
Polymers for biodegradable medical devicesII. Hydroxybutyrate-hydroxyvalerate copolymers: hydrolytic...
7
Polymers for biodegradable medical deuces II. Hy~~bu~a~-~~o~~era~ copol~e~: cyclic degradation studies S.J. Holland, AM. Jolly, M. Yasinand B.J. Tighe Speciality Polymer Group, Depanment of Molecular Sciences. Aston University, Birmingham 84 7iT UK (Rece/ved 17 November 1986; accepted 28 December 7986) The hydrolytic degradation of poly(hydro~butyrate) together with a series of hydro~butyrate-hydro~- valerate copolymers has been studied. The effects of copolymer composition and molecular weight are presented together with the results of varying pH and temperature on the degradation rate. ~eg~dation has been monitored by weight loss and water uptake measurements together with goniophotometric, surface energy and scanning electron microscopic studies. Some comparisons with the more widely used so-called ‘biodegradable’ polymers, poly(glycolic acid), poly(dioxanone) and the glycolic-lactic acid (90: 10) copolymers are presented together with the effect of blood plasma on the degradation process. Keywords: Polymers, poly(hydroxybutyrate), poly(hydroxyvalerate). bioerosion, biodegradation, goniophotometry Despite the growing interest in applications of polymers where degradation within the body is important, there are relatively few such polymers that are available and currently used for such purposes. Fu~hermore, the spectrum of degradation times and profiles that would be ideally required for applications ranging from multifilament and mono- filament sutures, through skin staples, fascia clips, bone pins, bone plates and other surgical fixation devices to drug delivery systems of various types is much wider than can be achieved with existing materials. In the previous part of this series we have reviewed the extensive literature on the so-called biodegradation of polyesters (which normally occurs by a simple hydrolytic mechanism) with particular emphasis on their possible use as controlled release systems for macromolecules’. The resultant attempt to bring together literature from various fields in which polyester degradation processes of this sort are important provides a good introduction to the further papers in this series in which the hydrolytic or biodegradation of some less common and purpose synthesized polyesters is described. The synthetic work has been concerned with novel ester-containing polymers and copolymers, in particular those containing pendant functional groups and those in which the structure of the copolymer series enables some control of the rate of hydrolytic degradation to be achieved. In addition the modification of degradation behaviour of these and other less common polyesters by blending and com- pounding with both synthetic and natural biod~radable polymers has been examined. In this paper we describe the hydrolytic degradation of a series of commercially available copolymers based on the hydroxybutyrate and hydroxy- valerate repeat units2-8. EXPERIMENTAL Materials Poly(hydroxybutyrate)-poly(hydrox~alerate) (PHB-PHV), co- polymers of O-20% hydroxyvalerate content and weight average molecular weights 36 000 to 800 000 used in these studies were obtained from Marlborough Biopolymers, ICI, Teeside, UK, and are known under the trade name ‘Biopol’. Samples were supplied in both non-nucleated and nucleated (boron nitride or Apatite) forms. Poly(glycolic acid) based sutures (Dexon ‘S’), were obtained from Davis Et Geck (Cyanamid of Gt Britain), Gosport, UK; both poly(gly~olic)/poly(iactic acid) 90/l 0 sutures (Vic~i), and ~ly(dioxanone) (PDS) sutures were obtained from Ethicon Ltd, Edinburgh, UK. Preparation of samples Films. Films were prepared by conventional solvent casting techniques from 13-20% (w/v) solutions of the PHB-PHV copolymers in chloroform using melinex sheets or glass petri dishes as casting surfaces. Melt pressed discs. Discs were prepared by pressing material in circular metal moulds in a heated electric press. The mould was preheated to the appropriate temperature for 2% min without pressure, and then for a further 2 min under pressure. The temperature at which the copolymers were pressed ranged from 185°C for the homopolymer to 130°C for the 20% copolymer. The mould was rapidly cooled under 0 1987 Butterworth Et Co (Publishers) Ltd. 0142-9612/87/040289-07$03.00 Biomaterials 1987, Vol 8 July 289
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of Polymers for biodegradable medical devicesII. Hydroxybutyrate-hydroxyvalerate copolymers: hydrolytic...

Polymers for biodegradable medical deuces II. Hy~~bu~a~-~~o~~era~ copol~e~: cyclic degradation studies
S.J. Holland, AM. Jolly, M. Yasin and B.J. Tighe Speciality Polymer Group, Depanment of Molecular Sciences. Aston University, Birmingham 84 7iT UK
(Rece/ved 17 November 1986; accepted 28 December 7986)
The hydrolytic degradation of poly(hydro~butyrate) together with a series of hydro~butyrate-hydro~- valerate copolymers has been studied. The effects of copolymer composition and molecular weight are presented together with the results of varying pH and temperature on the degradation rate. ~eg~dation has been monitored by weight loss and water uptake measurements together with goniophotometric, surface energy and scanning electron microscopic studies. Some comparisons with the more widely used so-called ‘biodegradable’ polymers, poly(glycolic acid), poly(dioxanone) and the glycolic-lactic acid (90: 10) copolymers are presented together with the effect of blood plasma on the degradation process.
Keywords: Polymers, poly(hydroxybutyrate), poly(hydroxyvalerate). bioerosion, biodegradation, goniophotometry
Despite the growing interest in applications of polymers where degradation within the body is important, there are relatively few such polymers that are available and currently used for such purposes. Fu~hermore, the spectrum of degradation times and profiles that would be ideally required for applications ranging from multifilament and mono- filament sutures, through skin staples, fascia clips, bone pins, bone plates and other surgical fixation devices to drug delivery systems of various types is much wider than can be achieved with existing materials.
In the previous part of this series we have reviewed the extensive literature on the so-called biodegradation of polyesters (which normally occurs by a simple hydrolytic mechanism) with particular emphasis on their possible use as controlled release systems for macromolecules’. The resultant attempt to bring together literature from various fields in which polyester degradation processes of this sort are important provides a good introduction to the further papers in this series in which the hydrolytic or biodegradation of some less common and purpose synthesized polyesters is described. The synthetic work has been concerned with novel ester-containing polymers and copolymers, in particular those containing pendant functional groups and those in which the structure of the copolymer series enables some control of the rate of hydrolytic degradation to be achieved. In addition the modification of degradation behaviour of these and other less common polyesters by blending and com- pounding with both synthetic and natural biod~radable polymers has been examined. In this paper we describe the hydrolytic degradation of a series of commercially available copolymers based on the hydroxybutyrate and hydroxy- valerate repeat units2-8.
EXPERIMENTAL
Materials
Poly(hydroxybutyrate)-poly(hydrox~alerate) (PHB-PHV), co- polymers of O-20% hydroxyvalerate content and weight average molecular weights 36 000 to 800 000 used in these studies were obtained from Marlborough Biopolymers, ICI, Teeside, UK, and are known under the trade name ‘Biopol’. Samples were supplied in both non-nucleated and nucleated (boron nitride or Apatite) forms.
Poly(glycolic acid) based sutures (Dexon ‘S’), were obtained from Davis Et Geck (Cyanamid of Gt Britain), Gosport, UK; both poly(gly~olic)/poly(iactic acid) 90/l 0 sutures (Vic~i), and ~ly(dioxanone) (PDS) sutures were
obtained from Ethicon Ltd, Edinburgh, UK.
Preparation of samples
Films. Films were prepared by conventional solvent casting techniques from 13-20% (w/v) solutions of the PHB-PHV copolymers in chloroform using melinex sheets or glass petri dishes as casting surfaces.
Melt pressed discs. Discs were prepared by pressing material in circular metal moulds in a heated electric press. The mould was preheated to the appropriate temperature for 2% min without pressure, and then for a further 2 min under pressure. The temperature at which the copolymers were pressed ranged from 185°C for the homopolymer to 130°C for the 20% copolymer. The mould was rapidly cooled under
0 1987 Butterworth Et Co (Publishers) Ltd. 0142-9612/87/040289-07$03.00
Biomaterials 1987, Vol 8 July 289

Degradation of PHB-PHV copolymers: S.J. Holland et al
pressure to room temperature before the discs were removed.
Injection moulded samples. Injection moulded specimens were prepared from both non-nucleated and nucleated (Apatite or boron nitride) materials. The copolymers were moulded using a bench top injection moulder (SPI, Small Power Machine Co. Ltd, Chippenham, UK), at barrel temperatures of 150°C for the 12% (3.5 X 105), and 140°C for the 20% (3 X 1 05) valerate copolymers respec- tively, using a heated mould (70-80°C).
Cold compressed discs’. Cold compressed discs of powder blends of 12% and 20% hydroxyvalerate copolymers [molecular weights (MW) 3.5 X 1 O5 and 3 X 1 05, respec- tively], with lactose and a high molecular weight dye (FITC 40), were prepared by manually loading 100 mg sieved (150 pm) samples into the die of a single punch tablet press (Manesty F3). fitted with 6 mm Normal Concave tooling and compressed at a pressure of 130 MPa. The tooling was prelubricated with a 2% solution of stearic acid in chloroform.
Degradation studies
Hydrolytic. Degradation of PHB-PHV copolymers was carried out in three aqueous buffer solutions (a) citric acid/ KzHP04,(b) KHzP04/K2HP04,and (c) NaHC03/NazC03.The corresponding pH’s of these buffers were 2.3, 7.4 and 10.6 at 37”C, respectively. Samples were placed in small bottles containing 50 ml of buffer and maintained at 37 f 0.1 “C or 70 f 0.1 “C, in water-baths. Samples were periodically removed, washed with distilled water, and placed between filter paper to remove the surface water. The samples were weighed wet and again after drying for several hours in a vacuum oven at 80°C.
Bloodplasma. Melt pressed discs were maintained in 3 ml of human blood plasma at 37 k 0.1 “C for successive periods of 15 h intervals. On removal the discs were washed and dried as above and subsequently returned to fresh plasma.
Oxygen plasma pretreatment. Samples were treated in an oxygen plasma for 30 min in a Nanotech Plastech 250, parallel plate plasma reactor, operating frequency 13.56 MHz at an RF power of 100 W at ambient tempera- ture. The oxygen flow rate was 10 ml min-’ and pressure in the reactor was nominally 0.3 torr.
Degradation monitoring techniques. Degradation of the PHB-PHV copolymers was followed conveniently by gravimetric methods (water uptake and dry weight loss). In addition surface erosion was monitored by goniophoto- metric analysis”, scanning electron microscopy (SEM) and surface energy measurements”.
Degradation ofsutures. Degradation of commercial sutures, Dexon ‘S’, (multifilament, grade 5 metric), Vicryl (multi- filament, grade 5 metric), and PDS (clear monofilament, grade 2 metric), was monitored by gravimetric methods.
Lengths of sutures (50 cm) were placed in 50 ml of the above buffers, at 37 + 0.1 “C and 70 + 0.1 “C. The sutures were removed periodically, washed thoroughly with distilled water and dried for several hours in a vacuum oven at 60°C before being weighed.
RESULTS AND DISCUSSION
Copolymer composition and molecular weight variations: weight loss studies
Initial hydrolysis studies on a series of hydroxybutyrate- hydroxyvalerate (PHB-PHV) copolymers were carried out on solvent cast polymer films in a pH 7.4 buffer at 70°C over a period of 2500 h. Figure 1 shows the weight loss profiles of these films as a function of degradation time. The depen- dence of the degradation rate on polymer molecular weight at the same copolymer composition is clearly shown by comparing the 3.6 X lo4 and 3 X lo5 molecular weight PHB profiles (both 20% PHV). The traces are characterized by an initial slow weight loss followed by a secondary enhanced degradation phase. The 3.6 X 1 O4 PHB sample is unrepresentative of the general trend of degradation rates since the physical strength associated with this low molecular weight material is not adequate for applications where mechanical properties are important.
The melting point and crystallinity of copolymers decreases with increasing hydroxyvalerate content and, in contrast to the poly(hydroxybutyrate) homopolymer, the hydroxyvalerate-hydroxybutyrate copolymers combine ease of processing, without appreciable degradation, with a reasonable spectrum of mechanical properties. These factors coupled with the adverse effect of decreasing molecular weight on physical strength led to the selection of 12% and 20% copolymers with weight average molecular weights in the range of 300 000 and above as the preferred materials for degradation studies.
The effect of specimen preparation technique on hydrolytic degradation was examined. The four techniques used (solvent casting, melt pressing, injection moulding and cold compression) were found to exert a marked effect on the degradation rates of the samples so produced. This is exemplified by the difference between Figure 1 (solvent cast films) and the results obtained with melt pressed discs shown in Figure 2.
In order to establish the importance of the effect of pH and temperature on the hydrolytic degradation of the specimens described above a series of experiments were carried out the results of which are summarized in Figure 3 (a andb). This information enables some links to be establish- ed between accelerated degradation at 7O”C, the rate at physiological temperatures, and additionally between the wide range of pH’s that may be encountered in possible extremes of degradation conditions. A useful comparison can be made by comparing times for 10% and 50% degradation as shown in Tables 1 and 2.
501 I \
I I I I I 0 IO 20 30 40 50 60 70
Time (d)
Figure 1 Progressive weight loss of solvent cast films (discs) of various PHB-PHv copolymers maintained in an aqueous environment at 70°C and
pH 7.4. Initial disc dimensions: 2 cm diam.; 0.15 mm thick. (+) 10% HV: 75OK (x) 20%. 3OOK; (a) 12%, 17OK (0) 12%, 1OOK (A) 20%. 36K
290 Biomaterials 1987, Vol 8 July

Degradation of PHB-PHV copolymers: S.J. Holland et al
Time Id)
Figure 2 Progressive weight loss of melt pressed discs of various PHB-PHV copolymers maintained in an aqueous environment at 70°C and
pH 7.4. Initial disc dimensions: 2 cm diam.; 0.15 mm thick. (+) 10% HV, 750 c (X) 12%. 350 K (A) 20%. 300 R (m) 12%. 170 K.
Tome (di
l2Or
0 20 40 60 80 100 120 Time id)
Figure 3 (a)Progressive weight loss of solvent cast films (discs) of 12% PHV copolymer (MW 3.5 X 1 05) under various conditions. Results for cold compressed tablet at 37°C and pH 10.6 shown for comparison. Initial disc dimensions:2cmdiam.;0.15mmthick. (+)?O”C,pH 7.4;(X) 70°C,2.3;(A) 37°C. 10.6; (m) 70°C. 10.6. (b) Progressive weight loss ofmelt-presseddiscs of 12% PHV copolymer (MW 3.5 X 705) and 20% PHV copolymer (MW 3.0 x 16)maintainedinanaqueousenvironmentat 70”CandatvarioospH values. Initial disc dimensions: 2 cm diam.; 0.15 mm thick. ( + ) 12%. pH 7.4; (X) 20%. 7.4; (A) 20%, 2.3; (m) 72%, 70.6; (A) 20%, 10.6.
From this data it can be seen that the various forms of
Biopol have differing stabilities to hydrolytic attack, the cold
compressed tablets being the least stable, with an increase
in stability for solvent cast films through melt pressed discs
up to the injection moulded pieces. This reflects the
combined effects of polymer compaction which in turn
affects the porosity of the matrix (most marked in the case of
cold compressed discs) together with the crystallinity of the
specimens which is a function of thermal history in the
fabrication. These two effects are not altogether easy to
distinguish but the relative crystallinity of the materials as
measured by d.s.c. is: cold compressed discs < solvent cast
films < melt pressed discs < injection moulded pieces.
An overview of the tabulated data indicates that
alteration of the copolymer ratio by increasing the valerate
composition enhances the degradation rate (probably due to
a fall in crystallinity levels with increasing valerate content).
Increasing alkalinity speeds up the degradation rate indicating
ester hydrolysis as the degradation mechanism. Additionally
a change from physiological temperature to 70°C generally
increases the degradation rate some 30 to 1 OO-fold.
The progressive degradation of various samples
studied was found to be marked by increased water uptake
as well as by a decrease in dry weight. Figure 4 shows the
increasing difference between the ‘wet’ (Equation 1) and
Table 1 Wetght loss for Biopol andcommeroal sutwe materials [t,o% and t50% at pH 7.4 (70°C) ]
Sample tl oyh (h) r5os (h)
VlCryl suture Dexon suture
PDS suture
PHB-PHV solvent cast films
MWx lo3 % PHV
36 20
100 12
170 20
300 20
350 12
750 10
Melt pressed discs
MWx lo3 96 PHV
10 65 15 70 80 140
200 400 825 2000 800 (37% at 2000)
1850 2300
1880 2900 1950 2400
300 20 2020 3080 350 12 2800 3125
InJectIon moulded plaques
MWX lo3 % PHV
300 20 2210 (15% at 2450)
350 12 1890 (22% at 2450)
Table 2 Weight loss for a melt pressed Biopol disc [MW 350 X ld (12% PHV) ]
Degradation conditions tlo% (h) t5ox (h)
pH 2.3 at 37°C (0.8% at 4590) h 5000
pH 7.4 at 37°C (1 .O% at 4724) h 5000
pH 10.6 at 37°C 3750 (16.4% at 4724)
pH 2.3 at 70°C 2800 (27.7% at 4500)
pH 7.4 at 70°C 2600 3125
pH 10.6 at 70°C 400 1226
60 I 1..
0 20 40 60 60 100 120
Time (d)
Figure 4 Comparison of progressive ‘wet’ and ‘dry’ weight loss of melt
pressed discs (+. X) and injection moulded (plaque) specimens (A, n ) at 70°C and pH 7.4. Initial disc dimensions; 2 cm diam.; 0.15 mm thick. Initial plaque dimensions 2 x 1.5 cm.
Biomaterials 1987, Vol8 July 291

Degradation of PHB-PHV copolymers: S.J. Holland et al
‘dry’ (Equation 2) weights of a melt pressed disc and injection moulded piece at the later stages of degradation (pH 7.4; 70’C). This indicates a large increase in the internal surface area and porosity of the disc samples as (disc) weight loss exceeds 20%.
‘Wet’ wt = Hydrated wt at time r
Dehydrated wt at time 0
‘Dry’ wt = Dehydrated wt at time t
Dehydrated wt at time 0
(1)
(2)
Surface studies
Although weight loss and water uptake measurements provide a convenient means of monitoring comparative erosion rates of polymers, valuable additional techniques are available that enable surface changes to be followed. SEM has the advantage of enabling detailed morphological investigation of the surfaces to be carried out but has the inherent disadvantage of sample destruction. Goniophoto- metryon the other hand is non-destructive and in some ways complementary to microscopic techniques. Thus it enables
-1
Angle of reflectance
d’f Id
Angle ot reflectance
J
1
Figure 5 (a) Goniophotometric curve for an injection mouldedspecimen of 12% PHVcopolymer (MW 3 X 16) before immersion in an aqueous buffer at 37°C and pH 7.4 showing intensity of reflected beam as a function of scattering angle and the position of diffuse intensity (Id), specular intensity (/,) and the peak width at half height (W ,,*). Parameters shown in Table 3.
(b) Goniophotometric curve for an injection mouldedspecimen of 12% PHV copolymer (M W 3.5 X 16) after immersion for5500 h in an aqueous buffer at 37°C and pH 7.4.
quantitative, as distinct from qualitative, information to be obtained but this is averaged from an area of between 0.5 and 100 mm2. As a result the fine resolution of individual surface features that SEM provides is absent.
Two typical goniophotometric curves are shown in Figure 5(a and 6) which relate to an injection moulded specimen of the 12% copolymer (A&V 3 X 1 05) and the same sample after immersion in aqueous pH 7.4 buffer at
‘37°C for 5500 h, respectively. Although the cumulative weight loss over this period was only a little over 1% the differences in the results of goniophotometric examination are very marked. The basic instrumentation involved in goniophotometrytogetherwith the principles involved in the interpretation of goniophotometric curves has been adequately described elsewhere”. Briefly, the technique involves the use of a collimated light beam impinging at an angle (normally 45”) on the surface of the sample in conjunction with a rotating photocell which records the intensity of scattered light as a function of scattering angle. The resultant scattering envelope (e.g. Figure 5f~) is characterized by three parameters. These are (i) the intensity of light scattered at the specular angle (I,), (ii) the diffuse reflectance (Id) and (iii) the peak width at a level midway between these two intensities, generally referred to as the half-height peak width (W,,2). These three factors are combined together in the so-called gloss factor, given by
(is-1d)N1/2. The specular angle (being the equivalent of the angle of incidence) is the angle of maximum reflectance for those surfaces that are not dominated by imperfections having dimensions similar to, or greater than, the wave- length of the incident light beam. Diffuse reflectance on the other hand is measured normal to the plane of specimen surface and is small for such surfaces. It is important to note that the reflectance curve is always amplified to something approaching a full scale display. The actual reflectance intensity values are given in Tab/e 3.
A relatively smooth surface, using the criteria described above, will give a sharp fairly symmetrical peak of the type shown in Figure 5 a. Development of surface imperfections of dimensions smaller than the wavelength of the light beam used (1. 0.5 pm) leads to the removal of energy from the beam and a reduction in peak intensity with little change in peak asymmetry. As imperfections in the region of 0.5 j.rrn or greater develop at the surface, however, the peak width at half-height (WI,2) increases together with the magnitude of I,,. This has occurred in moving from Figure 5a to 5 b. As degradation proceeds the changes in the goniophotometric curve become more marked as typified by Figure 6 which relates to a melt-pressed disc of 20% PHV copolymer (MW = 3 X 1 05) treated in the same pH 7.4 aqueous buffer as in Figure 5 b but at 70°C (2500 h). This sample has
Table 3 Goniophotometric and surface energy parameters of Figures 5-8
Correspond- Goniophotometric Surface energy + ing parameter* (mN m-l)
Figure ‘s ‘d WI/2 yF Yd YT
5a 37.6 0.59 3.5 9.0 39.0 48.0
5b 14.2 1.1 13.0 13.4 39.7 53.1
6 6.13 5.58 22.5 37.5 37.6 75.1
7 6.07 1.01 21.0 29.5 37.2 66.7
8 14.5 1.0 8.5 10.1 38.7 48.8
*Goniophotometric parameters: Is (specular intensity); Id (diffuse intensity); W,,, (peak width at half-peak height); intensity of incident beam 600 (arbitrary) units.
T Surface energy parameters: y, (polar component); yd (dispersive component); yT (total).
292 Biomaterials 1987, Vol8 July
V 112
‘\ -- u
Angle of reflectance
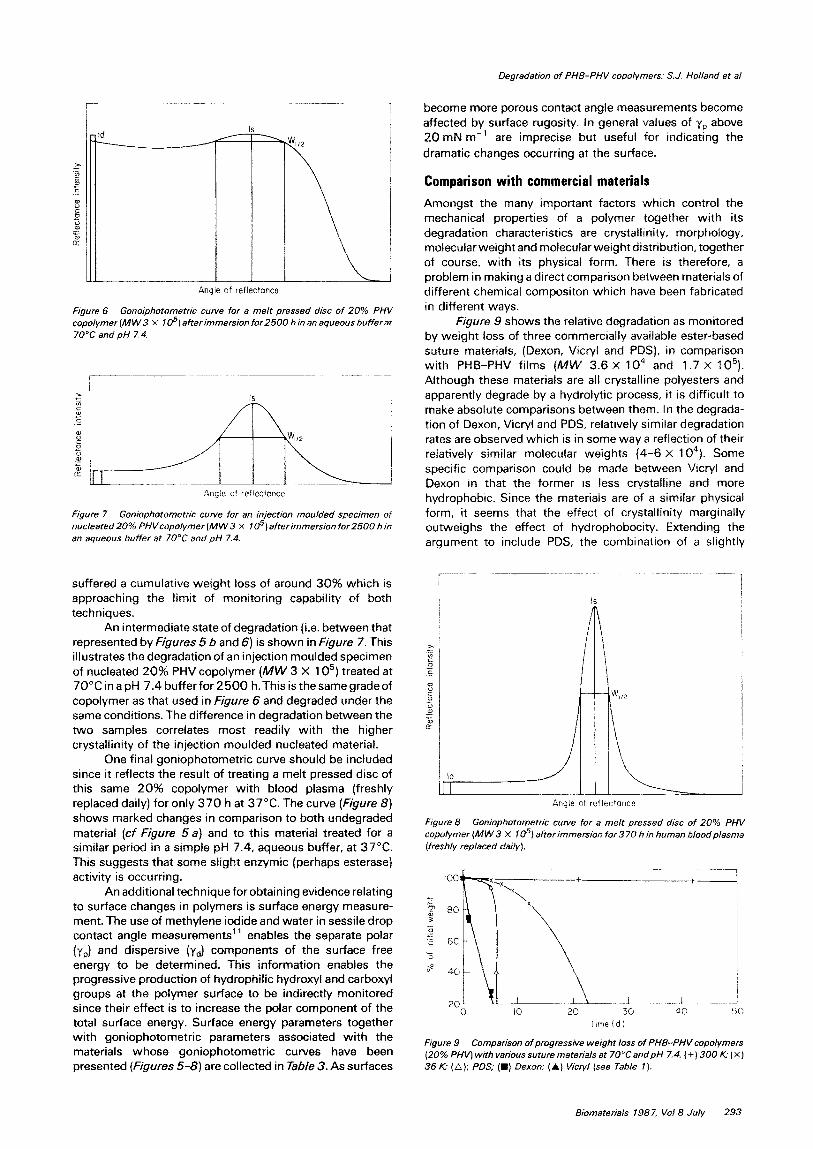
Figure 6 Gonoiphotometric curve for a melt pressed disc of 20% PHV copolymer (MW 3 X 1 05) atierimmersion for2500 h in en aqueous buffer at 70°C and pH 7.4.
Angie of ref!ectance
Figure 7 Goniophotometric cwva for an injection moulded specimen of nucleated20% PHVcopolymer(MW3 x Id)afterimmersionfor2500 hin an aqueous buffer at 70°C and pH 7.4.
suffered a cumulative weight loss of around 30% which is approaching the limit of monitoring capabili~ of both techniques.
An intermediate state of degradation (i.e. between that represented by Figures 5 b and 6) is shown in Figure 7. This illustrates the degradation of an injection moulded specimen of nucleated 20% PHV copolymer (MW 3 X 1 05) treated at 70”CinapH 7.4 bufferfor h.Thisisthesamegradeof copolymer as that used in Figure 6 and degraded under the same conditions. The difference in degradation between the two samples correlates most readily with the higher crystallinity of the injection moulded nucleated material.
One final goniophotometric curve should be included since it reflects the result of treating a melt pressed disc of this same 20% copolymer with blood plasma (freshly replaced daily) for only 370 h at 37°C. The curve (Figure 8) shows marked changes in comparison to both undegraded material (cf Figure 54 and to this material treated for a similar period in a simple pH 7.4, aqueous buffer, at 37°C. This suggests that some slight enzymic (perhaps esterase) activity is occurring.
An additional technique for obtaining evidence relating to surface changes in polymers is surface energy measure- ment. The use of methylene iodide and water in sessile drop contact angle measurements” enables the separate polar (yP) and dispersive (yd) components of the surface free energy to be determined. This information enables the progressive production of hydrophilic hydroxyl and carboxyl groups at the polymer surface to be indirectly monitored since their effect is to increase the polar component of the total surface energy. Surface energy parameters together with goniophotometric parameters associated with the materials whose goniophotometric curves have been presented (Figures 5-8) are collected in Table 3. As surfaces
Degradation of PHB-PHV copolymers: S.J. Holland et al
become more porous contact angle measurements become affected by surface rugosity. In general values of yP above 20 mN m-’ are imprecise but useful for indicating the
dramatic changes occurring at the surface.
Comparison with commercial materials
Amongst the many important factors which control the mechanical properties of a polymer together with its degradation characteristics are c~stallinity, morphology~ molecular weight and molecular weight distribution, together of course, with its physical form. There is therefore, a problem in making a direct comparison between materials of different chemical compositon which have been fabricated in different ways.
Figure 9 shows the relative degradation as monitored by weight loss of three commercially available ester-based suture materials, (Dexon, Vicryl and PDS), in comparison with PHB-PHV films (A&&’ 3.6 x 10” and 1.7 x f05). Although these materials are all crystalline polyesters and apparently degrade by a hydrolytic process, it is difficult to make absolute comparisons between them. In the degrada- tion of Dexon, Vicryl and PDS, relatively similar degradation rates are observed which is in some way a reflection of their relatively similar molecular weights (4-6 X 104). Some specific comparison could be made between Vicryl and Dexon in that the former is less crystalline and more hydrophobic. Since the materials are of a similar physical form, it seems that the effect of crystallinity marginally outweighs the effect of hydrophobocity. Extending the argument to include PDS, the combination of a slightly
.: Id
_
Angle of reflectance
Figure 8 Goniophotometric curve for a me/t pressed disc of 20% PHV copotymer(MW3 X 705) afterimmersion for370 h in human bloodplasma (fresh/y replaced daily)
-+- i
Time(d)
Figure 9 Comparison of progressive weight loss of PHB-PHV copolymers (20% PHV) with various suture materials at 70°C andpH 7.4. (f ) 300 K (X) 36 k (A): PDS; (m) Dexon; (A) Vicry/ (see Table I).
Biomateriais 1987, Vol 8 July 293

Degradation of PHB-PHV copolymers: S.J. Holland at al
higher molecular weight and a reduced concentration of The weight loss and buffer solution uptake studies ester groups in the backbone produces a somewhat slower show that degradation at elevated temperture and under degradation rate. Although these differences are difficult to alkaline conditions produces similar degradation profiles to discern in Figure 9, the scale of which has been adjusted to those obtained under conditions of physiological pH and give relative comparisons with the poly(hydroxybutyrate) temperature, but with compressed time scales. Of the many copolymers, they are apparent in experiments in which direct degradative options available the pH 10.6 and 37°C comparisons are made. Much greater difficulty, however is hydrolysis conditions are more suitable than pH 7.4 at 70°C encountered in comparing the group of PHB-PHV co- for predictive accelerated studies. Since the degradation rate polymers with this small group of commercially available increase over that obtained with in vitro physiol~ical suture materials. It is not possible from our experiments to conditions is less for the low temperature alkali than for the make direct comparisons in a situation where crystallinity, high temperature studies at pH 7.4 however, the point of molecular weight and physical form were similar for the two catastrophic matrix breakdown is more likelyto be pinpointed groups of materials. under the former set of conditions.
The effects of molecular weight and copolymer composition on degradation rates of the PHB-PHV copolymers have been discussed already. The most appropriate comparison that can be made with commercial sutures is that under conditions where molecular weight and composition of the PHB-PHV copolymers are chosen to produce acceptable mechanical properties the degradation rates are markedly slower than those of any of the commercial sutures examined in this study. This is dramatically illustrated in Figure 9, where even the mechanically inadequate 20% copolymer (MW 3.6 X 1 04) is seen to be less prone to degradation than even the most stable (i.e. PDS) of the commercially available suture materials studied.
We believe then, that the results of these studies provide some insight into the m~hanism of hydrol~ic degradation of PHB-PHV copolymers. In the first place the relative effects of pH and blood plasma give some indication that in biological environments the underlying hydrolytic mechanism is likely to prevail. Thus biodegradation in different body sites will certainly be dramatically influenced by the pH of the local environment. Additionally, as far as blood contact is concerned some low level esterase activity may be involved but this does not have a major effect on the overall rate of hydrolytic degradation. Results obtained elsewhere do indicate, however, that quite pronounced acceleration of degradation is observed in the presence of certain bacteria’. This suggests that biod~radation in certain bodily environments may be faster than the under- lying rate of hydrolytic degradation although such cases would appear to be the exception rather than the rule.
CONCLUSIONS
Investigation of the in vitro degradation of the PHB-PHV copolymer series has shown the influence of many experimental variables on the rate of matrix buffer hydrolysis, with molecular weight and copolymer ratio in addition to the physical form of the samples playing important roles in the degradation process.
The rate of degradation of the various physical forms of the PHV-PHV series in a range of aqueous buffer solutions increases with temperature in accordance with the proposed ester hydrolysis mechanism. The enhancement of degrada- tion rate as the hydrolytic medium becomes more alkaline is also evident which is possibly due in part to the increasing solubility of the polymer degradation products at higher pH as well as to the expected rise in hydrolysis rate.
Alteration of the physical form of the polymer matrix has a pronounced effect on the rate of degradation with the (non-nucleated) solvent cast films being more susceptible to hydrolysis than melt pressed discs or injection moulded samples, presumably due in part to the lower crystallinity of these samples. Cold compressed tablets are simple compacted physical blends of polymer powder with various excipients and the large increase in surface porosity and surface area (available for degradative attack) makes this form of the PHB-PHV copolymer series the least stable of all the prepared matrices. Two additional factors are sample molecular weight and copolymer composition. The consistent increase in the degradation rate of the higher valerate copolymers over the low valerate polymer samples again reflects the importance of sample c~stallin;ty which decreases with increasing valerate content. However polymer molecular weight is the dominant parameter, with large decreases in degradation rate occurring as polymer molecular weight increases from 3.6 X IO4 to 3 X IO5 although polymers having molecular weights below 2 X 1 O5 have mechanical properties that are inadequate for most purposes.
The combined dry and wet gravimetric results taken together with surface energy and goniophotometric measurements provide an interesting composite picture of the degradation process. It is apparent that in the initial stages of the degradation little change in the bulk or mass of the sample is detected. Despite this, subsequent events show that a gradual diffusion of water into the bulk occurs from the outset and this is obviously accompanied by progressive chain scission within the polymer matrix.
The most obvious initial events, however, are those occurring at the polymer surface. An increase in surface energy reflects the increase in the concentration of hydroxyl and carboxyl groups at the surface as a consequence of ester hydrolysis at the polymer-water interface. As this process progresses the resultant surface erosion produces changes in surface rugosity that in turn, produce noncurrent changes in the goniophotometric scattering envelope. Initially the changes in rugosity consist of an increasing concentration of irregularities that are smaller than the wavelength of light. These reduce the intensity of the scattered light beam without greatly affecting the dispersity of scattering and as a resultthevalueof l*dropswithout anycon~urrent increasein half-height peak width (W,~*).
The advancement of the surface erosion process leads to an increase in the size of the physical irregularities at the surface until this reaches and exceeds the wavelength of the incident light beam (2. 500 nm). When this occurs the asymmetry of the goniophotometr~c curves becomes more marked because W,,z increases together with the intensity of diffuse reflectance (Id) as a result of the greater dispersity of the scattered light beam. It is apparent that these progressive changes at the surface are occurring concurrently with a bulk erosional process that results from the diffusion from the matrix of the products of chain scission processes. Initially only very low molecular weight fragments are able to diffuse out but as the process proceeds the matrix becomes
294 Biomaterials 1987, Vol8 J&y

Figure 10 SEA& of degraded P#B-PHV copolymer (see Text) (a) x 56; (6) x 222; (c) x 7 7.X?.
Degradat/on of PHB-PHV copolymers. S.J Holland et a/
progressively more porous allowing an increasing loss of higher molecular weight degradation products. The increasing porosity of the matrix is reflected in the increasing difference between dry and wet weights and in the almost ‘autocatalytic’ profiles of the degradation curves showing, as they do, initial induction periods followed by quite marked accelerations in the degradation process.
The development of porosity as the degradation proceeds, together with the importance of crystallinity in the progress of the erosional process are illustrated in Figure 70.
This shows SEMs for a sample of 12% PHB-PHVcopolymer with an initial molecular weight of 350 000 degraded for 1400 h at 70°C and pH 10.6. In common with most reactions of this type amorphous regions have been preferentially attacked leaving a residual porous, pre- dominantly crystalline matrix. This fact, coupled with the role of other structural factors reported here in controlling degradation explains the relative stability of drawn PHB monofilament recently reported by Williams and Miller12.
The difference between rates of PHB-PHV copolymer hydrolysis and those of commercially exploited polyesters of biomedical interest has been highlighted in a previous section. It is apparent that the PH B-PHV copolymers provide a useful extension of these materials in that they are uniformly more resistant to hydrolysis whilst on the other hand being ultimately completely degraded to readily metabolized fragments. Whereas currently used absorbable surgical devices (clips, sutures, etc.} exploit the more rapid degradation behaviour of poly(glycolic acid) and related materials, there are a range of surgical and orthopaedic devices awaiting development that could utilize the somewhat greater stability of the PHB-PHV copolymer series. The results presented here, coupled with the known thermal sensitivity of the polymer2, do however, indicate the importance of careful control of processing conditions in such applications.
REFERENCES
1
2
3
10
11
12
Holland, S.J., Tighe, 8.J. and Gould. P.L., Polymers for bi~egradable medrcai devices (i): The potentral of polyesters as controlled macro-
molecular release systems, J. Controlled Release 1986.4. 155- 180 Holmes, P.A., Degradation and stability of poly hydroxyesters. Polymer Degradation Discussion Group Conf., The Understanding and Control of Polymer Degradation Processes, University of Glasgow. September 1 S-2 1, 1984 Rodgers, P.J., Polyhydroxybutyric acru and related copolymers - propenres and potential, Royal Sot. of Chemistw - Industrial Division. EI/otech. Subject Group Symp., London, December 12. 1985
Holmes. P.A., Wnght, L.F. and Collrns, S.H., Beta-hydroxybutyrate polymers. EU Patent No. 0 052 459, May 21, 1982 Holmes, P.A.. Collins, S.H. and Wright, L.F., 3-Hydroxybutyrate
polymers, US Patent No. 4 477 654, October 16, 1984 Barham. P.J.. Keller, A., Otun. EL. and Holmes, P.A.. Crystailization and morphol~ of a bacterial thermoplastic: Poly-3-hydroxybu~rate, 1 Mat. Sci. 1984. 19 (9). 2781-2794 Barham, P.J., Nucleation behavtourof pol~3-hydroxybutyrate,~. Mat Sci. 1984, 19 (f 2). 3826-3834 Holmes, P.A.. Applications of PHB-a mrcrobiaily produced bio- degradable thermoplasbc, fhys. Technol. 1985. 16, 32-36 Gould, P.L.. Holland, S.J. and Trghe, B.J.. Polymers for biodegradable medical devices (IV): Hydroxybutyrate-valerate copolymers as non-drsrntegrating matrices for controlled release oral dosage forms, lnt. J. Pharmaceutics (in Press) Trghe. B.J., Subjective and oblectrve assessment of surfaces, rn Polymer Surfaces, (Eds D.T. Clark and W.J. Feast), John Wiley B Sons, 1978 Ch. 14. pp 269-286 Owens, D.K. and Wendt, R.C.. Estimation of the surface free energy of
polymers, J. Appl. Folym. Sci. 1969, 13, 7 741-l 747 Williams, D.F. and Miller, ND.. The degradation of ~ly(hydroxy- butyrate) (PHB), presented at Europ. Congr. ~jomafe~;als, Bologna, Italy, September 14-l 7. 1986
Biomaterials 1987, Vol8 July 295




















