The patient experienced persistentdepressive symptoms despite this medica-tion regimen, and out of frustration, dis-continued all his psychotropic medications3 weeks before admission. Citalopram wasthe single medication that he associatedwith previous antidepressant benefit andthus, it was reinitiated at a dosage of 20 mgdaily. He consented to a concurrent trial ofbilateral electroconvulsive therapy (ECT).
Fourteen days after admission, que-tiapine was initiated at a dosage of 100 mgat bedtime for insomnia and to augmenttreatment with citalopram. On day 8 afterstarting quetiapine, 3 days after complet-ing his fifth ECT treatment, the patientbegan exhibiting bizarre and uncharacter-istic behavior, including picking at the airand continuously pacing around the wardunsettled. He disclosed psychotic symp-toms including auditory hallucinations andwas disoriented to place. He had neverbefore exhibited symptoms of psychosisor delirium. His disorganized speech andbehavior worsened through the day. Crea-tine kinase level on day 8 was 156 IU/L(reference range, 52Y175 IU/L). The nextday (day 9), the patient became agitated andincreasingly tremulous. Vital signs takenrepeatedly during the course of day 9 re-vealed persistent tachycardia (120 beatsper minute) and hypertension (158/104mm Hg), both absent on admission. Thepatient remained afebrile at all time points.Laboratory investigations showed a nor-mal complete blood cell count. Throughoutthe course of day 9 and into day 10, histremulousness became more pronounced,and he developed an increase in muscletone andmildmuscular rigidity in his upperlimbs. His rigidity was not deemed severebecause he was able to maintain a normalcomplete range of motion in all limbs.Intravenous hydration was initiated. Crea-
tine kinase levels taken repeatedly showeda rapid rise reaching 3781 IU/L. Allpsychotropic drugs were immediately dis-continued, and the patient was urgentlytransferred to an acute medical ward forongoing care. The patient received intrave-nous dantrolene at regular intervals fromday 10 to day 12 (7 doses of 200 mg andsubsequently 3 doses of 80 mg). His condi-tion resolved from day 12 to 15. His mentalstatus normalized, and the psychotic symp-toms subsided within 48 hours. His vitalsigns returned to reference ranges by day 14of treatment. His muscle rigidity subsided,and creatine kinase levels normalized byday 15. The patient was transferred back tothe psychiatric ward, citalopram was re-started, and ECTwas resumed.
DISCUSSIONThe Diagnostic and Statistical Man-
ual of Mental Disorders, Fourth EditionVText Revision (DSM-IV-TR) research cri-teria (Appendix B) for NMS (AmericanPsychiatric Association, 2000) present inthis case featured principally B criteria in-cluding diaphoresis, tremor, changes inlevel of consciousness, tachycardia, hyper-tension, and elevated creatine kinase levels.Criteria A features were absent: there wasno severe muscular rigidity and no hyper-thermia. Although the dose of quetiapinewas low, the temporal association impli-cated it as the inciting agent. Serotoninsyndrome was considered on differentialgiven that its significant symptom overlapwith NMS, as the patient was also oncitalopram. The absence of hyperreflexiaand myoclonus and the presence ofrhabdomyolysis led to it being dismissedas the probable diagnosis.
There has been a growing body ofliterature suggesting the existence ofatypical presentations of NMS, particular-ly in association with atypical antipsycho-tics.2 Atypical NMS differs from classicNMS by its minimal or absent DSM-IV-TRcriteria A features.2 Hyperthermia andsevere muscular rigidity are core featuresof usual NMS presentations; however, inseveral case reports of NMS with second-generation antipsychotics, extreme tem-perature elevations and/or extrapyramidaldysfunction have been absent.2,16,17
It remains unclear whether theseatypical presentations should be consid-ered as a separate diagnostic entity or ifthey instead represent early or partial signsof an impending typical presentation ofNMS.2 Typical cases of NMS encompass aheterogeneous collection of presentationsthat differ in the onset and progression ofsymptoms.18 It is possible that improvedrecognition and earlier treatment of NMS
has resulted in presentations that mayseem atypical because the full spectrumof usual symptoms has been prevented. Incontrast, if atypical NMS is indeed aseparate construct, it may require support-ive treatment and possibly adjunctivemedications at a point where they wouldnot be used in a usual case of NMS thathas more identifiable intervention points.Further studies examining progression andoutcomes of atypical NMS are neededbefore it can be determined whethertreatment approaches should differ fromtypical NMS.
To our knowledge, there have been 12English language case reports of possibleNMS associated with quetiapine. Eight ofthese cases featured presentations consis-tent with atypical NMS.4Y11 The remaining4 cases12Y15 described features of standardNMS consistent with DSM-IV-TR criteria.
Among case reports describing atyp-ical NMS, patients generally incurredmilder physical symptoms than character-istically observed in classic NMS. Of the8 case reports, 4 showed either minimal ortotal absence of muscle rigidity.4Y7 Othersnoted the lack of hyperthermia, with tem-peratures remaining below 38.0-C.7Y9
Most of the cases demonstrated an eleva-tion of creatine kinase level whether as-sociated with or without muscle rigidity.In general, however, the increase in crea-tine kinase level was not extreme.7Y10 All8 patients demonstrated significant men-tal status changes, and all except one7 hadautonomic instability of some form.
Of interest is the interaction betweenantipsychotics and selective serotoninreuptake inhibitors (SSRIs) in the patho-genesis of NMS. Serotonin further inhibitsdopamine release and thereby may worsena hypodopaminergic state induced byantipsychotics. Twenty-nine cases of NMShave been reported with combinations ofselective serotonin reuptake inhibitorsand atypical antipsychotics. Quetiapinewas implicated in 3 cases11,12,19; our casewould be the fourth. Clinicians should beaware that adjunctive use of selectiveserotonin reuptake inhibitors may increasethe risk of NMS in patients receivingsecond-generation antipsychotics.20
Taken together, these case reportsprovide evidence that quetiapine is asso-ciated with NMS and that it is most oftenNMS with an atypical presentation. Ourcase is consistent with an atypical presen-tation of NMS and adds to the growingnumber of case reports implicating que-tiapine as a causative agent in NMS. Des-pite these reports of NMS with quetiapine,it is important to recognize that it is a rareevent. The low dose of quetiapine in ourcase exemplifies the idiosyncratic nature
Letters to the Editors Journal of Clinical Psychopharmacology & Volume 29, Number 5, October 2009
498 www.psychopharmacology.com * 2009 Lippincott Williams & Wilkins
9Copyright @ 200 Lippincott Williams & Wilkins. Unauthorized reproduction of this article is prohibited.

of the syndrome. Clinicians need to beaware of atypical presentations of NMSbecause they may obscure diagnosing apotentially fatal condition and delay life-saving treatment.
ACKNOWLEDGMENTPreparation of this paper was sup-
ported by a Manitoba Health ResearchCouncil Operating Grant awarded toJames M. Bolton.
Sherief El-Gaaly, MDDepartment of PsychiatryUniversity of Manitoba
Winnipeg, Manitoba, Canada
Philip St. John, MD, MPH, FRCPCSection of Geriatrics
Department of Internal Medicineand Centre on Aging
University of ManitobaWinnipeg, Manitoba, Canada
Sara Dunsmore, MDDepartment of Internal Medicine
University of ManitobaWinnipeg, Manitoba, Canada
James M. Bolton, MD, FRCPCDepartment of PsychiatryUniversity of Manitoba
Winnipeg, Manitoba, [email protected]
REFERENCES
1. Caroff SN, Mann SC, Campbell EC. Atypicalantipsychotics and neuroleptic malignantsyndrome. Psychiatr Ann. 2000;30:314Y321.
2. Picard LS, Lindsay S, Strawn JR, et al.Atypical neuroleptic malignant syndrome:diagnostic controversies and considerations.Pharmacotherapy. 2008;28:530Y535.
3. Buckley PF, Hasan S. Atypical neurolepticmalignant syndrome and atypicalantipsychotics. Am J Psychiatry.1998;155:1633.
4. Solomons K. Quetiapine and neurolepticmalignant syndrome. Can J Psychiatry.2002;47:791.
5. Bourgeois J, Babine S, Meyerovich M, et al.A case of neuroleptic malignant syndromewith quetiapine. J Neuropsychiatry ClinNeurosci. 2002;14:87.
6. Whalley N, Diaz P, Howard J. Neurolepticmalignant syndrome associated with the useof quetiapine. Can J Hospital Pharmacy.1999;52:112.
7. Choi-Kain L, Pope H. BAtypical[neuroleptic malignant syndrome and thespectrum of malignant cerebrotoxicsyndromes. Harv Rev Psychiatry.2007;15:181Y186.
8. Stanley AK, Hunter J. Possible neurolepticmalignant syndrome with quetiapine. Br JPsychiatry. 2000;176:497.
9. Bora E, Gonul A, Akdeniz F, et al.Neurolepticmalignant-like syndrome inducedwith low-dose quetiapine treated withelectroconvulsive therapy. Eur Psychiatry.2003;18:322Y323.
10. Kobayashi A, Kawanishi C, Matsumura T,et al. Quetiapine-induced neurolepticmalignant syndrome in dementia withLewy bodies: a case report. ProgNeuropsychopharmacol Biol Psychiatry.2006;30:1170Y1172.
11. Grignon S, Brethes JI, Chamberland M, et al.Incipient neuroleptic malignant syndromewith quetiapine/paroxetine combinationtreatment: atypical presentation and early,successful rechallange with olanzapine.Int J Psychiatr Clin Pract. 2005;9:296Y298.
12. Matsumoto R, Kitabayashi Y, Nakatomi Y,et al. Neuroleptic malignant syndromeinduced by quetiapine and fluvoxamine.Am J Psychiatry. 2005;162:812.
13. Sing K, Ramaekers G, Van Harten P.Neuroleptic malignant syndrome andquetiapine. Am J Psychiatry. 2002;159:149Y150.
14. Al-Waneen R. Neuroleptic malignantsyndrome associated with quetiapine.Can J Psychiatry. 2000;45:764Y765.
15. Hatch CD, Lund BC, Perry PJ. Failedchallenge with quetiapine after neurolepticmalignant syndrome with conventionalantipsychotics. Pharmacotherapy. 2001;21:1003Y1006.
16. Farver DK. Neuroleptic malignantsyndrome induced by atypical antipsychotics.Expert Opin Drug Saf. 2003;2:21Y35.
17. Karagianis JL, Phillips LC, Hogan KP, et al.Clozapine-associated neuroleptic malignantsyndrome: two new cases and a review ofthe literature. Ann Pharmacother. 1999;33:623Y630.
18. Strawn JR, Keck PE Jr, Caroff SN.Neuroleptic malignant syndrome. Am JPsychiatry. 2007;164:870Y876.
19. Marlowe K, Schirgel D. Quetiapine andcitalopram: aetiological significance inserotonin syndrome. N Z Med J. 2006;119:2058Y2060.
20. Stevens DL. Association between selectiveserotonin-reuptake inhibitors,second-generation antipsychotics, andneuroleptic malignant syndrome. AnnPharmacother. 2008;42:1290Y1297.
Oral Urea Treatment forPolydipsia-HyponatremiaSyndrome in PatientsWith Schizophrenia
To the Editors:
A t least 20% of hospitalized schizo-phrenic patients demonstrate poly-
dipsia and polyuria; more than 25%polydipsic patients may eventually devel-op hyponatremia, which is called poly-dipsia-hyponatremia syndrome (PHS).1
An acute and severe hyponatremia due topolydipsia can lead to a serious clinicalcondition called water intoxication, whichmay cause loss of consciousness, seizures,or even death.1,2 Several pharmacologicapproaches have been tested to treat poly-dipsia in schizophrenic patients; however,none of them has been proven to be ef-fective, thus far.3 Clozapine, a prototypeof so-called atypical antipsychotic drugs,may reduce polydipsia in schizophrenicpatients.4 Unfortunately, it has seriousadverse effects and may not be prescribedfor all the patients with PHS; clozapine isnot yet approved for clinical use in Japan.
Another pharmacologic approach forthe management of PHS would be toincrease renal capacity to excrete water.Patients with PHS have reduced free waterclearance rate, which has generally beenlinked to a syndrome of inappropriatesecretion of antidiuretic hormone.5 Sev-eral researchers have examined the effectof a vasopressin receptor antagonist, deme-clocycline, on patients with PHS; how-ever, a carefully performed double blindplacebo-controlled trial failed to showany significant benefit.6 More recently,specific vasopressin V2 receptor antago-nist, tolvaptan, has been shown to elevateserum sodium levels in schizophrenicpatients with chronic hyponatremia.7 Theresult seems to be promising, althoughefficacy and safety of this drug should beconfirmed in a longer-term study.
Verhoeven et al8 reported that orallyadministered urea elevated serum sodiumlevels in 7 patients with mental illnessincluding 5 schizophrenic patients withPHS. Urea is one of the main componentsresponsible for urine osmolarity. Whenadministered orally, urea is quicklyabsorbed and excreted in urine, efficientlyproducing osmotic diuresis.9 In Japan,urea has previously been used to reduceedema in chronic heart failure or ascitesin liver cirrhosis.9 Urea has been reportedto reduce water retention efficiently andsafely in patients with syndrome of in-appropriate secretion of antidiuretic hor-mone.10 If disturbed free water clearanceis a key factor for the development of hy-ponatremia, it is likely that urea will re-duce water retention and prevent severehyponatremia in PHS. Therefore, we con-ducted a clinical trial to confirm the effectof oral urea treatment on PHS in schizo-phrenic patients.
Inclusion criteria for selecting sub-jects were as follows: (1) patients with acondition diagnosed as schizophrenia
Journal of Clinical Psychopharmacology & Volume 29, Number 5, October 2009 Letters to the Editors
* 2009 Lippincott Williams & Wilkins www.psychopharmacology.com 499
9Copyright @ 200 Lippincott Williams & Wilkins. Unauthorized reproduction of this article is prohibited.
(Diagnostic and Statistical Manual ofMental Disorders, Fourth Edition criteria)and hospitalized in our facility; (2) poly-dipsia of the patients had to be identifiedby (a) staff observations, (b) constant hypo-sthenuria, and (c) marked diurnal weightgain (DWG, 95%); (3) patients had toshow hyponatremia (G135 mEq/L) in rou-tine blood analysis at least once in the last6 months; and (4) patients had to have atleast one clear episode of water intoxica-tion during their hospitalization period.Patients with a history of renal disease,heart disease, or diabetes mellitus were ex-cluded. Seven male schizophrenic patientsmet the criteria and provided written in-formed consent to the study. Mean age ofthe subjects was 53.3 T 5.9 years (range,44Y63 years). Means of durations of ill-ness and hospitalization periods were31.3 T 7.0 (range, 21Y42) and 20.1 T 8.9(range, 5Y32) years, respectively. Subjectswere basically allowed free access to waterthroughout the study period except for se-veral hours of involuntary water-restrictionperiod occasionally imposed according to atarget-weight procedure.1,2 The water re-striction has not been done on the day ofblood and urine analyses. Subjects werenot allowed to take any diuretics and/orlithium during the study. The study proto-col was approved by the ethical committeesof both the Tsukuba University HospitalandMitsukaido-Kosei Hospital.
The ultrafine grade of urea (purity,999.0%) was purchased from the Taka-sugi Pharmaceutical Co, Ltd (Fukuoka,Japan). A starting dosage of urea, 30 g/d,was decided according to the previousreport8; the dosage was increased subse-quently as described later on. Because theurea tasted so bitter and salty, we dis-solved it in orange juice and added artifi-cial sweetener containing aspartame(Palsweet; Ajinomoto, Tokyo, Japan). Theurea/orange juice cocktail was divided into2 or 3 portions depending on the urea dos-age and served twice or thrice a day.
The blood and urine analyses werecarried out twice every month on the firstand the third Friday. Verhoeven et al8 re-ported the elevated morning serum sodi-um levels in their subjects after the ureatreatment. However, the serum sodiumlevels of the typical patients with PHSdecrease in the afternoon even if thosein the morning are normal.2 Thus, wedecided to collect the blood and urinesamples in the afternoon at 4 PM; se-rum sodium, potassium, chloride, creati-nine, and blood urea nitrogen levels weredetermined. In addition, morning (at 6 AM)blood samples were analyzed once witha given urea dosage; serum electrolytes,blood cell counts, liver enzymes, fastingblood glucose, total cholesterol, and trigly-ceride levels were determined. Urine sam-ples were taken twice every month at 4 PM
on the same day of the afternoon bloodanalysis. Urine specific gravity, osmolar-ity, and sodium and creatinine concentra-tions were determined. Morning urinesamples were also collected at 6 AM onthe same day; urine creatinine concen-trations were determined. Normalizeddiurnal body weight gain (NDWG) wascalculated every day for each subject aspreviously described.11 The blood andurine examinations were performed for3 months before the beginning of ureatreatment; means of the 3-month datawere regarded as baseline for each sub-ject. Changes of the data after oral ureatreatment were monitored and statisticallyanalyzed using 1-way analysis of variancewith repeated measures followed by Tukeymultiple comparison test.
When 30 g/d of urea was given to thesubjects for 2 months, their serum sodiumlevels did not seem to elevate. Therefore,we increased the dosage to 45 g/d and ob-served for additional 3 months. The meanserum sodium level of the subjects appar-ently elevated with this dosage; however, itstill remained below the reference range(Table 1). We decide to increase the dosageto 67.5 g/d, and the observation continuedfor an additional 2 months. As shown inTable 1, the means of serum sodium con-centrations at 4 PM were significantlyhigher than the baseline at daily doses of45 and 67.5 g, although a difference
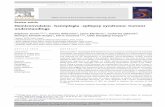
TABLE 1. Mean T SD Changes in Serum and Urine Data and NDWG After Oral Urea Treatment (n = 7)
Dosage of Urea, g/d
Baseline 30 45 67.5
Serum analysis at 4 PM
Na+, mEq/L 127.6 T 3.0 128.9 T 3.5 131.7 T 3.9*† 133.5 T 3.0‡§
K+, mEq/L 3.75 T 0.53 3.87 T 0.47 4.02 T 0.60|| 3.99 T 0.35||
Clj, mEq/L 90.0 T 4.5 91.0 T 4.2 93.0 T 5.3|| 94.4 T 3.6*Blood urea nitrogen level, mg/dL 7.7 T 4.3 20.1 T 11.6|| 26.5 T 14.0‡ 48.8 T 20.0‡§
Cre, mg/dL 0.84 T 0.26 0.81 T 0.28 0.83 T 0.26 0.84 T 0.28Urine analysis at 4 PM
Osmolarity, mOsm 92.0 T 36.0 132.8 T 63.4 171.1 T 67.8* 231.2 T 94.0‡¶
Na+, mEq/L 23.6 T 7.3 21.9 T 8.4 21.4 T 9.8 18.6 T 5.0Cre, mg/dL 15.2 T 8.4 14.9 T 9.6 14.0 T 7.2 14.3 T 5.8
Serum and urine analyses at 6 AM
Serum Na+, mEq/L 131.1 T 4.3 132.0 T 3.3 135.0 T 4.4|| 136.3 T 2.6*†
Urine Cre, mg/dL 17.4 T 4.6 22.4 T 10.2 26.6 T 9.8 37.6 T 14.1*†
NDWG, % 5.7 T 1.4 6.0 T 1.9 6.2 T 1.7 6.2 T 1.3
*P G 0.01 versus baseline.†P G 0.05 versus 30 g of urea; by 1-way analysis of variance with repeated measures followed by Tukey test.‡P G 0.001 versus baseline.§P G 0.001 versus 30 g of urea; by 1-way analysis of variance with repeated measures followed by Tukey test.||P G 0.05 versus baseline.¶P G 0.01 versus 30 g of urea; by 1-way analysis of variance with repeated measures followed by Tukey test.
Letters to the Editors Journal of Clinical Psychopharmacology & Volume 29, Number 5, October 2009
500 www.psychopharmacology.com * 2009 Lippincott Williams & Wilkins
9Copyright @ 200 Lippincott Williams & Wilkins. Unauthorized reproduction of this article is prohibited.

between 45 and 67.5 g was not significant.The means of morning serum sodium con-centrations were also significantly higherthan the baseline at doses of 45 and 67.5 g(Table 1). Mean concentrations of serumpotassium and chloride at 4 PM were alsosignificantly higher than the baseline atdoses of 45 and 67.5 g (Table 1). Noneof these variables showed any significantdifferences between the 45- and 67.5-gdoses. No particular adverse event wasobserved during the study.
The mean urine creatinine concen-tration at 6 AM was significantly elevatedwith 67.5-g dose of urea as compared withbaseline, suggesting that the subjects be-came capable of reducing water retentionby the morning. This may indicate thattheir renal capacity to excrete water hasimproved. On the other hand, the meanurine creatinine level at 4 PM of our sub-jects remained low despite the urea treat-ment (Table 1). Although Verhoeven et al8
reported that NDWG of their subjects sig-nificantly decreased after the urea treat-ment, our subjects failed to show anyreduction in NDWG (Table 1). It is sug-gested, therefore, that our subjects werestill polydipsic and experiencing water re-tention in the afternoon. This was, how-ever, somewhat puzzling because theserum sodium levels at 4 PM of our sub-jects significantly increased. A possibleexplanation for this discrepancy would bethat the elevated serum sodium after theurea treatment was not entirely related tothe reduction of water retention. Muschet al12 suggested that a solute loss, as wellas thewater-retention, plays a major role inpolydipsia-related hyponatremia. It maybe the case that the oral urea treatmentelevates serum sodium not only by reduc-ing water retention but also by preventingsodium loss in the urine. This notionremains to be clarified in future studies.
This study has some limitations. Thestudy was designed as an open naturalistictrial without a control group. The samplesize was small. The mean of serum sodiumconcentration with highest dosage used(67.5 g/d) was still below reference range,suggesting that oral urea treatment wouldnot be an entire solution for PHS.We couldnot find significant difference betweenmean serum sodium concentrations with45- and 67.5-g doses. The optimal ureadosage should be determined in futurestudies. Nevertheless, increased serum so-dium levels not only in themorning but alsoin the afternoon in schizophrenic patientswith PHS support the notion that the oralurea treatment possibly reduces the risk ofsevere hyponatremia. Further studies withlarger sample sizes and longer-term pro-tocols are required to evaluate beneficial
effects and safety of the oral urea treatmentfor schizophrenic patients with PHS.
AUTHOR DISCLOSUREINFORMATION
The authors declare no funding,relevant financial disclosures, or conflictsof interest with this study.
Nobutoshi Kawai, MD, PhDDepartment of Psychiatry
Institute of Clinical MedicineUniversity of Tsukuba
Tsukuba-shi, Japanand Department of Clinical Psychiatry
Mitsukaido-Kosei HospitalJoso-shi, Japan
Kazuhiro Ishikawa, MD
Kiyotaka Nemoto, MDDepartment of Psychiatry
Institute of Clinical MedicineUniversity of Tsukuba
Tsukuba-shi, Japan
Tsunahiro Katano, MDDepartment of Clinical Psychiatry
Mitsukaido-Kosei HospitalJoso-shi, Japan
Sho Takahashi, MD
Takafumi Hori, MD, PhD
Takashi Asada, MD, PhDDepartment of Psychiatry
Institute of Clinical MedicineUniversity of Tsukuba
Tsukuba-shi, Japan
REFERENCES
1. Verghese C, de Leon J, Josiassen RC.Problems and progress in the diagnosis andtreatment of polydipsia and hyponatremia.Schizophr Bull. 1996;22:455Y464.
2. Vieweg WVR. Overview. In: Schnur DB,Kirch DG, eds. Water Balance inSchizophrenia. Washington, DC: AmericanPsychiatric Press Inc; 1996:1Y42.
3. Lawson WB. Pharmacological approaches todisturbances in water regulation in severelymentally ill patients. In: Schnur DB, KirchDG, eds. Water Balance in Schizophrenia.Washington, DC: American Psychiatric PressInc; 1996:201Y210.
4. De Leon J, Verghese C, Stanilla JK, et al.Treatment of polydipsia and hyponatremia inpsychiatric patients; can clozapine be a newoption?Neuropsychopharmacology. 1995;12:133Y138.
5. Goldman MB. Pathophysiology of fluidbalance dysreguration in psychiatric patients.In: Schnur DB, Kirch DG, eds. WaterBalance in Schizophrenia. Washington, DC:American Psychiatric Press Inc; 1996:109Y123.
6. Alexander RC, Karp BI, Thompson S, et al.
A double blind, placebo-controlled trialof demeclocycline treatment ofpolydipsia-hyponatremia in chronicallypsychotic patients. Biol Psychiatry. 1991;30:417Y420.
7. Josiassen RC, Goldman M, Jessani M,et al. Double-blind, placebo-controlledtrial of a vasopressin V2-receptorantagonist in patients with schizophreniaand hyponatremia. Biol Psychiatry. 2008;64:1097Y1100.
8. Verhoeven A, Munsch W, Decaux G.Treatment of the polydipsia-hyponatreimasyndrome with urea. J Clin Psychiatry.2005;66:1372Y1375.
9. Society of Japanese Pharmacopoeia. TheJapanese Pharmacopoeia. 10th ed.Maebashi, Japan: Hirokawa Shoten Co. Ltd;1981. [In Japanese]
10. Decaux G, Prospert F, Penninckx R, et al.5-year treatment of the chronic syndrome ofinappropriate secretion of ADH with oralurea. Nephron. 1993;63:468Y470.
11. Kawai N, Baba A, Suzuki T. Risperidonefailed to improve polydipsia of theschizophrenic patients. Psychiatry ClinNeurosci. 2002;56:107Y110.
12. Musch W, Xhaet O, Decaux G. Solute lossplays a major role in polydipsia-relatedhyponatreima of both water drinkersand beer drinkers. Q J Med. 2003;96:421Y426.
Clozapine Monotherapy for66 Months in TreatmentResistant Bipolar Disorder
A Case Report
To the Editors:
In the last decade, there has been a rapidgrowth in treatment approaches for bi-
polar disorder. Most atypical antipsy-chotics are now licensed for maintenancetherapy in bipolar disorder. However,despite the use of anticonvulsants, moodstabilizers and atypical antipsychotics,many patients are resistant to treatment.
Clozapine has been shown to be use-ful in the treatment of acute mania1,2 andrefractory psychotic mania.3 Long-termstudies of clozapine as add-on or mono-therapy in treatment resistant bipolar dis-order have shown significant improvementin manic symptoms, decrease in hospita-lizations, and reduction in suicidality.4Y6
Two case reports and a case series havealso highlighted the beneficial effect ofclozapine add-on in treatment resistantbipolar disorder.7Y9
I describe the case of a young manwith treatment resistant bipolar disorder
Journal of Clinical Psychopharmacology & Volume 29, Number 5, October 2009 Letters to the Editors
* 2009 Lippincott Williams & Wilkins www.psychopharmacology.com 501
9Copyright @ 200 Lippincott Williams & Wilkins. Unauthorized reproduction of this article is prohibited.

who has remained well for a follow-upperiod of 66 months on clozapinemonotherapy.
CASEMr AB had bipolar disorder (first
episode at age 20 years) and 11 hospitalepisodes during 8 years (Table 1). Epi-sodes of depression often resulted in over-dose or suicidal thoughts. Manic episodeswere very disruptive and posed a substan-tial risk to himself and to others. Somehospitalizations were involuntary. Co-morbid substance abuse was present onlyin the first 2 years of illness. Compliancewas good because of the patient’s insight,good family support, and strong commu-nity mental health team involvement.
Eight years after his first episode, hewas started on clozapine. Clozapine wasinitially prescribed with lithium. However,lithium was stopped after 6 months. Sixand a half years after starting clozapine,the patient has had no further hospitaliza-tions. He has not had any further episodesof mania or depression. He has not hadweight gain, his blood glucose level iswithin limits, and he does not have si-alorrhea. However, he suffers from drymouth for which he uses a chewing gum,and he is on cholesterol levelYloweringtablets (atorvastatin).
DISCUSSIONIn open uncontrolled studies, cloza-
pine has demonstrated efficacy in treatment-resistant bipolar disorder,10,11 includingmixed episodes,12 and rapid cycling.13,14
Long-term studies of clozapine asadd-on or monotherapy have shown that itis useful in treatment-refractory symptomsof bipolar disorder.4,15 Follow-up periodsin reported studies have varied from 12to 48 months. Ciapparelli et al16 reporteda 48-month naturalistic follow-up andfound a sustained response to clozapinein treatment-resistant schizophrenia, schi-zoaffective disorder, or bipolar disorderwith psychotic features.
Most of these studies have lookedat clozapine as add-on treatment ratherthan monotherapy. In 1 retrospective study,monotherapy with clozapine showed 65%of the cohort had no further affective epi-sodes or hospitalization. The mean follow-up in that study was only 16 months.5
Clozapine has been suggested to havegreater antimanic properties than anti-depressant efficacy.5,17 However, 1 studyhad suggested an antidepressant effect,10
and 1 case report demonstrates the strongantisuicidal effect of clozapine add-on.8
Clozapine’s role in decreasing suicidalityin schizophrenia has been robustly re-ported,18 and it is licensed in the United
States for reducing the risk of suicidal be-havior in schizophrenia or schizoaffectivedisorder. This case report demonstrates
effectiveness in treating suicidal thoughtsin bipolar disorder for a 66-month follow-up period.
TABLE 1. Retrospective Chart Study Revealing Hospital Episodes and OutpatientAppointments Along With Diagnostic Information and Treatment Prescribed
Inpt Epi Admission Discharge Mental State Treatment, mg
1 November 1994YFebruary 1995 Li 800 and CPZ 400OPA, May and June 1995 Li 800 and CPZ 200
OPA, September 95 Li 800 and Lof 1402 October 1995 October 1995 Mod dep Li 800 and Flu 20
DV, September 1996 Cannabis/ecstasyand hypomania
Li 800 and Thio 300
3 October 1996YDecember 1996 BAD-mania Li 800 and Thio 600OPA, January 1997 Positive about future Li 800 and Thio 400OPA, March 1997 Feeling low Li 800
OPA, November 1997 Hypomania4 November 1997YFebruary 1998 BAD-mixed Li 800 and Zuclo 305 September 1998YJanuary 1999 BAD Val 2000 and
Droperidol 20January 1999YMarch 1999 BAD Val 2000 and Zuclo 20April 1999 April 1999 BAD Val 2000 and Zuclo 60
OPA, May 1999 Feeling low Val 2000, Zuclo 60,and Flu 20
OPA, August 1999 Fairly well Val 2000 and Flu 206 September 1999YDecember 1999 BAD-mania Li 800, CBZ 1200,
and Ola 20OPA, February 2000 Depressed Li 800, CBZ 800,
and Ola 10OPA, August 2000 Subj depressed Li 800 and CBZ 800
7 September 2000YDecember 2000 BAD-maniaJanuary 2001YMarch 2001 BAD-hypomania Li 1200, Val 1600,
and Ola 208 March 2001YApril 2001 BAD-overdose Li 1200, Val 1600,
and Ola 20OPA, August 2001 Well Li 1200, CBZ 400,
Val 1200, and Ola 10OPA, September 2001 Depressed Add Flu 20
9 September 2001YOctober 2001 BAD-dep Li 1200, CBZ 400,and Val 800
10 November 2001 November 2001 BAD-hypomania Li 400, Val 1600,CBZ 400, and Ola 10
November 2001YFebruary 2002 BAD-hypomania Li 1200, Val 1600,CBZ 400, and Ola 20
February 2002YApril 2002 BAD Li 1200, Val 2000,and Ola 10
OPA, May 2002 Mod dep/mixed Li, Val, and Ola 20OPA, July 2002 Mod dep Li 1200, Val 2000,
and Ola 1011 August 2002 August 2002 BAD-depressed Val 1000, Li 1200,
and Par 10September 2002YMarch 2003 BAD Clozapine 400
and Li 1200March 2003 March 2003 BAD Clozapine 400 and Li 800
OPA, July 2003 Asymptomatic Cloz 350 and Li 400OPA, OngoingVOctober 2008 Asymptomatic Cloz 350
BAD indicates bipolar affective disorder; CBZ, carbamazepine; Cloz, clozapine; CPZ, chlor-promazine; Dep, depression; DV, domiciliary visit/home visit; Flu, fluoxetine; Inpt Epi, inpatient episode;Li, lithium; Lof, lofepramine; Mod, moderate; Ola, olanzapine; OPA, outpatient appointment;Par, paroxetine; Subj, subjectively; Thio, thioridazine; Val, valproate semisodium; Zuclo, zuclopenthixol.
Letters to the Editors Journal of Clinical Psychopharmacology & Volume 29, Number 5, October 2009
502 www.psychopharmacology.com * 2009 Lippincott Williams & Wilkins
9Copyright @ 200 Lippincott Williams & Wilkins. Unauthorized reproduction of this article is prohibited.

The limitations of this report are thatit is retrospective and a single case. Thestrengths are that it presents monotherapywith clozapine in treatment-resistant bipo-lar disorder and a sustained improvementfor more than 5 years. With the presentfocus on atypical antipsychotics in bipolardisorder, it is imperative that clozapine hasa randomized controlled trial to look atits efficacy and tolerability in treatment-resistant bipolar disorder.
AUTHOR DISCLOSUREINFORMATION
No funding had been sought or pro-vided for this case report. Dr ManeeshGupta has accepted hospitality and remu-neration from various pharmaceuticalcompanies for speaking in events spon-sored by them. Dr Gupta holds a few sharesin Indian pharmaceutical companies.
Maneesh Gupta, MD, DNB, CCSTDepartment of Psychiatry
Mater HospitalBelfast, United Kingdom
REFERENCES
1. Barbini B, Scherillo P, Benedetti F, et al.Response to clozapine in acute mania is morerapid than that of chlorpromazine. Int ClinPsychopharmacol. 1997;12:109Y112.
2. Degner D, Bleich S, Muller P, et al. Clozapinein the treatment of mania. J NeuropsychiatryClin Neurosci. 2000;12:283.
3. Green AI, Tohen M, Patel JK, et al. Clozapinein the treatment of refractory psychoticmania. Am J Psychiatry. 2000;157:982Y986.
4. Suppes T, Webb A, Paul B, et al. Clinicaloutcome in a randomized 1-year trial ofclozapine versus treatment as usual forpatients with treatment-resistant illness and ahistory of mania. Am J Psychiatry. 1999;156:1164Y1169.
5. Zarate CA Jr, Tohen M, Baldessarini RJ.Clozapine in severe mood disorders. J ClinPsychiatry. 1995;56:411Y417.
6. Ciapparelli A, Dell’Osso L, Pini S, et al.Clozapine for treatment-refractoryschizophrenia, schizoaffective disorder, andpsychotic bipolar disorder: a 24 monthnaturalistic study. J Clin Psychiatry. 2000;61:329Y334.
7. Calabrese JR, Gajwani P. Lamotrigine andclozapine for bipolar disorder. Am JPsychiatry. 2000;157:1523.
8. Vangala VR, Brown ES, Suppes T. Clozapineassociated with decreased suicidality inbipolar disorder: a case report. BipolarDisord. 1999;1:123Y124.
9. Fehr BS, Ozcan ME, Suppes T. Low dosesof clozapine may stabilize treatment-resistant
bipolar patients. Eur Arch Psychiatry ClinNeurosci. 2005;255:10Y14.
10. Banov MD, Zarate CA Jr, Tohen M, et al.Clozapine therapy in refractory affectivedisorders: polarity predicts response inlong-term follow-up. J Clin Psychiatry.1994;55:295Y300.
11. Calabrese JR, Kimmel SE,WoyshvilleMJ, et al.Clozapine for treatment-refractory mania.Am J Psychiatry. 1996;153:759Y764.
12. Suppes T, McElroy SL, Gilbert J, et al.Clozapine in the treatment of dysphoricmania. Biol Psychiatry. 1992;32:270Y280.
13. Calabrese JR, Meltzer HY, Markowitz PJ.Clozapine prophylaxis in rapid cyclingbipolar disorder. J Clin Psychopharmacol.1991;11:396Y397.
14. Suppes T, Phillips K, Judd C. Clozapinetreatment of nonpsychotic rapid cyclingbipolar disorder. A report of three cases.Biol Psychiatry. 1994;3:338Y340.
15. Keck PE Jr, McElroy SL, Strakowski SM.Anticonvulsants and antipsychotics in thetreatment of bipolar disorder. J ClinPsychiatry. 1998;59(suppl 6):74Y81.
16. Ciapparelli A, Dell’Osso L, Bandettini diPoggio A, et al. Clozapine in treatment-resistantpatients with schizophrenia, schizoaffectivedisorder, or psychotic bipolar disorder: anaturalistic 48-month follow-up study. J ClinPsychiatry. 2003;64:451Y458.
17. Frye MA, Ketter TA, Altshuler LL, et al.Clozapine in bipolar disorder: treatmentimplications for other atypical antipsychotics.J Affect Disord. 1998;48:91Y110.
18. Meltzer HY, Alphs L, Green AI, et al. TheInterSePT Study Group. Clozapine treatmentfor suicidality in schizophrenia: InternationalSuicide Prevention Trial (InterSePT). ArchGen Psychiatry. 2003;60:82Y91.
Waxing-and-WaningCatatonia After IntermittentExposure to Aripiprazolein a Case of Autism and
Bipolar DisorderTo the Editors:
We report a case of a patient withautism and bipolar disorder who de-
veloped chronic waxing-and-waning cata-tonia coinciding with intermittent exposureto aripiprazole. Eventually, he presentedexacerbation of his chronic alternatingcatatonia that rapidly progressed to a con-dition impossible to distinguish betweenneuroleptic-induced malignant catatonia(NIMC) and neuroleptic malignant syn-drome (NMS). Neuroleptic-induced malig-nant catatonia is a severe extrapyramidalreaction produced by neuroleptics charac-terized by staring, mutism, negativism,
withdrawal, waxy flexibility, posturing,and muscular rigidity. Neuroleptic malig-nant syndrome presents a similar pictureand includes hyperthermia and autonomicdischarge. It has been postulated that thefirst is a stage in the progression towardthe second.1 Wing and Shah2 have calledattention to the marked overlap of the be-havioral features between autism and cata-tonia, raising questions about the natureof their relationship. To our knowledge,waxing-and-waning catatonia in responseto neuroleptic challenge has not been des-cribed in the literature.
CASE REPORTA 26-year-old male patient with a
history of autism, bipolar disorder, mod-erate mental retardation, and hypertensionwas brought to the emergency departmentby his mother, who reported that for thelast 2 days he had been Bout of it, notresponding, and not comprehending any-thing.[ He was first diagnosed withDiagnostic and Statistical Manual ofMental Disorders, Fourth Edition majordepression at age 20 years and was treatedwith sertraline 100 mg daily. A year later,he developed a manic episode and wasstarted on risperidone and oxcarbazepine.At age 22 years, because of weight gain,risperidone was replaced by aripiprazole5 mg daily. No catatonia-like features weredescribed before or during risperidonetreatment. Aripiprazole treatment contin-ued for 29 months, except for 3 interrup-tions lasting in total 18 months, butremained on this drug continuously for77 days before the onset of his presentillness. All 3 interruptions of aripiprazoletreatment were requested by his motherout of concern with Bsedation.[ Duringperiods of aripiprazole treatment, thepatient developed tremors and manner-isms and was spaced out, less communi-cative, touching his tongue repetitively,and not eating. These symptoms resolvedevery time aripiprazole was discontinued,only to be followed by relapse of physi-cally aggressive and hypersexual beha-viors (exposing himself, touching femalepeers, using foul language). On admis-sion to our psychiatric unit, hewas holdinghis arms rigidly in front, presented gegen-halten, mutism, negativism, rigidity, pos-turing, diaphoresis, grasp reflex, and fixedgaze. Temperature was 36.9-C; pulse rate,82 beats/min; respiratory rate, 22 breaths/min; blood pressure, 170/83 mm Hg;and white blood cell count, 13,200/KL.The patient scored 28 in the Bush-Francisrating scale for catatonia3 and had a po-sitive response to lorazepam challengetest. Two days later, he presented elevated
Journal of Clinical Psychopharmacology & Volume 29, Number 5, October 2009 Letters to the Editors
* 2009 Lippincott Williams & Wilkins www.psychopharmacology.com 503
9Copyright @ 200 Lippincott Williams & Wilkins. Unauthorized reproduction of this article is prohibited.

temperature (38.8-C); blood pressure, 154/84 mm Hg; pulse rate, 163 beats/min;respiratory rate, 54 breaths/min; whiteblood cell count, 15,100/KL; and creati-nine phosphokinase, 2656 IU/L. He scored18/36 in the Sachdev4 NMS rating scale.Aripiprazole was discontinued, and treat-ment was initiated with bromocriptine,lorazepam, and intravenous fluids. The pa-tient’s symptoms gradually abated, andcreatinine phosphokinase decreased to 57IU/L on day 13. The patient was dischargedto a nursing home where he continuedtreatment with clonazepam and topiramate.He was not rechallenged with antipsy-chotics. Residual catatonic symptoms re-mained and eventually disappeared after6 months. Medications were discontinued,and the patient has remained at his baselinestate for another 6 months.
DISCUSSIONAripiprazole is a dopamine-serotoninY
system stabilizer that seems to reduce therisk of extrapyramidal side effects. Nev-ertheless, acute dystonic reactions havebeen reported after initiating this agent.5
In this case, we suspect that a nonmalig-nant catatonia was induced after exposureto aripiprazole. Some authors argue thatcatatonia and NMS are a single entity,6
whereas others contend that NMS casescan be differentiated in catatonic andnoncatatonic pathological reactions to anti-psychotics.7 Still others regard NMS andcatatonia as 2 distinct disorders with partialpathophysiological overlapping.8 Ourpatient’s symptoms (drastically decreasedspeech, stereotypy, posturing, tremors, andnegativism) met, during the waxing-and-waning phase, the diagnostic catatoniacriteria for autistic spectrum disorders.9
He also met, during his present illness, theFink and Taylor’s10 proposed criteria forcatatonia and met Adityanjee et al researchdiagnostic criteria for NMS.11 No catatonicsymptoms were reported before aripipra-zole exposure (even during risperidonetreatment). Even more, symptoms resolvedafter discontinuation of the agent, at whichtimemood symptoms recurred, only to havecatatonic symptoms emerge again whenaripiprazole was reintroduced. We specu-late that our case exhibited symptoms ofantipsychotic-induced nonmalignant cata-tonia that waxed and waned, as aripiprazolewas interrupted and reintroduced, until theyworsened to the point of full-blown catato-nia that progressed in 48 hours to NIMC/NMS. A limitation of this case report is thatsymptoms correlating with aripiprazoletreatment were noted in retrospect by care-fully going trough the clinical records ofprevious clinicians raising doubts that theon-off, quasi-ABAB naturalistic case study
was clear-cut. The likelihood that catatonicsymptoms were not recognized during theBoff [ periods, leading to subsequent re-challengewith aripiprazole, is rather strong.Catatonia is generally underdiagnosedand frequently goes unrecognized whenit occurs in a waxing-and-waning mannerboth in autism12 and in the relapsing formof bipolar disorder.10 Although there seemsto be a temporal relationship between re-ceiving aripiprazole and symptoms’ emer-gence, we are not sure how much theadministration of this agent contributedin causing NIMC/NMS. Lausberg andHellweg13 postulate that catatonia underneuroleptic medication seems to be causedby the interaction of individual predispo-sition, morbigenous, and pharmacogenic-factors. Orbitofrontal cortical dysfunctionobserved inautism14mayhave facilitated, asproposed by Northoff,8 abnormal top-downmodulation overlapping with dopamineblockadeYinduced, bottom-up modulationin ventral striatum and midbrain/brainstemnuclei. Although aripiprazole has shown alow liability for extrapyramidal side effects,this case draws attention to the need forcaution when treating patients with autismand bipolar disorder with this agent.
AUTHOR DISCLOSUREINFORMATION
Dr De Leon received consultant feeslast year from Pfizer. Drs Shepherd andGarza have no conflict of interest.
Jonathan Shepherd, MD
Vıctor M. Garza, MD
Ovidio A. De Leon, MDDepartment of Psychiatry
University of Illinois at ChicagoChicago, [email protected]
REFERENCES
1. Woddbury MM, Woodbury MA.Neuroleptic-induced catatonia as a stage inthe progression toward neuroleptic malignantsyndrome. J Am Acad Child AdolescPsychiatry. 1992;31:1161Y1164.
2. Wing L, ShahA. Catatonia in autistic spectrumdisorders. Br J Psychiatry. 2000;176:357Y362.
3. Bush G, Fink M, Petrides G, et al. Catatonia:I: rating scale and standardized examination.Acta Psychiatr Scand. 1996;93:137Y143.
4. Sachdev PS. A rating scale for neurolepticmalignant syndrome. Psychiatry Res.2005;135:249Y256.
5. Varkula M, Dale R. Acute dystonic reactionafter initiating aripiprazole monotherapy in a20-year-old man. J Clin Psychopharmacol.2008;28:245Y247.
6. White AC. Catatonia and neuroleptic
malignant syndromeVa single entity?Br J Psychiatry. 1992;161:558Y560.
7. Lee JWY. Catatonic variants, hyperthermicextrapyramidal reactions, and subtypes ofneuroleptic malignant syndrome. Ann ClinPsychiatry. 2007;19:9Y16.
8. Northoff G. Catatonia and neurolepticmalignant syndrome: psychopathologyand pathophysiology. J Neural Transmission.2002;109:1453Y1467.
9. Dosssche DM, Shah A, Wing L. Blueprintsfor the assessment, treatment, and future studyof catatonia in autism spectrum disorders.Int Rev Neurobiol. 2006;72:267Y284.
10. Fink M, Taylor MA. Catatonia. Cambridge,UK: Cambridge University Press; 2003.
11. Adityanjee A, Mathews T, Aderibigbe YA.Proposed research diagnostic criteria forneuroleptic malignant syndrome. Int JNeuropsychopharmacol 1999;2:129Y144.
12. Kakooza-Mwesige A, Wachtel LE, DhosscheDM. Catatonia in autism: implications acrossthe life span. Eur Child Adolesc Psychiatry.2008;17:327Y335.
13. Lausberg H, Hellweg R. Katatones dilemma.Therapie mit lorazepam und clozapin. DerNervenarzt. 1998;69:818Y22.
14. Bachevalier J, Loveland KA. Theorbitofrontal-amygdala circuit andself-regulation of social-emotional behaviorin autism. Neurosci Biobehav Rev. 2006;30:97Y117.
AsymptomaticHyperamylasemia and
Hyperlipasemia AssociatedWith Aripiprazole
To the Editors:
CASE REPORTA 29-year-old white woman with
a psychotic mixed state was hospitalizedfor a severe suicide attempt by ammoniacdetergent ingestion. After a period spentin the emergency unit, she was transferredto our psychiatric ward as soon as her so-matic condition was stabilized, and a totalparenteral nutrition was started.
The onset of her psychiatric historydates back to when she was 18 years oldand experienced a major depressive epi-sode with psychotic symptoms includingparanoid delusions. In that episode, shewas prescribed risperidone and tricyclicantidepressants showing only a partialresponse. During the following years,depressive symptoms fully remitted, butconcurrent persecutory and reference de-lusions did not respond to risperidone andwere partially resistant to several othertrials of typical and atypical antipsychotic
Letters to the Editors Journal of Clinical Psychopharmacology & Volume 29, Number 5, October 2009
504 www.psychopharmacology.com * 2009 Lippincott Williams & Wilkins
9Copyright @ 200 Lippincott Williams & Wilkins. Unauthorized reproduction of this article is prohibited.

drugs as well. A good control of the symp-toms was early achieved with clozapine,but this drug was discontinued because ofthe occurrence of severe neutropenia. Thetherapeutic schema in the last monthsbefore hospitalization included lithium,valproate, paliperidone, and diazepam.
At psychiatric ward admission, thepatient presented mood instability, affectivelability, experiences and attitudes of per-plexity, high levels of anxiety, and persis-tent persecutory delusions. She had nohistory of medical illness, except for celiacdisease. In particular, no history of diabetesmellitus, pancreatitis, cholelithiasis, andsubstance abuse/dependence was present.Extensive blood tests before beginningpsychopharmacological treatments yieldedthe following fundamentally normal val-ues: fasting glucose, 77 mg/dL (referencerange, 65Y110 mg/dL); serum triglycer-ides, 91 mg/dL (reference range, 91 mg/dL); total cholesterol, 105mg/dL (referencerange, G200 mg/dL); calcium, 8.2 mg/dL(reference range, 8.0Y10.5 mg/dL); potas-sium, 3.35mEq/L (reference range, 3.5Y5.5mEq/L); sodium, 141 mEq/L (referencerange, 136Y142 mEq/L); creatinine, 0.82mg/dL (reference range, 0.50Y1.10mg/dL);bilirubin, 0.80 mg/dL (reference range,0.50Y1.10 mg/dL); aspartate aminotrans-ferase, 11 U/L (reference range, G50 U/L);alanine aminotransferase, 13U/L (referencerange, G50 U/L); amylase, 38 U/L (refer-ence range, 15Y53 U/L); and lipase, 20 U/L(reference range, G60 U/L). A minimalnormochromic anemia and a normal whiteblood cell count were detected; no anoma-lies in urinalysiswere found; and bodymassindex was 20.9 kg/cm2.
A psychopharmacological treat-ment with haloperidol at 5 mg/d, aripipra-zole at 2.5 mg/d, and diazepam at 4 mg/dwas started on the same day of hospitali-zation in our psychiatric ward. The patientalso received a gastroprotective therapy asadvised by the gastroenterologist, consist-ing of omeprazole at 80 mg/d and su-cralfate at 4 g/d plus a total parenteralnutrition (fat emulsion injections). Threedays after the beginning of the psycho-pharmacological treatment, the serum am-ylase level increased to 163 U/L and thelipase level to 62 U/L. From days 1 to 6,aripiprazole was titrated up to 12.5 mg/d,and on day 10 from the admission, theamylase reached the level of 225 U/L.After a further increase of aripiprazole upto 15 mg/d on day 18, amylase and lipasecontinued to increase, with the maximumlevels measured at 385 U/L (day 27) and424 U/L (day 28), respectively. On day 28,the patient underwent an ultrasound ofthe abdomen, which was negative for pan-creatitis and gallstones.
During all the aforementionedperiod, the subject did not report anypancreas-related complaints; all otherserum chemistries, including blood glu-cose levels, were repeated periodicallyand remained within the ranges of nor-mality. Moreover, an esophagogastroduo-denoscopy was performed, which did notshow any gastroenteric mucosal ulcerativelesions possibly related to an increase inserum amylase level. On day 32, aripipra-zole was discontinued, and the patientcontinued to receive only haloperidol anddiazepam at the same dosage. On day 33,the amylase and lipase levels were 291 U/Land 348 U/L, respectively (the pancreaticamylase isoenzyme level was 243 U/L). In10 days, pancreatic enzyme values werereduced to less than one third of themaximum values detected (118 U/L and132 U/L on day 43), and in 30 days, theywere definitively normalized.
In the absence of an adequate ther-apeutic response to pharmacological treat-ment, the patient was referred to a set of6 sessions of electroconvulsive therapyand experienced a progressive improve-ment of clinical picture. A month afterdischarge from the psychiatric ward, lab-oratory test results continued to be nega-tive, and the patient carried on with atherapy containing haloperidol and lith-ium salts.
DISCUSSIONTo the best of our knowledge, this is
the first published case of a subject intreatment with aripiprazole, presentingacute laboratory signs of pancreatitis with-out evidence of other predisposing andmediating factors.
Atypical antipsychotic agents havebeen repeatedly associated with the in-duction of acute pancreatitis, with mostcases involving clozapine and the struc-turally related compound olanzapine.1Y9
Antipsychotic-induced pancreatitis is usu-ally described as a rare adverse eventwhose pathogenesis is explained by theoccurrence of hyperglycemia, diabeticketoacidosis, and hyperlipidemia. Usually,timing of pancreatitis onset is within6 months after administration.5
Nevertheless, the exact process lead-ing to toxicity in the pancreatic islets is notyet fully understood. In fact, cases of pan-creatitis involving antipsychotics consid-ered at lower risk of inducing diabetes andketoacidosis have been reported as well.Gropper and Jackson10 have summarized3 cases related to the use of quetiapine: 2of the patients described were also takingvalproate, a drug known to induce pan-creatitis,11 but no evidence of hypergly-cemia and ketoacidosis was seen in any of
the 3 subjects. Koller et al,5 reviewing dataon this issue, found cases of pancreatitisassociated with the prescription of halo-peridol, even if in 50% of the patientsreceiving this drug, an atypical antipsy-chotic was coprescribed. Moreover, Gasseet al,12 in a recent population-based case-control study, found that current use oflow-potency conventional antipsychoticswas more frequently linked to hospital-ization for acute pancreatitis than bothhigh-potency conventional antipsychoticsand atypical antipsychotics.
In the case reported, the increase inpancreatic enzyme levels exceeded thethreshold of a 3-fold elevation and wasneither accompanied by pancreas-relatedcomplaints (eg, abdominal pain, nausea,loss of weight, etc) nor confirmed byultrasonography. The cause-and-effect re-lationship was suggested because of 2 dif-ferent considerations: (1) other possiblecommon (eg, gallstones and alcohol abuse)and uncommon (eg, abdominal trauma/surgery, hypercalcemia, and hyperlipid-emia) causes13 were excluded; and (2) thelevels of pancreatic enzymes increasedafter the introduction of aripiprazole, wors-ened further paralleling the uptitration ofthe drug, and normalized after aripiprazolewas stopped.
However, we do not have the finalconfirmation provided by rechallenge withthe drug, and we cannot entirely rule outthe possible role of ammoniac ingestionand concomitant treatments as aggravat-ing cofactors. Thus far, ziprasidone andaripiprazole are considered the safestsecond-generation antipsychotics con-cerningmetabolic dysregulation.14,15 Nev-ertheless, a subgroup of patients receivingaripiprazole could develop an increase inlipid and pancreatic enzyme levels andnew-onset hyperglycemia with diabeticketoacidosis.16Y19
Because our patient did not showdiabetes, ketoacidosis, or hyperlipidemia,we cannot rule out that aripiprazole causedpancreatic enzyme level elevation throughother mechanisms such as cytotoxic dam-age to the acinar cells or hypersensitivityreactions.20 Therefore, we recommendperiodical monitoring of serum amylaseand lipase in patients receiving aripipra-zole, particularly in subjects with predis-posing conditions.
AUTHOR DISCLOSUREINFORMATION
The authors declare no conflicts ofinterest.
Lorenzo Lattanzi, MDDivision of Psychiatry
University of PisaPisa, Italy
Journal of Clinical Psychopharmacology & Volume 29, Number 5, October 2009 Letters to the Editors
* 2009 Lippincott Williams & Wilkins www.psychopharmacology.com 505
9Copyright @ 200 Lippincott Williams & Wilkins. Unauthorized reproduction of this article is prohibited.

Francesco Casamassima, MDDivision of Psychiatry
University of PisaPisa, Italy
Department of PsychiatryMassachusetts General Hospital
Boston, [email protected]
Maurizia Brunetto, MD
Alessandro Tatulli, MD
Antonio Longobardi, MD
Elisa Schiavi, MD
Alessandra Danese, MD
Antonella Litta, MDDivision of Psychiatry
University of PisaPisa, Italy
Jonathan P. Stange, BADepartment of Psychiatry
Massachusetts General HospitalBoston, MA
Giovanni B. Cassano, MDDivision of Psychiatry
University of PisaPisa, Italy
REFERENCES
1. Frankenburg FR, Kando J. Eosinophilia,clozapine, and pancreatitis. Lancet. 1992;340(8813):251.
2. Gatto EM, Castronuovo AP, Uribe Roca MC.Clozapine and pancreatitis. ClinNeuropharmacol. 1998;21(3):203.
3. Cerulli TR. Clozapine-associated pancreatitis.Harv Rev Psychiatry. 1999;7(1):61Y63.
4. Bergemann N, Ehrig C, Diebold K, et al.Asymptomatic pancreatitis associated withclozapine. Pharmacopsychiatry. 1999;32(2):78Y80.
5. Koller EA, Cross JT, Doraiswamy PM, et al.Pancreatitis associated with atypicalantipsychotics: from the Food and DrugAdministration’s MedWatch surveillancesystem and published reports.Pharmacotherapy. 2003;23(9):1123Y1130.
6. Goldstein LE, Sporn J, Brown S, et al.New-onset diabetes mellitus and diabeticketoacidosis associated with olanzapinetreatment. Psychosomatics. 1999;40(5):438Y443.
7. Waage C, Carlsson H, Nielsen EW.Olanzapine-induced pancreatitis: a casereport. JOP. 2004;5(5):388Y391.
8. Doucette DE, Grenier JP, Robertson PS.Olanzapine-induced acute pancreatitis. AnnPharmacother. 2000;34(10):1128Y1131.
9. Hagger R, Brown C, Hurley P. Olanzapineand pancreatitis. Br J Psychiatry. 2000;177:567.
10. Gropper D, Jackson CW. Pancreatitisassociated with quetiapine use. J ClinPsychopharmacol. 2004;24(3):343Y345.
11. Binek J,HanyA,HeerM.Valproic-acidYinducedpancreatitis. Case report and review of theliterature. J Clin Gastroenterol. 1991;13(6):690Y693.
12. Gasse C, Jacobsen J, Pedersen L, et al. Riskof hospitalization for acute pancreatitisassociated with conventional and atypicalantipsychotics: a population-basedcase-control study. Pharmacotherapy. 2008;28(1):27Y34.
13. van Brummelen SE, Venneman NG, vanErpecum KJ, et al. Acute idiopathicpancreatitis: does it really exist or is it a myth?Scand J Gastroenterol Suppl. 2003;(239):117Y122.
14. Nasrallah HA, Newcomer JW. Atypicalantipsychotics and metabolic dysregulation:evaluating the risk/benefit equation andimproving the standard of care. J ClinPsychopharmacol. 2004;24(5 suppl 1):S7YS14.
15. Melkersson K, Dahl ML. Adverse metaboliceffects associated with atypical antipsychotics:literature review and clinical implications.Drugs. 2004;64(7):701Y723.
16. McQuade RD, Stock E, Marcus R, et al. Acomparison of weight change during treatmentwith olanzapine or aripiprazole: results froma randomized, double-blind study. J ClinPsychiatry. 2004;65(suppl 18):47Y56.
17. Church CO, Stevens DL, Fugate SE. Diabeticketoacidosis associated with aripiprazole.Diabet Med. 2005;22(10):1440Y1443.
18. Reddymasu S, Bahta E, Levine S, et al.Elevated lipase and diabetic ketoacidosisassociated with aripiprazole. JOP. 2006;7(3):303Y305.
19. Tolliver BK, McRae AL, Verduin ML, et al.Reversible elevation of triglycerides indual-diagnosis patients taking aripiprazole: acase series. J Clin Psychopharmacol.2008;28(4):464Y467.
20. Underwood TW, Frye CB. Drug-inducedpancreatitis.Clin Pharm. 1993;12(6):440Y448.
A Case ofAripiprazole-Associated
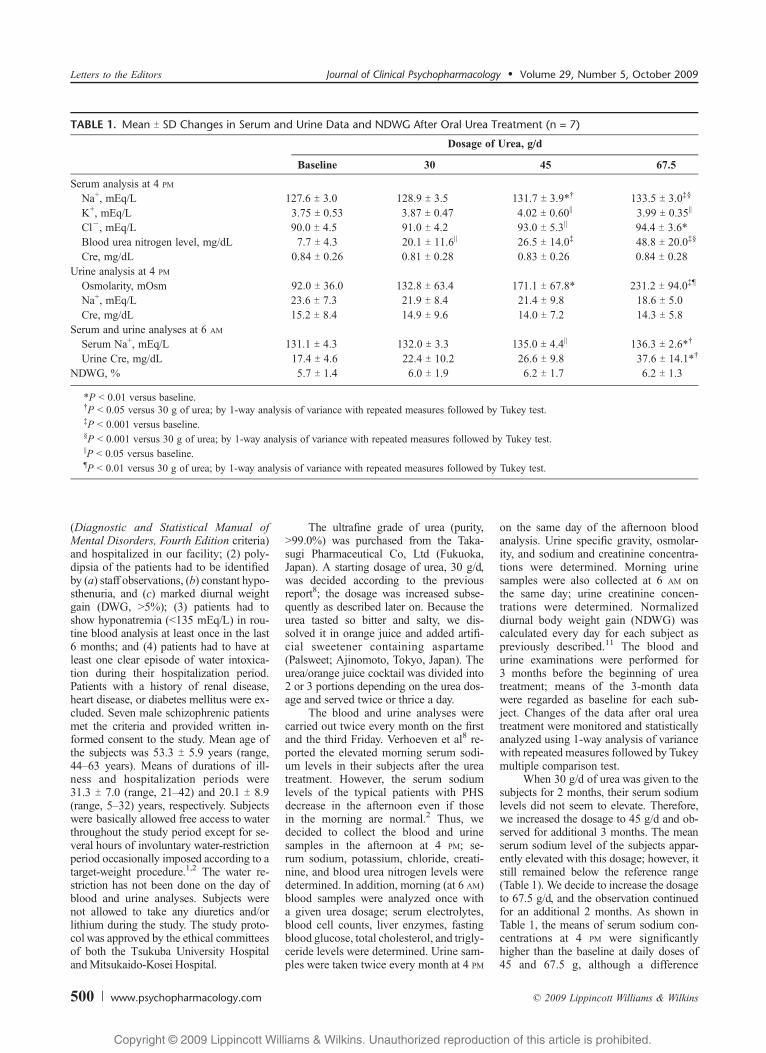
Paroxysmal SupraventricularTachycardia
To the Editors:
A ripiprazole is a relatively new antipsy-chotic drug, said to be the prototype
of a new third generation of antipsy-chotics, and has also unique propertiesshowing a combined partial agonist activ-ity at D2 and serotonin 1A receptors withan antagonism at serotonin 2A receptors.1
It has also been shown to be effective fortreatment of acute bipolar mania2 and hasa low liability for negative cardiovasculareffects.3 Nevertheless, some case reports
found that aripiprazole might be arrhyth-mogenic in special circumstances.4,5
Herein, we report a patient with bipolardisorder who developed paroxysmal sup-raventricular tachycardia (PSVT) afteraripiprazole treatment.
Mr A, a 19-year-old man, was hos-pitalized because of acute psychoticsymptoms manifested as disorganizedspeech, grandiose ideation, and irritability.There was no substance abuse history.Bipolar I disorder with psychotic featurewas diagnosed preliminarily. He hadneither taken any neuroleptics nor experi-enced any cardiovascular disease beforethe admission. Although his mood andpsychotic symptoms were prominent atadmission, his vital signs remained rela-tively stable during the first 3 days ofhospitalization except mild tachycardiawith pulse rates of 100, 104, and 104beats per minute (bpm), respectively. Thebaseline electrocardiogram (ECG) wasnormal. During hospitalization, risperi-done (2 mg/d) was prescribed for hispsychotic symptoms and discontinued thenext day because of oculogyric crisis.Aripiprazole was started at 10 mg/d, andlorazepam at 3 mg/d and biperidin at 6mg/d were combined.
He had complained of chest pain andchest tightness 3 days after aripiprazoleadministration and was found to have aheart rate of 178 bpm. His systolic anddiastolic blood pressure was 98 and 65mm Hg, respectively, with body temper-ature of 36.6-C and respiratory rate of 16breaths per minute. Physical examinationresults showed oriented consciousness andno signs of dehydration, rigidity of ex-tremities, abnormal neurological sign, anddisturbance of other autonomic systems.We did not perform blood biochemistryexamination considering that there was nospecific physical abnormality, and thedata, including glucose level, renal func-tion, hepatic function, and electrolytelevel, were normal 3 days before this at-tack. The electrocardiogram revealedPSVT that responded to propranolol at10 mg 60 minutes later (Fig. 1). Aripipra-zole was then shifted to flupentixol at 12mg/d, and the manic symptoms subsided1 month later. Neither chest pain nor chesttightness was experienced after the shift.There was no PSVT attack again duringadmission, and the follow-up ECG did notshow any abnormalities. In addition, hedid not show similar symptoms/signs untilnow, 9 months after his discharge. Re-viewing his history, his mother had ar-rhythmia and received regular medicaltreatment according to her self-report.
Many clinical conditions, such asdehydration, psychomotor agitation, or
Letters to the Editors Journal of Clinical Psychopharmacology & Volume 29, Number 5, October 2009
506 www.psychopharmacology.com * 2009 Lippincott Williams & Wilkins
9Copyright @ 200 Lippincott Williams & Wilkins. Unauthorized reproduction of this article is prohibited.
adverse effects of medication, may causetachycardia in a patient with acute psy-chosis. As for this patient, his intake wasquite well during admission, supported byincreased body weight of 2 kg in 1 week,and his psychomotor agitation seemed tobe independent of pulse rate because hisagitation persisted for more than 2 weeksbut only 1 episode of PSVT and relativelystable heart rate for the rest of the time.Although a high dose of anticholinergicagent, biperidin at 6 mg/d, may inducetachycardia,3 the dose remained the sameduring the whole hospitalization. Further-more, there were no symptoms/signs, suchas muscle rigidity, fever, other autonomicsymptoms, or conscious disturbance, in-dicating neuroleptic malignant syndrome.
Most studies emphasized that aripi-prazole has low incidence of ECG abnor-malities, such as corrected QT intervalprolongation.4 However, Egger et al5 re-ported that a case of schizophrenia devel-oped dose-dependent incomplete rightbundle-branch block when aripiprazolewas increased from 15 to 30 mg, and itdisappeared after discontinuation of med-ication. Torgovnick et al6 further reporteda case of aripiprazole-induced orthostatichypotension and cardiac arrhythmia. Inaddition, 1 clinical trial had revealed thatthe standing heart rate was increased tomore than 120 bpm, 15 bpm higher thanthose at baseline, in patients of aripipra-zole group.4 To our knowledge, althoughPSVT is thought to be randomly occurringcongenital anomalies, heredity may alsocontribute to its development.7 Paroxys-mal supraventricular tachycardia in Mr A,hereditary vulnerability with mild tachy-cardia at admission, may arise fromaripiprazole-induced sympathetic stimula-
tion, which is commonly used to facilitateinduction of this tachycardia.8 Becauserisperidone and its active metabolite, 9-OH-risperidone, have strong >-adrenergiceffects and long elimination half lives,particularly in those with poor CYP2D6activity,9 and CYP2D6 genetic poly-morphisms may also affect the metabo-lism of aripiprazole,10 potential drug-druginteraction and their possible influencescannot be totally excluded.
Although, we did not rechallenge thispatient by aripiprazole because of ethicalconsideration, the chronological presenta-tion of his PSVT, his family history of ar-rhythmia, and the aforementioned reportsmay support the possible association be-tween aripiprazole use and PSVT in thiscase. Thus, this case highlights the impor-tance of closely ECGmonitoring in patientstreated with aripiprazole if they have fam-ily history of cardiac conduction disease orcardiovascular disease themselves.
AUTHOR DISCLOSUREINFORMATION
The authors declare no conflicts ofinterest.
Pei-Chen Tsai, MDDepartment of Psychiatry
Taipei City Psychiatric CenterTaipei City Hospital
Taipei, Taiwan
Shih-Hung Hsiao, MDCardiovascular Center
Department of Internal MedicineKaohsiung Veterans General Hospital
Kaohsiung, Taiwanand School of Medicine
National Yang-Ming UniversityTaipei, Taiwan
Chih-Chiang Chiu, MDDepartment of Psychiatry
Taipei City Psychiatric CenterTaipei City Hospital
Taipei, Taiwanand Department of Psychiatry
College of MedicineTaipei Medical University
Taipei, [email protected]
REFERENCES
1. Taylor DM. Aripiprazole: a review of itspharmacology and clinical use. Int J ClinPract. 2003;63:49Y54.
2. Smith LA, Cornelius V, Warnock A, et al.Pharmacological interventions for acutebipolar mania: a systemic review ofrandomized placebo-controlled trials.Bipolar Disord. 2007;9:551Y560.
3. Ikawa M, Tabuse H, Ueno S, et al. Effectsof combination psychotropic drug treatmenton heart rate variability in psychiatricpatients. Psychiatry Clin Neurosci.2001;55(4):341Y345.
4. McEvoy JP, Daniel DG, Carson WH Jr, et al.A randomized, double-blind,placebo-controlled, study of the efficacyand safety of aripiprazole 10, 15 or 20 mg/dayfor the treatment of patients with acuteexacerbations of schizophrenia. J PsychiatrRes. 2007;41:895Y905.
5. Egger C, Rauscher A, Muehlbacher M, et al.A case of dose-dependent aripiprazoleinduced conduction disturbance. J ClinPsychopharmacol. 2006;26:436.
6. Torgovnick J, Sethi NK, Arsura E.Aripiprazole-induced orthostatic hypotensionand cardiac arrhythmia. Psychiatry ClinNeurosci. 2008;62:485.
7. Hayes JJ, Sharma PP, Smith PN, et al.
FIGURE 1. Lead II of ECG results (A) before PSVT, admission day 1; (B) PSVT, admission day 5; (C) and after PSVT, admission day 30(voltage, 5 mm/mV; ECG paper rate, 25 mm/s).
Journal of Clinical Psychopharmacology & Volume 29, Number 5, October 2009 Letters to the Editors
* 2009 Lippincott Williams & Wilkins www.psychopharmacology.com 507
9Copyright @ 200 Lippincott Williams & Wilkins. Unauthorized reproduction of this article is prohibited.

Familial atrioventricular nodal reentrytachycardia. Pacing Clin Electrophysiol.2004;27(1):73Y76.
8. Chiou CW, Chen SA, Kung MH, et al. Effectsof continuous enhanced vagal tone on dualatrioventricular node and accessorypathways. Circulation. 2003;107(20):2583Y2588.
9. Mannens G, Huang ML, Meuldermans W,et al. Absorption, metabolism, and excretionof risperidone in humans. Drug MetabDispos. 1993;21(6):1134Y1141.
10. Kim JR, Seo HB, Cho JY, et al.Population pharmacokinetic modelling ofaripiprazole and its active metabolite,dehydroaripiprazole, in psychiatric patients.Br J Clin Pharmacol. 2008;66(6):802Y810.
Possible Lamotrigine-InducedMania in a Child With
Autism Spectrum Disorderand Epilepsy
To the Editors:
Lamotrigine, a mood stabilizer and ananticonvulsant medication, has been
reported to induce manic reaction inyoung and adult subjects with mood dis-orders in whom lamotrigine was used asa mood stabilizer.1Y5 However, to ourknowledge, there is no report of lamotri-gine-induced mania in children who weretreated with lamotrigine for epilepsy. Inthis study, we present a 10-year-old boywith diagnosis of autism spectrum disor-der (ASD), familial hypomagnesemia, andepilepsy who developed a manic reactionduring lamotrigine treatment for his epi-lepsy. The mother had consented for thepublication of this report.
CASE REPORTThe subject is a 10-year-old boywho
presented with his mother because ofrelatively sudden onset behavioral changesfor the last month. The mother reported thathe started to display frequent laughingwith-out any reason, increased hyperactivity, ex-cessive talking and asking same questionstoo many times, decreased need for sleep,increased irritability, aggressive behaviors,talkingbillingsgate (abusive language), rub-bing his genitalia frequently, spending ordemanding more money than before, re-fusing to wear same clothes and insist-ing to have new and beautiful ones, andincreased religiosity (such as using thephrase bismillah [meaning with the nameof God] many times, awaking at dawn,and asking many questions about ezan,which is a call to pray in Islamic tradi-
tion). During the interview, he was notedto have hyperactivity, self-talking, distract-ibility, and smiling and laughing with anunusually happy affect. No psychoticsymptoms (hallucinations or thought dis-order) were reported or detected.
The mother reported that he startedto exhibit these behaviors after his anti-epileptic medication was changed fromcarbamazapine to lamotrigine. The motherreported that he first started to exhibit fre-quent laughing without any reason and in-creased self-talking at 100 mg/d dosage oflamotrigine. Lamotrigine was increased to150 mg/d, and the aforementioned manicsymptoms emerged after 1 week with arelatively sudden onset. The mother re-ported that he did not exhibit such beha-viors before lamotrigine treatment exceptasking the same question several times,self-talking (in the form of a verbal per-severation), and some level of irritability.They did not report any obvious rash.However, they reported some level ofdizziness, headache, and transient abdom-inal discomfort possibly related to lamo-trigine treatment.
He was born after a full term,uneventful pregnancy with abdominal de-livery. He had multiple generalized tonic-clonic seizures during the first year ofhis life. He was seizure-free between2 and 4 years of age but started to haveoccasional atonic or absence seizures dur-ing 4 to 9 years of age. His condition waseventually diagnosed with familial hypo-magnesemia at the age of 9 as the etiologyof his seizures. He has been on severalantiepileptic medications (phenobarbital,phenytoin, valproate, carbamazepine, andlamotrigine) for seizure control since hisinfancy and magnesium preparates for hisfamilial hypomagnesemia for the last year.He has been seizure-free for the last year.However, his antiepileptic medication waschanged from carbamazepine to lamotri-gine 4 months ago because of abnormalityin his sleep as shown in the electroenceph-alogram (EEG). He had a diagnosis ofASD and mild mental retardation at theage of 7. There is no family history ofmood or neurological disorders.
The child was examined by a neu-rologist before being referred to a psychi-atrist, but they did not consider thesymptoms as lamotrigine related. Howev-er, because we considered that these manicsymptoms could be related to lamotriginetreatment, we aimed to gradually decreaselamotrigine to 100 mg/d within 2 weeks.On the next visit, 3 weeks later, the motherreported that all manic symptoms showedmoderate to much improvement afterlamotrigine was decreased to 100 mg/d.No changewas reported in his neurological
status. We further decreased lamotrigine to75 mg/d within 2 weeks. On the next visit,3 weeks later, the mother reported and weobserved further decrease in manic symp-toms. During the next 3 months of follow-up, he remained seizure-free with almostno significant manic symptoms.
DISCUSSIONEmergence of manic symptoms with
lamotrigine treatment in a dose-dependentfashion and disappearance or improvementin these symptoms after lamotrigine wasdecreased may suggest enough causalitybetween manic symptoms and lamotriginetreatment. Naranjo causality scale revealeda score of 8 that signifies probable adversedrug reaction.6
Lamotrigine has been used as a moodstabilizer in subjects with bipolar disorderparticularly for subjects with bipolar de-pression.7,8 There are several reports oflamotrigine-induced mania or hypomaniain young or adult subjects with mood dis-orders who had been taking lamotrigineas a mood stabilizer.1Y5 In such cases, un-derlying mood disorder may be consid-ered as a risk factor for developing manicreaction during lamotrigine treatment.However, to our knowledge, there is noreport of lamotrigine-induced mania inchildren who used lamotrigine for epilep-sy. Furthermore, lamotrigine has also beenreported to be associated with forced nor-malization in subjects with epilepsy.9
Forced normalization was first introducedby Landolt (1953) as Ba phenomenon char-acterized by the fact that, with the occur-rence of psychotic states, the EEG becomesmore normal or entirely normal as com-pared with previous and subsequent EEGfindings.[9 Forced normalization can beobserved both in treated and untreatedepileptic patients during the course of ill-ness in that course of epilepsy could sud-denly change and the seizures somehowbe replaced by a behavioral disorder.9,10
He had been seizure-free for the last year.However, because his control EEG wasscheduled at a later time by a neurologist,it remains unclear whether this manic re-action was somewhat a forced normaliza-tion or an adverse drug reaction related tolamotrigine.
The exact mechanisms of action oflamotrigine are not well known as a moodstabilizer and antiepileptic medication.However, it has a well-documented effi-cacy for the treatment and/or prevention ofdepressive episodes in subjects with bipo-lar disorder.7,8 Lamotrigine is a phenyl-triazine anticonvulsant agent that differsstructurally from other currently availableanticonvulsant agents. Animal studies
Letters to the Editors Journal of Clinical Psychopharmacology & Volume 29, Number 5, October 2009
508 www.psychopharmacology.com * 2009 Lippincott Williams & Wilkins
9Copyright @ 200 Lippincott Williams & Wilkins. Unauthorized reproduction of this article is prohibited.

indicate that lamotrigine may stabilizeneuronal membranes (and be effectivein the management of tonic-clonic andpartial or absence seizures) by blockingvoltage-sensitive sodium channels. It mayalso inhibit the presynaptic excitatory re-lease of glutamate and aspartate, blockhigh-voltage activated N- and P-type cal-cium channels, block presynaptic seroto-nin reuptake, and prevent amygdala andcortical kindling. These properties, partic-ularly inhibition of presynaptic aspartateand glutamate release and serotonin re-uptake, have been proposed to be related tolamotrigine’s anticonvulsant and antide-pressant action.11
He developed manic symptoms in adose-dependent fashion and manic symp-toms resolved within several weeks againin a dose-dependent fashion as lamotri-gine was decreased from 150 to 75 mg/d.This may be consistent with the previousreports that lamotrigine-induced manicreaction in a dose-dependent fashion andsymptoms resolved within several weeksof discontinuation.1Y4 Besides havingepilepsy, this subject also had ASD andfamilial hypomagnesemia. To our knowl-edge, this is also the first report on the con-currence of familial hypomagnesemia anddrug-induced mania in children with ASD.It could be possible that these conditionsmay have rendered his brain more vul-nerable to develop such an adverse drugreaction.
In conclusion, clinicians should bealert to the possibility of manic reactionin young subjects not only with mooddisorders but also with seizure disorderstreated with lamotrigine.
AUTHOR DISCLOSUREINFORMATION
The authors warrant that they haveno conflict of interests in general or inconnection with the submitted paper andno financial relationships with any phar-maceutical company.
Murat Coskun, MD
Hasan Bozkurt, MD
Salih Zoroglu, MDChild and Adolescent Psychiatry Department
Istanbul Medical FacultyIstanbul University
Istanbul, [email protected]
REFERENCES
1. Desarkar P, Sinha VK. Lamotrigine-inducedsevere manic switch. Aust N Z J Psychiatry.2006;40:718.
2. Moor S, Luty S, Joyce P. Lamotrigine-induced
mania in adolescents. Aust N Z J Psychiatry.2007;41:1013Y1014.
3. Margolese HC, Beauclair L, Szkrumelak N,et al. Hypomania induced by adjunctivelamotrigine. Am J Psychiatry. 2003;160:183Y184.
4. Raskin S, Teitelbaum A, Zislin J, et al.Adjunctive lamotrigine as a possible maniainducer in bipolar disorder. Am J Psychiatry.2006;163:159Y160.
5. Selek S, Savas HA. Lamotrigine-inducedmanic switches have already been reported.Aust NZ J Psychiatry. 2007;41:195.
6. Naranjo CA, Busto U, Sellers EM, et al. Amethod for estimating the probability ofadverse drug reactions. Clin Pharmacol Ther.1981;30:239Y245.
7. Chang K, Saxena K, Howe M. An open-labelstudy of lamotrigine adjunct or monotherapyfor the treatment of adolescents with bipolardepression. J Am Acad Child AdolescPsychiatry. 2006;45:298Y304.
8. Calabrese JR, Bowden CL, Sachs GS, et al.A double-blind placebo controlled studyof lamotrigine monotherapy in outpatientswith bipolar I depression. Lamictal 602Study Group. J Clin Psychiatry. 1999;60:79Y88.
9. Clemens B. Forced normalisation precipitatedby lamotrigine. Seizure. 2005;14:485Y489.
10. Krisnamoorthy ES, Trimble MR. Forcednormalization: clinical and therapeuticrelevance. Epilepsia. 1999;40(suppl 10):S57YS64.
11. Davanzo P, McCracken J. Mood stabilizer:lithium and anticonvulsants. In: Andres M,Lawrance S, Dennis SC, James FL, eds.Pediatric Psychopharmacology. OxfordUniversity Press; New York, NY:2003:309Y327.
Valproic Acid ToxicityAssociated With Low Doseof Aspirin and Low Total
Valproic Acid LevelsA Case Report
To the Editors:
Valproic acid (VPA) is frequently usedas an anticonvulsant or as a mood
stabilizer. Valproic acid has a relativelynarrow therapeutic window or index;therefore, therapeutic drug monitoring isused to avoid adverse effects, especiallyneurological toxicity. Therapeutic drugmonitoring almost always involves mea-suring the total plasma concentration.Measuring the free VPA plasma concen-tration is rarely needed because total VPAlevels usually predict both toxicity andfree VPA levels quite well. Of the 2, free
VPA levels are more closely associatedwith neurological toxicity.1
Valproic acid is highly bound toplasmatic protein, including albumin. Itis believed that the rate is 85% to 95% atlow doses and 70% with high doses.1 FreeVPA increases relative to hypoalbumin-emia. Thus, in patients with hypoalbu-minemia, the same total concentrationsproduce higher therapeutic free VPAlevels2; hence, free VPA levels rather thantotal levels should bemeasured. Bauer et al3
also recommendmeasuring free VPA levelsin the elderly and suggest that although theelderly may have normal albumin concen-tration, they have altered binding.
Drug-drug interactions (DDIs) asso-ciated with protein-binding displacementconstitute a controversial issue4; very fewclinical cases have been published. Theo-retical reviews in the literature of the 1970sand 1980s5 suggested that protein bindingshould be relevant to DDI. More recentliterature suggests that protein binding israrely relevant to DDI and may not berelevant to VPA.6 Another recent reviewsuggests that protein-binding displacementDDIs are generally of minimal clinical sig-nificance, but this assumption is based onthe lack of evidence to the contrary.7
Protein-binding displacement and freeVPA levels may be important when aspirinis coprescribed. Aspirin can cause a DDIassociated with increased VPA-free con-centrations. It was first described in phar-macokinetic studies in children8Y10; then,three cases of serious toxicity were de-scribed in adults.11 Sandson et al2 de-scribed a patient who took and thendiscontinued aspirin and was monitoredby measuring free VPA concentrations.The mechanism explaining this DDI iscomplex because VPA is metabolized byseveral enzymes. The mitochondrial beta-oxidation pathway accounts for approxi-mately 40% of VPA metabolism1; aspirincan inhibit this pathway.10 Additionally,aspirin is very highly bound and can dis-place VPA from the albumin, causing amuch larger increase in free concentrationthan in total VPA concentration. Sandsonet al2 described in their case that 325 mg/dof aspirin caused an 8-fold increase in freeVPA level, whereas the total level increasedonly 1.8-fold. In children, Farrell et al8
suggested that antipyretic dosages of aspi-rin (12Y17 mg/kg per day) can produce a49% increase of free VPA with very smallchanges in total concentration.
The number of PubMed cases withclear VPA toxicity associated with aspirinDDIs is limited11; it is not known whetherlow aspirin doses may be safe in patientstaking VPA.3 Ours is a case of VPA tox-icity probably explained by an increase
Journal of Clinical Psychopharmacology & Volume 29, Number 5, October 2009 Letters to the Editors
* 2009 Lippincott Williams & Wilkins www.psychopharmacology.com 509
9Copyright @ 200 Lippincott Williams & Wilkins. Unauthorized reproduction of this article is prohibited.

in free VPA concentration, despite only81 mg of aspirin being used and relativelylow VPA dosages being prescribed (500Y1000 mg/d; Table 1).
Ms Awas a 59-year-old white womanwith a weight of 71.1 kg, a body massindex of 24.8 kg/m2, moderatemental retar-dation, and a diagnosis of schizoaffectivedisorder. She had severalmedical problems,including a history of seizures, hyperten-sion, diabetes mellitus type 2, hypercholes-terolemia, and hypothyroidism. Becauseshe had not had a recent seizure, a neuro-logist recommended that 800 mg/d ofcarbamazepine be discontinued by taperingoff for 2 months. The patient remained freeof seizures for 1.5 months after completediscontinuation. At the time of the seizure,she was taking the following: atenolol (50mg/d), benazepil (60 mg/d), and hydro-chlorothiazide (50 mg/d) for her hyperten-sion; glipizide (10 mg/d) for her diabetesmellitus; simvastatin (10 mg/d) and niacin(1500 mg/d) for her hyperlipidemia; 1aspirin (81 mg/d) for cardiovascular pre-vention; levothyroxine (25 mg/d) for herhypothyroidism; risperidone (2.5 mg/d) forher psychosis; bromocriptine (5 mg/d) forcorrecting risperidone adverse effects; rani-tidine (300 mg/d) for gastroesophagealreflux disease; and lactulose (30 mL/d) forconstipation. Supplements were calciumand vitamins D and E.
The patient had a 2-minute general-ized tonic-clonic seizure with urinaryincontinence at 1.5 months after carba-mazepine discontinuation. She was imme-diately treated with lorazepam (1 mg/d).After discussion with the psychiatrist, shewas started on enteric-coated divalproexsodium (500 mg twice a day) to controlseizures and to possibly have a positiveeffect on the schizoaffective disorder. Oneweek later, her trough total VPA levelwas 112 Kg/mL (recommended range,50Y125 Kg/mL). Six days later (13 daysafter starting VPA), the speech/language
pathologist observed that the patient wasconfused and was complaining of dizzi-ness. The next day, the patient’s sister alsothought that the patient was lethargic andher behavior was unusual (Bher person-ality had changed[). The dosage was de-creased to 750 mg/d (250 mg in themorning and 500 mg in the evening).
On the 15th day after starting VPA,the staff observed a hand tremor. On day17, the staff continued to see that the pa-tient was lethargic and incoherent. On day21, the total VPA was 87 Kg/mL. On day22, the dosage was decreased to 500 mg/d(250 mg twice a day). Despite all thesemeasures, the patient had hypotension anddeveloped hyperglycemia, leading to dis-continuation of all antihypertensives andthe addition of metformin (500 mg/d). Onday 33, her total VPA was 66 Kg/mL. Ondays 47 to 53, the hand tremor continuedto be obvious. On day 56, she had per-sistent hand tremors, and the total andthe free VPAs were measured. The totalVPA continued to be on the low side(64 Kg/mL), but the free VPA was high(13.1 Kg/mL; range, 4Y12 Kg/mL). There-fore, it was decided to discontinue theVPA and switch to carbamazepine.
This patient had signs compatible withdrug toxicity for almost 2 months. Confu-sion, dizziness, lethargy, hand tremor, andincoherent speech can be explained by drugtoxicity and could possibly have been at-tributed to risperidone, VPA, or their com-bination with the other drugs. However, thecause became clear after free VPA levelswere measured and then VPA was discon-tinued; this was associated with the disap-pearance of the symptoms. The toxicity waspresent even with low dosages (500 mg/d)and low total VPA (60 Kg/mL). The corre-sponding free VPAwas 13.1 Kg/mL, whichwas outside the recommended range. We donot have free VPA levels at the higherdosages, but it is easy to speculate thatduring the initial dosage (1000 mg/d), they
may have been at least twice as high(926 Kg/mL).
It seems unusual that very low dos-ages of aspirin (81 mg/d) would havecontributed to this DDI and to the VPAtoxicity. Two possible factors contributingto an explanation of this unusual casemay be mild hypoalbuminemia and poly-pharmacy. The albumin concentrationwas 3.4 g/dL (recommended range, 3.5Y5.0 g/dL). The total protein concentrationwas 6.2 g/dL (recommended range, 6.4Y8.3 g/dL). The mild hypoalbuminemiamay have modest effects on increasingthe VPA toxicity in this case.
The presence of other medicationswith very high protein binding may alsohave contributed to the displacement ofVPA from the plasmatic proteins. The pa-tient was taking 11 medications besidesaspirin when VPAwas started. Five of thesemedications have high protein binding,which may have contributed to the VPAdisplacement. The expected protein bind-ing12 is 99% for simvastatin (10 mg/d),92% to 99% for glipizide (10 mg/d), 90%to 96% for bromocriptine (5 mg/d), 77%to 90% for risperidone (2 mg/d), and 68%for hydrochlorothiazide (50 mg/d). Levo-thyroxine is very highly bound (999%),but the dosages are 1000 times lower thanthose of the other medications (25 Kg/d).When the free VPA was measured, thepatient was still taking 3 medications in themilligram-per-day range with high proteinbinding (10 mg/d of simvastatin, 10 mg/dof glipizide, and 5 mg/d of bromocriptine).
Despite the limitations of a case re-port format and the lack of some free VPAmeasures at the time of the highest VPAtotal levels, this case suggests that evenvery low aspirin doses may contribute toincreased free VPA levels. Finally, the mostimportant message for clinicians is that ifthere is unexpected neurological toxicityassociated with VPA, suggesting toxic lev-els in the presence of normal total VPAlevels, free VPA levels should bemeasured.
AUTHOR DISCLOSUREINFORMATION
No commercial organizations had anyrole in the writing of this paper forpublication. Dr de Leon is currently a co-investigator in a National Institutes ofHealth Small Business Innovation researchgrant awarded to Genomas, Inc. In the pastyear (since January 1, 2008), Dr de Leonhad no conflicts of interest related to theinformation described here. He has neverlectured for Abbott (marketer of Depakote).He has never been a consultant with Abbott(or other pharmaceutical companies) or
TABLE 1. Total and Free VPA Levels
Day
VPA
Dosage, mg/dTotal Level
(Range, 50Y125 Kg/mL)Free Level
(Range, 4Y12 Kg/mL)
1 10007 1000 11214 75021 750 8722 50033 500 6656 500 64 13.1
Letters to the Editors Journal of Clinical Psychopharmacology & Volume 29, Number 5, October 2009
510 www.psychopharmacology.com * 2009 Lippincott Williams & Wilkins
9Copyright @ 200 Lippincott Williams & Wilkins. Unauthorized reproduction of this article is prohibited.

owned any Abbott stocks. The otherauthorshave no conflicts of interest.
Jose de Leon, MDMental Health Research Center
Eastern State HospitalLexington, KY
John L. Kiesel, MD
Michael W. Fleming, MD
Bryan Strobl, RPhCentral State Intermediate Care Facility
for the Mentally RetardedLouisville, KY
REFERENCES
1. DeVane CL. Pharmacokinetics, druginteractions and tolerability of valproate.Psychopharmacol Bull. 2003;37(suppl 2):25Y42.
2. Sandson NB, Marcucci C, Bourke DL, et al.An interaction between aspirin and valproate:the relevance of plasma protein displacementdrug-drug interactions. Am J Psychiatry.2006;163:1891Y1896.
3. Bauer LA, Davis R, Wilensky A, et al.Valproic acid clearance: unbound fractionand diurnal variation in young and elderlyadults. Clin Pharmacol Ther. 1985;37:697Y700.
4. Levy RH, Shen DD, Abbott FS, et al.Valproic acid: chemistry, biotransformation,and pharmacokinetics. In: Levy RH,Mattson RH, Meldrum BS, et al, eds.Antiepileptic Drugs. 5th ed. Philadelphia,PA: Lippincott Williams & Wilkins;2002:780Y800.
5. Greenblatt DJ, Sellers EM, Koch-Weser J.Importance of protein binding for theinterpretation of serum or plasma drugconcentrations. J Clin Pharmacol. 1982;22:259Y283.
6. Benet LZ, Hoener B-A. Changes in plasmaprotein binding have little clinical relevance.Clin Pharmacol Ther. 2002;71:115Y121.
7. DeVane CL. Clinical significance of drugbinding, protein binding, and bindingdisplacement drug interactions.Psychopharmacol Bull. 2002;36:5Y21.
8. Farrell K, Orr JM, Abbott FS, et al. Theeffect of acetylsalicylic acid on serum freevalproate concentrations and valproateclearance in children. J Pediatr. 1982;101:142Y144.
9. Orr JM, Abbott FS, Farrell K, et al.Interaction between valproic acid and aspirinin epileptic children: serum protein bindingand metabolic effects. Clin Pharmacol Ther.1982;31:642Y649.
10. Abbott FS, Kassam J, Orr JM, et al. Theeffect of aspirin on valproic acid metabolism.Clin Pharmacol Ther. 1986;40:94Y100.
11. Goulden KJ, Dooley JM, Camfield PR, et al.Clinical valproate toxicity induced byacetylsalicylic acid. Neurology. 1987;37:1392Y1394.
12. Lacy CF, Armstrong LL, Goldman MP, et al.Drug Information Handbook. 15th ed.Hudson, OH: Lexi-Comp; 2007.
Combination Therapyof AcetylcholinesteraseInhibitor and Vitamin Ein Alzheimer Disease
To the Editors:
A lzheimer disease (AD) is a disablingdisease affecting approximately 25 to
29 million people worldwide. As a pos-sible therapeutic strategy, vitamin E hasbeen shown to prolong the duration oftime before hospitalization is required foralmost 1 year,1 presumably by trappingfree radicals and thus interrupting thechain reaction that subsequently leads tocellular damage.2 Cholinesterase inhibi-tors, including donepezil, rivastigmine,and galantamine, augment cholinergicfunction by increasing synaptic acetyl-choline concentration resulting in cognitivebenefits and behavioral improvement.3Y7
Cholinesterase inhibitors and vitamin E actthrough different mechanisms that wouldbe expected to have additive effects intreatment. On the other hand, it may bepossible that the substances interfere witheach other for unexpected reasons. It is thussurprising that to date, combination studiesare restricted to a retrospective chart re-view of donepezil and vitamin E, includinga comparison with a historical sample ofuntreated patients.8
In the uncontrolled retrospective studyreported here, comprising 49 patientsdiagnosed with mild to moderate ADaccording to the criteria of the NationalInstitute of Neurological and Communi-cative Disorders Association,9 the patientswere divided into 2 groups, one that re-ceived an acetylcholine-esterase inhibitoralone (29patients, 19males and10 females;mean [SD] of age, 67.7 years [8.3 years])and one where it was combined with high-dose vitamin E (20 patients, 9 males and11 females; mean [SD] of age, 70.1 years[6.8 years]). Daily doses of at least 5 mg ofdonepezil, 6 mg of rivastigmine, and 16mgofgalantaminewere taken.VitaminE intakewas at least 500 IU daily (mean, 1805 IUdaily). None of the subjects with adequatefollow-up was excluded for failure tocontinue treatment, but in 1 patient in eachgroup, rivastigmine was changed to donep-
ezil due to side effects. In the cholinesteraseinhibitor group, the dosage of rivastigmineperformed at that point was 12 mg daily,whereas in the combination group, it wasonly 6 mg daily. None received additionalAD medication, for example, memantine.No further adverse events were reportedduring follow-up.
The study was performed in accor-dance with the declaration of Helsinki.
Apolipoprotein E (ApoE) genotyp-ing was performed by isoelectric focusingand subsequent precipitation by specificpolyclonal antibodies and visualization byautomated silver staining in accordancewith Hackler et al.10
Age, sex, medical history, and medi-cations and their dosages were recorded.Mini-Mental State Examination (MMSE)11
was recorded at the initial and at the lastregistered visit. It was always rated by thesame clinician (DMB). Because the designwas retrospective, no special effect owingto group distribution can be assumed.
For group comparison between pa-tients treated with additional vitamin E asopposed to cholinesterase inhibitor alone,an independent t test was applied. Whendemographic datawere compared and therewas more than 1 category for a variable,a W
2 test was applied. To control forpossible confounding factors, a stepwiseregression analysis was performed. Thesignificance threshold was set at 0.05.
There were no differences in basicdemographic data between the 2 groupswith respect to age, sex, ApoE phenotype,type of cholinesterase inhibitor, or timeof follow-up. Mean (SD) time of follow-up for all patients was 14.6 months (8.8months) (mean [SD] of cholinesterase in-hibitor alone, 14.9 months [9.4 months];mean [SD] combination, 13.9 months [7.7months]). At initiation of treatment, al-though far from being significant, subjectsreceiving a cholinesterase inhibitor alonehad slightly better cognition (mean [SD],22.9 [4.0] vs 21.5 [4.7]). The averageMMSE of patients on cholinesterase in-hibitors and vitamin E declined at a sig-nificantly lower rate as compared with thegroup treated with a cholinesterase inhib-itor alone (combination, j0.4 vs cholin-esterase inhibitor j3.0; mean [SD]difference, j2.6 [1.0]; 95% confidenceinterval, j0.7 to j4.9; P = 0.01; Fig. 1).
We used a forward stepwise regres-sion approach in which independent var-iables are entered one after another toanalyze how much each one adds to theexplanation of the dependent variable(MMSE change in follow-up). In a signif-icant model (F = 5.4, P = 0.027) wheresex, age, initial MMSE, type of cholines-terase inhibitor, and ApoE were entered,
Journal of Clinical Psychopharmacology & Volume 29, Number 5, October 2009 Letters to the Editors
* 2009 Lippincott Williams & Wilkins www.psychopharmacology.com 511
9Copyright @ 200 Lippincott Williams & Wilkins. Unauthorized reproduction of this article is prohibited.
the treatment with vitamin E turned outalone to be a sufficient predictor (A =0.410, t = 2.334).
DISCUSSIONTo date, vitamin E is not recom-
mended in routine treatment of AD.However, our data provide some evidencethat combination of vitamin E and acholinesterase inhibitor may be reason-able. The effect remained after correctingfor initial MMSE, age, sex, and ApoEphenotype.
In AD, cholinesterase inhibitor thera-py and vitamin E, which likely act throughdifferent mechanisms, seem not to interferewith each other but to act synergisticallyand in combination to offer additionalbenefits. One possible explanation is theprevention of anticholinesterase-inducedoxidative injury attributed to cholinester-ase inhibitors.12 Although a strong limi-tation of our study is the small sample sizeand the retrospective design, nevertheless,it is the first time that a combination ofboth substances in comparison with acholinesterase inhibitor alone has beeninvestigated. Previously, only efficacy ofcombination therapy with vitamin E anddonepezil compared with controls hasbeen demonstrated.8 In a recent study,vitamin E and donepezil were compared inmild cognitive impairment, the diagnosisof which can be considered to indicate arisk of AD or preclinical AD,13 and someeffects of vitamin E on language andexecutive functioning could be observedat up to 18 months.14 However, these ef-ofects were only temporary and of smallsize. Furthermore, in another study, nopositive impact of vitamin E on progres-sive atrophy rate was observed.15
CONCLUSIONSThe strength of our study is the com-
parison of cholinesterase inhibitors aloneversus in combination with vitamin E,which to our knowledge has been con-ducted for the first time. It could be de-monstrated that combination therapy issuperior to cholinesterase inhibitor the-rapy alone and can be recommended inclinical practice. Age, sex, and ApoE phe-notype did not influence the course ofdisease in our sample. However, there areconcerns regarding the design in that itwas retrospective and not placebo con-trolled. Furthermore, the sample size wassmall. Larger, prospective, randomized,double-blind placebo-controlled trials withmore frequent follow-up ratings are war-ranted and seem to be promising. Al-though in our study no severe adverseevents occurred, vitamin E applicationmay be harmful. Although AD treatmentwith high-dose vitamin E decreased the ob-served risk of mortality,1 in a more recentmeta-analysis, high-dose application of vi-tamin E increased the risk of mortality withspecial respect to coronary artery disease.16
AUTHOR DISCLOSUREINFORMATION
The author received no funding forthis study. The author has received dona-tions for oral presentations by Novartisand Eisai.
Daniel M. Bittner, MDDepartment of NeurologyUniversity of Magdeburg
Magdeburg, Germanyand Department of Neurology
University of UlmUlm, Germany
REFERENCES
1. Sano M, Ernesto C, Thomas RG, et al. Acontrolled trial of selegiline, alpha-tocopherolor both as treatment for Alzheimer’s disease.N Engl J Med. 1997;336:1216Y1222.
2. Halliwell B, Gutteridge JMC. Oxygenradicals in the neuron system. TrendsNeurosci. 1985;8:22Y26.
3. Corey-Bloom J, Anand R, Veach J, et al. Arandomized trial evaluating the efficacy andsafety of ENA713 (rivastigmine tartrate), anew acetylcholinesterase inhibitor, in patientswith mild to moderately severe Alzheimer’sdisease. Int J Geriatr Psychopharmacol.1998;1:55Y65.
4. Raskind MA, Peskind ER, Wessel T, et al.Galantamine in AD: A 6-monthrandomized, placebo-controlled trial witha 6 month extension. The GalantamineUSA-1 Study Group. Neurology.2000;54:2261Y2268.
5. Rogers SL, Doody RS, Pratt RD. Long-termefficacy and safetyof donepezil in the treatmentof Alzheimer’s disease: final analysis of a USmulticenter open-label study. EurNeuropsychopharmacol. 2000;10:195Y203.
6. Rogers SL, Farlow MD, Doody RS, et al. A24-week, double-blind, placebo-controlledtrial of donepezil in patients with Alzheimer’sdisease. Neurology. 1998;50:136Y145.
7. Rosler M, Anand R, Cicin-Sain A, et al.Efficacy and safety of rivastigmine in patientswith Alzheimer’s disease: internationalrandomized controlled trial. BMJ. 1999;318:633Y638.
8. Klatte ET, Scharre DW, Nagaraja HN, et al.Combination therapy of donepezil andvitamin E in Alzheimer’s disease. AlzheimerDis Assoc Disord. 2003;17:113Y116.
9. McKhann G, Drachman D, Folstein M, et al.Clinical diagnosis of Alzheimer’s disease:report of the NINCDS-ADRDAWork Group
FIGURE 1. Difference in initial and follow-up MMSE (mean [SD]; P = 0.01), dependent on the different cholinesterase inhibitors andtreatment groups (P 9 0.05).
Letters to the Editors Journal of Clinical Psychopharmacology & Volume 29, Number 5, October 2009
512 www.psychopharmacology.com * 2009 Lippincott Williams & Wilkins
9Copyright @ 200 Lippincott Williams & Wilkins. Unauthorized reproduction of this article is prohibited.

under auspices of theDepartment ofHealth andHuman Services Task Force on Alzheimer’sDisease.Neurology. 1984;34:939Y944.
10. Hackler R, Schafer JR, Motzny S, et al. Rapiddetermination of apolipoprotein E phenotypesfrom whole plasma by automated isoelectricfocusing using PhastSystem andimmunofixation. J Lipid Res. 1994;35:153Y158.
11. Folstein MF, Folstein SE, McHugh PR.Mini-Mental State: a practical method forgrading the cognitive state of patients for theclinician. J Psychiatr Res. 1975;12:189Y198.
12. Milatovic D, Gupta RC, Aschner M.Anticholinesterase toxicity andoxidative stress.ScientificWorldJournal. 2006;6:295Y310.
13. Petersen RC, Smith GE, Waring SC, et al.Mild cognitive impairment: clinicalcharacterization and outcome. Arch Neurol.1999;56:303Y308.
14. Petersen RC, Thomas RG, Grundman M,et al. Vitamin E and donepezil for thetreatment of mild cognitive impairment.N Engl J Med. 2006;352:2379Y2388.
15. Jack CR Jr, Petersen RC, Grundman M,et al. Longitudinal findings from the vitaminE and donepezil treatment study for MCI.Neurobiol Aging. 2008;29:1285Y1295.
16. Miller ER III, Pastor-Barriuso R, Dalal D,et al. Meta-analysis: high-dosage vitamin Esupplementation may increase all-causemortality. Ann Intern Med. 2005;142:37Y46.
Treatment of UnipolarPsychotic Depression
An Open Study of LithiumAddition in RefractoryPsychotic Depression
To the Editors:
Psychotic depression is the most severeform of depression. Electroconvulsive
therapy is considered the most effectivetreatment of psychotic depression, butpharmacotherapy may also be regardedas a suitable treatment option. Most stud-ies report that psychotic depressivesrespond poorly to antidepressant mono-therapy.1,2 For patients with antidepres-sant-refractory nonpsychotic depression,addition of lithium to ongoing antidepres-sant treatment is the most-studied nextstep. Despite suggestions that lithium ad-dition might also be useful in patients withpsychotic depression,3 evidence for itsefficacy is still very limited. Becauserelapse after successful electroconvulsivetherapy is a major problem, there is aneed to investigate pharmacological treat-ment strategies for psychotic depression,
and lithium addition may prove to bean effective strategy for antidepressantnonresponders.
The present study was designed tocompare imipramine, venlafaxine, andvenlafaxine plus quetiapine (phase 1),followed by lithium addition (phase 2) forphase 1 nonresponders. The results of thephase 1 study comparing the efficacy ofimipramine, venlafaxine, and venlafaxine-quetiapine are presented elsewhere.4 Thepresent report focuses on attaining remis-sion during lithium addition.
PHASE 1: IMIPRAMINE VERSUSVENLAFAXINE VERSUSVENLAFAXINE PLUS
QUETIAPINE (7 WEEKS)This study is the second phase of a
randomized, double-blind, controlled trialwith 8 participating centers in the Nether-lands. The protocol and the results of thefirst phase have been described elsewhere.4
In short, 122 inpatients who met theDiagnostic and Statistical Manual of Men-tal Disorders, Fourth EditionVText Revi-sion criteria for psychotic major depressionwith a score of 18 or higher on theHamiltonRating Scale for Depression (HAM-D)5
and who were drug-free for at least 4 daysbefore the study were randomized 1:1:1 to7-week treatment with imipramine (withtarget plasma levels of 200Y300 Kg/L),6
venlafaxine (maximum dosage of 375 mg/d), or venlafaxine plus quetiapine (maxi-mum dosage of 375 and 600 mg/d, respec-tively). As concomitant psychotropicmedication, only benzodiazepines (maxi-mum dosage of 3 mg of lorazepam equiv-alent per day) were allowed. The study wasapproved by the ethical review boards of thecenters. All patients gave written informedconsent before enrollment. At baseline,diagnosis was confirmed with the Struc-tured Clinical Interview for Diagnostic andStatistical Manual of Mental Disorders,Fourth Edition Axis I disorders.7
Severity of depressive symptoms wasassessed at baseline and then weeklyusing the 17-item HAM-D.5 In addition,all individual psychotic features (includ-ing whether they were mood congruentor mood incongruent) were documentedat baseline and then weekly.
PHASE 2: LITHIUM ADDITION(4 WEEKS)
For patients who did not attainresponse (HAM-D reduction Q50% com-pared with baseline and final HAM-Dscore e14) at the end of phase 1, lithiumwas added for an additional 4 weeks.Patients entering phase 2 continued their
blinded study medication at the samedose, and lithium was started at an initialdosage of 600 mg at 8.00 P.M. Plasmalithium levels (12 hours after dose) weremeasured on day 7 and weekly thereafter.Doses were adjusted to attain a lithiumlevel of 0.6 to 1.0 mmol/L. Weekly as-sessments of the 17-item HAM-D and psy-chotic features continued during phase 2.After the 4-week study period, the codewasbroken. At 2 and 4months after completionof phase 2, patients had 2 follow-up visitsto assess whether response and remissionwere sustained.
Our a priori purpose was to comparethe efficacy of the three 2-step treat-ment strategies. However, because only15 patients participated in phase 2 (lithiumaddition), this analysis seemed to be im-possible. Therefore, the efficacy of lith-ium addition was evaluated for all patientsreceiving lithium, calculating remission(final HAM-D score e7), response(HAM-D reduction Q50% during phase2), and mean HAM-D reduction duringphase 2.
Adverse events were measured on a4-point scale (none, light, moderate, andsevere) and were considered relevant iftheir severity was at least moderate and ifthey had increased compared with the endof phase 1.
Unfortunately, it seemed to be difficultto enroll the patients for the phase 2 of thestudy. This was due to 2 reasons: (1) fewerpatients than anticipated participated inphase 1; and (2) the study protocol forphase 2 was completed substantially laterthan that for phase 1, approval from theethical review board was obtained approx-imately 18months after phase 1was started.During the 5-year study period of phase 1,122 patients were enrolled. Of those, 100patients completed phase 1, 41 werenonresponders and were eligible for lith-ium addition. However, 26 of them did notenter phase 2 because most (n = 16) hadcompleted phase 1 before May 2003 (theinitiation of phase 2). Thus, the total studysample consisted of 15 patients: 3 patientstaking imipramine (mean dosage, 275 mg/d;range, 225Y375 mg/d), 7 patients takingvenlafaxine (all with a dosage of 375 mg/d),and 5 receiving venlafaxine plus quetiapine(all with a dosage of 375 and 600 mg/d,respectively). The patients’demographic andclinical characteristics at start of lith-ium addition (week 7) are summarized inTable 1. During the 4-week study period,none of the patients dropped out. Allpatients obtained a lithium plasma levelwithin the therapeutic range with a mean(SD) plasma level of 0.68 (0.14) mmol/L ata mean (SD) dosage of 840 (445) mg/d.During phase 2, 8 (53%) of 15 patients used
Journal of Clinical Psychopharmacology & Volume 29, Number 5, October 2009 Letters to the Editors
* 2009 Lippincott Williams & Wilkins www.psychopharmacology.com 513
9Copyright @ 200 Lippincott Williams & Wilkins. Unauthorized reproduction of this article is prohibited.

1 to 3 mg of lorazepam equivalent per dayas concurrent medication. At the start ofphase 2, psychotic features were still pres-ent in 11 of 15 patients.
The mean HAM-D score decreased13.0 points during the 4-week period oflithium addition.
After 2 weeks of lithium addition, 4patients had achieved response and 2 re-mission; at the end of the lithium additionphase, 9 (60%) of 15 patients were respond-ers and the same number of patients metthe criteria for remission. During the ad-ditional 15-week follow-up period, allpatients, except 3 nonresponders, continuedtheir studymedication. For all 9 remitters, atthe end of a 4-week lithium addition, theirremission was sustained during the follow-up period.
Three patients received imipramine,mean dose 275 (range, 225Y375) mg; all7 patients on venlafaxine received 375mg;5 patients receiving combination treat-ment all used 375 mg of venlafaxine and600 mg of quetiapine. Plasma lithiumlevels are available for all 15 patients forwhom lithium was started. The mean (SD)lithium level after attainment of the targetlevel was 0.68 (0.14) mmol/L. Mean lith-ium dose after achieving the target levelwas 840 T 445 (range, 400Y120) mg.
No patients dropped out because ofadverse effects during phase 2, whereasonly 1 patient in whom lithium was addedto venlafaxine dropped out during follow-up (because of an allergic skin reaction).Compared with the last assessment beforelithium addition, 3 patients experiencedsevere adverse events: 2 patients receiv-ing venlafaxine and lithium experiencedsevere loss of libido and a severe tremor,and another patient receiving venlafaxine,quetiapine, and lithium had a severetremor.
DISCUSSIONOur main finding is that most phase 1
nonresponders (60%) responded to lith-ium addition. To our knowledge, there are
no previous studies on lithium addition inantidepressant-refractory psychotic de-pressives. Therefore, we consider ourfindings with impressive response andremission rates of 60% (albeit resultingfrom an open, uncontrolled study) to be ofclinical relevance. Our aim was to includein phase 2 approximately 50% of thepatients enrolled in phase 1. This assump-tion was based on previous 2-step treat-ment studies in depressed inpatientswhose sample consisted of both patientswith psychotic depression and patientswith nonpsychotic depression. In thesestudies, 57 (53%) of 107 and 71 (51%) of138 patients who entered phase 1 subse-quently received lithium addition.8,9 How-ever, we only succeeded in including 15patients, and because of this small sample,comparing the efficacy of the 3 treatmentstrategies was impossible. We thereforeconsider phase 2 as an open study. Be-cause our study lacks a placebo arm, ourfindings suggest but do not prove thataddition of lithium is effective as a sub-sequent treatment step for antidepressantnonresponders (with or without an anti-psychotic) with psychotic depression.Furthermore, the sustaining remissionduring the 15-week follow-up periodalso argues against a response because ofnonspecific factors. The sustaining remis-sion in patients during continuation treat-ment with antidepressants and lithium isin accordance with the results of previousstudies.10,11 Another possible explanationfor our finding is a delayed response to themedication of phase 1; however, becausethe mean HAM-D scores of the patients inour study had improved only 9.8 pointsduring phase 1 compared with 13.0 pointsin phase 2 and that most improvement inphase 2 did not occur in the first week (3.6points) but in the last 3 weeks (9.4 points),we consider this a rather unlikely expla-nation. In the present study, because theeffect of lithium addition gradually ap-peared between weeks 2 and 4, this raisesthe question whether a 4-week treatment
period is optimal for this strategy. In thepresent study, the combination of lithiumwith high doses of venlafaxine was toler-ated well with no indication for a sub-stantial risk of a serotonin syndrome, asdescribed previously during venlafaxine-lithium treatment.6
In conclusion, although our datasuggest that lithium addition is an effec-tive subsequent and well-tolerated step inthe treatment for patients with psychoticdepression, larger controlled studies oflithium addition in psychotic depressionare warranted.
ACKNOWLEDGMENTSThe authors thank the following for
their support: E.A. Wijnia, K. Nijssen, J.W.van de Maaskant of Julius Center; J.Verploegh and J. Leijtens, research nurses;N.P.J.T. van Schayk, I. Vosjan, I. van Geel,and I. Kop of AstraZeneca; andH.Kornaat,M. Kanters-van Buren, E. Ides, and M.Griekspoor of Wyeth.
This study was supported by grantsfrom AstraZeneca and Wyeth Pharmaceu-ticals, both of which also provided the studymedication. These companies had noinvolvement in study design, in the collec-tion, analysis, and interpretation of data,and in the decision to submit the paper forpublication. They were given the opportu-nity to comment on the manuscript.
AUTHOR DISCLOSUREINFORMATION
T.K. Birkenhager has received sup-port from Wyeth and received honoraria/speaker’s fees fromWyeth and Servier. W.W.van den Broek has received support fromHersenstichting and received honoraria/speaker’s fees from Servier and Wyeth. J.Wijkstra has received support from Astra-Zeneca and Wyeth and received honoraria/speaker’s fees from AstraZeneca andWyeth. J.A. Bruijn and E. van Os have noconflicts of interest to disclose. M.P.M.Boks has received support from ZonMw.R.J. Verkes has received support fromZonMw, AstraZeneca, Eli Lilly, Pfizer,Wyeth, and Organon, received honoraria/speaker’s fees from AstraZeneca, EliLilly, Lundbeck, Wyeth, and Organon,and participated in the advisory boards ofEli Lilly, Servier, Organon, and Lareb.J.G.E. Janzing has received support fromHersenstichting and received honoraria/speaker’s fees from Bristol-Myers Squibb.W.A. Nolen has received support from theNetherlands Organisation for Health Re-search and Development, Stanley MedicalResearch Institute, AstraZeneca, Eli Lilly,GlaxoSmithKline, and Wyeth, receivedhonoraria/speaker’s fees from AstraZeneca,
TABLE 1. Demographic and Clinical Characteristics of Patients Receiving LithiumAddition After 7 Weeks of Antidepressant Treatment (n = 15)
Characteristic
Age, mean (range), yr 53.9 (40Y63)Women, n (%) 11 (73.3)White, n (%) 15 (100)No. previous episodes, mean T SD 1.0 T 1.6Duration of index episode, mean T SD, wk 38.9 T 36.8HAM-D score at the start of lithium addition, mean T SD 22.1 T 5.8HAM-D score after 4 weeks of lithium treatment, mean T SD 9.1 T 8.0With hallucinations, n (%) 0 (0)With delusions, n (%) 11 (73)
Letters to the Editors Journal of Clinical Psychopharmacology & Volume 29, Number 5, October 2009
514 www.psychopharmacology.com * 2009 Lippincott Williams & Wilkins
9Copyright @ 200 Lippincott Williams & Wilkins. Unauthorized reproduction of this article is prohibited.

Eli Lilly, Pfizer, Servier, and Wyeth, andparticipated in the advisory boards ofAstraZeneca, Cyberonics, Eli Lilly, Glaxo-SmithKline, Pfizer, and Servier.
Tom K. Birkenhager, MD, PhD
Walter W. van den Broek, MD, PhDDepartment of PsychiatryErasmus Medical Center
Rotterdam, the [email protected]
Jaap Wijkstra, MDDepartment of Psychiatry
Rudolf Magnus Institute of NeuroscienceUniversity Medical Center Utrecht
Utrecht, the Netherlands
Jan A. Bruijn, MD, PhD
Erik van Os, MDDepartment of PsychiatryErasmus Medical Center
Rotterdam, the Netherlands
Marco Boks, MD, PhDDepartment of Psychiatry
Rudolf Magnus Institute of NeuroscienceUniversity Medical Center Utrecht
Utrecht, the Netherlands
Robbert J. Verkes, MD, PhD
Joost G.E. Janzing, MD, PhDDepartment of Psychiatry
Radboud University Nijmegen Medical CentreNijmegen, the Netherlands
Willem A. Nolen, MD, PhDDepartment of Psychiatry
University Medical Center GroningenUniversity of Groningen
Groningen, the Netherlands
REFERENCES
1. Wheeler Vega JA, Mortimer AM,Tyson PJ. Somatic treatment of psychoticdepression: review and recommendationsfor practice. J Clin Psychopharmacol.2000;20:504Y519.
2. Schatzberg AF, Rothschild AJ. Psychotic(delusional) depression: should it be includedas a distinct syndrome in DSM-IV? Am JPsychiatry. 1992;149:733Y745.
3. Bruijn JA, Moleman P, Mulder PG, et al.Treatment of mood-congruent psychoticdepression with imipramine. J Affect Disord.2001;66:165Y174.
4. Wijkstra J, Burger H, van den Broek WV,et al. Treatment of unipolar psychoticdepression. A randomized, double-blindstudy comparing imipramine, venlafaxine,and venlafaxine plus quetiapine. ActaPsychiatr Scand. In Press.
5. Hamilton M. A rating scale for depression.J Neurol Neurosurg Psychiatry. 1960;23:56Y62.
6. Bertschy G, Ragama_Pardos E, Ait-Ameur A,
et al. Lithium augmentation in venlafaxinenon-responders: an open study. Eur Psychiatry.2003;18:314Y317.
7. First MB, Spitzer RL, Gibbon M, et al.Structured Clinical Interview for DSM-IVAxis I Disorders. Dutch Translation. Lisse,The Netherlands: Swets & Zeitlinger; 1999.
8. Bruijn JA, Moleman P, Mulder PG, et al.Comparison of 2 treatment strategies fordepressed inpatients: imipramine andlithium addition or mirtazapine andlithium addition. J Clin Psychiatry.1998;59:657Y663.
9. Birkenhager TK, van den Broek WW,Mulder PG, et al. Comparison oftwo-phase treatment with imipramine orfluvoxamine, both followed by lithiumaddition, in inpatients with majordepressive disorder. Am J Psychiatry.2004;161:2060Y2065.
10. Bauer M, Bschor T, Kunz D, et al.Double-blind, placebo-controlled trial of theuse of lithium to augment antidepressantmedication in continuation treatment ofunipolar major depression. Am J Psychiatry.2000;157:1429Y1435.
11. Bschor T, Berghofer A, Strohle A, et al.How long should the lithium augmentationstrategy be maintained? A 1-year follow-upof a placebo-controlled study in unipolarrefractory major depression. J ClinPsychopharmacol. 2002;22:427Y430.
Journal of Clinical Psychopharmacology & Volume 29, Number 5, October 2009 Letters to the Editors
* 2009 Lippincott Williams & Wilkins www.psychopharmacology.com 515
9Copyright @ 200 Lippincott Williams & Wilkins. Unauthorized reproduction of this article is prohibited.