
Translation : quá trình dịch mã ( tổng hợp chuỗi polypeptide

This journal is©The Royal Society of Chemistry 2014 Chem. Commun., 2014, 50, 4971--4988 | 4971
Cite this:Chem. Commun., 2014,
50, 4971
Polypeptide films via N-carboxyanhydridering-opening polymerization (NCA-ROP):past, present and future
Steven Harris Wibowo, Adrian Sulistio, Edgar H. H. Wong, Anton Blencowe† andGreg G. Qiao*
The formation of surface-grafted polypeptide films and interfaces via N-carboxyanhydride ring-opening
polymerization (NCA-ROP) holds great potential for the facile preparation of next-generation
multifunctional and responsive materials with excellent biocompatibility, biodegradability, tunable
conformations and chemical functionalities. Therefore, the aim of this feature article is to provide a
topical review of developments in the area of polypeptide films derived through NCA-ROP. It is evident
that studies reported thus far have only started to unveil the full potential of peptide-based interfaces
and materials, and with continued advancements it is anticipated that the strategic combination of NCA-
ROP with modern synthetic chemistries will continue to yield versatile platforms for broader applications
in the fields of polymer therapeutics, tissue engineering, (bio)nanocoatings, (bio)chemosensors, catalysis
and separation technologies.
1. Introduction
Ultrathin polymeric films and interfaces are integral for thedevelopment of next-generation advanced functional materialsand nanotechnologies, with applications spanning from drug-delivery vectors,1–3 artificial cells,4–6 biomaterials,7–9 (micro)-electronic devices,10,11 stimuli-responsive surfaces,12–15 energy
Department of Chemical and Biomolecular Engineering, The University of
Melbourne, Parkville, VIC 3010, Australia. E-mail: [email protected]
Steven Harris Wibowo
Steven Harris Wibowo graduatedwith a B. Eng (First ClassHonours) in Chemical andBiomolecular Engineering fromThe University of Melbourne(Australia) in 2011. Asrecognition for his outstandingundergraduate research project,he was awarded the 2011 CRC-Polymers Prize by the RACIPolymer Division. Upongraduation, he commenced hisPhD studies under thesupervision of Prof. Greg Qiao
and was recently awarded the 2013 Treloar Prize by RACI. Hiscurrent research focuses on developing surface-driven controlledpolymerization technique to construct nanoscale (bio)materialswith unique architectures, diverse chemical functionalities andtunable physical characteristics.
Adrian Sulistio
Adrian Sulistio graduated withB. Eng (Chemical, Hons)/B. Commfrom The University of Melbournein 2006 before joining Prof. GregQiao’s group at The University ofMelbourne to pursue his PhD in2007, and was awarded hisdoctorate in 2011. He has sincecontinued working with Prof.Qiao as a postdoctoral fellowworking on an ARC Linkageproject in collaboration withindustry partner to developpolymer–drug implants for sus-
tained drug delivery for osteoarthritis and glaucoma treatment.His other research interest is in the development of functionalnanomaterials, bio-interface and self-assembly of polymers tocreate different molecular architectures for various applications.
† Present address: Mawson Institute, Division of Information Technology,Engineering and the Environment, University of South Australia, SA 5095,Australia.
Received 13th January 2014,Accepted 17th February 2014
DOI: 10.1039/c4cc00293h
www.rsc.org/chemcomm
ChemComm
FEATURE ARTICLE

4972 | Chem. Commun., 2014, 50, 4971--4988 This journal is©The Royal Society of Chemistry 2014
capture/storage16,17 to membrane and separation technologies.18,19
In particular, recent advancements in the (bio)nanotechnologyfield warrant the assembly of biocompatible and biodegradablefunctional materials derived from renewable resources (e.g.,amino acids). Given the large family of amino acids (bothnatural and non-natural), synthetic polypeptides can be engi-neered to display a diverse range of chemical and physicalfunctionalities via chemical derivatization protocols, whilemaintaining excellent biocompatibility and (bio)degradability.Furthermore, depending on the amino acid constituents, poly-peptides with well-ordered conformations (e.g., a-helices andb-sheets) can be prepared through non-covalent interactions, suchas hydrogen bonding, van der Waals forces and p–p stacking.20
This intrinsic capability to form higher-ordered structuresallows the construction of unique hierarchically-organizedmaterials not readily accessible by other organic (macro)mole-cules. These aspects make polypeptides an interesting andpromising class of building block for materials fabrication.
Conventionally, solid-phase peptide synthesis (SPPS) isemployed to produce synthetic polypeptides with precise struc-tures via the step-wise coupling of protected amino acids ontosolid resins, followed by cleavage of the polypeptides fromthe resin.21 The SPPS approach employs iterative coupling–wash–deprotection–wash cycles to sequentially add amino acidresidues onto the polypeptide chains, which allows for theformation of polypeptides with controlled sequences on alaboratory scale. However, SPPS is a costly process and theachievable length of polypeptides is limited by resin porosity(steric hindrance), incomplete coupling steps and side reac-tions.22,23 In comparison, controlled ring-opening polymeriza-tion of a-amino acid N-carboxyanhydrides (NCA-ROP) is aversatile technique for the facile preparation of high molecularweight synthetic polypeptides, although precise control overthe primary amino acid sequence has yet to be achieved.Significant advancements in regulating the polymerizationand reducing side reactions over the past 15 years have seenNCA-ROP emerge as a simple, robust and efficient techniquefor the scalable preparation of polypeptides with defined archi-tecture, stereochemistry and functionality.
As a result of the recent progress in the field of controlledNCA-ROP and the unique properties derived from syntheticpolypeptides, the utilization of polypeptides in materialsscience has expanded considerably. For instance, numerousgroups have investigated the application of polypeptides asadvanced functional coatings and interfaces on materials. Oneof the most robust and commonly utilised techniques forgenerating polypeptide films is the surface-grafting approach,which can be divided into the ‘grafting-to’ and ‘grafting-from’
Anton Blencowe
Anton Blencowe received hisMaster’s degree in chemistrywith honours from the Universityof Reading in 2002. He completedhis PhD under the supervision ofProfessor Wayne Hayes at theUniversity of Reading in 2006before working as a PostdoctoralFellow with Professor Greg Qiaoat The University of Melbourne.In 2009, he was awarded an ARCAustralian Postdoctoral Fellow-ship. Currently, he is a ResearchLeader at the Mawson Institute at
the University of South Australia. His research interests span fromfundamental studies to applied sciences and encompass the fieldsof macromolecular engineering and self-assembly, polymertherapeutics, biomaterials, biomimetics, and nanomaterials.
Greg G. Qiao
Prof. Greg G. Qiao received hisB. Eng in Polymer Engineeringfrom Donghua University in1982 before completing his PhDin synthetic organic chemistry atThe University of Queensland in1996. Since 2000, he has been theleader of the Polymer ScienceGroup at The University ofMelbourne. He is an ARC FutureFellow and is currently aProfessor of MacromolecularChemistry and Engineering,Assistant Dean (Research) in the
Melbourne School of Engineering, and the current Deputy Head ofthe Department of Chemical and Biomolecular Engineering. Hismain research focuses on novel macromolecular architectures,nanostructured materials, soft tissue engineering, biomaterialsand functional polymers for industrial applications.
Edgar H. H. Wong
Edgar Wong commenced his PhDstudies under the supervision ofProf. Christopher Barner-Kowollik (and jointly supervisedby Prof. Thomas Junkers andProf. Martina Stenzel) at TheUniversity of New South Wales(Australia) in 2008. En route tothe completion of his PhD in2011, Dr Wong has published 13peer-reviewed journal articles. Hehas been working as apostdoctoral researcher underthe supervision of Prof. Greg
Qiao and Prof. Frank Caruso at The University of Melbourne(Australia) since the submission of his PhD thesis. His researchinterests include mechanism and kinetics of polymerizationprocesses, modular conjugation reactions, thin films, self-assembly, bio- and nanostructured materials.
Feature Article ChemComm
Publ
ishe
d on
17
Febr
uary
201
4. D
ownl
oade
d by
The
Uni
vers
ity o
f M
elbo
urne
Lib
rari
es o
n 28
/11/
2014
21:
47:5
2.
View Article OnlineView Journal | View Issue

This journal is©The Royal Society of Chemistry 2014 Chem. Commun., 2014, 50, 4971--4988 | 4973
strategies. Whereas the grafting-to approach generally relies onthe coupling between end-functionalized peptide chains (pre-formed via NCA-ROP) with a complementary functionalizedsubstrate, the grafting-from approach involves surface initiatedring-opening polymerization of amino acid NCA derivativesfrom initiator functionalized substrates. Despite the relativeinfancy of this field, the number of reported studies usingsurface-grafting methodologies to form polypeptide films con-tinues to grow rapidly, with a wide range of applicationsdemonstrated in the fields of tissue engineering,24 macro-molecular recognition,25,26 separation technologies,27 (bio)-responsive materials,28–30 microdevices (e.g., biosensors),31 cata-lysis32 and stereospecific chemistry.33 While early studies focusedon the fundamentals of film formation, recent application-oriented studies have been of great interest to the broaderscientific community. Therefore, this review article highlightsthe milestones achieved in the peptide surface-grafting field inrelation to NCA-ROP. Current synthetic approaches and techno-logical limitations, as well as potential future developments andprospective practical applications will be discussed.
2. Ring-opening polymerization ofa-amino acid N-carboxyanhydrides(NCA-ROP): a brief historical overview
The synthesis of a-amino acid N-carboxyanhydride (NCA) deri-vatives was first reported by Leuchs in 1906.23 By the 1950s, thering-opening polymerization (ROP) of NCA monomers usingwater, alcohols and primary amine initiators had been reportedwith poor control over the polymerization which resulted in theformation of heterogeneous materials with ill-defined chain-length, composition, sequence and chain-end functionality(Scheme 1).22,23 Interestingly, despite the lack of control overpolymerization and the chemical properties of the resultingpolypeptides, they still displayed a-helix and b-sheet secondarystructures, similar to their natural counterparts.22
Traditionally, NCA monomers have been synthesized byeither the Leuchs method or the preferred Fuchs–Farthingmethod (Scheme 1).22,34 While the Leuchs approach involvesthe reaction of N-alkyloxycarbonylamino acids with halogenatingcompounds, the Fuchs–Farthing approach employs the directaddition of phosgene gas (acting as carbonyl source) to a-aminoacids. As a result of the toxicity of phosgene gas and the associatedstringent safety measures, stable phosgene substitutes such astriphosgene, diphosgene and di-tertbutyltricarbonate are nowalmost exclusively used. Following NCA formation, purificationand isolation are generally achieved via recrystallization, although
other methods such as flash column chromatography under inertconditions have also been employed.34,35 Also, depending on themethod of synthesis, impurities and common by-products such asHCl, HCl–amino acid salts and 2-isocyanatoacyl chlorides can bedetrimental to well-controlled NCA-ROP.
In 1997 Deming reported the pivotal work on metal-catalyzed NCA-ROP, which described the first ‘living’ polymer-ization of a-amino acid NCAs.36 This work provided access towell-defined high molecular weight homo or co-polypeptideswith excellent homogeneity and end-group fidelity. Since then,various studies have reported controlled NCA-ROP using novelinitiators (e.g., silazane derivatives37,38 and primary aminehydrochlorides39) and polymerization techniques (e.g., highvacuum polymerization40), as well as optimized reaction con-ditions (e.g., temperature and pressure).41,42 Recently, largescale synthesis of high molecular weight synthetic polypeptides(multigram scale) has also been reported using non-nucleophilic initiators based on tetrafluoroborate ammoniumsalts.43 However, one of the remaining challenges in NCA-ROPis to achieve control over the primary amino acid sequence,which would bring the scientific community a step closertowards emulating the elegant sequence control displayed bynature.
The wide spread interest in synthesizing polypeptides byNCA-ROP can be partially attributed to the large family ofamino acids that can be incorporated. Natural amino acidswith various hydrophobic, acidic and basic side chain function-alities (e.g., carboxylic acids, phenol, amines, thiol, indole,imidazole) are abundant in all living organisms.44 In addition,synthetic amino acid derivatives with specialized function-alities (e.g., azide, alkyne, allyl) are also readily available.29,45
This diverse range of functionalities not only allows for further(bio)chemical derivatization of polypeptides, but also contri-butes to the formation of higher-ordered structures, andresponsiveness towards external stimuli such as pH, tempera-ture, electrolytes, solvents and enzymes.34 These propertiesmake synthetic polypeptides a versatile class of organic macro-molecules for the preparation of drug-delivery devices,29,46–50
responsive polymeric brushes,51,52 core cross-linked stars (CCS)polymers44,53–55 and templates for inorganic oxides.56–58 Formore detailed information on the NCA-ROP mechanisms andvarious macromolecular architectures, the reader is referred toother excellent recent review articles.22,23,34,35
3. Surface-grafting of polypeptides:a general overview
The grafting-to and-from approaches have been identified to bethe most versatile techniques for the formation of ultrathinpolypeptide films. Central to this assembly process is theformation of polypeptide chains with one-end covalently teth-ered to a substrate surface (also commonly referred to aspolypeptide brushes). In most cases, covalent anchoring ispreferred as it provides greater film stability under differentenvironmental conditions.22Scheme 1 Synthesis of synthetic polypeptides by NCA-ROP.
ChemComm Feature Article
Publ
ishe
d on
17
Febr
uary
201
4. D
ownl
oade
d by
The
Uni
vers
ity o
f M
elbo
urne
Lib
rari
es o
n 28
/11/
2014
21:
47:5
2.
View Article Online

4974 | Chem. Commun., 2014, 50, 4971--4988 This journal is©The Royal Society of Chemistry 2014
Surface-grafting can involve either grafting-to or grafting-from strategies. In the grafting-to technique, pre-synthesizedpolypeptide chains are covalently linked or ‘end-grafted’ from asolution onto a complementary functionalized substrate sur-face (Scheme 2A). The main advantage of this approach is theopportunity to comprehensively characterize the polypeptidechains prior to grafting, leading to films with defined composi-tion. One of the important aspects of the grafting-to techniqueis the end-functionalization of the peptide chains with func-tional groups complementary to the surface-bound moieties.To date, the most practical controlled NCA-ROP has beeninitiated with primary amines. As such, functional end-groupscan be judiciously located at the C-terminus (a-carbon) or theN-terminus (propagating o-amino) (Scheme 2A). Functionalmoieties can be introduced at the C-terminus through use ofa functional (macro)initiator, while the N-terminus end-groupcan be attained from post-polymerization conversion of thepropagating o-amino group. However, as the NCA-ROPinitiated by primary amines can experience side reactions andpremature termination, the fidelity of the propagating aminogroup may be reduced, resulting in lower functionality.22,41,42
Furthermore, since the grafting-to technique relies on thediffusion of peptides chains onto the surface, diffusion and sterichindrance often result in the formation of non-homogenous filmswith low grafting densities.22,28
In comparison, the grafting-from approach involving surface-initiated NCA-ROP allows for the formation of films with highgrafting density. This method relies on the anchoring of suitableinitiators onto the substrate’s surface from which NCA-ROPoccurs to form polypeptide brushes (Scheme 2B). The immobi-lization of initiators onto the surface is crucial for the grafting-from approach and therefore, extensive work has been reportedon initiator immobilization using non-covalent and covalentmeans. Surface anchored primary amines have predominantlybeen utilized as initiators; however, controlling the molecularweight distribution of the grafted peptide chains remains a
challenge as a result of the difficulty in controlling the initiationand propagation rates from surface-bound initiators.22,34 Never-theless, this drawback is offset by the high density of polymerbrushes, which offers unique physical properties endowed by theinteraction between adjacent polypeptide chains.
Given the ease and versatility of both surface-graftingapproaches, synthetic polypeptides have been grafted on a widerange of organic and inorganic substrates, including silicananoparticles (nonporous and mesoporous),28,59 macroporouspolymeric templates,30 silicon wafers,60–63 gold substrates,64–68
glass slides,69 magnetite nanoparticles,32,70 microporous mem-branes,71,72 cotton fabrics69 and cellulose.69 In addition, thesecondary structure adopted by surface-grafted polypeptides andthe availability of chemical functionalities have been employed asplatforms for (bio)conjugation of proteins,28,30,72 and sugar mole-cules.29 The stimuli responsive behaviour of the grafted polypep-tides has also been used for (macro)molecular recognition,25,26,32
ion exchange,27,72 and drug release.28,30 Furthermore, improvedbiocompatibility and processability of nanoparticles and nano-tubes have been reported as a result of polypeptide grafting.31,70
More recently, the peptide grafting technique has also been usedto tailor surface hydrophilicity69 as well as for the formation offree-standing peptide architectures (i.e., capsules).73
4. Polypeptide films by the grafting-toapproach
One of the early works on the formation of polypeptide filmsby the grafting-to approach was reported by Samulski and
Scheme 2 Fabrication of polypeptide films by the (A) grafting-to and (B)grafting-from approaches.
Scheme 3 Surface grafting-to of a lipoic acid-modified poly(benzyl-L-glutamate) (PBLG-SS) on a gold substrate. Higher grafting densities wereachieved by applying an electrical field across the gold substrate, inducingelectrostatic interaction between the PBLG-SS and the negatively chargedsubstrate.
Feature Article ChemComm
Publ
ishe
d on
17
Febr
uary
201
4. D
ownl
oade
d by
The
Uni
vers
ity o
f M
elbo
urne
Lib
rari
es o
n 28
/11/
2014
21:
47:5
2.
View Article Online

This journal is©The Royal Society of Chemistry 2014 Chem. Commun., 2014, 50, 4971--4988 | 4975
co-workers (Scheme 3).74,75 In this study, the N-terminus of apoly(g-benzyl-L-glutamate) (PBLG) was modified with lipoic acid(containing a cyclic disulfide moiety) (PBLG-SS). Upon exposureto gold substrates, the disulfide moiety of the lipoic acid acts asa reactive end-group, forming a strong thiolate–gold inter-action, and thereby tethering the PBLG-SS chains onto thesurface. This chemisorption process was studied and comparedwith the physical adsorption of unmodified PBLG and PBLGmonolayers prepared by Langmuir–Blodgett (LB) deposition.From Fourier-transform infrared (FT-IR) spectroscopic analysisand water contact angle measurements it was determined thatthe unmodified PBLG lies in the plane with low graftingefficiency while PBLG-SS are tethered onto the gold surfacewith a higher grafting density. The grafted PBLG-SS chainsretain a characteristic a-helical rod-like structure and thesurface-coverage of the film depends on the solvent and extentof aggregation in solution. Further studies by Samulski et al.65
revealed the influence of electrostatic interactions on theorientation of the PBLG-SS grafts. By applying an electricalcurrent, the gold substrate surface is rendered negativelycharged (Scheme 3). Helical polypeptides such as PBLG areknown to display macroscopic dipole moments from theN-terminus to C-terminus in the presence of electrostaticpotentials. Driven by this macrodipole moment of PBLG-SS,the N-terminus of the PBLG-SS chains were electrostaticallyattracted to the gold substrate and allowed the lipoic acid end-group to efficiently form thiolate–gold interactions. By utilizingelectrostatic interactions, an improvement in the overall graft-ing efficiency was achieved, as indicated by the formation ofthicker PBLG-SS films.
In 1999, Higashi and co-workers demonstrated the reversi-ble assembly of single and double-layered PBLG on goldsubstrates based upon the helix–macrodipole interactions ofPBLG.68 In this study, the strong intermolecular dipole inter-actions between PBLG chains were utilized to assemble anti-parallel pairs of rod-like PBLG on a gold substrate. This workrevealed the importance of the helical structure towards theassembly of double-layered PBLG. For instance, in a helicogenicsolvent (e.g., CHCl3), PBLG retains its helical structure andtherefore, lead to the formation of the double-layered structure.However, in a non-helicogenic solvent (e.g., DMSO), only mono-layers were formed as a result of the lower helix–macrodipolemoment.
To further elucidate the principles governing the self-assembly of modified PBLG, Williams and Gupta adopted adifferent synthetic route towards disulfide-modified PBLG.76,77
Using N-lipoyl-1,3-diaminopropane as an initiator, NCA-ROP ofbenzyl-L-glutamate NCA (BLG-NCA) afforded disulfide-modifiedPBLG (SS-PBLG) with a free N-terminus (Scheme 4). In thisapproach, the C-terminus of the PBLG is located adjacent to thedisulfide moieties and therefore, the macrodipole moment is inthe opposite direction to the previously reported systems.76
Surface plasmon resonance spectroscopy revealed that the highmolecular weight polypeptides (Mw: 27 kDa, Mw/Mn: 1.22) self-assembled rapidly on gold substrates (within minutes), andthat the surface coverage was affected by the assembly time,
PBLG molecular weight and solvent.76,77 Their studies alsoconcluded that physisorbed PBLG (non-specific hydrophobicinteractions) creates a barrier to the overall assembly process.
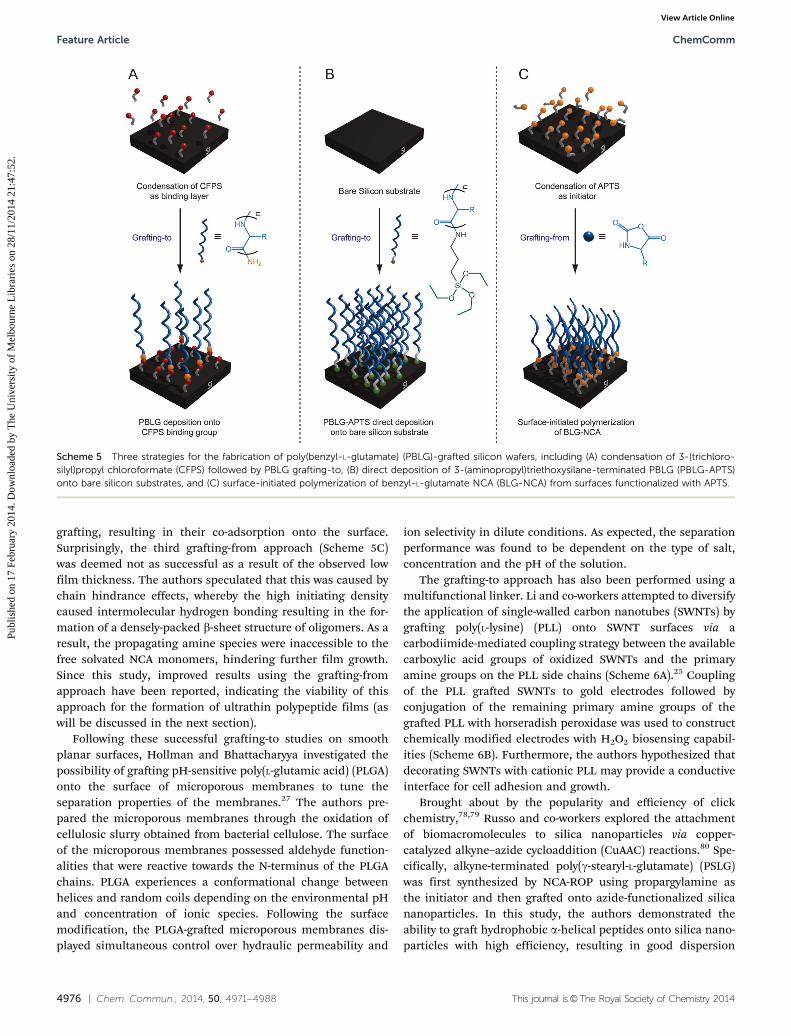
In comparison to the organosulfide compounds on gold,Chang and Frank utilized functional silanes on planar siliconsubstrates, and compared three strategies for the preparationof surface-grafted PBLG.61 In their first approach, 3-(trichloro-silyl)propyl chloroformate (CFPS) was attached to the substratethrough the reaction between the surface silanol moieties andchlorosilane groups to generate a chloroformate decoratedsurface. Subsequently, PBLG was grafted-to the surface throughreaction of the chloroformate groups with the terminal aminogroups of the PBLG (Scheme 5A). In the second approach,3-(aminopropyl)triethoxysilane (APTS) was utilized as an initia-tor in NCA-ROP to form a PBLG with APTS end-group (PBLG-APTS), which was then grafted-to silicon wafers (Scheme 5B).The third approach involved the functionalization of siliconwafers with APTS, followed by surface-initiated NCA-ROP ofBLG-NCA (Scheme 5C). Overall, all three approaches success-fully yielded PBLG brushes on the surfaces. However, elucida-tion of the nanostructures of the grafted polypeptide films wascomplicated by the presence of inter- and intramolecularinteractions. Their first approach (Scheme 5A) was significantlyaffected by substrate–polypeptide and intermolecular dipoleinteractions resulting from the PBLG chains lying parallel tothe surface, ultimately resulting in films with low graftingdensity. Although this orientation is similar to non-covalentfilms fabricated by the LB technique, the chemical stability ofthe films under ultrasonication in chloroform proved that thePBLG chains are covalently tethered to the surface. On the otherhand, the surface condensation of PBLG-APTS resulted in ahigher grafting density (Scheme 5B), which was attributed tothe oligomerization of the PBLG-APTS in solution prior to
Scheme 4 Synthetic route towards the formation of rod-like disulfide-containing PBLG (SS-PBLG) with a macrodipole moment.
ChemComm Feature Article
Publ
ishe
d on
17
Febr
uary
201
4. D
ownl
oade
d by
The
Uni
vers
ity o
f M
elbo
urne
Lib
rari
es o
n 28
/11/
2014
21:
47:5
2.
View Article Online
4976 | Chem. Commun., 2014, 50, 4971--4988 This journal is©The Royal Society of Chemistry 2014
grafting, resulting in their co-adsorption onto the surface.Surprisingly, the third grafting-from approach (Scheme 5C)was deemed not as successful as a result of the observed lowfilm thickness. The authors speculated that this was caused bychain hindrance effects, whereby the high initiating densitycaused intermolecular hydrogen bonding resulting in the for-mation of a densely-packed b-sheet structure of oligomers. As aresult, the propagating amine species were inaccessible to thefree solvated NCA monomers, hindering further film growth.Since this study, improved results using the grafting-fromapproach have been reported, indicating the viability of thisapproach for the formation of ultrathin polypeptide films (aswill be discussed in the next section).
Following these successful grafting-to studies on smoothplanar surfaces, Hollman and Bhattacharyya investigated thepossibility of grafting pH-sensitive poly(L-glutamic acid) (PLGA)onto the surface of microporous membranes to tune theseparation properties of the membranes.27 The authors pre-pared the microporous membranes through the oxidation ofcellulosic slurry obtained from bacterial cellulose. The surfaceof the microporous membranes possessed aldehyde function-alities that were reactive towards the N-terminus of the PLGAchains. PLGA experiences a conformational change betweenhelices and random coils depending on the environmental pHand concentration of ionic species. Following the surfacemodification, the PLGA-grafted microporous membranes dis-played simultaneous control over hydraulic permeability and
ion selectivity in dilute conditions. As expected, the separationperformance was found to be dependent on the type of salt,concentration and the pH of the solution.
The grafting-to approach has also been performed using amultifunctional linker. Li and co-workers attempted to diversifythe application of single-walled carbon nanotubes (SWNTs) bygrafting poly(L-lysine) (PLL) onto SWNT surfaces via acarbodiimide-mediated coupling strategy between the availablecarboxylic acid groups of oxidized SWNTs and the primaryamine groups on the PLL side chains (Scheme 6A).25 Couplingof the PLL grafted SWNTs to gold electrodes followed byconjugation of the remaining primary amine groups of thegrafted PLL with horseradish peroxidase was used to constructchemically modified electrodes with H2O2 biosensing capabil-ities (Scheme 6B). Furthermore, the authors hypothesized thatdecorating SWNTs with cationic PLL may provide a conductiveinterface for cell adhesion and growth.
Brought about by the popularity and efficiency of clickchemistry,78,79 Russo and co-workers explored the attachmentof biomacromolecules to silica nanoparticles via copper-catalyzed alkyne–azide cycloaddition (CuAAC) reactions.80 Spe-cifically, alkyne-terminated poly(g-stearyl-L-glutamate) (PSLG)was first synthesized by NCA-ROP using propargylamine asthe initiator and then grafted onto azide-functionalized silicananoparticles. In this study, the authors demonstrated theability to graft hydrophobic a-helical peptides onto silica nano-particles with high efficiency, resulting in good dispersion
Scheme 5 Three strategies for the fabrication of poly(benzyl-L-glutamate) (PBLG)-grafted silicon wafers, including (A) condensation of 3-(trichloro-silyl)propyl chloroformate (CFPS) followed by PBLG grafting-to, (B) direct deposition of 3-(aminopropyl)triethoxysilane-terminated PBLG (PBLG-APTS)onto bare silicon substrates, and (C) surface-initiated polymerization of benzyl-L-glutamate NCA (BLG-NCA) from surfaces functionalized with APTS.
Feature Article ChemComm
Publ
ishe
d on
17
Febr
uary
201
4. D
ownl
oade
d by
The
Uni
vers
ity o
f M
elbo
urne
Lib
rari
es o
n 28
/11/
2014
21:
47:5
2.
View Article Online

This journal is©The Royal Society of Chemistry 2014 Chem. Commun., 2014, 50, 4971--4988 | 4977
stability in organic solvents such as tetrahydrofuran, toluene andchloroform. FT-IR spectroscopic analysis was employed to verifythat the grafted PSLG chains retained their a-helical structure.
Similarly, Gupta and co-workers investigated the grafting ofPLL on silica nanoparticles for applications in gene delivery, andas antimicrobial agents, whereby the high density of cationiccharges on the PLL side chains could be utilized for suchpurposes.81 Following the modification of silica nanoparticleswith azidopropyltriethoxysilane (AzPTS), CuAAC-mediatedgrafting-to with a propargyl-terminated poly(e-carbobenzyloxy-L-lysine) (PZLL) was conducted to generate core–shell nano-particles. Similar to the previously discussed study,80 the graftedpeptides were shown to retain their a-helical structure. Notably,the ability to graft the amphiphilic block copolypeptide poly-(L-lysine-block-L-leucine) onto colloidal silica particles was alsodemonstrated, generating particles with antimicrobial propertiesagainst gram-positive and gram-negative bacteria.
Subsequently, Gupta and co-workers investigated the graft-ing of PLL onto mesoporous silica materials utilizing a similarCuAAC approach.82 This study revealed that a reasonablegrafting density could be achieved and that the resulting hybridmaterials retained their porosity after conjugation. This workrepresents an early example where the grafting-to techniquewas employed to modify the properties of mesoporous
materials; such hybrid materials are promising candidates forapplications in drug-delivery.
Gupta and co-workers have also reported the pH-dependentself-assembly of PLGA-grafted silica nanoparticles in combi-nation with an ice templating technique to form 3D macro-porous materials that possess interesting lamellar structureswith fishbone-type architectures (Scheme 7).24 The stiffness ofthe macroporous structures could be tuned by varying the PLGAmolecular weight and the size of the silica nanoparticles. Inter-estingly, the electrostatic repulsion between the carboxylate sidegroups of PLGA results in the disassembly of the macroporousscaffolds upon contact with water. To improve the mechanicalstability of these structures, the particles were stabilized by cross-linking with poly(ethyleneimine) (PEI). Hybrid macroporousmaterials of this type display potential for the development ofbiocompatible 3D scaffolds in the tissue engineering field.
5. Polypeptide films by the grafting-fromapproach
Although grafting-to is a versatile approach for the formation ofnumerous functional materials, this approach is usually limited bylow grafting efficiency resulting from inherent steric hindrances. Analternative approach towards polypeptide films is the grafting-fromapproach involving surface-initiated NCA-ROP. The grafting-fromapproach relies upon the anchoring of initiator (macro)moleculesonto substrates followed by NCA-ROP to form polypeptide brushes.
Scheme 6 (A) Dicyclohexylcarbodiimide (DCC)-mediated grafting ofpoly(L-lysine) (PLL) onto single-walled carbon nanotubes (SWNT) and (B)the surface immobilization of SWNT-PLL onto a gold electrode followedby 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI)-mediated cou-pling of horseradish peroxidase (HRP) to afford a H2O2 responsive electrode.
Scheme 7 Preparation of PLGA-based macroporous structure by graftingalkyne-terminated PLGA onto silica particles followed by ice templating(adapted with permission from r2011 American Chemical Society).
ChemComm Feature Article
Publ
ishe
d on
17
Febr
uary
201
4. D
ownl
oade
d by
The
Uni
vers
ity o
f M
elbo
urne
Lib
rari
es o
n 28
/11/
2014
21:
47:5
2.
View Article Online

4978 | Chem. Commun., 2014, 50, 4971--4988 This journal is©The Royal Society of Chemistry 2014
The high density of peptide brushes achievable via this approach isexpected to enhance intermolecular interactions and impartimproved properties to the resulting films. To date, amine-initiators or metal catalysts have been predominantly employedfor the grafting-from technique on both planar and nonplanarsurfaces to form unique hybrid materials.
5.1 Surface immobilization of initiators
Central to the grafting-from approach is the immobilization ofinitiators onto the surface of the substrate. Ideally, initiation sitesshould be located at controlled intervals to allow the graftedpolypeptide chains to adopt their secondary or higher-orderedstructure. Common methods for immobilizing initiators include:(a) self-assembled monolayers (SAMs) on gold and silicon-basedsubstrates; (b) plasma treatment of substrates (e.g., polymericmembranes), and; (c) direct chemical modification of substratesurfaces (e.g., silica, poly(styrene) and carbon nanotube).
A common theme between these immobilization techniquesis the introduction of primary amines as the initiating groups.While primary amines are good nucleophilic initiators, con-trolling the initiation and propagation remains a challenge dueto the coexisting pathways of NCA-ROP: the ‘normal aminemechanism’ (NAM) and the ‘activated monomer mechanism’(AMM).22,34 Initiation by NAM begins with the nucleophilicattack of the surface-bound amine initiator to an NCA ringwhich yield a free amine group to promote subsequent propa-gation (Scheme 8A). This mechanism has a fast initiation rateand offers good control over end group fidelity and molecularweight distribution. However, the molecular weight distribu-tion also depends on other factors such as solvent impurities,NCA purity, initiator nucleophilicity, temperature and thepressure at which the ROP is conducted.34 In general, thismechanism of NCA-ROP is favored for the grafting-fromapproach as it leads to the formation of polypeptides brushesthat are covalently tethered to the surface.
In the AMM, acidic N–H proton is abstracted from the NCAring under basic condition leading to the formation of NCA
anion (Scheme 8B).23 The nucleophilic ionized NCA then initiateROP to form polypeptide chains. Polymerizations following thismechanism experiences relatively lower initiation rate and typi-cally results in polypeptide chains with high molecular weightdistribution and regarded as ‘uncontrolled’.34 Furthermore,AMM leads to polymerization in solution which competes withthe polymerization occurring on the surface via NAM. Thus, thesuppression of the AMM is crucial to reduce the formation of‘non-grafted’ peptides. Unfortunately, since both AMM and NAMoccur simultaneously during grafting-from with primary amineinitiators, the suppression of solution polymerization is still oneof the greatest challenges facing the grafting-from approach. Formore detailed information on the NCA-ROP mechanisms thereader is referred to other recent review articles.22,23,34,35
The most common approach for immobilizing initiatorsinvolves the formation of SAMs, particularly with the use oforganosulfide/thiol and alkylsilane compounds on gold andsilicon-based substrates, respectively. However, as the monolayersare often densely packed, the area occupied by the initiating sitesis smaller than the required cross-sectional area of peptidehelices.22,64 Therefore, a number of studies have explored thepossibility of controlling the space between initiating sites toallow the grafting chains to adopt their native secondary structure.
Whitesell and Chang investigated the effect of spacingbetween initiating sites on the growth of poly(alanine) andpoly(phenylalanine) (PPA) brushes from gold and indium–tin–oxide substrates.64 The diameter of helical peptides (ca. 9 Å forpoly(alanine)) is typically larger than the interatomic space ofgold atoms (4.1 Å). As such, the ideal initiating sites needs to bespaced at least 9 Å apart to allow the peptide chains to adopthelical structures. Therefore, the authors designed the amino-trithiol tripod 1 (Scheme 9), with a cross-sectional area slightlyabove the 9 Å threshold, and tethered it to a gold surfacethrough thiolate–gold interactions. The surface immobilizedtripod 1 was found to effectively initiate the polymerization ofalanine-NCA (Ala-NCA) to afford poly(alanine) grafts with helicalstructure. The authors then postulated the need for initiating sites
Scheme 8 (A) ‘Normal amine mechanism’ (NAM) results in the formation of surface-grafted polypeptide while (B) ‘Activated monomer mechanism’(AMM) leads to the formation of non-grafted polypeptide in solution.
Feature Article ChemComm
Publ
ishe
d on
17
Febr
uary
201
4. D
ownl
oade
d by
The
Uni
vers
ity o
f M
elbo
urne
Lib
rari
es o
n 28
/11/
2014
21:
47:5
2.
View Article Online

This journal is©The Royal Society of Chemistry 2014 Chem. Commun., 2014, 50, 4971--4988 | 4979
with larger cross-sectional area (i.e., increased spacing) for thepolymerization of amino acids with longer/bulkier side chains(e.g., phenylalanine). To increase the space between initiatingsites, a tripod 1-immobilized gold surface was initially treatedwith chloroacetyl chloride to form tris-a-chloroamide, and thenexposed to more tripod 1 molecules to form a second layer oftripod 1 (as depicted in Scheme 9). The formation of this doublelayered system increased the distance between the initiatingamine groups to ca. 15 Å and allowed for the formation of graftedpoly(phenylalanine) with a high degree of helicity.
Similarly, Kinoshita and co-workers controlled the spatialarrangement of initiators on gold substrates by preparing mixedcomposition SAMs of 11-amino-1-undecanethiol (C11N) and butyl-disulfide (C4) at specified molar ratios.83 At a C11N : C4 ratio of1 : 4.5, the lateral distance between the primary amine groups wasdetermined to be ca. 20 Å and therefore, allowed for the formationof poly(leucine) grafts that adopted helical structures with lateraldimensions of ca. 15 Å. Using mixed composition SAMs, the sameauthors also fixed the number of initiating amine groups on goldnanoparticles for the polymerization of g-methyl-L-glutamate NCA(MLG-NCA).84 The copolymerization of MLG-NCA and BLG-NCAfollowed by solvent-dependent aggregation of the coated nano-particles was also demonstrated. Another excellent example ofmixed composition SAMs is the use of 1-bromo-11-(trichlorosilyl)-undecane and 1-(trichlorosilyl)undecane on silicon substratesreported by Menzel and co-workers.60 The bromo groups weresubstituted for azido groups and subsequently reduced to primaryamines before being used as NCA-ROP initiators.
A number of proposed applications for surface-grafted poly-peptide films such as optoelectronic devices, molecular electro-nics, and biosensors demand the ability to control thecomposition and pattern of polypeptide films in micrometerscale. This can be achieved through the controlled immobiliza-tion of initiators via the micropatterning SAM approach.
To date, patterned SAMs have been produced by using eitherconventional photolithography85 or microcontact printing(mCP).86 Braun and co-workers employed the mCP process ongold substrates to immobilized 12-mercaptododecylaminewithin well-defined areas (hexagonal motif).86 As analyzed bySEM and AFM, subsequent ROP of BLG-NCA allows for theformation of patterned PBLG films resembling the geometricfeatures introduced during the mCP process. The reported filmthickness by AFM was determined to be 32 nm and FT-IRconfirmed the a-helical secondary structure. More recently,Wang and Chang have also combined conventional photolitho-graphy and surface-initiated vapor deposition polymerization(SI-VDP) of NCAs to produce a multilevel pattern consisting ofPMLG and PMLG-b-PBLG (Fig. 1).87 In their synthetic strategy,
Scheme 9 Surface-initiated NCA-ROP of alanine-NCA and phenylalanine-NCA from single or double layer of surface-deposited aminotrithiol initiator.
Fig. 1 AFM (A) topographic image and (B) cross-sectional profile of patternedsurface-grafted PMLG and PMLG-b-PBLG prepared by vapor silanization ofAPTS and SI-VDP (adapted with permission from r2003 Wiley VCH).
ChemComm Feature Article
Publ
ishe
d on
17
Febr
uary
201
4. D
ownl
oade
d by
The
Uni
vers
ity o
f M
elbo
urne
Lib
rari
es o
n 28
/11/
2014
21:
47:5
2.
View Article Online

4980 | Chem. Commun., 2014, 50, 4971--4988 This journal is©The Royal Society of Chemistry 2014
the authors first deposit a photoresist layer (stripes 15 mm wide)on clean silicon wafers followed by the vapor silanization ofAPTS onto the remaining uncovered area. Upon removal of thephotoresist layer and subsequent ROP of MLG-NCA, patternedPMLG film was obtained. Then, the authors used the photoresiststripes to cover portions of the PMLG films while exposing theuncovered regions to BLG-NCA to allows for chain extension inthese regions. These studies thus demonstrate the feasibility offabricating patterned polypeptide interfaces via the strategicimmobilization of initiator and NCA-ROP.
For polymeric membranes, plasma treatment (also known asglow discharge treatment) has been a popular method for theintroduction of primary amine groups. For instance, Imanishiand co-workers functionalized a polymer membrane made upof poly(tetrafluoroethylene)/poly(ethylene) by plasma treatmentusing a high-frequency modulator in the presence of ammoniagas at 0.05 Torr.71 It was found that the number of aminegroups produced on the surface correlated to the plasmatreatment time. Similarly, Seta and co-workers functionalizedmicroporous poly(propylene) membranes by exposing the sur-face to ammonia plasma, and determined the increase innitrogen content (primary amine groups) with XPS.72 In addi-tion, the increase in hydrophilicity of the membrane wasdetermined by measuring the water contact angle.
Direct chemical modification of the native surface function-ality also serves as a valid approach to introduce NCA-ROPinitiators. For example, Brinson and co-workers reported twoprotocols for the modification of the carboxyl groups present onthe surface of oxidized SWNTs to primary amines.88 The firstprotocol involved the reaction of the carboxylic acid groups withethylenediamine to introduce amine groups via the formation ofamide bonds. In their second approach, the carboxyl groupswere first reduced to hydroxymethyl moieties before theirsubsequent conversion into aminomethyl groups. Chen andco-workers introduced free amine groups on multi-walled carbonnanotubes (MWNTs) by firstly creating carboxylic acid groups onthe surface through oxidation in the presence of concentratednitric acid and sulfuric acid.89 The carboxylic acid groups werethen converted into acylchlorides and eventually into primaryamines after the addition of excess ethylenediamine. Similarwork by Hua and co-workers used 1,6-diaminohexane in place ofethylenediamine.31
5.2 Surface-initiated NCA-ROP on planar surfaces
Early studies by Menzel and co-workers investigated the poly-merization of BLG-NCA from functionalized silicon wafers atroom temperature over 14 days.60 Using mixed compositionSAMs, the number of initiating amine groups on the surfacewas varied and a correlation was observed between initiatingamine groups and the surface morphology and the thickness ofthe resulting PBLG films. As a result, it was determined thatfilms with minimum surface roughness were obtained withSAMs composed of 40% initiator. Increasing the initiatorconcentration beyond this point only increased the surfaceroughness of the resultant films. Notably, since the orientationof the grafted polypeptides was yet to be determined during the
time of the study, the authors believed that interpreting theroughness values with respect to polypeptide orientationswould be speculative.
Jaworek and co-workers investigated the electromechanicalproperties of 15 nm thick PBLG films fabricated by the grafting-from technique on flat aluminum surfaces.90 By studying thechange in film thickness under the influence of an electricalfield, an inverse-piezoelectric effect was observed, indicatingthat the grafted PBLG chains adopt parallel arrangements.Furthermore, it was determined that despite the low inverse-piezoelectric coefficient of the grafted polypeptides, the voltagesensitivity exhibited is comparable to commercial piezoelectricmaterials, which highlights the potential to grow piezoelectric-active films directly on various electrodes and substrates.
Grafting-from in solution experiences extensive chain-transfer or termination side reactions, which resulted in poly-peptide grafts with low degree of polymerization and largeproportion of NCA monomer remains unreacted in solution.As an alternative, Wieringa and Schouten investigated thefeasibility and efficiency of melt surface-initiated NCA-ROP.91
In this study, silicon wafers were functionalized with APTS andthen a layer of MLG-NCA was deposited by spin-coating. Thecoated substrates were then heated to a temperature above themonomer’s melting point (i.e., 105 1C). The elevated tempera-tures increase the mobility of the monomers, allowing thesurface-bound amine groups to initiate ROP of the NCA mono-mers. However, NCA-monomers can undergo polymerizationvia thermal initiation at elevated temperatures. Therefore, eventhough complete monomer conversion was achieved within30 min, only 20% of the monomers are grafted onto the surface.Despite the low grafting efficiency, the grafted PMLG chainswere shown to adopt a purely helical structure while non-grafted thermally-initiated PMLG displayed both a-helical andb-sheet characteristics. In addition, control over the amount ofgrafted material was achievable by varying the frequency duringthe spin coating process.
Schouten and co-workers studied the surface-initiated ROPof BLG-NCA and MLG-NCA from APTS-functionalized siliconwafers and quartz slides prepared by vapor deposition.92–94 Forpolymerizations conducted at 40 1C in anhydrous N,N-dimethyl-formamide (DMF), polymer growth occurred in the first 5 h andthe initial monomer concentration was found to influence thefinal film thickness. High monomer concentrations and shortpolymerization periods appeared to favour the formation ofa-helical peptides oriented perpendicular to the surface. Theeffect of side-chain groups on the helix orientation was alsostudied, with grafted PMLG having a higher tilt angle (y) to thesubstrate as a result of the higher grafting density. This highergrafting density was attributed to the smaller MLG-NCA mole-cules (methyl side group), which allows for more grafted PMLGhelices to grow on the same surface area. The higher graftingdensity combined with steric hindrance and unfavorable polarinteractions was hypothesized to force the grafted PMLG helicesinto a more upright orientation. Notably, the authors alsoreported the formation of non-grafted materials that aggregatebetween and on top of the growing helices, inhibiting the
Feature Article ChemComm
Publ
ishe
d on
17
Febr
uary
201
4. D
ownl
oade
d by
The
Uni
vers
ity o
f M
elbo
urne
Lib
rari
es o
n 28
/11/
2014
21:
47:5
2.
View Article Online

This journal is©The Royal Society of Chemistry 2014 Chem. Commun., 2014, 50, 4971--4988 | 4981
propagation step. In addition, it was also demonstrated that thegrafted peptide films could be chain extended in a subsequentNCA-ROP to form surface-grafted block copolymers. For exam-ple, grafted diblock copolypeptides composed of PBLG as thefirst block and PMLG as the second block were prepared fromAPTS-functionalized silicon wafers.94 Initially, the BLG-NCApolymerization was conducted for a short period of time (2 h)to minimize the formation of non-grafted materials in solution,whilst ensuring the reactive amino end-groups are free fromundergoing any chain-transfer or termination reactions. Afterremoval of unreacted monomers, the grafted PBLG act asmacroinitiators for the subsequent polymerization of MLG-NCA. Interestingly, the importance of grafting density to theformation of block copolypeptide grafts was also highlighted.Based upon XPS analysis, it was found that within an hour ofBLG-NCA polymerization the maximum grafting density had yetto be achieved, and by introducing MLG-NCA at this stageresulted in the formation of PMLG helices in between thepreformed PBLG chains (initiated by the remaining availableAPTS amine groups on the surface).
Following these studies, Schouten and co-workers investi-gated the reversible manipulation of helix screw sense ofsurface-grafted polypeptides.95 The helical sense of certainpolypeptides, such as poly(aspartate) derivatives, depends onthe structure of the ester side chain as well as external stimulisuch as temperature and solvent. For example, polymerizationof poly(b-phenethyl-L-aspartate) (PPLA) from primary amine-functionalized silicon and quartz substrates results in theformation of grafted polypeptides with right-handed a-helicalconformations. Upon annealing at 150 1C for 30 min, a helixscrew inversion occurred and the grafted PPLA chains adopteda left-handed p-helical orientation. This thermally-inducedinversion remains stable in the solid-state, despite any furtherchanges in the external temperature. However, upon immer-sion of the films into a helicogenic solvent (i.e., CHCl3), thegrafted PPLA reverts back to its original right-handed a-helicalconformation. These helix sense inversion cycles were repeata-ble over successive annealing and solvent treatment steps. Topreserve the helical orientation of surface-grafted polypeptides,the same authors also devised a chemical cross-linking proce-dure which involves the ROP of g-4-vinylbenzyl-L-glutamateNCA. Subsequently, the 4-vinylbenzyl groups were polymerizedusing radical initiator and thereby cross-linking the surface-grafted peptide helices.33
Concerned with the low degree of polymerization and theformation of non-grafted materials as a result of solventimpurities, Chang and Frank developed a solvent-free techni-que based on vapor deposition-polymerization (VDP) of NCAmonomers on APTS-functionalized silicon substrates.62 Thesolvent-free nature of this technique reduced the oligomeriza-tion of NCA monomers normally resulting from solventimpurities. Meanwhile, the vacuum reaction condition (0.02–0.04 Torr) allowed for the NCA vaporization to occur at lowertemperatures. More importantly, in contrast to the melt poly-merization technique91 where NCA monomers are always indirect contact with the initiator layer, the vapor reactants in the
VDP technique remain mobile in the gaseous phase until theycondenses onto the surface. The VDP method potentially offersan easy approach to tune the resultant film properties byvarying the local NCA concentration (controllable by changingthe temperature, pressure, substrate–monomer separation dis-tance and reaction time). In a model study using vaporizedBLG-NCA, key findings include the ability to obtain films withhomogenous surface morphologies (surface roughness ofca. 3.5 nm), tunable thickness between 4 to 40 nm within 4 h,and grafted PBLG chains with pure a-helical conformations.
Further improvements in the VDP method were introducedin a follow-up study by Wang and Chang by redesigning thereaction chamber and optimizing reaction conditions, such asmonomer concentrations, substrate temperature and reactiontime.96 For instance, in the optimized system (95 1C monomerevaporating temperature, 0.1 Pa and 75 1C substrate temperature),187 nm PBLG films were fabricated in 30 min. The versatilityand robustness of this technique was further demonstratedby the formation of homopolypeptide and block copolypeptidethin films composed of PZLL, PMLG, PPA and poly(b-benzyl-L-aspartate) (PBLA). In later studies, the difference in surfacemorphology between chemisorbed (grafted) and physisorbed(non-grafted) films were investigated.92 These investigationsconcluded that the association between individual polymerchains is the most dominant factor influencing the morphologyof vapor-deposited poly(amino acid) films.
Higashi and co-workers described the surface-initiated poly-merization of BLG-NCA in the presence of pre-grafted PBLGchains on gold surfaces.67 Initially, modified PBLGs with dis-ulfide moieties at the C- and N-terminus (termed PBLG-C-SSand PBLG-N-SS, respectively) were synthesized (refer toSchemes 3 and 4) and then grafted-to the gold surfaces result-ing in PBLG films with opposite macrodipole moments(Scheme 10). Subsequently, mixed composition SAMs wereused to distribute free amine moieties (initiators) on theremaining uncovered surfaces. Upon exposure to BLG-NCA itwas found that the rate of grafting-from polymerization wassignificantly influenced by the direction of the macrodipolemoment of the pre-adsorbed PBLG helices. In the presence ofpre-grafted PBLG-N-SS, the rate of NCA-ROP increased by ten-fold when compared to that in the absence of pre-adsorbedPBLG. This increase is polymerization rate was attributed to thehelix macrodipole moment of PBLG-N-SS, which is antiparallelto the polymerization direction of PBLG chains from the sur-face and therefore, favours the formation of PBLG with a higherdegree of polymerization (DP: 95). In contrast, the macrodipolemoment of PBLG-C-SS is parallel to the growing PBLG chainsand creates an energetic barrier, which resulted in the for-mation of PBLG helices with low molecular weight (DP: 15).
Recently, Klok and co-workers investigated the suitability ofthe grafting-from approach to create non-fouling peptidefilms.26 In this study, oligo(ethyleneglycol) modified L-lysineNCA was polymerized from APTS-functionalized silicon, quartzand glass substrates at 50 1C (48–72 h) in a THF/DMF (5 : 1)solvent mixture. Successful polymerization was confirmed byXPS analysis, while circular dichroism measurements confirmed
ChemComm Feature Article
Publ
ishe
d on
17
Febr
uary
201
4. D
ownl
oade
d by
The
Uni
vers
ity o
f M
elbo
urne
Lib
rari
es o
n 28
/11/
2014
21:
47:5
2.
View Article Online

4982 | Chem. Commun., 2014, 50, 4971--4988 This journal is©The Royal Society of Chemistry 2014
the formation of stable a-helical conformations between pH 4and 9. The nonspecific adsorption of fluorescently labeledbovine serum albumin (BSA) and fibrinogen (FBG) onto thecoated substrates was then evaluated. After exposure to theseproteins, the fluorescent intensities of non-coated and coatedsubstrates were compared and the authors observed a significantreduction in fluorescence intensity (more than 85% reduction)indicative of the low-fouling behavior of the coated substrates.Notably, the authors also observed improvement of the non-fouling characteristics with longer oligo(ethyleneglycol) sidechains.
5.3 Surface-initiated NCA-ROP on non-planar surfaces
Given the success of surface-initiated NCA-ROP on planarsurfaces, the grafting-from technique has also been employedon a range of non-planar surfaces including silica-based (col-loidal) particles, carbon nanotubes (CNTs), magnetite nano-particles and mesoporous substrates.
The earliest example of peptide grafting from colloidal silicaparticles was reported by Hamann and co-workers in 1974,97
whereby silica particles with covalently-bound aminophenylgroups were employed to initiate NCA-ROP of L-alanine NCA(Ala-NCA) and L-leucine NCA (Leu-NCA) at 15 1C in dioxane.Their studies concluded that most of the polymer content wascovalently bound to the silica particles and the separation ofthe grafted polymers from the underlying particles was notpossible. Sone and co-workers reported the polymerization ofMLG-NCA from carbon black surfaces functionalized withdifferent amines.98 Initially, amino, methylamino or dimethyl-amino groups were introduced through reaction of the corre-sponding ethylenediamine derivatives with acyl azide groupson the carbon black surface. Polymerization of MLG-NCA wasthen conducted at 40 1C in dioxane for up to 4 days. It wasfound that as a result of better initiation, higher graftingefficiencies were observed for carbon black functionalized withprimary amine initiators. These studies conclusively provedthat covalently-bonded peptides could be obtained by the sur-face grafting-from technique. However, as a result of thelimited synthetic tools and characterization methods available,the uniformity of particles could not be determined.
Placing greater emphasis on particle uniformity and char-acterization, Fong and Russo employed the classical Stobermethod to prepare monodisperse silica particles (Scheme 11).59
After silanization with APTS, the amine-functionalized silicaparticles were used to initiate polymerization of BLG-NCA toproduce a polypeptide shell. The resulting PBLG-coated nano-particles were then analyzed by electron microscopy, dynamiclight scattering (DLS), infrared spectroscopy and thermogravi-metry (TGA). Electron microscopy images revealed aggregatesof coated particles and therefore prohibited size determination.However, based upon DLS analysis, the average radius of baresilica particles were determined to be ca. 97 nm. After poly-merization, the radius increases to 175 nm, indicating a
Scheme 10 The influence of pre-grafted PBLG chains on the surface-initiated NCA-ROP of BLG-NCA. As a result of the macrodipole moment,adsorbed PBLG-N-SS accelerated the polymerization of BLG-NCA whilePBLG-C-SS created an energetic barrier against NCA-ROP.
Scheme 11 Surface-initiated NCA-ROP of BLG-NCA from APTS-functionalized silica nanoparticle. After extensive washing in DMF, peptidechains were degrafted from the silica support.
Feature Article ChemComm
Publ
ishe
d on
17
Febr
uary
201
4. D
ownl
oade
d by
The
Uni
vers
ity o
f M
elbo
urne
Lib
rari
es o
n 28
/11/
2014
21:
47:5
2.
View Article Online

This journal is©The Royal Society of Chemistry 2014 Chem. Commun., 2014, 50, 4971--4988 | 4983
polymer shell thickness of 78 nm. Furthermore, infrared spec-tra revealed that the grafted PBLG chains adopted an a-helicalconformation, while thermogravimetric analysis showed thatca. 20% of the total mass can be ascribed to grafted PBLG.Notably, the authors concluding remarks suggest that greatercontrol over shell thickness required lower surface density ofinitiators and reduction in non-immobilized initiators. In thisstudy, the authors also described the preparation of silicacolloidal particles coated with PZLL and show the crystallineorder of the resultant core–shell particles.
Russo and co-workers reported a method that allows for thepreparation of core–shell silica–peptide hybrid materials withvariable and controllable shell growth.99 Central to this methodis the incorporation of surface passivation agent to control thedensity of amine moieties on the silica particles (i.e., initiatordensity). This was achieved by functionalizing silica particleswith a mixture of 3-(aminopropyl)trimethoxysilane (APTMS)and non-initiating methyltrimethoxysilane (MTMS), in a simi-lar approach to preparing mixed composition SAMs.60 Fromthese silica particles, sequential polymerization of BLG-NCAand carbobenzyloxy-L-lysine NCA (ZLL-NCA) was performed toprovide control over the shell thickness, as analyzed by FT-IR,XPS, DLS and transmission electron microscopy (TEM).
Building upon the fundamental knowledge accumulatedover the years of peptide grafting, Heise and co-workersreported the formation of pH-responsive polypeptide core–shellsilica nanoparticles as responsive materials, and for bioconju-gation purposes.28 In their studies, PLBG, PZLL and s-tert-butylprotected polycysteine (PtBLC) grafted shells were preparedfrom APTS-functionalized silica nanoparticles with high initiat-ing rates and grafting densities. After deprotection, these pep-tide shells bear either acid, amine or thiol functional groups,respectively, making them stimuli responsive and available forfurther chemical modifications for targeted applications. Inthis study, the authors emphasized the importance of polymer-ization parameters in relation to the size and uniformity of theresultant films. At lower temperatures (i.e., 0 1C), the relation-ship between monomer feed and the resultant film thicknessconverged towards linearity, suggesting better control overthe grafting process. This observation is in agreement withresults obtained in solution NCA-ROP.28,41,42 Furthermore, thepH-responsive behaviour of the PBLG peptide shell was demon-strated by deprotecting the benzyl ester protecting groups toobtain the pendant carboxylic acid groups of poly(L-glutamicacid) (PLGA). When the solution pH was raised from pH 2 to 10,the PLGA chains experience a conformational change fromhelices to random-coil as a result of the increased electrostaticrepulsion between the deprotonated pendant acid moieties.Based on this mechanism, the authors also demonstrated thepH-responsive behavior of the core–shell architecture by show-ing the release of entrapped rhodamine B molecules intosolution. Other than physical entrapment, the authors alsoexplored the feasibility of bioconjugation of green fluorescentprotein (GFP).
While studies have been predominantly performed on non-porous silica particles, Lunn and Shantz prepared organic–inorganic
nanocomposites composed of grafted polypeptides on orderedmesoporous silica (OMS) particles.98 OMS particles are ofinterest as a result of their controllable pore sizes (2–15 nm)and mechanical and thermal stability. In this study, nano-particle composites were formed by functionalizing OMS withAPTMS followed by the polymerization of ZLL-NCA or Ala-NCA.The porosity of the grafted nanocomposites was found to bedramatically lower than the untreated OMS as a result of thehigh grafting density of peptide chains. Furthermore, thegrafting density was tunable by varying the surface-initiatorloading, pore size, pore topology and monomer type.
Other than using mesoporous silica templates, recent workby Heise and co-workers demonstrated the grafting of BLG-NCAand ZLL-NCA from macroporous templates as a platform forbioconjugation (Scheme 12).30 Macroporous polymeric mono-liths is an interesting class of materials with high surface areaand excellent mass transport properties, which make themsuitable for applications in tissue engineering, 3D cell culture,supported organic chemistry and column filtration/separa-tion.30 In this study, high internal phase emulsion (HIPE)composed of styrene, divinylbenzene and 4-vinylbenzylaminewas first prepared in water by the aid of surfactant Span 80.After thermal polymerization at 70 1C and subsequent washingsteps, porous polymeric monoliths functionalized with primaryamine groups were obtained (polyHIPE-NH2). These primaryamine moieties were then employed for the surface-initiatedNCA-ROP of BLG-NCA and ZLL-NCA to form dense homo-genous coatings of PBLG and PZLL, respectively, as confirmedby SEM and FT-IR analysis. After deprotection of the graftedchains, the polypeptide-coated polyHIPE surfaces were pH
Scheme 12 Bioconjugation of PLL-grafted polyHIPE with enhancedgreen fluorescent protein (eGFP) to afford fluorescent macroporousmonoliths (adapted from complete citation r2012 American ChemicalSociety).
ChemComm Feature Article
Publ
ishe
d on
17
Febr
uary
201
4. D
ownl
oade
d by
The
Uni
vers
ity o
f M
elbo
urne
Lib
rari
es o
n 28
/11/
2014
21:
47:5
2.
View Article Online

4984 | Chem. Commun., 2014, 50, 4971--4988 This journal is©The Royal Society of Chemistry 2014
responsive. Furthermore, the high density of functional groupson the surface (e.g., acid groups for PLGA coatings) wasemployed for the covalent conjugation of enhanced greenfluorescent protein (eGFP) and fluorescein isocyanate (FITC).
Surface-initiated NCA-ROP has also been used to decoratemultiwalled carbon nanotubes (MWNTs). Hua and co-workersconverted oxidized MWNTs decorated with carboxylic acid groupsto primary amine-functionalized MWNTs (MWCNT-NH2) groupsthrough reaction with excess 1,6-diaminohexane.31 Followingpolymerization of ZLL-NCA from the amine functionalized MWNTand deprotection of the grafted polypeptides, the PLL-modifiedMWNT were found to be dispersible in water. This approach ishighly applicable towards improving the processability of CNTs,which are of great interest for the fabrication of molecular wires,sensors, probes and other (bio)electronic devices.
Further developments in peptide grafting from non-planarsurfaces have moved beyond the polymerization fundamentalsand started to build multi-functional materials. For example,magnetic nano- and micro-spheres possess unique qualitiessuch as superparamagnetism, high field irreversibilities, highsaturation fields and large specific surface areas. These uniqueproperties have garnered considerable attention for applicationin biomedical devices. Specifically, magnetic–silica hybrid micro-spheres have been studied for their chemical and physicalstability, as well as magnetic responsiveness. To further impartunique characteristics, Yang and co-workers prepared PBLGgrafted magnetite–silica nanoparticles (Scheme 13) via thegrafting-from approach.100 Firstly, magnetite (Fe3O4) nano-particles were prepared via a solvothermal process and thenencapsulated within a silica shell through a sol–gel reactionwith tertraethoxysilane (TEOS). Amine groups were then intro-duced via silanization with APTS followed by the surface-initiated
NCA-ROP of BLG-NCA to afford the desired hybrid materials. Oneof the key findings reported is the improvement in the dispersionof the magnetite nanoparticles following encapsulation in silicaand PBLG grafting. Despite the reported colloidal stability (morethan 3 days), separation of the magnetic nanoparticles fromsolution could be achieved in the presence of an applied magnetfield (t = 10 s), indicating the good processability of the system.
One of the drawbacks of silica coatings on magnetite parti-cles is the reduction in magnetic properties. To avoid thisproblem, Fernandez-Garcıa and co-workers devised a newstrategy for the functionalisation of magnetite nanoparticles.70
In particular, the authors employed self-polymerization ofdopamine as a bio-inspired adhesive to introduce aminegroups onto the magnetite nanoparticles, which were employedfor surface-initiated NCA-ROP of BLG-NCA. Following deprotec-tion of the grafted polypeptides, the resultant pH sensitivenanoparticles were then conjugated with the model drug pro-caine to study their potential application as contrast agents formagnetic resonance imaging (MRI) and drug delivery. Usingthe same strategy, magnetic–peptide–metal hybrid nano-particles have also been prepared.32 After the preparation ofPBLG-coated magnetite nanoparticles, catechol groups wereintroduced into the PBLG shell through aminolysis reactionwith dopamine (mediated by 4-dimethylaminopyridine). Thecatechol moieties were then used to interact with gold ionsbefore the addition of sodium borohydride (reductant) whichlead to the formation of gold nanoparticles in the PBLG shell.Subsequently, the catalytic activity of the gold-decorated hybridmagnetic nanoparticles to reduce 4-nitrophenolate ions to4-aminophenol was studied. Notably, high catalytic efficiencywas reported (conversion up to 95% in 25 min), and the hybridmaterials could be easily separated and reused over eightcatalytic cycles without significant loss of activity. This exampledemonstrates how grafted peptide chains can act as efficientplatforms for catalyst immobilization, and separation whencombined with magnetic nanoparticles.
Recently, Heise and co-workers combined polypeptidegrafting-from and click chemistry to fabricate Fe3O4 nano-particles decorated with a high surface density of mono-saccharides (i.e., galactose).29 The monodisperse glycosylatedmagnetic nanoparticles were prepared via the NCA-ROP ofg-propargyl glutamate NCA from APTS-functionalized Fe3O4
nanoparticles followed by CuAAC with azido-modified galac-tose. The resultant nanoparticles were found to be biocompa-tible and highly crystalline, making them good candidates asMRI contrast agents. It was also shown that the galactosemoieties endowed the nanoparticles with specificity towardslectin, demonstrating the potential of this synthetic strategy toproduce functional nanoparticles for targeted therapeuticdelivery.
Predominantly, studies on peptide grafting-from particleshave been focused on the formation of core–shell structures;however, free-standing architectures such as peptide capsulesare also attractive as biomedical devices. In general, peptidefilms fabricated by either the grafting-to or grafting-fromapproaches lack mechanically stable cross-linked structures,
Scheme 13 (A) Surface-initiated NCA-ROP of BLG-NCA monomers fromAPTS-functionalized hybrid magnetite–silica nanoparticles. TEM images ofthe (B) untreated magnetic hybrid nanoparticles and (C) after graftingPBLG-helices (adapted from complete citation r2011 Elsevier).
Feature Article ChemComm
Publ
ishe
d on
17
Febr
uary
201
4. D
ownl
oade
d by
The
Uni
vers
ity o
f M
elbo
urne
Lib
rari
es o
n 28
/11/
2014
21:
47:5
2.
View Article Online

This journal is©The Royal Society of Chemistry 2014 Chem. Commun., 2014, 50, 4971--4988 | 4985
which prevent the formation of free-standing architectures. Forthese reasons, Qiao and Caruso et al. developed a syntheticstrategy that employs cross-chain termination reactionsbetween growing polypeptide brushes as a new in situ cross-linking mechanism (Scheme 14).73 In this assembly system,hyperbranched PEI was immobilized onto silica particles byelectrostatic interactions to introduce free amine groups ontothe surface. BLG-NCA was then polymerized from the surface,during which the propagating amine groups undergo nucleo-philic attack at the carbonyl carbon of the benzyl-ester side-groups on adjacent polymer chains (cross-chain terminationreactions). This event led to the formation of inactive amidegroups, consequently cross-linking the grafted polymer chains.Following the deprotection of remaining benzyl protectinggroups and dissolution of the silica template (via hydrofluoricacid treatment), the cross-linked peptide chains formed free-standing architectures in the form of thin polypeptide capsules.Whereas the thickness of the capsules could be tuned byvariation of the polymerization time and initial monomerconcentration, the composition and functionality could bemodified by using a combination of different amino acidNCA derivatives. Furthermore, despite the highly cross-linkedstructure, the capsules were found to be biodegradable in thepresence of enzymes, making them good candidates for bio-medical applications.
More recently, we have also demonstrated that surface-initiated NCA-ROP is a versatile method to form polypeptidenanocoatings with tailored hydrophilicities on various organic(cotton and cellulose) and inorganic substrates (glass).69 Asbefore, PEI was deposited onto the substrates and employed asa hyperbranched multifunctional initiator. Through conscien-tious selection of hydrophobic/hydrophilic NCA monomers forthe grafting-from process, the surface wetting characteristics ofthe substrates could be tailored. Confirmed by water contactangle measurements, PLL coatings displayed hydrophilic beha-viour while poly(L-valine) coatings increased the surface hydro-phobicity of the substrates. Notably, by varying thepolymerization time of valine NCA on porous materials (i.e.,cellulose-based filter paper), the surface hydrophobicity andwater absorption rate of the materials could be tuned effec-tively, providing opportunities for the development of mem-branes with selective separation capabilities.
5.4 Surface-initiated NCA-ROP with transition metal
Living transition-metal catalyzed NCA-ROP in solution hasenabled the formation of well-defined synthetic polypep-tides.22,23,36 However, the use of transition metal catalyst insurface-initiated NCA-ROP has yet to gain popularity. Never-theless, Witte and Menzel demonstrated the feasibility ofsurface-initiated NCA-ROP using organonickel-complex initia-tors.101 In this study, commercially-available polystyrene (PS)nanoparticles were used as spherical template with either theblock co-polymerization or the alloc-amide approach (Scheme 15).In the block copolymerization approach, PS nanoparticles werefirst functionalized with NCA monomers (Scheme 15A). Thesurface-anchored NCA monomers were then reacted with anactivated Ni amido–amidate complex to create living chain ends.Excess Ni-complex was washed from the surface before theaddition of BLG-NCA to allow surface-initiated NCA-ROP. Thegrafted polypeptides were then cleaved from the resin by photo-lysis of the methylphenacyl group (UV at 365 nm) and analysed viasize exclusion chromatography, which revealed PBLG with amolecular weight of 22 kDa and polydispersity index (PDI) of1.52. This is a relatively low PDI value for a surface graftingprocess and indicates good control over the polymerization.However, this block copolymerization approach also sufferedfrom the significant formation of non-grafted polymers insolution (ca. 82%), which was attributed to the presence ofphysisorbed initiator and the potential byproducts (bromideand hydroxyl groups) formed during the initial surfacefunctionalisation steps.
To circumvent this problem, an alloc-amide approach wasadopted, in which alloc-amide moieties were introduced ontothe surface via the reaction between the N-hydroxysuccinimideester of leucine-alloc-amide with commercially available aminofunctionalized PS resin (Scheme 15B).101 Following exposure toexcess bis(cyclooctadiently)-nickel(0) (Ni(COD)2) and 1,10-phenantroline as ligand, the activated surface was then usedto polymerize BLG-NCA. This approach resulted in well-definedinitiators on surfaces and reduced the formation of free-polymer in solution (45–50%).
Scheme 14 (A) Assembly strategy for the formation of free-standingPLGA-films via the synergistic combination of surface-deposited hyper-branched macroinitiators, the grafting-from approach and cross-chaintermination reactions. (B) Differential interference contrast (DIC) micro-scopy image and (C) AFM image of PLGA-capsules obtained after 96 hpolymerization (adapted with permission from r2013 Wiley VCH). Scalebar for DIC microscope and AFM images are 5 mm and 2 mm, respectively.
ChemComm Feature Article
Publ
ishe
d on
17
Febr
uary
201
4. D
ownl
oade
d by
The
Uni
vers
ity o
f M
elbo
urne
Lib
rari
es o
n 28
/11/
2014
21:
47:5
2.
View Article Online

4986 | Chem. Commun., 2014, 50, 4971--4988 This journal is©The Royal Society of Chemistry 2014
Recently, Patton and co-workers also reported the surface-initiated NCA-ROP using the alloc-amide approach on siliconwafers.63 Silicon wafers serve as a model substrate configu-ration for biosensor platforms and other thin film applications.Central to their study was the reaction between APTS and alloc-L-leucine to form alloc-L-leucine-3-aminopropyltriethoxysilane(Aloc-ATPS). Following functionalisation of the silicon waferwith Aloc-APTS, the Ni catalyst was immobilized on the surface,and the NCA-ROP of a range of NCA monomers in DMF wasconducted. In addition to grafted homopeptides, the formationof block- and clickable copolypeptides was also demonstratedvia this nickel-mediated grafting-from approach. Notably, XPSresults indicated that the Ni species remains attached to theend-groups of the grafted polymers, which is important for theformation of grafted multiple block copolypeptides. However,the cytotoxic effects of these organonickel moieties are yet to beassessed, which may limit the potential application of thisapproach, particularly in the biomedical field.
6. Concluding remarks and futureoutlook
It is clear that surface grafting techniques hold significant potentialfor the development of nanoscale (hybrid) peptide-based
stimuli-responsive materials with tunable conformations,chemical functionalities and 3D nanostructures. These aspectsincrease the applicability of grafted peptides and are likely tolead to new avenues of research. Thus far, the breadth of thestudy in this field has been focused on the preparation ofsurface-grafted peptide films and their characterization. Cur-rent challenges in the field, such as the suppression of sidereactions and the formation of non-grafted materials, are stillto be optimized and resolved. A facile synthetic strategy toenable control over the primary sequence of grafted peptides insurface-initiated NCA-ROP would also lead to significantadvances in this field as well as biomimetics and proteomics.Although there is still a need to conduct fundamental studies toimprove and understand certain aspects, advances so far haveprovided the synthetic tools to engineer grafted polypeptidebased materials with excellent properties and foreseeable appli-cations to drug delivery, catalysis, separation technologies andtheranostics.
Given the limited number of studies conducted to date, thefull potential of peptide surface-grafting has yet to be fullyrealized. For instance, many of the naturally (and non-natural)occurring amino acids have not been investigated as buildingblocks in thin film fabrication. In addition, the secondarystructure of the polypeptide films could be harnessed tocreate new functional and hierarchically structured materials.
Scheme 15 Organonickel-mediated surface-initiated NCA-ROP of BLG-NCA from PS resin via (A) the block copolymerization and (B) alloc-amideapproaches.
Feature Article ChemComm
Publ
ishe
d on
17
Febr
uary
201
4. D
ownl
oade
d by
The
Uni
vers
ity o
f M
elbo
urne
Lib
rari
es o
n 28
/11/
2014
21:
47:5
2.
View Article Online

This journal is©The Royal Society of Chemistry 2014 Chem. Commun., 2014, 50, 4971--4988 | 4987
Also, there are still numerous unexplored applications whereimplementation of grafted polypeptides may be beneficial,such as the construction of biofriendly interfaces that mediatecellular growth and differentiation. Crucial elucidation of thefate of polypeptide films in biological environments, theirdegradation and metabolism, and potential toxicity are yet tobe reported. In addition, advanced functional hybrid filmsconsisting of synthetic polymer–peptide mixtures/conjugatesare also yet to be demonstrated. Therefore, in the future,advancements in the field will undoubtedly exploit novel aminoacids and extend the potential functionalities of the surface-grafted materials to form new materials with unique structuresand properties. Undoubtedly, peptide grafting approaches havea bright future with great potential in the fields of bionano-technology, regenerative nanomedicine, tissue engineering,(bio)sensors, (bio)nanocoatings, diagnostic devices, catalysisand separation technologies.
References1 A. L. Becker, A. P. R. Johnston and F. Caruso, Small, 2010, 6,
1836–1852.2 P. T. Hammond, Mater. Today, 2012, 15, 196–206.3 B. B. Hsu, S. Y. Wong, P. T. Hammond, J. Chen and A. M. Klibanov,
Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 61–66.4 R. Chandrawati, L. Hosta-Rigau, D. Vanderstraaten, S. A. Lokuliyana,
B. Stadler, F. Albericio and F. Caruso, ACS Nano, 2010, 4, 1351–1361.5 B. Stadler, R. Chandrawati, A. D. Price, S.-F. Chong, K. Breheney,
A. Postma, L. A. Connal, A. N. Zelikin and F. Caruso, Angew. Chem.,Int. Ed., 2009, 48, 4359–4362.
6 L. Hosta-Rigau, B. Stadler, Y. Yan, E. C. Nice, J. K. Heath,F. Albericio and F. Caruso, Adv. Funct. Mater., 2010, 20, 59–66.
7 M. M. Stevens and J. H. George, Science, 2005, 310, 1135–1138.8 G. M. Whitesides, Nat. Biotechnol., 2003, 21, 1161–1165.9 T. Boudou, T. Crouzier, K. Ren, G. Blin and C. Picart, Adv. Mater.,
2010, 22, 441–467.10 P. K. H. Ho, D. S. Thomas, R. H. Friend and N. Tessler, Science,
1999, 285, 233–236.11 Y. Xia and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37,
550–575.12 D. Mertz, J. Hemmerle, F. Boulmedais, J.-C. Voegel, P. Lavalle and
P. Schaaf, Soft Matter, 2007, 3, 1413–1420.13 P. Lavalle, C. Gergely, F. J. G. Cuisinier, G. Decher, P. Schaaf,
J. C. Voegel and C. Picart, Macromolecules, 2002, 35, 4458–4465.14 L. A. Connal, Q. Li, J. F. Quinn, E. Tjipto, F. Caruso and G. G. Qiao,
Macromolecules, 2008, 41, 2620–2626.15 M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Muller, C. Ober,
M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban,F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010,9, 101–113.
16 J. Y. Kim, K. Lee, N. E. Coates, D. Moses, T.-Q. Nguyen, M. Danteand A. J. Heeger, Science, 2007, 317, 222–225.
17 A. S. Arico, P. Bruce, B. Scrosati, J.-M. Tarascon and W. vanSchalkwijk, Nat. Mater., 2005, 4, 366–377.
18 H. Zou, S. Wu and J. Shen, Chem. Rev., 2008, 108, 3893–3957.19 T. Ikai and Y. Okamoto, Chem. Rev., 2009, 109, 6077–6101.20 R. Jones, Nat. Nanotechnol., 2008, 3, 699.21 R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149–2154.22 N. Hadjichristidis, H. Iatrou, M. Pitsikalis and G. Sakellariou,
Chem. Rev., 2009, 109, 5528–5578.23 H. R. Kricheldorf, Angew. Chem., Int. Ed., 2006, 45, 5752–5784.24 M. Kar, M. Pauline, K. Sharma, G. Kumaraswamy and S. Sen Gupta,
Langmuir, 2011, 27, 12124–12133.25 Y. Zhang, J. Li, Y. Shen, M. Wang and J. Li, J. Phys. Chem. B, 2004,
108, 15343–15346.26 J. Wang, M. I. Gibson, R. Barbey, S.-J. Xiao and H.-A. Klok,
Macromol. Rapid Commun., 2009, 30, 845–850.
27 A. M. Hollman and D. Bhattacharyya, Langmuir, 2002, 18,5946–5952.
28 T. Borase, M. Iacono, S. I. Ali, P. D. Thornton and A. Heise, Polym.Chem., 2012, 3, 1267–1275.
29 T. Borase, T. Ninjbadgar, A. Kapetanakis, S. Roche, R. O’Connor,C. Kerskens, A. Heise and D. F. Brougham, Angew. Chem., 2013,125, 3246–3249.
30 F. Audouin, M. Fox, R. Larragy, P. Clarke, J. Huang, B. O’Connorand A. Heise, Macromolecules, 2012, 45, 6127–6135.
31 J. Li, W.-D. He, L.-P. Yang, X.-L. Sun and Q. Hua, Polymer, 2007, 48,4352–4360.
32 G. Marcelo, A. Munoz-Bonilla and M. Fernandez-Garcıa, J. Phys.Chem. C, 2012, 116, 24717–24725.
33 J. Luijten, D. Y. Groeneveld, G. W. Nijboer, E. J. Vorenkamp andA. J. Schouten, Langmuir, 2007, 23, 8163–8169.
34 J. Huang and A. Heise, Chem. Soc. Rev., 2013, 42, 7373–7390.35 H. Lu, J. Wang, Z. Song, L. Yin, Y. Zhang, H. Tang, C. Tu, Y. Lin and
J. Cheng, Chem. Commun., 2014, 50, 139–155.36 T. J. Deming, Nature, 1997, 390, 386–389.37 H. Lu and J. Cheng, J. Am. Chem. Soc., 2007, 129, 14114–14115.38 H. Lu and J. Cheng, J. Am. Chem. Soc., 2008, 130, 12562–12563.39 I. Dimitrov and H. Schlaad, Chem. Commun., 2003, 2944–2945.40 T. Aliferis, H. Iatrou and N. Hadjichristidis, Biomacromolecules,
2004, 5, 1653–1656.41 G. J. M. Habraken, M. Peeters, C. H. J. T. Dietz, C. E. Koning and
A. Heise, Polym. Chem., 2010, 1, 514–524.42 G. J. M. Habraken, K. H. R. M. Wilsens, C. E. Koning and A. Heise,
Polym. Chem., 2011, 2, 1322–1330.43 I. Conejos-Sanchez, A. Duro-Castano, A. Birke, M. Barz and
M. J. Vicent, Polym. Chem., 2013, 4, 3182–3186.44 A. Sulistio, P. A. Gurr, A. Blencowe and G. G. Qiao, Aust. J. Chem.,
2012, 65, 978–984.45 A. C. Engler, H.-i. Lee and P. T. Hammond, Angew. Chem., Int. Ed.,
2009, 48, 9334–9338.46 S. J. Lam, A. Sulistio, K. Ladewig, E. H. H. Wong, A. Blencowe and
G. G. Qiao, Aust. J. Chem., 2014, DOI: 10.1071/CH13525.47 A. Mata, Y. Geng, K. J. Henrikson, C. Aparicio, S. R. Stock,
R. L. Satcher and S. I. Stupp, Biomaterials, 2010, 31, 6004–6012.48 C. W. Bond, N. Angeloni, D. Harrington, S. Stupp and
C. A. Podlasek, J. Sex. Med., 2013, 10, 730–737.49 R. C. Claussen, B. M. Rabatic and S. I. Stupp, J. Am. Chem. Soc.,
2003, 125, 12680–12681.50 M. Byrne, D. Victory, A. Hibbitts, M. Lanigan, A. Heise and
S.-A. Cryan, Biomater. Sci., 2013, 1, 1223–1234.51 M. Takaki, R. Asami, Y. Hanada and N. Ochiai, Polym. Bull., 1987,
18, 105–110.52 J. K. Kammeyer, A. P. Blum, L. Adamiak, M. E. Hahn and
N. C. Gianneschi, Polym. Chem., 2013, 4, 3929–3933.53 A. Sulistio, A. Blencowe, A. Widjaya, X. Zhang and G. Qiao, Polym.
Chem., 2012, 3, 224–234.54 A. Sulistio, J. Lowenthal, A. Blencowe, M. N. Bongiovanni, L. Ong,
S. L. Gras, X. Zhang and G. G. Qiao, Biomacromolecules, 2011, 12,3469–3477.
55 A. Sulistio, A. Widjaya, A. Blencowe, X. Zhang and G. Qiao, Chem.Commun., 2011, 47, 1151–1153.
56 K. M. Hawkins, S. S. S. Wang, D. M. Ford and D. F. Shantz, J. Am.Chem. Soc., 2004, 126, 9112–9119.
57 J. S. Jan and D. F. Shantz, Adv. Mater., 2007, 19, 2951–2956.58 J.-S. Jan and D. F. Shantz, Chem. Commun., 2005, 2137–2139.59 B. Fong and P. S. Russo, Langmuir, 1999, 15, 4421–4426.60 A. Heise, H. Menzel, H. Yim, M. D. Foster, R. H. Wieringa,
A. J. Schouten, V. Erb and M. Stamm, Langmuir, 1997, 13, 723–728.61 Y.-C. Chang and C. W. Frank, Langmuir, 1996, 12, 5824–5829.62 Y.-C. Chang and C. W. Frank, Langmuir, 1998, 14, 326–334.63 B. J. Sparks, J. G. Ray, D. A. Savin, C. M. Stafford and D. L. Patton,
Chem. Commun., 2011, 47, 6245–6247.64 J. K. Whitesell and H. K. Chang, Science, 1993, 261, 73–76.65 C. G. Worley, R. W. Linton and E. T. Samulski, Langmuir, 1995, 11,
3805–3810.66 N. Higashi, T. Koga and M. Niwa, Langmuir, 2000, 16, 3482–3486.67 M. Niwa, Y. Kuwagaki, S. Yamaguchi and N. Higashi, Angew. Chem.,
Int. Ed., 2003, 42, 1839–1841.68 M. Niwa, T. Murata, M. Kitamastu, T. Matsumoto and N. Higashi,
J. Mater. Chem., 1999, 9, 343–344.
ChemComm Feature Article
Publ
ishe
d on
17
Febr
uary
201
4. D
ownl
oade
d by
The
Uni
vers
ity o
f M
elbo
urne
Lib
rari
es o
n 28
/11/
2014
21:
47:5
2.
View Article Online

4988 | Chem. Commun., 2014, 50, 4971--4988 This journal is©The Royal Society of Chemistry 2014
69 S. H. Wibowo, E. Wong, A. Sulistio, A. Blencowe and G. G. Qiao,Aust. J. Chem., 2014, DOI: 10.1071/CH13519.
70 G. Marcelo, A. Munoz-Bonilla, J. Rodrıguez-Hernandez andM. Fernandez-Garcıa, Polym. Chem., 2013, 4, 558–567.
71 Y. Ito, Y. Ochiai, Y. S. Park and Y. Imanishi, J. Am. Chem. Soc., 1997,119, 1619–1623.
72 Z.-M. Liu, Z.-K. Xu, J.-Q. Wang, Q. Yang, J. Wu and P. Seta, Eur.Polym. J., 2003, 39, 2291–2299.
73 S. H. Wibowo, E. H. H. Wong, A. Sulistio, S. N. Guntari,A. Blencowe, F. Caruso and G. G. Qiao, Adv. Mater., 2013, 25,4619–4624.
74 E. P. Enriquez, K. H. Gray, V. F. Guarisco, R. W. Linton, K. D. Marand E. T. Samulski, Seattle, Washington, USA, 1992.
75 E. P. Enriquez, K. H. Gray, V. F. Guarisco, R. W. Linton, K. D. Marand E. T. Samulski, J. Vac. Sci. Technol., A, 1992, 10, 2775–2782.
76 A. J. Williams and V. K. Gupta, J. Phys. Chem. B, 2001, 105,5223–5230.
77 A. J. Williams and V. K. Gupta, Thin Solid Films, 2003, 423, 228–234.78 F. Himo, T. Lovell, R. Hilgraf, V. V. Rostovtsev, L. Noodleman,
K. B. Sharpless and V. V. Fokin, J. Am. Chem. Soc., 2004, 127,210–216.
79 J. E. Hein, J. C. Tripp, L. B. Krasnova, K. B. Sharpless andV. V. Fokin, Angew. Chem., Int. Ed., 2009, 48, 8018–8021.
80 S. S. Balamurugan, E. Soto-Cantu, R. Cueto and P. S. Russo,Macromolecules, 2010, 43, 62–70.
81 M. Kar, P. S. Vijayakumar, B. L. V. Prasad and S. S. Gupta,Langmuir, 2010, 26, 5772–5781.
82 M. Kar, B. Malvi, A. Das, S. Panneri and S. S. Gupta, J. Mater. Chem.,2011, 21, 6690–6697.
83 M. Higuchi, T. Koga, K. Taguchi and T. Kinoshita, Chem. Commun.,2002, 1126–1127.
84 M. Higuchi, K. Ushiba and M. Kawaguchi, J. Colloid Interface Sci.,2007, 308, 356–363.
85 S. Britland, E. Perez-Arnaud, P. Clark, B. McGinn, P. Connolly andG. Moores, Biotechnol. Prog., 1992, 8, 155–160.
86 T. Kratzmuller, D. Appelhans and H.-G. Braun, Adv. Mater., 1999,11, 555–558.
87 Y. Wang and Y. C. Chang, Adv. Mater., 2003, 15, 290–293.88 T. Ramanathan, F. T. Fisher, R. S. Ruoff and L. C. Brinson, Chem.
Mater., 2005, 17, 1290–1295.89 Y. Yao, W. Li, S. Wang, D. Yan and X. Chen, Macromol. Rapid
Commun., 2006, 27, 2019–2025.90 T. Jaworek, D. Neher, G. Wegner, R. H. Wieringa and
A. J. Schouten, Science, 1998, 279, 57–60.91 R. H. Wieringa and A. J. Schouten, Macromolecules, 1996, 29,
3032–3034.92 R. H. Wieringa, E. A. Siesling, P. F. M. Geurts, P. J. Werkman,
E. J. Vorenkamp, V. Erb, M. Stamm and A. J. Schouten, Langmuir,2001, 17, 6477–6484.
93 R. H. Wieringa, E. A. Siesling, P. J. Werkman, H. J. Angerman,E. J. Vorenkamp and A. J. Schouten, Langmuir, 2001, 17, 6485–6490.
94 R. H. Wieringa, E. A. Siesling, P. J. Werkman, E. J. Vorenkamp andA. J. Schouten, Langmuir, 2001, 17, 6491–6495.
95 J. Luijten, E. J. Vorenkamp and A. J. Schouten, Langmuir, 2007, 23,10772–10778.
96 Y. Wang and Y.-C. Chang, Langmuir, 2002, 18, 9859–9866.97 V. E. Dietz, N. Fery and K. Hamann, Angew. Makromol. Chem., 1974,
35, 115–129.98 N. Tsubokawa, K. Kobayashi and Y. Sone, Polym. J., 1987, 19,
1147–1155.99 E. Soto-Cantu, S. Turksen-Selcuk, J. Qiu, Z. Zhou, P. S. Russo and
M. C. Henk, Langmuir, 2010, 26, 15604–15613.100 D. Liu, Y. Li, J. Deng and W. Yang, React. Funct. Polym., 2011, 71,
1040–1044.101 P. Witte and H. Menzel, Macromol. Chem. Phys., 2004, 205,
1735–1743.
Feature Article ChemComm
Publ
ishe
d on
17
Febr
uary
201
4. D
ownl
oade
d by
The
Uni
vers
ity o
f M
elbo
urne
Lib
rari
es o
n 28
/11/
2014
21:
47:5
2.
View Article Online
Copyright © 2022 FDOKUMEN




















