
Dispersion Polymerization of Methyl Methacrylate in Supercritical CO2

Poly( ethylene oxide) / Poly( ethyl methacrylate) Blends: Crystallization, Melting
Behavior, and Miscibility
SOSSIO CIMMINO, EZIO MARTUSCELLI, and CLARA SILVESTRE, Istituto di Ricerche su Tecnologiu dei Polimeri e Reologia, C.N.R. Via Toiano 2, Arco Felice, Napli , Italy, and
MAURIZIO CANE"I'1, CLAUDIA DE LALLA, and ALBERT0 SEVES, S&zione Sperimentule per la Cellulosa, Cartu e Fibre Tessili,
Vegetali ed Artifkid, Piazza Leonardo da Vinci 26, Milano, Italy
Synopsis
Results of an investigation of isothermal crystallization and thermal behavior of poly(ethy1ene oxide)/poly(ethyl methacrylate) (PEO/PEMA) blends are reported. The blend composition and the crystallization temperature strongly influence the crystallization process from the melt and the melting temperature of PEO. T h e addition of PEMA to PEO causes a depression in the spherulite growth rate, in the overall kinetic crystallization constant, and in the melting tempera- ture. Experimental data on the radial growth rate G and overall kinetic rate constant K, are analyzed by means of the latest kinetic theory. From this analysis it emerges that the crystalliza- tion of pure PEO and PEO in the blend conforms to the regime I process of surface secondary nucleation. The depression of the melting temperature cannot be explained only in terms of a diluent effect due to the compatibility of the two components in the melt. Annealing and morphological effects, dependent on composition and time, must also be taken into account.
INTRODUCTION
Blends of poly(ethy1ene oxide) (PEO) with poly(methy1 methacrylate) (PMMA) or poly(viny1acetate) (PVAc) have been the subject of recent pa- pers.'-12 These studies have shown clear evidence of miscibility in the melt of PEO with PMMA or PVAc. An interesting feature is that PEO is able to crystallize, even from mixtures with a very high content of the noncrystalliz- able component.
The crystallization process in polymeric material is governed by the degree of undercooling and by the resistance to segmental motions, and i t is possible only at temperatures between the melting temperature T, and the glass transition temperature Tg. Blending two polymers can have important effects on both T, and Tg. For the blends reported above, Tg of the mixture increases upon adding the amorphous diluent, whereas the T, of the crystallizing component decreases. These effects retard molecular motions, reduce the interval T, - Tg, and in general tend to restrict crystallization.
In previous papers', lo we reported the results of investigations concerning mainly the crystallization thermal behavior of PEO/PMMA and PEO/PVAc blends. By studying the influence of blend composition and crystallization conditions on the overall kinetics of crystallization, the radial growth rate of
Journal of Polymer Science: Part B: Polymer Physics, Vol. 27, 1781-1794 (1989) 0 1989 John Wiley & Sons, Inc. CCC 0887-6266/89/091781-14$04.00

1782 CIMMINO ET AL.
spherulites, and the observed and equilibrium melting temperatures, it was found that information about the melt compatibility of blend components and phase structure could be obtained.
It is the purpose of the present paper to report the results of a similar study on PEO/PEMA blends. To our knowledge, no such studies have been re- ported on this system.
EXPERIMENTAL
Materials and Spherulite Growth Rate Measurements
Blends of poly(ethy1ene oxide) ( M , = 2.0 X lo4) and poly(ethy1 methacry- late) ( M , = 1.50 X lo5) were prepared by dissolving the two components in the desired proportions in chloroform and by evaporation of the solvent at 50°C under vacuum. Thin films were prepared by melt-squeezing the blend powder between two microscope glasses. Before crystallization the samples were kept at 75°C for 10 min, the temperature was then rapidly lowered to T, (43-54"C), and the blends were allowed to crystallize isothermally.
The radial growth rates, G = dR/dt (where R is the radius of the spherulite, and t is the time), were calculated by measuring the size of PEO spherulites as a function of time during isothermal crystallization. An optical polarizing microscope fitted with an automatized hot stage (precision k0.2"C) was used. From plots of R against t , the radial growth rate G was calculated as the slope of the resulting straight lines. After crystallization, the samples were heated 3"C/min until birefringence patterns disappeared. The corresponding temperature was taken as the observed optical melting point of PEO(TA).
Calorimetric Measurements
The overall kinetics of crystallization and the thermal properties of the homopolymer and blends were analyzed by differential scanning calorimetry (Perkin-Elmer DSC 4).
The isothermal crystallization process was studied using the following procedure: samples were heated up to 75°C and kept at this temperature for 10 min. The samples were rapidly cooled to the desired crystallization temper- ature T,, and the heat cEH/dt evolved during the isothermal overall crystal- lization was recorded as a function of time. The weight fraction X, of material crystallized at time t was calculated from the ratio of the heat generated at time t to the total heat corresponding to the completion of crystallization.
The observed calorimetric melting temperatures T; and the apparent en- thalpies of melting AH* of the isothermally crystallized samples were ob- tained from the maxima and the area of the melting peaks, respectively. The samples were heated directly from T, to the melting point at a scanning rate of 5"K/min.
The glass transition temperature of the pure materials were obtained by heating the samples (about 10 mg) from - 100 to 100°C at 10"C/min and by recording the heat evolved during the scanning process as a function of temperature. The 2'' of the sample was taken as the temperature correspond- ing to 50% of the transition.

PEO/PEMA BLENDS 1783
TABLE I Glass Transition Temperature Tg (K), Equilibrium Melting Temperature T, (K),
Free Energies of Folding Calculated by Using Eq. 6 ( u ~ ) ~ , and Eq. 2 ( Q ) ~
Tm ( d l ( % ) 2
(erg/cm2 ) (erg/cm2 Blend composition Tg a
PEO/PEMA (K) (K)
1oo:o 9o:lO 80:20 70:30
218 348 75 226 344 42 234 342 34 243 340 29
58 28 24 14
'T, for blends calculated using Eq. (1) bThe error in the T, values has been calculated as 5 2 K
'The resulting values of Tg for the PEO and PEMA were 218 and 344 K, respectively. The Tg of the blends Tg(blend) were calculated by using the Fox equation13
where W(pm,, Tg(pEO) and W(PEMA), Tg(pEm) are the weight fractions and the glass temperatures of plain PEO and PEMA, respectively.
This theoretical relation assumes random mixing at the segmental level. The calculated values of the Tgs for all the blends studied are reported in Table I.
RESULTS AND DISCUSSION
Spherulite Radial Growth Rate
Neat PEO and PEO in the blends crystallize prevailingly in a spherulite morphology (see Fig. 1). A study of the influence of PEMA addition on the PEO spherulitic texture will be the subject of a forthcoming paper. After the completetion of the crystallization, the samples are completely filled with spherulites truncated by impingement. Plots of the radius of the spherulites R against time t for all the samples result in straight lines for any T, and composition investigated. These results together with those relative to the dependence of G on composition (see next section) seem to indicate, in agreement with Keith and Padden,14 that the concentration of the PEMA at the growth front is probably constant during the entire crystallization process and consequently that the PEMA phase is incorporated in the PEO spherulites. From the slope of R versus t the isothermal radial growth rate G is obtained. In Figure 2, G is reported as a function of T, for all the compositions investigated. It can be observed that the addition of PEMA to PEO causes a depression in the G values at a given T,. As is shown by Figure 2 for a given T, this effect is composition dependent, (the reduction in G increases with the increase of PEMA in the blends.) In the case of a polymer-diluent system, the theory developed by Lauritzen and Hoffman and recently modified by

1784 CIMMINO ET AL.
(b)
Fig. 1. Optical micrographs of thin films of pure PEO (a) and PEO/PEMA 80:20 blend (b) crystallized at T, = 320 K.
Hoffmm15 for homopolymers must be refined to take into account: (1) the entropic contribution to the free energy of formation of a nucleus of critical dimensions that represents the probability of selecting the required number of crystalline sequences from a mixture with volume fraction +2 of crystallizable polymer, as discussed by Boon and Azcue;16 (2) the energy for transport of the amorphous component from the growth front into interlamel- lar region^;^',^* (3) the influence of the diluent on Tg and T,.

PEO/PEMA BLENDS 1785
11
9
7
5
3
1
a i16 32 0 324 328 Tc( K )
Fig. 2. Variation of the spherulite radial growth rate with crystallization temperature for all the samples examined.
Thus the equation that describes the growth rate of spherulites of a crystallizable polymer in a one-phase melt containing a second polymer acting as a diluent can be derived in the following f ~ r m : ' ~ - ' ~
where Go is the preexponential factor that includes all terms that are taken as effectively independent of temperature. The term (Ul* + U,*)/R(Tc - T,) contains the contribution of diffusion processes of the amorphous and crystal- lizable materials to the growth rate; Ul* + U2* = U* is the sum of activation energies for the motions in the melt of noncrystallizable and crystallizable molecules, and T, (T, = Tg - C) is the temperature below which such mo- tions stop. T, is the glass transition of the blend (see Table I). (p2 is the volume fraction obtained using the mass densities of the pure polymers at the crystallization temperature?*34 The factor f is a correction term that takes into account the dependence of AH" on T, and is given empirically as f = 2T,/(T,O + TC);l5s2' the term K , is equal to
where u and a, are the lateral-surface and fold-surface free energies, and n is

1786
15.
13-
11,
9
CIMMINO ET AL.
4 8 12 16 20
[l/Tc(AT)f] x105
Fig. 3. Plots of a, versus l / [ T , ( A T ) f ] for eq. ( 2 ) with U' = 1500 cal/mol and C = 51.6 K.
a variable that can assume values of 2 or 4 according to the regime of crystallization:? n = 4 when the crystallization conforms to regime I and regime I11 kinetics, whereas n = 2 when the crystallization process occurs according to regime 11; b, is the thickness of a macromolecular layer, and k is the Boltman constant. In the present work we exclude, a priori, a spherulite growth rate belonging to regime I11 kinetics because of the low undercoolings in~estigated.'~
The equation was used to fit the experimental data both for the homopoly- mer and the blends using a wide range of U* and C values (namely C = 30, 40, 51.6 K and U* from 1000 to 4000 in steps of 100, and 4129 cal/g). I t is to be noted that U* = 4120 cal/g and C = 51.6K are the values of the WLF constants used in the traditional expression for the transport term.lg For any value of C and U* used, eq. (2) always gave a good fit of the experimental G data. Some examples of fitting are shown in Figure 3. Hence, our crystalliza- tion data do not seem to provide a sensitive test of the applicability of the various transport parameters in eq. (2), probably because our growth rate data fall in the region where the formation of a nucleus of critical size controls the
+The regimes of behavior described in detail by Hoffman15 are determined by the mechanism of nucleation and growth of molecular layers on the surface of a lamellar crystal and are mainly dependent on the undercooling AT. At low AT the rate of molecular nucleation is sufficiently small that the formation of a surface secondary nucleus is followed by rapid completion of the substrate (regime I). The overall growth rate is hence proportional to the rate of molecular nucleation and n in eq. (3) is 4.
Similarly at low temperatures (high AT) the surface nuclei form in such large numbers that they have little, if any, room to spread laterally. Under these conditions the growth rate is again proportional to the rate of nucleation, and hence n is still equal to 4 (regime 111).
Finally, a t intermediate temperatures, the growth behavior is intermediate between regimes I and 111: nucleation now occurs a t higher rates than in regime I, so that adjacent nuclei must compete in spreading laterally on the crystal substrate; a t the same time, nucleation is not so dense as to hinder such lateral growth. In this range (regime II), the overall crystallization rate is proportional to the square root of the nucleation rate, and n is 2.

PEO/PEMA BLENDS 1787
growth rate. This seems to lead to the hypothesis that the crystallization conforms to regime I, where the rate of molecular nucleation is sufficiently small that adsorbed molecules can freely spread by chain folding, along the width of a lamellar substrate before a new nucleation event occurs.15
This hypothesis is confirmed by the application of the Lauritzen 2 test2' to our blends. 2 is a quantity defined by2'
X 2
z = 103( &) exp - (4)
[ L is the lamellar width, and a, is the chain width (in our case a, = b,)]. According to this test, if, by substitution for X in eq. (4) of values of K ,
derived from eq. (2), results in 2 < 0.01, then crystallization conforms to regime I. On the contrary, if substitution of 2 K , for X gives 2 > 1, then crystallization follows regime II.'o,22,23
On the basis of the 2 test if crystallization for our blends conforms to regime I, the lamellar width L of PEO should range between a few tens up to some hundreds of Angstroms for all the samples, whereas if crystallization conforms to regime 11, L values would be always greater than lo4 A. It seems realistic therefore to assume that the crystallization of pure PEO and of PEO from PEO/PEMA blends probably occurs according to regime I.
From the slope of the straight lines obtained by using eq. (2), the values of K , and a, can be obtained by setting u = O.lb, AHf and b,, = 4.65 A.2' I t is to be noted that K , varies only slightly with the U* and C values used in eq. (2) and that average values of K , are used in the calculation of ue. The values of ue for any blend composition are given in Table I.
It can be observed that a, decreases monotonically with the increase of PEMA in the blend. Such a depression may be accounted for by assuming that the presence of PEMA induces an increase in the surface entropy of folding. Consequently PEO lamellar crystals with a less regular fold surface are obtained when the PEO is allowed to crystallize isothermally from PEO/PEMA blends. Similar conclusions were also reached in the cases of PEO/PVAc" and PEO/PMMA systems.'
Isothermal Bulk Crystallization Kinetics
Typical crystallization isotherms, obtained by plotting X, (weight fraction of crystallizable material crystallized at time t ) against time,25*26 are reported in Figure 4, for 80:20 PEO/PEMA blends at different T,s. From these curves the half-time of crystallization defined as the time for half the crys- tallinity to develop, is obtained and plotted against T, for different blend compositions in Figure 5. From these curves it emerges that for a given T, the addition of PEMA causes a drastic reduction in the overall rate of crystalliza- tion and that such a depression increases with increasing PEMA content in the blend.
The Avrami equation24 was used to analyze the bulk kinetics of crystalliza- tion:
log( - h(1 - X , ) ) log K , + n log t (5)

1788 CIMMINO ET AL.
1.0
0.5
n -
PEO~PEMA 8012 0
XT
-
-
1700.
1300
9 0 0 .
5 0 0 .
100
319 K 32OK 321K 322 K
-
- 100 300 500 700 900 1100 1300
Fig. 4. Typical crystallization isotherm for the PEO/PEMA 80:20 blends crystallized at different T,.
( n is the Avrami index, and K , is the overall kinetic rate constant). The experimental data also fit the Avrami equation well a t very high time of crystallization, indicating that no observable secondary crystallization occurs. From plots of log( - ln(1 - X,) versus log t (always linear as in Fig. S), values of K , and n determined by the intercept and the slope are obtained (see Table 11) for all crystallization temperatures and compositions.
In almost all cases, the values of n are nonintegral, contrary to the theoretical ~red ic t ion .~~ These nonintegral values are generally accounted for by mixed growth and/or surface nucleation modes and secondary crystalliza-
Fig. 5.
I t,, ( r e c )

PEO/PEMA BLENDS
- 1.2
1789
L
LOG t -
EO/PEMA
80120 I 0.8
- 0.4 2
f L ao 2
r - Q
- 0.4
-0a
324
TABLE I1 Values of the Avrami Index n and of the Overall Kinetic Rate Constant K , as Functions of Crystallization Temperature T, and Blend Composition
PEO/PEMA (1OO:O) 320 321 322 323 324 325 326 327
319 320 32 1 322 323 324
319 320 32 1 322 323 324
316 317 318 319 320
PEO/PEMA (9O:lO)
PEO/PEMA (80:20)
PEO/PEMA (70:30)
2.1 2.0 2.2 2.0 2.3 2.3 2.4 2.3
2.4 2.5 2.5 2.1 2.2 2.3
2.5 2.4 2.5 2.8 2.5 2.3
2.8 2.4 2.1 2.2 2.4
4.171 X 1.714 X 1.338 X 6.144 X 2.665 x 1 0 - ~ 1.111 x 1 0 - ~ 2.840 x 1.067 X
6.197 X
1.134 X 4.614 X
2.068 x 1 0 - 6
1.005 x 1 0 - ~ 3.306 x 10-8
1.442 X 5.082 X 2.008 x 1 0 - ~
2.110 x 10-8 7.686 x lo-@
4.823 X lo-'
2.497 X
5.829 X lo-' 2.740 X 1.315 X
1.384 x 1 0 - ~

1790 CIMMINO ET AL.
tion (but this last process seems not to occur in our case, as we specified above). Recently Grenier and Prud’h~mme~~ found that experimental factors such as erroneous determination of the “zero” time and of the enthalpy of melting of the polymer at a given time can cause n to be nonintegral.
For a polymer-diluent system, the temperature dependence of the overall kinetic rate constant K , may be expressed by the
This equation also fits well the experimental data for a large range of U* and T values. In fact, irrespective of the U* and T values used, plots of a2 versus l / (Tc ATf ) always result in straight lines. From the slopes of such lines values of a, dependent on blend composition are obtained (see Table I). The composi- tion dependence of ue derived by the overall kinetics of crystallization is in agreement with that obtained from radial growth rate data, even though comparatively larger values are observed (compare data in Table I).
Melting Behavior and Melting Point Depression
The variation of the optically observed melting temperature T,,!, with the crystallization temperature T, for pure PEO and for PEO crystallized from the blends is shown in Figure 7. For a given T,, T,,!, values related to the blends are always lower than those for pure PEO, and the depression increases with increasing (PEMA) content. In the range of T, examined, the T,,!, of the samples increases linearly with T, according to the equation of Hoffman and weeks: 28
where Tm is the equilibrium melting temperature, and y is the ratio of the lamellar thickness Z to the lamellar thickness of the critical nucleus Z* at T,.
0
/
316 324 332 340 348 Tc(K)
Fig. 7. Variation of the observed melting temperature with T,.

PEO/PEMA BLENDS 1791
On the basis of eq. (7) the extrapolation of the straight lines to the intersection with the T; = T, line yields the equilibrium melting points, reported in Table I as a function of composition. The equilibrium melting temperature of PEO is lowered by the addition of the PEMA, and the depression is proportional to the PEMA content of the blend.
Generally the melting point depression of a crystalline polymer observed in blends is a result of kinetic, morphological, and thermodynamic factors. The kinetic effects result because crystals are formed at temperatures below the equilibrium melting point and can be avoided using equilibrium melting temperatures derived from Hoffman-Weeks plots.28
Melting point depressions caused by morphological effects29-32 are associ- ated with changes in crystal perfection or geometry and with different thermal histories of the sam~1es.l~ In fact Paul et aL3' have found depression of the melting points of poly(vinylidene fluoride)/polystyrene (PVFJPS) immiscible blends. Moreover, as Bernstein et aL3' reported, poly( ecaprolac- tone) (PCL) causes a depression of the PVF, melting point of about 6 or 7°C in blends containing 40% PVF,. The PVFJPCL system is known to be immiscible, at least in the molten state. Finally the melting point depression can be due to the decrease of the chemical potential of the crystallizable polymer caused by the addition of the miscible diluent. The expression of melting point depression due only to thermodynamic effects is given according to the Flory-Huggins theory
(8) 1
1 - -[- RV, h+, + (; - a)+' + xl&] T, T," AHoVl m,
The subscripts 1 and 2 represent the noncrystallizable and the crystallizable polymer, respectively, xlz is the polymer-polymer interaction parameter, V is the molar volume of the polymer repeat unit at the equilibrium melting temperature, +i is the volume fraction of component i in the blend, AHo is the perfect-crystal heat of fusion of the crystallizable polymer, m is the degree of polymerization, R is the universal gas constant, and T," and T, are the equilibrium melting temperatures of the pure crystallizable component and of the blend, respectively.
By rearranging the terms of eq. (8), one gets
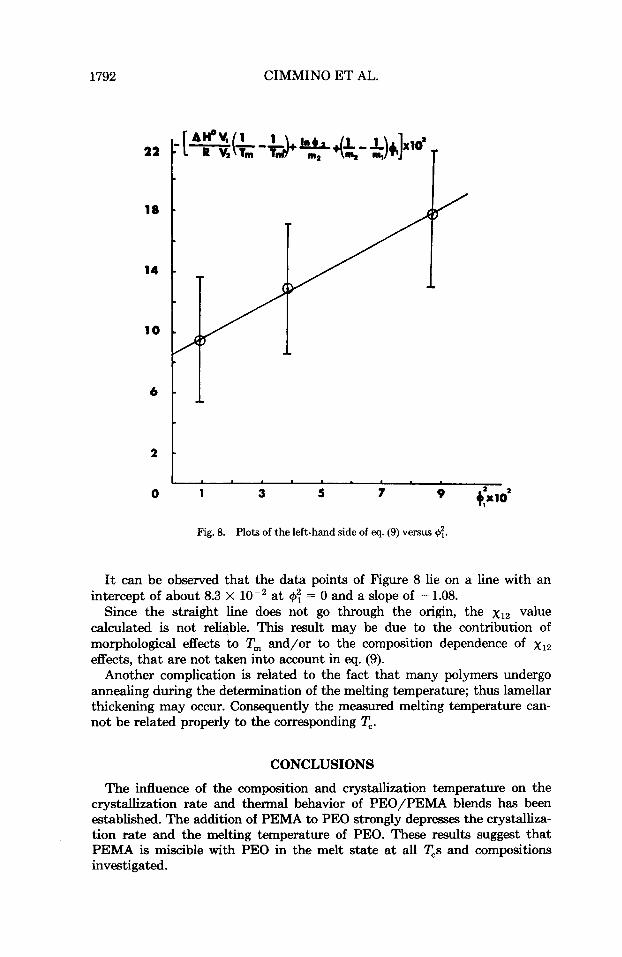
A plot of the left-hand side of eq. (9) versus should give a straight line passing through the origin if the interaction parameter can be assumed to be composition independent. The slope of the straight line gives xlz directly.
With the values of T," and the T,s reported in Table I, the plot shown in Figure 8 is obtained. The error bars are asS0ciat.d with the experimental error in T;. The following parameter values have been used in our calculations: AHo = 1980 (cal mol-'), V, = 102 (an3 mol-'): V, = 40.6 (cm3 mol-'),34 m, = 1316, and m2 = 454.
1792 CIMMINO ET AL.
22
18
14
10
6
2
0 1 3 5 7
Fig. 8. Plots of the left-hand side of eq. (9) versus +;.
It can be observed that the data points of Figure 8 lie on a line with an intercept of about 8.3 X lo-' at $I: = 0 and a slope of - 1.08.
Since the straight line does not go through the origin, the xI2 value calculated is not reliable. This result may be due to the contribution of morphological effects to T, and/or to the composition dependence of xlz effects, that are not taken into account in eq. (9).
Another complication is related to the fact that many polymers undergo annealing during the determination of the melting temperature; thus lamellar thickening may occur. Consequently the measured melting temperature can- not be related properly to the corresponding T,.
CONCLUSIONS
The influence of the composition and crystallization temperature on the crystallization rate and thermal behavior of PEO/PEMA blends has been established. The addition of PEMA to PEO strongly depresses the crystalliza- tion rate and the melting temperature of PEO. These results suggest that PEMA is miscible with PEO in the melt state at all T,s and compositions investigated.

PEO/PEMA BLENDS 1793
TABLE 111 Comparison of Spherulite Radial Growth Rate G and the Half-time of Crystallization to.5
for PEO/PVAc, PEO/PMMA, Systems at different Crystallization Temperatures and Compositions
320 32 1 322 323 324 325 326
512 387 305 244 181 139 105
0.5 0.8 0.9 1.2 1.8 2.5 4.7
T, PEO/PEMA PEO/PVAc PEO/PMMA PEO/PEMA PEO/PVAc PEO/PMMA
PEO I: 90:lO 320 32 1 322 323 324 325
320 321 322 323 324 325
PEO I: 80:20
468 358 270 210 154 -
326 255 210 150 123 -
325 180 135 110 90 60
160 120 95 70 45 -
126 122 84 46 28
53.9 42.2 28.3 34.2 19.1 12.3
4.2 5.5 8.1
15.7 30.5
4.7 6.9
10.1 16.9 30.5
1.5 1.7 1.7 3.4 5.7 8.7
1.8 1.7 2.5 3.7 6.2 7.9
3.2 3.2 4.4 4.5 6.6
11.1
6.5 8.5
10.5 15.9 21.6 -
Evaluation of the Flory-Huggins interaction parameter by analysis of the melting point depression is not possible, perhaps because of the influence on T, of kinetic and morphological factors associated with the annealing process and/or maybe because xI2 of the PEO/PEMA blend is composition depen- dent. It is noteworthy that influence of composition and T, upon kinetic and thermodynamic quantities found for the PEO/PEMA blends is like that observed by us for PEO/PMMA' and PEO/PVAc" blends.
On the other hand for the same blend composition and the same T, (see Table 111), the values of G and to,, of PEO depend on the kind of amorphous material added to PEO (for example G p E O p E m > GPEO,PVAc > G p E O p M m
at a given T, and composition). These differences result surely from a complex balance among:
1. Degree of miscibility and mobility of the crystallizable and noncrystalliz-
2. influence of the amorphous material on the nucleation of PEO. 3. influence of noncrystallizable material on the secondary nucleation pro-
cess, i.e., on the crystallization regime. In this regard it is worth mentioning that pure PEO and PEO/PEMA blends crystallize according to regime I, whereas the crystallization of PEO/PMMA and PEO/PVAc systems con- forms to regime 11, at least in the range of composition and T, investigated previously in our laboratory. Therefore the secondary nucleation process of
able components in the melt state.

1794 CIMMINO ET AL.
PEO is dependent on the type of amorphous component present in the blend, as is confirmed also by morphological analysis (see Refs. 7 and 10).
A careful analysis is in progress in order to understand the predominance of one of the factors indicated above with respect to the others and therefore to explain the differences in crystallization rates here reported for the three systems.
This work has been partially supported by C.N.R. "Progetto Finalizzato Chimica Fine e Secondaria."
References 1. E. Martuscelli and G. B. Demma, in Polymeric Blenak: Processing M o p h b g y and
Properties, E. Martuscelli, R. Palumbo, and M. Kryszewski, Eds., Plenum Press, New York, 1980; E. Martuscelli, M. Pracella, W. P. Yue, Polymer, 25, 1097 (1984).
2. R. L. Imken, D. R. Paul, and J. W. Barlow, Polym. Eng. Sci., 16, 593 (1976). 3. E. Martuscelli, M. Canetti, L. Vicini, and A. Seves, Polymer, 23, 331 (1982). 4. E. Martuscelli, G. Demma, E. R&, and A. L. Sege, Polym. Commun., 24, 266 (1983). 5. E. Martuscelli, L. Vicini, and A. Seves, Makromol. Chem., Rapid Commun. 5, 255 (1984). 6. G. Ramana Rao, C. Castiglioni, M. Gussoni, E. Martuscelli, and G. Zerbi, Polymer, 26, 811
7. E. Martuscelli, C. Silvestre, M. L. Addoniio, and L. Amelino, Makromol. Chem., 187, 1557
8. M. L. Addoniio, E. Martuscelli, and C. Silvestre, Polymer, 28, 183 (1987). 9. C. Silvestre, S. Cimmino, E. Martuscelli, F. E. Karasz, and W. W. Macknight, Polymer, 28,
(1985).
(1986).
1191 (1987). 10. E. Martuscelli, C. Silvestre, and G. Gismondi, Makromol. Chem., 187, 2161 (1985). 11. I. C. Sanchez, in Polymer Blends, D. R. Paul and S. Newman, eds., Academic Press, New
12. N. Kalfoglou, J. Polym. Sci. Polym. Phys. Ed., 20, 1259 (1982). 13. T. G. Fox, Bull. Am. Phys. SOC., 2, 123 (1956). 14. H. D. Keith and F. P. Padden, J. Appl. Phys., 535, 1270 (1964). 15. J. D. Hoffman, Polymer, 24, 3 (1982). 16. J. Boon and J. M. Azcue, J. Polym. Sci., Part A-2, 6, 885 (1968). 17. E. Martuscelli, Polym. Eng. Sci., zd, 563 (1984). 18. G. C. Alfonso and T. P. Russell, Macromolecules, 19, 1143 (1986). 19. M. L. Williams, R. F. Landel, and J. D. Ferry, J. Am. Chem. SOC., 77, 3701 (1975). 20. J. I. Lauritzen Jr., J. Appl. Phys., 44, 4353 (1973). 21. G. Vidotto, D. L. Levy, and A. J. Kovacs, Kollold 2.2. Polymer, 230,289 (1969). 22. J. I. Lauritzen, Jr. and J. D. Hoffman, J. Appl. Phys., 44, 4340 (1973). 23. T. T. Wang and T. Nishi, Macromolecules, 10, 421 (1977). 24. M. Avrami, J. Chem. Phys., 7 , 1103 (1939). 25. D. Grenier and E. R. Prud'homme, J. Polym. Sci. Polym. Phys. Ed., 18, 1655 (1980). 26. B. Wunderlich, M a c r o m l e c h Physics, Academic, New York, 1976, Vol. 2. 27. L. Mandelkem, Crystallization of Polymers, McGraw-Hill, New York, 1964. 28, J. D. Hoffman and J. J. Weeks, J. Chem. Phys, 37, 172 (1962). 29. D. R. Paul, J. W. Barlow, R. E. Bemstein, and D. C. Wahrmund, Polym. Eng. Sci., 18,
30. R. E. Bemstein, D. C. Wahrmund, J. W. Barlow, and D. R. Paul, Polym. Eng. Sci., 18,
31. I. R. Harrison and J. Runt, J. Polym. Sci. Polym. Phys. Ed., 18, 2257 (1980). 32. J. P. Runt, Macromolecules, 14, 420 (1981). 33. P. J. Flory, Binciph of Polymer Chemisby, Comell University Press, Ithaca, New York,
34. T. Nishi and T. T. Wang, Macromolecules, 8, 909 (1975). 35. D. W. Van Krevelen, Properties of Polymers, Elsevier, Amsterdam, 1972.
York, 1978, Vol. 1, Chap. 3.
1225 (1978).
1220 (1978).
1953.
Received April 1, 1987 Accepted April 15, 1988
Copyright © 2022 FDOKUMEN




















