
Physical geography: fundamentals of the physical environment
Upload
independentCategory
view
0download
0
ELSEVIER Surface Science Reports 24 (1996) 223-287
surface science reports
Physical and chemical properties of bimetallic surfaces
Jos6 A. Rodriguez *'~
Department of Chemistry, Brookhaven National Laboratory, P.O. Box 5000, Upton, N Y I 1973, USA
Manuscript received in final form 4 March 1996
Abstract
Recent studies dealing with the structural, electronic, chemical and catalytic properties of well-defined bimetallic surfaces are reviewed. LEED and STM show that two metals interacting on a surface can form compounds with structures not seen in bulk alloys. Many novel phenomena related to the kinetics of growth of metals on metals have been discovered. The knowledge gathered in this area provides a solid basis for the synthesis of new materials with applications in areas of catalysis, electro-chemistry and microelectronics. In many cases, the formation of a surface bimetallic bond induces large changes in the band structure of the metals. For surfaces that contain transition or s,p metals, the strongest metal-metal interactions occur in systems that combine a metal with a valence band almost fully occupied and a metal in which the valence band is almost empty. A very good correlation is found between the electronic perturbations in a bimetallic system and its cohesive energy. Bimetallic bonds that display a large stability usually involve a significant redistribution of charge around the metal centers. The electronic perturbations affect the reactivity of the bonded metals toward small molecules (CO, NO, Hz, Ca, S2, CzH4, CH3OH, etc.). For supported monolayers of Ni, I'd, Pt and Cu a correlation is observed between the shifts in surface core-level binding energies and changes in the desorption temperature of CO from the metal adlayers. Examples are provided which demonstrate the utility of single-crystal studies for understanding the role of "ensemble" and "ligand" effects in bimetallic catalysts.
Keywords: Catalysis; Chemisorption; Epitaxy; Faceting; Growth; Surface chemical reaction; Surface electronic phenom- ena; Alloys; Metal-metal interfaces
1. Introduction
In recent years there has been a substantial effort in the surface science community to investigate the structural, electronic and chemical properties of bimetallic systems. A major goal is to identify phenomena that accompany the formation ofheteronuclear metal-metal bonds, and determine how the properties of these bonds depend on the interacting metals and on the geometrical structure of the surface. A basic understanding of the nature of the bimetallic bond is a prerequisite for a scientific
* E-mail: [email protected]. 1 Supported by the Department of Energy, Office of Basic Energy Sciences, Division of Chemical Sciences.
Published by Elsevier Science B.V. PII: SO 167-5729(96)00004-0

224 J.A. Rodriouez/Surface Science Reports 24 (1996) 223-287
Contents
1. Introduction 2. Atomic structure of bimetallic surfaces
2.1. Well-defined surfaces of bulk alloys 2.1.1. Surface vs bulk geometry 2.1.2. Surface segregation in bulk alloys
2.2. Deposition of metals on single-crystal metal surfaces 2.2.1. Growth modes of metal films 2.2.2. Admetal induced reconstruction of a metal substrate: Faceting 2.2.3. Alloy formation through metal-on-metal deposition
3. Electronic properties of bimetallic surfaces 3.1. Group 8 and 9 admetals 3.2. Group-10 admetals 3.3. Group-l l admetals 3.4. Group- 12 admetals 3.5. Summary
4. Chemical properties of bimetallic surfaces 4.1. CO chemisorption 4.2. NO chemisorption 4.3. Chemisorption of ethylene, acetylene and benzene 4.4. Chemisorption of"large" hydrocarbons 4.5. H, chemisorption 4.6. Interaction with oxygen 4.7. Interaction with sulfur
5. Catalytic properties of bimetallic surfaces 5.1. CO oxidation 5.2. CO hydrogenation 5.3. Alkane hydrogenolysis 5.4. Cyclohexane dehydrogenation 5.5. Olefin hydrogenation 5.6. Cyclotrimerization of acetylene
6. Conclusion
Acknowledgements References
225 226 226 226 228 229 229 237 239 241 241 242 249 255 256 256 256 261 262 262 263 264 266 268 269 270 272 275 275 276 278
278 278

Physical and chemical properties of bimetallic surfaces
J.A. Rodriguez Department of Chemistry, Brookhaven National Laboratory, Upton, NYl1973, USA
ELSEVIER
Amsterdam-Lausanne-New York-Oxford-Shannon-Tokyo

226 J.A. Rodriouez/Surface Science Reports 24 (1996) 223-287
design of mixed-metal compounds with industrial applications in areas of catalysis, electrochemis- try, microelectronics and materials science.
For a long time it has been known that a bimetallic surface can exhibit chemical and catalytic properties that are very different from those of the surfaces of the individual metals. Research on alloy catalysts started in the late 1940s [1-3] with the purpose of establishing links between the electronic and catalytic properties of a surface. However, due to the lack of adequate techniques for the preparation and characterization of the surface of alloys, no real progress was made at an experimental level. In the 1960s and 1970s the development of bimetallic catalysts for hydrocarbon refo~ing in the petrochemical industry increased the need for a fundamental understanding of the behavior of bimetallic surfaces, and renewed the interest in catalysis by alloys [4-7]. This effort provided the basis for the concepts of"ensemble" and "ligand or electronic" effects [8], which are commonly used to rationalize the superior activity or selectivity of bimetallic catalysts [4-7]. Finally, in the last two decades, the development of new and reliable techniques for surface characterization [9-11] has made feasible a systematic study of the properties of bimetallic surfaces. It is now possible to explore relationships between the structural, electronic and chemical properties of a bimetallic surface in detail. Experiments performed using the modern techniques of surface science have given exciting insights into phenomena responsible for the behavior of bimetallic surfaces.
In this article, we will examine the properties of bimetallic surfaces that contain transition and s,p metals. A number of reviews have appeared in the literature dealing with various aspects of this subject [4-7,12,13]. It is not our intention to cover the literature already examined in these reviews. We will focus our attention on recent studies, making emphasis on works that illustrate new concepts or provide results that lead to a general and coherent picture for the behavior of bimetallic surfaces. The review begins with a discussion of the structural properties of well-defined bimetallic surfaces. Next, the results ofworks examining bimetallic bonding and the electronic interactions that accompany surface metal-metal bonds are presented. Finally, we show studies concerned with the chemical and catalytic properties of bimetallic surfaces.
2. Atomic structure of bimetallic surfaces
In the study of bimetallic surfaces, one can prepare well-defined systems by cutting a sample of a bulk alloy into a particular orientation, or by vapor depositing a metal onto a single-crystal face of a second metal. By far, the second methodology is more frequently used. It allows for the preparation of a large diversity of systems, including surface intermetallic compounds that are not stable in bulk alloys.
2.1. Well-defined surfaces of bulk alloys
2.1.1. Surface vs bulk geometry The surfaces of fcc alloys with a AB 3 composition (AuCu3, TiPt3, AINi 3 and SnPt3) and a L12
supeflattice have been the subject of a large series of structural studies I" 14-22]. Most of the attention has been focused on the AB3(0 01) surface, for which there are two possible terminations, both

J.A. Rodriguez/Surface Science Reports 24 (1996) 223-287 227
corresponding to bulk truncation [17,20]. In one case, the outermost plane is composed only of B atoms and in the other of a 1:1 mixed ordered "AB" plane (see Fig. 1) 120]. LEED analysis indicates that the TiPt3(001) surface has a surface composition corresponding to the "all Pt" termination [20]. In contrast, the (001) surfaces of AuCu 3 ['17"], AINi 3 E 18] and SnPt 3 E21] exhibit mixed AuCu, AINi and SnPt terminations. For SnPt 3 (0 01) [21 ], LEED results indicate that the Sn atoms in the outermost plane are displaced upward by about 0.2 A [21]. In the case of AINi3(001), LEED reveals a small contraction of 2.8% for the first interlayer spacing (bulk value 1.78 ~), with the AI atoms slightly farther out (0.02_+0.03 ~) than the Ni atoms [18]. For these systems, the structure of the surface is close to that expected for ideal truncation of the bulk crystal structure of the alloy. The same is valid for the AINi3(1 1 1) [18] and SnPt3(1 1 1) [21] surfaces. However, for the TiPt3(l 1 I) system [22], there is segregation of Pt toward the surface, and only Pt atoms are present in the topmost layer.
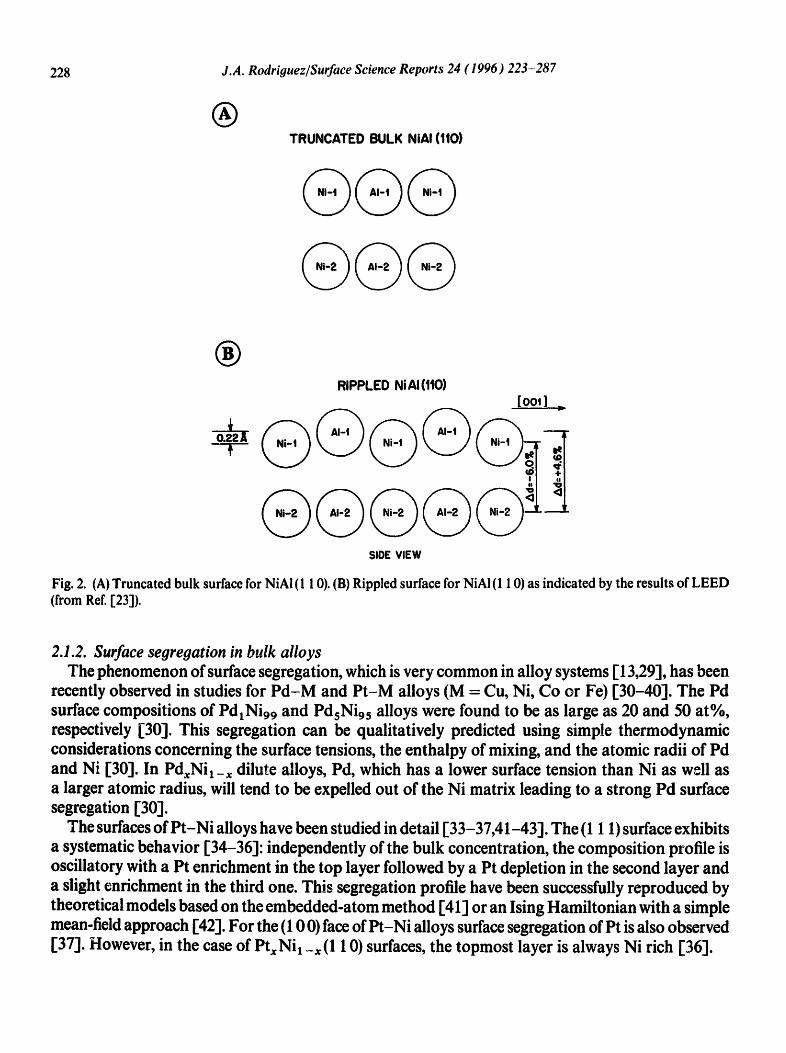
Quantitative LEED analysis have been done for low-index surfaces of NiAl, FeAI and TiAI [23-27]. Ordered NiAl has a CsCI structure, so its hypothetical truncated bulk (1 10) surface consists of composite Ni-AI layers parallel to the surface, with each layer containing half Ni sites and half AI sites which are exactly coplanar (see Fig. 2) [23]. The LEED results reveal that the actual NiAI (1 10) surface has a large rippled relaxation (see Fig. 2), with the AI sites of the outermost composite layer being 0.22 A above the Ni sites [23]. This phenomenon has been attributed to charge transfer effects [28].
The (0 01) planes in the bulk of a AB alloy with the CsCl structure alternate between a whole A and a whole B plane [26]. There are two possible ways to terminate a single crystal of NiAI or FeAI on a (0 01) plane, namely, one way with a Ni or Fe layer, the other way with an AI layer. The NiAI (0 01) [23,24] and FeAI(001) [26] systems are both terminated by an AI layer, and in both cases the surface relaxations involve contraction of the first (da 2) and expansion of the second (d23) interlayer spacing. For NiAI(001) [23,24], there is a contraction of d~2 by 8.5% and an expansion of d23 by 4%. In FeAI(001) [26a], d~2 is contracted by 14.6%, while d23 is expanded by 4%.
A bulk TiAI alloy has the tetragonal structure of AuCu I. In this structure, (001) planes are alternatively 100% Ti ~nd 100% AI, whereas (010) planes are all 50% Ti and 50% Al [27]. A quantitative LEED analysis for TiAI (010) found that the surface is chemically reconstructed: Ti and AI atoms in the first and second layer exchange places, so that the first layer is all AI and the second is all Ti [27].
Fig. 1. Schematic model for the structure of AB3 alloys (AuCu3, TiPt3, NiAI3, SnPt3). Open circles: B atoms, full circles: A atoms (from Ref. 120"1).
228 J.A. Rodrittuez/Surface Science Reports 24 (1996) 223-287
® TRUNCATED BULK NiAI (110)
GGG
®
T
RIPPLED NiAI(110} [ o o ~ ] _
SlOE VIEW
Fig. 2. (A) Truncated bulk surface for NiAI (1 10). (B) Rippled surface for NiAI (1 10) as indicated by the results of LEED (from Ref. [231).
2.1.2. Surface seoreoation in bulk alloys The phenomenon of surface segregation, which is very common in alloy systems [13,291, has been
recently observed in studies for Pd-M and Pt-M alloys (M = Cu, Ni, Co or Fe) [30-40]. The Pd surface compositions of PdlNi99 and PdsNi9s alloys were found to be as large as 20 and 50 at%, respectively [30]. This segregation can be qualitatively predicted using simple thermodynamic considerations concerning the surface tensions, the enthalpy of mixing, and the atomic radii of Pd and Ni [30]. In PdxNi 1 _x dilute alloys, Pd, which has a lower surface tension than Ni as well as a larger atomic radius, will tend to be expelled out of the Ni matrix leading to a strong Pd surface segregation [30].
The surfaces of Pt-Ni alloys have been studied in detail [33-37,41-43]. The (1 1 1) surface exhibits a systematic behavior [34-36]: independently of the bulk concentration, the composition profile is oscillatory with a Pt enrichment in the top layer followed by a Pt depletion in the second layer and a slight enrichment in the third one. This segregation profile have been successfully reproduced by theoretical models based on the embedded-atom method [4 l'l or an Ising Hamiltonian with a simple mean-field approach [42]. For the (10 0) face of Pt-Ni alloys surface segregation of Pt is also observed [37]. However, in the case of PtxNit_~(1 10)surfaces, the topmost layer is always Ni rich [36].

J.A. Rodriguez/Surface Science Reports 24 (1996) 223-287 229
A comparison of the structural properties of PtsoFe2o(1 1 1) [39a], Pt8oCo2o(1 1 1) [40] and Pt 7 s Ni 2 2 (1 1 1) [ 34] surfaces reveals that in all these systems the first layer is essentially pure Pt, and the composition profile damps out to the bulk value within four layers. However, important differences have been observed for the behavior of the second and third layers [40]. Pt8oCo2o(1 1 1) and Pt78Ni22(1 1 1) show a clear oscillation in the concentration of Pt, with the first and third layers being Pt rich, and the second layer Pt poor. On the other hand, PtsoFe2o(1 1 I) exhibits a monotonic decrease in the Pt concentration when going from the first layer (pure Pt) to the fourth layer (bulk composition) of the system. The changes in the composition profile (oscillations for PtNi and PtCo, and monotonous variation for PtFe) strongly correlate with changes in the Pt-Pt interatomic distance in the top layer of the alloys [40]. Oscillatory layer dependent segregation has also been found in MoxRel _x(100) surfaces (x = 0.75, 0.85 and 0.95) [43]. Structural studies for a PtCu3(1 1 1) alloy showed a surface composition of 80%Cu-20%Pt in the first layer, while the second layer was 69%Cu-31%Pt [391>]. In the first layer of this system, the Pt and Cu atoms were coplanar [39b]. In Pt-Rh(100) alloys, there is a Pt-enriched topmost layer and a Pt-depleted second layer [39d].
The phenomenon of surface segregation in alloys has been addressed using several theoretical models [29,30,41-45]. In general, the surface segregation of an alloy component depends on the entlialpy of mixing, the atomic sizes of the metals, and the surface free energies (which are proportional to the heats of sublimation).
2.2. Deposition of metals on single-crystal metal surfaces
2.2.1. Growth modes of metal films A large number of crystalline bimetallic systems have been prepared by vapor-depositing one
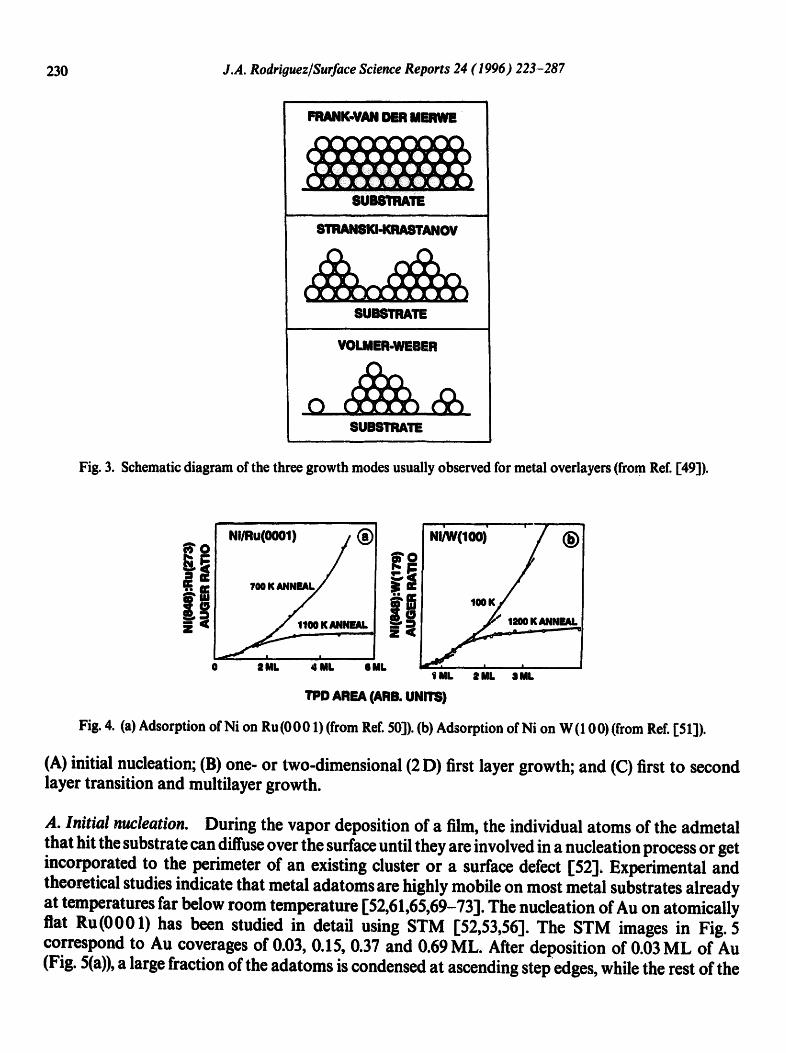
metal onto the surface of a second metal. Experimental evidence indicates that an admetal can exhibit a large diversity of spatial arrangements, varying from randomly adsorbed individual atoms to densely packed overlayers with approximately bulk structure. From a thermochemical viewpoint, three different growth modes can be expected [46,47]: layer-by-layer or Frank-van der Merwe (FV) growth, layer-plus-island or Stranski-Krastanov (SK) growth, and finally island or Volmc, r-Weber (VW) growth. Fig. 3 shows a schematic ofthe topology of these modes. The growth mode adopted by a metal film depends largely upon the relative surface free energies of the pure admetal (~A), the pure substrate ('/s), and the interface (~A-S) [46-48]. The ideal layer-by-layer or FV mode can only be expected when A~'=~'A + ?A-S--~'S <0. The VW or island mode is usually found when A~ = ~A + ~'A-S-- ~'S > 0. The ~A-S term depends strongly on the nature of the bimetallic bond. This term, both in sign and magnitude, can play an important role in determining the growth behavior at surfaces. Fig. 4 shows AES and TPD results for Ni overlayers on Ru (0 0 0 1) [ 50] and W (10 0) [ 51]. On these substrates, Ni grows layer-by-layer at 100 K. Layers in excess of the first monolayer are metastable, forming three-dimensional (3 D) islands at temperatures above 1000 K [50,511. This type of behavior (FV growth at ~ 100 K, SK growth at much higher temperatures) has been observed for many bimetallic systems [12,13,49].
The analysis presented above for the growth modes of a metal film is based on thermodynamic properties of the admetal-substrate system, and is correct only if thermodynamic equilibrium is established [52]. In many cases, however, film growth proceeds under conditions far from equilib- rium 152]. Many novel phenomena related to the kinetics of growth have been discovered in recent studies [52-68!. In general, the growth of a metal film can be divided in three different stages [52]:
230 J.A. Rodriouez/Surface Science Reports 24 (1996) 223-287
i | , l
FRANK.VAN Dim MERWE
s u m n ~
STRANSKI-KRASTANOV
SUBb'TRATE
VOLMER.WESER
8UBSTRATE
Fig. 3. Schematic diagram of the three growth modes usually observed for metal overlayers (from Ref. [49]).
t ML 2 ML $ ML
TPD AREA (ARB. UNITS)
Fig. 4. (a) Adsorption of Ni on Ru(0001) (from Ref. 50]). (b) Adsorption of Ni on W (100) (from Ref. [51]).
(A) initial nucleation; (B) one- or two-dimensional (2 D) first layer growth; and (C) first to second layer transition and multilayer growth.
A. Initial nucleation. During the vapor deposition of a film, the individual atoms of the admetal that hit the substrate can diffuse over the surface until they are involved in a nucleation process or get inco~orated to the perimeter of an existing cluster or a surface defect [52], Experimental and theoretical studies indicate that metal adatoms are highly mobile on most metal substrates already at temperatures far below room temperature [52,61,65,69-73]. The nucleation of Au on atomically fiat Ru(0001) has been studied in detail using STM [52,53,56]. The STM images in Fig. 5 correspond to Au coverages of 0.03, 0.15, 0.37 and 0.69 ML. After deposition of 0.03 ML of Au (Fig. 5(a)), a large fraction of the adatoms is condensed at ascending stepedges, while the rest of the

J.A. Rodriouez/Surface Science Reports 24 (1996) 223-287 23 !
(a)
(c)
(
. . . . . . . .
Fig. 5. STM images of four submonolayer coverages of Au on Ru (0001) deposited at 300 K with a flux of 1.8 ML/min. (a) 0Au-0.03ML, 0.70pm×0.60pm. (b) 0Au---0.15ML, 1.31pm× 1.20pm. (c) 0A~-0.37ML, 1.13pm×0.96pm. (d) 0^~ = 0.69 ML, 1.32 pm × 1.13 pm (from Ref. [53]).
Au is accommodated in a few islands. The islands are exclusively formed on the large, central terrace. With increasing Au coverage (Figs. 5(b)-5(d)) more islands are formed and at the same time they increase in size. In the central areas of large terraces these islands are evenly distributed, indicating a homogeneous nucleation mechanism 152"1.
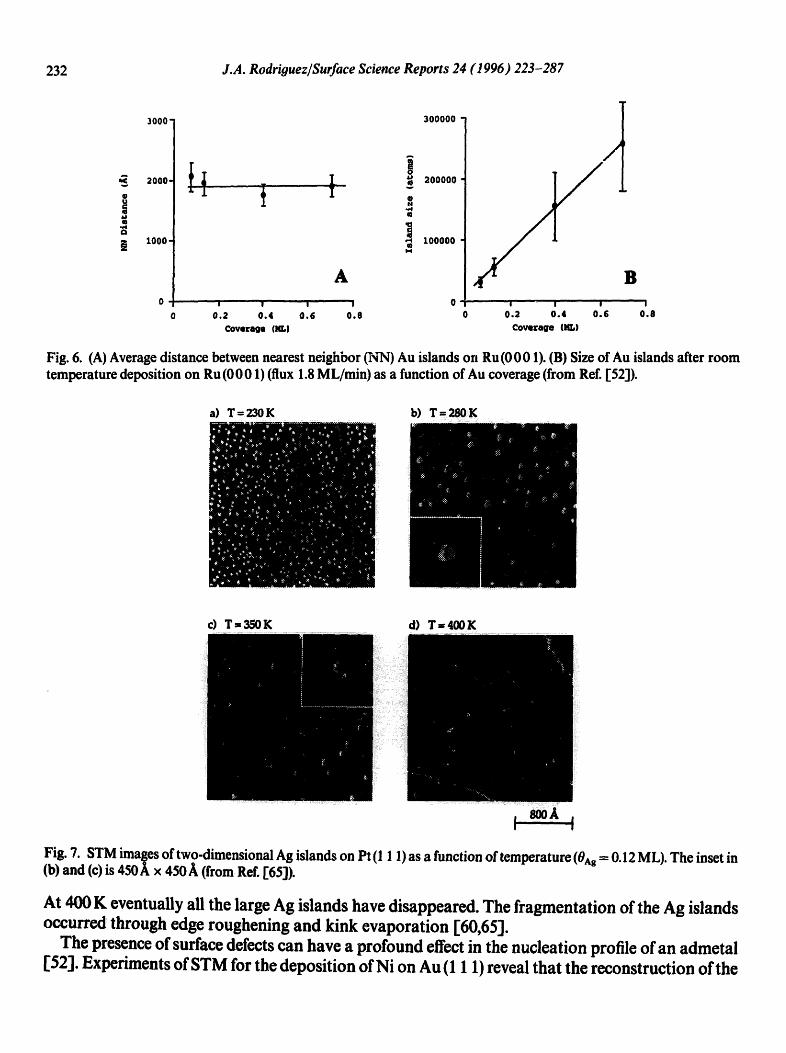
The results of a quantitative evaluation of the island sizes and island densities are shown in Fig. 6 [52]. One can see a continuous increase in the size of the islands with Au coverage. The island density (and the average distance between islands) reaches a constant value around 0.1-0.15 ML. Additional Au deposits are almost exclusively accommodated by island growth rather than by nucleation of additional islands. These observations can be rationalized in terms of a simple picture [ 52]. Au atoms impinging on the R u (0 0 01) substrate diffuse freely over its surface, forming a 2D gas of mobile adatoms whose density increases with deposition and decreases with the formation of small Au clusters. Once a cluster is formed, it can dissolve back to individual adatoms. Since the dissociation rate of adatoms from a cluster depends on the cluster size, there is a minimum size for clusters to survive ("critical cluster size"). The "critical cluster size" is a complex function of the deposition rate, the substrate temperature and cluster interactions energies [52].
Fig. 7 illustrates the effects of temperature on the stability of Ag clusters deposited on Pt (1 1 1) [65]. The islands in Fig. 7(a) were grown by dimer nucleation at 50K (0Ag =0.12 ML) and subsequent annealing to 230 K, giving a narrow island size distribution with an average cluster size of about 230 Ag atoms ['61,65]. Annealing to 280 K increases the average island size to about 1000 atoms [65]. Around 350 K, one can see dramatic changes in the morphology of the system, and the surviving Ag islands coexist with a 2D lattice-gas of Ag adatoms (not see'n in the STM images) [65].
232 J.A. Rodriguez/Surface Science Reports 24 (1996) 223-287
3000"
2000-
1000 -
~-I { I
A ' I | I l
0 0 . 2 0 .4 0 . 6 0 .8
Coverage (I~[,!
300000
200000
100000
0 ' ' I 0 0 .2 0 . 4 0 . 6 0 .8
Coverage ( ! ~ )
Fig. 6. (A) Average distance between nearest neighbor (NN) Au islands on Ru (0 0 01). (B) Size of Au islands after room temperature deposition on Ru (0001) (flux 1.8 ML/min) as a function of Au coverage (from Ref. [52]).
a) T= 230K b) T=280K
c) T=3~0K .... . . . . d) T=400K
Fig. 7. STM images of two-dimensional Ag islands on Pt (1 1 1) as a function of temperature (0^g -- 0.12 ML). The inset in (b) and (c) is 450 ~ x 450 A (from Ref. [65]).
At 400 K eventually all the large Ag islands have disappeared. The fragmentation of the Ag islands occurred through edge roughening and kink evaporation [60,65].
The presence of surface defects can have a profound effect in the nucleation profile of an admetal [52]. Experiments of STM for the deposition of Ni on Au (1 1 1) reveal that the reconstruction of the

J.A. Rodriouez/Surface Science Reports 24 (I 996) 223-287 233
Au substrate determines the local nucleation probability of Ni [54]. The Ni islands grow following the lattice dislocations induced by the Au(1 1 1) "herringbone" reconstruction [54,76]. A similar phep~menon is observed for the deposition of Fe, Co and Pd (see Fig. 8) [74]. A comparison of the growth of different metals on Au (1 1 1) [74a] shows that for Au and Ag on Au (1 1 1) the nucleation occurs predominantly at the gold step edges [75,76]. On the other hand, admetals that have a larger misfit (Fe [74a], Co [74a] and Ni [54,56])nucleate predominantly at the kinks ofthe Au reconstruc- tion pattern forming polygonal islands [74a].
B. One- or two-dimensional first layer growth. In the FV and SK growth modes, an admetal can grow at submonolayer coverages forming 1D chains or 2D islands. Elementary thermodynamics asserts that 1D crystals of infinite length cannot exist in an isotropic environment [77]. However, a single crystal substrate can have an anisotropic atomic distribution and, therefore, stabilize a 1D chain against decay into a 2D island or atoms in a 2D gas [77,78]. One-dimensional chains of admetals have been observed on a few metal substrates at relatively low temperatures [77-83]. The results of LEED and FIM indicate that Ir, Rh, Pt, Pd and Ni form one-dimensional chains on W (1 10) [77,78,81-83]. In the case of Rh, the individual chains tend to order into a chain lattice forming a (3 x 1) superstructure [78]. A 1D ~ 2D cluster transition occurs at ~ 490 K [78]. For the deposition of Cu on Pd (1 10) at 300 K, the results of STM experiments show the formation of long monatomic Cu chains along the (1 i0) direction that exhibit lengths up to several hundred ~ngstr/Sm [73c]. The formation of the chains is possible due to the difference in the Cu diffusion barriers along the longitudinal (li0) and transverse (001) directions [73b,73c]. The monatomic Cu chains are metastable nanostructures. Upon annealing to 350 K, only 2D copper islands are observed on the surface [73c].
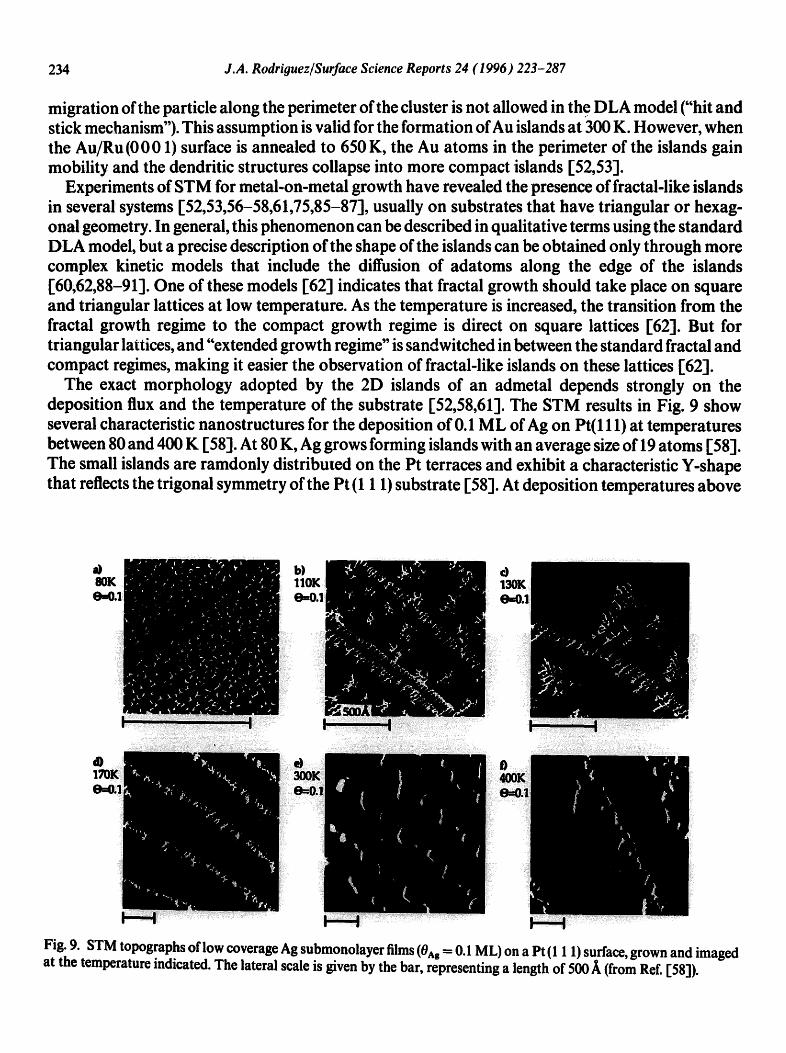
The STM images in Fig. 5 indicate that Au grows on Ru(0001) at 300 K forming one layer 2D islands of dendritic shape [52,53]. The dendritic islands exhibit a fractal character, and a dimen- sional analysis yields a fractal dimension of 1.72 + 0.07 [53]. The dendritic islands resemble island structures obtained in simulations based on the "diffusion-limited aggregation (DLA) model" [84]. In this model particles are added, one at a time, to a growing cluster of identical particles via random walk paths that start at "infinity". When a particle foflowing a random walk or Brownian path hits the growing cluster, it is incorporated at the position in which it first contacts the cluster. Later
Fig. 8. (a) Reconstructed Au (1 1 1)surface (730 × 730 ~2) with domains forming an ordered zigzag pattern. (b)0.3 ML Co coverage on Au(1 1 1) (1600 x 1600,~z); 2 ML-high polygonal Co islands nucleate at the kinks of the Au(1 1 1) zigzag reconstruction (from Ref. [74a]).
234 J.A. Rodriguez/Surface Science Reports 24 (1996) 223-287
migration of the particle along the perimeter of the cluster is not allowed in th e DLA model ("hit and stick mechanism"). This assumption is valid for the formation of Au islands at 300 K. However, when the Au/Ru(0001) surface is annealed to 650 K, the Au atoms in the perimeter of the islands gain mobility and the dendritic structures collapse into more compact islands [52,53].
Experiments of STM for metal-on-metal growth have revealed the presence of fractal-like islands in several systems [52,53,56-58,61,75,85-87], usually on substrates that have triangular or hexag- onal geometry. In general, this phenomenon can be described in qualitative terms using the standard DLA model, but a precise description of the shape of the islands can be obtained only through more complex kinetic models that include the diffusion of adatoms along the edge of the islands [60,62,88-91]. One of these models [62] indicates that fractal growth should take place on square and triangular lattices at low temperature. As the temperature is increased, the transition from the fractal growth regime to the compact growth regime is direct on square lattices [62]. But for triangular lattices, and "extended growth regime" is sandwitched in between the standard fractal and compact regimes, making it easier the observation of fractal-like islands on these lattices [62].
The exact morphology adopted by the 2D islands of an admetal depends strongly on the deposition flux and the temperature of the substrate [52,58,61]. The STM results in Fig. 9 show several characteristic nanostructures for the deposition of 0.1 ML of Ag on Pt(111) at temperatures between 80 and 400 K [58]. At 80 K, Ag grows forming islands with an average size of 19 atoms [58]. The small islands are ramdonly distributed on the Pt terraces and exhibit a characteristic Y-shape that reflects the trigonal symmetry of the Pt (1 1 1) substrate [58]. At deposition temperatures above
Fig. 9. STM topographs of low coverage Ag submonolayer films (0Ag = 0.1 ML) on a Pt (1 1 1) surface, grown and imaged at the temperature indicated. The lateral scale is given by the bar, representing a length of 500 A (from Ref. [58]).

J.A. Rodriguez/Surface Science Reports 24 (1996) 223-287 235
110 K the pre-existing steps of the Pt (1 1 1) surface become increasingly important. The Pt step edges act as sinks for Ag adatoms moving on the Pt terraces, and Ag islands nucleate preferentially at the Pt step edges from where they propagate onto the lower terraces. This is evident in Figs. 9(b)-9(d), covering the temperature range from 110 to 170 K. The Ag islands nucleating and growing on terraces at deposition temperatures between 100 and 150 K have a highly dendritic form [58]. Above ~ 160 K very large and compact Ag islands grow away from the substrate steps [58]. Finally, for deposition temperatures higher than 300 K, the STM images reveal a two-phase equilibrium ofa 2D solid (large condensed Ag islands) and a 2D gas (small Ag clusters plus Ag adatoms) [58].
For the deposition of Co on Ru (0 0 01) at 300 K, the admetal forms compact triangular islands (see Fig. 10(a)) [52,56]. The shape of the adlayer seam changes between subsequent terraces. This change originates in the ABAB stacking of Ru (000 1) and the preferential formation of steps with fcc (100)- and (111 )-like microfacets [52,56]. Kinetic limitations in the migration of the Co adatoms lead to the formation of the triangular islands. When the system is annealed to 600 K, one sees the formation of thermodynamically favored, highly compact hexagonal Co islands (see Fig. 10(b)) [52,56].
C. First to second layer transition and multilayer growth. In the FV and SK growth modes, the transition from first to second layer marks the beginning of 3D growth. The term "pseudomorphic growth" refers to a situation where the first metal overlayer adopts a lattice constant which differs from its bulk crystal structure but which matches coherently the lattice of the underlying substrate. This phenomenon has been observed in a very large number of bimetallic systems [12,13,491. In general, only the first layer grows pseudomorphically, while subsequent layers tend to present lattice constants that are closer to the crystal structure of the admetal. The strain induced by the substrate on the adatoms in a pseudomorphic monolayer affects the growth mode of the second layer [52]. Hero, we will discuss only two systems: one that shows a compression in the first layer, Ag/Pt (1 1 1) [63], and one in which there is an expansion, Ni/Ru(0001) [66].
Fig. 10. STM images of a 0.17ML Co film deposited on Ru(0001). (a) After deposition (flux 0.11 ML/min) at 300K (750nm x 930rim). (b) After subsequent flash annealing to 600 K (560nm x 570nm) (from Ref. [52]).

236 J.A. Rodriouez/Surface Science Reports 24 (1996) 223-287
For Ag/Pt (1 1 1), the first monolayer grows in a (1 x 1) fashion at equilibrium, and therefore is is.tropically compressed by 8.9% in atomic density with respect to the Ag (1 1 1) bulk plane [63]. The strain in the compressed Ag monolayer on Pt (1 1 1) is relieved in the bilayer by the formation of a trigonal network of domain wafts that separate fcc and hcp regions 163]. The STM image in Fig. 11 shows two subsequent atomic terraces, both covered with two Ag layers [63]. A small area on the right-hand side still displays the commensurate first layer and the position of a former substrate step [63]. On the second layer two almost parallel sets of domain walls rotated 120 ° are clearly visible. According to the atomic model in Fig. 1 l(b), the atomic density of the second layer is reduced by 7% with respect to that of the first layer [63]. The ratio of fcc to hcp sites is 2.8 [63].
For Ni/Ru(0001), the atoms in the pseudomorphic monolayer show a Ni-Ni separation ~ 8 %
larger than in Ni (1 1 1) [66]. After completion of the first monolayer, incorporation of additional Ni adatoms leads to a reconstruction of that layer to form a denser phase with a periodic arrangement of triangular domain boundaries between fcc and hcp areas [66]. This structure is shown in Fig. 12(a). The white lines represent Ni atoms located on slightly higher bridge sites, while in the other areas the Ni atoms reside on lower fcc and hcp-type threefold hollow sites [66]. Similar structures have been observed for Cu films on Ru(0001) [92]. For bilayer and thicker films, hexagonal moir6 structures are formed (Figs. 12(b) and (c) [66]). The atomic spacings of 2.5 ,~, the distance between the long-range corrugation maxima of 32 A, and the fact that these maxima are oriented along atomic rows, all agree with a model where a nondistorted hexagonal Ni lattice
• ~ • e . o • e . o . o . o , o , o , o . e . q , . . . . . ~ o . e , o , o . o . o • e - o -
• O • • • • • • • • • ~ O O O e O e O e O e O • • • • • • • O~.a%,eee.O • • • • • • • • • • • • o . o O • • • • • • • • p g e o e o O o I o e o e 0 O • • • • •
• oO~O0~ ~ • • • • • • • • . o_~ . • . 0000 OoO O•O • • • • • • ,, tOeO.O• . •O , ,~ . . , , , . • • • O B B O O O 0 . . . . ~ 0 ' ~0 "~ ~ V t m t eO
• O 'O " " • " " " PO 'O 'O 'O 'O•O 'O•O 'O " " • • ° ' OOOO . O .O • • t I • O 'O • • ~ " O .O 'O 'O•O 'O•O 'O 'O 'O • • • . • o o o • o o o o o : ; ; , ; , ~ - . -..- ;/.........-o-...:.'.'N~. • ; -.,,
• oOo .o .o - • - o - - - a , o •o .o •o .o .o .o •o .o .qp .o . o - . - . - . - • v i i g i g
" o ' o ' o ' o ' e - ; . - ; e ' o ' o ' o ' o ' o ' e ' o ' e ' o ' o ' e ' o ' o -~-'o" . '~:'.~';.,.,.,.,.,.,.,.,.,.".,. ',. '~:~.,. • o . o . o •o .o • . o , o -o , , o •o .o •o .o .o .o .o •o .o .o • . o • • ........... ~............................-...).,~.... . . . . . . . . . , . ; , . ; , . ; , / . •
' . ' .Co " • • " o ' o ' oOo°oeo ' o •oeo ' o ' o " " " • go i v
; " , ' o ' o ' o ' o ' , ' o " ~ ; • . • ~oo_ o o o o o
• . oeoOoeo , , oeo • • • • • , • • • . o . o . o •oeo •o . o . o / . . . . . . %o.e.o.o.o.o.o.o.o~' . . . . .
Fig. 11. (a) STM image of the trigonai network constituting the equilibrium structure for the second Ag layer on Pt (1 1 1), 520,~ x 520 ~. Inset: Model for the trigonal network, where domain walls (dark lines) separate fcc and hcp (hatched) areas• (b) Atomic model of the trigonal incommensurate phase (from Ref. 163"1).
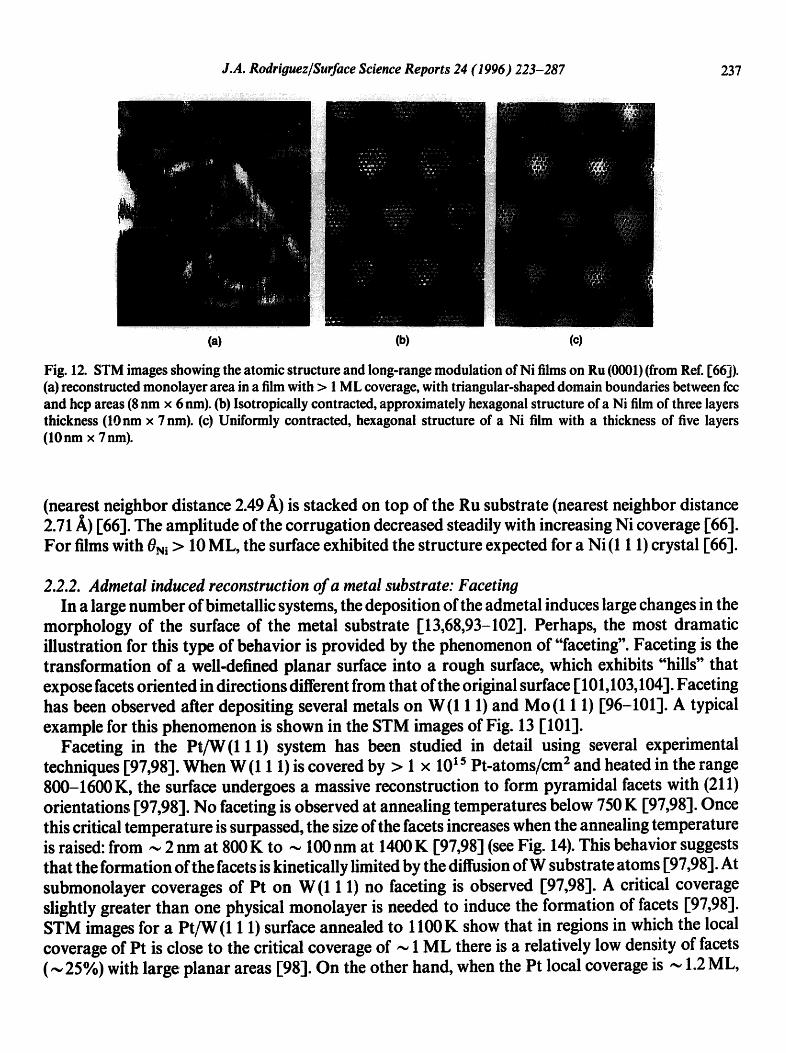
J.A. Rodriguez/Surface Science Reports 24 (1996) 2'23-287 237
(a) (b) (c)
Fig. 12. STM images showing the atomic structure and long-range modulation of Ni films on Ru (0001) (from Ref. [66]). (a) reconstructed monolayer area in a film with > 1 ML coverage, with triangular-shaped domain boundaries between fcc and hcp areas (8 nm x 6 nm). (b) Isotropicaily contracted, approximately hexagonal structure of a Ni film of three layers thickness (10nm x 7nm). (c) Uniformly contracted, hexagonal structure of a Ni film with a thickness of five layers (10nm x 7 nm).
(nearest neighbor distance 2.49 A,) is stacked on top of the Ru substrate (nearest neighbor distance 2.71 A) [66]. The amplitude of the corrugation decreased steadily with increasing Ni coverage [66]. For films with 0Ni > 10 ML, the surface exhibited the structure expected for a Ni (1 1 1) crystal [66].
2.2.2. Admetal induced reconstruction of a metal substrate: Faceting In a large number of bimetallic systems, the deposition of the admetal induces large changes in the
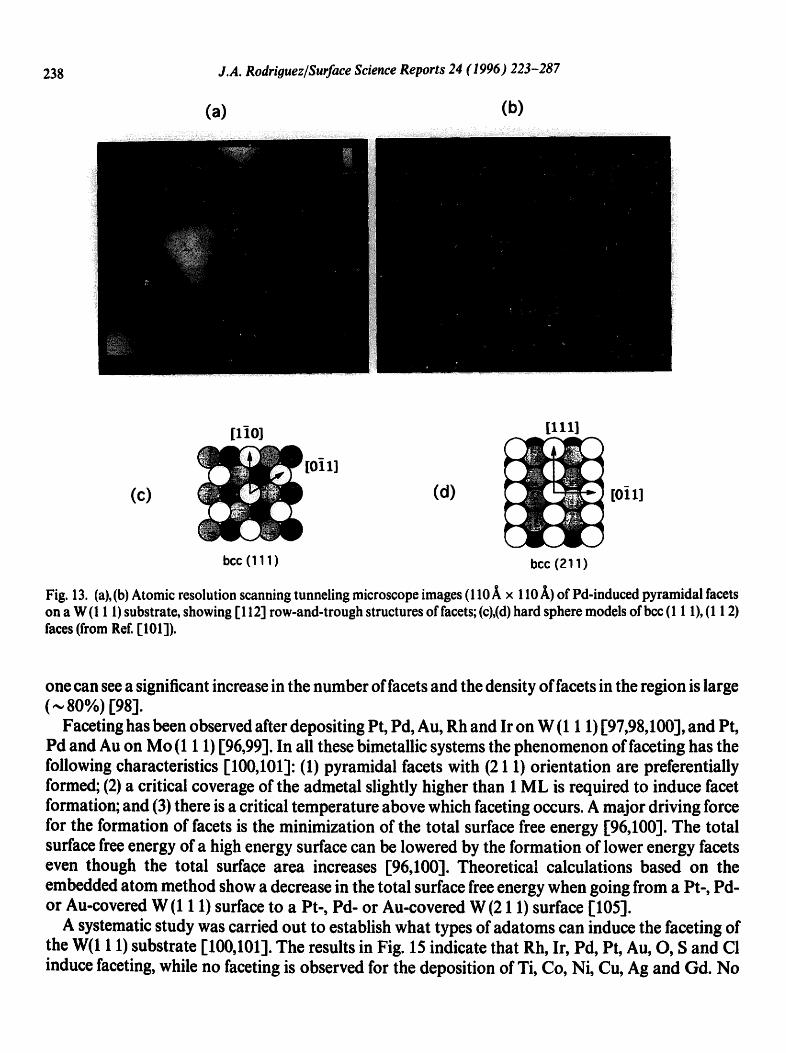
morphology of the surface of the metal substrate [13,68,93-102]. Perhaps, the most dramatic illustration for this type of behavior is provided by the phenomenon of"faceting". Faceting is the transformation of a well-defined planar surface into a rough surface, which exhibits "hills" that expose facets oriented in directions different from that of the original surface [ 101,103,104]. Faceting has been observed after depositing several metals on W(1 1 1) and Mo(1 1 1) [96-101]. A typical example for this phenomenon is shown in the STM images of Fig. 13 ] 101l.
Faceting in the Pt/W(1 1 1) system has been studied in detail using several experimental techniques [97,98]. When W (1 1 1) is covered by > 1 x 10 I5 Pt-atoms/cm 2 and heated in the range 800-1600 K, the surface undergoes a massive reconstruction to form pyramidal facets with (211) orientations [97,98]. No faceting is observed at annealing temperatures below 750 K [97,98]. Once this critical temperature is surpassed, the size of the facets increases when the annealing temperature is raised: from ~ 2 nm at 800 K to ~ 100nm at 1400 K [97,98] (see Fig. 14). This behavior suggests that the formation of the facets is kinetically limited by the diffusion ofW substrate atoms [97,98]. At submonolayer coverages of Pt on W (1 1 1) no faceting is observed [97,98]. A critical coverage slightly greater than one physical monolayer is needed to induce the formation of facets [97,98]. STM images for a Pt/W (1 1 1) surface annealed to 1100 K show that in regions in which the local coverage of Pt is close to the critical coverage of ~ 1 ML there is a relatively low density of facets (,-, 25%) with large planar areas [98]. On the other hand, when the Pt local coverage is ~ 1.2 ML,
238 J.A. Rodriouez/Surface Science Reports 24 (I 996) 223-287
(a) (b)
(c)
[11o]
(d)
[111]
~ [Oil1
bcc (111 ) bcc (211 )
Fig. 13. (a),(b) Atomic resolution scanning tunneling microscope images (110 • x 110 ~) of Pal-induced pyramidal facets on a W (1 1 l) substrate, showing [ 112] row-and-trough structures of facets; (c),(d) hard sphere models of bcc (1 1 1), (1 1 2) faces (from Ref. [101]).
one can see a significant increase in the number of facets and the density of facets in the region is large (~ 80%) [98].
Faceting has been observed after depositing Pt, Pd, Au, Rh and Ir on W (1 1 1) [97,98,100], and Pt, Pd and Au on Mo (1 1 1) [96,99]. In all these bimetallic systems the phenomenon of faceting has the following characteristics [100,101]: (1) pyramidal facets with (21 1) orientation are preferentially formed; (2) a critical coverage of the admetal slightly higher than 1 ML is required to induce facet formation; and (3) there is a critical temperature above which faceting occurs. A major driving force for the formation of facets is the minimization of the total surface free energy [96,100]. The total surface free energy of a high energy surface can be lowered by the formation of lower energy facets even though the total surface area increases [96,100]. Theoretical calculations based on the embedded atom method show a decrease in the total surface free energy when going from a Pt-, Pd- or Au-covered W (1 1 1) surface to a Pt-, Pd- or Au-covered W (21 1) surface [105].
A systematic study was carried out to establish what types of adatoms can induce the faceting of the W(I 1 1) substrate [100,101]. The results in Fig. 15 indicate that Rh, Ir, Pd, Pt, Au, O, S and CI induce faceting, while no faceting is observed for the deposition of Ti, Co, Ni, Cu, Ag and Gal. No

J.A. Rodriguez/Surface Science Reports 24 (1996) 223-287 239
' ~!?!i:!!!ill !!i~i iiii~!ii!i!!~!i~!i! i iii~!~!!~i~! i!i ~ !i!ii i !!~ i~ ~ ~III~SI!!I~ ~i! !i! !i i~i!! ̧ ~ ? ? ~ i~ ̧ i ~ ? i! ̧̧ ~ ~ .......
Fig. 14. STM images for a Pt/W (1 1 1) surface annealed to 880 (top), 1200(middle) and 1400 K (bottom). The images are all top views with tbe same lateral scale, 350rim x 350nm. The Pt coverage in each case is between 1.5 and 2 ML. The gray scale is related to the vertical height, and is different for each image (from Ref. [98]).
direct correlation was found between the ability of an admetal to induce faceting and its atomic size or surface-free energy [ 1130]. A very good correlation was found between faceting and the Pauling electronegativity of the adsorbed species [100]: Adatoms with a Pauling electronegativity larger than 2.0 cause faceting while those with a value below 2.0 do not (see Fig. 15). This suggests that electronic interactions between the adsorbate and W substrate play a role in the formation of the facets [100]. A more detailed discussion of this issue is presented in Ref. [ 101].
2.2.3. Alloy formation through metal-on-metal deposition In Sections 2.2.1 and 2.2.2, we have discussed growth modes that are usually observed when the
admetal and the substrate have a low miscibility, and no interdiffusion of the metals occurs. More complicated situations can arise when the components of the bimetallic system present a large

240 J.A. Rodriguez/Surface Science Reports 24 (1996) 223-287
Li. Be
Elect ronogst ivit y Element
~ e s
Na Ng
K Ca S c ~ /
2.b Sz"
n c t w / ~ F se
Ca Ba
m
i
V C r t~te~ F e Z n Ga Ge A e [ $ e D r K r
m R S ' I Y Zz" Nb No Te Ru: Cd I n 8 n 8 b T o I" Xe
a e Ta W 7 n e om MigNilmlll x g Tx P b a, I ,o
Fig. 15. Periodic Table listing the Pauling electronegativity and whether or not faceting occurs for the elements surveyed (from Ref. 1"1001).
miscibility. In those cases substrate atoms can migrate into the film, forming alloys of variable composition and crystal structure as a function of annealing temperature. An interesting phenom- enon has been observed for Rh/Ag(100) [106] and Fe/Au (100) [107]. The components of these systems have very limited miscibilities. However, their equilibrium configurations do not match any of the growth modes described above. In these systems, the equilibrium structure is that of a Ag-Rh-Ag or Au-Fe-Au sandwich. The formation of these "sandwiches" is thermodynamically driven by the differences in surface free energies between Ag and Rh or Fe and Au [106].
In 1968, the formation of a well-defined surface alloy was observed upon the deposition of Au on Cu (100)[ 108]. Recently, a large number ofmetal-on-metal systems have been explored which show the formation of ordered surface alloys [109-127]. The deposition of 0.5 ML of Au [109], Pd [111] or Mn [121] on Cu(100) leads to the formation surface alloys that exhibit a c(2 × 2) structure. In these systems, the first layer consists of an ordered 50-50% mixture of the admetal and Cu, with subsequent layers containing only Cu atoms. The structure of the Cu (10 0)-c(2 × 2)Pal surface is almost planar, with the Pd atoms located 0.02 +_ 0.03 ,~ outwards from the Cu atoms [111]. In contrast, the Cu (10 0)-c(2 × 2)Mn surface shows a pronounced corrugation in which the Mn atoms are displaced outwards by 0.30 _+ 0.02 ,~ [121]. Ordered 2D alloys with a c(2 × 2) structure have also been observed after depositing 0.5 ML of Mn [121] or Sn [126] on Ni(100). The surface of these alloys is corrugated with the atoms of the admetal displaced outwards by 0.25 _ + 0.02 A in the case of Mn [ 121], and 0.44 _+ 0.05 ~ in the case of Sn [ 126]. A systematic study of the interaction of Sn with the (1 1 1) surfaces of Cu, Ni and Pt shows the formation of ordered 2D alloys in these bimetallic systems [116]. At a Sn coverage of 0.33 ML a p(v/3 × w/3)-R30 ° superstructure is observed on all the substrates [ 116]. In this structure, the Sn atoms are not coplanar with the atoms in the first layer of the substrate, showing an outward buckling [116]. The magnitude of the buckling was linearly correlated to the lattice constant of the substrates, varying from 0.22,~, in Sn/Pt to 0.39 ~ in Sn/Cu, and 0.46.~ in Sn/~i [116].

J.A. Rodriouez/Surface Science Reports 24 (1996) 223-287 241
Experiments of STM indicate that Au deposited on a Ni (1 10) surface alloys in the first layer despite the fact that Au and Ni show absolutely no tendency for alloying in the bulk [128]. This phenomenon has been explained by the results of total energy calculations within the effective- medium theory [ 128]. A Au atom dissolved among the atoms at the Ni(1 10) surface provides extra electron density to the "electron-deficient" Ni surface atoms stabilizing the system. On the other hand, when a Au atom is substituted into bulk Ni, the 12 Ni atoms surrounding the Au atom cannot accommodate the extra electron density and no bulk alloy is formed [128]. Monolayer-confined mixing has also been observed for the Ag/Pt (1 1 1) system [ 129]. STM images reveal that the growth of Ag on Pt (1 1 1) at temperatures above 620 K results in the formation of a 2D alloy consisting of Ag clusters dissolved in the Pt layer (0Ag<0.SML) and Pt clusters in the Ag matrix layer (0.5 ML < 0As < 1 ML) [129]. The embedded clusters have a narrow size distribution centered around 10,~ [129]. The second silver layer does not dissolve into the interface, but grows layerwise on top [129].
3. Electronic properties of bimetallic surfaces
In general, metals are elements characterized by the presence of a large number of valence orbitals that can donate or accept electrons. The formation of a surface metal-metal bond can produce large perturbations in the electronic properties of a metal. In many cases, the phenomena responsible for these perturbations have not been identified, and the problem of understanding the electronic properties of bimetallic surfaces is currently attracting a great deal of theoretical and experimental work [130-132]. Extensive studies have been carried out investigating the behavior of bimetallic systems formed by depositing elements from Groups 8-12 of the Periodic Table onto single-crystal faces of transition or s,p metals. By analyzing the results of experimental techniques (photoemission, L-edge X-ray absorption fine structure, work function measurements, etc) and theoretical calcula- tions (ab initio Hartree-Fock methods, first-principles local-density functional theory), one can obtain a general idea of the nature of the bimetallic bond in these systems.
3.1. Group 8 and 9 admetals
Bimetallic systems that combine films of ferromagnetic metals on top of noble metals have received a lot of attention due to their magneto-optical and magneto-resistive properties [ 133-143]. Two-dimensional magnetic systems are predicted to have enhanced magnetic moments as a result of a reduction in dimensionality and coordination number [144,145]. However, this may not be observed due to electronic interactions between the admetal and metal substrate [143,146], and difficulties in avoiding interlayer mixing between the admetal and metal substrate [147,148].
Fe/Ag(100) is one of the most extensively studied bimetallic systems [134,137-140,142, 144,145,147]. Theoretical calculations for a monolayer of Fe on Ag(100) predict an enhancement of the magnetic moment of the admetal from 2.2/~e (bulk bcc Fe) to 3.0/~B [144]- In general, one would expect an increase of the magnetic moment when diluting the Fe atoms, since this element has a large moment of 4/~B in the atomic limit due to Hund's rule [134]. The verification of this hypothesis at an experimental level is difficult since in many situations Fe grows on Ag (100) neither layer-by-layer nor in a Stranski-Krastanov mode [147,148], with interdiffusion at the Fe/Ag(100) interface.

242 J.A. Rodriouez/Surface Science Reports 24 (1996) 223-287
The empty electronic states of Fe and Co adlayers deposited on Cu (10 0), Ag (10 0) and Au (10 0) surfaces have been examined using inverse photoemission [134,137,142,149-151]. The empty 3d bands of the supported Fe and Co monolayers exhibit magnetic-exchange splittings that are very different from those of the bulk admetals. Quantum-weU states have been observed for Fe and Co films embedded in Au surfaces [135,150].
3.2. Group-lO admetals
Bimetallic catalysts that contain Group- 10 metals are ideal for many applications: isomerization of hydrocarbons, olefin hydrogenation, CO oxidation, alcohol synthesis, acety!ene trimerization, etc. This fact has motivated many studies examining the electronic properties of the Group- 10 metals in bimetallic surfaces. In principle, the behavior of Pt, Pd and Ni in a bimetallic surface can be different due to several factors. In the atomic state, the valence electronic configurations of these elements are not the same: Ni, dSs2; Pd, dl°s°; and Pt, d9s 1 [184]. For each element, the role of the d and s,p orbitals in bimetallic bonding can differ. There are also significant variations in the electron affinities of Ni (156 kJ/mol [184]), Pd (98 kJ/mol [184]) and Pt (247 kJ/mol [184]). Among the transition metals Pt has the largest electron affinity [184], and it should be a much better electron acceptor than Pd or Ni when present in a bimetallic surface.
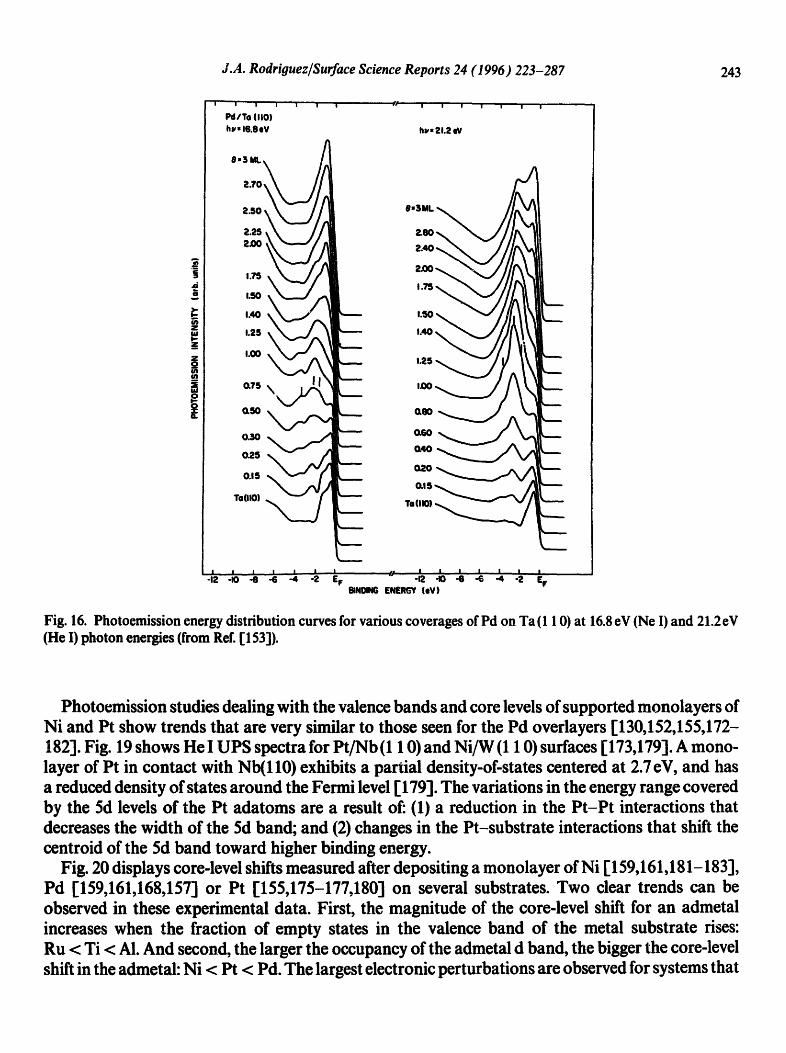
A Pd atom deposited on the surtace of a second metal can exhibit large changes in its electronic properties [132,152-168]. The valence photoemission spectra shown in Fig. 16 illustrate this phenomenon [153]. In early studies examining the interaction of Pd with Nb(1 10) [154], it was found that the supported Pd monolayer (ML) had a relatively narrow 4d band which exhibited a low density of states (DOS) around the Fermi level (EF). In contrast, Pd multilayers and bulk Pd show emission spectra characterized by a large DOS at EF. More recent photoemission studies for a Pd layer in contact with Ta (1 10) [153] (Fig. 16), W (1 10) [158], W (100) [158], AI(1 1 1) r167a] or Zn (0001) [165,166] also show a narrow Pd(4d) band with a centroid shifted toward higher binding energy.
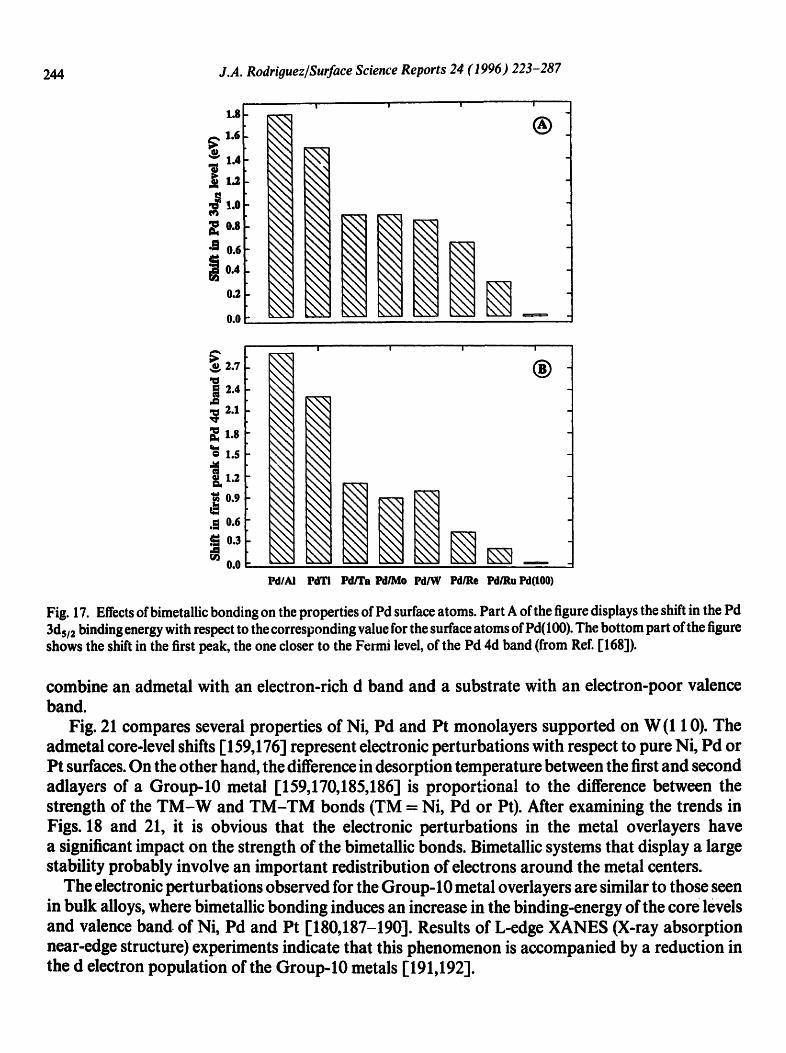
In general, Pd adatoms exhibit core and valence levels shifted toward higher binding energy with respect to those of pure Pd [132,168]. The magnitude of the binding-energy shift depends on the position of the metal substrate in the Periodic Table. Fig. 17 displays the electronic pertm bations observed for Pd in surface alloys (PdTi [157] and PdAl [167b,168]) and Pd monolayers supported on several metals (Ta(1 10) [153,161], Mo(1 10) [160,171], W(1 10) [158,159], Re(0001) [161], Ru (0 0 01) [ 161 ] and AI ( 1 1 1) r 167a]). The experimental results are ordered according to the group in the Periodic Table of the metal bonded to Pd. The electronic perturbations found after bonding Pd to a s,p metal like AI are as large as those found for Pd bonded to early-transiti0n metals, and much bigger than those found when Pd is bonded to late-transition metals. In general, the magnitude of the shift in the core and valence levels of Pd increases when the fraction of empty states in the valence band of the metal substrate rises [132,168].
For supported monolayers of Pd, a correlation has been found between the electronic perturba- tions in the Pd 3ds/z core level and the strength of the bimetallic bond [130,161]. Desorption temperatures of Pd monolayers from several metal substrates (Ta (1 10) [ 169a], W (1 10) [ 170], Mo (110) [161], Re(0001) [161], Ru(0001) [161] and Rh(1 1 1) [171]) are presented in Fig. 18(A). In going from a Rh to a Ta substrate, there is an increase of ~ 150 K in the desorption temperature, which indicates an enhancement of ~10 kcal/mol in the strength of the Pd-substrate bond. The larger the electronic perturbations in the Pd atoms, the stronger the bimetallic bond.
J.A. Rodriguez/Surface Science Reports 24 (1996) 223-287 243
I I I I I I I J'P I ' l I l I I I
Pd/To (110) ha,, 16.8 iV ha,, 21.2 IN
030
025
0.15
1"o(110)
- , , , - , , r : ' ' ' ' ' . , -12 -tO -0 4 -4 -2 E F -12 40 -O -6 -4 -2 £1r
BINDING ENERGY (or)
Fig. 16. Photoemission energy distribution curves for various coverages of Pd on Ta(l 10) at 16.8 eV (Ne I) and 21.2eV (He I) photon energies (from Ref. [153]).
Photoemission studies dealing with the valence bands and core levels of supported monolayers of Ni and Pt show trends that are very similar to those seen for the Pd overlayers [130,152,155,172- 182]. Fig. 19 shows He I UPS spectra for Pt/Nb (1 10) and Ni/W (1 10) surfaces [ 173,179]. A mono- layer of Pt in contact with Nb(110)exhibits a partial density-of-states centered at 2.7 eV, and has a reduced density of states around the Fermi level [179]. The variations in the energy range covered by the 5d levels of the Pt adatoms are a result of: (1) a reduction in the Pt-Pt interactions that decreases the width of the 5d band; and (2) changes in the Pt-substrate interactions that shift the centroid of the 5d band toward higher binding energy.
Fig. 20 displays core-level shifts measured after depositing a monolayer of Ni[ 159,161,181-183], Pd [159,161,168,157] or Pt [155,175-177,180] on several substrates. Two clear trends can be observed in these experimental data. First, the magnitude of the core-level shift for an admetal increases when the fraction of empty states in the valence band of the metal substrate rises: Ru < Ti < AI. And second, the larger the occupancy of the admetal d band, the bigger the core-level shift in the admetal: Ni < Pt < Pd. The largest electronic perturbations are observed for systems that
244 J.A. Rodriguez/Surface Science Reports 24 (1996) 223-287
1.8
1.6
0.8 .El o.¢
~ 0 . 4
0.2
0.0
I
~ J
%%%1
% % % 1 %.%~% \ ~ % 1 % % %
% ~ % 1 % % %
. . . . . % % %
% % % ,
%%'% %%.%
% % %
% % %
I i
®
%.,%.,%
% % % % % % , % % %
% ' % % % % . %
T
~2.7
2.4 .~ 2.1
1.8 o 1.5
~ 1 . 2
~ 0.9
.H o.6 , ~0 .3
r~ 0.0 Pd/AI
I" I" I
®
PdTi Pdfra PdlMo Pd/W Pdnite PdlRu Pd(lO0)
Fig. 17. Effects of bimetallic bonding on the properties of Pd surface atoms. Part A of the figure displays the shift in the Pd 3ds/2 binding energy with respect to the corresponding value for the surface atoms of Pd(100). The bottom part of the figure shows the shift in the first peak, the one closer to the Fermi level, of the Pd 4d band (from Ref. [168]).
combine an admetal with an electron-rich d band and a substrate with an electron-poor valence band.
Fig. 21 compares several properties of Ni, Pd and Pt monolayers supported on W (1 10). The admetal core-level shifts [ 159,176] represent electronic perturbations with respect to pure Ni, Pd or Pt surfaces. On the other hand, the difference in desorption temperature between the first and second adlayers of a Group-10 metal [159,170,185,186] is proportional to the difference between the strength of the TM-W and TM-TM bonds (TM - Ni, Pd or Pt). After examining the trends in Figs. 18 and 21, it is obvious that the electronic perturbations in the metal overlayers have a significant impact on the strength of the bimetallic bonds. Bimetallic systems that display a large stability probably involve an important redistribution of electrons around the metal centers.
The electronic perturbations observed for the Group- 10 metal overlayers are similar to those seen in bulk alloys, where bimetallic bonding induces an increase in the binding-energy of the core levels and valence band of Ni, Pd and Pt [180,187-190]. Results of L-edge XANES (X-ray absorption near-edge structure) experiments indicate that this phenomenon is accompanied by a reduction in the d electron population of the Group-10 metals [191,192].
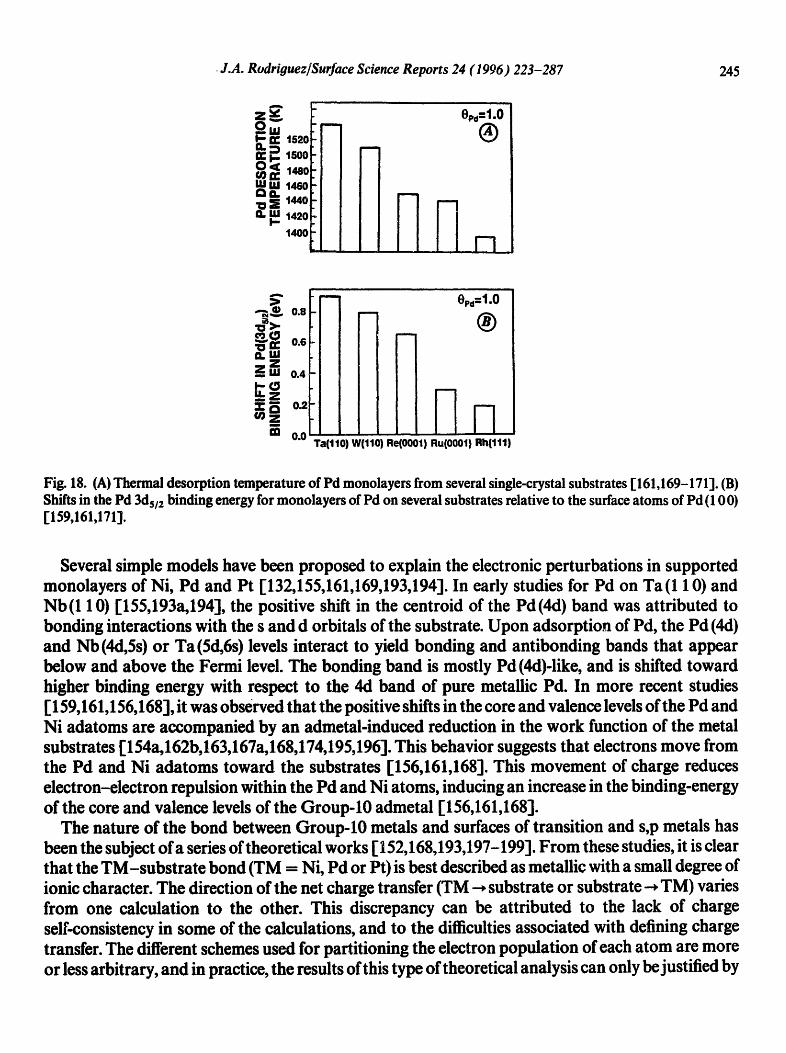
• J.A. Rodriouez/Surface Science Reports 24 (1996) 223-287 245
Z ~ ~w ~- n- 1520 L ~ I~e I.. 1500 O < 148o cn~e ILIm 1460
, . o
1420
N ~ o.s
• o a : o.6 L m
UJZ 0.4
0.0 Ta(110) W(110) Re(0001) Ru(0001) Rh(111)
Fig. 18. (A) Thermal desorption temperature of Pd monolayers from several single-crystal substrates [161,169-171]. (13) Shifts in the Pd 3ds/2 binding energy for monolayers of Pd on several substrates relative to the surface atoms of Pd (10 0) [159,161,171].
Several simple models have been proposed to explain the electronic perturbations in supported monolayers of Ni, Pd and Pt [132,155,161,169,193,194]. In early studies for Pd on Ta(1 10) and Nb(1 10) [155,193a,194], the positive shift in the centroid of the Pd(4d) band was attributed to bonding interactions with the s and d orbitals of the substrate. Upon adsorption of Pd, the Pd (4d) and Nb (4d, Ss) or Ta (Sd,6s) levels interact to yield bonding and antibonding bands that appear below and above the Fermi level. The bonding band is mostly Pd (4d)-like, and is shifted toward higher binding energy with respect to the 4d band of pure metallic Pal. In more recent studies [ 159,161,156,168], it was observed that the positive shifts in the core and valence levels of the Pd and Ni adatoms are accompanied by an admetal-induced reduction in the work function of the metal substrates [154a,162b,163,167a,168,174,195,196]. This behavior suggests that electrons move from the Pd and Ni adatoms toward the substrates [156,161,168]. This movement of charge reduces electron-electron repulsion within the Pd and Ni atoms, inducing an increase in the binding-energy of the core and valence levels of the Group-10 admetal [156,161,168].
The nature of the bond between Group-10 metals and surfaces of transition and s,p metals has been the subject of a series of theoretical works [ 152,168,193,197-199]. From these studies, it is clear that the TM-substrate bond (TM = Ni, Pd or Pt) is best described as metallic with a small degree of ionic character. The direction of the net charge transfer (TM -, substrate or substrate-, TM) varies from one calculation to the other. This discrepancy can be attributed to the lack of charge self-consistency in some of the calculations, and to the difficulties associated with defining charge transfer. The different schemes used for partitioning the electron population of each atom are more or less arbitrary, and in practice, the results of this type of theoretical analysis can only be justified by
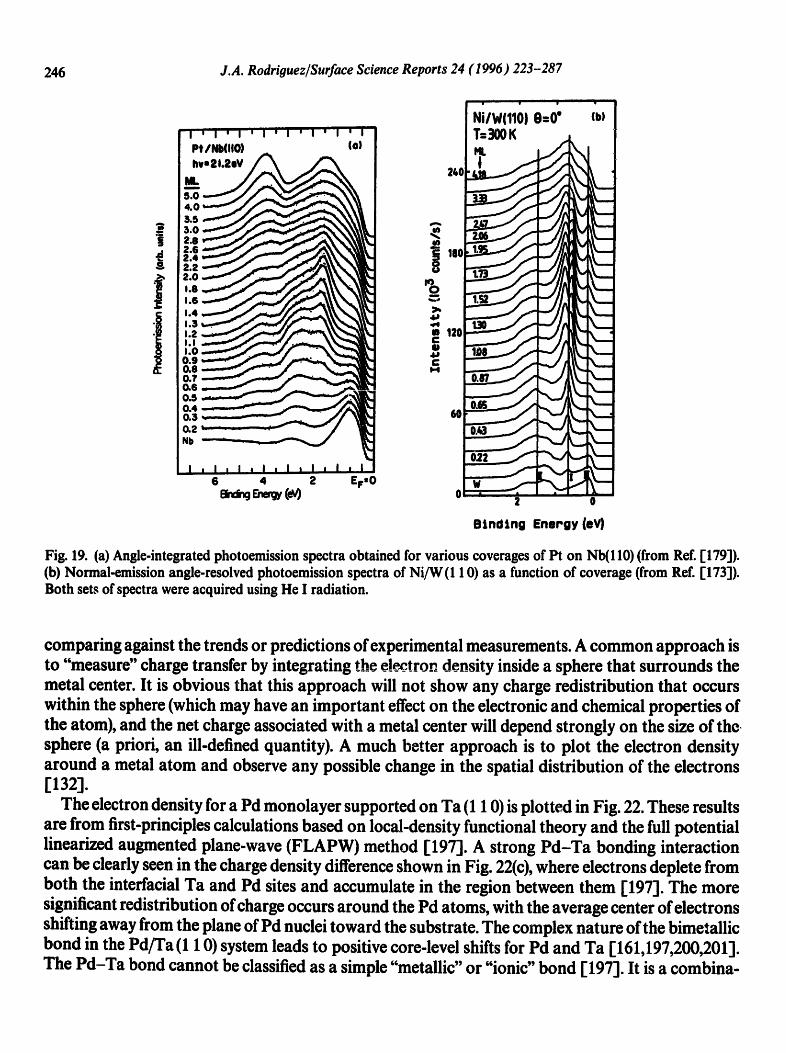
246 J.A. Rodriguez/Surface Science Reports 24 (1996) 223-287
I ' I ' I ' I ' I ' '1 ' I ' I
P t / N b ( l l O ) (0)
24(
i
J -,
J a c ip
c
10|
120
60
6 4 2 ( F , O e~r~ ~ (e~ o
2 0
Binding Energy (eV~
Fig. 19. (a) Angle-integrated photoemission spectra obtained for various coverages of Pt on Nb(110) (from Ref. [179]). (b) Normal-emission angle-resolved photoemission spectra of Ni/W (1 10) as a function of coverage (from Ref. [173]). Both sets of spectra were acquired using He I radiation.
comparing against the trends or predictions of experimental measurements. A common approach is to "measure" charge transfer by integrating the electron density inside a sphere that surrounds the metal center. It is obvious that this approach will not show any charge redistribution that occurs within the sphere (which may have an important effect on the electronic and chemical properties of the atom), and the net charge associated with a metal center will depend strongly on the size of the sphere (a priori, an ill-defined quantity). A much better approach is to plot the electron density around a metal atom and observe any possible change in the spatial distribution of the electrons [132].
The electron density for a Pd monolayer supported on Ta (1 10) is plotted in Fig. 22. These results are from first-principles calculations based on local-density functional theory and the full potential linearized augmented plane-wave (FLAPW) method [197]. A strong Pd-Ta bonding interaction can be clearly seen in the charge density difference shown in Fig. 22(c), where electrons deplete from both the interracial Ta and I'd sites and accumulate in the region between them [197]. The more significant redistribution of charge occurs around the Pd atoms, with the average center of electrons shifting away from the plane of Pd nuclei toward the substrate. The complex nature of the bimetallic bond in the Pd/Ta (1 10)system leads to positive core-level shifts for Pd and Ta [161,197,200,201]. The Pd-Ta bond cannot be classified as a simple "metallic" or "ionic" bond [197"!. It is a combina-
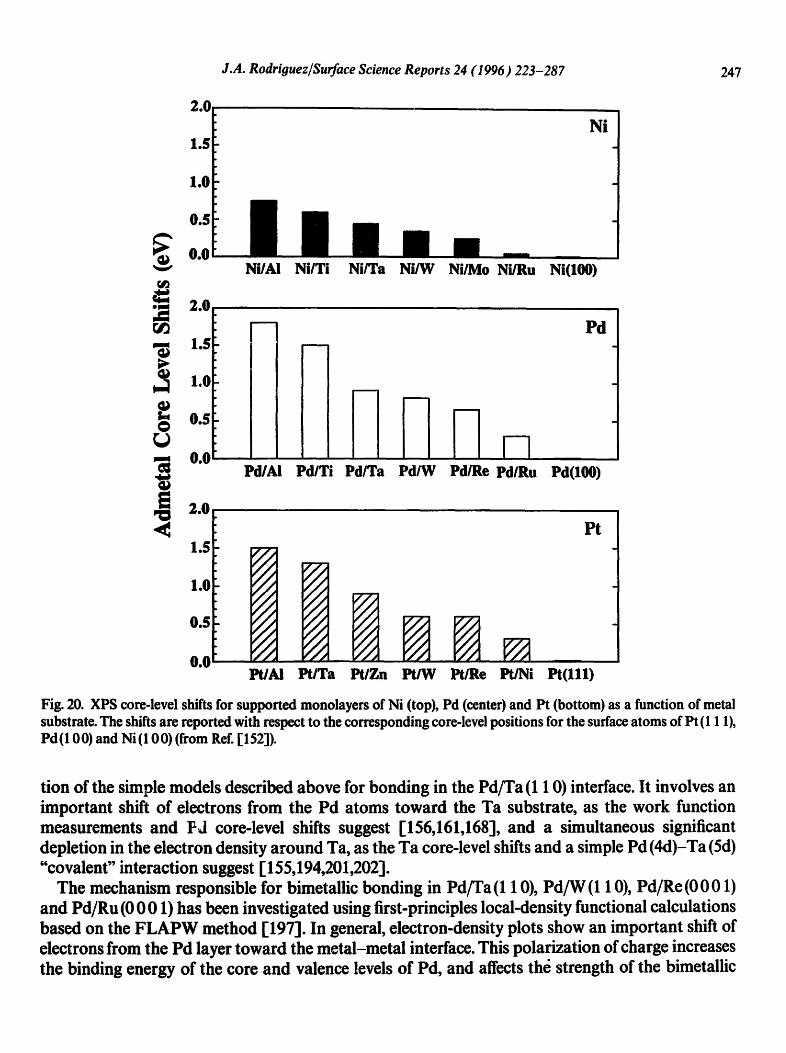
J.A. Rodriguez/Surface Science Reports 24 (1996) 223-287 247
2.0,
1"$ I
1.01-
Ni
0.$
0.0 Ni/A! Ni/Ti Nifra Ni/W Ni/Mo Ni/Ru Ni(100)
r ~
2.0
~ 0.5
. . 0.0 ~ Pd/AI Pd/Ti PdlTa Pd/W Pd/Re Pd/Ru Pd(100)
2.0i
Pt/A! Pt/Ta Pt/Zn Pt/W Pt/Re Pt/Ni Pt ( l l l )
Pd
Pt
Fig. 20. XPS core-level shifts for supported monolayers of Ni (top), Pd (center) and Pt (bottom) as a function of metal substrate. The shifts are reported with respect to the corresponding core-level positions for the surface atoms of Pt (111), ed(100) and Ni (10 0) (from Ref. 1152"i).
tion of the simple models described above for bonding in the Pd/Ta (1 10) interface. It involves an important shift of electrons from the Pd atoms toward the Ta substrate, as the work function measurements and Pd core-level shifts suggest [156,161,168], and a simultaneous significant depletion in the electron density around Ta, as the Ta core-level shifts and a simple Pd (4d)-Ta (5d) "covalent" interaction suggest [155,194,201,202].
The mechanism responsible for bimetallic bonding in Pdfl'a(1 10), PdfW (1 10), Pd/Re(0001) and Pd/Ru (0 0 01) has been investigated using first-principles local-density functional calculations based on the FLAPW method [197]. In general, electron-density plots show an important shift of electrons from the Pd layer toward the metal-metal interface. This polarization of charge increases the binding energy of the core and valence levels of Pd, and affects thd strength of the bimetallic
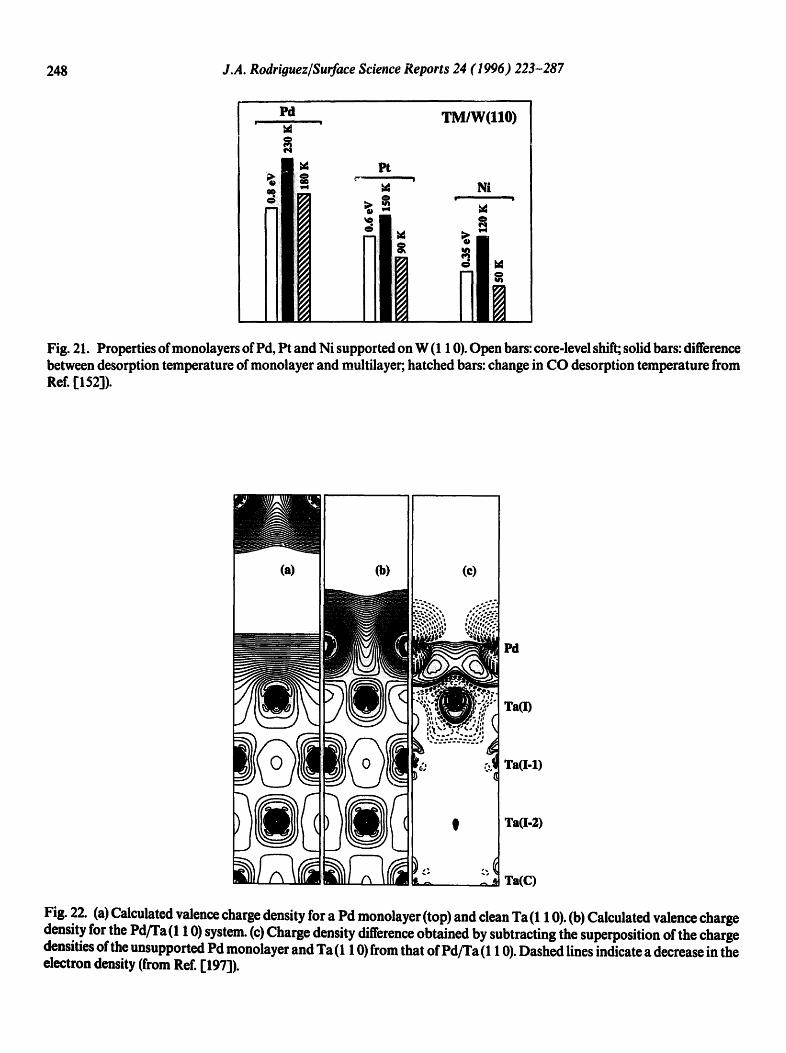
248 J.A. Rodriguez/Surface Science Reports 24 (1996) 223-287
Pd TMIW(110)
m: )::'~ " ~ Ni ~.~ - ~ -
Fig. 21. Properties of monolayers of Pd, Pt and Ni supported on W (1 10). Open bars: core-level shift; solid bars: difference between desorption temperature of monolayer and multilayer; hatched bars: change in CO desorption temperature from Ref. [1523).
" II (b) (c)
/,::ii:,:/,:/'.j
c-~ Ta(l-l)
Fig. 22. (a) Calculated valence charge density for a Pd monolayer (top) and clean Ta (1 10). (b) Calculated valence charge density for the Pd/Ta (1 10) system. (c) Charge density difference obtained by subtracting the superposition of the charge densities of the unsupported Pd monolayer and Ta (1 10) from that of Pd/Ta (1 10). Dashed lines indicate a decrease in the electron density (from Ref. [197]).
J.A. Rodriouez/Surface Science Reports 24 (1996) 223-287 249
bonds [197]. Ab initio self-consistent-field (SCF) calculations for the deposition of Group-10 atoms on clusters modeling surfaces of transition and s,p metals (Ti, W, Mo, Ru, Rh, Zn and AI) also show a redistribution of charge around Ni, Pd and Pt [ 152,168,171,203]. In these cases, bimetallic bonding induces a TM(d)-, TM(s,p) rehybridization that shifts d electrons from around the Group- 10 metal into the metal-metal interface, producing an accumulation of electrons around the bimetallic bonds [152,168,203]. The change in the d population of a Group-10 element increases in the following order: Ni < Pt < P d [152]. This sequence agrees with the relative occupancy of the d shell in the isolated metals: Ni < Pt < Pd [184]. L2,3-edge XANES measurements for a large series of bulk compounds also show that Pd exhibits a bigger tendency to shift d electrons through chemical bonding than Pt [192]. The trends in the experimental results displayed in Figs. 18 and 21 reflect changes in the d electron population of the Group-10 metals [152]. The larger the movement of TM(d) electrons toward the substrate, the bigger the positive binding-energy shift in the core levels of the Group-10 metal, and the larger the accumulation of charge in the interface region with a stronger admetal-substrate bond [152].
3.3. Group-l l admetals
During the past two decades, there has been a large interest in the electronic properties of intermetallic compounds that contain noble metals [132,204-207]. In early models, the participa- tion of the noble-metal d band in metal-metal bonding was neglected. Recent experimental and theoretical studies for bulk alloys show that the d bands of Cu, Ag and Au are active in bimetallic bonding [192,204-206].
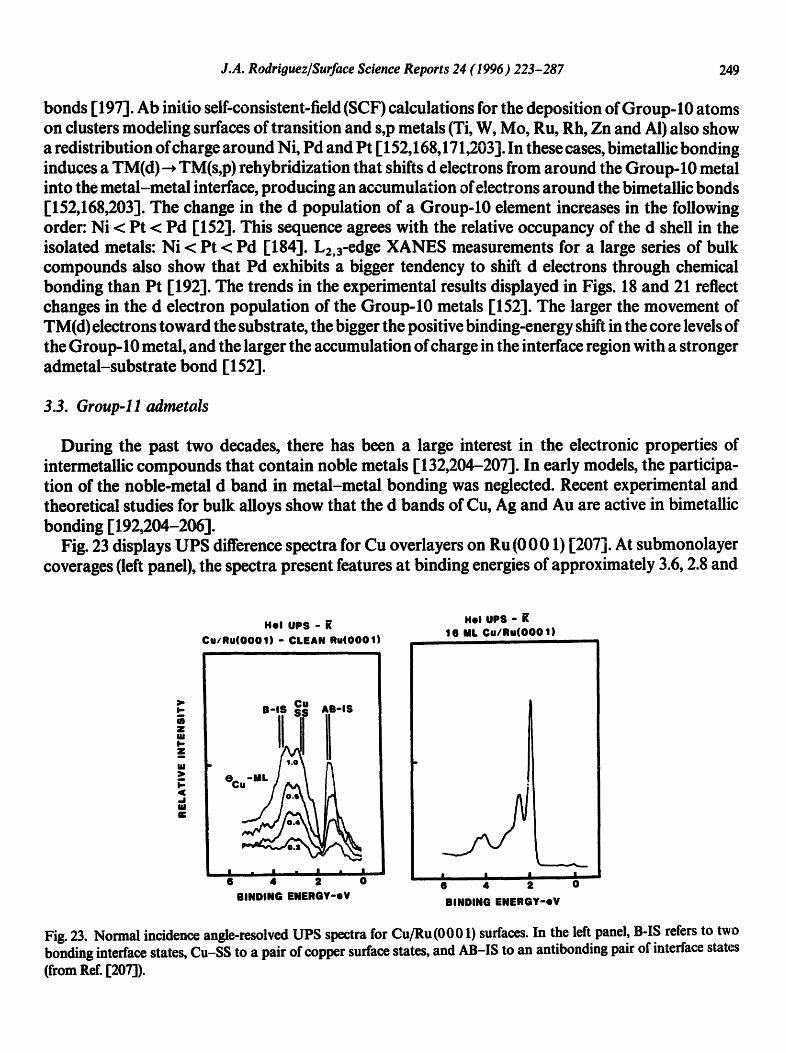
Fig. 23 displays UPS difference spectra for Cu overlayers on Ru (0001) [207]. At submonolayer coverages (left panel), the spectra present features at binding energies of approximately 3.6, 2.8 and
I
Hel UPS - K" Hel U P S - K 1 • ML C u / R u ( O 0 0 1 )
Cu /Ru (O001 ) - CLEAN Ru(OOOl)
| z_
z
B 4 2 0 6 4 2 BINDING ENERGY-eV BINDING ENERGY-eV
Fig. 23. Normal incidence angle-resolved UPS spectra for Cu/Ru(0001) surfaces. In the left panel, B-IS refers to two
bonding interface states, Cu-SS to a pair of copper surface states, and AB-IS to an antibonding pair of interface states (from Ref. [207]).
250 2.A. Rodriouez/Surface Science Reports 24 (1996) 223-287
1.SeV. Theoretical calculations utilizing the linearized augmented plane-wave (LAPW) method [207] indicate that the structures at 3.6 and 1.5 eV correspond to two bonding and antibonding interface states, respectively. These interface states are mainly a consequence of the interaction between the Cu (3d) and Ru (4(1) levels [207]. It is clear from the spectra in Fig. 23 that the electronic prope~ies of Cu films with submonolayer coverage are very different from those of Cu multilayers.
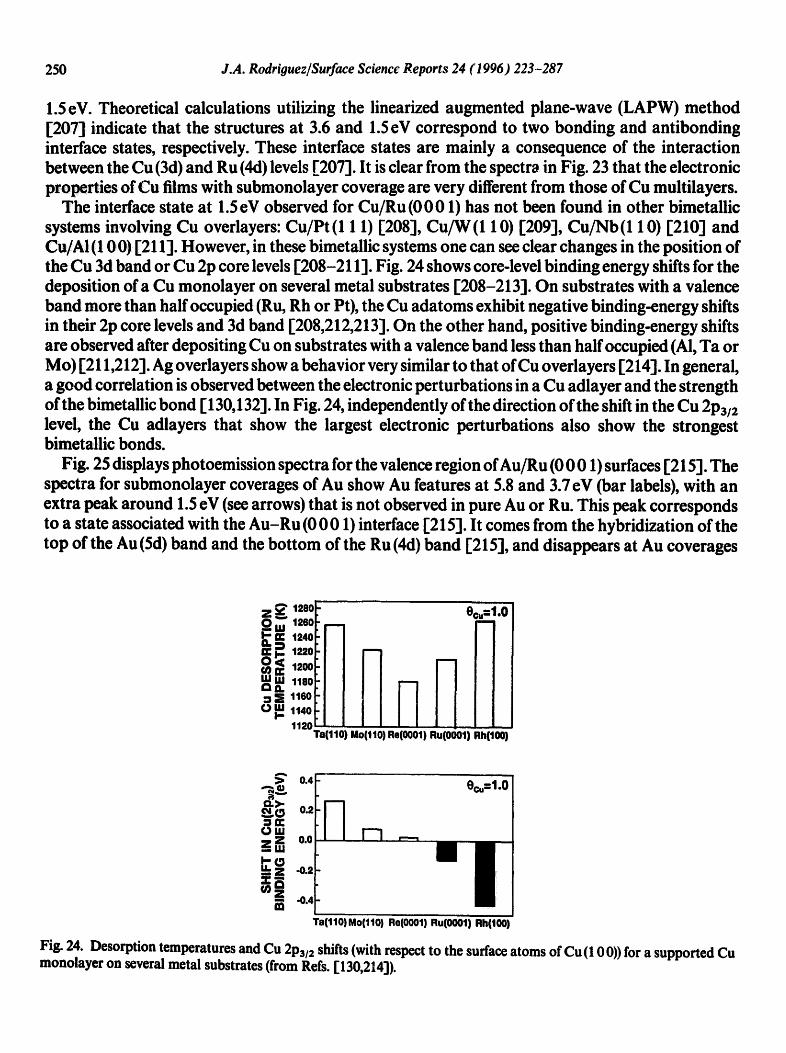
The interface state at 1.5 eV observed for Cu/Ru (0 0 01) has not been found in other bimetallic systems involving Cu overlayers: Cu/Pt(1 1 1) [208], CufW(1 10) [209], Cu/Nb(1 10) [210] and Cu/AI (100)[211 ]. However, in these bimetallic systems one can see clear changes in the position of theCu 3d band or Cu 2p core levels [208-211]. Fig. 24 shows core-level binding energy shifts for the deposition of a Cu monolayer on several metal substrates [208-213]. On substrates with a valence band more than half occupied (Ru, Rh or Pt), the Cu adatoms exhibit negative binding-energy shifts in their 2p core levels and 3d band [208,212,213]. On the other hand, positive binding-energy shifts are observed after depositing Cu on substrates with a valence band less than half occupied (AI, Ta or Mo) [211,212]. Ag overlayers show a behavior very similar to that of Cu overlayers [214]. In general, a good correlation is observed between the electronic perturbations in a Cu adlayer and the strength of the bimetallic bond [130,132]. In Fig. 24, independently of the direction of the shift in the Cu 2P3/2 level, the Cu adlayers that show the largest electronic perturbations also show the strongest bimetallic bonds.
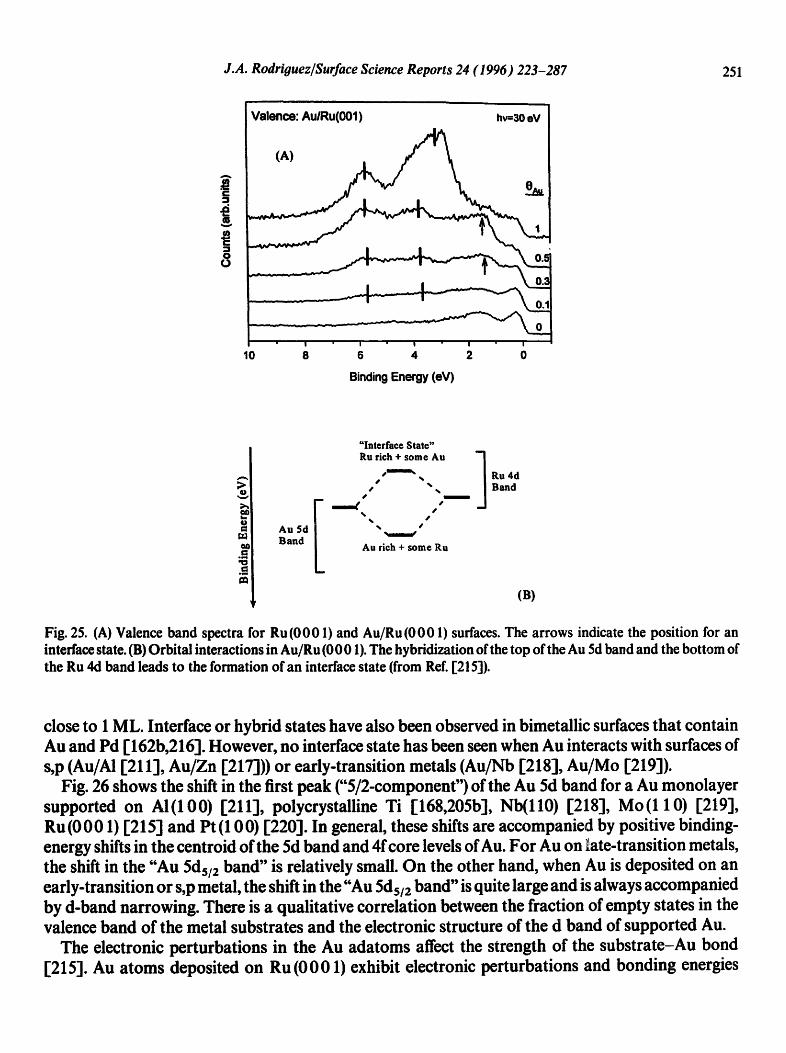
Fig. 25 displays photoemission spectra for the valence region of Au/Ru (0 0 01) surfaces [215]. The spectra for submonolayer coverages of Au show Au features at 5.8 and 3.7 eV (bar labels), with an extra peak around 1.5 eV (see arrows) that is not observed in pure Au or Ru. This peak corresponds to a state associated with the A u- R u (0 0 01) in terrace [ 215]. I t comes from the hybridization of the top of the Au (Sd) band and the bottom of the Ru (4d) band [215], and disappears at Au coverages
O W' 12601-
~ ' ~ , = o F - I I ~ I I
==S1160[-J J m I I I I I I I
Ta(110) Mo(110)Re(0001) R.(0001) Rh(100)
A
Q.>.
:_,,:, I - (3
0.4
0.2
0.0
0c.=1.0
~ .v.q[- m J Ta(110) Mo(110) Re(0001) Ru(0001) Rh(t00)
Fig. 24. Desorption temperatures and Cu 2P3/2 shifts (with respect to the surface atoms of Cu (10 0)) for a supported Cu monolayer on several metal substrates (from Refs. [130,214]).
J.A. Rodriouez/Surface Science Reports 24 (1996) 223-28 ? 251
i 0
Valence: Au/Ru(001) hv=30 eV
I " I " I
Binding Energy (eV)
"Interface State"
• j
A u 5d • B a n d
¢m Au rich + s o m e Ru
.=_
Ru 4d B a n d
(B)
Fig. 25. (A) Valence band spectra for Ru(0001) and Au/Ru(0001) surfaces. The arrows indicate the position for an interface state. (B) Orbital interactions in Au/Ru (0001). The hybridization of the top of the Au 5d band and the bottom of the Ru 4d band leads to the formation of an interface state (from Ref. 1"215]).
close to 1 ML. Interface or hybrid states have also been observed in bimetallic surfaces that contain Au and Pd [162b,216]. However, no interface state has been seen when Au interacts with surfaces of s,p (Au/AI [211], Au/Zn [217]))or early-transition metals (Au/Nb [2181, A u/Mo 1-219]).
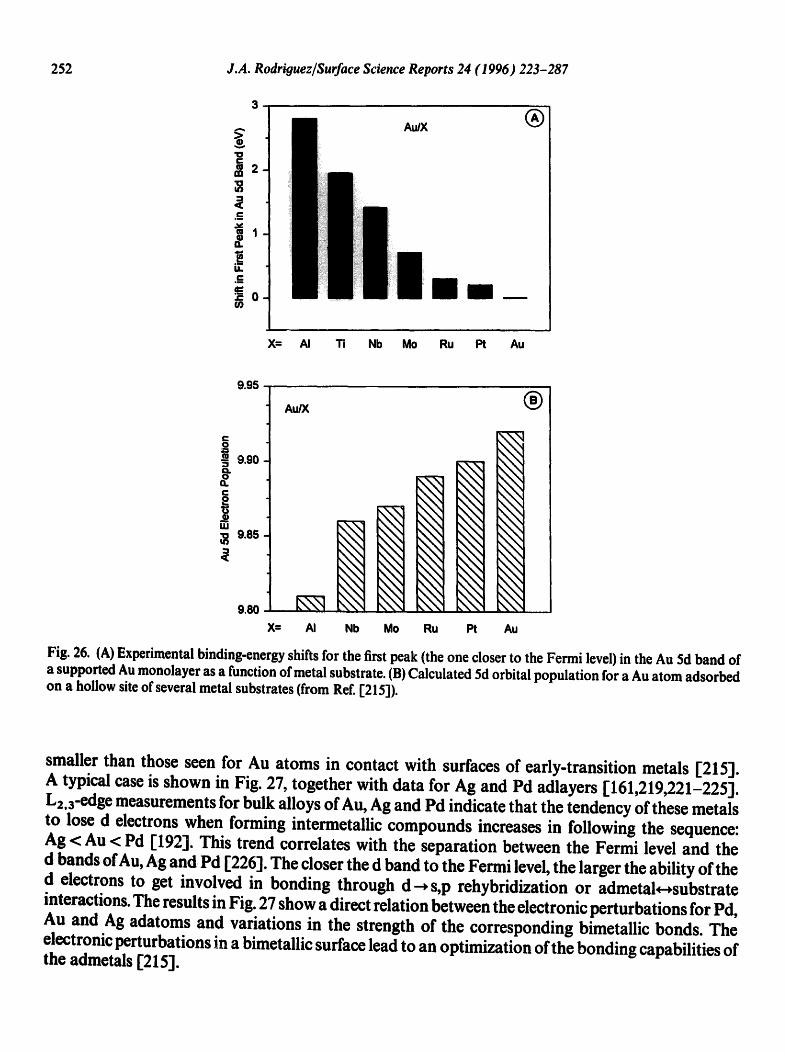
Fig. 26 shows the shift in the first peak C5/2-component") of the Au 5d band for a Au monolayer supported on AI(100) [211], polycrystalline Ti 1"168,205b], Nb(ll0) [218"1, MoO 10) [219], Ru (0 001) [215] and Pt (10 0) [220]. In general, these shifts are accompanied by positive binding- energy shifts in the centroid of the 5d band and 4fcore levels of Au. For Au on late-transition metals, the shift in the "Au 5ds/2 band" is relatively small. On the other hand, when Au is deposited on an early-transition or s,p metal, the shift in the"Au 5ds/2 band" is quite large and is always accompanied by d-band narrowing. There is a qualitative correlation between the fraction of empty states in the valence band of the metal substrates and the electronic structure of the d band of supported Au.
The electronic perturbations in the Au adatoms affect the strength of the substrate-Au bond 1215]. Au atoms deposited on Ru(0001) exhibit electronic perturbations and bonding energies
252 J.A. Rodr~3uez/Surface Science Reports 24 (1996) 223-287
" o ¢:
. . ,2
.¢_
M.
.¢_
~ o BI m
®
X= AI Ti Nb Mo Ru Pt Au
9.95 -
e- o
9.90 o~
UJ ;X 9.s5
9.80
Au/X ®
X= AI Nb Mo Ru Pt Au
Fig, 26. (A) Experimental binding-energy shifts for the first peak (the one closer to the Fermi level) in the Au 5d band of a supported Au monolayer as a function of metal substrate. (B) Calculated 5d orbital population for a Au atom adsorbed on a hollow site of several metal substrates (from Ref. [215]).
smaller than those seen for Au atoms in contact with surfaces of early-transition metals [215]. A typical case is shown in Fig. 27, together with data for Ag and Pd adlayers [161,219,221-225]. L2.3-edge measurements for bulk alloys of Au, Ag and Pd indicate that the tendency of these metals to lose d electrons when forming intermetallic compounds increases in following the sequence: Ag < Au < Pd [192]. This trend correlates with the separation between the Fermi level and the d bands ofAu, Ag and Pd [226]. The closer the d band to the Fermi level, the larger the ability of the d electrons to get involved in bonding through d-,s ,p rehybridization or admetal,-,substrate interactions. The results in Fig. 27 show a direct relation between the electronic perturbations for Pd, Au and Ag adatoms and variations in the strength of the corresponding bimetallic bonds. The electronic perturbations in a bimetallic surface lead to an optimization of the bonding capabilities of the admetals [215].
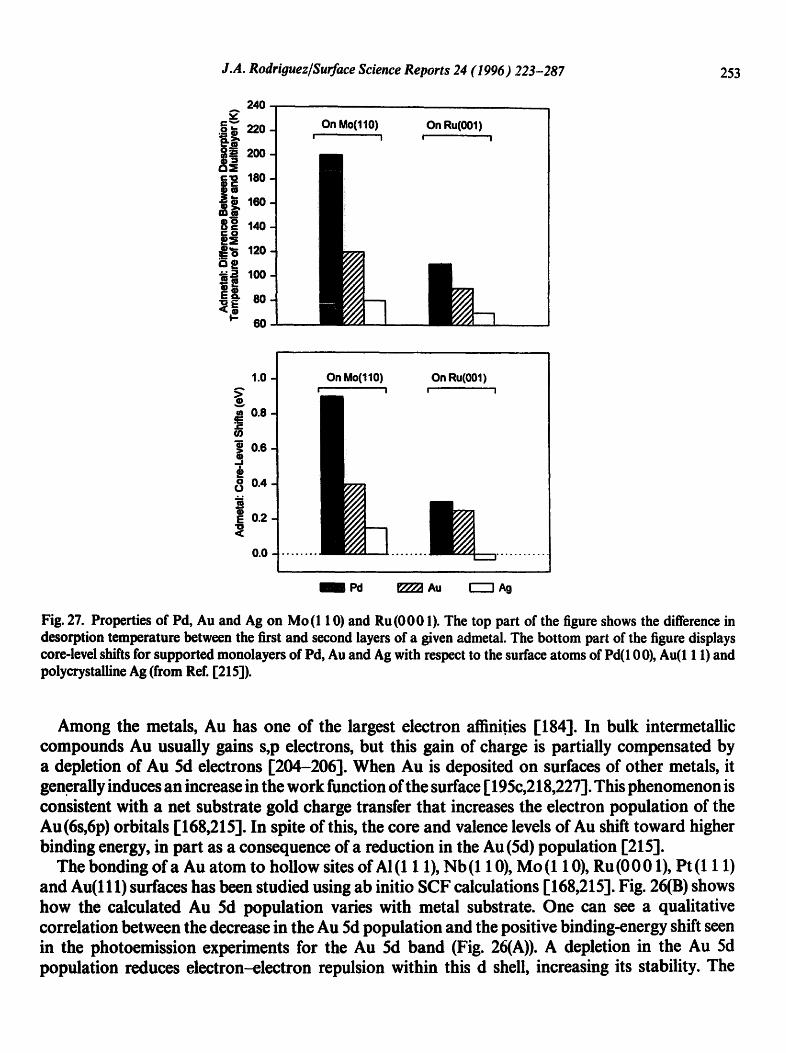
J.A. Rodriouez/Surface Science Reports 24 (I 996) 223-287 253
240 A
~ 220- ~ 2oo- cqo 180-
I° 8~ 14o r - o
. ~6 120 c~s
~ .o
60
On Mo(110) On Ru(O01) I I i ..... I
1 . 0 -
.~ 0 .8 - e.-
u)
~ 0 .6 -
o 0 .4 - (.,I
E 0 .2 -
0.0
On Mo(110) On Ru(O01) I I I I
Pd FT"/~ Au I I Ag
Fig. 27. Properties of Pd, Au and Ag on Mo(1 10) and Ru(0001). The top part of the figure shows the difference in desorption temperature between the first and second layers of a given admetal. The bottom part of the figure displays core-level shifts for supported monolayers of Pd, Au and Ag with respect to the surface atoms of Pd(100), Au(l 1 1) and polycrystalline Ag (from Ref. 1"215]).
Among the metals, Au has one of the largest electron affinit.ies [184]. In bulk intermetallic compounds Au usually gains s,p electrons, but this gain of charge is partially compensated by a depletion of Au 5d electrons [204-206"1. When Au is deposited on surfaces of other metals, it generally induces an increase in the work function of the surface [ 195c,218,227]. This phenomenon is consistent with a net substrate gold charge transfer that increases the electron population of the Au(6s,6p) orbitals [168,215]. In spite of this, the core and valence levels of Au shift toward higher binding energy, in part as a consequence of a reduction in the Au (Sd) population [2151.
The bonding ofa Au atom to hollow sites of AI(1 1 1), Nb(1 10), Mo(1 10), Ru(0001), Pt(1 1 1) and Au(111 ) surfaces has been studied using at) initio SCF calculations [ 168,215]. Fig. 26(B) shows how the calculated Au 5d population varies with metal substrate. One can see a qualitative correlation between the decrease in the Au 5d population and the positive binding-energy shift seen in the photoemission experiments for the Au 5d band (Fig. 26(A)). A depletion in the Au 5d population reduces electron-electron repulsion within this d shell, increasing its stability. The

254 J.A. Rodriguez/Surface Science Reports 24 (1996) 223-287
behavior seen for the Au adatoms is similar to that found for Au in bulk compounds [206]. Linear-augmented Slater-type-orbital (LASTO) calculations for binary alloys containing Au and a 5d-transition metal show a decrease in the Au 5d electron popul~tion when the fraction of empty states in the valence bands of the transition metal increases [206]. Au L~,~-edge XANES experiments indicate a net loss of electrons from the Au 5d orbitals upon the formation of b~,~:k AuAI, AuTi and AuTa alloys [205]. Hybridization of the electron-rich 5d band of Au with the electron-poor valence bands of Al or an early-transition metal leads to a loss of Au 5d character in the occupied states ofthe system.
Cu and Ag have electron affinities that are much smaller than that of Au [184], and can behave as net electron donors when present on a metal surface. The bonding interactions in the Cu/Pt(1 1 1) and Ag/Pt(1 1 1) systems have been examined using ab initio SCF methods [214] (cluster models) and first-principles local-density functional calculations [70] (slab models). The results of these theoretical studies indicate that there is a net movement of electrons from the noble-metal adatoms toward the Pt substrate, as work function measurements suggest [214]. This is illustrated by the electron-density plot in Fig. 28 [70]. One can see a clear shift of electrons from Ag toward the Pt substrate. The redistribution of charge in these bimetallic systems produces a reduction in the population of the Cu (4s,4p) and Ag(Ss, Sp) orbitals [214]. This is accompanied by an increase in the Cu 3d and Ag 4d populations, and a depletion in the Pt 5d population [214]. The changes in the d populations have a strong impact on the core-level shifts of Pt and the noble metals [214].
t
i ~ . , ~ "
. . . .
]:1 ..,.
:!. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . O I I l 4 6 I ? 8 IP tO U UB 13 N IS UI
Fig. 28. Bond-charge density for Ag adsorption on a two-fold bridge of Pt(l 1 1). The vacuum is at the top of the plot. Contour labels are in units of 10- 3 e/bohr 3. Solid contours correspond to an increase in electron density relative to the sum of clean surface and isolated Pt atom pseudo-charges; dashed contours correspond to a decrease. Heavy dots indicate the positions of Pt nuclei. Inset: the geometry of the surface plus adsorbate. The Ag adatom is a solid square. Substrate nuclei are circles. The solid circles and solid square define the plane of the contour plot (from Ref. [70]).
J.A. Rodriguez/Surface Science Reports 24 (1996) 223-287 255
3.4. Group-12 admetals
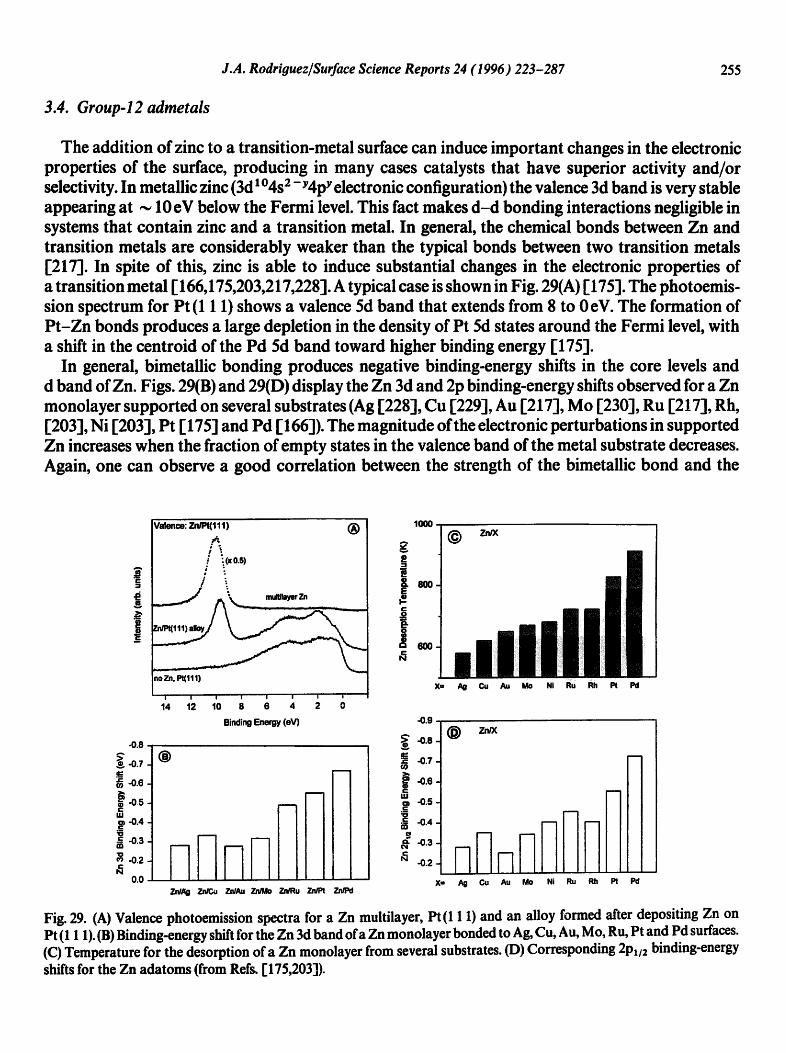
The addition of zinc to a transition-metal surface can induce important changes in the electronic properties of the surface, producing in many cases catalysts that have superior activity and/or selectivity. In metallic zinc (3d i O4s2 - Y4pY electronic configuration) the valence 3d band is very stable appearing at ~ 10 eV below the Fermi level. This fact makes d-d bonding interactions negligible in systems that contain zinc and a transition metal. In general, the chemical bonds between Zn and transition metals are considerably weaker than the typical bonds between two transition metals [217]. In spite of this, zinc is able to induce substantial changes in the electronic properties of a transition metal I 166,175,203,217,228]. A typical case is shown in Fig. 29(A) r 175]. The photoemis- sion spectrum for Pt (1 1 1) shows a valence 5d band that extends from 8 to 0 eV. The formation of Pt-Zn bonds produces a large depletion in the density of Pt 5d states around the Fermi level, with a shift in the centroid of the Pd 5d band toward higher binding energy [175].
In general, bimetallic bonding produces negative binding-energy shifts in the core levels and d band of Zn. Figs. 29(B) and 29(D) display the Zn 3d and 2p binding-energy shifts observed for a Zn monolayer supported on several substrates (Ag [228], Cu [229], Au [217], Mo [230], Ru [217], Rh, [203], Ni [203], Pt I175] and Pd I 166]). The magnitude ofthe electronic perturbations in supported Zn increases when the fraction of empty states in the valence band of the metal substrate decreases. Again, one can observe a good correlation between the strength of the bimetallic bond and the
-0 .8 -
-0.7
-0.e~ ~ - 0 . 5
~ -0.4 ~ -0.3 ~ -0.2 N
0.0
vate.=e: ZnIpt(111) ®
.~ "lxo.~
i -
noZn, Pt(1111
I / I I I I I t
14 12 10 8 6 4 2 0
Binding Energy (eV)
®
: 3
r.-
,v,
! i O
1000
800
6OO
X a Ag Co Au Mo Ni Ru Rh R
Znl~ Z n ~ Zn/Au Znmlo Z ~ u Zn/Pt Zr~Pd
-0.9
-0.7 ¢D
-0.6 g
-0.4
-0.3
• .o.2 [-"] X= Ag Cu Au Mo Ni Ru Rh R
Fig. 29. (A) Valence photoemission spectra for a Zn multilayer, Pt (1 1 1) and an alloy formed after depositing Zn on Pt (1 1 1). (B) Binding-energy shift for the Zn 3d band ofa Zn monolayer bonded to Ag, Cu, Au, Mo, Ru, Pt and Pd surfaces. (C) Temperature for the desorption of a Zn monolayer from several substrates. (D) Corresponding 2pl/2 binding-energy shifts for the Zn adatoms (from Refs, [175,203]).

256 J.A. Rodriouez/Surface Science Reports 24 (1996) 223-287
magnitude of the electronic perturbations in the admetal. In Fig. 29(C), the overall change in the Zn adsorption energy is close to 20 kcal/mol [203]. In a simple model based on the results of ab initio SCF calculations, the negative 2p and 3d shifts for Zn can be attributed to a "spillover" of electrons from the metal substrate into the region around the bimetallic bonds [175,203]. The electron density around a Zn-substrate bond is larger than that near a Zn-Zn bond. This extra electron density is "felt" by the Zn 2p and 3d electrons. The bigger the accumulation of electrons in the metal-metal interface, the stronger the bimetallic bond and the larger the destabilization of the Zn 3d and 2p levels [175,203].
3.5. Summary
From the experimental and theoretical results described above, we can conclude that in bimetallic surfaces that contain metals from Groups 8-12 the bonds are best described as "metallic" with a small degree of ionic character. Bimetallic bonds that display a large stability usually involve a significant redistribution ofelectrons around the metal centers. This may be viewed as a result ofan optimization of the bonding capabilities of the metals. In general, the strongest metal-metal interactions occur in systems that combine a metal with a valence band almost fully occupied and a metal in which the valence band is almost empty.
4. Chemical properties of bimetallic surfaces
The studies discussed in the previous section indicate that bimetallic bonding can induce significant changes in the band structure of a metal, introducing in this way the possibility for new and unique chemical properties. The results of many works dealing with the chemisorption of simple molecules (H 2 [154,170,231,232], CO [13,130,154,156,233], NO [234], C2H 4 r235], etc.)on well-defined bime- tallic surfaces show novel and interesting changes in the chemical reactivity of the bonded metals.
4.1. CO chemisorption
The interaction of CO with bimetallic surfaces has been the subject of many works in recent years [13,130-132,161-164,166-172, 175,179,199,203,212,231,233,236-247]. CO is an ideal probe mol- ecule to investigate the chemisorption properties of bimetallic surfaces. There is extensive informa- tion about the surface chemistry of this molecule in monometallic substrates, and the bonding mechanism is much better known for CO than for any other simple molecule. In addition, CO is involved in many catalytic processes of industrial importance.
Fig. 30(A) displays CO-thermal desorption spectra acquired after adsorbing the molecule on Pd(100), and on a Pd monolayer supported on Ta(1 10) [169a], W(1 10) [170], Re(0001) [:161] and Ru(0001) [161]. For the Pd/Ta (1 10) and Pd/W (1 10) systems, the large decrease in the CO desorption temperature (180-230 K) indicates that there is a big weakening in the strength of the Pd-CO bond. The isosteric heat of adsorption of CO on the supported Pd monolayers is 15-20 kcal/mol smaller than on Pd(l 1 1) (Fig. 30(B)) [246,247].
The valence photoemission spectra in Fig. 3 I(A) were acquired after dosing CO to a Pd(111)-like thick film and a Pd monolayer supported on Ta (1 10) [248]. The spectrum for the thick palladium
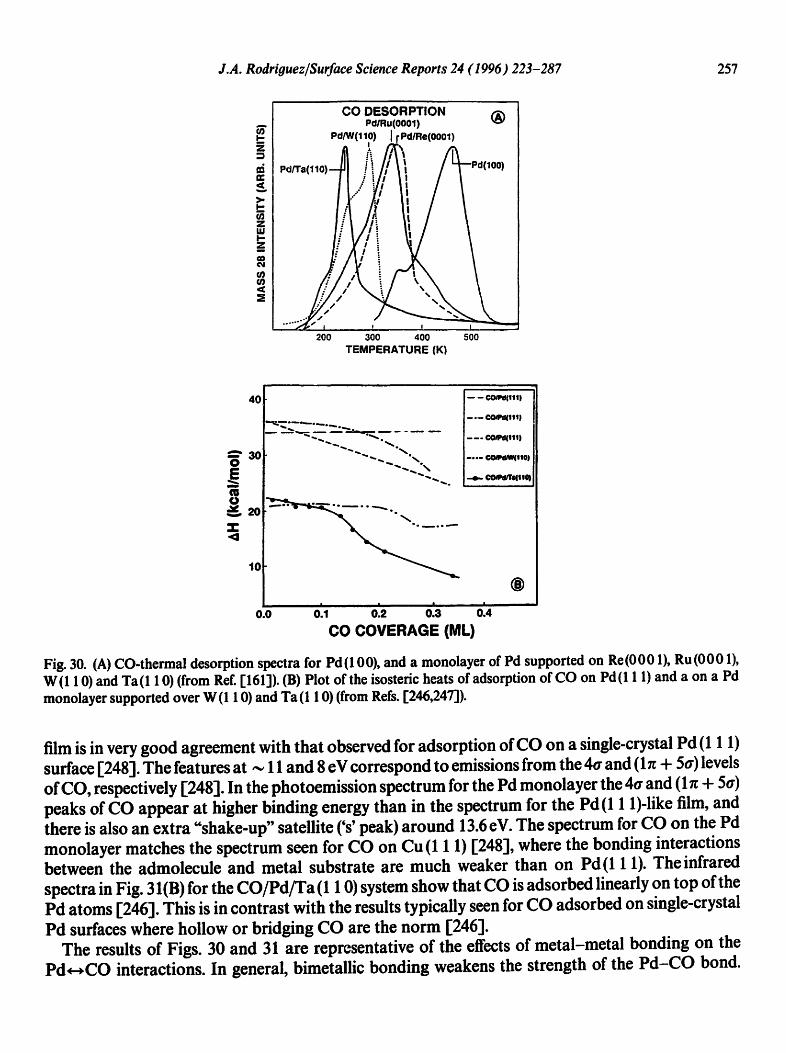
J.A. Rodriouez/Surface Science Reports 24 (1996) 223-287 257
u~ k- in Z
d
>. p. n c~ Z w !-
CO DESORPTION Pd/R.(0001)
Pal/W(110)
Pd/ra(110)-- . ;
1
1
-" j
. . . . o-" ]
2OO
TEMPERATURE (K)
® ~Pd/Re(0001)
~ i / ~ P d ( 1 0 0 ) I I
L ,
/
300 400 500
40
3O
m ¢B
• O 20 =<
10
"~ . . .~ .~ ~ ' ~
0.0 0'.1 0'.2 0:3 CO COVERAGE (ML)
- - - r. ,on,dl~lW)
- . - ¢ 0 o ~ ' . + 1
- - - ¢ O + l ~ r . )
. . . . ¢ O O 4 M l l t O )
. - e - ¢ O + l ~ n ' a l S I0)
® I
0 . 4
Fig. 30. (A) CO-thermal desorption spectra for Pd(100), and a monolayer of Pd supported on Re(0001), Ru(0001), W(I 10) and Ta(l 10) (from Ref. 1"161"1). (B) Plot of the isosteric heats of adsorption of CO on Pd(l I 1) and a on a Pd monolayer supported over W (1 10) and Ta (1 10) (from Refs. 1"246,247]).
film is in very good agreement with that observed for adsorption of CO on a single-crystal Pd (1 1 1) surface [248]. The features at ~ 11 and 8 eV correspond to emissions from the 4~ and (17c + 5¢r) levels of CO, respectively [248]. In the photoemission spectrum for the Pd monolayer the 4~ and (1 ~ + 5~) peaks of CO appear at higher binding energy than in the spectrum for the Pd (1 1 1)-like film, and there is also an extra "shake-up" satellite ('s' peak) around 13.6eV. The spectrum for CO on the Pd monolayer matches the spectrum seen for CO on Cu (1 1 1) [248], where the bonding interactions between the admolecule and metal substrate are much weaker than on Pd (1 1 1). The infrared spectra in Fig. 3 I(B) for the CO/Pd/Ta (1 10) system show that CO is adsorbed linearly on top of the Pd atoms [246]. This is in contrast with the results typically seen for CO adsorbed on single-crystal Pd surfaces where hollow or bridging CO are the norm [246].
The results of Figs. 30 and 31 are representative of the effects of metal-metal bonding on the Pd~-,CO interactions. In general, bimetallic bonding weakens the strength of the Pd-CO bond.
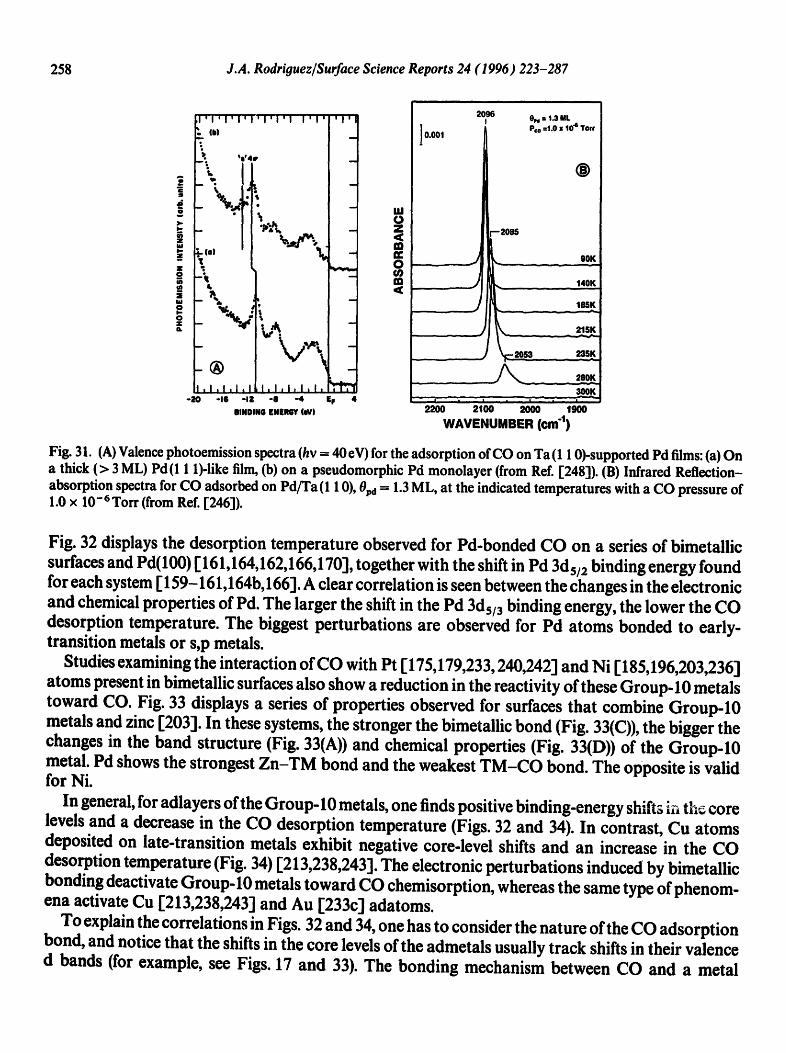
258 J.A. Rodriguez/Surface Science Reports 24 (1996) 223-287
,,m
|
-ZO -16 -12 -8 - 4 E F 4
BINOING ENERGY (W)
2096 e ~ : 1.3 ML
o.ool p. =,.o.,o~~,,
~ ~t~2085
. . . . . . ~ _ _ 280K - - 1
~ t00 ~ 7 - ~ 2200 21 2000 1900
WAVENUMBER (cm "1) Fig. 31. (A) Valence photoemission spectra (hv = 40 eV) for the adsorption of CO on Ta (1 10)-supported Pd films: (a) On a thick (> 3 ML) Pd(l l l)-like film, (b) on a pseudomorphic Pd monolayer (from Ref. [248]). (B) Infrared Reflection- absorption spectra for CO adsorbed on Pd/Ta (1 10), 0pd -- 1.3 ML, at the indicated temperatures with a CO pressure of 1.0 x 10-~Torr (from Ref. [246]).
Fig. 32 displays the desorption temperature observed for Pd-bonded CO on a series of bimetallic surfaces and Pd(100) [ 161,164,162,166,170], together with the shift in Pd 3ds/2 binding energy found for each system [159-161,164b,166]. A clear correlation is seen between the changes in the electronic and chemical properties of Pd. The larger the shift in the Pd 3ds/3 binding energy, the lower the CO desorption temperature. The biggest perturbations are observed for Pd atoms bonded to early- transition metals or s,p metals.
Studies examining the interaction of CO with Pt [ 175,179,233, 240,242] and Ni [ 185,196,203,236] atoms present in bimetallic surfaces also show a reduction in the reactivity of these Group-10 metals toward CO. Fig. 33 displays a series of properties observed for surfaces that combine Group-10 metals and zinc [203]. In these systems, the stronger the bimetallic bond (Fig. 33(C)), the bigger the changes in the band structure (Fig. 33(A)) and chemical properties (Fig. 33(D)) of the Group-10 metal. Pd shows the strongest Zn-TM bond and the weakest TM-CO bond. The opposite is valid for Ni.
In general, for adlayers ofthe Group- 10 metals, one finds positive binding-energy shifts [~ the core levels and a decrease in the CO desorption temperature (Figs. 32 and 34). In contrast, Cu atoms deposited on late-transition metals exhibit negative core-level shifts and an increase in the CO desorption temperature (Fig. 34) [213,238,243]. The electronic perturbations induced by bimetallic bonding deactivate Group-10 metals toward CO chemisorption, whereas the same type of phenom- ena activate Cu [213,238,243] and Au [233c] adatoms.
To explain the correlations in Figs. 32 and 34, one has to consider the nature of the CO adsorption bond, and notice that the shifts in the core levels of the admetals usually track shifts in their valence d bands (for example, see Figs. 17 and 33). The bonding mechanism between CO and a metal
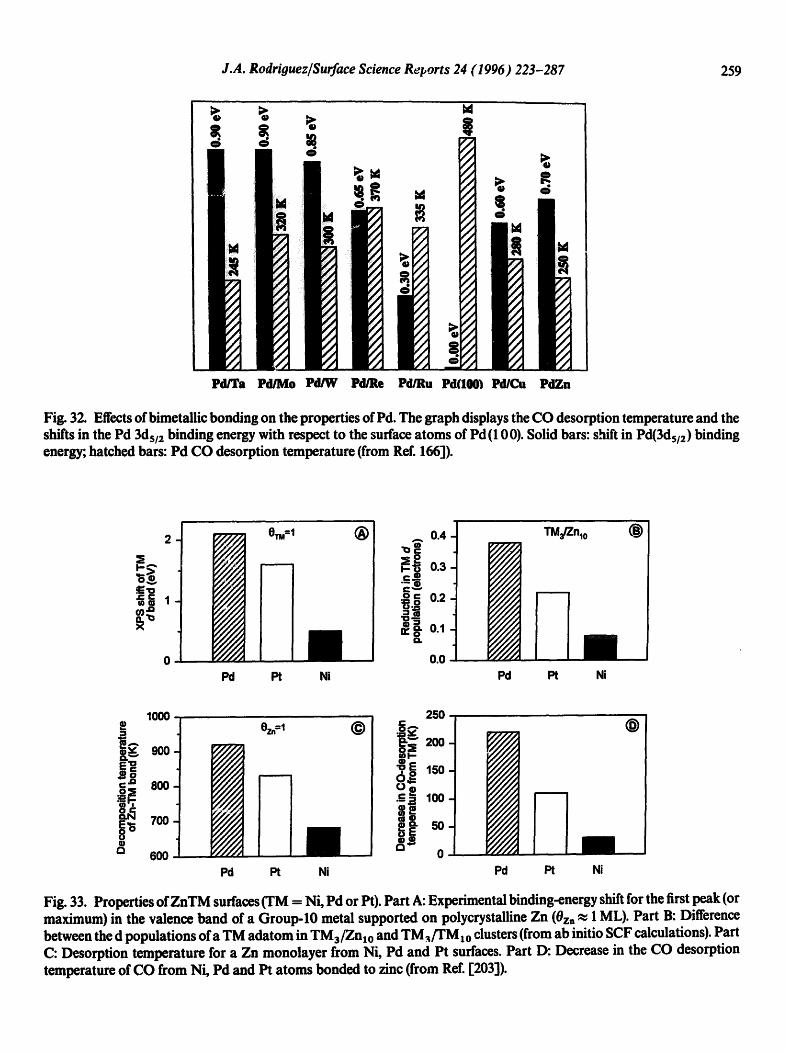
J.A. Rodriguez/Surface Science Reports 24 (1996) 223-287 259
t .
POTa Pd/Mo Pd/W Pd/ge Pd/Ru Pd(1O0) Pd/Cu PdZn
Fig. 32. Effects of bimetallic bonding on the properties of Pd. The graph displays the CO desorption temperature and the shifts in the Pd 3ds/2 binding energy with respect to the surface atoms of Pd (10 0). Solid bars: shift in Pd(3ds/2) binding energy; hatched bars: Pd CO desorption temperature (from Ref. 166]).
2 - OTM=I
~= 1
.
i
o ; Pd ~ Ni
® 0.4
:Ze ~:! 0.3 ",7.~ o = 0.2 ~o
i"0.1 0.0
TM3IZn,0 t'~
Pd lit Ni
1000 250
I~ 9o0 .,_oSi 200
! I #,= o 0,~ 150
ii, .oo
8 0 50
soo 8 - o Pd Pt Ni Pd Pt Ni
Fig. 33. Properties of ZnTM surfaces (TM = Ni, Pd or Pt). Part A: Experimental binding-energy shift for the first peak (or maximum) in the valence band of a Group-10 metal supported on polycrystalline Zn (0z, ~ 1 ML). Part B: Difference between the d populations ofa TM adatom in TM3/Znlo and TM~frMlo clusters (from ab initio SCF calculations). Part C: Desorption temperature for a Zn monolayer from Ni, Pd and Pt surfaces. Part D: Decrease in the CO desorption temperature of CO from Ni, Pd and Pt atoms bonded to zinc (from Ref. 1"203]).

260 J.A. Rodriouez/Surface Science Reports 24 (1996) 223-287
l~ l ' a . -150 K I +1.30 eV b , \ \ \ \ \ \ ~ \ \ \ \ \ \ \ \ \ ' q
i~Zn, -110 K [ +0.80 eV Ib,NNNN\NNNN'q
It /W. -90 K I +0.60 eV ~\\\ \ \NN
PUMo, -120 K [
m ' ~ , -50 K ] +0.3S eV ~\ \ 'q
NI/Mo, -30 K +0.25 eV
I + 50 K, Ni~[~u +O.OS eV
I +2S K, CutRe +0.02 eV
~ +30 K, CufRu • 4).13 eV
~ . _ . J +70 K, Cu/Pt -0.27 eV
I +70 K, Cu/Rh k\\\\X~l -0.43 eV |
Fig. 34. Correlation between shifts in surface core-level binding energy (crossed bars) and shifts in CO desorption temperature (empty bars). The properties of the Pt, Ni and Cu monolayers are compared with the corresponding values for the pure metals (from Refs. [132,152]).
involves electron transfer from the CO(5a) orbital into the empty bands of the metal a donation, and electron transfer from the occupied bands of the metal into the CO(2~r*) orbitals, ~ backdonation [249,250]. From a thermochemical viewpoint, ~ the backdonation is energetically more important than the ~r donation [250]. For metals, the valence d orbitals are much better for bonding interactions with CO than the s,p orbitals [250b,251]. In the formalism of crystal-orbital perturba- tion theory [252], the heat of adsorption of CO on a metal atom, Qco, should be roughly proportional to the occupancy of the metal d band, N d, and inversely proportional to the separation between the centroid of the metal d band and the CO(27c*) orbitals, namely
Qco oc Na/(Eco~z,o- EMma)).
The electron-density plots in Fig. 22 for the Pd/Ta (1 10) system [ 197] show a clear movement of electrons from the top of Pd toward the metal-metal "interface". A similar shift of electrons is observed in first-principles local-density functional calculations for Pd/W (1 10), Pd/Re (0 0 01 ) and Pd/Ru(0001) [197]. These electrons are not available for 7r-backdonation toward CO. In general, the interaction between Pd and a metal substrate moves the Pd (4d) orbitals toward higher binding energy (away from the CO(2~*) orbitals [253]), and reduces the electron population of these d orbitals by a Pd(4d)-, Pd(Ss,Sp)rehybridization [152,168]. The combination of these phenomena leads to a reduction in the Pd(4d).-~CO(2~*) bonding interactions. The weakening of the Pt-CO and Ni-CO bonds in Figs. 33 and 34 can be explained ina similar way [152,199,203,252c,254]. On the other hand, for Cu atoms in contact with late-transition metals, an increase in the Cu(3d) population causes a shift in the Cu 3d band toward lower binding energy and an enhancement in the Cu(3d)~CO(21c*) bonding interactions [214]. The very good correlations found between the

J.A. Rodriouez/Surface Science Reports 24 (1996) 223-28 7 261
changes in the core levels of an admetal and variations in its ability to adsorb CO arise from the fact that both properties are very sensitive to perturbations in the valence d levels of the admetal [132,152,214,252c].
In most of the studies discussed above the changes in the ability of a metal to adsorb CO are mainly a consequence of the electronic interactions within the bimetallic bond, while variations in the structure of the surface play only a secondary role. For example, a pseudomorphic monolayer of Pd on Re (0 0 01) has an atomic density that is almost identical to that of Pd (111), but the electronic and chemical properties of the Pd atoms in these systems are very different [161]. For a Pd monolayer on Mo(100) the strain is larger than on Ta(1 10) or Nb(1 10) [169b]. Nevertheless, the reduction in the CO desorption temperature is smaller on Pd/Mo(100) than on Pd/Ta(1 10) or Pd/Nb(l 10) [169b]. These studies highlight the importance of the Pd-substrate electronic interactions. On the other hand, for the interaction of CO with (x/~ x x/~)R30°-Sn/Ni (1 1 1) and (x/~ x x/~)R30°-Sn/Pt(1 1 1) surface alloys, variations in the geometrical structure of the surface determine the reactivity of the Group-10 metal toward CO [261]. For the Sn-Ni alloy, the electronic perturbations on Ni are negligible, but there is a large decrease ( ~ 15 kcal/mol) in the CO adsorption energy as a result of repulsive Sn-CO interactions at the Ni-CO distance required for chemisorp- tion of the molecule [261]. The differences in the CO chemisorption properties of the Sn-Ni and Sn-Pt alloys can be rationalized by considering the different sizes of the surface unit cells and the location of Sn with respect to the surface plane (i.e. the Sn buckling distance) [261].
The C-O stretch frequency in the IR spectrum is very sensitive to changes in the morphology of a metal ovedayer [255-259]. Disorder-order transitions of Ni, Cu and Co films on several substrates have been monitored with CO chemisorption and IR spectroscopy [255-259]. For a Ni monolayer supported on Mo (1 10), a change in the morphology of the adlayer from disordered to an ordered (7 x 2) structure is accompanied by a reduction of ~ 30 cm- 1 in the stretching frequency of adsorbed CO [256]. A similar phenomenon has been observed for Cu overlayers on Ru (0001) [257], Rh(100) [259] and Pt(1 1 1) [255]. The vibrational frequency of CO adsorbed on small Cu clusters is much higher (25-40cm-1) than that of CO bonded to well-ordered 2D islands of Cu, which are pseudomorphic to the metal substrate. One possible cause for this phenomenon is an increase in the packing density of the metal adatoms during the phase transition [255,260].
4.2. NO chemisorption
NO is isoelectronic with CO-, and the additional electron in the antibonding 27r* orbital alters the adsorption behavior of NO compared to CO !234a]. This is illustrated by the interaction of NO with a (x/~ x x/~)R30°-Sn/Pt (1 1 1) surface alloy [234]. In this bimetallic system the presence of Sn induces only a small reduction (1-4 kcal/mol) on the CO adsorption energy over Pt [234,262]. In contrast, the adsorption energy of NO is reduced by 8-10 kcal/mol compared to Pt(1 1 1), with the sticking coefficient of the molecule decreasing from 0.9 on the pure Pt single-crystal to 0.6 on the (x//3 x ~/3)R30°-Sn/Pt(l 1 1) surface alloy [234a].
Studies for the interaction of NO with Pt-Rh (100) surface alloys [234c] show dissociation of the molecule. The adsorbed N atoms have equal affinity for the Pt and Rh atoms present in the surface. On the other hand, adsorbed oxygen makes a stronger bond with the Rh atoms so that Rh atom segregation onto the surface is included [234c].

262 J.A. Rodriouez/Surface Science Reports 24 (1996) 223-287
4.3. Chemisorption of ethylene, acetylene and benzene
Monolayers of Au and Cu supported on Ru(0001) exhibit C2H¢ adsorption energies that are larger than those of surfaces of pure Au and Cu [263]. Thus whereas pure Au is inactive as an adsorbent, it wifi ~r-adsorb ethylene when activated by Ru [263]. At 130 K, ethylene adsorbs molecularly on clean and Cu-covered Ru (0001) surfaces [263]. Results of HREELS indicate that on Cu < 1 .o /Ru (00 01) , ethylene is ~r-bonded to the Cu atoms, but all-C-bonded to the Ru sites [263]. The ethylene attached to Ru dissociates upon heating to 230 K, whereas no decomposition and desorption is observed for the Cu-bonded C2H4 [263,290].
Ethylene is reversible adsorbed on p(2 × 2)-Sn/Pt(1 1 1)and (~/~ x ~//3)R30°-Sn/Pt (1 1 1) surface alloys [262b]. As the Sn concentration in the surface alloy is increased, there is a marked decrease in the ethylene desorption temperature from 285 K on Pt (1 1 1) to 240 K on the p(2 × 2) alloy, and to 184 K on the (V/3 × ~/3)R30 ° alloy [262b]. This results from a significant electronic effect induced by Sn on the Pt-ethylene chemisorption bond strength [262b]. Electronic perturbations also reduce the adsorption energy of ethylene on a Pd monolayer supported on Me(100) [235]. Ethylene is weakly chemisorbed on the Pd monolayer (desorption temperature ~ 250 K against ~ 290 K on pure Pal), and the adsorbed species is much less rehybridized from sp 2 in the gas phase toward sp 3 o n
this surface compared to C e l l a chemisorbed on the (100) face of pure Pd [235]. On Pd/Mo (10 0), acetylene chemisorption is not affected like C2H4 chemisorption, and C2H2 is
strongly rehybridized from sp in the gas phase toward sp 3 o n the Pd monolayer as it is on bulk Pd (10 0) [235]. The C2H 2-Pd interaction is probably strong enough to rehybridize the Pd atoms in the supported monolayer back toward their normal bulk electronic structure in order to have the properties necessary for a large heat of adsorption [235]. A pseudomorphic monolayer of Pd on Me(100) has a structural configuration that disfavors the formation of benzene from acetylene ['235]. On the other hand, Pd overlayers on Au (1 1 1) efficiently catalyze the trimerization of C2H2 to C~H~ [74b,264"!. The extent of acetylene conversion and the binding energy of the resulting benzene depend on the atomic composition, morphology and electronic properties of the surface [162c,264]. The presence of Sn atoms at the surface of p(2 × 2)- and (~/~ × ~/~)R30°-Sn/Pt (1 1 1) surface alloys stron0y suppresses the decomposition of acetylene (C2D2) to deuterium and adsorbed carbon, opening an effective reaction pathway for the formation of benzene [265].
Biand Sn adatoms effectively block the chemisorption ofbenzene on Pt (1 1 1) [266,267], inducing a substantial reduction in the desorption temperature of this aromatic molecule. When benzene is coadsorbed with Bi or Sn adatoms, the competition between dehydrogenation and molecular desorption is strongly influenced [266,267]. Dehydrogenation is almost completely suppressed by 0Bi ~ 0.15 ML, with a corresponding increase in molecular desorption [266]. Since the activation energies for desorption and dehydrogenation are not strongly influenced at such low Bi coverages, this result can be attributed to the steric blocking by Bi of free Pt sites needed for the dehydrogena- tion of benzene [266].
4.4. Chemisorption of"large" hydrocarbons
Platinum is a well-known catalyst for hydrocarbon reforming. This fact has motivated several works examining the interaction of n-butane [268], isobutane [268], cyclopentane [269], cyc-
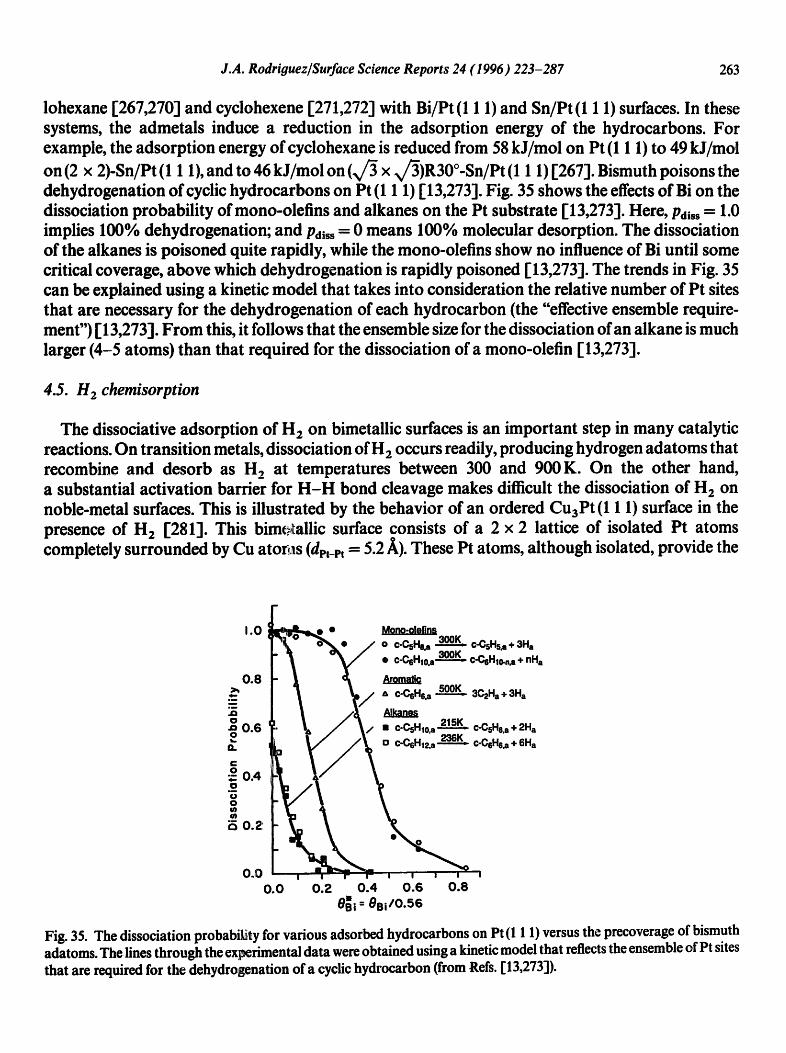
J.A. Rodriguez/Surface Science Reports 24 (1996) 223-287 263
lohexane [267,270] and cyclohexene [271,272] with Bi/Pt (1 1 1) and Sn/Pt (1 1 1) surfaces. In these systems, the admetals induce a reduction in the adsorption energy of the hydrocarbons. For example, the adsorption energy of cyclohexane is reduced from 58 kJ/mol on Pt (1 1 1) to 49 kJ/mol on (2 x 2)-Sn/Pt (1 1 1), and to 46 kJ/mol on (x/~ x x/~)R30°-Sn/Pt (1 1 1) [267]. Bismuth poisons the dehydrogenation of cyclic hydrocarbons on Pt (1 1 1) [13,273]. Fig. 35 shows the effects of Bi on the dissociation probability of mono-olefins and alkanes on the Pt substrate [13,273]. Here, Pdi, = 1.0 implies 100% dehydrogenation; and Pdi, = 0 means 100% molecular desorption. The dissociation of the alkanes is poisoned quite rapidly, while the mono-olefins show no influence of Bi until some critical coverage, above which dehydrogenation is rapidly poisoned [13,273]. The trends in Fig. 35 can be explained using a kinetic model that takes into consideration the relative number of Pt sites that are necessary for the dehydrogenation of each hydrocarbon (the "effective ensemble require- ment") [13,273]. From this, it follows that the ensemble size for the dissociation of an alkane is much larger (4-5 atoms) than that required for the dissociation of a mono-olefin [13,273].
4.5. H 2 chemisorption
The dissociative adsorption of H 2 on bimetallic surfaces is an important step in many catalytic reactions. On transition metals, dissociation of H2 occurs readily, producing hydrogen adatoms that recombine and desorb as H 2 at temperatures between 300 and 900K. On the other hand, a substantial activation barrier for H - H bond cleavage makes difficult the dissociation of H2 on noble-metal surfaces. This is illustrated by the behavior of an ordered Cu3Pt(1 1 1) surface in the presence of H 2 [281]. This bim¢~,~allic surface consists of a 2 x 2 lattice of isolated Pt atoms completely surrounded by Cu ator,~s (dpt-pt - 5.2 J~). These Pt atoms, although isolated, provide the
1.0 • • Mono.olefins o • o c-CsI'Is~ 30OK. c-CsHs~ + 3Ha
• c-C-,sHI0.a 30OK. c-CsHIo.,.a+ nHa
~:~ 0 . 8 Aromatic A c'CsHs.a 500K, 3C2H a + 3Ha
~0.6 Alkanes • c-CsI'Ilo,a 215K- c-CsHs.a + 2Ha
n o c'CsHI2.a 236K. c.Csl.ls ~ + 6Ha
~ 0.4
i5 O.2:
0.,0 0.0 0.2 0.4 0.6 0.8
or3~ = Oe i /o .ss
Fig. 35. The dissociation probabiEty for various adsorbed hydrocarbons on Pt (1 1 1) versus the precoverage of bismuth adatoms. The lines through the experimental data were obtained using a kinetic model that reflects the ensemble of Pt sites that are required for the dehydrogenation of a cyclic hydrocarbon (from Refs. [13,273]).

264 3.,4. Rodriguez/Surface Science Reports 24 (1996) 223-287
only reaction channel available for the dissociative adsorption of H 2 on the CuaPt(1 1 1) surface [281]. Works examining the adsorption of H2 on Ru(0001) and Re(0001) surfaces precovered with submonolayer coverages of Cu show a "spiUover" of hydrogen atoms, Ha, from the transition metals to the noble metal [285,286,290]. In these systems, Cu acts as a "sink" for atoms produced by the dissociation of H2 on Ru or Re [285,286,290]. This phenomenon has important implications in the use of H2 adsorption to selectively quantify the amount of transition-metal sites on the surface of {Cu + transition metal} bimetallic catalysts [285].
Bimetallic bonding can produce interesting changes in the reactivity of a metal toward hydrogen [13,154,170,262,274-293]. Experimental evidence indicates that hydrogen dissociatively chemisorbs to Pd/Nb(1 10) at ~ 300 K when 01,d > 1 ML, but no (or little) hydrogen adsorbs when 0pd = 1 ML [154]. A similar behavior is observed for the interaction o f H 2 with Pd/Ta (1 10) [287], Pd/Mo (100) [288] and Fe/Rh (100) [282]. In these systems, the supported monolayers of Pd and Fe exhibit positive binding-energy shifts in their d-bands with a large reduction in the density of states around the Fermi level (for example, see Fig. 17) [154,282,287]. These modifications in the band structure lead to a decrease in the reactivity of Pd and Fe toward H 2 (i.e. "noble metal-like behavior") [154,282,287,291a]. Electronic perturbations also induce a significant weakening in the strength of the Pt-H bond on PtaoFe2o (1 1 1)and PtsoNiso(1 1 1)surfaces [274,277,291b,294].
4.6. Interaction with oxygen
Surface segregation of one metal due to oxygen chemisorption or oxidation is a common phenomenon in bimetallic alloys [13]. In general, the metal enriched at the surface is that which forms the most stable oxide or Oa species [13]. For example, the oxidation of nickel-aluminum alloys leads to the formation of ultrathin films ofaluminum oxide [295-299]. Highly ordered films of 7-A120 3 can be grown epitaxially on NiAI (1 10) by adsorbing oxygen on this bimetallic surface [297-299].
Adsorbed atomic oxygen induces segregation of Cu on W (1 10) [300] and Rh (10 0) [301], Ni on W(I 10)[302], Au on Mo (1 10) [303] and Ru (0 0 01) [ 56,304], and Ag on Ru (0 0 01) [305,306]. On these surfaces there is a large O-induced weakening in the strength of the bimetallic bonds, and islands of Oa coexist with three-dimensional clusters of the admetal. For the coadsorption of submonolayer coverages of O and N i on W (1 10) [ 302] or O and Zn on R u (0 0 01 ) [307] there is no evidence for the formation of NiOx or ZnOx on the surface, in spite of very exothermic heats of formation for the bulk oxides of the admetals. Theoretical calculations for the { Zn + O)/Ru (0 0 01) system show a strong hybridization of the Zn(4s,4p) or O(2s,2p) orbitals that maximizes the strength of the Ru~Zn or Ru,-,O interactions and prevents any significant lateral bonding between Zna and
[307]. Fig. 36 shows Ag-TDS spectra acquired after depositing this noble metal on Ru(0001),
O/Ru (00 01) and 0t-Al20~ (1 12 0)surfaces [305]. For Ag/Ru (0 0 01), one can see that the first layer peak (above 1000 K) saturates first, and is then followed by growth of the second layer peak and eventually by the multilayer (bulk-like) desorption peak [305]. Thus, there is clear wetting of the Ru surface by the Ag atoms. For the oxygen-modified Ru surface (Fig. 36(b)), the TDS data do not indicate any high-temperature feature characteristic for desorption of the first layer in direct contact with Ru. In these cases, the Ag desorption spectra show highly asymmetric peaks with common leading edges that are governed by zero-order desorption kinetics [305,306]. This implies that Ag
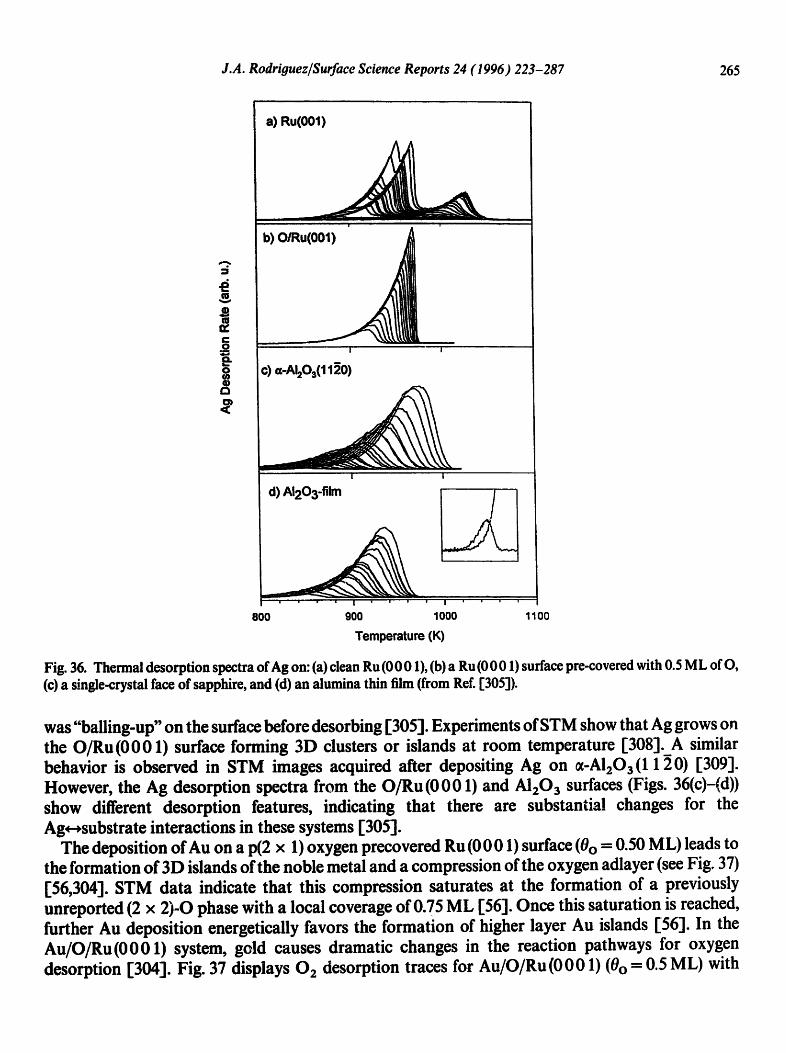
3.A. Rodriguez/Surface Science Reports 24 (1996) 223-287 265
A
¢1
m
t IX:
t -
.o_ E" o o a
a) Ru(O01)
b) O/Ru(O0
I I
c) a-A~03(11~.O)
I !
d) AI203-film
800 g00
LZ ' I ' ' ' i
1000 1100
Temperature (K)
Fig. 36. Thermal desorption spectra of Ag on: (a) clean Ru (00 01), (b) a Ru (0 0 01) surface pre-covered with 0.5 ML of O, (c) a single-crystal face of sapphire, and (d) an alumina thin film (from Ref. 1"305"1).
was"bailing-up" on the surface before desorbing [305]. Experiments of STM show that Ag grows on the O/Ru(0001) surface forming 3D clusters or islands at room temperature [308]_A similar behavior is observed in STM images acquired after depositing Ag on 0c-A1203(1 120) [309]. However, the Ag desorption spectra from the O/Ru(0001) and AI20 3 surfaces (Figs. 36(c)-(d)) show different desorption features, indicating that there are substantial changes for the Ag,-,substrate interactions in these systems [305].
The deposition of Au on a p(2 × 1)oxygen precovered Ru (0001) surface (0o = 0.50 ML) leads to the formation of 3D islands of the noble metal and a compression of the oxygen adlayer (see Fig. 37) [56,304]. STM data indicate that this compression saturates at the formation of a previously unreported (2 × 2)-0 phase with a local coverage of 0.75 ML [56]. Once this saturation is reached, further Au deposition energetically favors the formation of higher layer Au islands [56]. In the Au/O/Ru(0001) system, gold causes dramatic changes in the reaction pathways for oxygen desorption [304"1. Fig. 37 displays 02 desorption traces for Au/O/Ru(0001) (0o = 0.5 ML) with
266 3.A. Rodriguez/Surface Science Reports 24 (1996) 223-287
14500
A
== 95OO
i "- 4500 E
900 1000 1100 1200 1300 1400 1500 TEMPERATURE ( K )
Fig. 37. Left panel: (a) STM topograph of a 0.1 ML Au deposition at room temperature on Ru(0001) precovered with a 0.5 ML p(2 × 1) oxygen adlayer (flux rate 0.15 ML/min, 850 ~ × 850 ~). (b) STM topograph of the same film after annealing to 600 K (500 ~ × 500 ~). Right panel: 02 TDS traces (m/e--- 32) from Ru(0001) saturated with oxygen and varying amounts ofcoadsorbed Au (in ML): (a) 0, (b) 0.67, (c) 1.3, (d) 2.0, and (e) 4.0. Inset: total 0 2 TDS area as a function of 0^u (from Refs. 1"56] and 1"304]).
0Au as a parameter [304]. As the Au coverage increases, one can see a big reduction in the 02 desorption temperature and an enhancement in the amount of oxygen that desorbs from the surface. The compression of the O adlayer by Au reduces the adsorption energy of oxygen on Ru(0001), favoring the "pairing" of oxygen adatoms and desorption of 02, and preventing in this way the diffusion of O into the bulk of Ru [304].
4.7. Interaction with sulfur
The understanding of the interaction of sulfur with bimetallic surfaces is an important issue in several areas of heterogeneous catalysis. Catalysts that combine noble and late-transition metals are very sensitive to sulfur poisoning [310,311]. On the otherhand, Mo- and W-based bimetallic catalysts are frequently used for hydrodesulfurization (HDS) processes in oil refineries [312,313]. A good knowledge of the factors that control the interaction between sulfur and a bimetallic system can have a significant impact in the design of new catalysts. This fact has motivated many studies investigating the adsorption of sulfur on well-defined bimetallic surfaces prepared by the deposition of a metal (Co, Ni, Cu, Ag, Au or Zn) onto a single-crystal face of another metal (Mo, Ru, Pt, W or Re) 1'31~328].
These studies have allowed a clear identification of several phenomena that can occur when sulfur interacts with a bimetallic surface. For some systems [317,320,322,325], one can observe the

J.A. Rodriguez/Surface Science Reports 24 (1996) 223-287 267
formation of bimetallic sulfides that exhibit chemical properties very different from those of the pure metals. In another type of systems E316,318,326,328], the interaction between sulfur and one of the metals is repulsive, with sulfur inducing a weakening of the bimetallic bonds and reducing the "mixing" of the metals. And finally, one can have bimetallic systems in which one of the metals increases or promotes the reactivity of the other toward sulfur E319-321,323,324,327]. In some situations, this phenomenon accelerates the poisoning of catalysts [319,321,328], while in anothers, the effect is beneficial enhancing the activity of catalysts for HDS processes [323,327].
Works examining the coadsorption of sulfur and gold on Mo(100) [318], Mo(1 1 O) E326], Ru(O001) [316,329], Rh(1 1 1) [326] and Pt(1 1 1) [328] show strong repulsive interactions between these adsorbates. Sulfur reduces the adsorption energy of Au. In some cases, the weakening of the gold-transition metal bonds is so large (5-7 kcal/mol) that the Au adatoms "ball up" on the surface instead of "wetting" the metal substrates. A S adatom modifies the chemical behavior of adjacent atoms in a transition-metal substrate by reducing their contribution to the density of states around the Fermi level of the system [326,330-332]. S and Au compete for making bonds with the metal substrates [326,328]. To minimize the effects of this competition they move apart on the surface, forming 2D islands of S with a high local coverage and 2D or 3D islands of Au [318,329]. In the Au/S/Ru (0 0 01) and Au/S/Rh (1 1 1) systems E316, 326], $2 desorbs at much lower temperatures (100-250K) than in S/Ru(O001) and S/Rh(1 1 1). For Au/S/Mo(1 1 O) E326], the presence of Au favors desorption of S 2 instead of migration of S into the bulk of the sample. These results are consistent with a "model" in which Au compresses the S overlayer into islands ofhigh local coverage, reducing in this way its stability and favoring the "pairing" of sulfur atoms and desorption of $2 [316,326].
Fig. 38 displays XPS spectra acquired after dosing $2 to a series of bimetallic surfaces. In the left panel, the Pt 4f core levels are shown for X/Pt(1 1 1) and clean Pt (1 1 1) exposed to $2 at 550 K [319,321]. The right panel shows Mo 3d core levels for X/Mo (1 1 O) systems and clean Mo(1 1 O) exposed to $2 at 700 K [320,323,324,327]. The exposure of Pt (1 1 1) and Mo (1 1 O) to large amounts of $2 produces only a chemisorbed layer of sulfur, without forming bulklike sulfides which are thermodynamically very stable (PtS2, AGf = - 24 kcal/mol; MoS2, AGf = - 54kcal/mol [333]). This is due to the high cohesive energies of the Pt(1 1 1) and Mo(1 1 O) substrates and the large difference in surface free energies between sulfur (0.08 J m- 2 [334]) and the substrates (2.69 J m- 2 for Pt and 2.88 J m- 2 for Mo [334]), both of which prevent sulfur migration into the bulk of Pt or Mo [320,321 ]. The addition of a second metal can promote the sulfidation of Pt (1 1 l)and M o (1 10), see Fig. 38, by overcoming the barrier for S penetration into the bulk E320,321]. Only in bimetallic systems in which the admetal forms sulfides that are less stable than those formed by the substrate can one see admetal-promoted sulfidation of a metal substrate [327]. Once that this condition is satisfied, the magnitude of the "promotional effect" depends on several factors E320,323,327]. An admetal can promote the formation of molybdenum and platinum sulfides by changing the structure of the surface (making it easier for the penetration of S into the bulk of the sample), or by modifying the electronic structure of Mo and Pt (favoring in this way the formation of Mo-S and Pt-S bonds) [320,323,327].
Fig. 39 compares trends seen in the HDS activity of a series of XSy/MoS2 catalysts (X = Zn, Cu, Fe, Co or Ni) E335] with trends found for the sulfidation of Mo in X/Mo (1 1 O) surfaces E320,323,324, 327]. In general, a good correlation is observed between the changes in the two properties The presence of Ni leads to a significant enhancement in the MowS interactions and very large HDS
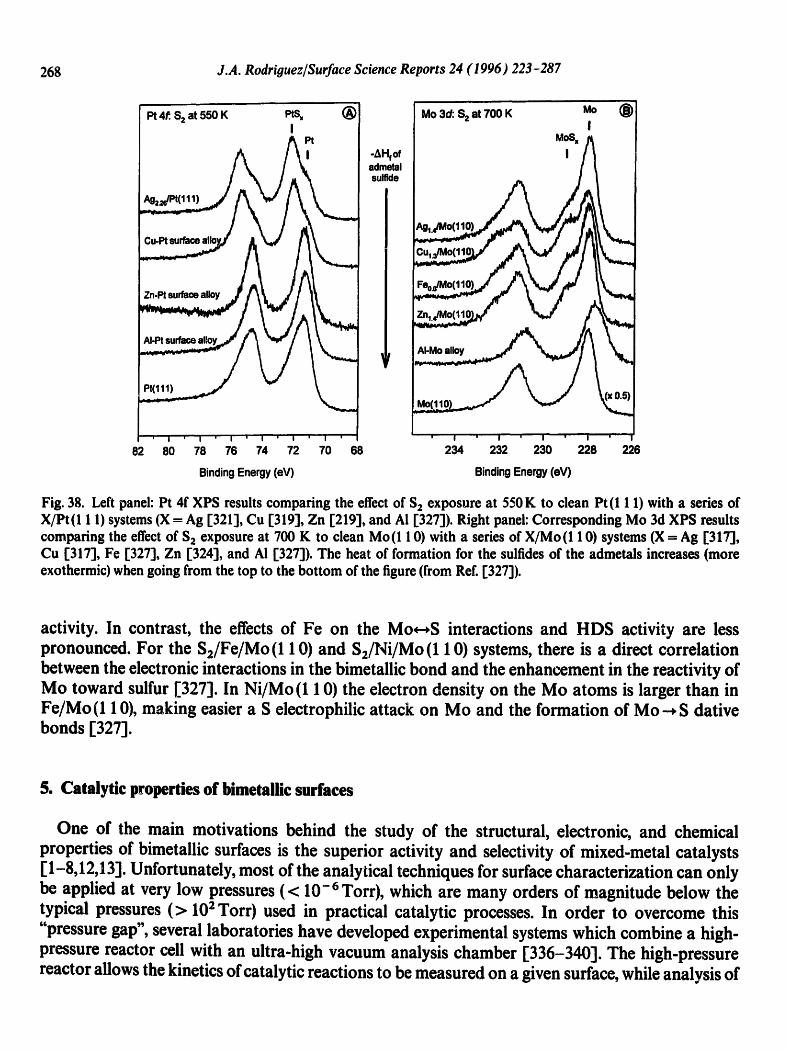
268 J.A. Rodriguez/Surface Science Reports 24 (1996) 223-287
Pt 4~ S z at 550 K PtS, (~ l I I
-AHfof admetal sulfide
Mo 3d: B 2 at 700 K Mo (~)1 I I
82 80 78 76 74 72 70 68 234 232 230 228 226
Binding Energy (eV) Binding Energy (eV)
Fig. 38. Left panel: Pt 4f XPS results comparing the effect of Sz exposure at 550K to clean Pt(l 1 1) with a series of X/Pt(I 1 1) systems (X = Ag [321,1, Cu [319,1, Zn [219,1, and AI [327,1). Right panel: Corresponding Mo 3d XPS results comparing the effect of S 2 exposure at 700 K to clean Mo(l 10) with a series of X/Mo(1 10) systems (X = Ag [317,1, Cu [317,1, Fe [327"1, Zn [324,1, and A! [327,1). The heat of formation for the sulfides of the admetals increases (more exothermic) when going from the top to the bottom of the figure (from Ref. [327,1).
activity. In contrast, the effects of Fe on the Mo,-,S interactions and HDS activity are less pronounced. For the Sz/Fe/Mo(1 10) and S2/Ni/Mo(1 10) systems, there is a direct correlation between the electronic interactions in the bimetallic bond and the enhancement in the reactivity of Mo toward sulfur [327]. In Ni/Mo(1 10) the electron density on the Mo atoms is larger than in Fe/Mo(1 10), making easier a S electrophilic attack on Mo and the formation of Mo--, S dative bonds [327].
5. Catalytic properties of bimetallic surfaces
One of the main motivations behind the study of the structural, electronic, and chemical properties of bimetallic surfaces is the superior activity and selectivity of mixed-metal catalysts [1-8,12,13"1. Unfortunately, most of the analytical techniques for surface characterization can only be applied at very low pressures (< 10-6Torr), which are many orders of magnitude below the typical pressures (> 102Torr) used in practical catalytic processes. In order to overcome this "pressure gap", several laboratories have developed experimental systems which combine a high- pressure reactor cell with an ultra-high vacuum analysis chamber [336-340]. The high-pressure reactor allows the kinetics of catalytic reactions to be measured on a given surface, while analysis of
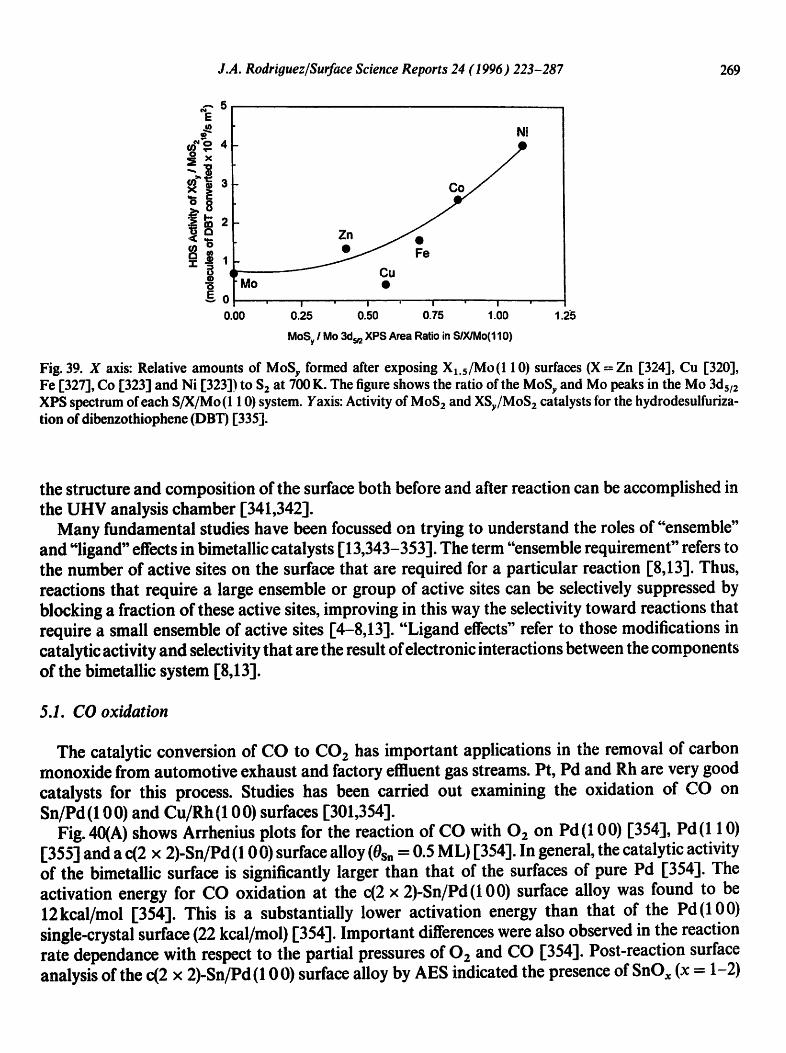
J.A. Rodriguez/Surface Science Reports 24 (1996) 223-287 269
~ 5
N[
L Zn / e F.
~ 4 o x
× = > 3 15=
0.00 0.25 0.50 0.75 1.00 1.25
MoS v I Mo 3din XPS Area Ratio in SIX/Mo(110)
Fig. 39. X axis: Relative amounts of MoSy formed after exposing Xa.s/Mo(l 10) surfaces (X = Zn [324], Cu [320], Fe [327], Co [323] and Ni [3231) to Sz at 700 K. The figure shows the ratio of the MoSy and Mo peaks in the Mo 3ds/z XPS spectrum of each S/X/Mo0 10) system. Yaxis: Activity of MoS2 and XSy/MoS z catalysts for the hydrodesulfuriza- tion of dibenzothiophene (DBT) [335].
the structure and composition of the surface both before and after reaction can be accomplished in the UHV analysis chamber [341,342].
Many fundamental studies have been focussed on trying to understand the roles of "ensemble" and "ligand" effects in bimetallic catalysts [ 13,343- 353]. The term "ensemble requirement" refers to the number of active sites on the surface that are required for a particular reaction [8,13]. Thus, reactions that require a large ensemble or group of active sites can be selectively suppressed by blocking a fraction of these active sites, improving in this way the selectivity toward reactions that require a small ensemble of active sites [4-8,13]. "Ligand effects" refer to those modifications in catalytic activity and selectivity that are the result of electronic interactions between the components of the bimetallic system [8,13].
5.1. CO oxidation
The catalytic conversion of CO to CO2 has important applications in the removal of carbon monoxide from automotive exhaust and factory effluent gas streams. Pt, Pd and Rh are very good catalysts for this process. Studies has been carried out examining the oxidation of CO on Sn/Pd(100) and Cu/Rh(100) surfaces [301,354].
Fig. 40(A) shows Arrhenius plots for the reaction of CO with O, on Pd(100) [354], Pal(1 10) [355] and a c(2 x 2)-Sn/Pd (100) surface alloy (0s, = 0.5 ML) [354]. In general, the catalytic activity of the bimetallic surface is significantly larger than that of the surfaces of pure Pd [354]. The activation energy for CO oxidation at the c(2 x 2)-Sn/Pd(100) surface alloy was found to be 12kcal/mol [354]. This is a substantially lower activation energy than that of the Pal(100) single-crystal surface (22 kcal/mol) [354]. Important differences were also observed in the reaction rate dependance with respect to the partial pressures of 02 and CO [354]. Post-reaction surface analysis of the c(2 x 2)-Sn/Pd (10 0) surface alloy by AES indicated the presence of SnOx (x = 1-2)
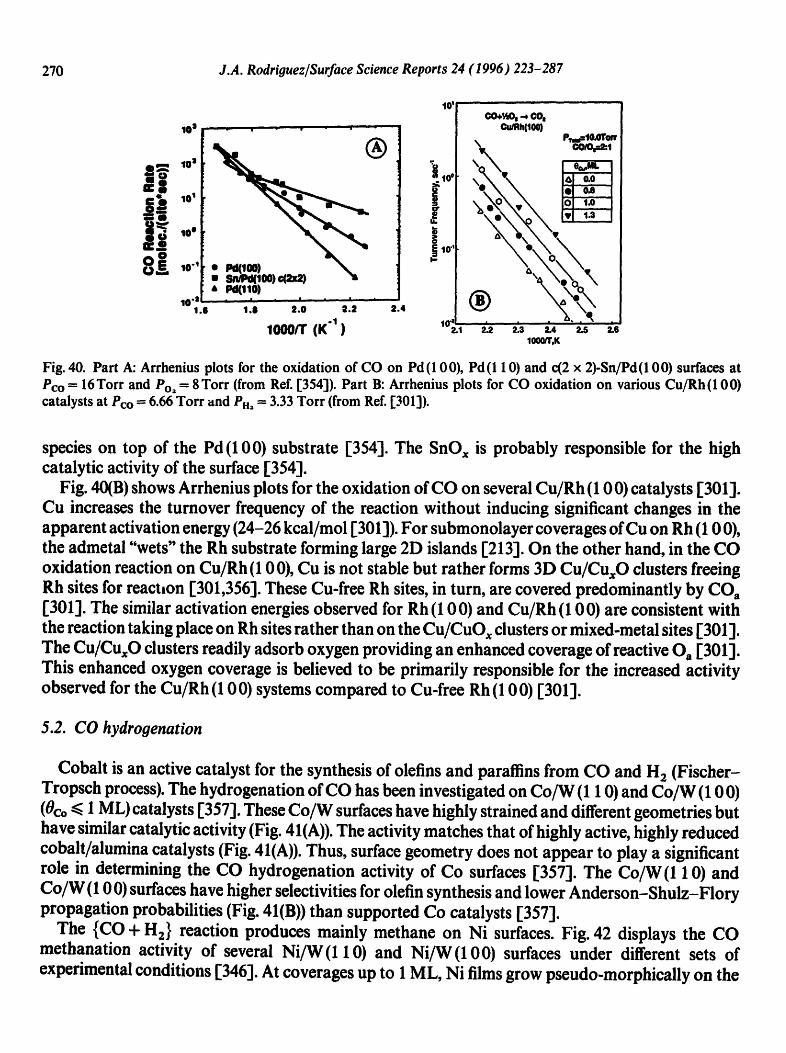
270 J.A. Rodriguez/Surface Science Reports 24 (1996) 223-287
101 ,,~ ~, ,, .
j ~ 10 ~
, ° '
J---- l O e
ol,o.,
®
• P d ( l m • SeOd(100) c(2x2) • p ~ ( . o )
10 .1 , • , , I 1 . 8 1.11 2 . 0 2 . 2 2 . 4
IO00/T (K "1 )
t 0 !
j 0
IE IO "1
107..1
Ou/Rh(lOO) P . r ~ l O . O l " o n ,
I o=,,.U I- I -g % - - " , , ~ \ i . i o.o i
\ ~ \ . \ Iol t.o 1
-\-',"o
I I • 2.2 2.3 2.4 2.5 2.6
1000/T,K
Fig. 40. Part A: Arrhenius plots for the oxidation of CO on Pd(100), Pd(l 10) and c(2 × 2)-Sn/Pd(100) surfaces at Pco = 16Torr and Po, = 8 Torr (from Ref. ['354]). Part B: Arrhenius plots for CO oxidation on various Cu/Rh(100) catalysts at Pco = 6.66 Torr and PH, -- 3.33 Torr (from Ref. [:301]).
species on top of the Pd(100) substrate [354]. The SnO~ is probably responsible for the high catalytic activity of the surface [354].
Fig. 40(B) shows Arrhenius plots for the oxidation of CO on several Cu/Rh (100) catalysts [301]. Cu increases the turnover frequency of the reaction without inducing significant changes in the apparent activation energy (24-26 kcal/mol [301 ]). For submonolayer coverages of Cu on Rh (10 0), the admetal "wets" the Rh substrate forming large 2D islands [213]. On the other hand, in the CO oxidation reaction on Cu/Rh (10 0), Cu is not stable but rather forms 3D Cu/CuxO clusters freeing Rh sites for react,on [:301,356]. These Cu-free Rh sites, in turn, are covered predominantly by COa [301]. The similar activation energies observed for Rh (! 00) and Cu/Rh (100) are consistent with the reaction taking place on Rh sites rather than on the Cu/CuO~ clusters or mixed-metal sites [301]. The Cu/Cu~O clusters readily adsorb oxygen providing an enhanced coverage of reactive Oa [301]. This enhanced oxygen coverage is believed to be primarily responsible for the increased activity observed for the Cu/Rh (100) systems compared to Cu-free Rh (100) [301:].
5.2. CO hydrogenation
Cobalt is an active catalyst for the synthesis of olefins and paraffins from CO and H~ (Fischer- Tropsch process). The hydrogenation of CO has been investigated on Co/W (1 10) and Co/W (10 0) (0co ~< 1 ML) catalysts [:357]. These Co/W surfaces have highly strained and different geometries but have similar catalytic activity (Fig. 41(A)). The activity matches that of highly active, highly reduced cobalt/alumina catalysts (Fig. 41(A)). Thus, surface geometry does not appear to play a significant role in determining the CO hydrogenation activity of Co surfaces [357]. The Co/W (1 10) and Co/W (10 0)surfaces have higher selectivities for olefin synthesis and lower Anderson-Shulz-Flory propagation probabilities (Fig. 41(B)) than supported Co catalysts [357].
The {CO + H2} reaction produces mainly methane on Ni surfaces. Fig. 42 displays the CO methanation activity of several Ni/W(1 10) and Ni/W(100) surfaces under different sets of experimental conditions [346]. At coverages up to 1 ML, Ni films grow pseudo-morphically on the
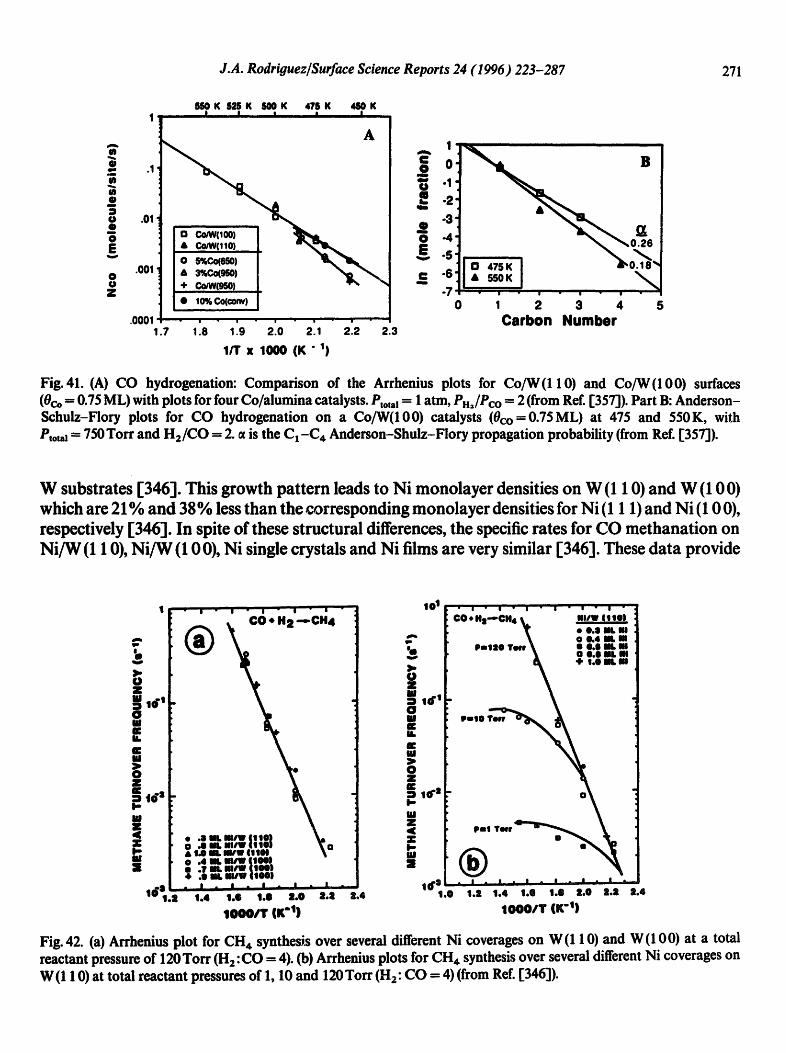
J.A. Rodriguez/Surface Science Reports 24 (1996) 223-287 271
A
m o
4 m m
m o
m : l 0
0 E
0 o Z
S60K S2SK 600K 47S K dJSOK I I I I I
A 1
.o~ •
o - .ool • ~ . o ) - ~ ~ _=
+ c ~ , w ( s s o ) . 7 ~
• :o~cc~conv) 0 ICarb: u3m / s .OOOl n N be
~.7 lls ~'., 2:o 21, ~12 2.3 1/T X 1000 (K " 1)
Fig.41. (A) CO hydrogenation: Comparison of the Arrhenius plots for Co/W(I 10) and Co/W(100) surfaces (0co = 0.75 ML) with plots for four Co/alumina catalysts./)tot,, = I arm, Ps,/Pco = 2 (from Ref. [357]). Part B: Anderson- Schulz-Flory plots for CO hydrogenation on a Co/W000) catalysts (0co=0.75ML) at 475 and 550K, with Ptot,l = 750Torr and H=/CO = 2. • is the C~-C4 Anderson-Shulz-Flory propagation probability (from Ref. [357]).
W substrates [346]. This growth pattern leads to Ni monolayer densities on W (1 10) and W (100) which are 21% and 38% less than the corresponding monolayer densities for Ni (1 1 1) and Ni (10 0), respectively [346]. In spite of these structural differences, the specific rates for CO methanation on Ni/W (1 10), Ni/W (100), Ni single crystals and Ni films are very similar [346]. These data provide
1 ~ - , - , - , - , - . " " 1 0 1
| i " Z
o J I l L m ~ I 11101 o
f o .4 I . mew Isooj i I I • .? i l l . m e w I l l l )
/ • . I m , ~ I l lO l [ t . , • , , . t 1C~
s o s . l s.4 s.s s.o z.o I . z z.4 1
IO00 /T (K "~)
T • e . 8 i t m ~ i ~ o o .4 I I . m
Pmll tO T~rr I I O.IJ Mi. Il l o O.O IlL OR
. ~ ~ + 1.o m. m
P t l O
[ k t® = . I • g , I . I .
1 , 0 l . I 1 . 4 I . I l . t t , 0 I , I i . 4
I O 0 0 1 T (K "~1
Fig. 42. (a) Arrhenius plot for CH 4 synthesis over several different Ni coverages on W(1 10) and W(100) at a total reactant pressure of 120Torr (H2 :CO = 4). (b) Arrhenius plots for CH, synthesis over several different Ni coverages on W(1 10) at total reactant pressures of 1, 10 and 120Torr (H2" CO - 4) (from Ref. [346]).
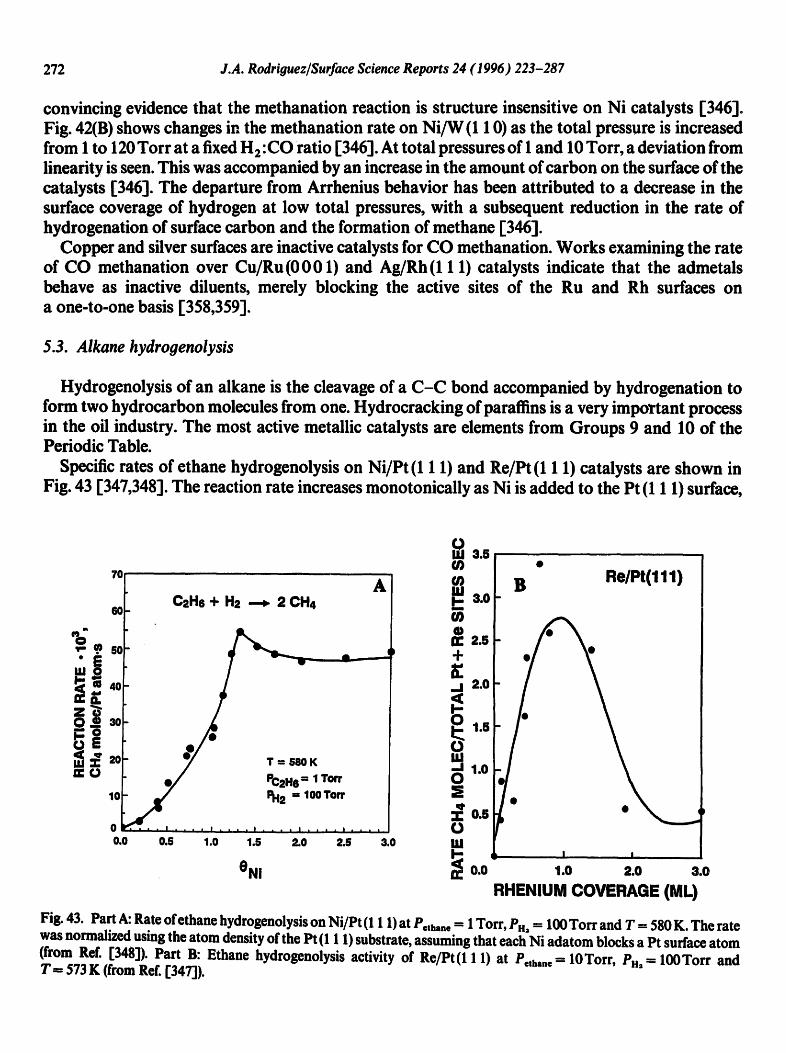
272 J.A. Rodriouez/Surface Science Reports 24 (1996) 223-287
convincing evidence that the methanation reaction is structure insensitive on Ni catalysts [346]. Fig. 42(B) shows changes in the methanation rate on Ni/W (1 10) as the total pressure is increased from 1 to 120 Torr at a fixed H2 :CO ratio [346]. At total pressures of 1 and 10 Torr, a deviation from linearity is seen. This was accompanied by an increase in the amount of carbon on the surface of the catalysts [346]. The departure from Arrhenius behavior has been attributed to a decrease in the surface coverage of hydrogen at low total pressures, with a subsequent reduction in the rate of hydrogenation of surface carbon and the formation of methane [346].
Copper and silver surfaces are inactive catalysts for CO methanation. Works examining the rate of CO methanation over Cu/Ru(0001) and Ag/Rh(1 1 1) catalysts indicate that the admetals behave as inactive diluents, merely blocking the active sites of the Ru and Rh surfaces on a one-to-one basis [358,359].
5.3. Alkane hydrogenolysis
Hydrogenolysis of an alkane is the cleavage of a C-C bond accompanied by hydrogenation to form two hydrocarbon molecules from one. Hydrocracking of paraffins is a very important process in the oil industry. The most active metallic catalysts are elements from Groups 9 and 10 of the Periodic Table.
Specific rates of ethane hydrogenolysis on Ni/Pt (1 1 1) and Re/Pt (1 1 1) catalysts are shown in Fig. 43 [347,348]. The reaction rate increases monotonically as Ni is added to the Pt (1 1 1) surface,
~3"5 1 • 70 A s.o ~ B Re/Pt(11 1)
C=Hs + H2 .-.-~ 2 CHA I so 2He r2 4
e / PC2.6: 1 Torr [
. . . . l O 1,'i;°°'°i ro eNi
<C 2"0
0 1.s
~0 .0 1.0 2.0 3.0 RHENIUM COVERAGE (ML)
Fig. 43. Part A: Rate of ethane hydrogenolysis on Ni/Pt (1 1 1) at/)ethane f f i 1Torr, Ps, ffi 100 Torr and T = 580 K. The rate was normalized using the atom density of the Pt (1 1 1) substrate, assuming that each Ni adatom blocks a Pt surface atom (from Ref. 1"348]). Part B: Ethane hydrogenolysis activity of Re/Pt(l I 1) at P®than® = 10Torr, Pn~= 100Torr and T ffi 573 K (from Ref. 1"347]).
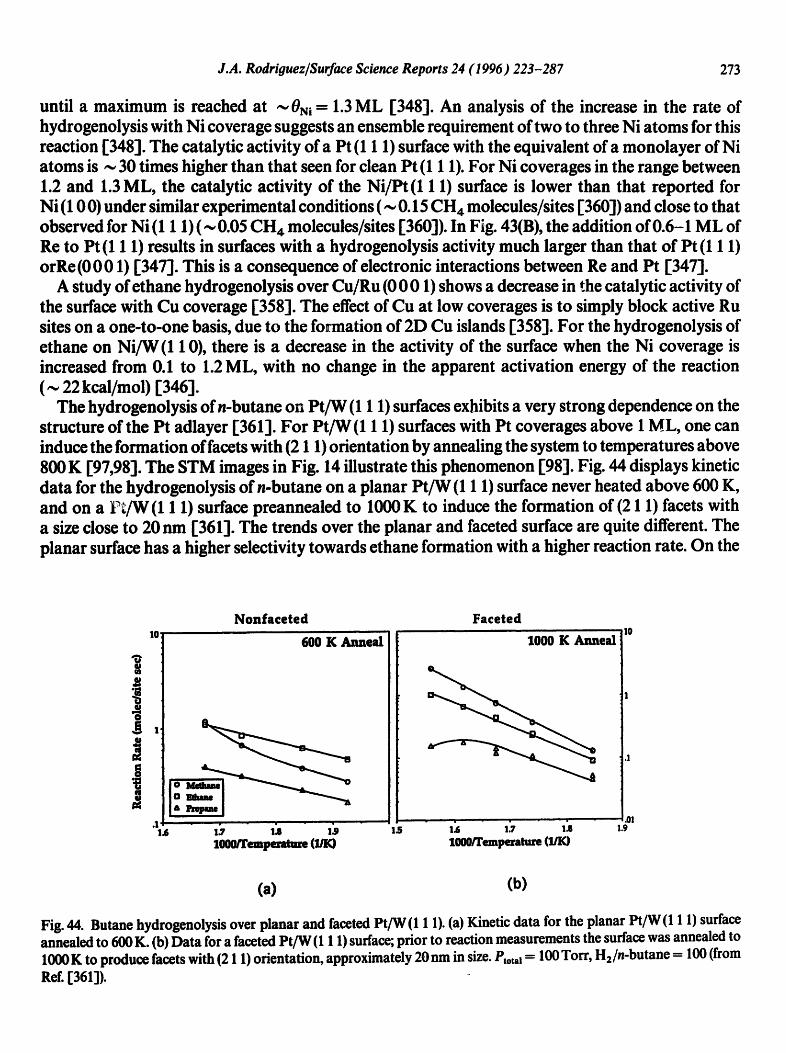
J.A. Rodriguez/Surface Science Reports 24 (1996) 223-287 273
until a maximum is reached at ~0Ni = 1.3 ML [348]. An analysis of the increase in the rate of hydrogenolysis with Ni coverage suggests an ensemble requirement of two to three Ni atoms for this reaction [:348]. The catalytic activity of a Pt (1 1 1) surface with the equivalent of a monolayer of Ni atoms is ~ 30 times higher than that seen for clean Pt (1 1 1). For Ni coverages in the range between 1.2 and 1.3 ML, the catalytic activity of the Ni/Pt(1 1 1) surface is lower than that reported for Ni (10 0) under similar experimental conditions ( ~ 0.15 CH4 molecules/sites [360]) and close to that observed for Ni (1 1 1) (~0.05 CH 4 molecules/sites [360]). In Fig. 43(B), the addition of 0.6-1 ML of Re to Pt (1 1 1) results in surfaces with a hydrogenolysis activity much larger than that of Pt (1 1 1) orRe(0001) [347]. This is a consequence of electronic interactions between Re and Pt [347].
A study of ethane hydrogenolysis over Cu/Ru (0 0 01) shows a decrease in the catalytic activity of the surface with Cu coverage [:358]. The effect of Cu at low coverages is to simply block active Ru sites on a one-to-one basis, due to the fo~a t ion of 2D Cu islands [358]. For the hydrogenolysis of ethane on Ni/W (1 10), there is a decrease in the activity of the surface when the Ni coverage is increased from 0.1 to 1.2 ML, with no change in the apparent activation energy of the reaction (~ 22 kcal/mol) [346].
The hydrogenolysis of n-butane on Pt/W (1 1 1) surfaces exhibits a very strong dependence on the structure of the Pt adlayer [361]. For Pt/W (1 1 1) surfaces with Pt coverages above 1 ML, one can induce the formation offacets with (21 1) orientation by annealing the system to temperatures above 800 K [97,98]. The STM images in Fig. 14 illustrate this phenomenon [98]. Fig. 44 displays kinetic data for the hydrogenolysis of n-butane on a planar Pt/W (1 1 1) surface never heated above 600 K, and on a F~/W (1 1 1) surface preannealed to 1000 K to induce the formation of (21 1) facets with a size close to 20 nm [361]. The trends over the planar and faceted surface are quite different. The planar surface has a higher selectivity towards ethane formation with a higher reaction rate. On the
10'
I J
g
1
Nonfaceted Faceted 10
1000 K Anneal
!
.1
600 K .Anneal
.Ol • 11.6 1.5 1~ 1~? 1~ 1.9
l O 0 0 / T e m p m t m (1/ I0
(a) (b)
Fig. 44. Butane hydrogenolysis over planar and faceted Pt/W (1 1 1). (a) Kinetic data for the planar Pt/W (1 1 1) surface annealed to 600 K. (b) Data for a faceted Pt/W (1 1 1) surface; prior to reaction measurements the surface was annealed to 1000 K to produce facets with (21 I) orientation, approximately 20 nm in size. Ptot,~ = 100 Torr, Ha/n-butane - 100 (from Ref. [361]).
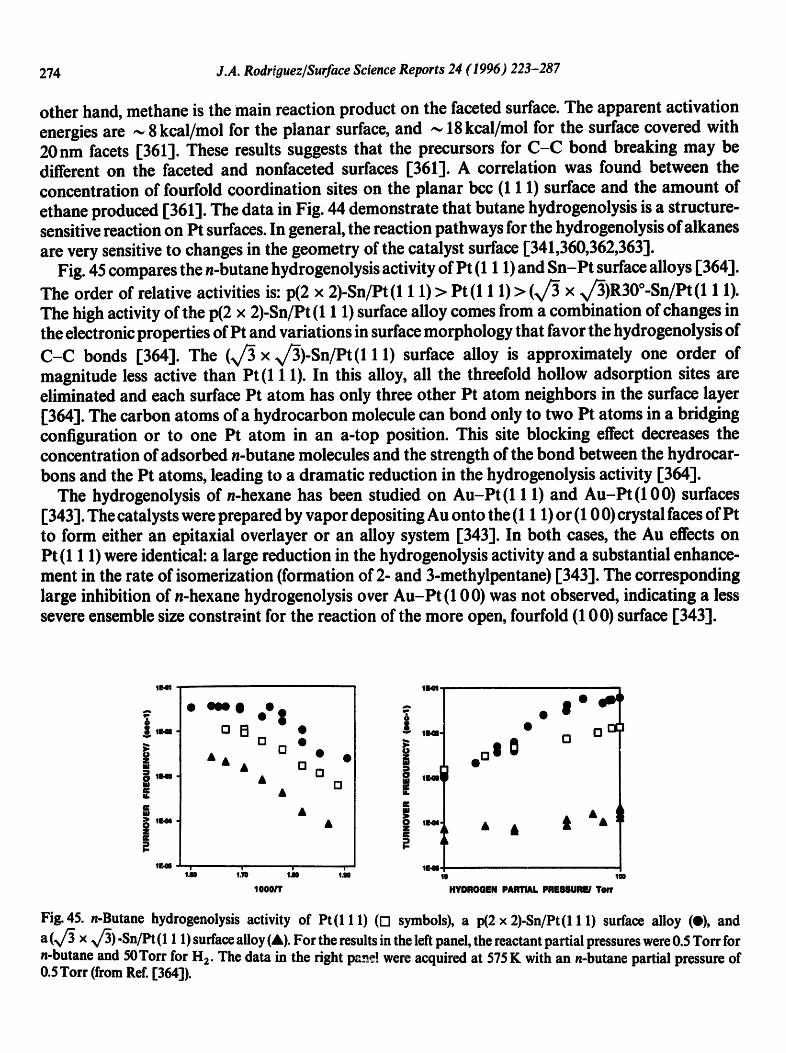
274 J.A. Rodriguez/Surface Science Reports 24 (1996) 223-287
other hand, methane is the main reaction product on the faceted surface. The apparent activation energies are ~ 8 kcal/mol for the planar surface, and ~ 18 kcal/mol for the surface covered with 20rim facets [361]. These results suggests that the precursors for C-C bond breaking may be different on the faceted and nonfaceted surfaces [361]. A correlation was found between the concentration of fourfold coordination sites on the planar bcc (1 1 1) surface and the amount of ethane produced [361]. The data in Fig. 44 demonstrate that butane hydrogenolysis is a structure- sensitive reaction on Pt surfaces. In general, the reaction pathways for the hydrogenolysis of alkanes are very sensitive to changes in the geometry of the catalyst surface [341,360,362,363].
Fig. 45 compares the n-butane hydrogenolysis activity of Pt (1 1 1) and Sn-Pt surface alloys [364]. The order of relative activities is: p(2 x 2)-Sn/Pt (1 1 1)> Pt (1 1 1)> (x//] x ~/3)R30°-Sn/Pt(1 1 1). The high activity of the p(2 x 2)-Sn/Pt (1 1 1) surface alloy comes from a combination of changes in the electronic properties of Pt and variations in surface morphology that favor the hydrogenolysis of C-C bonds [364]. The (x/~ x x/~)-Sn/Pt(1 1 1) surface alloy is approximately one order of magnitude less active than Pt (1 1 1). In this alloy, all the threefold hollow adsorption sites are eliminated and each surface Pt atom has only three other Pt atom neighbors in the surface layer [364]. The carbon atoms of a hydrocarbon molecule can bond only to two Pt atoms in a bridging configuration or to one Pt atom in an a-top position. This site blocking effect decreases the concentration of adsorbed n-butane molecules and the strength of the bond between the hydrocar- bons and the Pt atoms, leading to a dramatic reduction in the hydrogenolysis activity [364].
The hydrogenolysis of n-hexane has been studied on Au-Pt (1 1 1) and Au-Pt (100) surfaces [343]. The catalysts were prepared by vapor depositing Au onto the ( 1 1 1) or ( 10 0) crystal faces of Pt to form either an epitaxial overlayer or an alloy system [343]. In both cases, the Au effects on Pt (1 1 1) were identical: a large reduction in the hydrogenolysis activity and a substantial enhance- ment in the rate of isomerization (formation of 2- and 3-methylpentane) [343]. The corresponding large inhibition of n-hexane hydrogenolysis over Au-Pt (100) was not observed, indicating a less severe ensemble size constrvint for the reaction of the more open, fourfold (100) surface [343].
II,M
i lI,m
g
I If,
I I
z E I-
tl,n
• " . . o m ;8 ,', ,-, • A _ G •
• Q Q
• 0
II,Ol
P,
n _
I III ~_ ,,,..,. I
II,,01
o l ° ~ ,~ • r l ~
oo l 0 o
• I t AA
I I I
! 1.10 IJO 1.110 10 IQO
1000r r HYDROGEN PARTIAL Pl I IBIURF.J Tolrr
Fig.45. n-Butane hydrogenolysis activity of Pt(l I 1) (D symbols), a p(2 x 2)-Sn/Pt(1 1 1) surface alloy (O), and a (~fJ x v/j) -Sn/Pt (1 1 1) surface alloy (A). For the results in the left panel, the reactant partial pressures were 0.5 Torr for n-butane and 50 Torr for H2. The data in the right panel were acquired at 575 K with an n-butane partial pressure of 0.5 Torr (from Ref. [364]).
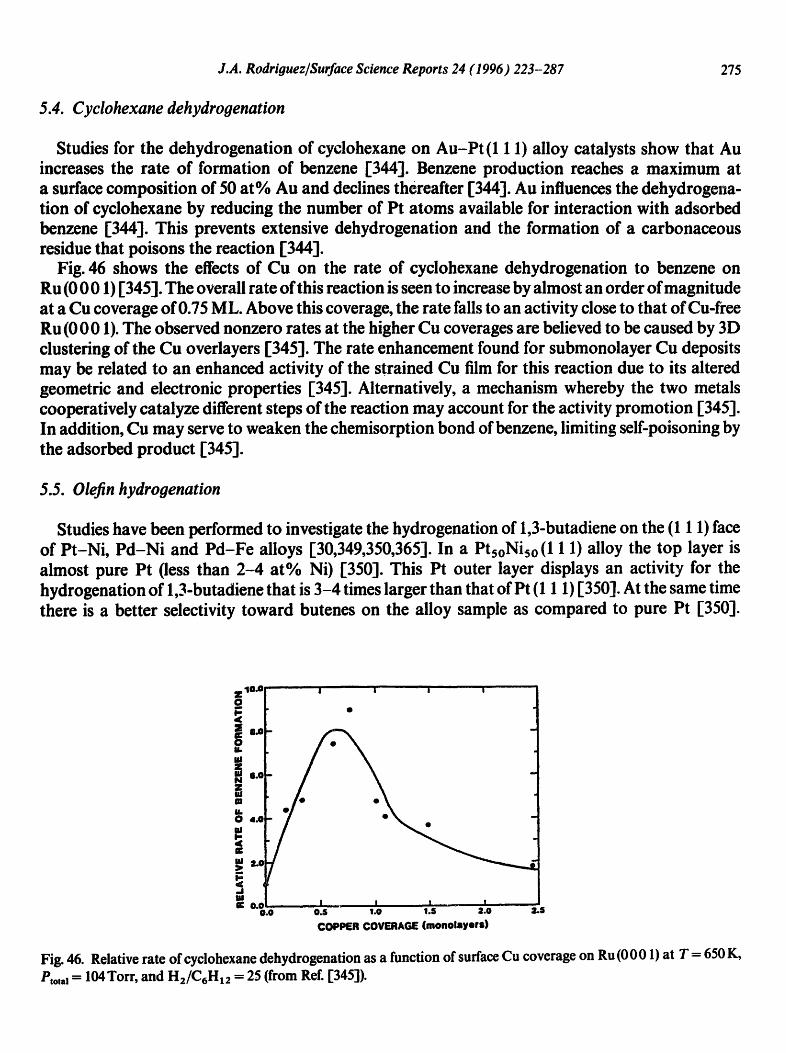
J.A. Rodriouez/Surface Science Reports 24 (1996) 223-287 275
5.4. Cyclohexane dehydrooenation
Studies for the dehydrogenation of cyclohexane on Au-Pt (1 1 1) alloy catalysts show that Au increases the rate of formation of benzene [344]. Benzene production reaches a maximum at a surface composition of 50 at% Au and declines thereafter [344]. Au influences the dehydrogena- tion of cyclohexane by reducing the number of Pt atoms available for interaction with adsorbed benzene [344]. This prevents extensive dehydrogenation and the formation of a carbonaceous residue that poisons the reaction [344].
Fig. 46 shows the effects of Cu on the rate of cyclohexane dehydrogenation to benzene on Ru (0 0 01) [345]. The overall rate of this reaction is seen to increase by almost an order of magnitude at a Cu coverage of 0.75 ML. Above this coverage, the rate falls to an activity close to that of Cu-free Ru(0001). The observed nonzero rates at the higher Cu coverages are believed to be caused by 3D clustering of the Cu overlayers [345]. The rate enhancement found for submonolayer Cu deposits may be related to an enhanced activity of the strained Cu film for this reaction due to its altered geometric and electronic properties [345]. Alternatively, a mechanism whereby the two metals cooperatively catalyze different steps of the reaction may account for the activity promotion [345]. In addition, Cu may serve to weaken the chemisorption bond of benzene, limiting self-poisoning by the adsorbed product [345].
5.5. Olefin hydrooenation
Studies have been performed to investigate the hydrogenation of 1,3-butadiene on the (1 1 1) face of Pt-Ni, Pd-Ni and Pd-Fe alloys [30,349,350,365]. In a PtsoNiso (1 1 1) alloy the top layer is almost pure Pt (less than 2-4 at% Ni) [350]. This Pt outer layer displays an activity for the hydrogenation of 1,3-butadiene that is 3-4 times larger than that of Pt (1 1 1) [350]. At the same time there is a better selectivity toward butenes on the alloy sample as compared to pure Pt [350].
| i I i O
o
Ul
m ee
,.I Ul E O.
0.0 I I IIS. I
2 .0 O.S 1.0
COPPER COVERAGE ( m o n o l x y e r s )
Z 10.@
Fig. 46. Relative rate of cyclohexane dehydrogenation as a function of surface Cu coverage on Ru (0 0 01) at T = 650 K, Ptotal = 104 Torr, and H2/CeHx2 - 25 (from Ref. [345]).

276 J.A. Rodriguez/Surface Science Reports 24 (I 996) 223-287
Electronic interactions between the metals probably contribute to these changes in the catalytic properties of Pt [277,291b,294,350].
For PdlX99 and PdsX9s alloys (X = Fe or Ni), the Pd enrichment at the surface reaches values as large as 65 at% (see Table 1)[30,349]. On all these catalysts the selectivity toward the synthesis of butenes (C4H6 + H2 -,C4Hs) is close to unity (i.e. there is no production of butane until all the 1,3-butadiene is transformed into butenes) [30,349]. The hydrogenation activity for the PdlFe99 and PdsFe9s ~loys is of the same order of magnitude as for (1 1 1) and (1 10) Pd single crystal faces (Table 1) [349]. The PdsNigs alloy displays an activity much bigger than that of pure Pd or Ni systems (Table 1) [30]. In the PdsNi9s alloy the active sites probably consist of small ensembles of Pd atoms (two or a little more Pd surface atoms), electronically modified by the surrounding Ni atoms [30]. The electronic modifications on these active sites act in such a way that they are more effective at olefin hydrogenation than active sites located in a pure Pd matrix [30].
5.6. Cyclotrimerization of acetylene
Under UHV conditions the cyclization of acetylene to benzene occurs on Pd (1 1 1) at tempera- tures below 250 K [366,367]. Pd/Au (1 1 I) surfaces are also effective catalysts for the trimerization of acetylene [ 162c,264]. On Au (1 1 1), pseudomorphic islands of Pd nucleate and grow uniformly from the partial surface dislocations of the "herringbone" reconstruction (see Fig. 47) [741)]. The activity of the surface for the production of benzene scales linearly with the coverage of the Pd islands up to ~0.6 ML (see Fig. 47) [74b]. Beyond this point, concurrent growth of higher layers leads to a roughened surface, the appearance of new binding sites for the formed benzene, and ultimately the suppression of all cyclization activity, thus demostrating the extreme structure-sensitivity of this reaction [74b].
For the interaction of C2H2 with Sn/Pt(1 1 1) surfaces under an UHV environment, the Sn adatoms block the active sites for the dissociation of acetylene, and one sees an improvement in the selectivity for the production ofbenzene from acetylene [265]. The di- and trimerization reactions of acetylene were studied over Pt (1 1 1) and (~/~ × ~/~)R30°-Sn/Pt (1 1 1) catalysts at moderate
Table 1 Activity of Pd-based bimetallic catalysts in the hydrogenation of 1,3-butadiene a
Sample Pd at% at the Activity surface ( × 10 Is molecules
c m - 2 S -1)
Selectivity into butenes
PdlFe99 55 2.1 1 PdsFe9s 65 9.3 1
PdlNi99 20 5.0 1 PdsNigs 50 30 1
Pd(1 1 1) 100 1.5 1 Pd(i 10) 100 7.1 1 Ni(l 1 1) 100 0.3 1
a From Refs. 1"30,349"1. The values are given for T - 300 K, Pu~ -- 35 Torr and PH~/PNc ,~ 10.
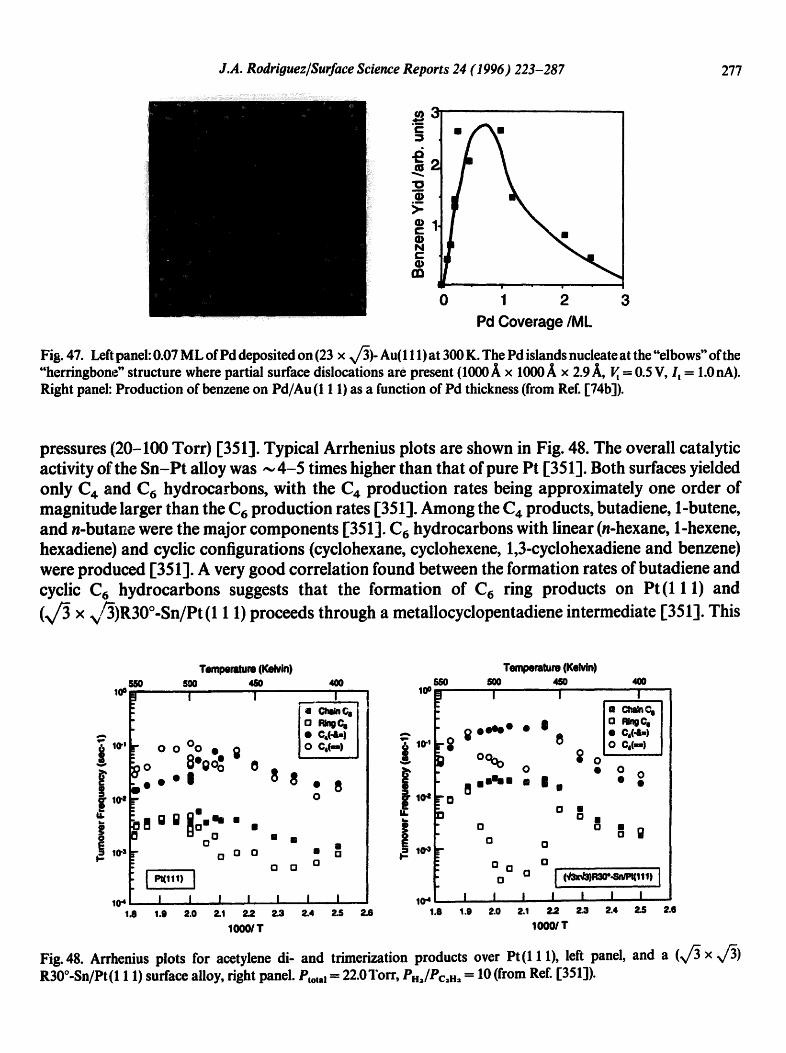
J.A. Rodriouez/Surface Science Reports 24 (1996) 223-287 277
, . 3
~2 .a
0
4) N e-
m
1 2 3
Pd Coverage/ML
Fig. 47. Left panel: 0.07 ML of I'd deposited on (23 x x/~)- Au(l I 1) at 300 K. The Pd islands nucleate at the "elbows" of the "herringbone" structure where partial surface dislocations are present (1000 ~, x 1000 ~, x 2.9 A, V t = 0.5 V, It - 1.0 nA). Right panel: Production of benzene on Pd/Au (1 1 1) as a function of I'd thickness (from Ref. [74b]).
pressures {20-100 Torr) [351]. Typical Arrhenius plots are shown in Fig. 48. The overall catalytic activity of the Sn-Pt alloy was ~ 4-5 times higher than that of pure Pt [351]. Both surfaces yielded only C4 and C6 hydrocarbons, with the C4 production rates being approximately one order of magnitude larger than the C6 production rates [351]. Among the C4 products, butadiene, 1-butene, and n-butane were the major components [351]. C6 hydrocarbons with linear (n-hexane, 1-hexene, hexadiene) and cyclic configurations (cyclohexane, cyclohexene, 1,3-cyclohexadiene and benzene) were produced [351]. A very good correlation found between the formation rates of butadiene and cyclic C6 hydrocarbons suggests that the formation of C6 ring products on Pt(1 1 1) and (x/~ x ~/3)R30°-Sn/Pt (1 1 1) proceeds through a metallocydopentadiene intermediate [351]. This
Temperiiture (Kolvin) Temperature (Kelvin) m soo 480 4o0 sso 5o0 48o 4o0
a R~C, II a n~c,
i i ° ',i'°'o'-: ' 10" O O Oo • O O,I I I ~ 10" ~ O C,(--)
8"oo o 0% - - - -
0
~ o • • O • 8 B = " = = = = l
1 1o~ o 1oa
~-i I P I g g ~ a : ~ • a a a a g
D a a 0
I ° ° I ~o-. I I I I I i i i 10"* I I I I I i i
~ ~.9 2.0 ~.~ 2.2 u P_, P..s 2.6 ~.e ~.9 zo 2.1 ~ P.a 2.4 ~ 2.e 10001T 10001 T
Fig. 48. Arrhenius plots for acetylene di- and trimerization products over Pt(l I 1), left panel, and a (x/~ x x/~) R30°-Sn/Pt(1 1 1) surface alloy, fight panel. Ptot.~-- 22.0 Torr, Pa, /Pc. . . = 10 (from Ref. [351]).

278 J.A. Rodriouez/Surface Science Reports 24 (1996) 223-287
species can either be hydrogenated off from the catalyst surfaces to produce butadiene or be reacted with a third acetylene molecule to form ring C6 hydrocarbons [351]. A similar reaction pathway is proposed for the cyclotrimerization of acetylene on Pd (1 1 1) and Pd/Au (1 1 1) surfaces [ 162c,264].
6. Conclusion
Experiments performed using modern techniques of surface science have given detailed and exciting information about the physical and chemical properties of bimetallic surfaces. Many new interesting phenomena have been discovered, and as a result models traditionally used to describe the properties of bimetallic surfaces have been modified. The knowledge gathered for the growth of metals on metals provides a solid basis for the synthesis of new materials with unique applications in areas of catalysis, electrochemistry and microelectronics. In many cases, the formation of a surface metal-metal bond induces large changes in the band structure of the metals producing systems with novel chemical and catalytic properties. As a result of experimental and theoretical studies, a fundamental understanding ofthe electronic and structural properties responsible for the chemical behavior of bimetallic surfaces is beginning to evolve. The progress in this area has been very impressive, but clearly much more research is necessary to produce the type of knowledge that is necessary for a rational design of bimetallic catalysts.
Acknowledgements
In the past years a great number ofpeople have been helpful in discussing the problems associated with the behavior of bimetallic surfaces. Special thanks to D.W. Goodman for many thought- provoking conversations. I am also grateful to C.T. Campbell, J. Hrbek, T.E. Madey, T.K. Sham and M. Strongin for helpful discussions. This work was carried out at Brookhaven National Laboratory under Contract DE-AC02-76CH00016 with the US Department of Energy, Office of Basic Energy Sciences, Chemical Science Division.
References
[11 [21 [31 [41 Is1 [61 ['I3 [81 1"91
1IO3
[I11 [121
G.M. Schwab, Disc. Faraday Soc. 8 (1950) 166. A. Couper and D.D. Eley, Disc. Faraday Soc. 8 (1950) 172. D.A. Dowden and P. Reynolds, Disc. Faraday Soc. 8 (1950) 184. .I.H. Sinfelt, Acc. Chem. Res. 10 (1977) 15. J.K.A. Clarke, Chem. Rev. 75 (1975) 291. W.M.H. Sachtler and R.A. van Santen, Adv. Catal. 26 (1977) 69. V. Ponec, Adv. Catai. 32 (1983) 149. W.H.M. Sachtler, Faraday Disc. Chem. Soc. 72 (1981) 7. G. Ertl and J. Kiippers, Low Energy Electrons and Surface Chemistry (VCH, Weinheim, 1985). D.P. Woodruff and T.A. Delchar, Modern Techniques of Surface Science (Cambridge University Press, New York, 1986). G.A. Somorjai, Introduction to Surface Chemistry and Catalysis (Wiley, New York, 1994). E. Bauer, in: The Chemical Physics of Solid Surfaces and Heterogeneous Catalysis, Vol. 3, Eds. D.A. King and D.P. Woodruff" (Elsevier, Amsterdam, 1984).

J.A. Rodriouez/Surface Science Reports 24 (1996) 223-287 279
[13] [14] [15] [16] [17] [183 [19] [203
[211 [22] [231 [24]
[25] [26]
[27] [28] [29]
[3o] [31]
E32] [33] [34] [35] [36] Dr] [38]
[39]
[40] [41] [421
D31
[44] [45]
[46] [47] [48] [49] [5o] [51]
C.T. Campbell, Annu. Rev. Phys. Chem. 41 (1990) 775. V.S. Sundaram, R.S. Alben and W.D. Robertson, Surf. Sci. 46 (1974) 653. H.C. Potter and J.M. Blakely, J. Vac. Sd. Technol. 12 (1975) 635. J.M. McDavid and S.C. Fain, Surf. S,A. 52 (1975) 161. T.M. Buck, G.H. Wheatley and L. Marchut, Phys. Rev. Lett. 51 (1983) 43. D. Sondericker, F. Jona and P.M. Marcus, Phys. Rev. B 33 (1986) 900, 6770. D. Sondericker, F. Jona, V.L. Moruzzi and P.M. Marcus, Solid State Commun. 53 (1985) 175. A. Atrei, L. Pedo~hi, U. Bardi, G. Rovida, M. Torrini, E. Zanazzi, M.A. Van Hove and P.N. Ross, Surf. Sci. 261 (1992) 64. A. Atrei, U. Bardi, G. Rovida, M. Torrini, E. Zanazzi and P.N. Ross, Phys. Rev. B 46 (1992) 1649. W. Chen, L. Severin, M. G6thelid, M. Hammar, S. Cameron and J. Paul, Phys. Rev. B 50 (1994) 5620. H.L. Davis and J.R. Noonan, Phys. Rev. Lett. 54 (1985) 566; J. Vac. Sd. Technol. A 3 (1985) 1507. H.L. Davis, in: The Structure of Surfaces II, Eds. J.F. van der Veen and M.A. van Hove (Springer, New York, 1988) p. 152. S.H. Overbury, D.R. Mullins and J.F. Wendelken, Surf. Sd. 236 (1990) 122. (a) C.P. Wang, F. Jona, N.R. Gleason, D.R. Strongin and P.M. Marcus, Surf. Sd. 298 (1993) 114; (b) H. Graupner, L. Hammer, K. Miiller and D.M. Zehner, Surf. Sci. 322 (1995) 103. S.K. Kim, F. Jona and P.M. Marcus, Phys. Rev. B 51 (1995) 5412; and references therein. J.I. Lee, C.L. Fu and A.J. Freeman, Phys. Rev. B 36 (1987) 9318. (a) W.M.H. Sachtler, Appl. Surf. Sci. 19 (1984) 167; (b) A.R. Miedema and J.W.F. Dorleijn, Surf. Sd. 95 (1980) 447; (c) A.R. Miedema, Z. Metallkd. 69 (1978) 287, 455. P. Miegge, J.L. Rousset, B. Tardy, J. Massardier and J.C. Bertolini, J. Catal. 149 (1994) 404, and references therein. J.C. Bertolini, J.L. Rousset, P. Miegge, J. Massardier, Tardy, Y. Samson, B.C. Khanra and C. Creemers, Surf. Sci. 281 (1993) 102. J.C. Bertolini, J.L. Rousset, P. Miegge, J. Massardier and B. Tardy, Surf. Sci. 287/288 (1993) 346. D. Fargues, J.J. Ehrhardt, M. Abort and J.C. Bertolini, Surf. Sci. 194 (1988) 149. Y. Gauthier, Y. Joly, R. Baudoing and J. Rundgren, Phys. Rev. B 31 (1985) 6216. R. Baudoing, Y. Gauthier, M. Lundberg and J. Rundgren, J. Phys. C 19 (1986) 2825. Y. Gauthier, R. Baudoing, M. Lundberg and J. Rundgren, Phys. Rev. B 35 (1987) 7867; 40 (1989) 1500. Y. Gauthier, W. Hoffmann and M. Wuttig, Surf. Sci. 233 (1990) 239. U. Bardi, A. Atrei, G. Rovida, E. Zanaz~i and P.N. Ross, Surf. Sci. 211/212 (1989) 441; Vacuum 41 (1990) 437. (a) P. Becxat, Y. Gauthier, R. Baudoing-Savois and J.C. Bertolini, Surf. Sci. 238 (1990) 105; (b) Y.G. Shen, D.J. O'Connor, K. Wander and R.J. MacDonald, Surf. Sci. 328 (1995) 21; (c) U. Schneider, G.R. Castro, H. Busse, T. Janssens and K. WandeR, Surf. Sci. 269/270 (1992) 316; (d) F.C.M.J.M. van Delft, A.D. van Langeveld and B.E. Nieuwenhuys, Surf. Sd. ~ 89/190 (1987) 1129. Y. Gauthier, R. Baudoing-Savois, J.M. Bugnard, U. Bardi and A. Atrei, Surf. Sci. 276 (1992) 1. M. Lundberg, Phys. Rev. B 36 (1987) 4692. (a) G. Tr~glia and B. Legrand, Phys. Rev. B 35 (1987) 4338; (b) B. Legrand, G. Trt~glia and F. Ducastelle, Phys. Rev. B 41 (1990) 4423. (a) R. D61, M. Kottcke, K. Heinz, L. Hammer, K. Miiller and D.M. Zehner, Surf. Sci. 307-309 (1994) 434; (b) S.H. Overbury, R.J.A. van der Oetelaar and D.M. Zehner, Phys. Rev. B 48 (1993) 1718; (c) H.L. Davis and B. Dotsch, Bull. Am. Phys. Soc. 36 (1991) 705. S.M. Foiles, J. Vac. Sci. Technol. A 4 (1986) 762. (a) L. Yang and A.E. DePristo, J. Catal. 148 (1994) 575; (b) A.D. van Langeveld and J.W. Niemantsverdriet, Surf. Sci. 178 (1986) 880. E. Bauer, Z. Krist. 110(1958) 372, 395. E. Bauer and J.H. van der Merwe, Phys. Rev. B 33 (1986) 3657. A. Zangwill, Physics at Surfaces (Cambridge Press, New York, 1988). J.A. Rodriguez and D.W. Goodman, J. Phys. Chem. 95 (1991) 4196. P.J. lkrlowitz, J.E. Houston, J.M. White and D.W. Goodman, Surf. Sci. 205 (1988) 1. P.J. Berlowitz and D.W. Goodman, Surf. Sci. 187 (1987) 463.

280 J.A. Rodriouez/Surface Science Reports 24 (1996) 223-287
C52]
[533 [54] [55] [56] [57] [583 [59] [6O3 [61] [62] [63] [64] [65] [66] [67] [68]
C69] C7o] C713 C723 C73]
C74]
[75] [76] [773 [783
C. Gfinther, S. Giinther, E. Kopatzaki, R.Q. Hwang, J. Schr6der, J. Vrijmoeth and R. Behm, Ber. Bunsenges. Phys. Chem. 97 (1993) 522. R.Q. Hwang, J. Schr6der, C. Gtintber and RJ. Behm, Phys. Rev. Lett. 67 (1991) 3279. D.D. Chambliss, R.J. Wilson and S. Chiang, Phys. Rev. Lett. 66 (1991) 1721. £A. Stroscio, D.T. Pierce, R.A. Dragoseta and P.N. First, J. Vac. Sci. Technol. A 10 (1992) 1981. R.Q. Hwang, C. Giinther, J. Schr6der, S. Giinther, E. Kopatzki and R.J. Behm, J. Vac. Sci. Technol. A 10 (1992) 1970. H. R6der, R. Schuster, H. Brune and K. Kern, Phys. Rev. Lett. 71 (1993) 2086. H. Ridder, H. Brune, J.-P. Bucher and K. Kern, Surf. Sci. 298 (1993) 121. J.A. Stroscio, D.T. Pierce and R.A. Dragoset, Phys. Rev. Lett. 70 (1993) 3615. P. Blandin, C. Massobrio and P. Ballone, Phys. Rev. Lett. 72 (1994) 3072; 73 (1994) 2144. H. Brune, C. Romainczyk, H. R6der and K. Kern, Nature 369 (1994) 469. Z. Zhang, X. [hen and M. Lagally, Phys. Rev. Lett. 73 (1994) 1829. H. Brune, H. R6der, C. Boragno and K. Kern, Phys. Rev. B 49 (1994) 2997. H.J. Ernst, F. Fabre and J. Lapujoulade, Phys. Rev. B 46 (1992) 1929. H. R6der, H. Brune and K. Kern, Phys. Rev. Lett. 73 (1994) 2143. J.A. Meyer, P. Schmid and R. Behm, Phys. Rev. Lett. 74 (1995) 3864. K. Bromann, H. Brune, H. R6der and K. Kern, Phys. Rev. Lett. 75 (1995) 677. J. Jacobsen, L.P. Nielsen, F. Besenbacher, I. Stensgaard, E. Laegsgaard, T. Rasmussen, K.W. Jacobsen and J.K. Nzrskov, Phys. Rev. Lett. 75 (1995) 489. G. Ehrlich, Surf. Sci. 246 (1991) 1. P.J. Feibelman, Surf. Sci. 313 (1994) L801. G.L. Kellogg, Surf. Sci. Rep. 21 (1994) 1. L.S. Perkins and A.E. DePristo, Surf. Sci. 317 (1994) L1152; 319 (1994) 225. (a) T. Raeker and A. DePristo, Surf. Sci. 317 (1994) 283; (b) C. Massobrio and P. Fernandez, J. Chem. Phys. 102 (1995) 605; (c) H. Roeder, E. Hahn, H. Brune, J.P. Bucher and K. Kern, Nature 366 (1993) 141. (a) B. Voigtlander, G. Meyer and N.B. Amer, Surf. Sci. 255 (1991) L529; Phys. Rev. B 44 (1991) 10354; (b) C.J. Baddeley, R.M. Ormerod, A.W. Stephenson and R.M. Lambert, J. Phys. Chem. 99 (1995) 5146. D.D. Chamblis and R.J. Wilson, J. Vac. Sci. Technol. B 9 (1991)928. D.D. Chamblis, R.J. Wilson and S. Chiang, J. Vac. Sci. Technol. B 9 (1991) 933. E. Bauer, Ber. Bunsenges. Phys. Chem. 95 (1991) 1315. J. Kolaczkiewicz and E. Bauer, Phys. Rev. B 44 (1991) 5779.
[793 T.T. Tsong, Pep. Prog. Phys. 51 (1988) 759; Surf. Sci. Rep. 8 (1988) 127. [80] P,R. Schwoebel and G.L. Kellogg, Phys. Rev. B 38 (1988) 5326. [813 G.L. Kellogg, Surf. Sci. 187 (1987) 153. [82]
C833 C84] C853 C86] C87] C88] C893 C9O3 C9q [923 [933 [943 [953 [96] [97]
(3. Ehrlich, in: Chemistry and Physics of Solid Surfaces, Vol. 5, Eds. R. Vanselow and R. Howe (Springer, Berlin, 1984). D.W. Bassett, Surf. Sci. 21 (1970) 181. T.A. Witten and L.M. Sander, Phys. Rev. Lett. 47 (1981) 1400. A. Brodde, (3. Wilhelmi, D. Badt, H. Wengelnik and H. Neddermeyer, J. Vac. Sci. TechnoL A 9 (1991) 920. T. Michely, M. Hohage, M. Bott and G. Comsa, Phys. Rev. l.~tt. 70 (1993) 3943. M. Bott, T. Michely and (3. Comsa, Surf. SCi. 272 (1992) 256. P. Meakin, Heterogeneous Chem. Rev. I (1994) 99. J. Tersoff, A.W. Denier and R.M. Tromp, Phys. Rev. Lett. 72 (I 994) 266. J.(3. Amar and F. Family, Phys. Rev. Lctt. 74 (1995) 2066. K. Morgenstern, (3, Rosenfeld, B. Poelsema and (3. Comsa, Phys. Rev. Lett. 74 (1995) 2058. (3. P6tschke, J. Schr6der, C. (3iinther, R.Q. Hwang and R.J. Behm, Surf. Sci. 251/252 (1991) 592. P.J. Schmitz, W.Y. Lcung, (3.W. Graham and P.A. Thiel, Phys. Rev. B 40 (1989) 1147% S.D. Bader and E.R. Moog, J. Appl. Phys. 61 (1987) 3729. J. Camarero, L. Sl~ndeler, (3. Schmidt, K. Heinz, JJ. de Miguel and R. Miranda, Phys. Rev. Lett. 73 (1994) 2448. K.-J. Song, J.C. Lin, M.Y. Lai and Y.L. Wang, Surf. Sci. 327 (1995) 17. (a) T.E. Madey, K.-J. Song, C.-Z. Dong and R.A. Demmin, Surf. Sci. 247 (1991) 175; (b) T.E. Madey, J. (3uan, C.Z. Dong and S.M. Shivaprasad, Surf. Sci. 287/288 (1993) 826.

J.A. Rodriouez/$urface Science Reports 24 (1996) 223-287 281
[98,1 C99,1
C1oo,1 CLO1,1 [1o2,1 C1O3,1
[104,1 [105"1 [106] C107-1 CIOS,1 C109,1
CLIO] [111]
[112,1 C113,1 Cl14,1 Cl15,1 Cl16] Cl17-1 C118,1 Cl19,1 C120,1 C121,1 C122,1 C123,1 C124,1 C125,1 C126,1 C12"rJ
C128,1
C129,1 C13O,1 C131,1
[132,1 [133,1 [134,1 [135,1 [136,1 C1373 C138,1 [139,1 C140,1 C141I [142,1 [143,1
C.Z. Dong, S.M. Shivaprasad, K.-J. Song and T.E. Madey, J. Chem. Phys. 99 (1993) 9172. J. Guan, R.A. Campbell and T.E. Madey, J. Vac. Sci. Technol. A 13 (1995) 1484. J. Guan, R.A. Campbell and T.E. Madey, Surf. Sci. 341 (1995) 311. T.E. Madey, J. Guan, C.-H. Nien, C.-Z. Dong, H.-S. Tao and R.A. Campbell, Surf. Rev. Lett., submitted. T.J. Raeker and A.E. DePristo, J. Vac. Sci. Technol. A 10 (1992) 2396. (a) G.A. Somorjai and M.A. Van Hove, Prog. Surf. Sci. 30 (1989) 201; (b) E.D. Williams and N.C. Bartelt, Uitramicroscopy 31 (1989) 36. E.H. Conrad, Prog, Surf. Sci. 39 (1992) 65. S.P. [hen, Surf. Sci. 274 (1992) L619. P.J. Schmitz, W.Y. Leung, G.W. Graham and P. Thiel, Phys. Rev. B 40 (1989) 11477. S.D. Bader and E.R. Moog, J. Appl. Phys. 61 (1987) 3729. P.W. Palmberg and T.N. Rhodin, J. Chem. Phys. 49 (1968) 134. S.H. Lu, Z.Q. Wang, S.C. Wu, C.K.C. Lok, J. Quinn, Y.S. Li, D. Tian, F. Jona and P.M. Marcus, Phys. Rev. B 37 (1988) 4296. G.W. Graham, Surf. Sci. 184 (1987) 137. S.C. Wu, S.H. Lu, Z.Q. Wang, C.K.C. Lok, J. Quinn, Y.S. Li, D. Tian, F. Jona and P.M. Marcus, Phys. Rev. B 38 (1988) 5363. G.W. Graham, P.J. Schmitz and P.A. Thiel, Phys. Rev. B 41 (1990) 3353. T.D. Pope, K. Grififiths and P.R. No,on, Surf. Sci. 306 (1994) 294. M.T. Paffett and R.G. Windham, Surf. Sci. 208 (1989) 34. S.H. Overbury, D.R. Mullins, M.F. Paffett and B.E. Koel, Surf. Sci. 254 (1991) 45. S.H. Overbury and Y.-S. Ku, Phys. Rev. B 46 (1992) 7868. Y.-S. Ku and S.H. Overbury, Surf. Sci. 273 (1992) 353. J. Kudrnovsky, S.K. Bose and V. Drchal, Phys. Rev. Lett. 69 (1992) 308. J.E. Black, Phys. Rev. B 46(1992) 4292. D. Naumovic, A. Stuck, T. Greber, J. Osterwalder and L. Schlapbach, Surf. Sci. 269/270 (1992) 719. M. Wuttig, C.C. Knight, T. Flores and Y. Gauthier, Surf. Sci. 292 (1993) 189. T.D. Pope, G.W. Anderson, K. GrHtiths, P.R. Norton and G.W. Graham, Phys. Rev. B 44 (1991) 11518. P.H. Bu, A. Wander, L. Morales, M.P. Bessent and D.A. King, Surf. Sci. 286 (1993) L542. W.J. Wytenburg, R.M. Ormerod and R.M. Lambert, Surf. Sci. 282 (1993) 205. D. Rouyer, C. Krembel, M.C. Hanf, D. Bolmont and G. Gewinner, Appl. Surf. Sci. 65/66 (1993) 54. Y.D. Li, L.Q. Jiang and B.E. Koel, Phys. Rev. B 49 (1994) 2813. (a) J. Yao, Y.G. Shen, DJ. O'Connor and B.V. King, J. Vac. Sci. Technol. A 13 (1995) 1443; (b) A. Atrei, U. Bardi, M. Galeotti, G. Rovida, M. Torrini and E. Zanazzi, Surf. Sci. 339 (1995) 323. L.P. Nielsen, F. Besenbacher, I. Stensgaard, E. Laegsgaard, C. Engdahl, P. Stolze, K.W. Jacobsen and J.K. Nzrskov, Phys. Rev. Lett. 71 (1993) 754. H. R6der, R. Schuster, H. Brune and K. Kern, Phys. Rev. Lett. 71 (1993) 2086. J.A. Rodriguez and D.W. Goodman, Science 257 (1992) 897. (a) M.W. Ruckman and M. Strongin, Act:. Chem. Res. 27 (1994) 250; (b) J.A. Rodriguez and D.W. Goodman, Acc. Chem. Res. 28 (1995) 477. J.A. Rodriguez, Heterogeneous Chem. Rev. 3 (1996) 17. Magnetism in Ultrathin Films, Ed. D. Pescia, Appl. Phys. A 49 (1989) (special issue). F.J. Hlmpsel, Phys. Rev. Lett. 67 (1991) 2363. R. Mamy, Surf. Sci. 322 (1995) 337. S.S. Parkin, Phys. Rev. Lett. 71 (1993) 1641. J.E. Ortega and F.J. Himpsel, Phys. Rcv. B 47 (1993) 16441. J. [hen, M. Drakaki and J.L. Erskine, Phys. Rev. B 45 (1992) 3636. D.P. Pappas, C.R. Brundle and H. Hopster, Phys. Rev. B 45 (1992) 8169. Z.Q. Qiu, J. Pearson and S.D. Bader, Phys. Rev. Lett. 70 (1993) 1006. K. Le Dang, P. Veillet and E. Velu, AppL Phys. Lett. 63 (1993) 108. G. Chiaia, S. De Rossi, L. Mazzolari and F. Ciccacci, Phys. Rev. B 48 (1993) 11298. N.B. Brookes, Y. Chang and P.D. Johnson, Phys. Rev. Lett. 67 (1991) 354.

282 J.A. Rodriguez/Surface Science Reports 24 (1996) 223-287
[144] [145] [146] [147] [148] [149] [150] [151] [152] [1533 [154]
[155] [156] [157] [158] [159] [160] [i61] [162]
[163] [164]
[165] [166] [167]
[168] [169]
[170] [171] [1723 [173] [174] [1753 [1763 [177] [178] [179] [1803 [1813 [182] [183] [184] [185] [1863 [187] [188] [189]
C.L. Fu, A.J. Freeman and T. Oguchi, Phys. Rev. Lett. 54 (1985) 2700. J.G. Gay and R. Richter, Phys. Rev. Lett. 56 (1986) 2728. J.E. Ortega and F. Himpsei, Phys. Rev. Lett. 69 (1992) 844. S. De Rosi and F. Ciccacci, Surf. Sci. 307-309 (1994) 496. Li, Y.S. Li, J. Quinn, D. Tian, J. Sokolov, F. Jona and P. Marcus, Phys. Rev. Lett. 42 (1990) 9195. W. Heinen, C. Carbone, T. Kachel and W. Gudat, J. Electron Spectrosc. Rel. Phen. 51 (1990) 701. F.J. Himpsel, Phys. Rev. B 44 (1991) 5966. H. Glatzel, R. Schneider, T. Fauster and V. Dose, Z. Phys. B 88 (1992) 53. J.A. Rodriguez, Surf. Sci. 345 (1996) 347. M.W. Ruckman, V. Murgai and M. Strongin, Phys. Rev. B 34 (1986) 6759. (a) S.-L. Weng and M. EI-Batanouny, Phys. Rev. Lett. 44 (1980) 612; (b) M. EI-Batanouny, M. Strongin, G.P. Williams and J. Colbert, Phys. Rev. Lett. 46 (1981) 269. M.W. Ruckman and M. Strongin, Phys. Rev. B 29 (1984) 7105; 35 (1987) 487. J.A. Rodriguez, R.A. Campbell and D.W. Goodman, Surf. Sd. 307-309 (1994) 377. P. Miku~ik and Z. Bastl, Phys. Scr. 41 (1990) 130. G.W. Graham, J. Vac. Sd. Technol. A 4 (1986) 760. R.A. Campbell, J.A. Rodriguez and D.W. Goodman, Surf. Sd. 240 (1990) 71. J.A. Rodriguez, R.A. Campbell and D.W. Goodman, J. Phys. Chem. 95 (1991) 5716. R.A. Campbell, J.A. Rodriguez and D.W. Goodman, Phys. Rev. B 46 (1992) 7077. (a) B.E. Koe|, A. Sellidj and M.T. Paffett, Phys. Rev. B 46 (1992) 7846; (b) A. Sellidj and B.E, Keel, Phys. Rev. B 49 (1994) 8367; (c) C.J. Baddeley, C.J. Barnes, A. Wander, R.M. Ormerod, D.A. King and R.M. Lambert, Surf. Sci. 314 (1994) 1. G.C. Smith, C. Norris, C. Binns and H.A. Padmore, J. Phys. C (Solid State Phys.) 15 (1982) 6481. (a) G.W. Graham, Surf. Sci. 171 (1986) L432; (b) G.W. Graham, P.J. Schmitz and P.A. Thiel, Phys. Rev. B 41 (1990) 3353; (c) T.D. Pope, K. Grifliths and P.R. Norton, Surf. Sci. 306 (1994) 294. A. Fasana and L. Braicovich, Surf. Sci. 120 (1982) 239. J.A. Rodriguez, J. Phys. Chem. 98 (1994) 5758. (a) B. Frick and K. Jacobi, Phys. Rev. B 37 (1988) 4408; (b) L.Q. Jiang, M.W. Ruckman and M. Strongin, Phys. Rev. B 39 (1989) 1564. J.A. Rodriguez, Surf. Sci. 318 (1994) 253; 303 (1994) 366.. (a) B.E. Koel, R.J. Smith and P.J. Berlowitz, Surf. Sci. 231 (1990) 325; (b) J.M. Heitzinger, S.C. Gebhard and B.E. Koel, Surf. Sci. 275 (1992) 209. P.J. Berlowitz and D.W. Goodman, Langmuir 4 (1988) 1091. J.A. Rodriguez and M. Kuhn, Surf. Sci., in press (1996). J.E. Houston, J.M. White, P.J. Feibeiman and D.R. Hamann, Phys. Rev. B 38 (1988) 12164. C. Koziol, G. Lilienkamp and E. Bauer, Phys. Rev. B 41 (1990) 3364. J. Kolaczkiewicz and E. Bauer, Surf. Sci. 144 (1984)495. J.A. Rodriguez and M. Kuhn, J. Chem. Phys. 102 (1995) 4279. R.W. Judd, M.A. Reichelt, E.G. Scott and R.M. Lambert, Surf. Sci. 185 (1987) 515; 198 (1988) 26. M. Alnot, A. Cassuto, J.J. Ehri~ardt, A. Slavin and B. Weber, Appl. Surf. Sci. 10 (1982) 85. M. Romeo, J. Majerus, P. L6gar6, N.J. Castellani and D.B. Leroy, Surf. Sci. 238 (1990) 163. X.-H. Pan, M.W. Ruckman and M. Strongin, Phys. Rev. B 35 (1987) 3734. J.A. Rodriguez and M. Kuhn, Chem. Phys. Lett. 240 (1995)435. R.A. Campbell, J.A. Rodriguez and D.W. Goodman, Surf. Sci. 256 (1991) 272. M. Kuhn, J.A. Rodriguez and J. Hrbek, in preparation. S.-C. Liu, J.W. Davenport, E.W. Plummer, D.M. Zehner and G.W. Fernando, Phys. Rev. B 42 (1990) 1582. J. Emsley, The Elements (Clarendon, Oxford, 1990) pp. 125, 137, 141 and 231. P.J. Berlowitz and D.W. Goodman, Surf. Sci. 187 (1987) 463. S.M. Shivaprasad, R.A. Demin and T.E. Madey, Thin Solid Films 163 (1988) 393. J.C. Fuggle, F.U. Hillebrecht, R. Zeller, Z. Zolnierek, P.A. Bennett and C. Freiburg, Phys. Rev. B 27 (1983) 2145. F.U. Hillebrecht, J.C. Fuggle, P.A. Bennett, Z. Zolnierek and C. Freiburg, Phys. Rev. B 27 (1983) 2179. G. Wertheim, D. Buchanan and J. Wernick, Phys. Rev. B 40 (1989) 5319.

J.A. Rodriguez/Surface Science Reports 24 (I 996) 223-287 283
[190] [191] [192] [193]
[194] [195]
[196] [197] [198] [199] [2oo] [201]
[2023 [203] [2O4] [2O5]
[206] [2o7] [2o8] [209] [210] [2113 [212] [213] [214] [215] [216]
[217] [218] [219] [220] [221] [222] [223] [224] [225] [226] [227]
[228] [229] [23O] [231] [232]
P.N. Ross, J. Vac. Sci. Technol. A I0 (1992) 2546. T.-K. Sham, Phys. Rev. B 31 (1985) 1903. Y. Jeon, J. Chen and M. Croft, Phys. Rev. B 50 (1994) 6555. (a) V. Kumar and K.H. Bennemann, Phys. Rev. B 28 (1983) 3138; (b) S. Pick and P. Miku~ik, Chem. Phys. Lett. 208 (1993) 97; 215 (1993) 319. A. Zangwill, Physics at Surfaces (Cambridge University Press, New York, 1988). (a) C. Park, E. Bauer and H. Poppa, Surf. Sci. 154 (1985) 371; (b) W. Schlenk and E. Bauer, Surf. Sci. 93 (1980) 9; (c) C. Park, Surf. Sci. 203 (1988) 395; (d) D.R. Baer, C.W. Hubbard and R.L. Gordon, J. Vac. Sci. Technol. A 10 (1992) 2391. P.J. Berlowitz, J.E. Houston, J.M. White and D.W. Goodman, Surf. Sci. 205 (1988) 1. (a) R. Wu, Chem. Phys. Lett. 238 (1995) 99; (b) R. Wu and A.J. Freeman, Phys. Rev. B 52 (1995) 12419. M. Weinert and R.E. Watson, Phys. Rev. B 51 (1995) 17168. P. Maciejewski, W. Wurth, S. K6stlmeier, G. Pacchioni and N. R6sch, Surf. Sci. 330 (1995) 156. M.W. Ruckman, L.Q. Jiang and M. Strongin, J. Vac. Sci. Technol. A 10 (!992) 2551; 11 (1993) 466. M. Strongin, M.W. Ruckman, M. Weinert, R.E. Watson and J.W. Davenport, in: Metallic Alloys: Experimental and Theoretical Perspectives, Proc. 1993 NATO Advanced Workshop in Alloys (Boca Raton, FL, July 1993). M. Strongin, private communication. J.A. Rodriguez and M. Kuhn, J. Phys. Chem. IOO (1996) 381. T.K. Sham, M.L. Perlman and R.E. Watson, Phys. Rev. B 19 (1979) 539. (a) T.K. Sham, Y.M. Yiu, M. Kuhn and K.H. Tan, Phys. Rev. B 41 (1990) 1188; (b) A. Bzowski and T.K. Sham, J. Vac. Sci. Technol. A 11 (1993) 2153; (c) M. Kuhn and T.K. Sham, Phys. Rev. B 49 (1994) 1647; (d) R. Sammynaiken, M. Kuhn and T.K. Sham, Physica B 208/209 (1995) 371. R.E. Watson, J.W. Davenport and M. Weinert, Phys. Rev. B 35 (1987) 508. J.E. Houston, C.H.F. Peden, P.J. Feibelman and D.R. Hamann, Surf. Sci. 192 (1987) 457. M.L. Shek, P.M. Stefan, I. Lindau and W.E. Spicer, Phys. Rev. B 27 (1983) 7277; 27 (1983) 7301. G. Lilienkamp, C. Koziol and E. Bauer, Surf. Sci. 226 (1990) 358. M. EI-Batanouny and M. Strongin, Phys. Rev. B 31 (1985) 4798. (a) W.F. Egelhoff, J. Vac. Sci. Technol. 20 (1982) 668; (b) W.F. Egelhof, Appl. Surf. Sci. 11/12 (1982) 761. W.K. Kuhn, R.A. Campbell and D.W. Goodman, J. Phys. Chem. 97 (1993)446. J.A. Rodriguez. R.A. Campbell and D.W. Goodman, J. Phys. Chem. 95 (1991) 2477. J.A. Rodriguez and M. Kuhn, J. Phys. Chem. 98 (1994) 11251. M. Kuhn, J.A. Rodriguez, J. Hrbek, A. Bzowski and T.K. Sham, Surf. Sci. 341 (1995) L1011. D.L. Weissman-Wenocur, P.M. Stefan, B.B. pate, M.L. Shek, I. Lindau and W.E. Spicer, Phys. Rev. B 27 (1983) 3308. J.A. Rodriguez and J. Hrbek, J. Chem. Phys. 97 (1992) 9427. M.W. Ruckman and L.-Q. Jiang, Phys. Rev. B 38 (1988) 2959. J.A. Rodriguez and M. Kuhn, Surf. Sci. 330 (1995) L657. M. Salmeron, S. Ferrer, M. JazTar and G.A. Somorjai, Phys. Rev. B 28 (1983) 1158. A. Pavlovska and E. Bauer, Surf. Sci. 175 (1986) 369. J.W. Niemantsverdriet, P. Dolle, K. Markert and K. WandeR, J. Vac. Sci. Technol. A 5 (1987) 875. J.A. Rodriguez, R.A. Campbell and D.W. Goodman, J. Phys. Chem. 95 (1991) 5716. J.A. Rodriguez and M. Kuhn, J. Phys. Chem. 99 (1995) 9567. J.A. Rodriguez, Surf. Sci. 296 (1993) 149. Y. Baer, P.F. Hed6n, J. Hedman, M. Klasson, C. Nordling and K. Siegbahn, Solid State Commun. 8 (1970) 517. (a) A. Pavlovska, M. Paunov and E. Bauer, Thin Solid Films 126 (1985) 129; (b) J. Kolaczldewicz and E. Bauer, Surf. Sci. 160 (1985) 1; (c) S.J.T. Coles and P.l. Jones, Surf. Sci. 68 (1977) 312. J.A. Rodriguez and J. Hrbek, Surf. Sci. 312 (1994) 345. J.A. Rodriguez and J. Hrbek, Chem. Phys. Lett. 220 (1994) 486. M. Kuhn and J.A. Rodriguez, Surf. Sci. 336 (1995) 1. P.J. Berlowitz and D.W. Goodman, Surf. Sci. 187 (1987) 463. A. Atli, M. Abon, J.C. lkrtolini, Y. Boudeville, M. Fallavier, M. Benmansour and J.P. Thomas, J. Phys. Chem. 98 (1994) 4895.

284 J.A. Rodriguez/Surface Science Reports 24 (1996) 223-287
[233]
[234]
[235] [236] [2373 [238] [239] [24O] [2413 [242] [2433 [2443 [245] [246] [247] [2483
[249] [250]
[251]
[252]
[253]
[254] [255] [256] [257] [258] [259] [260] [261] [262]
[263] [264]
[265] [266]
[267] [268] [269] [270]
(a) A.K. Santra and C.N.R. Rao, J. Phys. Chem. 98 (1994) 5962; (b) G.R. Castro, U. Schneider, H. Busse, T. Janssens and K. Wandelt, Surf. Sci. 269/270 (1992) 321; tc) P.J. Schmitz, H.C. Kang, W.-Y. Leung and P.A. Thiel, Surf. Sci. 248 (1.991) 287. (a) C. Xu and B.E. Koel, Surf. Sci. 310 (1994) 198; (b) C. Xu and B.E. Koel, J. Chem. Phys. 100 (1994) 664; (c) H. Hirano, T. Yamada, K. Tanaka, J. Siera and B.E. Nieuwenhuys, Surf. Sci. 222 (1989) L804. J.M. Hei~nger, S.C. Gebhard and B.E. Koel, J. Phys. Chem. 97 (1993) 5327. J.-W. He, W.-L. Shea, X. Jiang and D.W. Goodman, J. Vac. Sci. Technol. A 8 (1990) 2435. R.A. Campbell, J.A. Rodriguez and D.W. Goodman, Surf. Sci. 256 (1991) 272. J.A. Rodriguez, R.A. Campbell and D.W. Goodman, J. Vac. Sci. Technol. A 10 (1992) 2540. X. Jiang and D.W. Goodman, Surf. Sci. 255 (1991) 1. R.A. Demmin, S.M. Shivaprasad and T.E. Madey, Langmuir 4 (1988) 1104. P. Ross, J. Vac. Sci. Technol. A 10 (1992) 2546. A. Linsebigler, G. Lu and J.T. Yates, Surf. Sci. 294 (1993) 284. M.T. Paffett, C.T. Campbell, T.N. Taylor and S. Srinivasam, Surf. Sci. 154 (1985) 284. M.T. Paffett, C.T. Campbell and T.N. Taylor, Langmuir 1 (1985) 741. Y.-W. Yang, J.C. Lin and T. Engel, Surf. Sci. 289 (1993) 267. W.K. Kuhn, J. Szanyi and D.W. Goodman, Surf. Sci. 303 (1994) 377. Y.B. Zhao and R. Gomer, Surf. Sci. 239 (1990) 189. (a) M.W. Ruckman, P.D. Johnson and M. Strongin, Phys. Rev. B 31 (1985) 3405; (b) M.W. Ruckman and M. Strongin, Phys. Rev. B 29 (1984) 7105. G. Blyholder, J. Phys. Chem. 68 (!964) 2772; 79 (1975) 756. (a) K. Hermann, P.S. Bagus and C.J. Nelin, Phys. Rev. B 35 (1987) 9467; (b) E.R. Davidson, K.L. Kunze, F.B.C. Machado and S.J. Chakravorty, Acc. Chem. Res. 26 (1993) 628. (a) For CO on transition metal surfaces, the valence d orbitals are actively involved in bonding and the heat of adsor- ption of the molecule varies between 90 and 20 kcal/mol. On the other hand, for CO on noble metals, the participa- tion of the valence d orbitals in bonding is weak, and the heat of adsorption of CO is relatively small (< 20 kcal/mol) [251(b)]; (b) G.A. Somorjai, Introduction to Surface Chemistry and Catalysis (Wiley, New York, 1994) p. 310. (a) E. Shustorovich, R.C. Baetzold and E.L. Muetterties, J. Phys. Chem. 81 (1984) 1966; (b) E. Shustorovich and R.C. Baetzold, Science 227 (1985) 876; (c) B. Hammer, Y. Morikawa and J.K. NOrskov, Phys. Rev. Lett. 76 (1996) 2141. (a) The CO(21t*) orbitals are located at ~ 1.8 eV above the vacuum level [253(b),(c)]; (b) E. Linhdholm and J. Li, J. Phys. Chem. 92 (1988) 1731; (c) M. Tronc, R. Azria and Y. Lecoat, J. Phys. B 13 (1980) 2327. (a) S. Pick, Chem. Phys. Lett. 239 (1995) 84; (b) R.M. Ferullo and N.J. Castellani, Langmuir 12 (1996) 70. J.A. Rodriguez, C.M. Truong and D.W. Goodman, J. Chem. Phys. 96 (1992) 7814. J.W. He, W.K. Kuhn and D.W. Goodman, J. Am. Chem. Soc. 113 (1991) 6416. F.M. Hoffmann and J. Paul, J. Chem. Phys. 86 (1987) 2990; 87 (1987) 1857. W.K. Kuhn, J.-W. He and D.W. Goodman, J. Vac. Sci. Technol. A 10 (1992) 2477. J.-W. He, W.K. Kuhn, L.-W.H. Leung and D.W. Goodman, J. Chem. Phys. 93 (1990) 7463. E. Kampshoff, E. Hahn and K. Kern, Phys. Rev. Lett. 73 (1994) 704. C. Xu and B.E. Koel, Surf. Sci. 327 (1995) 38. (a) M.T. Paffett, S.C. Gebhard, R.G. Windham and B.E. Koel, J. Phys. Chem. 94 (1990) 6831; (b) M.T. Paffett, S.C. Gebhard, R.G. Windharwem and B.E. Koel, Surf. Sci. 223 (1989) 449. B. Sakakini, A.J. Swift, J.C. Vickerman, C. Harendt and K. Christmann, J.Chem. Soc. Faraday Trans. I, 83 (1987) 1975. (a) R.M. Ormerod, C.J. Baddeley and R.M. Lambert, Surf. Sci. 259 (1991) L709; (b) G. Pacchioni and R.M. Lambert, Surf. Sgi. 304 (1994) 208. C. Xu, J.W. Peck and B.E. Koel, J. Am. Chem. Soc. 115 (1993) 751. (a) J.M. Campbell, S. Seimanides and C.T. Campbell, J. Phys. Chem. 93 (1989) 815; (b) C.T. Campbell, J.A. Rodriguez, F.C. Henn, J.M. Campbell, P.J. Dalton and S. Seimanides, J. Chem. Phys. 88 (1988) 6585. C. Xu, Y.-L. Tsai and B.E. Koel, J. Phys. Chem. 98 (1994) 585. C. Xu, B.E. Koei and M.T. Paffett, Langmuir 10 (1994) 166. F.C. Henn, P.J. Dalton and C.T. Campbell, J. Phys. Chem. 93 (1989) 836. (a) J.A. Rodriguez and C.T. Campbell, J. Phys. Chem. 93 (1989) 826; (b) M.E. Bussell, F.C. Henn and C.T. Campbell, J. Phys. Chem. 96 (1992) 5978; (c) M.E. Domagala and C.T. Campbell, J. Vac. Sci. Technol. A 11 (1993) 2128.

J.A. Rodriouez/Surface Science Reports 24 (1996) 223-287 285
[271]
[272] [273]
[274] [275] [276]
[277]
[278] [279] [28o] [281] [282] [283] [284] [285] [286] [287] [288] [289] [29O] [291]
[292] [293] [294] [295] [296] [297]
[298]
[299] [3OO3 [301] [302] [303] [3043 [3o53 [3o6] [307] [308] [3o9]
[310]
[311] [312] [313]
(a) J.A. Rodriguez and C.T. Campbell, J. Catal. 115 (1989) 500; (b) M.B. Hugenschmidt, A.L. Diaz and C.T. Campbell, J. Phys. Chem. 96 (1992) 5974. C. Xu and B.E. Koel, Surf. Sci. 304 (1994) 249. C.T. campbell, J.M. Campbell, P.J. Dalton, F.C. Henn, J.A. Rodriguez and S.G. Seimanides, J. Phys. Chem. 93 (1989) 806. A. Atli, M. Alnot, J.J. Ehrhardt, J.C. Bertolini and M. Abon, Surf. Sci. 269/270 (1992) 365. A. Rochefort, M. Abon, P. Delich6re and J.C. Bertolini, Surf. Sci. 294 (1993) 43. M. FaUavier, M. Benmansour, B. Hjrvarsson, J.P. Thomas, M. Abon, H.A. Atli and J.C. Bertolini, Surf. Sci. 311 (1994) 24. A. Atli, M. Abon, J.C. Bertolini, Y. Bouderville, M. Fallavier, M. Benmansour and J.P. Thomas, J. Phys. Chem. 98 (1994) 4895. S. Sieben and H. Bonzel, Surf. Sci. 282 (1993) 246. H. Onischi, T. Aruga and Y. Iwasawa, Surf. Sci. 283 (1993) 213. U. Schneider, G.R. Castro and K. Wandelt, Surf. Sci. 287/288 (1993) 146. R. Linke, U. Schneider, H. Busse, C. Becker, U. Schr'6der, G.R. Castro and K. Wandelt, Surf. Sci. 307-309 (1994) 407. C. Egawa, S. Oki and Y. Murata, Surf. Sci. 304 (1994) L488. J.E. Whitten and R. Gomer, Surf. Sci. 316 (1994) 23. J.T. Yates, C.H.F. Peden and D.W. Goodman, J. Catal. 94 (1985) 576. D.W. Goodman and C.H.F. Peden, J. Catal. 95 (1986) 321. J.A. Rodriguez, R.A. Campbell and D.W. Goodman, Surf. Sci. 244 (1991) 211. J. Heitzinger, A. Avoyan and B.E. Koel, Surf. Sci. 294 (1993) 251. J.M. Heitzinger, S.C. Gebhard and B.E. Koel, Chem. Phys. Lett. 200 (1992) 65. P. Lenz and K. Christmann, J. Catal. 139 (1993) 611. J.A. Rodriguez, R.A. Campbell, J.S. Corneille and D.W. Goodman, Chem. Phys. Lett. 180 (1991) 139. (a) ~]. Pick, Chem. Phys. Lett. 225 (1994) 62; (b) ~]. Pick, P. Miku~ik and C. Demangeat, J. Phys.: Condens. Matter 7 (1995) 2673. A. Noordermeer, G.A. Kok and B.E. Nieuwenhuys, Surf. Sci. 165 (1986) 375. M.T. Paffett, C.T. Campbell, R.G. Windham and B.E. Koel, Surf. Sci. 207 (1989) 274. N.J. Castellani and P. Legar6, Surf. Sci. 320 (1994) 39; J. Phys. Chem. 98 (1994) 9606. U. Bardi, A. Atrei and G. Rovida, Surf. Sci. 239 (1990) L511. U. Bardi, A. Atrei and G. Rovida, Surf. Sci. 268 (1992) 87. R.M. Jaeger, H. Kuhlenbeck, H.-J. Freund, M. Wittig, W. Hoffmann, R. Franchy and H. Ibach, Surf. Sci. 259 (1991) 235. H.-J. Freund, B. Dillmann, D. Ehdich, M. Hassel, R.M. Jaeger, H. Kuhlenbeck, C.A. Ventrice, F. Wilkelmann, S. Wohlrab, C. Xu, T. Bertrams, A. Brodde and H. Neddermayer, J. MoL Catal. 82 (1993) 143. J. Libuda, M. Biiumer and H.-J. Freund, J. Vac. Sci. Technol. A 12 (1994) 2259. J.C. Lin, N. Shamir and R. Gomer, Surf. Sci. 206 (1988) 61, 86. J. Szanyi and D.W. Goodman, J. Catal. 145 (1994) 508. J.E. Whitten and R. Gomer, Surf. Sci. 316(1994) 1. A. Pavlovska and E. Bauer, Surf. Sd. 175 (1986) 369. I. Malik and J. Hrbek, J. Phys. Chem. 95 (1991) 2466. D.G. Van Campen and J. Hrbek, J. Phys. Chem. 99 (1995) 16389. H. Bludau, M. Skottke, B. Pennenmann, P. Mrozek and K. Wandelt, Vacuum 41 (1990) 1106. J.A. Rodriguez, J. Phys. Chem. 97 (1993)6509. J. Hrbek, A.K. Schmid and R.Q. Hwang, in preparation. G. Beitel, K. Markert, J. Wiechers, J. Hrbek and R.J. Behm, in: Adsorption on Ordered Surfaces of Ionic Solids and Thin Films, Eds. J. Umbach and H.-J. Freund (Springer, Berlin, 1993) pp. 71-82. (a) C.H. Bartholomew, P.K. Agrawal and J.R. Katzer, Adv. Catal. 31 (1982) 135; (b) J. Oudar and H. Wise, Eds., Deactivation and Poisoning of Catalysts (Dekker, New York, 1985). J.K.A. Clarke, Chem. Rev. 75 (1975) 291. R. Prins, V.H.J. de Beer and G.A. Somorjai, Catal. Rev.-Sci. Eng. 31 (1989) 1. R.R. Chianelli, M. Daage and M.J. Ledoux, Adv. Catal. 40 (1994) t77.

286 J.A. Rodriouez/Surface Science Reports 24 (1996) 223-287
[314]
[315] [316] [317] [318] [319] [320] [321] [3223 [323] [3243 [325] [326] [327] [328] [329] [330] [331] [332] [333] [334] [335] [336] [337] [338] [339] [3403 [341] [342] [343]
[3443 [345] [346] [347] [348] [349] [350] [3513 [3523 [353] [354] [3553 [356] [357] [358] [359] ~[36o] [361] [362]
(a) C.C. Knight and G.A. Somorjai, Surf. Sci. 240 (1990) 101; (b) D.A. Chen, C.M. Friend and H. Xu, Langmuir 12 (1996) 1528. M. Kuhn, J.A. Rodriguez and J. Hrbek, Surf. Sci. 314 (1994) L897. M. Kuhn and J.A. Rodriguez, Chem. Phys. Lett. 231 (1994) 199. M. Kuhn and J.A. Rodriguez, J. Phys. Chem. 98 (1994) 12059. J.C. Dunphy, C. Chapvlier, D.F. Ogletree and M.B. Salmeron, J. Vac. Sci. T~hnol. B 12 (1994) 1742. M. Kuhn and J.A. Rodriguez, Catal. Lett. 32 (1995) 345. J.A. Rodriguez and M. Kuhn, J. Phys. Chem. 99 (1995) 9567. M. Kuhn and J.A. Rodriguez, J. Catal. 154 (1995) 355. F.H. Ribeiro, A.L. Bonivardi, C. Kim and G.A. Somorjai, J. Catal. 150 (1994) 186, and references therein. M. Kuhn and J.A. Rodriguez, Surf. Sci., 355 (1996) 85. M. Kuhn and J.A. Rodriguez, Surf. Sci. 336 (1995) 1. W.K. Kuhn, J.-H. He and D.W. Goodman, J. Vac. Sci. Technol. A 10 (1992) 2477. J.A. Rodriguez, M. Kuhn and J. Hrbek, J. Phys. Chem. 100 (1996) 3799. M. Kuhn, J.A. Rodriguez and J. Hrbek, Surf. Sci., in press (1996). J.A. Rodriguez, M. Kuhn and J. Hrbek, submitted. 3. Hrbek, J.L. Stevens and R.Q. Hwang, submitted. (a) P.J. Feibelman and D.R. Hamann, Surf. Sci. 149 (1985) 48; (b) P.J. Feibelman, Phys. Rev. B 43 (1991) 9452. D.R. Mullins, P.F. Lyman and S.H. Overbury, Surf. Sci. 277 (1992) 64. J.A. Rodriguez, M. Kuhn and J. Hrbek, Chem. Phys. Lett. 251 (1996) 13. Lange's Handbook of Chemistry, 13th ed. (MacGraw-Hill, New York, 1985) Table 9'1. L.Z. Mezey and J. Giber, Jpn. J. Appl. Phys. 21 (1982) 1569. S. Harris and R.R. Chianelli, 3. Catal. 98 (1986) 17. D.R. Kahn, E.E. Petersen and G.A. Somorjai, J. Catal. 34 (1974) 294. B.A. Sexton and G.A. Somorjai, J. Catai. 46 (1977) 167. D.W. Goodman, R.D. Kelley, T.E. Madey and 3.T. Yates, J. Catal. 63 (1980) 226. H.P. Bonzel and H.J. Krebs, Surf. Sci. 91 (1980) 499. C.T. Campbell and M.T. Paffett, Surf. Sci. 139 (1984) 396. J.A. Rodriguez and D.W. Goodman, Surf. Sci. Rep. 14 (1991) 1. C.T. Campbell, Adv. Catal. 36 (1989) 1. (a) 3.W.A. Sachtler and G.A. Somorjai, J. Catai. 81 (1983) 77; (b) R.C. Yeates and G.A. Somorjai, J. Catal. 103 (1987) 208. 3.W.A. Sachtler and G.A. Somorjai, J. Catal. 89 (1984) 35. C.H.F. Peden and D.W. Goodman, 3. Catal. 104 (1987) 347. C.M. Greenlief, P.M. Berlowitz, D.W. Goodman and 3.M. White, J. Phys. Chem. 91 (1987) 6669. D.3. Godbey, F. Garin and G.A. Somorjai, J. Catal. 117 (1989) 144. 3.A. Rodriguez and D.W. Goodman, J. Phys. Chem. 94 (1990) 5342. 3.C. Ikrtolini, 3.L. Rousset, P. Miegge, J. Massardier and B. Tardy, Surf. Sci. 287/288 (1993) 346. 3. Massardier and 3.C. Bertolini, 3. Catal. 90 (1984) 358. 3. Szanyi and M.T. Paffett, 3. Am. Chem. Soc. 117 (1995) 1034. P. Miegge, J.L. Rousset, B. Tardy, 3. Massardier and 3.C. Bertolini, J. Catal. 149 (1994) 404. J. Szanyi and M.T. Paffett, Catal. Lett. 29 (1994) 133. A.D. Logan and M.T. Paffett, J. Catal. 133 (1992) 179. P.3. Berlowitz, C.H.F. Peden and D.W. Goodman, J. Phys. Chem. 92 (1988) 5213. 3. Szanyi and D.W. Goodman, Catal. Lett. 14 (1992) 27. B.G. Johnson, C.H. Bartholomew and D.W. Goodman, J. Catal. 128 (1991) 231. C.H.F. Peden and D.W. Goodman, Ind. Eng. Chem. Fundam. 25 (1986) 58. D.W. Goodman, in: Proc. IUCCP Conf. on Heterogeneous Catalysis (Texas A&M University, 1984). D.W. Goodman, Surf. Sci. 123 (1982) L679. R.A. Campbell, J. Guan and T.E. Madey, Catal. Lett. 27 (1994) 273. J.R. Engstrom, D.W. Goodman and W.H. Heinberg, 3. Am. Chem. Soc. 110 (1988) 8305; 3. Phys. Chem. 94 (1990) 396.

J..4. Rodriguez/Surface Science Reports 24 (1996) 223-287 287
[363] A.K. Datye, B.F. Hegarty and D.W. Goodman, Faraday Disc. Chem. Soc. 87 (1989) 18. [364] J. Szanyi, S. Anderson and M.T. Paffett, J. Catal. 149 (1994) 438. [365] J.C. Bertolini and J. Massardier, Catal. Lett. 9 (1991) 183. [366] C.H. Patterson, J.M. Muldenar, P.Y. Timbrell, A.J. Gellman and R.M. Lambert, J. Phys. Chem. 92 (1988) 1266. [367] C.H. Patterson and R.M. Lambert, J. Phys. Chem. 92 (1988) 1266.
Copyright © 2022 FDOKUMEN




















