
Photocatalytic reactions over TiO2 supported on porcelain spheres
Upload
independentCategory
view
3download
0
Journal of Chemical Technology and Biotechnology J Chem Technol Biotechnol 81:1787–1796 (2006)
Photocatalytic ozonationof gallic acid in waterFernando J Beltran,∗ Olga Gimeno, Francisco J Rivas and Marıa CarbajoDepartamento de Ingenierıa Quımica y Energetica, Universidad de Extremadura, 06071 Badajoz, Spain
Abstract: Aqueous solutions of gallic acid have been treated with five different oxidation-radiation processes: visibleand ultraviolet A radiation (VUVA), TiO2 adsorption, ozonation, VUVA/TiO2 photocatalysis and VUVA/O3/TiO2
photocatalytic ozonation. With the exception of VUVA radiation and TiO2 adsorption, ozone and photolyticprocesses allow for the total removal of gallic acid in a period between 50 and 90 min. The time taken toachieve 100% gallic acid conversion depends on the oxidation process applied, photocatalytic ozonation beingthe most effective technique. Throughout the process, oxalic and formic acids were identified as byproducts.Some other unidentified compounds probably related to pyruvic, malonic and maleic acids were also detected.The appearance of these compounds can be justified from direct reactions of both hydroxyl radical and ozonein water. Only photocatalytic ozonation leads to total mineralisation of the organic matter in less than 90 min.The photocatalyst used, TiO2, showed good activity and stability (no leaching was observed) after five consecutivephotocatalytic ozonation runs with the same semiconductor-catalyst mass. 2006 Society of Chemical Industry
Keywords: gallic acid; photocatalytic ozonation; mineralisation; water treatment
INTRODUCTIONGallic acid is a common polyphenol present indifferent agroindustrial wastewaters.1 With the aimof collecting data for wastewater remediation, itis usually taken as a model compound to studythe reactivity of polyphenols subjected to differentoxidation processes, and the literature reports studiesof the ozonation alone2 and catalytic ozonation withactivated carbon3 of gallic acid in water. As with manyother phenolic compounds, gallic acid reacts rapidlywith ozone. Moreover, owing to the dissociatingcharacter of phenols, the rate constant betweenozone and gallic acid increases with increase in pH.4
Consequently, the application of combined ozonation(ozone combined with other agents such as hydrogenperoxide or activated carbon) to eliminate gallic acidfrom water is mainly aimed at mineralisation of theorganic content of the water rather than the studyof gallic acid removal. The use of these processesis mainly intended to mineralise ozone refractorygallic acid oxidation end-products. Gallic acid israpidly degraded by ozone to yield first unsaturatedcarboxylic acids. The latter are further converted tosaturated carboxylic acids and aldehydes. Becausethese compounds do not react with ozone, theyfinally tend to accumulate in water. Accordingly, theformation of the latter compounds and the evolutionof total organic carbon (TOC) are the two parametersthat really matter in combined ozonation studies ofphenols.
Photocatalytic ozonation is another possible tech-nique to remove and mineralise organic contaminantsfrom water. Photocatalytic ozonation is a recentadvanced oxidation process applied to increase theconcentration of hydroxyl radicals in water. Hydroxylradicals are the principal oxidant species allowing forthe mineralisation of the organic matter.5–7 It is a pro-cess that combines the positive effects of ozonation andphotocatalytic oxidation. The mechanism relies on thesemiconductor properties of titania and the chemicalproperties of ozone to produce free radicals.8,9 Pho-tocatalytic oxidation is based on the absorption oflight energy by the semiconductor surface to exciteelectrons from the valence band to the conductionband. In this process, very oxidant positive holes inthe valence band leading to hydroxyl radical forma-tion are generated. However, this system presents thedrawback of electron–positive hole recombination thatinhibits the oxidation route.8 To avoid this recombi-nation, the presence of some oxidant species, usuallyoxygen, to trap the electrons is necessary. Applica-tion of ozone to this process not only can enhancethe removal of electrons from the conduction bandbut also produces ozonide ion radicals (O•−
3 ) thatrapidly lead to hydroxyl radicals.10 Thus, photocat-alytic ozonation is a technique that could lead tohydroxyl radicals through two ways: from the positivehole:
h+ + OH− −−−→ HO•(1)
∗ Correspondence to: Fernando J Beltran, Departamento de Ingenierıa Quımica y Energetica, Universidad de Extremadura, Avenida de Elvass/n, 06071Badajoz, SpainE-mail: [email protected]/grant sponsor: CICYTContract/grant sponsor: European Feder Funds; contract/grant number: PPQ2003/00554(Received 10 February 2006; revised version received 24 May 2006; accepted 25 May 2006)Published online 2 October 2006; DOI: 10.1002/jctb.1605
2006 Society of Chemical Industry. J Chem Technol Biotechnol 0268–2575/2006/$30.00

FJ Beltran et al.
and from the ozone–electron reaction:
e− + O3 −−−→ O−•3
H+−−−−−→ O2 + HO•
(2)
The main purpose of this work was, therefore, tocheck the synergistic effect of the combination of ozoneand TiO2 photocatalysis on the mineralisation of gallicacid in water.
MATERIALS AND METHODSGallic, formic and oxalic acids were of either reagent oranalytical grade purchased from Sigma (St Louis, MO,USA) and used as received. Organic solvents wereof HPLC grade obtained from Panreac (Barcelona,Spain).
A commercial sample of TiO2, Degussa (Frankfurt,Germany) P25 (70% anatase and 30% rutile), wasused as catalyst with average particle size of 30 nm anda BET surface area of 50 m2 g−1.
Ultra-pure water provided by a Milli-Q Academicsystem (Millipore, Bedford, MA, USA) was usedthroughout.
Figure 1 shows the experimental set-up used. A1-L capacity tubular borosilicate glass photoreactor(450 mm long, 80 mm diameter), depicted in aprevious paper,11 was used in all experiments. Thereactor walls were first covered with aluminiumfoil and then with an insulating material to avoidrelease of radiation and heat to the surroundings.The degradation of gallic acid was carried out atatmospheric pressure in 0.85 L of aqueous solutioncontaining approximately 170 mg L−1 of gallic acidat a constant stirring rate. Aqueous solutions wereprepared at their natural pH (3.5) and werecomparatively degraded by O3, visible and ultravioletA radiation (VUVA), O3/VUVA, O2/TiO2/VUVAand O3/VUVA/TiO2 systems.
In ozonation experiments, ozone containing oxygenwas bubbled continuously into the solution througha diffuser placed at the bottom of the reactor andflowed upwards in the annular section between the
Figure 1. Experimental set-up: 1, oxygen cylinder; 2, ozonegenerator; 3, ozone analyser; 4, thermostatic bath; 5, gas flow ratemeter; 6, gas feeding system; 7, oxidation contactor; 8, samplingpoint; 9, electric power system for the VUVA lamp.
UVA–visible lamp well and the reactor wall. Ozonewas produced from pure oxygen using a SanderLaboratory Ozonator (Ingetec SA, Barcelona, Spain).The gas flow was maintained constant at 50 L h−1 withan ozone concentration of 5 mg L−1.
In experiments with VUVA radiation, the aqueoussolution was irradiated with a high-pressure mercurylamp (Heraeus, Madrid, Spain, TQ 718, 700 W)immersed in a glass cooling well placed in the middleof the reactor. The lamp bandwidth was in the range238–579 nm. However, UVB radiation was cut offbecause of the presence of the glass well. Then, thetwo main wavelengths were 313 and 366 nm withincident radiations of 0.189 and 0.331 einstein h−1,respectively, according to the manufacturer.
When TiO2 was used, the solid was maintained insuspension by magnetic stirring with a concentrationof 1.5 g L−1 in all the experiments. Prior to theanalysis, the solid was removed from samples by firstcentrifuging in micro-Eppendorf tubes for 6 min at12 000 rpm (Eppendorf 5415D centrifuge; Madrid,Spain) and finally by filtration through Millex-HAfilters (Millipore, 0.45 µm). This applied both toadsorption alone and to photocatalytic oxidationexperiments.
Gallic acid was determined by high-performance liq-uid chromatography (HPLC) (Series 1100, Hewlett-Packard, Palo Alto, CA, USA) with a Kromasil C18
column and UV detector. The analysis was performedin the isocratic mode with acetonitrile–water (10:90)containing 0.1% phosphoric acid as the mobile phase.Gallic acid was detected at 268 nm.
The concentrations of by-products, oxalic, formic,pyruvic, maleic and malonic acids, were also moni-tored by HPLC using a Supelcogel C-610H column(Supelco, Bellefonte, PA, USA) and UV detector. Themobile phase was water containing 0.1% phosphoricacid at a flow-rate of 0.75 mL min−1. The wavelengthof the UV detector was set at 210 nm.
The dissolved ozone was measured before solidfiltration with the method proposed by Baderand Hoigne12 based on the decoloration of 5,5,7-indigotrisulfonate. For that purpose, a Thermo Spec-tronic Helios (Madrid, Spain) α spectrophotometerwas used. Additionally, ozone in the gas phase wasmonitored by means of an Anseros (Madrid, Spain)Ozomat ozone analyser. The analysis was based on theabsorbance at 254 nm.
The total organic carbon (TOC) content ofthe samples was measured using a TOC analyser(OI Analytical TOC-1010, Microbeam, Barcelona,Spain). The pH of the reaction solution was measuredwith a Crison (Barcelona, Spain) 507 pH-meter.
RESULTS AND DISCUSSIONIn a first attempt to establish the importance ofphotocatalytic ozonation of gallic acid, some exper-iments with different oxidising processes were carried
1788 J Chem Technol Biotechnol 81:1787–1796 (2006)DOI: 10.1002/jctb

Photocatalytic ozonation of gallic acid in water
0 50 100 150 2000
0.2
0.4
0.6
0.8
1
1.2
Time (min)
C/C
o
Figure 2. Time distribution curves of remaining gallic aciddimensionless concentration corresponding to oxidation–radiationexperiments in water. Conditions: gallic acid initial concentration,10−3 mol L−1; pH, 3.5; T0 = 20 ◦C; CO3inlet = 10−4 mol L−1; gasflow-rate, 50 L h−1; CTiO2 , 1.5 g L−1, intensity of radiation, 0.189 and0.366 einstein h−1 at 313 and 366 nm, respectively. Symbols: °,VUVA radiation; �, adsorption; ž, ozonation; �, O3/VUVA; �,O2/VUVA/TiO2; �, O3/VUVA/TiO2.
out. Figure 2 shows the evolution of the remain-ing dimensionless concentration of gallic acid withtime corresponding to VUVA radiation, ozonationalone (O3), TiO2 adsorption, photocatalytic oxidation(VUVA/TiO2/O2), ozone photolysis (VUVA/O3) andphotocatalytic ozonation (VUVA/TiO2/O3). It can beseen that under the experimental conditions appliedin this work, all processes, except TiO2 adsorption
and VUVA radiation, allow for high degradation ratesof gallic acid, reaching complete elimination of thispolyphenol in the interval 50–90 min, depending onthe oxidation system applied. Among oxidation pro-cesses, photocatalytic ozonation is the process allowingfor the highest degradation rate with complete elimi-nation of gallic acid in 50 min.
Formation of intermediates: comparison resultsDuring the reaction period, several intermediatesproducts are formed. Figure 3(a)–(d) depicts theintermediates detected in the different oxidation pro-cesses applied (intermediates from VUVA radiationare not shown since this process hardly removesgallic acid from water). Two intermediates wereidentified: oxalic and formic acids. In addition, thechromatograms clearly showed other peaks that couldnot be assigned to any specific compound. In anycase, their retention times were close to those ofmaleic, pyruvic and malonic acids (Fig. 4). Therefore,concentrations of these unknown intermediates weremeasured as maleic, pyruvic and malonic acids. Theintermediates detected can then be classified into twotypes: those that are formed and removed after somereaction time (formic acid and the other unidentifiedcompounds), and oxalic acid, which, in all cases butone (when VUVA/TiO2/O3 is applied), is present inwater after the total reaction time of 240 min has beenreached. In Fig. 3 the evolution of intermediate con-centration for oxalic and formic acids and unidentifiedcompounds (measured as maleic, pyruvic and malonic
0 50
(a)
100 150 200 2500
20
40
60
80
100
120
Time (min)
C (m
g L
-1)
0
10
20
30
40
C (m
g L
-1)
(b)
00 50 100 150 200 250
20
40
60
80
Time (min)
C (m
g L
-1)
0
5
10
15
20
25
30
C (m
g L
-1)
(c)
0 40 60 800
10
20
30
40
Time (min)
C (m
g L
-1)
0
2
4
6
8
10
12
14
C (m
g L
-1)
(d)
0 100 150 200 2500
20
40
60
80
Time (min)
C (m
g L
-1)
0
2
4
6
8
10
C (m
g L
-1)
Figure 3. Time distribution of intermediate concentrations during the oxidation–radiation processes of gallic acid in water. Conditions as in Fig. 2.(a) Ozonation; (b) O3/VUVA; (c) O3/VUVA/TiO2; (d) O2/VUVA/TiO2. Symbols: °, oxalic acid; �, formic acid; ž, intermediate measured as malonicacid; ♦, intermediate measured as maleic acid; �, intermediate measured as pyruvic acid.
J Chem Technol Biotechnol 81:1787–1796 (2006) 1789DOI: 10.1002/jctb

FJ Beltran et al.
C C
O
OHHO
O
Oxalic acid
H C
O
OH
Formic acid
C C
O
HO
O
CH3
Pyruvic acid
C CH2
HO
O
C
Malonic acid
OH
O
C CH
HO
O
CH C
O
OH
Maleic acid
Figure 4. Structures of intermediates detected or tentativelydetected.
acids) is shown after 20–50 min from the start of oxi-dation. It should be pointed out that quantitative orqualitative analysis of these compounds with the useof the analytical procedure available was not possiblewhile gallic acid at moderate concentration remainedin water. All intermediates are formed within the first20–40 min of reaction, giving an idea of the very fastoxidation of gallic acid (see Fig. 2). From Fig. 3, twomain conclusions can be drawn: (1) oxalic acid is thelast intermediate formed in the oxidation processes and(2) photocatalytic processes, especially when ozoneis present, allow for the highest intermediate oxida-tion rates with complete removal of these compounds(except oxalic acid) in 60 min. In ozonation alone andozone/VUVA processes, intermediates are present inwater for longer times. Thus, a maleic acid-relatedintermediate is present in water up to 200 min in theseprocesses. Differences in the reaction rates of inter-mediates are probably due to the different pathwaysof the oxidation processes, especially when ozone ispresent. Therefore, in photocatalytic ozonation thereare more ways to produce hydroxyl radicals, the mainoxidising species in these systems (see later), than inozonation alone or the ozone/VUVA system. Hencethe oxidation rates will be higher in photocatalyticozonation.
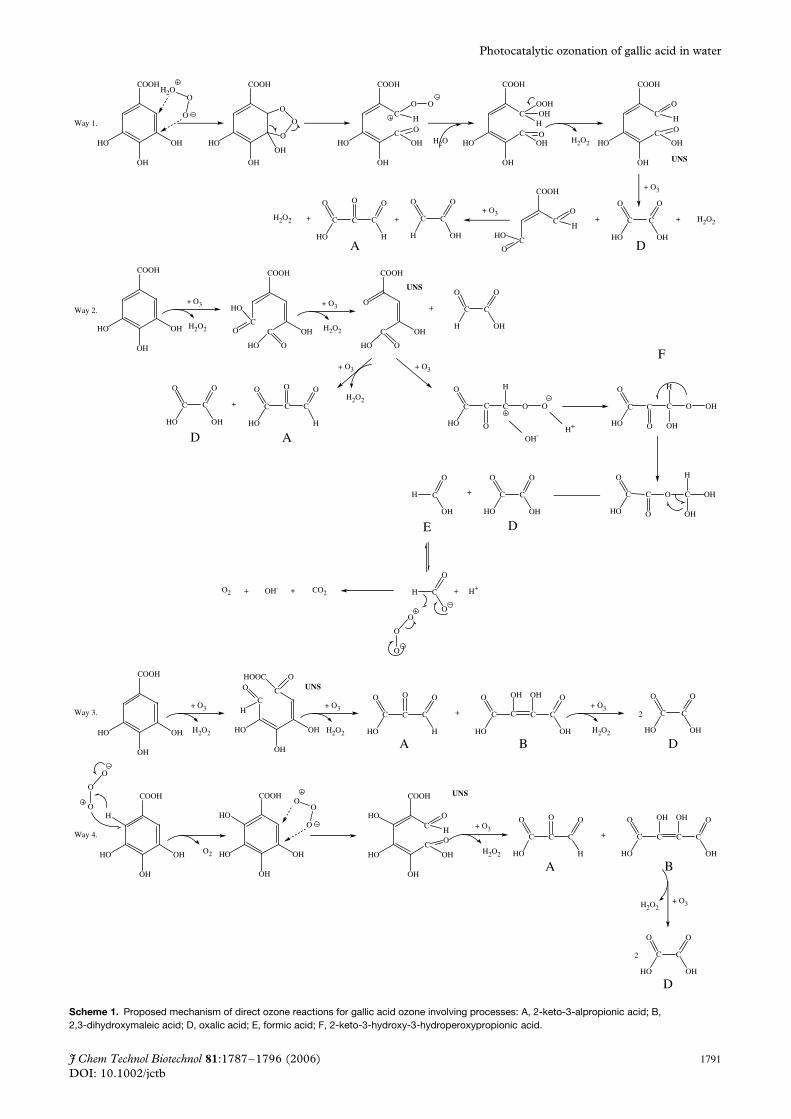
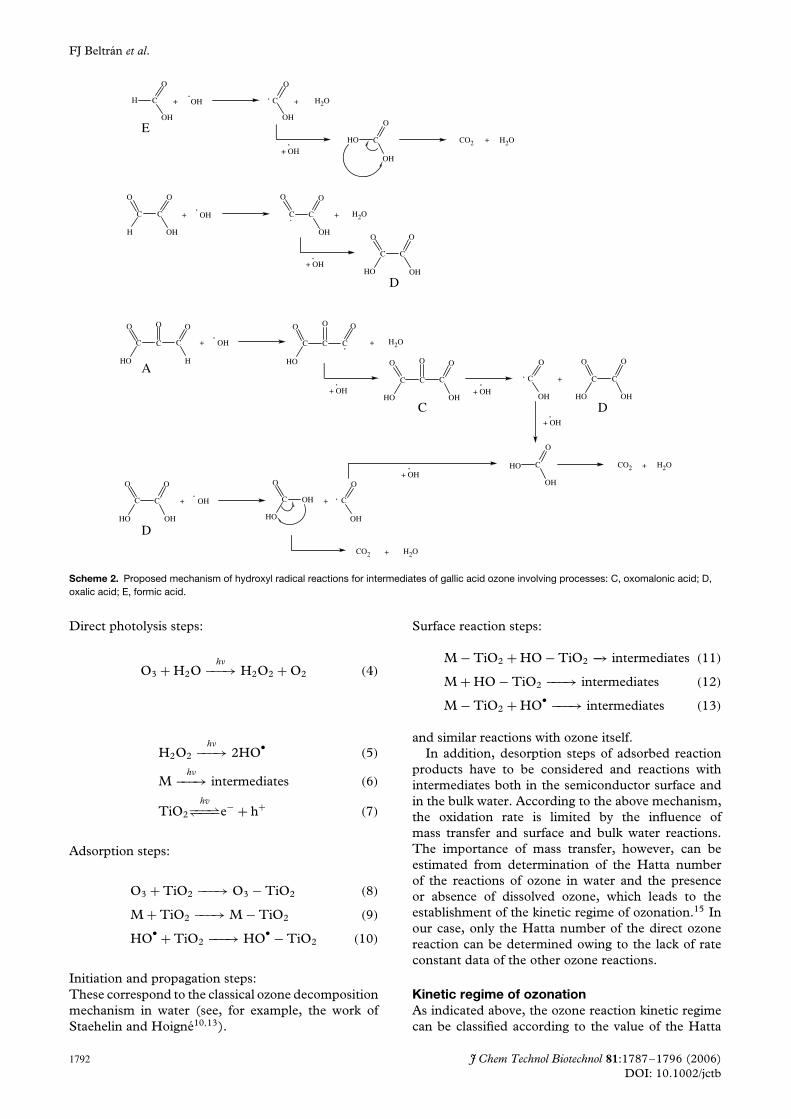
Mechanisms of reactionsFormation of the intermediates detected in this workcan be justified from classical ozone reactions inwater, both from direct reactions and from reac-tions with hydroxyl free radicals formed in ozone andphotocatalytic processes. Schemes 1 and 2 show theproposed reaction mechanisms for gallic acid withozone and hydroxyl radicals accounting for the forma-tion of intermediates and mineralisation, respectively.Scheme 1 shows the formation mechanism of unsat-urated and saturated carboxylic acids (some of themdetected in this work) from ozone direct reactions (aro-matic dipolar cycloaddition and double bond additionreactions).9 From this mechanism, it is seen that com-pounds such as 2-ketopropionaldehyde (A) and 2,3-dihydroxymaleic acid (B) can be formed from directreactions of ozone with gallic acid and first unsatu-rated carboxylic acids (marked UNS in Scheme 1).As shown in Scheme 2, the former compound can
lead to oxomalonic acid (C) after hydroxyl radicaloxidation. Both compounds A or C and B could beresponsible for peaks formed at retention times closeto those of malonic, pyruvic and maleic acids, respec-tively, although confirmation with other techniques isneeded. Also, oxalic and formic acids can be formedby different pathways. Thus, oxalic acid (D), which isthe most abundant intermediate detected in all pro-cesses studied, can be formed both from ozone andhydroxyl radical reactions as shown in Schemes 1 and2. Formic acid (E), on the other hand, can be formedfrom abnormal ozonolysis of hydroxyhydroperoxide-type compounds (F) and decompose to carbon dioxideand water through reactions with ozone (Scheme 1) orhydroxyl radicals (Scheme 2). Oxalic acid, however, ismainly mineralised through hydroxyl radical reactions(Scheme 2). As also observed from Scheme 1, directreactions of ozone to break the aromatic ring or doublebonds lead to hydrogen peroxide although at the pHof the reactions (3.5) it is not expected that the reac-tion between hydrogen peroxide and ozone13 will yieldfree radicals. These results are in good agreement withthe fast reactions of ozone with phenols and unsatu-rated carboxylic acids (probably first intermediates notdetected in this work) that, at the pH investigated, haverate constants between 103 and 105 L mol−1 s−1.2,4 Onthe other hand, rate constants of ozone with saturatedcarboxylic acids can vary from 100 L mol−1 s−1 (inthe case of the formic acid–ozone reaction) and lessthan 4 × 10−2 L mol−1 s−1 (in the case of the oxalicacid–ozone reaction)4. In any case, these values sug-gest that direct reactions of ozone with the byproductsdetected cannot afford the removal of low molecu-lar weight saturated carboxylic acids, especially oxalicacid, from water at the reaction times observed inFig. 3. In addition, it is known that hydroxyl freeradicals are formed in ozonation (and photocatalytic)processes when no fast direct ozone reactions develop,i.e. when the direct reactions between ozone andorganics (gallic acid in this case) are in the moder-ate or low kinetic regime of absorption.9,14 This wasalso confirmed in this work, as shown below.
The presence of hydroxyl radicals implies thedevelopment of a free radical mechanism of reactionsstarting with the initiation reactions. Also, in ozoneprocesses, mass transfer resistance in the liquid filmclose to the gas–water interface has to be considered.In fact, under the conditions investigated, especiallyduring the first 40–60 min of reaction, where thedissolved ozone concentration is low, the oxidationrate is probably mass transfer controlled. To have anidea about the complex mechanism of these processes,the case of photocatalytic ozonation is considered,since it involves all possible reactions present in theother processes studied. A simplified mechanism ofreactions would involve, at least, the following steps:Mass transfer step:
O3(g)kLa−−−→ O3 (3)
1790 J Chem Technol Biotechnol 81:1787–1796 (2006)DOI: 10.1002/jctb
Photocatalytic ozonation of gallic acid in water
COOH
OH
OH
HO
COOH
OH
OH
HO
O
O
O C
C
COOH
OH
OH
HO
O
OH
C
C
COOH
OH
OH
HO
OH
OH
OOH
C
C
COOH
OH
OH
HO
OH
OO
C
C
COOH
HOH
O
O
C C
O
OHHO
O
H2O2C C
O
OHH
O
H2O2
+ O3
+ O3+ + + +
H2O H2O2
COOH
OH
OH
HO
+ O3
CC
COOH
OH
HO
O
HO
O
O
C
COOH
OH
HO O
C C
O
OHH
O
C C
O
OHHO
O O
HO
O
CCC
O
H OHO
O
CCC O
H
O
OHO
O
CCC O
H
OH
OH
C C
OHO
O
O C OH
H
OH
CC C
O
OHHO
O
H
O
OH
H2O2
+ O3
H2O2
+
+ O3+ O3
H2O2+
+
OH-H+
COOH
OH
OH
HO
CC
HOOC
OH
OH
HO
OO
H
O
HO
O
CCC
O
H HO
O
CCC
OH OH
C
O
OH
C C
O
OHHO
O
2++ O3
H2O2
+ O3
H2O2
+ O3
H2O2
Way 1.
Way 2.
Way 3.
H2OO
O
AD
DE
A B
O
HO
O
CCC
O
HA
H C
O
O
H+CO2OH-O2 + + +
O
O
O
COOH
OH
OH
HO
Way 4.
COOH
OH
OH
HO
H
O
O
O
HO
OO
O
C
C
COOH
OH
OH
HO
HO O
HO C C
O
HO
O
C
O
H
C C
HO
O
C
OH OH
C
O
OH
+
A B
C C
O
OHHO
O
2
O2 H2O2
+ O3
H2O2+ O3
D
D
D
F
UNS
UNS
UNS
UNS
Scheme 1. Proposed mechanism of direct ozone reactions for gallic acid ozone involving processes: A, 2-keto-3-alpropionic acid; B,2,3-dihydroxymaleic acid; D, oxalic acid; E, formic acid; F, 2-keto-3-hydroxy-3-hydroperoxypropionic acid.
J Chem Technol Biotechnol 81:1787–1796 (2006) 1791DOI: 10.1002/jctb
FJ Beltran et al.
H C
O
OH
OH+ C
O
OH
+ H2O
HO C
O
OH
CO2 + H2O
C C
O
OHHO
O
OH+
+ OH
C OH
HO
O
C
O
OH
+
CO2 + H2O
C
O
HO
O
C C
O
H
OH+
O
HO
O
CCC
O
+ H2O
O
HO
O
CCC
O
OH
C C
O
OHH
O
OH+ C C
OO
H2O+
C C
O
OHHO
O
+ OH
+ OH
HO C
O
OH
CO2 + H2O
C
O
OH
C C
O
OHHO
O
+
+ OH
OH
+ OH
+ OH
D
C
A
D
D
E
Scheme 2. Proposed mechanism of hydroxyl radical reactions for intermediates of gallic acid ozone involving processes: C, oxomalonic acid; D,oxalic acid; E, formic acid.
Direct photolysis steps:
O3 + H2Ohν−−−→ H2O2 + O2 (4)
H2O2hν−−−→ 2HO•
(5)
Mhν−−−→ intermediates (6)
TiO2
hv−−−⇀↽−−−e− + h+ (7)
Adsorption steps:
O3 + TiO2 −−−→ O3 − TiO2 (8)
M + TiO2 −−−→ M − TiO2 (9)
HO• + TiO2 −−−→ HO• − TiO2 (10)
Initiation and propagation steps:These correspond to the classical ozone decompositionmechanism in water (see, for example, the work ofStaehelin and Hoigne10,13).
Surface reaction steps:
M − TiO2 + HO − TiO2 −→ intermediates (11)
M + HO − TiO2 −−−→ intermediates (12)
M − TiO2 + HO• −−−→ intermediates (13)
and similar reactions with ozone itself.In addition, desorption steps of adsorbed reaction
products have to be considered and reactions withintermediates both in the semiconductor surface andin the bulk water. According to the above mechanism,the oxidation rate is limited by the influence ofmass transfer and surface and bulk water reactions.The importance of mass transfer, however, can beestimated from determination of the Hatta numberof the reactions of ozone in water and the presenceor absence of dissolved ozone, which leads to theestablishment of the kinetic regime of ozonation.15 Inour case, only the Hatta number of the direct ozonereaction can be determined owing to the lack of rateconstant data of the other ozone reactions.
Kinetic regime of ozonationAs indicated above, the ozone reaction kinetic regimecan be classified according to the value of the Hatta
1792 J Chem Technol Biotechnol 81:1787–1796 (2006)DOI: 10.1002/jctb

Photocatalytic ozonation of gallic acid in water
number.15 The square of this number represents theratio between chemical reaction and mass transferrates and is defined as follows:
Ha =√
kDO3CB
kL(14)
where k, DO3, CB and kL represent the rate constantof the ozone–B direct reaction, the ozone diffusivityin water, the concentration of compound B thatreacts with ozone and the liquid-phase mass transfercoefficient, respectively. The condition for the kineticregime be moderate or low is Ha < 3. Accordingto Eqn (14) and literature data for the values of k,DO3 and kL,2,16 this Hatta value corresponds to aconcentration of gallic acid of 1.3 × 10−3 mol L−1.Since the starting concentration of gallic acid inthe experiments of this work was 10−3 mol L−1 it isdeduced that from the beginning of the ozonationsthe kinetic regime of ozone absorption was moderate.The low dissolved ozone concentration corroboratesthis situation. Also, this means that the formationof any intermediates, even mineralisation, from bothdirect ozone reactions and hydroxyl radical reactions,was possible just from the start of ozonation. Thisjustifies that for some intermediates their maximumconcentration is achieved at reaction times shorterthan 50 min as deduced from Fig. 3. Although notobserved in Fig. 3 (because of analytical interferences),one can deduce that concentrations of intermediatesincrease rapidly, reach a maximum value in manycases and then even decrease during the first 50 minof reaction.
It should be noted that both the moderate kineticregime and low dissolved ozone concentration suggestthat mass transfer has a great influence on the processrate and, hence, an increase in variables such asgas flow-rate would improve the removal rate ofcontaminants regardless of the different pathwaysof oxidation (see Schemes 1 and 2 and reactionsin the free radical mechanism above). However,for longer reaction times, the ozone concentrationbecomes higher and the process starts to be chemicallycontrolled. Then, under these conditions, processessuch as photocatalytic ozonation are more appropriatethan single oxidation processes due to the developmentof more pathways of organic removal (see reactionmechanism above). This is corroborated in Fig. 3,where it is observed that end compounds such as oxalicacid show the highest removal rate in photocatalyticozonation.
MineralisationMineralisation is the second important matter todeal with in combined ozonation processes. Inthis work, different levels of TOC removal wereachieved depending on the oxidation process applied.Figure 5 illustrates the evolution of TOC with timecorresponding to the processes studied, adsorptionalone included. It can be seen that VUVA radiation
0 50 100 150 200 2500
0.2
0.4
0.6
0.8
1
1.2
Time (min)
TO
C/T
OC
o
Figure 5. Time distribution curves of dimensionless remained TOCcorresponding to oxidation–radiation experiments in water.Conditions and symbols as in Fig. 2.
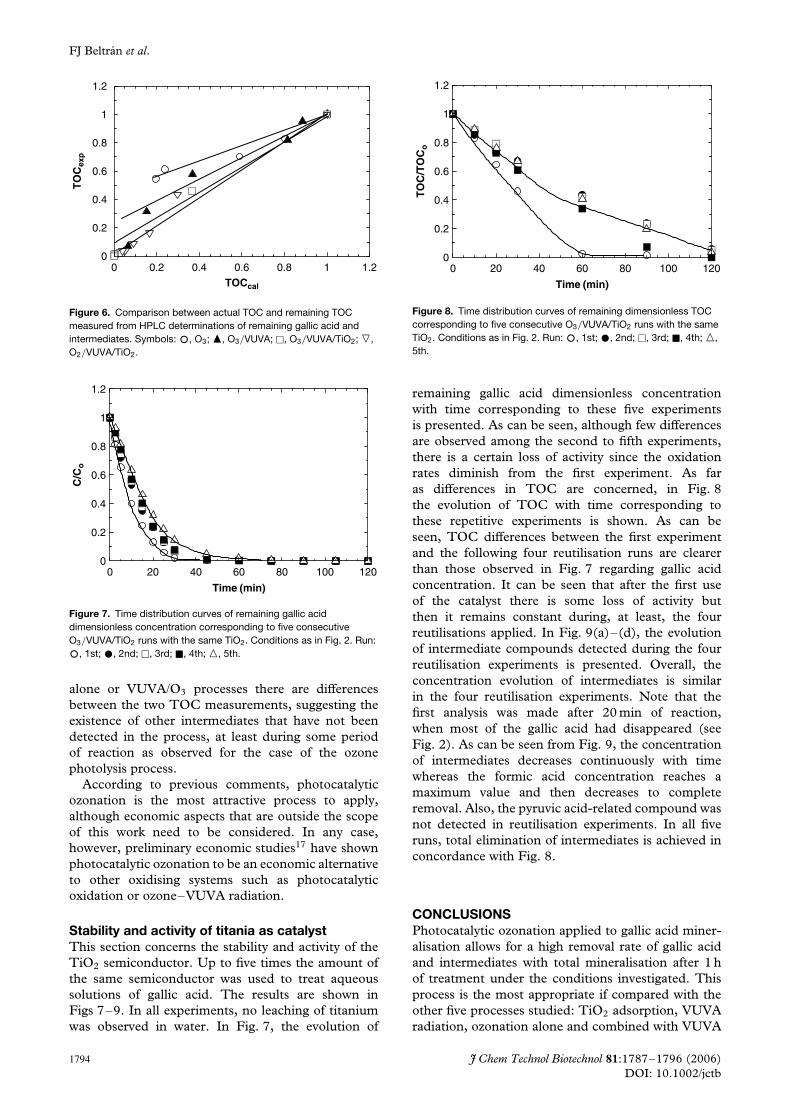
does not lead to any TOC reduction, and adsorptionaccounts for only about 12% of TOC eliminationin 4 h. Photocatalytic (O2/VUVA/TiO2) oxidationallows for almost complete TOC removal in the sameperiod of time while ozonation processes lead to TOCreductions of 55, 95 and 100% when ozone alone orcombined with VUVA radiation or the photocatalyticprocess is used. In spite of the similar TOC reductionsachieved in both photocatalytic processes applied, animportant difference is observed. The photocatalyticoxidation process has an induction period of about40 min, probably needed in order to generate asignificant concentration of hydroxyl radicals in themedium to trigger the oxidation rate. In photocatalyticozonation, this induction period is not observed andthe oxidation rate seems to stay constant during thefirst 40 min, where roughly 80% TOC reductionis reached. In other words, after approximately60 min of reaction, complete mineralisation is achievedwith photocatalytic ozonation whereas only 60%is reached in the photocatalytic oxidation (withoutozone) process. Ozone photolysis also presents aconstant TOC oxidation rate although lower thanthat in photocatalytic ozonation. In the ozone aloneprocess, a rapid oxidation rate is observed at the startof the process, which is followed by a continuousdecrease in oxidation rate during the rest of theexperiment that allows for only a 55% reduction inTOC, probably owing to the low concentration offree radicals that can be generated in this system.Both intermediates and TOC aspects are related inFig. 6 where, for the oxidation processes studied,a plot of the TOC measured with the carbonanalyser (which represents the actual TOC) againstthe TOC determined from the remaining gallic acidand intermediates (oxalic and formic acids and theother three unidentified compounds measured asmalonic, pyruvic and maleic acids) is presented. Ascan be seen, only in the oxidation systems involvingphotocatalysis is the carbon balance complete, at leastat the end of the process, which means that allintermediates formed have been detected. In ozone
J Chem Technol Biotechnol 81:1787–1796 (2006) 1793DOI: 10.1002/jctb
FJ Beltran et al.
0 0.2 0.4 0.6 0.8 1 1.20
0.2
0.4
0.6
0.8
1
1.2
TOCcal
TO
Cex
p
Figure 6. Comparison between actual TOC and remaining TOCmeasured from HPLC determinations of remaining gallic acid andintermediates. Symbols: °, O3; �, O3/VUVA; �, O3/VUVA/TiO2; �,O2/VUVA/TiO2.
0 20 40 60 80 100 1200
0.2
0.4
0.6
0.8
1
1.2
Time (min)
C/C
o
Figure 7. Time distribution curves of remaining gallic aciddimensionless concentration corresponding to five consecutiveO3/VUVA/TiO2 runs with the same TiO2. Conditions as in Fig. 2. Run:
°, 1st; ž, 2nd; �, 3rd; �, 4th; �, 5th.
alone or VUVA/O3 processes there are differencesbetween the two TOC measurements, suggesting theexistence of other intermediates that have not beendetected in the process, at least during some periodof reaction as observed for the case of the ozonephotolysis process.
According to previous comments, photocatalyticozonation is the most attractive process to apply,although economic aspects that are outside the scopeof this work need to be considered. In any case,however, preliminary economic studies17 have shownphotocatalytic ozonation to be an economic alternativeto other oxidising systems such as photocatalyticoxidation or ozone–VUVA radiation.
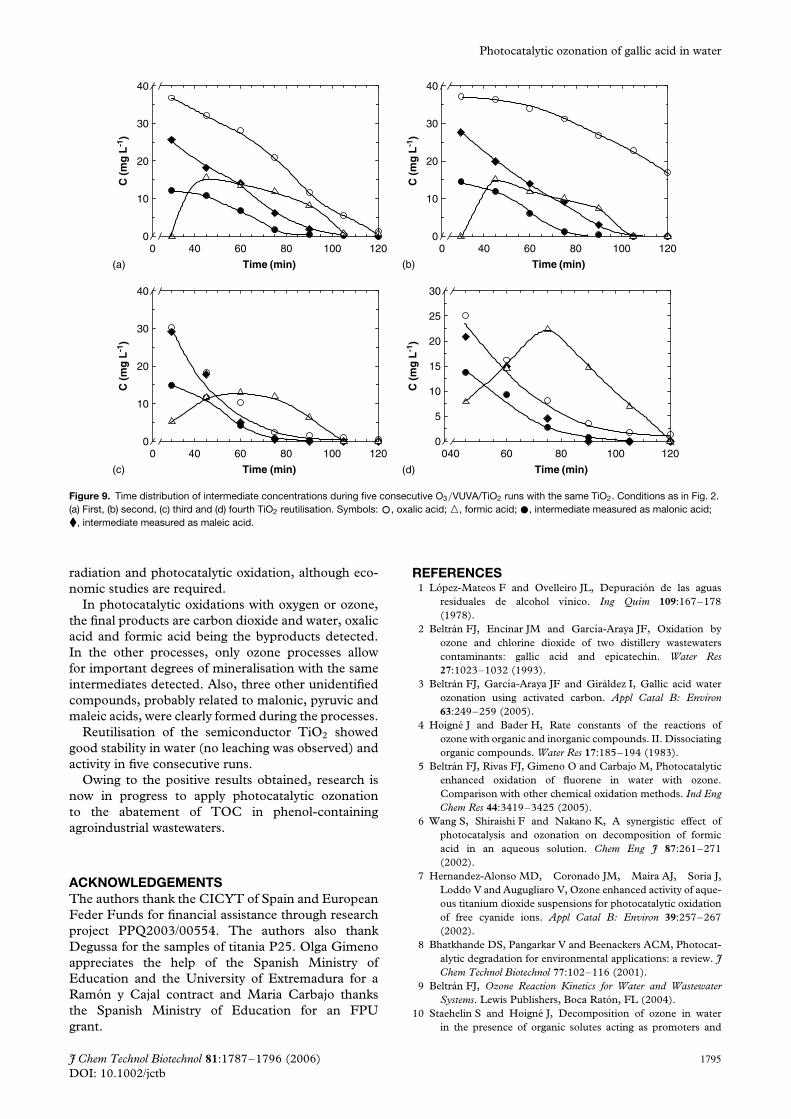
Stability and activity of titania as catalystThis section concerns the stability and activity of theTiO2 semiconductor. Up to five times the amount ofthe same semiconductor was used to treat aqueoussolutions of gallic acid. The results are shown inFigs 7–9. In all experiments, no leaching of titaniumwas observed in water. In Fig. 7, the evolution of
0 40 60 80 100 1200
0.2
0.4
0.6
0.8
1
1.2
Time (min)
TO
C/T
OC
o
20
Figure 8. Time distribution curves of remaining dimensionless TOCcorresponding to five consecutive O3/VUVA/TiO2 runs with the sameTiO2. Conditions as in Fig. 2. Run: °, 1st; ž, 2nd; �, 3rd; �, 4th; �,5th.
remaining gallic acid dimensionless concentrationwith time corresponding to these five experimentsis presented. As can be seen, although few differencesare observed among the second to fifth experiments,there is a certain loss of activity since the oxidationrates diminish from the first experiment. As faras differences in TOC are concerned, in Fig. 8the evolution of TOC with time corresponding tothese repetitive experiments is shown. As can beseen, TOC differences between the first experimentand the following four reutilisation runs are clearerthan those observed in Fig. 7 regarding gallic acidconcentration. It can be seen that after the first useof the catalyst there is some loss of activity butthen it remains constant during, at least, the fourreutilisations applied. In Fig. 9(a)–(d), the evolutionof intermediate compounds detected during the fourreutilisation experiments is presented. Overall, theconcentration evolution of intermediates is similarin the four reutilisation experiments. Note that thefirst analysis was made after 20 min of reaction,when most of the gallic acid had disappeared (seeFig. 2). As can be seen from Fig. 9, the concentrationof intermediates decreases continuously with timewhereas the formic acid concentration reaches amaximum value and then decreases to completeremoval. Also, the pyruvic acid-related compound wasnot detected in reutilisation experiments. In all fiveruns, total elimination of intermediates is achieved inconcordance with Fig. 8.
CONCLUSIONSPhotocatalytic ozonation applied to gallic acid miner-alisation allows for a high removal rate of gallic acidand intermediates with total mineralisation after 1 hof treatment under the conditions investigated. Thisprocess is the most appropriate if compared with theother five processes studied: TiO2 adsorption, VUVAradiation, ozonation alone and combined with VUVA
1794 J Chem Technol Biotechnol 81:1787–1796 (2006)DOI: 10.1002/jctb
Photocatalytic ozonation of gallic acid in water
0
(a)
(c)
(b)
(d)
40 60 80 100 1200
10
20
30
40
Time (min)
C (m
g L
-1)
0 40 60 80 100 1200
10
20
30
40
Time (min)
C (m
g L
-1)
0 40 60 80 100 1200
10
20
30
40
Time (min)
C (m
g L
-1)
040 60 80 100 1200
5
10
15
20
25
30
Time (min)
C (m
g L
-1)
Figure 9. Time distribution of intermediate concentrations during five consecutive O3/VUVA/TiO2 runs with the same TiO2. Conditions as in Fig. 2.(a) First, (b) second, (c) third and (d) fourth TiO2 reutilisation. Symbols: °, oxalic acid; �, formic acid; ž, intermediate measured as malonic acid;�, intermediate measured as maleic acid.
radiation and photocatalytic oxidation, although eco-nomic studies are required.
In photocatalytic oxidations with oxygen or ozone,the final products are carbon dioxide and water, oxalicacid and formic acid being the byproducts detected.In the other processes, only ozone processes allowfor important degrees of mineralisation with the sameintermediates detected. Also, three other unidentifiedcompounds, probably related to malonic, pyruvic andmaleic acids, were clearly formed during the processes.
Reutilisation of the semiconductor TiO2 showedgood stability in water (no leaching was observed) andactivity in five consecutive runs.
Owing to the positive results obtained, research isnow in progress to apply photocatalytic ozonationto the abatement of TOC in phenol-containingagroindustrial wastewaters.
ACKNOWLEDGEMENTSThe authors thank the CICYT of Spain and EuropeanFeder Funds for financial assistance through researchproject PPQ2003/00554. The authors also thankDegussa for the samples of titania P25. Olga Gimenoappreciates the help of the Spanish Ministry ofEducation and the University of Extremadura for aRamon y Cajal contract and Maria Carbajo thanksthe Spanish Ministry of Education for an FPUgrant.
REFERENCES1 Lopez-Mateos F and Ovelleiro JL, Depuracion de las aguas
residuales de alcohol vınico. Ing Quım 109:167–178(1978).
2 Beltran FJ, Encinar JM and Garcıa-Araya JF, Oxidation byozone and chlorine dioxide of two distillery wastewaterscontaminants: gallic acid and epicatechin. Water Res27:1023–1032 (1993).
3 Beltran FJ, Garcıa-Araya JF and Giraldez I, Gallic acid waterozonation using activated carbon. Appl Catal B: Environ63:249–259 (2005).
4 Hoigne J and Bader H, Rate constants of the reactions ofozone with organic and inorganic compounds. II. Dissociatingorganic compounds. Water Res 17:185–194 (1983).
5 Beltran FJ, Rivas FJ, Gimeno O and Carbajo M, Photocatalyticenhanced oxidation of fluorene in water with ozone.Comparison with other chemical oxidation methods. Ind EngChem Res 44:3419–3425 (2005).
6 Wang S, Shiraishi F and Nakano K, A synergistic effect ofphotocatalysis and ozonation on decomposition of formicacid in an aqueous solution. Chem Eng J 87:261–271(2002).
7 Hernandez-Alonso MD, Coronado JM, Maira AJ, Soria J,Loddo V and Augugliaro V, Ozone enhanced activity of aque-ous titanium dioxide suspensions for photocatalytic oxidationof free cyanide ions. Appl Catal B: Environ 39:257–267(2002).
8 Bhatkhande DS, Pangarkar V and Beenackers ACM, Photocat-alytic degradation for environmental applications: a review. JChem Technol Biotechnol 77:102–116 (2001).
9 Beltran FJ, Ozone Reaction Kinetics for Water and WastewaterSystems. Lewis Publishers, Boca Raton, FL (2004).
10 Staehelin S and Hoigne J, Decomposition of ozone in waterin the presence of organic solutes acting as promoters and
J Chem Technol Biotechnol 81:1787–1796 (2006) 1795DOI: 10.1002/jctb

FJ Beltran et al.
inhibitors of radical chain reactions. Environ Sci Technol19:1206–1212 (1985).
11 Beltran FJ, Rivas FJ and Gimeno O, Comparison betweenphotocatalytic ozonation and other oxidation processes forthe removal of phenols from water. J Chem Technol Biotechnol80:973–984 (2005).
12 Bader H and Hoigne J, Detemination of ozone in water by theindigo method. Water Res 15:449–456 (1981).
13 Staehelin S and Hoigne J, Decomposition of ozone in water:rate of initiation by hydroxide ions and hydrogen peroxide.Environ Sci Technol 16:666–681 (1982).
14 Beltran FJ, Theoretical aspects of the kinetics of competitiveozone reactions in water. Ozone Sci Eng 17:163–181(1994).
15 Charpentier JC, Mass transfer rates in gas liquid absorbers andreactors, Adv Chem Eng 11:3–133 (1981).
16 Johnson PN and Davis RA, Diffusivity of ozone in water. J ChemEng Data 41:1485–1487 (1996).
17 Gimeno O, Carbajo M, Beltran FJ and Rivas FJ, Phenol andsubstituted phenols AOPs remediation. J Hazard Mater B119:99–108 (2005).
1796 J Chem Technol Biotechnol 81:1787–1796 (2006)DOI: 10.1002/jctb
Copyright © 2022 FDOKUMEN




















