
Shape and size of micelles in the sodium dodecyl sulfate-formamide system

Ab
GF
a
ARRAA
KASSPMH
1
CsBaatrcatfifiaccm1bfR
0h
International Journal of Biological Macromolecules 51 (2012) 351– 363
Contents lists available at SciVerse ScienceDirect
International Journal of Biological Macromolecules
jo u r n al hom epa ge: ww w.elsev ier .com/ locate / i jb iomac
nticancer effect of atorvastatin nanostructured polymeric micellesased on stearyl-grafted chitosan
eorge M. Mekhail, Amany O. Kamel, Gehanne A.S. Awad, Nahed D. Mortada ∗
aculty of Pharmacy, Department of Pharmaceutics and Industrial Pharmacy, Ain Shams University, Cairo, Egypt
r t i c l e i n f o
rticle history:eceived 12 April 2012eceived in revised form 11 May 2012ccepted 17 May 2012vailable online 29 May 2012
eywords:
a b s t r a c t
The purpose of this study was to develop a new therapeutic approach for atorvastatin (ATV) adopt-ing nanostructured polymeric micelles for its controlled delivery to the cancer cells. Amphiphilic blockcopolymers of stearyl chitosan (SC) and sulfated stearyl chitosan (S-SC) that could self assemble to formpolymeric micelles with different degree of substitution (DS) were synthesized and characterized. Thesynthesized chitosan derivatives were able to self assemble and form micelles encapsulating ATV withcritical micellar concentrations ranging from 6.9 to 21 �g/ml, drug-loading ranging from 40% to 84.1%
torvastatinynthesistearyl-grafted chitosanolymeric micellesCF 7
and encapsulation efficiency ranging from 10.4% to 35%. ATV caused a significant decrease in particlesize and zeta potential of both SC and S-SC micelles. Micelles encapsulating ATV exhibited a sustainedrelease and more cytotoxic activity against MCF 7 and HCT 116 cell lines than ATV alone. The 50% cellulargrowth inhibition (IC50%) of the drug decreased from 10.4 to 3.7 in case of MCF 7 and from 9.4 to 3.4 incase of HCT 116 after its loading in micelles. These results indicate that SC ATV polymeric micelles can
sing s
CT 116 be considered as a promi. Introduction
Statins are 3-hydroxy-3-methylglutaryl coenzyme A (HMG-oA) reductase inhibitors, the rate-limiting enzyme in mevalonateynthesis, which is the precursor for cholesterol biosynthesis.eyond their cholesterol lowering effect, statins stabilize plaquend prevent strokes through anti-inflammatory and other mech-nisms [1]. Moreover, they have been shown to be useful inreating aortic aneurysms [2], inflammatory colitis [3], chronicenal disease [4], nasal polyp disease [5] and chronic lympho-ytic leukemia [6]. It has also been proven that statins have annticancer effect due to suppression of the mevalonate biosyn-hesis which is a precursor for isoprenoid intermediates, such asarnesyl pyrophosphate and geranylgeranyl pyrophosphate, themportant lipid moieties added during post-translational modi-cation of a variety of proteins such as G-protein subunits Rasnd Rho. These proteins are involved in cell cycle progression,ell signaling, and membrane integrity [7–9]. Atorvastatin cal-ium (ATV) ([R-(R*,R*)]-2-(4-fluorophenyl)-�,�-dihydroxy-5-(1-ethylethyl)-3-phenyl-4-[(phenylamino) carbonyl]-1H-pyrrole-
-heptanoic acid) is a high M.wt (1209.4), lipid-lowering agent that
elongs to the statin group. It showed an anticancer effect on dif-erent cell lines. Collission et al. [10] proved that ATV inhibitedho activation and reverted the metastatic phenotype of human∗ Corresponding author. Tel.: +20 1222125688; fax: +20 2 24051106.E-mail address: ndsm [email protected] (N.D. Mortada).
141-8130/$ – see front matter © 2012 Elsevier B.V. All rights reserved.ttp://dx.doi.org/10.1016/j.ijbiomac.2012.05.026
ystem for site specific controlled delivery of ATV to tumor cells.© 2012 Elsevier B.V. All rights reserved.
melanoma cells viz. A375M, CHL, SK-Mel-28, and WM 166-4 in vitro.Moreover, they proved that, at plasma levels comparable to thoseused to treat hypercholesterolemia, it inhibited in vivo metastasis ofmelanoma cells in mice over expressing RhoC. In another study, Caf-fori et al. [11] found that ATV suppressed T, B lymphoblastoid andmyeloma cells when incubated with Human T cell lymphoblast-like cell line (Jurkat and CEM), lymphocyte cell line (human IM9),and myeloma cell line (U266). In addition, they postulated thatATV could be useful in combination with traditional chemothera-peutic agents such as doxorubicin since it suppressed doxorubicinresistant plasma cell line (MCC-2). Furthermore, ATV decreased cel-lular proliferation in Caco-2 colorectal cancer cell lines [12] andinduced growth inhibition of human pancreatic cell lines (Panc-1and MIA PaCa-2 cells) [13]. However, being a poorly water-soluble,hydrophobic drug, it will suffer from problems during therapeuticapplications such as poor absorption and bioavailability, moreover,drug aggregation-related complications such as embolism mightoccur [14]. Hence, drug encapsulation in amphiphilic copolymersvia polymeric micellization would be advantageous.
Polymeric micelles have been shown to be more effective insolid tumor targeting compared to nanoparticles, liposomes, andlipid-based drug delivery systems [15]. They have proven theireffectiveness preclinically and clinically and it is crucial that thepolymers used be biocompatible and biodegradable [15].
Among the various materials used for preparing polymericmicelles, chitosan has gained increasing attention as a non-toxic,biocompatible, and biodegradable polymer. By being, hydrophilicand cationic chitosan can be chemically grafted with hydrophobic

352 G.M. Mekhail et al. / International Journal of Biolo
Tab
le
1Pa
rtic
le
size
, pol
ydis
per
sity
ind
ices
and
zeta
pot
enti
al
of
un
load
ed
and
ATV
load
ed
pol
ymer
ic
mic
elle
s.
Poly
mer
typ
e
Poly
mer
cod
eC
hit
osan
tost
eari
can
hyd
rid
em
olar
rati
o
Deg
ree
ofac
ylat
ion
(%)
CA
C(�
g/m
l)Pa
rtic
le
size
(nm
) ±
SD
(PD
I)Ze
ta
pot
enti
al
±
SD
Un
load
ed
Load
ed
at
dif
fere
nt
ph
ase
volu
me
rati
osU
nlo
aded
Load
ed
at
dif
fere
nt
ph
ase
volu
me
rati
os
1:1
1:2
1:1
1:2
Stea
ryl c
hit
osan
SC1
1:1
1.7
11
160.
8
±
2.2
(0.2
3)
139
±
2.2(
0.16
)
132.
3
±
1.2(
0.23
)
−26.
4
± 0.
7 −1
6.1
±
1.02
−15
±
2.7
SC2
1:1.
5
4
9.3
151.
7
±
2.9
(0.2
4)
136.
1
±
2.1(
0.13
)
129
±
1.7(
0.20
1)
−29.
8 ±
0.6
−15.
2
±
1.1
−14
±
2.5
SC3
1:2
17.3
7.7
128
±
3.1
(0.1
9)
116.
8
±
1.9(
0.21
)
97.1
9
±
0.8(
0.23
)
−33.
1 ±
0.7
−14.
8
±
1.4
−8.2
7
±
1.78
SC4
1:3
18.8
6.9
121.
8
±
2.81
(0.2
7)
113.
6
±
1.88
(0.1
8)
91.2
±
1.09
(0.2
5)
−36.
8 ±
0.5
−17.
1
±
1.88
−16.
3
±
3.9
Sulf
ated
stea
ryl c
hit
osan
S-SC
1
1:1
1.7
21
174
±
2.33
(0.2
9)
143.
7
±
2.5(
0.27
)
131.
3
±
2.2(
0.36
)
−30.
8
±
0.4
−17.
9
±
1.96
−21.
2
±
1.4
S-SC
21:
1.5
4
11.5
162
±
2.4
(0.2
6)
135.
8
±
2.8(
0.31
)
126.
5
±
2.43
(0.3
2)
−33.
3
±
0.5
−11.
4
±
1.3
−17.
2
±
1.2
S-SC
31:
2
17.3
9
147
±
4.2
(0.3
3)
121.
60
±
2.8(
0.29
)
103.
1
±
2.8
(0.2
9)
−36.
6
±
0.7
−9.2
±
1.2
−14.
2
±
1.1
S-SC
41:
3
18.8
7.5
140
±
3.87
(0.2
9)
111
±
3.1(
0.29
)
101.
8
±
2.1(
0.32
) −3
9.7
±
1.1
−9
±
1.03
−14
±
1.03
gical Macromolecules 51 (2012) 351– 363
moieties forming amphiphilic copolymers which can self-assemblein aqueous media forming modified chitosan-based self-assemblednanoparticles [16]. These self-assembled nanoparticles can circu-late in the bloodstream for a relatively long time without beingrecognized by macrophages, moreover, it can easily accumulate atthe leaky vasculature throughout the enhanced permeability reten-tion (EPR) effect [17].
In light of the above, our goal was to develop a new therapeuticapproach for ATV by adopting nanostructured polymeric micellesfor its controlled delivery to the cancer cells. Amphiphilic blockcopolymers of stearyl chitosan (SC) and sulfated stearyl chitosanwhich could self assemble to form polymeric micelles were syn-thesized and characterized. The in vitro antitumor activity of theprepared micelles against human breast adenocarcinoma cell line(MCF 7) and human colon carcinoma cell line (HCT 116) as modeltumor cells was evaluated.
2. Experimental
2.1. Materials
Chitosan low molecular weight, stearic anhydride, dimethyl sul-foxide (DMSO), sulfur trioxide N,N-dimethyl formamide, pyrene,and phosphotungestic acid were purchased from Sigma–Aldrich,California, USA. Sodium hydroxide, acetone, ethanol, diethyl ether,N,N-dimethyl formamide, and chloroform were purchased from ElNasr Pharmaceutical Company, Cairo, Egypt. Atorvastatin calciumwas kindly supplied by Amoun Pharmaceutical Company, Cairo,Egypt. Trypsin, trypan blue, trichloroacetic acid, sulforhodamine-B, acetic acid, and tris base (tris(hydroxymethyl)aminomethane)were purchased from Sigma Chemical Co., St. Louis, MO, USA.
2.2. Cell line
Human breast adenocarcinoma cell line (MCF) and human coloncarcinoma cell line (HCT) were obtained frozen in liquid nitrogen(−180 ◦C) from the American Type Culture Collection (Manassas,VA, USA).
2.3. Synthesis of stearyl and sulfated stearyl chitosan derivatives
Four grades of amphiphilic stearyl chitosan co-polymers (SC)were prepared by acylation of chitosan with stearic anhydrideadopting the synthetic approach described earlier [18]. Purifiedchitosan was obtained by dissolving 0.006 moles of the polymerin 100 ml 0.1 M acetic acid, reprecipitated with 100 ml 0.2 M NaOH,collected by filtration and left overnight in a desiccator. The puri-fied chitosan was dispersed in 100 ml DMSO with magnetic stirringand left for one night to allow swelling. Different moles of stearicanhydride (M.wt = 550.95) viz. 0.006, 0.009, 0.012 and 0.018 wereadded to the chitosan dispersion so as chitosan: acetic anhydridemolar ratios were 1:1, 1:1.5, 1:2 and 1:3 respectively. The mixtureswere heated at 60 ◦C for 8 h under magnetic stirring, poured intoacetone to precipitate the stearyl chitosan which was collected byfiltration and washed five times with acetone and diethyl ether. Theobtained products were dried at 60 ◦C for 48 h.
Sulfation of the prepared stearyl chitosan powders was carriedout by the method described earlier by Schweiger and Yoshiokaet al. [19,20]. The prepared stearyl chitosan derivative powderswere left in 40 ml N,N-dimethyl formamide overnight for swellingthen cooled by refrigeration for another 24 h. A fixed amount of0.5 g sulfur trioxide N,N-dimethyl formamide was then added and
mixed using a magnetic stirrer for 3 h, in an ice bath, till com-plete dissolution of stearyl chitosan. The solutions were neutralizedwith NaOH (0.2 M) then dialyzed against distilled water using acellophane membrane with molecular weight cut off 12,000 to
f Biolo
gsulMscs
2
2
eodh
2
3wwCi
mt
2
tE
ff
D
2
ra(wr
2
mUwostTTrFompc
G.M. Mekhail et al. / International Journal o
et rid of the unreacted sulfating agent. The end point of dialy-is was checked by the absence of a white precipitate of BaSO4pon addition of BaCl2 solution to the receptor medium. The dia-
yzed solution was freeze dried (Laboratory Freeze Dryer Alpha 1-4,artin Christ Gefriertrocknungsanlagen GmbH, Germany), and the
ulfated stearyl chitosan (S-SC) was obtained as a white solid. Theomposition and codes of synthesized SC and S-SC derivatives arehown in Table 1.
.4. Characterization of the prepared SC and S-SC polymers
.4.1. FT-IR spectrometryFT-IR spectra were recorded on a Nicolet 670 FT-IR spectrom-
ter (Thermo Nicolet, USA) in potassium bromide discs. Samplesf 2–3 mg of the prepared polymers were ground with 100 mg ofry potassium bromide powder and compressed into a disc with aydrostatic press. The scanning range was 450–4000 cm−1.
.4.2. 1H NMR spectrometry1H NMR spectra were performed on a Varian Mercury VX-
00 NMR spectrometer (Agilent Technologies, USA). 1H spectraere run at 300 MHz using deuterated DMSO (DMSO-d6). Chitosanas dissolved in the deuterated mixture consisting of D2O andD3COOD. While the prepared chitosan derivatives were dissolved
n D2O and DMSO-d6 mixture.The degree of substitution or acylation (DS) was then deter-
ined by using the ratio of acyl methyl protons CH3(ı = 0.85 ppm)o H-2 protons (ı = 2.89 ppm) of chitosan.
.4.3. Elemental analysisThe sulfur content of the S-SC derivatives was determined using
he automatic elemental analyzer: Automated Vario EL III CHNSlemental Analyzer (Elementar, Germany).
The degree of sulfation, defined as the average number of sul-ate groups attached to a glucose unit was then calculated by theollowing formula [21]:
egree of sulphation = 162 × S%/32100 − ((80/32) × S%)
.4.4. X-ray diffractionX-ray diffraction (XRD) patterns of the prepared samples were
ecorded on an X-ray diffractometer with area detector operatingt a voltage of 40 kV and a current of 30 mA using Cu Ka radiationk = 0.154 nm), and X-ray tube PW3373/00 CuLFF. The scanning rateas 1.2◦/min and the scanning scope of 2� was from 50 to 400 at
oom temperature.
.4.5. Determination of critical aggregation concentration (CAC)The CAC of the synthesized SC and S-SC copolymers was deter-
ined by fluorescence spectroscopy (Edinburgh Instruments Ltd.,K) using pyrene as a hydrophobic fluorescence probe. First pyreneas dissolved in acetone for quantitation, which was then evap-
rated. A volume of 25 ml of aqueous solution of each of theynthesized modified chitosan co-polymers with different concen-rations ranging from 3.2 �g/ml to 32 �g/ml was added to pyrene.he final concentration of pyrene was controlled at 6.0 × 10−7 M.he solutions were water-bath sonicated for 30 min, and the fluo-escence spectra recorded on a fluorometer at room temperature.rom the pyrene emission spectra, the intensity ratio of first peak
f pyrene in water (I1, 372 nm) to its third peak in hydrophobicedium (I3, 383 nm) was analyzed. The apparent CAC of the co-olymers was obtained from the plot of the I1/I3 ratio against theorresponding co-polymers logarithmic concentration.
gical Macromolecules 51 (2012) 351– 363 353
2.5. Preparation of ATV loaded polymeric micelles
ATV loaded chitosan derivatives polymeric micelles wereprepared using oil-in-water emulsion method [22]. Chitosanderivatives were dispersed and ultrasonicated in distilled water toobtain polymeric micellar solutions of final concentrations equiv-alent to triple their CAC. The ratio of organic to aqueous phasewas changed to optimize the preparation method in terms of drugloading (DL%) and EE%. ATV was dissolved in different volumes ofchloroform to obtain final ratios of organic to aqueous phase viz. 1:1,1:2 and 1:3. The organic drug solution was then added drop wiseto the aqueous phase, mixed on a magnetic stirrer at 1000 rpm tillcomplete evaporation of chloroform, then left for equilibrium for24 h.
2.6. Characterization of the prepared polymeric micelles
2.6.1. Morphological examinationThe morphology of polymeric micelles was observed by TEM
using a JEM-2010 electron microscope (Jeol Jem 1230, Tokyo, Japanwith Semafore program ver. 4.01) operating at an accelerating volt-age of 20–100 kV. A drop of sample solutions was placed ontoa 200 mesh copper grid coated with carbon, taped with a filterpaper to remove surface water and air dried for 5 min. These self-aggregates were deposited on the grid, followed by the applicationof phosphotungestic acid negative staining.
2.6.2. Encapsulation efficiency and drug loadingThe ATV-loaded micelles solution was centrifuged at 3000 rpm
for 15 min after being placed in Nanosep® centrifugal unit (Pall LifeSciences, USA), to separate the free drug from the loaded micelles.The ATV content in filtrate (Cf) was measured spectrophotomet-rically at predetermined �max. ATV-loaded micelle solution weredissolved in 90% DMSO and the total drug content (Ct) was deter-mined. The drug content incorporated into micelles was obtainedfrom Ct − Cf. The drug encapsulation efficiency (EE%) and drug load-ing (DL) % were then calculated by the following equations:
EE% = Ct − Cf
Amount of drug used in formula× 100
DL% = Ct − Cf
Amount of encapsulated drug + amount of polymer used in formula× 100
2.6.3. Particle size and size distributionAverage particle diameter (Z-average), and size distribution
(PDI) of drug-free and ATV-loaded micelles in aqueous mediawere measured by dynamic light scattering using a Zetasizer nanoZS with 4 mW 633 nm He–Ne lasers at 25 ◦C (Malvern Zetasizer,Malvern Instruments Ltd., Malvern, UK).
2.6.4. Zeta potentialZeta potential of drug-free and ATV-loaded micelles in aqueous
media were measured using a Zetasizer nano ZS with 4 mW 633 nmHe–Ne lasers at 25 ◦C (Malvern Zetasizer, Malvern Instruments Ltd.,Malvern, UK).
2.6.5. In vitro release studyThe in vitro release studies were done adopting the dialysis
membrane diffusion technique [23]. The dialysis membrane (Spec-tra/Por Molecular weight cut off size 12,000 Da) was used forretaining polymeric micelles and allowing free drug passage into
the release media. Briefly, an amount of drug loaded polymericmicelles equivalent to 1200 �g of ATV were transferred to a dialy-sis bag having the length of 5 cm and diameter of 2.5 cm presoakedovernight in distilled water. The dialysis bag was placed in 50 ml
354 G.M. Mekhail et al. / International Journal of Biological Macromolecules 51 (2012) 351– 363
san, (B
pcwtass
2
iuic0eJcaa
Fig. 1. FTIR spectra of (A) chito
hosphate buffer saline pH 6.8. This volume provided complete sinkonditions for the drug. The entire system was kept at 37 ◦C ± 0.5 ◦Cith continuous magnetic stirring at 100 rpm. At predetermined
ime intervals (0.5, 1, 2, 3, 4, 6, 8, 12, 24, 48, 72 and 96 h); anliquot (1 ml) of the release medium was withdrawn and analyzedpectrophotometrically at 242 nm for drug content. Withdrawnamples were replaced by fresh buffer.
.6.6. Sulforhodamine-B (SRB) assay of cytotoxic activityThe cytotoxicity of both blank micelles and ATV-loaded micelles
n comparison with free ATV against MCF-7 and HCT-116 cell linessing SRB method was evaluated. This assay was carried out accord-
ng to the method of Skehan et al. [24]. Cells were used when 90%onfluence was reached; adherent cell lines were harvested with.025% trypsin and the viability was determined by trypan bluexclusion using the inverted microscope (Olympus 1x70, Tokyo,
apan). The cells were seeded in 96-well micro titer plates at a con-entration of 5 × 104–105 cell/well in a fresh medium and left tottach to the plates for 24 h. Then the cells were incubated with theppropriate concentration ranges (0, 5, 12.5, 25, 50 �g/ml) of free) SC1, (C) SC2, (D) SC3, (E) SC4.
ATV, SC3 unloaded polymeric micelles and SC3 loaded polymericmicelles, completed to total of 200 �l volume/well using freshmedium and incubation was continued for 48 and 72 h. Controlcells were treated with vehicle alone. For each drug concentration,4 wells were used. Following 48 and 72 h treatment, the cells werefixed with 50 �l cold 50% trichloroacetic acid for 1 h at 40 ◦C. Wellswere washed 5 times with distilled water and stained for 30 minat room temperature with 50 �l 0.4% SRB dissolved in 1% aceticacid. The wells were then washed 4 times with 1% acetic acid. Theplates were air-dried and the dye was solubilized with 100 �l/wellof 10 mM tris base (pH 10.5) for 5 min on a shaker at 1600 rpm. Theoptical density (O.D.) of each well was then measured spectropho-tometrically at 564 nm using an ELIZA microplate reader (Metertech. � 960, USA). The mean background absorbance was automati-cally subtracted and the mean value of each drug concentration wascalculated.
The percentage of cell survival was calculated as follows [25]:
Survival fraction = O.D. (treated cells)O.D. (control cells)
.

G.M. Mekhail et al. / International Journal of Biological Macromolecules 51 (2012) 351– 363 355
(B) S-
cttg
2
t(t
3
3
3
1vbaab
Fig. 2. FTIR spectra of (A) chitosan,
Graphical presentations of survival fraction versus different con-entrations of the drugs at different time intervals were plotted andhe IC50 values (the concentrations of the anticancer drug requiredo produce 50% inhibition of cell growth) were obtained from theraph. The experiment was repeated 3 times for each cell line.
.7. Statistical analysis
All the presented data are the mean of three determinations andhe results were compared using One-Way Analysis Of VarianceANOVA) test followed by Tukey–Kramer multiple comparisonsest using Graph Pad InStat version 3.06 software.
. Results and discussion
.1. Characterization of the prepared chitosan derivatives
.1.1. FT-IR spectroscopyFig. 1B–E shows that after N-acylation, the absorption at
656.8 cm−1 of chitosan (Fig. 1A), ascribed to the N H bendingibration mode of nonacylated � aminoglucose amine was replaced
y prominent bands at 1650 cm−1 and 1550 cm−1, respectivelyssigned to the carbonyl stretching vibration mode of secondarymide (amide band I) and the bending vibration mode of the amideand II. Also bands at 2850 cm−1 and 2920 cm−1 appeared afterSC1, (C) S-SC2, (D) S-SC3, (E) S-SC4.
acylation assigned to the aliphatic C H stretching of the stearylgroup [26]. No absorptions in the range of 1710–1760 cm−1 wasobserved in the spectra of the N-stearyl chitosans, confirming theabsence of O-stearyl ester groups [27]. The acylated chitosans herein are different from those reported by other researchers [28] whocoupled acyl chloride with soluble chitosan or swollen chitosan,and thus obtained N, O-acyl chitosan with higher DS. In our work,N-acylation was intended to allow further sulfation of the preparedSC derivatives using the unsubstituted hydroxyl groups of chitosan.
The S-SC derivatives shown in Fig. 2B–E reveal specific absorp-tion bands of sulfate groups in the area of 1200–1260 cm−1
and 1060 cm−1, representing asymmetric valence fluctuations andsymmetric valence fluctuations of SO2 respectively. Also valencefluctuations of C O S appeared in the area of 900 cm−1 which arecharacteristic to sulfo groups [29]. Furthermore, the OH stretchingbroad band of chitosan at 3500 cm−1 (Fig. 2A) disappeared indicat-ing 3 and 6 O-sulfation.
3.1.2. H NMR spectroscopyAccording to Fig. 3 the peaks in the 1H NMR spectrum of the d-
acetyl glucosamine unit of chitosan are nearly identical to those in
previous reports [30] and could be assigned as follows: ı 1.9 ( CH3,acetyl group), ı 4.7 (1-H), ı 2.9(2-H), ı 3.4–3.8 (3-H–6-H) and ı 8.23to N H. Compared to chitosan, these proton assignments accordingto the 1H NMR spectra of the prepared SC derivatives (Fig. 4), are
356 G.M. Mekhail et al. / International Journal of Biolo
a(ıoNt
3.1.4. X-ray diffraction (XRD)
Fig. 3. H NMR spectrum of chitosan.
s follows: ı 0.85 = CH3 (stearyl), ı 1.23 = CH2 (stearyl), ı 1.49 = CH2stearyl deshielded by carbonyl, � to carbonyl), ı 2.08 = CH3 (acetyl),
2.88 to a H-2 proton, ı 3.34–3.71 to the ring protons (H-3, 4,5,6,6′)
verlap with H2O, ı 4.95–5.44 to the H-1 protons while ı 8.23 toH disappeared. These results confirm the N-acylation of chi-osan.
Fig. 4. H NMR spectra of SC
gical Macromolecules 51 (2012) 351– 363
Table 1 shows the calculated DS of the different prepared SCderivatives using MestReC 4.9.9.6 software. It is obvious that theDS of SCs increases along with increasing the amount of stearicanhydride where SC1, SC2, SC3 and SC4 prepared with the respec-tive molar ratios of chitosan to acetic anhydride 1:1, 1:1.5, 1:2 and1:3 exhibited 1.7%, 4%, 17.3% and 18.8% DS respectively [18,26].As the concentration of acetic anhydride increases, the frequencyof the molecules colliding increased, striking each other more fre-quently by being in closer contact at any given point in time, henceincreasing the degree of acylation.
3.1.3. Elemental analysisElemental analysis performed on the sulfated chitosan deriva-
tives shows no great variation in the percent of elemental sulfuramong the different sulfated polymers prepared. The degree ofsulfation varied from 0.102 to 0.162 for the four tested polymers.
Fig. 5 shows the XRD patterns of chitosan and SC derivatives withdifferent degrees of substitution. Chitosan shows two characteris-tic peaks around 2� = 10.3◦ and 20.1◦ indicating its high degree
1, SC2, SC3 and SC4.
G.M. Mekhail et al. / International Journal of Biological Macromolecules 51 (2012) 351– 363 357
F
o2tXtrB22anctcl[
3
aie2ttawF
Fs
ig. 5. X-ray diffraction pattern of (A) chitosan, (B) SC1, (C) SC2, (D) SC3, (E) SC4.
f crystallinity as previously reported [31]. The reflection fall at� = 10.3◦ was assigned to crystal forms I and strongest reflec-ion appeared at 2� = 20.1◦ corresponding to crystal forms II. TheRD patterns of SC derivatives are significantly different from chi-
osan, they show a strong reflection at about 2� = 21◦ and a weakereflection at around 2� = 6◦ and another reflection at 2� = 23◦.y increasing the DS from 1.7% to 18.8%, the reflection at around� = 21◦ becomes acute and the reflection at around 2� = 23◦ and� = 9◦ increases. Although SC has hindered the formation of inter-nd extra-molecular hydrogen bonds, they could have formed aew kind of crystallinity because of the perfect arrangement afterhemical modification. The crystallinity of SC might be ascribed tohe presence of long chain residue. The increase in number of longhains and the improvement of arrangement of the long chainseads to the increased crystallinity as evidenced in SC4 (trace E)32]. The XRD of S-SC was similar to SC (figure not shown).
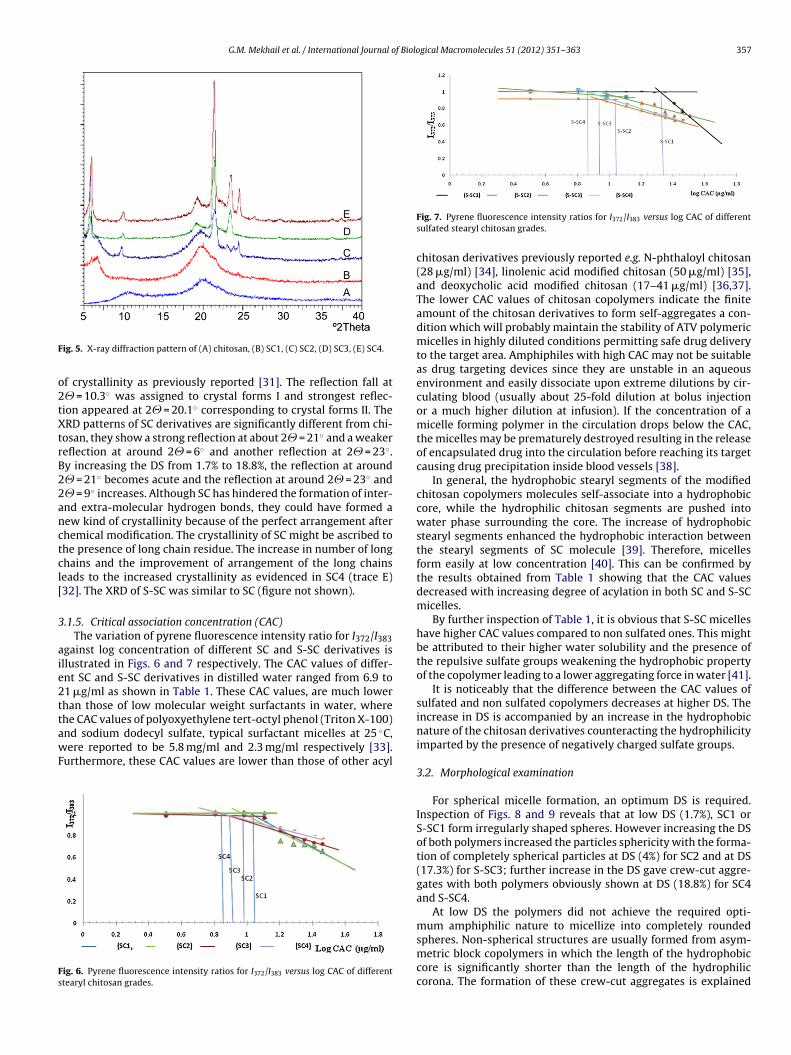
.1.5. Critical association concentration (CAC)The variation of pyrene fluorescence intensity ratio for I372/I383
gainst log concentration of different SC and S-SC derivatives isllustrated in Figs. 6 and 7 respectively. The CAC values of differ-nt SC and S-SC derivatives in distilled water ranged from 6.9 to1 �g/ml as shown in Table 1. These CAC values, are much lowerhan those of low molecular weight surfactants in water, wherehe CAC values of polyoxyethylene tert-octyl phenol (Triton X-100)
nd sodium dodecyl sulfate, typical surfactant micelles at 25 ◦C,ere reported to be 5.8 mg/ml and 2.3 mg/ml respectively [33].urthermore, these CAC values are lower than those of other acyl
ig. 6. Pyrene fluorescence intensity ratios for I372/I383 versus log CAC of differenttearyl chitosan grades.
Fig. 7. Pyrene fluorescence intensity ratios for I372/I383 versus log CAC of differentsulfated stearyl chitosan grades.
chitosan derivatives previously reported e.g. N-phthaloyl chitosan(28 �g/ml) [34], linolenic acid modified chitosan (50 �g/ml) [35],and deoxycholic acid modified chitosan (17–41 �g/ml) [36,37].The lower CAC values of chitosan copolymers indicate the finiteamount of the chitosan derivatives to form self-aggregates a con-dition which will probably maintain the stability of ATV polymericmicelles in highly diluted conditions permitting safe drug deliveryto the target area. Amphiphiles with high CAC may not be suitableas drug targeting devices since they are unstable in an aqueousenvironment and easily dissociate upon extreme dilutions by cir-culating blood (usually about 25-fold dilution at bolus injectionor a much higher dilution at infusion). If the concentration of amicelle forming polymer in the circulation drops below the CAC,the micelles may be prematurely destroyed resulting in the releaseof encapsulated drug into the circulation before reaching its targetcausing drug precipitation inside blood vessels [38].
In general, the hydrophobic stearyl segments of the modifiedchitosan copolymers molecules self-associate into a hydrophobiccore, while the hydrophilic chitosan segments are pushed intowater phase surrounding the core. The increase of hydrophobicstearyl segments enhanced the hydrophobic interaction betweenthe stearyl segments of SC molecule [39]. Therefore, micellesform easily at low concentration [40]. This can be confirmed bythe results obtained from Table 1 showing that the CAC valuesdecreased with increasing degree of acylation in both SC and S-SCmicelles.
By further inspection of Table 1, it is obvious that S-SC micelleshave higher CAC values compared to non sulfated ones. This mightbe attributed to their higher water solubility and the presence ofthe repulsive sulfate groups weakening the hydrophobic propertyof the copolymer leading to a lower aggregating force in water [41].
It is noticeably that the difference between the CAC values ofsulfated and non sulfated copolymers decreases at higher DS. Theincrease in DS is accompanied by an increase in the hydrophobicnature of the chitosan derivatives counteracting the hydrophilicityimparted by the presence of negatively charged sulfate groups.
3.2. Morphological examination
For spherical micelle formation, an optimum DS is required.Inspection of Figs. 8 and 9 reveals that at low DS (1.7%), SC1 orS-SC1 form irregularly shaped spheres. However increasing the DSof both polymers increased the particles sphericity with the forma-tion of completely spherical particles at DS (4%) for SC2 and at DS(17.3%) for S-SC3; further increase in the DS gave crew-cut aggre-gates with both polymers obviously shown at DS (18.8%) for SC4and S-SC4.
At low DS the polymers did not achieve the required opti-mum amphiphilic nature to micellize into completely rounded
spheres. Non-spherical structures are usually formed from asym-metric block copolymers in which the length of the hydrophobiccore is significantly shorter than the length of the hydrophiliccorona. The formation of these crew-cut aggregates is explained
358 G.M. Mekhail et al. / International Journal of Biological Macromolecules 51 (2012) 351– 363
ryl ch
bcchtn
mmhm
3
r
piSoSa1ohd
te
Fig. 8. TEM photomicrographs of stea
y a force balance effect between the degree of stretching of theore-forming blocks, the interfacial energy between the micelleore and the solvent, and the interaction between corona-formingydrophilic chains [42–44]. On the contrary at the highest DS [45]he amphiphilic molecules face a steric hindrance due to their bulkyature giving smaller micelles with less sphericity.
Micellar sphericity was achieved with stearyl chitosan copoly-er at lower DS (4%) than sulfated derivative DS (17.3%). Thisight be due to the sulfate group which increases the polymer
ydrophilicity, hence a higher DS is required to give sphericalicelles.
.3. Encapsulation efficiency and drug loading
Figs. 10 and 11 show that EE% and DL% of ATV-SC and ATV-S-SCanged from 10.4% to 35% and from 40% to 84.1% respectively.
It is obvious from Fig. 10 that increasing the DS of the acylatedolymers at all organic to aqueous phase ratios leads to a signif-
cant increase in the EE% with an optimum value at DS 17.3% inC3 (P < 0.05). This might be due to the hydrophobic interactionf the drug with the hydrophobic core [26]. However, in case ofC4 with DS 18% the EE% decreased significantly (P < 0.01) in 1:1nd 1:2 ratios while no significant change was observed in case of:3 ratio. The core crystallinity in case of SC4 might be at the basef this unexpected low EE%. Moreover the core crystallinity mightave decreased the accessible volume for drug loading and hence
ecreased the EE% [46].On the other hand the EE% of ATV is almost not affected byhe variation in DS of S-SC at all organic to aqueous phase ratios,specially for the sulfated polymers (S-SC1, S-SC2 and S-SC3).
itosan derivatives (bar scale: 0.5 �m).
It can be deduced that the presence of the hydrophilic divalentSO4—group had minimized the hydrophobic effect due to acylation.The hydrophilicity of the sulfated moiety which retards the micellesformation with a compactly arrayed corona might lead after thisnon significant difference in EE%, to almost an equal values for S-SC1and S-SC2. As previously proved by TEM, S-SC3 showed micelleswith compact corona and encapsulated the maximum amount ofATV (34%).
The optimum organic to aqueous phase ratio with all ATV-SCpolymeric micelles is 1:2. Changing the ratio of organic to aque-ous phase from 1:1 to 1:2 leads to a significant increase in theEE% and DL% (P < 0.001). With the decrease in the volume of theorganic phase the polymer concentration increases forming morecompact micelles with higher EE% [47]. Further decrease in theorganic volume (in the ratio 1:3) causes a significant decrease inthe EE% and DL% (P < 0.001), which might be due to inadequateemulsification.
On the contrary, in case of ATV-sulfated stearyl chitosan micellesand at all DS (Figs. 10 and 11), the most appropriate organic to aque-ous phase ratio was found to be 1:1; further decrease in the organicphase volume causes a significant decrease in the EE% and DL%(P < 0.001). The lower aqueous phase needed for polymer micelliza-tion, hence drug encapsulation, is probably due to more hydrophilicnature of the S-SC. However, in case of SC derivatives, with morehydrophobic nature, higher water amounts were needed to facili-tate polymer migration to the aqueous phase, a prerequisite step
for polymer self assembly accompanying drug encapsulation.ATV loaded polymeric micelles prepared using organic to aque-ous phase volume ratio 1:3 was excluded from further studies dueto their lower DL and EE percentage.
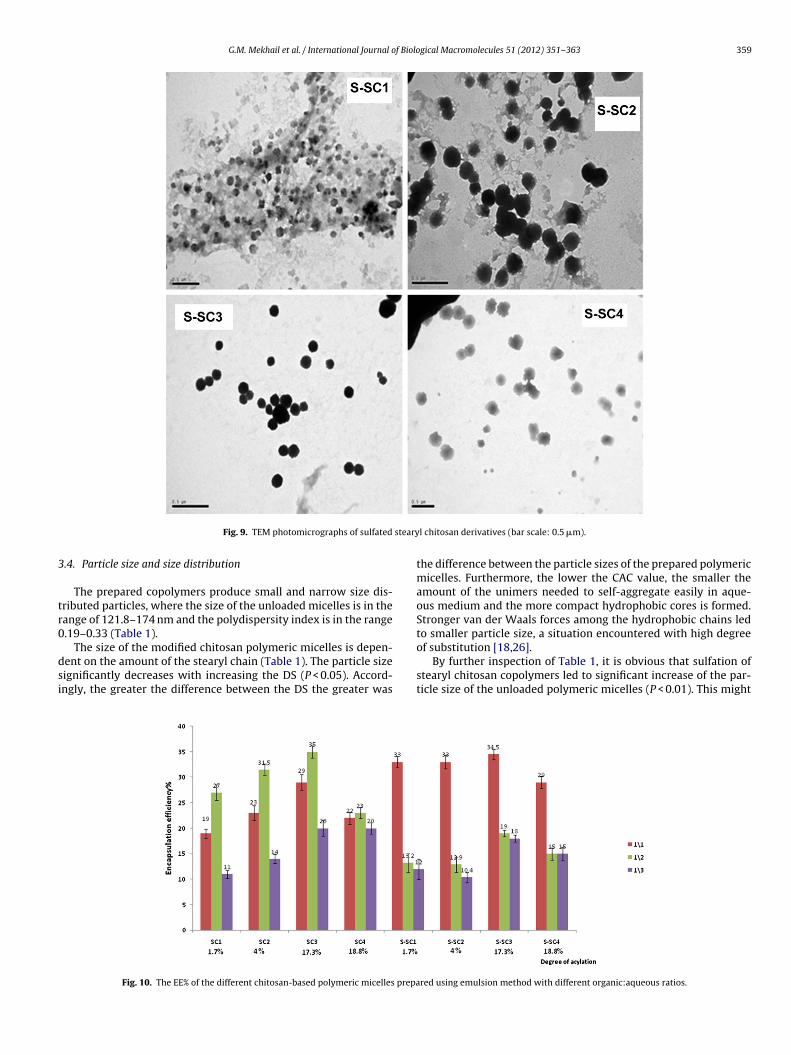
G.M. Mekhail et al. / International Journal of Biological Macromolecules 51 (2012) 351– 363 359
steary
3
tr0
dsi
Fig. 9. TEM photomicrographs of sulfated
.4. Particle size and size distribution
The prepared copolymers produce small and narrow size dis-ributed particles, where the size of the unloaded micelles is in theange of 121.8–174 nm and the polydispersity index is in the range.19–0.33 (Table 1).
The size of the modified chitosan polymeric micelles is depen-ent on the amount of the stearyl chain (Table 1). The particle sizeignificantly decreases with increasing the DS (P < 0.05). Accord-ngly, the greater the difference between the DS the greater was
Fig. 10. The EE% of the different chitosan-based polymeric micelles prepa
l chitosan derivatives (bar scale: 0.5 �m).
the difference between the particle sizes of the prepared polymericmicelles. Furthermore, the lower the CAC value, the smaller theamount of the unimers needed to self-aggregate easily in aque-ous medium and the more compact hydrophobic cores is formed.Stronger van der Waals forces among the hydrophobic chains ledto smaller particle size, a situation encountered with high degree
of substitution [18,26].By further inspection of Table 1, it is obvious that sulfation ofstearyl chitosan copolymers led to significant increase of the par-ticle size of the unloaded polymeric micelles (P < 0.01). This might
red using emulsion method with different organic:aqueous ratios.

360 G.M. Mekhail et al. / International Journal of Biological Macromolecules 51 (2012) 351– 363
prepa
boomdStlii
pam
acbhb
3
csamcf
ipat
trac1tr
Fig. 11. The DL% of the different chitosan-based polymeric micelles
e due to the higher CAC of SC derivatives requiring larger numberf polymer molecules to form the micelles. Moreover, the presencef sulfate group, as previously discussed under results of drug EE%,inimized the hydrophobic effect of stearyl radical. This was evi-
enced by the smaller difference in the particle size of unloaded-SC2 and S-SC3 (15 nm) on one hand and SC2 and SC3 (23 nm) onhe other hand. An observation which was amplified after optimallyoading the drug (S-SC2 and S-SC3 at 1:1 ratio with 14 nm differencen particle size while SC2 and SC3 at 1:2 ratio with 31 nm differencen particle size).
Furthermore, increasing polymer concentration in the organichase, by decreasing the phase volume, in case of 1:2 organic toqueous phase ratio might have decreased the aggregation time oficellization forming more compact nanoparticles [47].Finally, Table 1 shows that for both types of polymers and at
ll organic to aqueous phase ratios, loading the micelles with ATVaused a significant (P < 0.01) decrease in particle size. This mighte due to increased van der Waals forces manifested by strongydrophobic interaction between the hydrophobic drug in com-ination with the stearyl molecule of the core [48].
.5. Zeta potential
Table 1 shows that loaded and unloaded polymeric micellesarry a negative charges simply referring to the negatively chargedtearyl and sulfate groups which hinders the positive charge of themino groups of chitosan. The negative charge of the polymericicelles, either loaded or unloaded, contribute to the stability of the
olloidal micellar solution as a result of the electrostatic repulsiveorces between the micelles.
It is obvious from Table 1 that in case of unloaded micelles,ncreasing the degree of acylation significantly increases the zetaotential (P < 0.001). Moreover, due to the presence of sulfate group,ll sulfated polymers exhibit a significant higher zeta potential thanheir corresponding stearyl chitosan copolymers (P < 0.001).
After loading, all polymers show significantly lower zeta poten-ial values either in 1:1 or 1:2 organic to aqueous phase volumeatios. This may be attributed to partial neutralization of the neg-tive charge of modified polymers with the positive charge of the
alcium ion of the ATV. For stearyl chitosan, the loaded particles of:1 organic to aqueous group show an apparent higher zeta poten-ial value than those prepared with the 1:2 organic to aqueousatio. The reverse was observed in case of sulfated stearyl chitosan.red using emulsion method with different organic:aqueous ratios.
A matter which could be explained based on the EE% of variousmicelles where higher EE% showed lower zeta potential.
3.6. In vitro release study
ATV-stearyl chitosan and ATV-sulfated stearyl chitosan poly-meric micelles, prepared using organic to aqueous phase volumeratio 1:2 and 1:1 respectively were subjected to in vitro releasestudy due to their optimum EE% and particle size.
Fig. 12 shows that after 8 h, a sustained release profile was seenwith SC2, SC3 and S-SC3, an observation which suggests that themicelle might have acted as a barrier against the release of ATVand that the hydrophobic core of the micelle strongly restrictedthe migration of the drug to the media [23].
All the loaded micelles which show a sustained effect in theirrelease profiles have shown a high degree of compactness intheir TEM photomicrograph where their optimum amphiphilicityallowed optimum core-corona packing delaying the release rateof the drug from the micellar core. On the contrary, the enhancedrelease of the drug from all other loaded micelles might be dueto their weak compactness where the crew-cut appearance of themicelles facilitates the rapid release of the drug from the micel-lar core. Comparing SC2 with SC3, the increase in the amount ofthe stearyl chain resulted in a decrease in the release rate of ATV.This might be due to the more hydrophobic domain of the polymerthat make stronger hydrophobic interactions with the hydrophobicdrug [49].
Based on the micelles characterization, SC3 loaded micelles pre-pared using organic to aqueous phase ratio 1:2, having the slowestrelease rate, optimum particle size (97 nm), the lowest negativezeta potential (−8.27), the maximum EE%, sufficiently low CACvalue and high packing pattern were chosen for further cytotoxicstudies.
3.7. Cytotoxic activity
The cell survival of human breast adenocarcinoma cell line (MCF7) and human colon carcinoma cell line (HCT 116) as model tumorcells against ATV, unloaded and loaded SC3 polymeric micelles wasevaluated using SRB method.
Figs. 13 and 14 show that ATV gives a cytotoxic effect againstboth MCF 7 and HCT 116 cells. This cancer prevention activity ofATV is believed to be due to its inhibition of the isoprenylation ofG-proteins and the subsequent alteration of downstream signaling

G.M. Mekhail et al. / International Journal of Biological Macromolecules 51 (2012) 351– 363 361
Fig. 12. Release profiles of ATV from ATV-stearyl chitosan and ATV-sulfated stearyl chitosan polymeric micelles in phosphate buffer pH 6.8.
F cells
pftrcHca
ntna
ls6
t
polymeric micelles were negatively charged, yet the bioadhesionof chitosan due to its polysaccharide nature would have facilitatedthe adhesion of particles to the cell membrane. Cell internalization
Table 2IC50 values of free ATV and ATV-SC3 loaded polymeric micelles at different timeintervals for MCF-7 and HCT-116 cell lines.
Formulation Cell line 48 h 72 h
ig. 13. Fraction of surviving MCF 7 cells against different ATV concentrations after
athways [50]. Moreover, other mechanisms might be responsibleor this antitumor effect, such as induction of apoptosis, inhibi-ion of cell growth, angiogenesis and enhancement of immuneesponse [51]. HMG-CoA reductase, the rate limiting enzyme in theholesterol biosynthetic pathway responsible for the conversion ofMG-CoA to mevalonate [52], was known to be augmented in mostolonic tumors at both gene and protein expression levels as wells in its activity [53,54].
Figs. 13 and 14 show that unloaded SC3 polymeric micelles doesot show any cytotoxicity on both cell lines even at the highestested concentration. This suggests that SC polymeric micelles areon-cytotoxic and good candidates for the potential application as
drug delivery carrier.However, after 48 h incubation in either MCF 7 or HCT 116 cell
ines, ATV-loaded SC3 polymeric micelles showed a slightly lower
urvival fraction than free ATV with respective IC50% 4.4 and 4.9 &.1 and 6.8 in MCF and HCT 116 respectively (Table 2).After 72 h ATV-loaded SC3 showed higher cytotoxicity, in bothypes of cells, than free ATV. Besides, in comparison with 48 h
incubation with free ATV, unloaded and loaded SC3 micelles for 48 and 72 h.
incubation, the cytotoxicity increased in case of loaded SC3 while itdecreased in case of the free ATV. The respective calculated IC50%values were 3.7 and 10.4 in case of MCF 7 and 3.4 and 9.4 with HCT116 (Table 2).
Nanoparticles uptake could be considered as an adhesion fol-lowed by an internalization process [55]. Although the prepared
Free drug MCF-7 4.9 ± 0.2 10.4 ± 0.5ATV-loaded micelles MCF-7 4.4 ± 0.2 3.7 ± 0.2Free drug HCT-116 6.8 ± 1.2 9.4 ± 0.8ATV-loaded micelles HCT-116 6.1 ± 0.8 3.4 ± 0.2

362 G.M. Mekhail et al. / International Journal of Biological Macromolecules 51 (2012) 351– 363
F ter ce
oi
gsswtsAncit[
tidaccS
ace
4
mfT(sotm
[
[
[
[[
[
[
[
[
[[
[
[
[
[
ig. 14. Fraction of surviving HCT-116 cells against different ATV concentrations af
ccurs mainly by adsorptive endocytosis initiated by nonspecificnteractions between NPs and cell membranes [56].
Moreover, the few cationic sites on the cell membrane [57] sug-est the binding possibility of negatively charged particles to theseites. The negatively charged polymeric micelles bind at the cationicites in the form of clusters because of the repulsive interactionsith the large negatively charged areas of the cell surface. In addi-
ion a reduction in the charge density due to NP adhesion to cellurface may have favored adsorption of other free particles [58].lthough the mechanism for cellular uptake of negatively chargedanoparticles needs further study, the adsorption of the negativelyharged particles at the positively charged sites, by electrostaticnteraction, with localized neutralization, can lead to bending ofhe membrane which in turn favors endocytosis for cellular uptake57].
The higher cytotoxic activity of ATV loaded micelles comparedo the free ATV might be attributed to the enhancement of fluid-ty of the cell membrane induced by SC polymer, as a result, morerug could be easily taken up into the cells. Moreover, the smallermount of drug leakage from the micelles before being uptaken byells might have enhanced the drug amount internalized into theells [59]. These results are in agreement with the release profile ofC3.
Finally, the noted increase in the cytotoxic effect of loaded SC3fter 72 h compared to free drug suggests that stearyl modifiedhitosan polymeric micelles enhanced and sustained the cytotoxicffect of ATV.
. Conclusion
It can be concluded that, the prepared SC3 ATV loaded polymericicelles can be considered as a promising drug delivery system
or tumor targeting due to its suitable size and zeta potential.he small particle size (97.19 nm) and the slight negative charges−8.27 ± 1.78 mv) is expected to minimize the recognition and non-
pecific uptake by macrophages in the liver due to the expected lowpsonization rate as well as electrostatic repulsion and accordingo cell culture study, the particles will tend to accumulate in tumorsore efficiently.
[
lls incubation with free ATV, unloaded and loaded SC3 micelles for 48 and 72 h.
References
[1] A.S. Wierzbicki, R. Poston, A. Ferro, Pharmacology & Therapeutics 99 (2003)95–112.
[2] M.M. McNally, S.C. Agle, F.M. Parker, W.M. Bogey, C.S. Powell, M.C. Stoner,Journal of Vascular Surgery 51 (2010) 1390–1396.
[3] J.Y. Lee, J.S. Kim, J.M. Kim, N. Kim, H.C. Jung, I.S. Song, InternationalImmunopharmacology 7 (2007) 241–248.
[4] K.I. Paraskevas, A.A. Tzovaras, V. Stathopoulos, D.P. Mikhailidis, InternationalUrology and Nephrology 42 (2010) 711–713.
[5] C. Folli, D. Descalzi, S. Bertolini, A.M. Riccio, F. Scordamaglia, C. Gamalero, M.Barbieri, G. Passalacqua, G.W. Canonica, European Annals of Allergy and ClinicalImmunology 40 (2008) 84–89.
[6] D.R. Friedman, L.A. Magura, H.A. Warren, J.D. Harrison, L.F. Diehl, J.B. Weinberg,Leukemia and Lymphoma 51 (2010) 2295–2298.
[7] J.L. Goldstein, M.S. Brown, Nature 343 (1990) 425–430.[8] K.K. Chan, A.M. Oza, L.L. Siu, Clinical Cancer Research 9 (2003) 10–19.[9] P.J. Casey, Science 268 (1995) 221–225.10] E.A. Collisson, C. Kleer, M. Wu, A. De, S.S. Gambhir, S.D. Merajver, M.S. Kolodney,
Molecular Cancer Therapeutics 2 (2003) 941–948.11] P. Cafforio, F. Dammacco, A. Gernone, F. Silvestris, Carcinogenesis 26 (2005)
883–891.12] Z. Yang, H. Xiao, H. Jin, P.T. Koo, D.J. Tsang, C.S. Yang, International Journal of
Cancer 126 (2010) 852–863.13] O. Mistafa, U. Stenius, Biochemical Pharmacology 78 (2009) 1115–1126.14] C.A. Lipinski, F. Lombardo, B.W. Dominy, P.J. Feeney, Advanced Drug Delivery
Reviews 46 (2001) 3–26.15] V.P. Torchilin, A.N. Lukyanov, Z. Gao, B. Papahadjopoulos-Sternberg, Proceed-
ings of the National Academy of Sciences of the United States of America 100(2003) 6039–6044.
16] J.H. Park, G. Saravanakumar, K. Kim, I.C. Kwon, Advanced Drug Delivery Reviews62 (2010) 28–41.
17] R. Gref, Y. Minamitake, M.T. Peracchia, V. Trubetskoy, V. Torchilin, R. Langer,Science 263 (1994) 1600–1603.
18] G.B. Jiang, D. Quan, K. Liao, H. Wang, Carbohydrate Polymers 66 (2006)514–520.
19] R.G. Schweiger, Carbohydrate Research 21 (1972) 219–228.20] H. Yoshioka, K. Nonaka, K. Fukuda, S. Kazama, Bioscience, Biotechnology, and
Biochemistry 59 (1995) 1901–1904.21] Y. Lin, L. Zhang, L. Chen, Y. Jin, F. Zeng, J. Jin, B. Wan, P.C.K. Cheung, International
Journal of Biological Macromolecules 34 (2004) 231–236.22] T. Ngawhirunpat, N. Wonglertnirant, P. Opanasopit, U. Ruktanonchai, R. Yok-
san, K. Wasanasuk, S. Chirachanchai, Colloids and Surfaces B: Biointerfaces 74(2009) 253–259.
23] X. Xiangyang, L. Ling, Z. Jianping, L. Shiyue, Y. Jie, Y. Xiaojin, R. Jinsheng, Colloidsand Surfaces B: Biointerfaces 55 (2007) 222–228.
24] P. Skehan, R. Storeng, D. Scudiero, A. Monks, J. McMahon, D. Vistica, J.T. Warren,
H. Bokesch, S. Kenney, M.R. Boyd, Journal of the National Cancer Institute 82(1990) 1107–1112.25] A. Monks, D. Scudiero, P. Skehan, R. Shoemaker, K. Paull, D. Vistica, C. Hose, J.Langley, P. Cronise, A. Vaigro-Wolff, Journal of the National Cancer Institute 83(1991) 757–766.

f Biolo
[
[
[
[
[
[[[
[
[
[
[[
[[
[
[[
[
[
[
[
[
[
[
[
[[[
[
[
[
G.M. Mekhail et al. / International Journal o
26] G.B. Jiang, D. Quan, K. Liao, H. Wang, Molecular Pharmaceutics 3 (2006)152–160.
27] S. Wu, F. Zeng, H. Zhu, Z. Tong, Journal of the American Chemical Society 127(2005) 2048–2049.
28] M.E.I. Badawy, E.I. Rabea, T.M. Rogge, C.V. Stevens, G. Smagghe, W. Steurbaut,M. Hefte, Biomacromolecules 5 (2004) 589–595.
29] G. Vikhoreva, G. Bannikova, P. Stolbushkina, A. Panov, N. Drozd, V. Makarov, V.Varlamov, L. Gal’braikh, Carbohydrate Polymers 62 (2005) 327–332.
30] S.C. Chen, Y.C. Wu, F.L. Mi, Y.H. Lin, L.C. Yu, H.W. Sung, Journal of ControlledRelease 96 (2004) 285–300.
31] H. Zhang, S.H. Neau, Biomaterials 22 (2001) 1653–1658.32] G. Ma, D. Yang, J.F. Kennedy, J. Nie, Carbohydrate Polymers 75 (2009) 390–394.33] A. Harada, H. Togawa, K. Kataoka, European Journal of Pharmaceutical Sciences
13 (2001) 35–42.34] P. Opanasopit, T. Ngawhirunpat, T. Rojanarata, C. Choochottiros, S. Chirachan-
chai, Colloids and Surfaces B: Biointerfaces 60 (2007) 117–124.35] C.G. Liu, K.G. Desai, X.G. Chen, H.J. Park, Journal of Agricultural and Food Chem-
istry 53 (2005) 437–441.36] K.Y. Lee, W.H. Jo, I.C. Kwon, Y.H. Kim, S.Y. Jeong, Macromolecules 31 (1998)
378–383.37] K.Y. Lee, W.H. Jo, I.C. Kwon, Y.H. Kim, S.Y. Jeong, Langmuir 14 (1998) 2329–2332.38] K. Letchford, H. Burt, European Journal of Pharmaceutics and Biopharmaceutics
65 (2007) 259–269.39] D.W. Lee, K. Powers, R. Baney, Carbohydrate Polymers 58 (2004) 371–377.40] M. Huo, Y. Zhang, J. Zhou, A. Zou, D. Yu, Y. Wu, J. Li, H. Li, International Journal
of Pharmaceutics 394 (2010) 162–173.41] Z. Sezgin, N. Ynksel, T. Baykara, European Journal of Pharmaceutics and Bio-
pharmaceutics 64 (2006) 261–268.42] L. Zhang, A. Eisenberg, Science 268 (1995) 1728–1731.43] L. Zhang, K. Yu, A. Eisenberg, Science 272 (1996) 1777–1779.
[
[
gical Macromolecules 51 (2012) 351– 363 363
44] L. Zhang, A. Eisenberg, Journal of the American Chemical Society 118 (1996)3168–3181.
45] C. Allen, Y. Yu, D. Maysinger, A. Eisenberg, Bioconjugate Chemistry 9 (1998)564–572.
46] Z.L. Tyrrell, Y. Shen, M. Radosz, Progress in Polymer Science 35 (2010)1128–1143.
47] H.M. Aliabadi, S. Elhasi, A. Mahmud, R. Gulamhusein, P. Mahdipoor, A. Lavasan-ifar, International Journal of Pharmaceutics 329 (2007) 158–165.
48] Y.Q. Ye, F.L. Yang, F.Q. Hu, Y.Z. Du, H. Yuan, H.Y. Yu, International Journal ofPharmaceutics 352 (2008) 294–301.
49] Y.Z. Du, L. Wang, H. Yuan, X.H. Wei, F.Q. Hu, Colloids and Surfaces B: Biointer-faces 69 (2009) 257–263.
50] O. Fromigue, E. Hay, D. Modrowski, S. Bouvet, A. Jacquel, P. Auberger, P.J. Marie,Cell Death and Differentiation 13 (2006) 1845–1856.
51] K. Gauthaman, C.Y. Fong, A. Bongso, Journal of Cellular Biochemistry 106 (2009)975–983.
52] G. Fritz, International Journal of Oncology 27 (2005) 1401–1409.53] M.G. Caruso, M. Notarnicola, Anticancer Research 25 (2005) 3393–3397.54] M. Notarnicola, C. Messa, M. Pricci, V. Guerra, D.F. Altomare, S. Montemurro,
M.G. Caruso, Anticancer Research 24 (2004) 3837–3842.55] J.L. West, N.J. Halas, Annual Review of Biomedical Engineering 5 (2003)
285–292.56] O. Harush-Frenkel, N. Debotton, S. Benita, Y. Altschuler, Biochemical and Bio-
physical Research Communications 353 (2007) 26–32.57] C. Wilhelm, C. Billotey, J. Roger, J.N. Pons, J.C. Bacri, F. Gazeau, Biomaterials 24
(2003) 1001–1011.58] S. Patil, A. Sandberg, E. Heckert, W. Self, S. Seal, Biomaterials 28 (2007)
4600–4607.59] Z. Liu, Z. Zhang, C. Zhou, Y. Jiao, Progress in Polymer Science 35 (2010)
1144–1162.
Copyright © 2022 FDOKUMEN




















