
Fiber-like Micelles via the Crystallization-Driven Solution Self-Assembly of Poly(3-hexylthiophene)-...
10
Fiber-like Micelles via the Crystallization-Driven Solution Self- Assembly of Poly(3-hexylthiophene)-block-Poly(methyl methacrylate) Copolymers Joe B. Gilroy, † David J. Lunn, † Sanjib K. Patra, † George R. Whittell, † Mitchell A. Winnik,* ,‡ and Ian Manners* ,† † School of Chemistry, University of Bristol, Cantock’s Close, Bristol BS8 1TS, United Kingdom ‡ Department of Chemistry, University of Toronto, Toronto, Ontario M5S 3H6, Canada * S Supporting Information ABSTRACT: The solution self-assembly of block copolymers with a π-conjugated, crystalline, core-forming block represents a facile strategy toward the preparation of semiconducting nanowires with potential for high-tech applications. In this study, two asymmetric block copolymers based on regioregular poly(3-hexylthiophene) (P3HT) and poly(methyl methacry- late) (PMMA), namely P3HT 40 -b-PMMA 520 (6a) and P3HT 40 -b-PMMA 1100 (6b) (block ratios = 1:13 and 1:27.5, respectively) were prepared via atom transfer radical polymer- ization (ATRP) from a P3HT macroinitiator. The solution self-assembly of the P3HT-b-PMMA block copolymers was subsequently studied under a variety of experimental conditions. Short, fiber-like micelles resulted when THF (common solvent for P3HT and PMMA) solutions of the block copolymer were dialyzed against ethyl acetate and n-butyl acetate (selective solvents for PMMA). The electronic properties of the fiber-like micelles obtained coupled with wide-angle X-ray scattering studies confirmed that the cores of the aggregates were crystalline and suggested that growth occurs via a crystallization-driven pathway. The average lengths of fiber-like micelles were shown to increase relative to those obtained from dialysis versus the PMMA selective solvent, when THF was slowly evaporated from mixtures containing n-butyl acetate and P3HT-b-PMMA unimers, thereby limiting the rate of P3HT aggregation. Furthermore, the formation of only relatively short (mainly under 200 nm, always <1 μm) fiber-like micelles in these studies, even when the ratio of THF/alkyl acetate was controlled carefully via dialysis or evaporation, indicated that homogeneous nucleation of P3HT-b-PMMA block copolymers is relatively facile. This behavior differs significantly from that detected for other block copolymers such as polyferrocenylsilane-based materials that undergo crystallization-driven self-assembly to form cylinders with lengths of up to 10 μm under analogous conditions. ■ INTRODUCTION The solution self-assembly of amphiphilic block copolymers in solution has emerged as a promising strategy for the realization of a wide range of functional core-shell nanostructures and depends on a number of parameters, including the solubility properties, molecular weights, and relative volume fractions of the constituent blocks. 1,2 In solvents selective for one of the blocks, toroids, 3 vesicles, 4 disks, 5 pointed ovals, 6 helices, 7 and more complex structures 8,9 have been shown to form. Fiber-like micelles 10-12 (nanofibrils or cylindrical micelles) formed in solution by the self-assembly of block copolymers have attracted significant interest as a result of their utility in drug delivery applications, 13 as strength-enhancers in plastic manufacturing, 14 as templates for inorganic nanoparticles, 15 and as precursors to nanopatterned ceramics. 16 Such structures, however, can only be obtained from all-amorphous block copolymers when the corona-forming block is short, and these materials must often be covalently cross-linked within the core or corona in order to preserve their micelle architectures and to limit their concentration-dependent properties. 17,18 We have found that employing block copolymers containing a semicrystalline core-forming block [e.g., poly- (ferrocenyldimethylsilane) (PFDMS)] permits the formation of fiber-like micelles with lengths between 500 nm and 10 μm over a wide compositional range. 10 Furthermore, as a consequence of the lattice energy associated with the favorable formation of the semicrystalline PFDMS core, 19 these kineti- cally trapped structures persist in dilute solution without disassembly to unimers, even in the absence of cross-linking. Moreover, we have been able to show that under certain conditions this type of crystallization-driven self-assembly can behave as a “living” process, and added block copolymer unimers will grow from the ends of shorter seed micelles. This seeded growth method relies on epitaxial crystallization of the core-forming block and has allowed access to cylindrical block comicelles with narrow length distributions, 19-21 spatially Received: April 22, 2012 Revised: June 18, 2012 Published: July 12, 2012 Article pubs.acs.org/Macromolecules © 2012 American Chemical Society 5806 dx.doi.org/10.1021/ma3008114 | Macromolecules 2012, 45, 5806-5815
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Fiber-like Micelles via the Crystallization-Driven Solution Self-Assembly of Poly(3-hexylthiophene)-...

Fiber-like Micelles via the Crystallization-Driven Solution Self-Assembly of Poly(3-hexylthiophene)-block-Poly(methylmethacrylate) CopolymersJoe B. Gilroy,† David J. Lunn,† Sanjib K. Patra,† George R. Whittell,† Mitchell A. Winnik,*,‡
and Ian Manners*,†
†School of Chemistry, University of Bristol, Cantock’s Close, Bristol BS8 1TS, United Kingdom‡Department of Chemistry, University of Toronto, Toronto, Ontario M5S 3H6, Canada
*S Supporting Information
ABSTRACT: The solution self-assembly of block copolymerswith a π-conjugated, crystalline, core-forming block representsa facile strategy toward the preparation of semiconductingnanowires with potential for high-tech applications. In thisstudy, two asymmetric block copolymers based on regioregularpoly(3-hexylthiophene) (P3HT) and poly(methyl methacry-late) (PMMA), namely P3HT40-b-PMMA520 (6a) andP3HT40-b-PMMA1100 (6b) (block ratios = 1:13 and 1:27.5,respectively) were prepared via atom transfer radical polymer-ization (ATRP) from a P3HT macroinitiator. The solution self-assembly of the P3HT-b-PMMA block copolymers wassubsequently studied under a variety of experimental conditions. Short, fiber-like micelles resulted when THF (common solventfor P3HT and PMMA) solutions of the block copolymer were dialyzed against ethyl acetate and n-butyl acetate (selectivesolvents for PMMA). The electronic properties of the fiber-like micelles obtained coupled with wide-angle X-ray scatteringstudies confirmed that the cores of the aggregates were crystalline and suggested that growth occurs via a crystallization-drivenpathway. The average lengths of fiber-like micelles were shown to increase relative to those obtained from dialysis versus thePMMA selective solvent, when THF was slowly evaporated from mixtures containing n-butyl acetate and P3HT-b-PMMAunimers, thereby limiting the rate of P3HT aggregation. Furthermore, the formation of only relatively short (mainly under 200nm, always <1 μm) fiber-like micelles in these studies, even when the ratio of THF/alkyl acetate was controlled carefully viadialysis or evaporation, indicated that homogeneous nucleation of P3HT-b-PMMA block copolymers is relatively facile. Thisbehavior differs significantly from that detected for other block copolymers such as polyferrocenylsilane-based materials thatundergo crystallization-driven self-assembly to form cylinders with lengths of up to 10 μm under analogous conditions.
■ INTRODUCTION
The solution self-assembly of amphiphilic block copolymers insolution has emerged as a promising strategy for the realizationof a wide range of functional core−shell nanostructures anddepends on a number of parameters, including the solubilityproperties, molecular weights, and relative volume fractions ofthe constituent blocks.1,2 In solvents selective for one of theblocks, toroids,3 vesicles,4 disks,5 pointed ovals,6 helices,7 andmore complex structures8,9 have been shown to form.Fiber-like micelles10−12 (nanofibrils or cylindrical micelles)
formed in solution by the self-assembly of block copolymershave attracted significant interest as a result of their utility indrug delivery applications,13 as strength-enhancers in plasticmanufacturing,14 as templates for inorganic nanoparticles,15 andas precursors to nanopatterned ceramics.16 Such structures,however, can only be obtained from all-amorphous blockcopolymers when the corona-forming block is short, and thesematerials must often be covalently cross-linked within the coreor corona in order to preserve their micelle architectures and tolimit their concentration-dependent properties.17,18
We have found that employing block copolymers containinga semicrystal l ine core-forming block [e.g. , poly-(ferrocenyldimethylsilane) (PFDMS)] permits the formationof fiber-like micelles with lengths between 500 nm and 10 μmover a wide compositional range.10 Furthermore, as aconsequence of the lattice energy associated with the favorableformation of the semicrystalline PFDMS core,19 these kineti-cally trapped structures persist in dilute solution withoutdisassembly to unimers, even in the absence of cross-linking.Moreover, we have been able to show that under certainconditions this type of crystallization-driven self-assembly canbehave as a “living” process, and added block copolymerunimers will grow from the ends of shorter seed micelles. Thisseeded growth method relies on epitaxial crystallization of thecore-forming block and has allowed access to cylindrical blockcomicelles with narrow length distributions,19−21 spatially
Received: April 22, 2012Revised: June 18, 2012Published: July 12, 2012
Article
pubs.acs.org/Macromolecules
© 2012 American Chemical Society 5806 dx.doi.org/10.1021/ma3008114 | Macromolecules 2012, 45, 5806−5815
segregated coronas22,23 or cores,9 hierarchical scarf-like micellesand micelle brushes,9,18 and the random and controlledformation of networks of cylindrical micelles.24 These recentadvances build on earlier studies on several crystalline-coilblock copolymers in which platelet-like micelles form insolvents selective for the amorphous block25 and have alsobeen extended to the self-assembly of fiber-like micelles basedon block copolymers with poly(ε-caprolactone),26 polyacrylo-nitrile,27 poly(ethylene oxide),28 polyethylene,12,29 and enan-tiopure polylactide30,31 crystalline blocks.The solution processing of π-conjugated, conducting
hompolymers to yield conducting fibers has been widelyexplored,32 but systems based on this strategy are oftenhampered by poor solubility/colloidal stability and unfavorablefiber aggregation. Significant advances have been made towardlimiting aggregation and controlling the structure of aggregatesby employing rod−rod block copolymers based on twodifferent poly(3-alkylthiophene)s (P3ATs),33 affording one-dimensional fibers with cross-linked shells,34 vesicles,35 andhelical structures.36 In contrast, the solution self-assembly ofamphiphilic rod−coil block copolymers containing a semi-crystalline, π-conjugated core-forming block to yield fiber-likemicelles has not been developed to the same extent.37−39
Continued development of solution self-assembly strategies forthe realization of fiber-like micelles based on rod−coil blockcopolymers containing a highly crystalline π-conjugated,conducting block is desirable as their inherent crystallinitymay significantly enhance device performance. This enhance-ment has been demonstrated in photovoltaic devices based oncrystalline P3HT homopolymer fibers prepared in solution,which outperform devices made from annealed thin films of thesame composition.40
Recently we reported preliminary studies of a P3HT blockcopolymer with a polydimethylsiloxane (PDMS) coronaforming block.38 We were able to demonstrate thatcrystallization-driven self-assembly of P3HT leads to theformation of cylindrical micelles with a P3HT core. However,attempts to control the lengths of the cylinders by seededgrowth met with only moderate success and was not possiblebeyond values of ca. 250 nm. Clearly, if this approach is toadvance, a more detailed understanding of the crystallization-driven self-assembly process for rod−coil block copolymerscontaining a π-conjugated P3HT core-forming block is needed.As a step toward this target, in this paper we describe thesynthesis and solution self-assembly of two P3HT-b-poly-(methyl methacrylate) (PMMA) block copolymers in selectivesolvents for PMMA.
■ RESULTS1. Synthesis of P3HT-b-PMMA Copolymers 6a and 6b.
a. Synthesis of a P3HT Atom Transfer Radical Polymer-ization (ATRP) Macroinitiator (5). A number of syntheticstrategies toward semiconducting polymers with controlledmolecular weights and narrow polydispersity indices (PDIs)exist.41,42 In this study we employed Grignard metathesis(GRIM) protocols to afford regioregular vinyl-terminatedP3HT.43 2,5-Dibromo-3-hexylthiophene (1) was first treatedwith tert-butylmagnesium chloride in order to generate a 3-hexylthiophene-based Grignard species. The Grignard was thenpolymerized for 10 min at 20 °C using [1,3-bis-(diphenylphosphino)propane]dichloronickel(II) [Ni(dppp)-Cl2] as a polymerization catalyst, before vinylmagnesiumchloride was added to quench the reaction, affording P3HT 2
with a vinyl group at the α-position (GPC: Mn = 9750 g mol−1,PDI = 1.18, Scheme 1, Table 1).44 Comparison of the 1H NMR
integrals for the geminal protons of the vinyl group with thethiophene proton of the regioregular polymer backbonerevealed a number-average degree of polymerization (DPn) of40 (1H NMR: Mn = 6750 g mol−1, Table 1).45 In order toeliminate the potential for undesired side reactions associatedwith the bromo substituent at the ω-position, α-vinyl-ω-bromoP3HT 2 was further reacted with phenylmagnesium bromideand Ni(dppp)Cl2 according to literature procedures to afford α-vinyl-ω-phenyl P3HT 3 (1H NMR: Mn = 7600 g mol−1, GPC:Mn = 9650 g mol−1, PDI = 1.24, Scheme 1, Table 1).46
Hydroboration of the vinyl end group using 9-borabicyclo[3.3.1]nonane (9-BBN) followed by oxidativework-up with NaOH/H2O2 introduced alcohol functionalityand afforded α-hydroxyethyl-ω-phenyl P3HT 4 (1H NMR: Mn= 8450 g mol−1, GPC: Mn = 7450 g mol−1, PDI = 1.43,Scheme 2, Table 1).47 Broadening of the molecular weightdistribution in P3HT 4 relative to its precursors may arise as aresult of undesired oxidative coupling reactions duringtreatment of the boryl intermediate with hydrogen peroxide.The alcohol functionality in 4 was then employed in theproduction of ATRP macroinitiator 5 (1H NMR: Mn = 6900 gmol−1, GPC: Mn = 7750 g mol−1, PDI = 1.40, Scheme 2, Table1), which was produced in quantitative yield via reaction of thealcohol group with α-bromoisobutyryl bromide in the presenceof triethylamine. The presence of the α-bromoisobutyrylfunctionality was confirmed by 1H NMR analysis of 5. Theresonances associated with the ethylene moiety originatingfrom the alcohol end group shifted from 3.88 ppm (d of t) and3.03 ppm (t) in 4 to 4.36 ppm (t) and 3.14 ppm (t) in 5.Furthermore, the appearance of a new resonance associatedwith the methyl groups of the α-bromoisobutyryl group at 1.96ppm (s) was consistent with the structure of the ATRPmacroinitiator 5.The physical properties of ATRP macroinitiator 5 were
studied in solution and the solid state. When dissolved in THF,5 exhibits a strong absorption in its electronic spectrum at λmax= 445 nm (π−π*), which is characteristic of regioregular π-conjugated P3HTs (Figure S2a).34,48 The photoluminescent
Scheme 1. Synthesis of α-Vinyl-ω-phenyl P3HT
Table 1. Molecular Weight Data for Homopolymers 2−5
polymer Mn (g mol−1)a Mw (g mol−1)a PDIa Mn (g mol−1)b DPnb
2 9750 11 550 1.18 6750 403 9650 11 900 1.24 7600 454 7450 10 600 1.43 8450 505 7750 10 800 1.40 6900 40
aDetermined by GPC (conventional calibration vs polystyrenestandards). bDetermined by comparing the 1H NMR integration ofthe resonances associated with the end group and the thiopheneproton. For refractive index (RI) and UV GPC traces see Figure S1.
Macromolecules Article
dx.doi.org/10.1021/ma3008114 | Macromolecules 2012, 45, 5806−58155807

properties of 5 in THF were also characteristic of regioregularP3HTs with a maximum observed emission intensity (λem) of575 nm (Figure S2b).49
In the solid state, P3HT hompolymers are semicrystallinedue to π-stacking interactions and favorable interdigitation ofthe hexyl side chains.50 In order to probe the crystallization ofhomopolymer 5, wide-angle X-ray scattering (WAXS) anddifferential scanning calorimetry (DSC) measurements wereconducted. The WAXS pattern obtained for 5 contained threedifferent peaks associated with three-dimensional orderingrelating to interdigitation of the hexyl side chains: 2θ = 5.6°, d =1.60 nm (100); 2θ = 11.1°, d = 0.080 nm (200); and 2θ =16.8°, d = 0.53 nm (300) and a lone signal associated with π-stacking between neighboring polythiophene chains at 2θ =23.0°, d = 0.38 nm (010) (Figure S3). DSC analysis (scan rate= 10 °C min−1) of 5 in the solid state revealed both a meltendotherm (Tm = 207 °C) and a crystallization exotherm (Tc =185 °C) (Figure S4). Integration of the melt exotherm affordedan enthalpy of fusion of 12.0 J g−1 for the as-prepared sample,which was ca. 12% of the value expected for an ideal crystal.51
The characterization data collected in solution and the solid-state for 5 confirmed the regioregular, π-conjugated nature ofthe homopolymer backbone and also the semicrystallinity ofbulk samples. With these factors in mind, ATRP macroinitiator5 was considered to be an ideal precursor to block copolymersexpected to undergo crystallization-driven self-assembly insolution.b. Synthesis of Regioregular P3HT-b-PMMA Copolymers
6a and 6b. P3HT homopolymers have been employed asmacroinitiators for a number of controlled radical polymer-izations [e.g., nitroxide-mediated polymerization (NMP),radical addition−fragmentation chain transfer (RAFT), andATRP] to afford a diverse range of block copolymers, includingP3HT-b-poly(methyl acrylate) (PMA),47,52,53 P3HT-b-poly-(N,N-dimethylamino-2-ethyl methacrylate) (DMAEMA),54
P3HT-b-poly(tert-butyl acrylate) (Pt-BuA),47 P3HT-b-polyiso-prene (PI),55 P3HT-b-polystyrene (PS),52,55 P3HT-b-PMMA,56 P3HT-b-poly(4-vinylpyridine) (P4VP),57 andP3HT-b-(vinylphenyl oxadiazole).58 In this work, we used an
α-bromoisobutyryl group as the initiatior for ATRP of methylmethacrylate. P3HT macroinitiator 5 was combined with equalamounts of THF and methyl methacrylate (5 mL of each) andN,N,N′,N′,N″-pentamethyldiethylenetriamine (PMDETA).The entire mixture was subjected to three freeze−pump−thaw cycles and the freshly degassed solution combined withcopper(I) bromide before it was rapidly warmed to 75 °C withconstant stirring (Scheme 3). After 30 min of stirring at 75 °Capproximately half of the reaction mixture was removed, filteredthrough a plug of neutral alumina to remove copper salts, andprecipitated into rapidly stirred methanol. The polymer wasrecovered, dissolved in THF, and the precipitation repeated.The polymer was collected and dried in vacuo to afford P3HT-b-PMMA block copolymer 6a as a glassy, dark purple solid. Theremainder of the reaction mixture was allowed to stir for afurther 30 min at 75 °C and subjected to the same purificationprocedure described above to yield P3HT-b-PMMA 6b, whichwas a less intense purple color compared to 6a as a result of therelatively higher PMMA content.P3HT-b-PMMA block copolymers 6a and 6b were purified
by preparative size exclusion chromatography (SEC) in orderto remove low molecular weight species (i.e., P3HThomopolymer, coupled P3HT homopolymer, and prematurelyterminated P3HT-b-PMMA). The results of the SECpurifications were dramatic with a decrease in PDI from 1.74to 1.28 for 6a (Figure 1) and from 2.01 to 1.27 for 6b(FigureS5). 1H NMR integration of unique signals (e.g., P3HTthiophene =CH− and PMMA −OCH3) in each of the blocksallowed for determination of the block ratio in each material[6a: P3HT40-b-PMMA520, Mn (1H NMR) = 58 650 g mol−1;6b: P3HT40-b-PMMA1100, Mn (
1H NMR) = 116 650 g mol−1].The properties of block copolymers 6a and 6b in the solid
state have some similarities to those of macroinitiator 5. TheWAXS patterns for 6a and 6b are dominated by an amorphoushalo associated with the predominant PMMA blocks between2θ = 7°−25°. However, reflections centered at 2θ = 5.5°, d =1.72 nm for both 6a and 6b were observed (Figure S6). Asthese signals are generally associated with the (100) reflectionin P3HT hompolymers, we interpret their presence as an
Scheme 2. Synthesis of P3HT-Based ATRP Macroinitiator 5
Scheme 3. Synthesis of P3HT-b-PMMA Block Copolymers from P3HT Macroinitiator 5
Macromolecules Article
dx.doi.org/10.1021/ma3008114 | Macromolecules 2012, 45, 5806−58155808
indication that the P3HT block is semicrystalline in bulksamples of 6a and 6b. DSC analysis (scan rate = 10 °C min−1)of block copolymers 6a and 6b revealed glass transitiontemperatures (Tg) of 126 °C associated with the PMMA blocks.Both copolymers showed endotherms due to melt transitionsassociated with the semicrystalline P3HT block. For blockcopolymer 6a, this feature was centered at Tm = 201 °C and theenthalpy of fusion (2.0 J g−1) corresponded to ca. 20% of thevalue expected for an ideal crystal (Figure S7).51 Similarly,copolymer 6b melted at Tm = 198 °C and had a similarenthalpy of fusion (1.2 J g−1, ca. 24% of ideal, Figure S8).51 For6a a small exotherm associated with crystallization of the P3HTblock was observed at Tc = 141 °C. However, for 6b nocrystallization exotherm was observed. This observation mayrelate to decreased mobility, and thus ability to crystallize, ofthe P3HT block as the relative volume fraction of PMMA isincreased or to the fact that the magnitude of the exotherm wastoo small to be observed.2. Solution Self-Assembly of P3HT-b-PMMA Copoly-
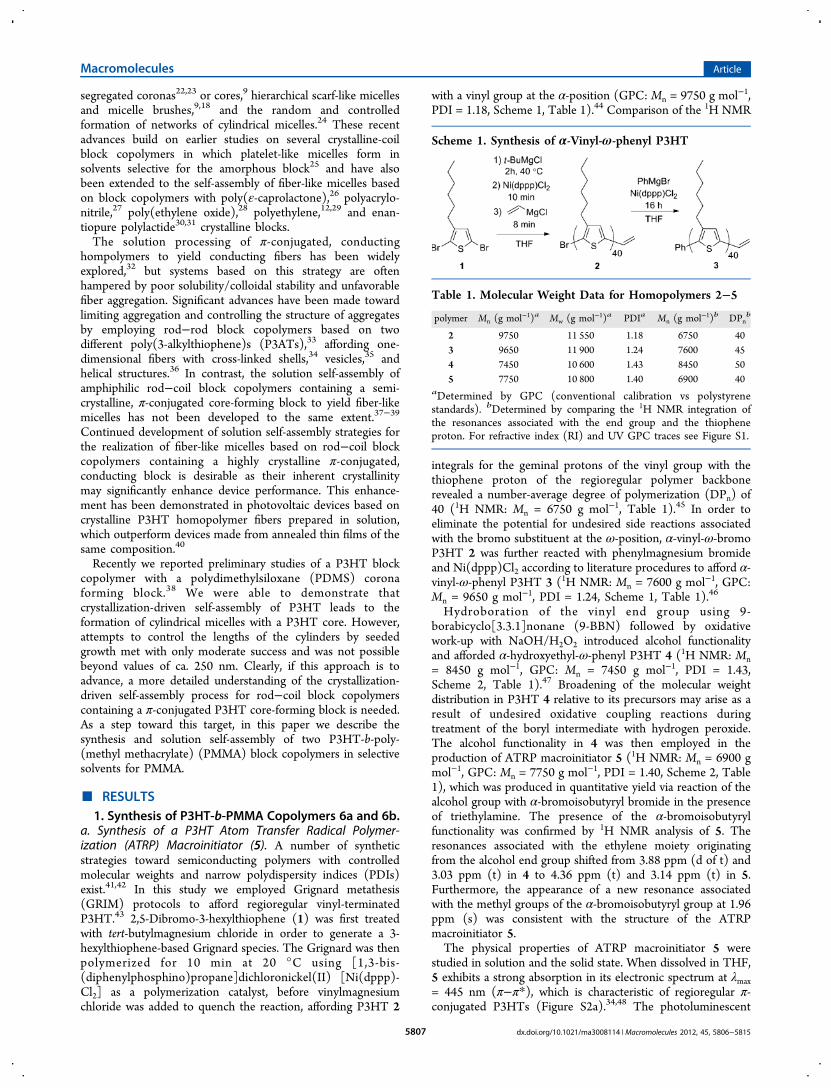
mers 6a and 6b. In order to observe the aggregation behaviorof the P3HT-b-PMMA block copolymers described in thisstudy, dialysis experiments were conducted. The slowintroduction of a solvent selective for the corona formingblock to a solution of common solvent has been widelyexplored as a method for preparing a wide variety of micellearchitectures.1 This method allows for the amount of commonsolvent, and thus free block copolymer (i.e., unimer) in solutionto be controlled. To this end, 0.5 mg mL−1 solutions of 6a and6b in THF were dialyzed against ethyl acetate (EtOAc) for 16 hto remove the common solvent and to allow for the aggregationand crystallization of the core-forming P3HT block. Thevolumes of the resulting solutions were adjusted by dilution tore-establish the original concentration before further analysiswas conducted. The introduction of a selective solvent forPMMA (i.e., EtOAc) was accompanied by a marked colorchange from yellow to dark red, which is commonly associatedwith the formation of crystalline P3HT aggregates in solution(Figure 2).34,48,50
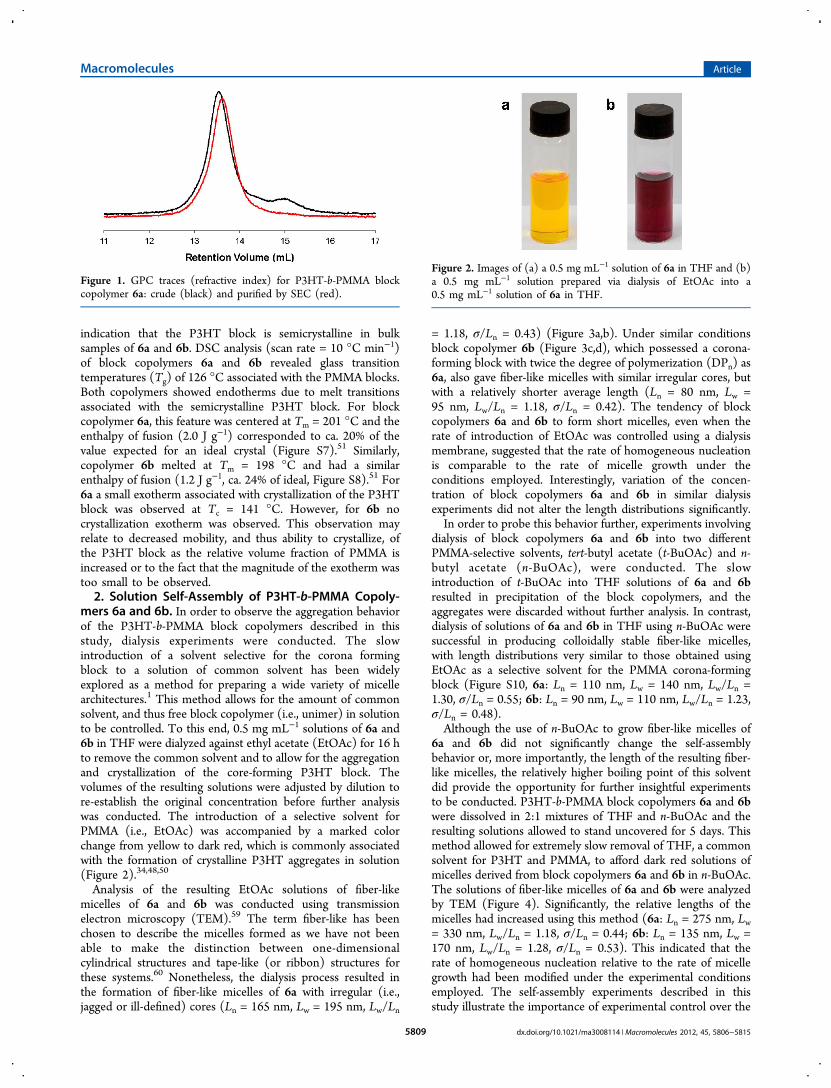
Analysis of the resulting EtOAc solutions of fiber-likemicelles of 6a and 6b was conducted using transmissionelectron microscopy (TEM).59 The term fiber-like has beenchosen to describe the micelles formed as we have not beenable to make the distinction between one-dimensionalcylindrical structures and tape-like (or ribbon) structures forthese systems.60 Nonetheless, the dialysis process resulted inthe formation of fiber-like micelles of 6a with irregular (i.e.,jagged or ill-defined) cores (Ln = 165 nm, Lw = 195 nm, Lw/Ln
= 1.18, σ/Ln = 0.43) (Figure 3a,b). Under similar conditionsblock copolymer 6b (Figure 3c,d), which possessed a corona-forming block with twice the degree of polymerization (DPn) as6a, also gave fiber-like micelles with similar irregular cores, butwith a relatively shorter average length (Ln = 80 nm, Lw =95 nm, Lw/Ln = 1.18, σ/Ln = 0.42). The tendency of blockcopolymers 6a and 6b to form short micelles, even when therate of introduction of EtOAc was controlled using a dialysismembrane, suggested that the rate of homogeneous nucleationis comparable to the rate of micelle growth under theconditions employed. Interestingly, variation of the concen-tration of block copolymers 6a and 6b in similar dialysisexperiments did not alter the length distributions significantly.In order to probe this behavior further, experiments involving
dialysis of block copolymers 6a and 6b into two differentPMMA-selective solvents, tert-butyl acetate (t-BuOAc) and n-butyl acetate (n-BuOAc), were conducted. The slowintroduction of t-BuOAc into THF solutions of 6a and 6bresulted in precipitation of the block copolymers, and theaggregates were discarded without further analysis. In contrast,dialysis of solutions of 6a and 6b in THF using n-BuOAc weresuccessful in producing colloidally stable fiber-like micelles,with length distributions very similar to those obtained usingEtOAc as a selective solvent for the PMMA corona-formingblock (Figure S10, 6a: Ln = 110 nm, Lw = 140 nm, Lw/Ln =1.30, σ/Ln = 0.55; 6b: Ln = 90 nm, Lw = 110 nm, Lw/Ln = 1.23,σ/Ln = 0.48).Although the use of n-BuOAc to grow fiber-like micelles of
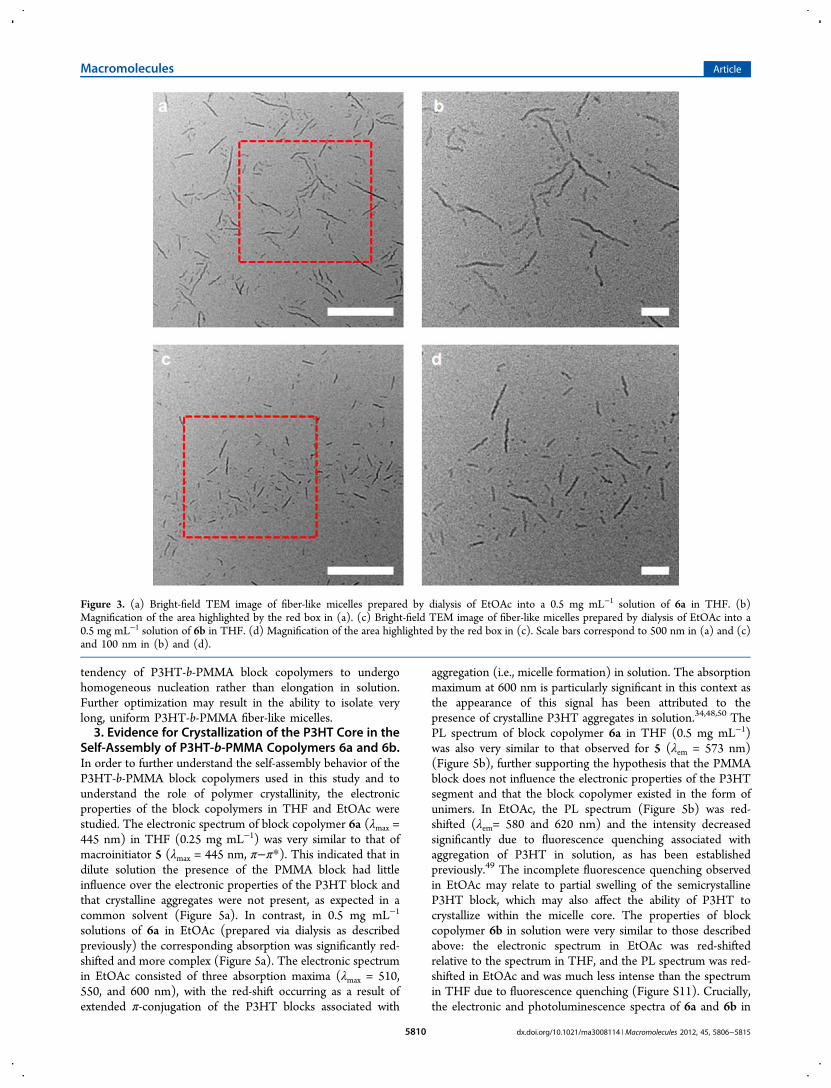
6a and 6b did not significantly change the self-assemblybehavior or, more importantly, the length of the resulting fiber-like micelles, the relatively higher boiling point of this solventdid provide the opportunity for further insightful experimentsto be conducted. P3HT-b-PMMA block copolymers 6a and 6bwere dissolved in 2:1 mixtures of THF and n-BuOAc and theresulting solutions allowed to stand uncovered for 5 days. Thismethod allowed for extremely slow removal of THF, a commonsolvent for P3HT and PMMA, to afford dark red solutions ofmicelles derived from block copolymers 6a and 6b in n-BuOAc.The solutions of fiber-like micelles of 6a and 6b were analyzedby TEM (Figure 4). Significantly, the relative lengths of themicelles had increased using this method (6a: Ln = 275 nm, Lw= 330 nm, Lw/Ln = 1.18, σ/Ln = 0.44; 6b: Ln = 135 nm, Lw =170 nm, Lw/Ln = 1.28, σ/Ln = 0.53). This indicated that therate of homogeneous nucleation relative to the rate of micellegrowth had been modified under the experimental conditionsemployed. The self-assembly experiments described in thisstudy illustrate the importance of experimental control over the
Figure 1. GPC traces (refractive index) for P3HT-b-PMMA blockcopolymer 6a: crude (black) and purified by SEC (red).
Figure 2. Images of (a) a 0.5 mg mL−1 solution of 6a in THF and (b)a 0.5 mg mL−1 solution prepared via dialysis of EtOAc into a0.5 mg mL−1 solution of 6a in THF.
Macromolecules Article
dx.doi.org/10.1021/ma3008114 | Macromolecules 2012, 45, 5806−58155809
tendency of P3HT-b-PMMA block copolymers to undergohomogeneous nucleation rather than elongation in solution.Further optimization may result in the ability to isolate verylong, uniform P3HT-b-PMMA fiber-like micelles.3. Evidence for Crystallization of the P3HT Core in the
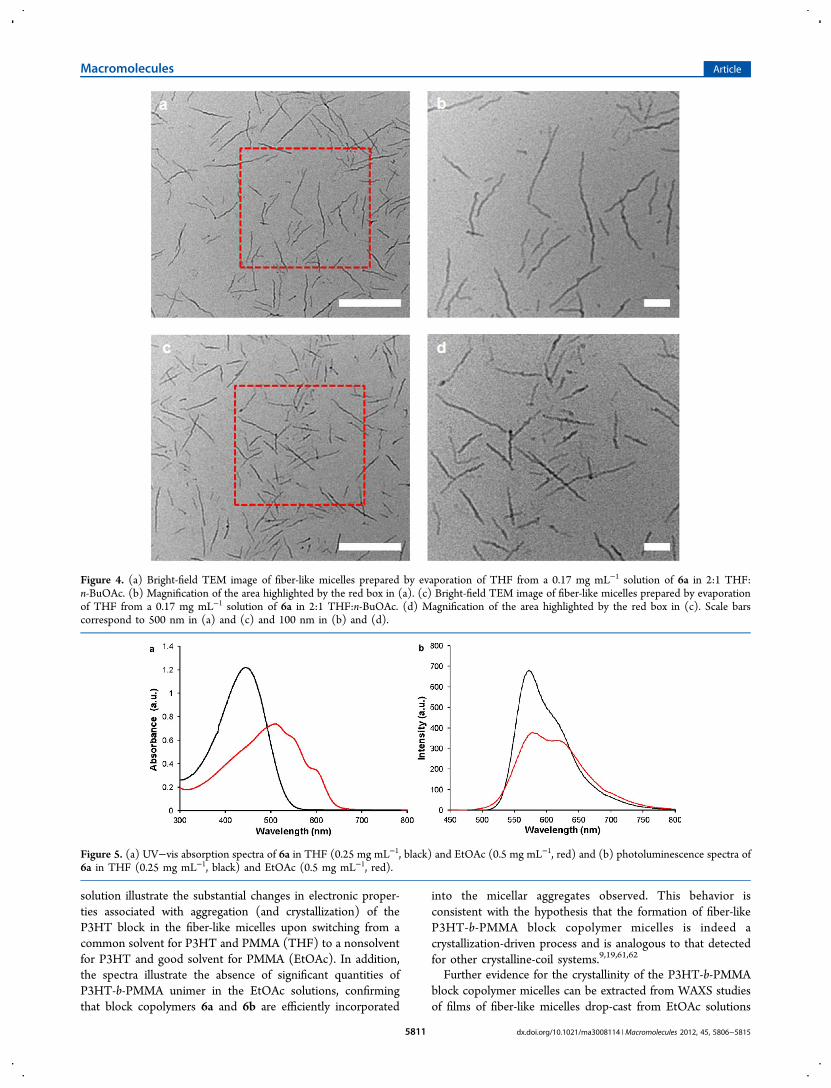
Self-Assembly of P3HT-b-PMMA Copolymers 6a and 6b.In order to further understand the self-assembly behavior of theP3HT-b-PMMA block copolymers used in this study and tounderstand the role of polymer crystallinity, the electronicproperties of the block copolymers in THF and EtOAc werestudied. The electronic spectrum of block copolymer 6a (λmax =445 nm) in THF (0.25 mg mL−1) was very similar to that ofmacroinitiator 5 (λmax = 445 nm, π−π*). This indicated that indilute solution the presence of the PMMA block had littleinfluence over the electronic properties of the P3HT block andthat crystalline aggregates were not present, as expected in acommon solvent (Figure 5a). In contrast, in 0.5 mg mL−1
solutions of 6a in EtOAc (prepared via dialysis as describedpreviously) the corresponding absorption was significantly red-shifted and more complex (Figure 5a). The electronic spectrumin EtOAc consisted of three absorption maxima (λmax = 510,550, and 600 nm), with the red-shift occurring as a result ofextended π-conjugation of the P3HT blocks associated with
aggregation (i.e., micelle formation) in solution. The absorptionmaximum at 600 nm is particularly significant in this context asthe appearance of this signal has been attributed to thepresence of crystalline P3HT aggregates in solution.34,48,50 ThePL spectrum of block copolymer 6a in THF (0.5 mg mL−1)was also very similar to that observed for 5 (λem = 573 nm)(Figure 5b), further supporting the hypothesis that the PMMAblock does not influence the electronic properties of the P3HTsegment and that the block copolymer existed in the form ofunimers. In EtOAc, the PL spectrum (Figure 5b) was red-shifted (λem= 580 and 620 nm) and the intensity decreasedsignificantly due to fluorescence quenching associated withaggregation of P3HT in solution, as has been establishedpreviously.49 The incomplete fluorescence quenching observedin EtOAc may relate to partial swelling of the semicrystallineP3HT block, which may also affect the ability of P3HT tocrystallize within the micelle core. The properties of blockcopolymer 6b in solution were very similar to those describedabove: the electronic spectrum in EtOAc was red-shiftedrelative to the spectrum in THF, and the PL spectrum was red-shifted in EtOAc and was much less intense than the spectrumin THF due to fluorescence quenching (Figure S11). Crucially,the electronic and photoluminescence spectra of 6a and 6b in
Figure 3. (a) Bright-field TEM image of fiber-like micelles prepared by dialysis of EtOAc into a 0.5 mg mL−1 solution of 6a in THF. (b)Magnification of the area highlighted by the red box in (a). (c) Bright-field TEM image of fiber-like micelles prepared by dialysis of EtOAc into a0.5 mg mL−1 solution of 6b in THF. (d) Magnification of the area highlighted by the red box in (c). Scale bars correspond to 500 nm in (a) and (c)and 100 nm in (b) and (d).
Macromolecules Article
dx.doi.org/10.1021/ma3008114 | Macromolecules 2012, 45, 5806−58155810
solution illustrate the substantial changes in electronic proper-ties associated with aggregation (and crystallization) of theP3HT block in the fiber-like micelles upon switching from acommon solvent for P3HT and PMMA (THF) to a nonsolventfor P3HT and good solvent for PMMA (EtOAc). In addition,the spectra illustrate the absence of significant quantities ofP3HT-b-PMMA unimer in the EtOAc solutions, confirmingthat block copolymers 6a and 6b are efficiently incorporated
into the micellar aggregates observed. This behavior isconsistent with the hypothesis that the formation of fiber-likeP3HT-b-PMMA block copolymer micelles is indeed acrystallization-driven process and is analogous to that detectedfor other crystalline-coil systems.9,19,61,62
Further evidence for the crystallinity of the P3HT-b-PMMAblock copolymer micelles can be extracted from WAXS studiesof films of fiber-like micelles drop-cast from EtOAc solutions
Figure 4. (a) Bright-field TEM image of fiber-like micelles prepared by evaporation of THF from a 0.17 mg mL−1 solution of 6a in 2:1 THF:n-BuOAc. (b) Magnification of the area highlighted by the red box in (a). (c) Bright-field TEM image of fiber-like micelles prepared by evaporationof THF from a 0.17 mg mL−1 solution of 6a in 2:1 THF:n-BuOAc. (d) Magnification of the area highlighted by the red box in (c). Scale barscorrespond to 500 nm in (a) and (c) and 100 nm in (b) and (d).
Figure 5. (a) UV−vis absorption spectra of 6a in THF (0.25 mg mL−1, black) and EtOAc (0.5 mg mL−1, red) and (b) photoluminescence spectra of6a in THF (0.25 mg mL−1, black) and EtOAc (0.5 mg mL−1, red).
Macromolecules Article
dx.doi.org/10.1021/ma3008114 | Macromolecules 2012, 45, 5806−58155811

(Figure S12). The WAXS patterns for the micelles of blockcopolymers 6a and 6b were very similar to those collected forfilms of block copolymers 6a and 6b drop-cast from THF. Thepatterns, which were again complicated by the presence of anamorphous halo associated with the PMMA blocks, possesspeaks potentially related to the (100) reflection in P3HThomopolymers at 2θ = 5.5°, d = 1.72 nm that arise due to hexylchain interdigitation.63 The presence of these signals in theWAXS patterns obtained for films of fiber-like P3HT-b-PMMAblock copolymer micelles, which are unlikely to contain anyresidual unimer (vide supra), again supports the hypothesis thatthe aggregates formed upon changing the solvent from THF toEtOAc possess a semicrystalline core and that the mechanismfor their formation is crystallization-driven.4. Studies of Homopolymer Fibers Derived from End-
Functionalized P3HTs 5 and 4. The fiber-like micellesformed in this study possess jagged, ill-defined P3HT cores. Inorder to probe whether this structural feature arises due to thepresence of the PMMA corona-forming block, a controlexperiment involving the growth of homopolymer fibers ofATRP macroinitiator 5 by slow cooling a 1:1 THF:EtOAcsolution (ca. 0.13 mg mL−1) was conducted. The resultinghomopolymer fibers were also irregular in shape (Figure 6),
suggesting that the presence of the PMMA corona-formingblock does not cause the nonuniformity of the micelle cores.Similar nanostructured materials were observed when homo-polymer fibers of 5 were grown by slow cooling cyclohexaneand n-decane solutions (Figure S13).64 A further experimentwas conducted in order to ensure that the nonplanar end groupin 5 was not playing an adverse role in the formation of thehomopolymer fibers in this study. Fibers grown by slow coolingof a solution of alcohol-terminated P3HT 4 in n-decane werevery similar (Figure S14), confirming that the end group didnot significantly affect the structure of the homopolymer fibers.
■ DISCUSSIONBlock copolymer micelles containing a semicrystalline PFDMScore have very uniform core morphologies with smooth edgesparallel to their long axis. In addition, the rate of homogeneousnucleation of PFDMS-containing block copolymers (e.g.,polyisoprene (PI)637-b-PFDMS53) in solution is slow compared
to the rate of growth from the ends of existing micelles,allowing for monodisperse micelles of controlled length to berealized (Figure 7).19,20,22 However, fiber-like micelles withirregular core structure have been observed when the core-forming PFDMS block is replaced by PFDES (poly-(ferrocenyldiethylsilane)).62 This observation has been ration-alized previously by considering the enthalpy of crystallizationof both polymers, which suggested that PFDES has a higherdegree of crystallinity compared to PFDMS.62 This enhancedcrystallinity may play a role as it is likely to affect the initialreversibility of micelle formation in solution and, as a result,prevent reorganization of the polymer chains to more regularcore morphologies.Homopolymer fibers and fiber-like block copolymer micelles
based on P3HT with core structures similar to those observedabove have been reported under a variety of conditions,36,38,39
confirming that the P3HT cores of the micelles formed fromP3HT-b-PMMA block copolymers in alkyl acetate solvents arenot unique with respect to their irregular cores. The structuralirregularities observed may relate to the highly crystallinenature of the P3HT polymers and the potential irreversibilityassociated with their aggregation in solution, as hypothesizedfor systems based on PFDES. This effect may be enhanced inlow molecular weight P3HT-based systems, where thecrystalline block behaves as a rigid rod, compared tocrystalline-coil systems where chain-folding within the crystal-line micelle cores occurs more readily.12,19,31
It is also important to consider the possible relationshipbetween the ill-defined structures observed in these studies andour inability to grow long fiber-like micelles. The short fiber-like micelles observed appear to be a consequence of thecompetition between an unusually favorable homogeneousnucleation mechanism and growth of P3HT-b-PMMA blockcopolymers at the ends of preformed micelles. These resultsdiffer from previous examples where very long (>5 μm) P3HThomopolymer fibers and fiber-like block copolymer micelleswith similar ill-defined cores have been reported.36,39 Therelationship between the length of the fiber-like micellesrealized in this study and the length of the corona-formingblock of each polymer was therefore considered (EtOAcdialysis: 6a, Ln = 165 nm; 6b, Ln = 80 nm. n-BuOAc dialysis:6a, Ln = 110 nm; 6b, Ln = 90 nm. n-BuOAc evaporation: 6a, Ln
= 275 nm; 6b, 135 nm). The inverse relationship between thelength of the fiber-like micelles and the length of the corona-forming block implies that, in addition to preventingaggregation of the fiber-like micelles and enhancing colloidalstability, the corona-forming block may prevent efficient growthfrom the ends of preformed micelles. This conclusion isconsistent with recent results published by Kamps, Fryd, andPark,39 where a similar relationship was described between thelength of the corona-forming block and the length of fiber-likemicelles formed from a range of P3HT-b-PEO blockcopolymers. Considering each of the factors discussed above,it appears that the presence of the relatively long PMMAcorona-forming blocks in the block copolymers employed inthese studies, rather than the ill-defined core structure, is likelyto inhibit growth at the ends of performed micelles. This resultsin a situation which allows for the usually unfavorablehomogeneous nucleation process to dominate, thus limitingmicelle length.
Figure 6. Bright-field TEM image of homopolymer fibers prepared byaging a 0.13 mg mL−1 solution of homopolymer 5 in 50:50EtOAc:THF for 48 h. Scale bar corresponds to 100 nm.
Macromolecules Article
dx.doi.org/10.1021/ma3008114 | Macromolecules 2012, 45, 5806−58155812
■ SUMMARYWe have synthesized two P3HT-b-PMMA block copolymersusing a regioregular P3HT ATRP macroinitiator. The macro-initiator was synthesized via a GRIM protocol followed byconversion of the vinyl end group to an alcohol and subsequentintroduction of an α-bromoisobutyryl group, a common ATRPinitiator. The P3HT macroinitiator was then used as aprecursor to two different P3HT-b-PMMA block copolymers,which were purified by preparative size exclusion chromatog-raphy. The resulting materials, P3HT40-b-PMMA520 (6a) andP3HT40-b-PMMA1100 (6b), self-assembled into fiber-likemicelles in solvents selective for the PMMA block (e.g.,EtOAc and n-BuOAc). Comparison of the electronic andphotoluminescence spectra of unimers in solution with themicelle aggregates confirmed that the micelles possessedsemicrystalline cores. This is consistent with their formationby a crystallization-driven growth mechanism, which wasfurther supported by WAXS studies. Furthermore, theobservation that homogeneous nucleation of P3HT-b-PMMAunimers competed with their growth from the ends ofpreformed micelles in these studies was particularly significantas it limited the length of the micelles observed. If conductingnanowires of controlled aspect ratio and more complexarchitectures such as block comicelles9,22 are to be realizedvia a living crystallization-driven self-assembly pathway, growthmust occur selectively at the end of preformed micelles, as hasbeen observed previously in PFDMS-based systems. Futurework in this area will focus on promoting this process andlimiting the tendency of P3HT block copolymers to undergohomogeneous nucleation in solution.
■ ASSOCIATED CONTENT*S Supporting InformationExperimental details, electronic and photoluminescence spec-tra, WAXS, DSC, GPC, and DLS data, and additionalmicroscopy figures. This material is available free of chargevia the Internet at http://pubs.acs.org.
■ AUTHOR INFORMATIONCorresponding Author*E-mail: [email protected] (M.A.W.), [email protected] (I.M.).
NotesThe authors declare no competing financial interest.
■ ACKNOWLEDGMENTSThis work was funded by the European Research Council(Advanced Investigator Grant to I.M.). We thank the NSERCof Canada (J.B.G.) and the EU Marie Curie program (J.B.G.and S.K.P.) for postdoctoral fellowships. D.J.L. thanks theBristol Chemical Synthesis Doctoral Training Centre, fundedby EPSRC (EP/G036764/1), AstraZeneca, GlaxoSmithKline,Novartis, Pfizer, Syngenta, and the University of Bristol, for theprovision of a Ph.D. studentship. M.A.W. thanks the NSERC ofCanada for financial support. I.M. also thanks the EU for aMarie Curie Chair, a Reintegration Grant, and the RoyalSociety for a Wolfson Research Merit Award. The authors alsothank Dr. Paul A. Rupar for helpful discussions and Mr. F. M.Maximilian Erhard for taking the photographs presented in thispaper.
■ REFERENCES(1) Cameron, N. S.; Corbierre, M. K.; Eisenberg, A. Can. J. Chem.1999, 77, 1311−1326.(2) (a) Zhulina, E. B.; Adam, M.; LaRue, I.; Sheiko, S. S.; Rubinstein,M. Macromolecules 2005, 38, 5330−5351. (b) Hayward, R. C.; Pochan,D. J. Macromolecules 2010, 43, 3577−3584. (c) Du, J.; O’Reilly, R. K.Chem. Soc. Rev. 2011, 40, 2402−2416. (d) Moughton, A. O.; Hillmyer,M. A.; Lodge, T. P. Macromolecules 2012, 45, 2−19.(3) Pochan, D. J.; Chen, Z.; Cui, H.; Hales, K.; Qi, K.; Wooley, K. L.Science 2004, 306, 94−97.(4) Discher, D. E.; Eisenberg, A. Science 2002, 297, 967−973.(5) (a) Li, Z.; Chen, Z.; Cui, H.; Hales, K.; Qi, K.; Wooley, K. L.;Pochan, D. J. Langmuir 2005, 21, 7533−7539. (b) Schleuss, T. W.;Abbel, R.; Gross, M.; Schollmeyer, D.; Frey, H.; Maskos, M.; Berger,R.; Kilbinger, A. F. M. Angew. Chem., Int. Ed. 2006, 45, 2969−2975.(6) Presa Soto, A.; Gilroy, J. B.; Winnik, M. A.; Manners, I. Angew.Chem., Int. Ed. 2010, 49, 8220−8223.(7) (a) Cornelissen, J. J. L. M.; Fischer, M.; Sommerdijk, N. A. J. M.;Nolte, R. J. M. Science 1998, 280, 1427−1430. (b) Dupont, J.; Liu, G.;Niihara, K.-i.; Kimoto, R.; Jinnai, H. Angew. Chem., Int. Ed. 2009, 48,6144−6147.(8) (a) Yu, K.; Zhang, L.; Eisenberg, A. Langmuir 1996, 12, 5980−5984. (b) Zhang, L.; Bartels, C.; Yu, Y.; Shen, H.; Eisenberg, A. Phys.Rev. Lett. 1997, 79, 5034−5037. (c) Stewart, S.; Liu, G. Angew. Chem.,Int. Ed. 2000, 39, 340−344. (d) Li, Z.; Kesselman, E.; Talmon, Y.;Hillmyer, M. A.; Lodge, T. P. Science 2004, 306, 98−101.
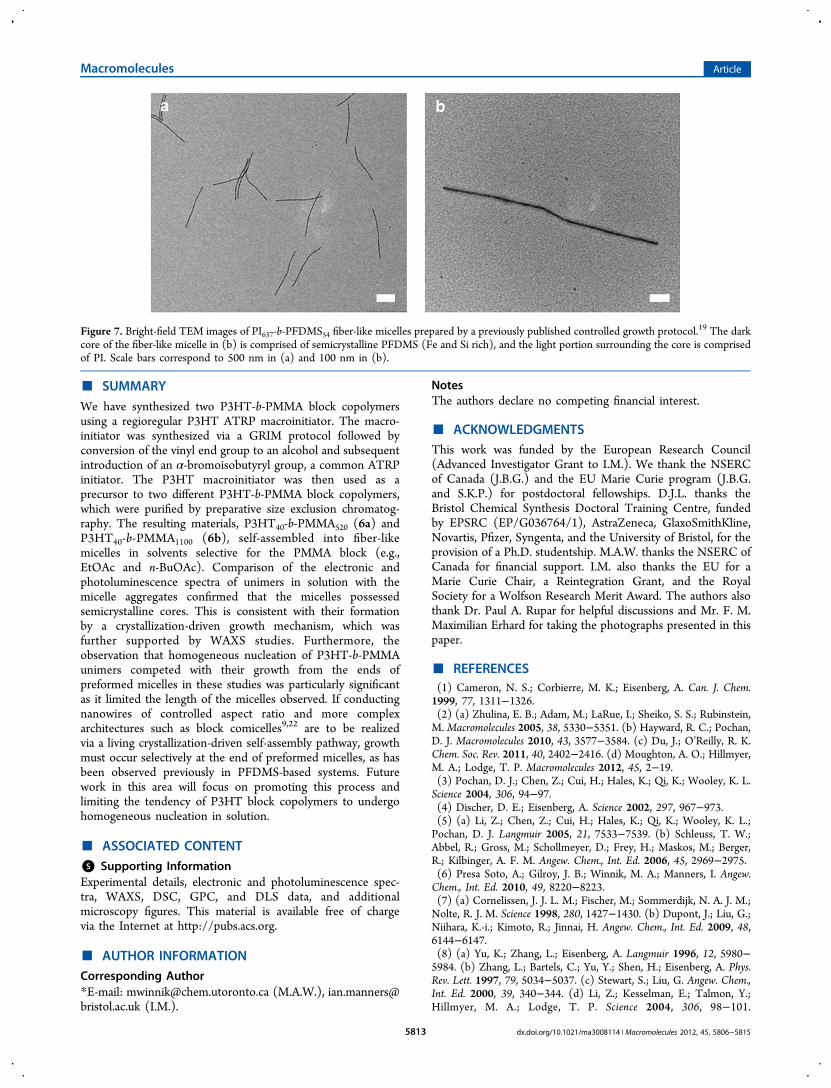
Figure 7. Bright-field TEM images of PI637-b-PFDMS54 fiber-like micelles prepared by a previously published controlled growth protocol.19 The dark
core of the fiber-like micelle in (b) is comprised of semicrystalline PFDMS (Fe and Si rich), and the light portion surrounding the core is comprisedof PI. Scale bars correspond to 500 nm in (a) and 100 nm in (b).
Macromolecules Article
dx.doi.org/10.1021/ma3008114 | Macromolecules 2012, 45, 5806−58155813

(e) Kubowicz, S.; Baussard, J.-F.; Lutz, J.-F.; Thunemann, A. F.; vonBerlepsch, H.; Laschewsky, A. Angew. Chem., Int. Ed. 2005, 44, 5262−5265. (f) Voets, I. K.; de Keizer, A.; de Waard, P.; Frederik, P. M.;Bomans, P. H. H.; Schmalz, H.; Walther, A.; King, S. M.; Leermakers,F. A. M.; Cohen Stuart, M. A. Angew. Chem., Int. Ed. 2006, 45, 6673−6676. (g) Walther, A.; Andre, X.; Drechsler, M.; Abetz, V.; Muller, A.H. E. J. Am. Chem. Soc. 2007, 129, 6187−6198. (h) Saito, N.; Liu, C.;Lodge, T. P.; Hillmyer, M. A. Macromolecules 2008, 41, 8815−8822.(i) Walther, A.; Drechsler, M.; Rosenfeldt, S.; Harnau, L.; Ballauff, M.;Abetz, V.; Muller, A. H. E. J. Am. Chem. Soc. 2009, 131, 4720−4728.(9) Gadt, T.; Ieong, N. S.; Cambridge, G.; Winnik, M. A.; Manners, I.Nat. Mater. 2009, 8, 144−150.(10) Qian, J.; Zhang, M.; Manners, I.; Winnik, M. A. TrendsBiotechnol. 2010, 28, 84−92.(11) (a) Zhang, L.; Eisenberg, A. Science 1995, 268, 1728−1731.(b) Spatz, J. P.; Moßmer, S.; Moller, M. Angew. Chem., Int. Ed. 1996,35, 1510−1512. (c) Liu, G.; Ding, J.; Qiao, L.; Guo, A.; Dymov, B. P.;Gleeson, J. T.; Hashimoto, T.; Saijo, K. Chem.Eur. J. 1999, 5, 2740−2749. (d) Won, Y.-Y.; Davis, H. T.; Bates, F. S. Science 1999, 283,960−963. (e) Jain, S.; Bates, F. S. Science 2003, 300, 460−464. (f) Jain,S.; Bates, F. S. Macromolecules 2004, 37, 1511−1523. (g) Korczagin, I.;Hempenius, M. A.; Fokkink, R. G.; Cohen Stuart, M. A.; Al-Hussein,M.; Bomans, P. H. H.; Frederik, P. M.; Vancso, G. J. Macromolecules2006, 39, 2306−2315. (h) Wurm, F.; Hilf, S.; Frey, H. Chem.Eur. J.2009, 15, 9068−9077. (i) Li, Z.; Ma, J.; Lee, N. S.; Wooley, K. L. J.Am. Chem. Soc. 2011, 133, 1228−1231. (j) Gao, Y.; Li, X.; Hong, L.;Liu, G. Macromolecules 2012, 45, 1321−1330.(12) Schmelz, J.; Karg, M.; Hellweg, T.; Schmalz, H. ACS Nano 2011,5, 9523−9534.(13) (a) Dalhaimer, P.; Engler, A. J.; Parthasarathy, R.; Discher, D. E.Biomacromolecules 2004, 5, 1714−1719. (b) Geng, Y.; Dalhaimer, P.;Cai, S.; Tsai, R.; Tewari, M.; Minko, T.; Discher, D. E. Nat.Nanotechnol. 2007, 2, 249−255.(14) Dean, J. M.; Verghese, N. E.; Pham, H. Q.; Bates, F. S.Macromolecules 2003, 36, 9267−9270.(15) (a) Yan, X.; Liu, G.; Haeussler, M.; Tang, B. Z. Chem. Mater.2005, 17, 6053−6059. (b) Wang, H.; Lin, W.; Fritz, K. P.; Scholes, G.D.; Winnik, M. A.; Manners, I. J. Am. Chem. Soc. 2007, 129, 12924−12925. (c) Wang, H.; Wang, X.; Winnik, M. A.; Manners, I. J. Am.Chem. Soc. 2008, 130, 12921−12930.(16) Wang, X.; Liu, K.; Arsenault, A. C.; Rider, D. A.; Ozin, G. A.;Winnik, M. A.; Manners, I. J. Am. Chem. Soc. 2007, 129, 5630−5639.(17) (a) Thurmond, K. B., II; Kowalewski, T.; Wooley, K. L. J. Am.Chem. Soc. 1996, 118, 7239−7240. (b) Tao, J.; Liu, G.; Ding, J.; Yang,M. Macromolecules 1997, 30, 4084−4089. (c) Ding, J.; Liu, G.Macromolecules 1998, 31, 6554−6558. (d) O’Reilly, R. K.; Hawker, C.J.; Wooley, K. L. Chem. Soc. Rev. 2006, 35, 1068−1083. (e) Read, E. S.;Armes, S. P. Chem. Commun. 2007, 3021−3035. (f) van Nostrum, C.F. Soft Matter 2011, 7, 3246−3259.(18) Rupar, P. A.; Cambridge, G.; Winnik, M. A.; Manners, I. J. Am.Chem. Soc. 2011, 133, 16947−16957.(19) Gilroy, J. B.; Rupar, P. A.; Whittell, G. R.; Chabanne, L.; Terrill,N. J.; Winnik, M. A.; Manners, I.; Richardson, R. M. J. Am. Chem. Soc.2011, 133, 17056−17062.(20) Gilroy, J. B.; Gadt, T.; Whittell, G. R.; Chabanne, L.; Mitchels, J.M.; Richardson, R. M.; Winnik, M. A.; Manners, I. Nat. Chem. 2010, 2,566−570.(21) Qian, J.; Guerin, G.; Lu, Y.; Cambridge, G.; Manners, I.; Winnik,M. A. Angew. Chem., Int. Ed. 2011, 50, 1622−1625.(22) Wang, X.; Guerin, G.; Wang, H.; Wang, Y.; Manners, I.; Winnik,M. A. Science 2007, 317, 644−647.(23) He, F.; Gadt, T.; Manners, I.; Winnik, M. A. J. Am. Chem. Soc.2011, 133, 9095−9103.(24) Mohd Yusoff, S. F.; Gilroy, J. B.; Cambridge, G.; Winnik, M. A.;Manners, I. J. Am. Chem. Soc. 2011, 133, 11220−11230.(25) (a) Vilgis, T.; Halperin, A. Macromolecules 1991, 24, 2090−2095. (b) Gast, A. P.; Vinson, P. K.; Cogan-Farinas, K. A.Macromolecules 1993, 26, 1774−1776. (c) Lin, E. K.; Gast, A. P.Macromolecules 1996, 29, 4432−4441. (d) Richter, D.; Schneiders, D.;
Monkenbusch, M.; Willner, L.; Fetters, L. J.; Huang, J. S.; Lin, M.;Mortensen, K.; Farago, B. Macromolecules 1997, 30, 1053−1068.(e) Ramzi, A.; Prager, M.; Richter, D.; Efstratiadis, V.; Hadjichristidis,N.; Young, R. N.; Allgaier, J. B. Macromolecules 1997, 30, 7171−7182.(f) Chen, W. Y.; Li, C. Y.; Zheng, J. X.; Huang, P.; Zhu, L.; Ge, Q.;Quirk, R. P.; Lotz, B.; Deng, L.; Wu, C.; Thomas, E. L.; Cheng, S. Z.D. Macromolecules 2004, 37, 5292−5299. (g) Cao, L.; Manners, I.;Winnik, M. A. Macromolecules 2002, 35, 8258−8260.(26) (a) Du, Z.-X.; Xu, J.-T.; Fan, Z.-Q. Macromolecules 2007, 40,7633−7637. (b) Du, Z.-X.; Xu, J.-T.; Fan, Z.-Q. Macromol. RapidCommun. 2008, 29, 467−471.(27) Lazzari, M.; Scalarone, D.; Vazquez-Vazquez, C.; Lopez-Quintela, M. A. Macromol. Rapid Commun. 2008, 29, 352−357.(28) (a) Mihut, A. M.; Chiche, A.; Drechsler, M.; Schmalz, H.; DiCola, E.; Krausch, G.; Ballauff, M. Soft Matter 2009, 5, 208−213.(b) Mihut, A. M.; Drechsler, M.; Moller, M.; Ballauff, M. Macromol.Rapid Commun. 2010, 31, 449−453. (c) Mihut, A. M.; Crassous, J. J.;Schmalz, H.; Drechsler, M.; Ballauff, M. Soft Matter 2012, 8, 3163−3173.(29) Schmalz, H.; Schmelz, J.; Drechsler, M.; Yuan, J.; Walther, A.;Schweimer, K.; Mihut, A. M. Macromolecules 2008, 41, 3235−3242.(30) Portinha, D.; Boue, F.; Bouteiller, L.; Carrot, G.; Chassenieux,C.; Pensec, S.; Reiter, G. Macromolecules 2007, 40, 4037−4042.(31) Petzetakis, N.; Dove, A. P.; O’Reilly, R. K. Chem. Sci. 2011, 2,955−960.(32) (a) Lim, J. A.; Liu, F.; Ferdous, S.; Muthukumar, M.; Briseno, A.L. Mater. Today 2010, 13, 14−24. (b) Kim, F. S.; Ren, G.; Jenekhe, S.A. Chem. Mater. 2011, 23, 682−732.(33) Wu, P.-T.; Ren, G.; Li, C.; Mezzenga, R.; Jenekhe, S. A.Macromolecules 2009, 42, 2317−2320.(34) Hammer, B. A. G.; Bokel, F. A.; Hayward, R. C.; Emrick, T.Chem. Mater. 2011, 23, 4250−4256.(35) Kim, J.; Song, I. Y.; Park, T. Chem. Commun. 2011, 4697−4699.(36) Lee, E.; Hammer, B.; Kim, J.-K.; Page, Z.; Emrick, T.; Hayward,R. C. J. Am. Chem. Soc. 2011, 133, 10390−10393.(37) (a) Jenekhe, S. A.; Chen, X. L. Science 1998, 279, 1903−1907.(b) Leclere, P.; Calderone, A.; Marsitzky, D.; Francke, V.; Geerts, Y.;Mullen, K.; Bredas, J. L.; Lazzaroni, R. Adv. Mater. 2000, 12, 1042−1046. (c) Wang, H.; Wang, H. H.; Urban, V. S.; Littrell, K. C.;Thiyagarajan, P.; Yu, L. J. Am. Chem. Soc. 2000, 122, 6855−6861.(d) Urban, V.; Wang, H. H.; Thiyagarajan, P.; Littrell, K. C.; Wang, H.B.; Yu, L. J. Appl. Crystallogr. 2000, 33, 645−649. (e) Wang, H.; You,W.; Jiang, P.; Yu, L.; Hau Wang, H. Chem.Eur. J. 2004, 10, 986−993.(f) Mori, T.; Watanabe, T.; Minagawa, K.; Tanaka, M. J. Polym. Sci.,Part A: Polym. Chem. 2005, 43, 1569−1578. (g) Tung, Y.-C.; Wu, W.-C.; Chen, W.-C. Macromol. Rapid Commun. 2006, 27, 1838−1844.(h) Lin, C.-H.; Tung, Y.-C.; Ruokolainen, J.; Mezzenga, R.; Chen, W.-C. Macromolecules 2008, 41, 8759−8769. (i) Tung, Y.-C.; Chen, W.-C.React. Funct. Polym. 2009, 69, 507−518. (j) Tian, Y.; Chen, C.-Y.; Yip,H.-L.; Wu, W.-C.; Chen, W.-C.; Jen, A. K.-Y. Macromolecules 2010, 43,282−291. (k) Park, S.-J.; Kang, S.-G.; Fryd, M.; Saven, J. G.; Park, S.-J.J. Am. Chem. Soc. 2010, 132, 9931−9933.(38) Patra, S. K.; Ahmed, R.; Whittell, G. R.; Lunn, D. J.; Dunphy, E.L.; Winnik, M. A.; Manners, I. J. Am. Chem. Soc. 2011, 133, 8842−8845.(39) Kamps, A. C.; Fryd, M.; Park, S.-J. ACS Nano 2012, 6, 2844−2852.(40) Berson, S.; De Bettignies, R.; Bailly, S.; Guillerez, S. Adv. Funct.Mater. 2007, 17, 1377−1384.(41) Osaka, I.; McCullough, R. D. Acc. Chem. Res. 2008, 41, 1202−1214.(42) Okamoto, K.; Luscombe, C. K. Polym. Chem. 2011, 2, 2424−2434.(43) Jeffries-El, M.; Sauve, G.; McCullough, R. D. Macromolecules2005, 38, 10346−10352.(44) Mass spectrometry studies have previously shown that polymersprepared by this method have either hydrogen or bromine substituentsat the ω position. For details, see ref 43.
Macromolecules Article
dx.doi.org/10.1021/ma3008114 | Macromolecules 2012, 45, 5806−58155814

(45) The degree of polymerization value determined for α-vinyl-ω-bromo P3HT 2 was used throughout the article, as this material wasassumed to contain minimal amounts of coupled polymers, potentiallygenerated during subsequent reactions.(46) Dai, C.-A.; Yen, W.-C.; Lee, Y.-H.; Ho, C.-C.; Su, W.-F. J. Am.Chem. Soc. 2007, 129, 11036−11038.(47) Iovu, M. C.; Jeffries-EL, M.; Sheina, E. E.; Cooper, J. R.;McCullough, R. D. Polymer 2005, 46, 8582−8586.(48) Rughooputh, S. D. D. V.; Hotta, S.; Heeger, A. J.; Wudl, F. J.Polym. Sci., Part B: Polym. Phys. 1987, 25, 1071−1078.(49) Xu, B.; Holdcroft, S. Macromolecules 1993, 26, 4457−4460.(50) Chen, T.-A.; Wu, X.; Rieke, R. D. J. Am. Chem. Soc. 1995, 117,233−244.(51) The enthalpy of fusion for an ideal P3HT crystal was 99 J g−1.For details of the determination of this value, see: Malik, S.; Nandi, A.K. J. Polym. Sci., Part B: Polym. Phys. 2002, 40, 2073−2085.(52) (a) Liu, J.; Sheina, E.; Kowalewski, T.; McCullough, R. D.Angew. Chem., Int. Ed. 2002, 41, 329−332. (b) Kowalewski, T.;McCollough, R. D.; Matyjaszewski, K. Eur. Phys. J. E 2003, 10, 5−16.(53) Lee, Y.; Fukukawa, K.-I.; Bang, J.; Hawker, C. J.; Kim, J. K. J.Polym. Sci., Part A: Polym. Chem. 2008, 46, 8200−8205.(54) Nguyen, H. T.; Coulembier, O.; De Winter, J.; Gerbaux, P.;Crispin, X.; Dubois, P. Polym. Bull. 2011, 66, 51−64.(55) Iovu, M. C.; Craley, C. R.; Jeffries-EL, M.; Krankowski, A. B.;Zhang, R.; Kowalewski, T.; McCullough, R. D. Macromolecules 2007,40, 4733−4735.(56) Lee, Y. J.; Kim, S. H.; Yang, H.; Jang, M.; Hwang, S. S.; Lee, H.S.; Baek, K.-Y. J. Phys. Chem. C 2011, 115, 4228−4234.(57) (a) Palaniappan, K.; Hundt, N.; Sista, P.; Nguyen, H.; Hao, J.;Bhatt, M. P.; Han, Y.-Y.; Schmiedel, E. A.; Sheina, E. E.; Biewer, M. C.;Stefan, M. C. J. Polym. Sci., Part A: Polym. Chem. 2011, 49, 1802−1808.(b) Lohwasser, R. H.; Thelakkat, M. Macromolecules 2012, 45, 3070−3077.(58) Fang, Y.-K.; Liu, C.-L.; Li, C.; Lin, C.-J.; Mezzenga, R.; Chen,W.-C. Adv. Funct. Mater. 2010, 20, 3012−3024.(59) Dynamic light scattering (DLS) studies of the P3HT-b-PMMAfiber-like micelles in solution were complicated by absorption/fluorescence of the P3HT block near the wavelength of the incidentradiation (633 nm). An example of the data obtained is shown inFigure S9.(60) Preliminary atomic force microscopy (AFM) studies haveproved inconclusive in determining the specific morphology of thefiber-like micelles formed in this study.(61) Massey, J. A.; Temple, K.; Cao, L.; Rharbi, Y.; Raez, J.; Winnik,M. A.; Manners, I. J. Am. Chem. Soc. 2000, 122, 11577−11584.(62) Gadt, T.; Schacher, F. H.; McGrath, N.; Winnik, M. A.;Manners, I. Macromolecules 2011, 44, 3777−3786.(63) The structure of the semicrystalline core of block copolymermicelles with a P3HT core-forming block has not been determined todate. We feel that core−corona interactions may play an importantrole in the structure of the core, potentially affecting the overallsymmetry. We have therefore described the structure of thesemicrystalline core of our P3HT-b-PMMA micelles in relation tothe previously determined structure of semicrystalline P3HThomopolymers, rather than indexing our WAXS data directly, untilsuch time that suitable structural studies have taken place.(64) This procedure has been employed previously to yield highlyuniform P3HT homopolymer fibers (Mn = 29 000 g mol−1, PDI =3.8). For details, see: Ihn, K. J.; Moulton, J.; Smith, P. J. Polym. Sci.,Part B: Polym. Phys. 1993, 31, 735−742.
Macromolecules Article
dx.doi.org/10.1021/ma3008114 | Macromolecules 2012, 45, 5806−58155815






![A novel diblock copolymer of (monomethoxy poly [ethylene glycol]-oleate) with a small hydrophobic fraction to make stable micelles/polymersomes for curcumin delivery to cancer cells](https://static.fdokumen.com/doc/165x107/6344914403a48733920af291/a-novel-diblock-copolymer-of-monomethoxy-poly-ethylene-glycol-oleate-with-a.jpg)













