
Photocatalytic degradation and drug activity reduction of Chloramphenicol
9
Available at www.sciencedirect.com journal homepage: www.elsevier.com/locate/watres Photocatalytic degradation and drug activity reduction of Chloramphenicol A. Chatzitakis a , C. Berberidou a , I. Paspaltsis b , G. Kyriakou c , T. Sklaviadis b , I. Poulios a, a Laboratory of Physical Chemistry, Department of Chemistry, Aristotle University of Thessaloniki, 54124 Thessaloniki, Greece b Prion Disease Research Group, Laboratory of Pharmacology, School of Pharmacy, Aristotle University of Thessaloniki, 54124 Thessaloniki, Greece c Laboratory of Inorganic Chemistry, Department of Chemical Engineering, Aristotle University of Thessaloniki, 54124 Thessaloniki, Greece article info Article history: Received 1 May 2007 Received in revised form 26 June 2007 Accepted 18 July 2007 Available online 27 July 2007 Keywords: Photocatalysis Chloramphenicol Photo-oxidation Drugs ZnO TiO 2 abstract The photocatalytic degradation of Chloramphenicol, an antibiotic drug, has been investigated in aqueous heterogeneous solutions containing n-type oxide semiconductors as photocatalysts. The disappearance of the organic molecule follows approximately a pseudo-first-order kinetics according to the Langmuir–Hinshelwood model. It was observed that, with TiO 2 P-25 as photocatalyst, quantitative degradation of the organic molecule occurs after 4 h of illumination. During this time, the dechlorination of the substrate is complete, while the organic nitrogen was recovered in the form of nitrate and ammonium ions. The effect of temperature on the degradation rate of Chloramphenicol shows similar apparent activation energies for both TiO 2 P-25 and ZnO photocatalysts. The initial apparent photonic efficiency (z 0 ) of the photo-oxidation and the mineralization under various experimental conditions have been calculated, while the Kirby–Bauer disc diffusion method showed a 100% reduction of the drug activity after 90 min of photocatalytic treatment. & 2007 Elsevier Ltd. All rights reserved. 1. Introduction Although pharmaceuticals have been consumed for many decades, only during the last few years their fate and release in the aquatic environment have been recognized as one of the most urgent questions of environmental chemistry. Current drinking water standards do not even require testing for any of the more than 7000 pharmaceutical compounds being prescribed. Pharmaceutically active compounds such as analgestics, antibiotics, steroid, hormones, etc., have been detected in several public water systems in Europe, USA and Australia as a result of human, animal and plant activities (Ajit et al., 2006; Kolpin et al., 2002). Mass balance of the input and output of pharmaceuticals in sewage treatment plants indicates that during sewage treatment not all pharmaceu- ticals are removed quantitatively (Heberer, 2002), and in surface and underground water, they are detected at times. Their environmental presence gains importance because they are related abnormal physiological processes in reproduction, increased incidences of cancer, development of antibiotic- resistant bacteria and potential increased toxicity of chemical mixtures. For many substances, the potential effects on hu- mans and aquatic ecosystems are not clearly understood (Halling-Sorensen et al., 1998). As a consequence, several efforts are being made to find out ways of inactivating or eliminating this class of substances in surface or wastewater. Among the so-called advanced oxidation processes, homo- geneous and heterogeneous photocatalytic oxidation have shown, recently, great promise in the treatment of industrial wastewater, groundwater and contaminated air. ARTICLE IN PRESS 0043-1354/$ - see front matter & 2007 Elsevier Ltd. All rights reserved. doi:10.1016/j.watres.2007.07.030 Corresponding author. Tel.: +30 2310 997785; fax: +30 2310 997784. E-mail address: [email protected] (I. Poulios). WATER RESEARCH 42 (2008) 386– 394
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Photocatalytic degradation and drug activity reduction of Chloramphenicol

ARTICLE IN PRESS
Available at www.sciencedirect.com
WA T E R R E S E A R C H 4 2 ( 2 0 0 8 ) 3 8 6 – 3 9 4
0043-1354/$ - see frodoi:10.1016/j.watres
�Corresponding autE-mail address: p
journal homepage: www.elsevier.com/locate/watres
Photocatalytic degradation and drug activity reduction ofChloramphenicol
A. Chatzitakisa, C. Berberidoua, I. Paspaltsisb, G. Kyriakouc, T. Sklaviadisb, I. Pouliosa,�
aLaboratory of Physical Chemistry, Department of Chemistry, Aristotle University of Thessaloniki, 54124 Thessaloniki, GreecebPrion Disease Research Group, Laboratory of Pharmacology, School of Pharmacy, Aristotle University of Thessaloniki, 54124 Thessaloniki,
GreececLaboratory of Inorganic Chemistry, Department of Chemical Engineering, Aristotle University of Thessaloniki, 54124 Thessaloniki, Greece
a r t i c l e i n f o
Article history:
Received 1 May 2007
Received in revised form
26 June 2007
Accepted 18 July 2007
Available online 27 July 2007
Keywords:
Photocatalysis
Chloramphenicol
Photo-oxidation
Drugs
ZnO
TiO2
nt matter & 2007 Elsevie.2007.07.030
hor. Tel.: +30 2310 [email protected] (I.
a b s t r a c t
The photocatalytic degradation of Chloramphenicol, an antibiotic drug, has been
investigated in aqueous heterogeneous solutions containing n-type oxide semiconductors
as photocatalysts. The disappearance of the organic molecule follows approximately a
pseudo-first-order kinetics according to the Langmuir–Hinshelwood model. It was observed
that, with TiO2 P-25 as photocatalyst, quantitative degradation of the organic molecule
occurs after 4 h of illumination. During this time, the dechlorination of the substrate is
complete, while the organic nitrogen was recovered in the form of nitrate and ammonium
ions. The effect of temperature on the degradation rate of Chloramphenicol shows similar
apparent activation energies for both TiO2 P-25 and ZnO photocatalysts. The initial
apparent photonic efficiency (z0) of the photo-oxidation and the mineralization under
various experimental conditions have been calculated, while the Kirby–Bauer disc diffusion
method showed a 100% reduction of the drug activity after 90 min of photocatalytic
treatment.
& 2007 Elsevier Ltd. All rights reserved.
1. Introduction
Although pharmaceuticals have been consumed for many
decades, only during the last few years their fate and release
in the aquatic environment have been recognized as one of
the most urgent questions of environmental chemistry.
Current drinking water standards do not even require testing
for any of the more than 7000 pharmaceutical compounds
being prescribed. Pharmaceutically active compounds such as
analgestics, antibiotics, steroid, hormones, etc., have been
detected in several public water systems in Europe, USA and
Australia as a result of human, animal and plant activities
(Ajit et al., 2006; Kolpin et al., 2002). Mass balance of the input
and output of pharmaceuticals in sewage treatment plants
indicates that during sewage treatment not all pharmaceu-
r Ltd. All rights reserved.
; fax: +30 2310 997784.Poulios).
ticals are removed quantitatively (Heberer, 2002), and in
surface and underground water, they are detected at times.
Their environmental presence gains importance because they
are related abnormal physiological processes in reproduction,
increased incidences of cancer, development of antibiotic-
resistant bacteria and potential increased toxicity of chemical
mixtures. For many substances, the potential effects on hu-
mans and aquatic ecosystems are not clearly understood
(Halling-Sorensen et al., 1998). As a consequence, several efforts
are being made to find out ways of inactivating or eliminating
this class of substances in surface or wastewater.
Among the so-called advanced oxidation processes, homo-
geneous and heterogeneous photocatalytic oxidation have
shown, recently, great promise in the treatment of industrial
wastewater, groundwater and contaminated air.

ARTICLE IN PRESS
WAT E R R E S E A R C H 4 2 ( 2 0 0 8 ) 3 8 6 – 3 9 4 387
The photocatalytic decomposition of organic compounds of
environmental concern (e.g. pesticides, dyes, etc.) has been
studied extensively during the last 20 years and it has been
demonstrated that heterogeneous photocatalysis using TiO2
(anatase) as catalyst can be an alternative to conventional
methods for the removal of organic pollutants from water and
air (Blake, 2001). Additionally, an advantage of the photo-
catalytic process is its mild operating conditions and the fact
that it can be powered by sunlight, thus reducing significantly
the electric power required and, therefore, operating costs
(Blanco and Malato, 2003). Recently, a number of research
groups have dealt with the homogeneous and heterogeneous
photocatalytic decomposition of various types of drugs in the
presence of near-UV (UV-A) or solar light with very encoura-
ging results (Andreozzi et al., 2003; Ravina et al., 2002).
The current study provides results describing the hetero-
geneous photocatalytic oxidation of the antibiotic Chloram-
phenicol, also known with the commercial name
Chloromycetin over semiconducting powders, such as TiO2
and ZnO, under various experimental conditions.
Chloramphenicol is a broad-spectrum antibiotic exhibiting
activity against both Gram-positive and Gram-negative bac-
teria, as well as other groups of microorganisms. It exerts its
action through microorganism protein synthesis inhibition
and it is effective in the treatment of several infectious
diseases. This, together with its low cost and ready avail-
ability, has made it extensively used since the 1950s in the
treatment of domestic animals all over the world (Ajit et al.,
2006). However, Chloramphenicol is, in certain susceptible
individuals, associated with serious toxic effects in humans
including bone marrow depression, particularly severe in the
form of fatal aplastic anemia (Woodward, 2004). Since this
condition is dose independent, Chloramphenicol has been
banned for use in food-producing animals in many countries
including EU. As a consequence, Chloramphenicol is included
in Annex IV of Council Decision 2077/90 (EEC, 1990), which
comprise the drugs with an established zero-tolerance level
in edible tissues. Its structure is presented below
O2N CH2O CH
NHCOCHCl2
CH2OH
Chloramphenicol, CAS 56-75-3.
2. Experimental
2.1. Materials
Chloramphenicol (C11H12O5N2Cl2, 2,2-dichloro-N-[2-hydroxy-
1 (hydroxyl-methyl)-2(4-nitrophenyl)ethyl]acetamide, MW
322.96) is a product of Sigma Chemie GmbH company and
was used as received.
As catalysts, for all photocatalytic experiments, TiO2 P-25
(anatase/rutile ¼ 3.6/1, surface area 56 m2 g�1, nonporous),
TiO2 (A) (Tronox McGee, BET 10.5 m2 g�1, 100% anatase), TiONa
(100% anatase) and ZnO (Merck, BET 10 m2 g�1) were used. All
other chemicals were purchased through commercial com-
panies and were used without further purification.
2.2. Procedures
Experiments were performed in a closed Pyrex cell of 300 mL
capacity (6 cm diameter and 15 cm height). In all cases during
the experiments, 250 mL of the Chloramphenicol solution
containing the appropriate quantity of the semiconductor
powder was magnetically stirred, before and during the
illumination, while the solution was purged with CO2� free
air. At specific time intervals samples of 4 mL were with-
drawn. To remove the TiO2 particles, the solution was filtered
through a 0.45mm filter (Schleicher and Schuell). The suspen-
sion was left for 15 min in the dark in order to achieve
maximum adsorption of the drug onto the semiconductor
surface. The reaction vessel was fitted with a central 9 W
lamp Osram Dulux S 9W/78 and had inlet and outlet ports for
bubbling the desired (CO2 free) gas under which the reaction
was taking place. The spectral response of the irradiation
source according to the producer, ranged between 320 and
400 nm with a maximum at 365 nm, a wavelength suitable for
the activation of the semiconductors.
The photon flow per unit volume of the incident light was
determined by chemical actinometry using potassium fer-
rioxalate (Braun et al., 1991). The initial light flux, under
exactly the same conditions as in the photocatalytic experi-
ments, was evaluated to be 1.12�10�4 Einstein min�1.
2.3. Analytical procedures
Changes in the concentration of Chloramphenicol were
observed from its characteristic absorption band at 276.5 nm
using a UV–Visible spectrophotometer (Perkin-Elmer, Lamba
3, UV–VIS Spectrophotometer). Since a linear dependence
between the initial concentration of the drug and the
absorption at 276.5 nm is observed, during the experimental
procedure the photodecomposition was monitored spectro-
photometrically at this wavelength.
In order to determine the reduction of the dissolved organic
carbon, a TOC analyzer (Shimandzu 5000) was used to
measure the organic content.
The ions were detected using a DIONEX–DX 100 ANION-
S–CATIONS Analyzer (Dionex, CA, USA) equipped with cation
and anion micromembrane suppressors and a DIONEX
electrical conductivity detector. The anions were analyzed
using an anion pre-column (Dionex AG4A-SC), an anion
separator column (Dionex AS4A-SC), a supressor column
(Dionex ASRS-ULTRA II) and a conductivity detector (Su-
pressed Conductivity meter). The mobile phase (flow rate
2 mL min�1) was 1.8 mM Na2CO3–1.7 mM NaHCO3 and the
suppressor solution was 12.5 mM H2SO4. The cations were
analyzed using a cation pre-column (Dionex CG 12), a cation
separator column (Dionex AS4A-SC), a supressor column
(Dionex CSRS-ULTRA) and a conductivity detector (Supressed
Conductivity meter). The mobile phase (flow rate 1 mL min�1)
was 18 mM methanesulfonic acid and the suppressor solution
was 12.5 mM H2SO4.

ARTICLE IN PRESS
0 20 40 60 80 1000
10
20
30
40
50
C (
mgL
-1)
Illumination time (min)
Fig. 1 – Photodegradation of 50 mg L�1 Chloramphenicol as a
function of irradiation time in the presence of (’) 1 g L�1
TiO2 P-25, (K) 1 g L�1 TiO2 (A), (m) 1 g L�1 ZnO, (.) 1 g L�1
TiONa and (E) without catalyst.
WA T E R R E S E A R C H 4 2 ( 2 0 0 8 ) 3 8 6 – 3 9 4388
All photocatalytic experiments were carried out at an initial
pH value of 5.170.2 and the reaction temperature was kept
constant at 2570.2 1C.
Some photocatalytic experiments were repeated three
times in order to check the reproducibility of the experi-
mental results. The accuracy of the optical density values was
within 75%, while the corresponding ones of TOC and
inorganic ions were within 710%.
2.4. Initial apparent photonic efficiency
The photonic efficiency (z) describing the effectiveness of
light conversion to product in heterogeneous, light scattering
systems, is directly proportional to the quantum yield, and
may be considered to be the lower limit of this more
commonly used measure of light efficiency (Muneer and
Bahnemann, 2002).
In our case of photo-oxidation, the following equation was
used for the calculation of the initial apparent photonic
efficiency:
z0ð%Þ ¼ r0ð323Ihv103Þ�1� 100, (1)
where r0 is the initial reaction rate of the photo-oxidation
(mg L�1 min�1), 323 is the molecular weight of Chloramphe-
nicol and Ihv the incident photon flux (mol photons
L�1 min�1¼ Einstein min�1).
For the mineralization process, the initial apparent photo-
nic efficiency, zDOC, per carbon atom can be calculated using
the following equation (Muneer and Bahnemann, 2002):
zDOCð%Þ ¼ rDOCð12 Ihv 103Þ�1� 100, (2)
where rDOC is the initial rate of dissolved organic carbon
reduction (mg L�1 min�1), while for the inorganic nitrogen
production, expressed as a sum of nitrogen in NO�3 ; NHþ4 , the
initial apparent photonic efficiency per nitrogen atom, zInN,
can be calculated using the following equation:
zInNð%Þ ¼ rInNð14Ihv103Þ�1� 100, (3)
where rInN is the initial rate of inorganic nitrogen production
(mg L�1 min�1).
In a similar way, the initial photonic efficiency zCl� (%) of
the Cl� release can be calculated using
zCl�ð%Þ ¼ rCl�ð35:5Ihv103Þ�1� 100. (4)
2.5. Antimicrobial susceptibility test
In order to determine the inhibition of Chloramphenicol’s
antibiotic activity caused by the photocatalytic treatment, the
Kirby–Bauer disk diffusion method was used (Bauer et al.,
1966). About 50 mg L�1 of the antibiotic was degraded photo-
catalytically in the presence of 1 g L�1 TiO2. Samples of 1 mL
were taken in time intervals during the photocatalytic
treatment, they were added on 25 mm diameter disks
(0.45mm Millipore filters) and were dried at 37 1C for 1 h.
The plates were prepared as following: 75 mL of LB agar (5 g
NaCl, 5 g yeast extract, 10 g tryptone and 15 g agarose per 1 L)
was added in Petri dishes (145�20 mm, Greiner). They were
then left to cool down at room temperature and stored at 4 1C.
E. coli top 10 bacteria were used as the test organism. Briefly,
150mL from an overnight culture were added in 6 mL of top
agarose (10 g tryptone, 5 g yeast extract, 5 g NaCl and 7 g of
agarose for 1 L) which was kept at 42 1C. After a short stir, the
mixture was added on top of the agar plates and was allowed
to cool for 5 min at room temperature. The filters containing
the test substrates were placed on top of the plate, which was
incubated overnight at 37 1C. A titration curve was prepared
by measuring the zones of inhibition of the bacterial growth
caused by known amounts of Chloramphenicol.
3. Results and discussion
3.1. Photodecomposition kinetics of Chloramphenicol
Results of the photolysis of a 50 mg L�1 (1.55�10�4 M)
Chloramphenicol solution containing 1 g L�1 TiO2 P-25, TiO2
(A), TiONa and ZnO are shown in Fig. 1. The amount of the
organic molecule present in the supernatant is plotted as a
function of the irradiation time.
Under the given experimental conditions, ZnO followed by
TiO2 P-25 appears to be the best catalysts leading after 90 min
of illumination to almost complete (�90%) degradation of the
antibiotic. The photocatalytic oxidation of Chloramphenicol,
on the other hand, in the presence of the commercial anatase
forms, TiO2 (A) and TiONa, is a slower process and, after
90 min of illumination, only 37% and 23%, respectively, of the
drug have been degraded. The superiority of TiO2 P-25 may be
attributed to the morphology of crystallites, which was
proposed to be one of the most critical properties for the
photocatalytic efficiency of P-25 among various grades of
TiO2. Recently, Hurum et al. (2003) had shown that in the case
of TiO2 P-25 a transfer of the photogenerated electrons from
rutile to anatase particles takes place leading to stabilization
of the charge separation and, therefore, lowering of recombi-
nation of the photogenerated carriers, which determine the
efficiency of most photocatalysts. Additionally, the small size
of the rutile particles in this formulation and their close
ARTICLE IN PRESS
0.0 0.5 1.0 1.5 2.0 2.5 3.0 3.5 4.0 4.50.0
0.2
0.4
0.6
0.8
1.0
r 0 (m
g L
-1 m
in-1
)
Fig. 2 – Dependence of the initial reaction rate, r0, on the
concentration of the TiO2 P-25 for constant Chloramphenicol
concentration (50 mg L�1).
0 20 40 60 800.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
1.6
1.8
0.00 0.02 0.04 0.06 0.08 0.100.40.60.81.01.21.41.61.82.0
r 0 (m
g L
-1 m
in-1
)
Ceq
(mg L-1)
Fig. 3 – Plot of r0 vs. Ceq at different Chloramphenicol
concentrations for constant catalyst concentration at 1 g L�1,
(’) TiO2 P-25 and (K) ZnO. Inset: linear transform of r0�1 vs.
WAT E R R E S E A R C H 4 2 ( 2 0 0 8 ) 3 8 6 – 3 9 4 389
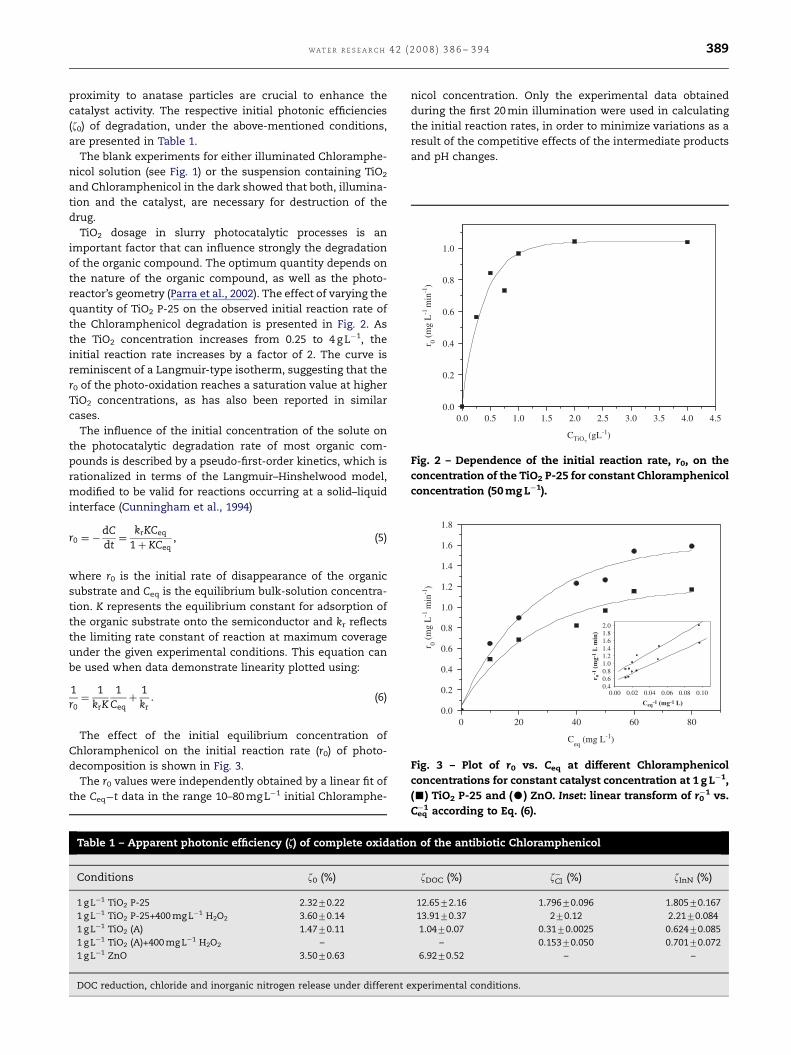
proximity to anatase particles are crucial to enhance the
catalyst activity. The respective initial photonic efficiencies
(z0) of degradation, under the above-mentioned conditions,
are presented in Table 1.
The blank experiments for either illuminated Chloramphe-
nicol solution (see Fig. 1) or the suspension containing TiO2
and Chloramphenicol in the dark showed that both, illumina-
tion and the catalyst, are necessary for destruction of the
drug.
TiO2 dosage in slurry photocatalytic processes is an
important factor that can influence strongly the degradation
of the organic compound. The optimum quantity depends on
the nature of the organic compound, as well as the photo-
reactor’s geometry (Parra et al., 2002). The effect of varying the
quantity of TiO2 P-25 on the observed initial reaction rate of
the Chloramphenicol degradation is presented in Fig. 2. As
the TiO2 concentration increases from 0.25 to 4 g L�1, the
initial reaction rate increases by a factor of 2. The curve is
reminiscent of a Langmuir-type isotherm, suggesting that the
r0 of the photo-oxidation reaches a saturation value at higher
TiO2 concentrations, as has also been reported in similar
cases.
The influence of the initial concentration of the solute on
the photocatalytic degradation rate of most organic com-
pounds is described by a pseudo-first-order kinetics, which is
rationalized in terms of the Langmuir–Hinshelwood model,
modified to be valid for reactions occurring at a solid–liquid
interface (Cunningham et al., 1994)
r0 ¼ �dCdt¼
krKCeq
1þ KCeq, (5)
where r0 is the initial rate of disappearance of the organic
substrate and Ceq is the equilibrium bulk-solution concentra-
tion. K represents the equilibrium constant for adsorption of
the organic substrate onto the semiconductor and kr reflects
the limiting rate constant of reaction at maximum coverage
under the given experimental conditions. This equation can
be used when data demonstrate linearity plotted using:
1r0¼
1krK
1Ceqþ
1kr
. (6)
The effect of the initial equilibrium concentration of
Chloramphenicol on the initial reaction rate (r0) of photo-
decomposition is shown in Fig. 3.
The r0 values were independently obtained by a linear fit of
the Ceq�t data in the range 10–80 mg L�1 initial Chloramphe-
Table 1 – Apparent photonic efficiency (f) of complete oxidatio
Conditions z0 (%)
1 g L�1 TiO2 P-25 2.3270.22
1 g L�1 TiO2 P-25+400 mg L�1 H2O2 3.6070.14
1 g L�1 TiO2 (A) 1.4770.11
1 g L�1 TiO2 (A)+400 mg L�1 H2O2 –
1 g L�1 ZnO 3.5070.63
DOC reduction, chloride and inorganic nitrogen release under different e
nicol concentration. Only the experimental data obtained
during the first 20 min illumination were used in calculating
the initial reaction rates, in order to minimize variations as a
result of the competitive effects of the intermediate products
and pH changes.
n of the antibiotic Chloramphenicol
zDOC (%) z�Cl (%) zInN (%)
12.6572.16 1.79670.096 1.80570.167
13.9170.37 270.12 2.2170.084
1.0470.07 0.3170.0025 0.62470.085
– 0.15370.050 0.70170.072
6.9270.52 – –
xperimental conditions.
Ceq�1 according to Eq. (6).

ARTICLE IN PRESS
0 100 200 300 400 500 600 7000.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
1.6
initi
al r
eact
ion
rate
, ro
(mg
L-1
min
-1)
WA T E R R E S E A R C H 4 2 ( 2 0 0 8 ) 3 8 6 – 3 9 4390
In both cases, the curves are reminiscent of a Langmuir-
type isotherm, for which the rate value of photodecomposi-
tion first increases and then reaches a saturation value at
higher concentrations of Chloramphenicol.
As indicated in the inset of Fig. 3, the plot of the reciprocal
initial rate r0 as a function of the reciprocal of the initial
concentration Ceq yields for constant concentration of TiO2
P-25 and ZnO at 1 g L�1 a straight line. The kr and K values
calculated according to Eq. (6) from the slope of each straight
line and from the intercept with the 1/r0 axis were,
respectively, in the case of TiO2 P-25, 1.34 mg L�1min�1
(4.15�10�6 M min�1) and 0.057 mg�1 L (1.76�104 M�1) and,
respected in the case of ZnO, 1.87 mg L�1 min�1
(5.79�10�6 M min�1) and 0.050 mg L�1 (1.55�104 M�1).
Fig. 4 – Effect of H2O2 concentration on the initial rate of
photodegradation of 50 mg L�1 Chloramphenicol in the
presence of 1 g L�1 TiO2 P-25.
3.2. Effect of H2O2 on the photodecomposition ofChloramphenicol
The addition of other powerful oxidizing species, such as
hydrogen peroxide and potassium peroxydisulfate, to TiO2
suspensions is a well-known and extensively studied proce-
dure and in many cases leads to an acceleration of the rate of
the photocatalytic degradation (Wolfrum and Ollis, 1994; Sioi
et al., 2006).
In our case the photocatalytic oxidation of 50 mg L�1
Chloramphenicol in the presence of 1 g L�1 TiO2 P-25 has
been studied at different H2O2 concentrations. The reaction
kinetics were similar to those observed without the oxidants.
H2O2 is considered to have two functions in the process of
photocatalytic degradation. It accepts a photogenerated
electron from the conduction band and thus promotes the
charge separation
H2O2 þ e� ! OH� þ dOH; (7)
and it also forms dOH radicals according to
H2O2 þO�2 ! OH� þ dOHþO2. (8)
However, a possible reaction between the H2O2 with the
photogenerated intermediates cannot be excluded. In the
presence of excess H2O2, it may act as a hole or dOH scavenger
or react with TiO2 to form peroxy compounds, which are
detrimental to the photocatalytic action. In addition, it can
also compete with the organic compound for the adsorption
sites on the semiconductor’s surface, resulting in a ‘‘chroma-
tographic peaking effect’’ of the pollutant concentration in
the solution during the initial stages of the photocatalytic
process (Sauer et al., 2002; Dionysiou et al., 2004). This
explains the need for an optimal concentration of H2O2 for
the maximum effect.
The influence of the H2O2 concentration on the initial rate
of photo-oxidation of Chloramphenicol is shown in Fig. 4. The
photocatalytic efficiency increases as the concentration of
H2O2 increases and it reaches the optimum in the area of
300–400 mg L�1. Consequently, it decreases as the concentra-
tion of H2O2 increases beyond the optimum. Under these
conditions, the presence of H2O2 at an initial pH value of 5.1,
and at concentrations of 50–400 mg L�1, results in a small
increase in the rate of the reaction (�1.5 of the initial one),
quite lower than that observed in other cases (Sioi et al., 2006).
3.3. Effect of temperature on the photocatalytic process
Although the temperature in a chemical reaction plays an
important role, very little information concerning the tem-
perature effect on the heterogeneous photocatalytic degrada-
tion of pollutants in aqueous solutions is available. This is due
to the fact that such reactions are usually not very tempera-
ture sensitive, because the forbidden band-gap energy of the
semiconductive catalysts is too high (�3.0 eV) to be overcome
by the thermal activation energy (kT ¼ 0.026 eV at room
temperature). Increasing the reaction temperature may
increase the oxidation rate of organic compounds at the
interface between the catalyst and the solution, but it also
reduces the adsorptive capacities associated with the organ-
ics and dissolved oxygen (Herrmann, 1999).
In the present paper, the Chloramphenicol degradation was
studied as a function of the reaction temperature under the
same operating conditions for both illuminated TiO2 P-25 and
ZnO systems. The results concerning the dependence of the
rate constant k of the photocatalytic degradation of Chlor-
amphenicol from the reaction temperature, in the range
3–57 1C, are shown in Fig. 5. By increasing the temperature, an
increase of the apparent reaction rate constant k is achieved
for both photocatalytic systems until 45 1C, while tempera-
tures higher than 45 1C lead to a decrease of the photocata-
lytic activity. A possible explanation in the case of the
temperatures above 45 1C could be the lower saturation value
of oxygen, a factor of great importance because it regulates
the photocatalytic mechanism by capturing the photogener-
ated electrons, as well as the increase of the desorption of the
reactants from the catalyst surface (Mills and Le Hunte, 1997;
Chen and Ray, 1998; Herrmann, 1999). The higher tempera-
ture dependence in the case of ZnO could be a result of its
higher donor density (lower resistivity) in comparison to TiO2
P-25. Rising the temperature then leads to higher amounts of
photogenerated carriers reaching the surface of the catalyst
and a reaction occured.
By plotting the natural logarithm of k as a function of the
reciprocal absolute temperature (T), a linear behavior was

ARTICLE IN PRESS
270 280 290 300 310 320 3300.00
0.01
0.02
0.03
0.04
0.05
3.1 3.2 3.3 3.4 3.5 3.6 3.7
-5
-4
-3
initi
al r
eact
ion
cons
tant
, k (
min
-1)
reaction temperature, T (K)
Fig. 5 – Initial reaction constant of 50 mg L�1 Chloram-
phenicol degradation vs. reaction temperature, (K) 0.5 g L�1
TiO2 P-25 and (’) 0.5 g L�1 ZnO. Inset: Dependence of the
natural logarithm of the reaction’s rate constant k from the
inverse of the absolute temperature.
0 50 100 150 200 2500
5
10
15
20
DO
C (
mgL
-1)
Illumination time (min)
Fig. 6 – Dissolved organic carbon (DOC) reduction as a
function of irradiation time during the photocatalytic
degradation of 50 mg L�1 Chloramphenicol: (’) 1 g L�1 ZnO,
(K) 1 g L�1 TiO2 P-25, (m) 1 g L�1 TiO2 (A) and (.) 1 g L�1 TiO2
P-25+400 mg L�1 H2O2.
WAT E R R E S E A R C H 4 2 ( 2 0 0 8 ) 3 8 6 – 3 9 4 391
obtained for both TiO2 P-25 and ZnO catalytic systems as
shown in the inset of Fig. 5. The activation energy (Ea)
determined according to Arrhenius equation (Eq. (9)) gives
33.375.4 and 27.274.4 kJ mol�1 for TiO2 P-25 and ZnO,
respectively:
ln kr ¼ ln A�Ea
R1T
. (9)
3.4. Photodegradation products
The photodecomposition of Chloramphenicol, like all the
other organic pollutants, is a complicated process with many
intermediary products. These intermediates are of great
significance in water treatment because they can be proved
more toxic than the parent compounds. Thus, it is of vital
importance to succeed complete mineralization of the
organic pollutants via the photocatalytic method. For this
reason, the extent of mineralization of Chloramphenicol was
studied under various experimental conditions.
The overall equation, valid after a long irradiation time in
the presence of excess oxygen, which describes the photo-
catalytic mineralization of Chloramphenicol, is
C11H12O5N2Cl2 þ 13:5O2 þ hvo400 nm
�!TiO2
11CO2 þ 2HNO3 þ 2HClþ 4H2O. ð10Þ
General description of heterogeneous photocatalysis under
artificial or solar irradiation is presented in several excellent
review articles (Fujishima et al., 2000; Hoffman et al., 1995;
Blanco and Malato, 2003). Therefore, the various stages of the
photocatalytic process will not be further presented here.
Fig. 6 shows the extent of the dissolved organic content
(DOC) reduction vs. illumination time of an air-saturated
solution containing 50 mg L�1 Chloramphenicol in the pre-
sence of 1 g L�1 TiO2 P-25, TiO2 (A), ZnO, as well as in the
presence of 1 g L�1 TiO2 P-25+400 mg L�1 H2O2, while in Table 1
the initial apparent photonic efficiencies (zDOC) of complete
oxidation of the antibiotic, calculated according to Eq. (2), are
given.
In the presence of TiO2 P-25, 88% of the initial carbon
content of Chloramphenicol was converted to CO2 after
120 min of illumination, while at the same time the decom-
position was almost complete, showing the presence of
intermediates. Full oxidation under the given experimental
conditions requires longer illumination times. The addition of
400 mg L�1 H2O2 in a suspension of TiO2/Chloramphenicol
does not alter significantly the efficiency of the photominer-
alization process and this agrees with the results of the
degradation experiments in the presence of various concen-
trations of H2O2 (Fig. 4).
In the case of TiO2 (A), as in the degradation experiments
(Fig. 1), the photomineralization proceeds with lower effi-
ciency compared to the one when TiO2 P-25 has been used
and after 240 min of illumination only 50% of the initial DOC
in the Chloramphenicol molecule has been converted to CO2.
In the case of ZnO despite the fact that it appears to be a
better catalyst than TiO2 P-25 (Fig. 1), same is not the case
with complete mineralization of the organic content of the
Chloramphenicol molecule. After 120 min of illumination,
44% of the initial carbon content of the antibiotic is remaining
in the solution followed by a retardation of the photominer-
alization rate toward a steady-state value. The dissolution as
well as the photodissolution of ZnO as a result of the self-
oxidation of the semiconductor by the photogenerated holes
could be a possible explanation for the decrease of its
photocatalytic activity (Pleskov and Gurevich, 1986; Evgen-
idou et al., 2005).
In Figs. 7 and 8, the conversion of organic chlorine and
nitrogen in the Chloramphenicol molecule to the respective
inorganic ions is shown, while in Table 1 the initial photonic
efficiencies of these conversions, calculated according to
Eqs. (3) and (4) respectively, under various experimental
conditions, are given.
The organic chlorine conversion to Cl� is an efficient
process in the case of TiO2 P-25, with and without the

ARTICLE IN PRESS
0 50 100 150 200 2500
20
40
60
80
100
Cl- r
elea
se (
%)
illumination time (min)
Fig. 7 – Cl� formation as a function of irradiation time during
the photocatalytic degradation of 50 mg L�1 Chloram-
phenicol: (’) 1 g L�1 TiO2 P-25, (K) 1 gL�1 TiO2 P-25+
400 mgL�1 H2O2, (m) 1 g L�1 TiO2 (A) and (.) 1 g L�1 TiO2 (A)+
400 mgL�1 H2O2.
0 50 100 150 200 2500
20
40
60
80
100
tota
l ino
rgan
ic n
itrog
en (
%)
illumination time (min)
Fig. 8 – Total inorganic nitrogen formation as a sum of
nitrogen in NHþ4 and NO�3 ions vs. irradiation time during
the photocatalytic degradation of 50 mg L�1 Chloram-
phenicol: (’) 1 g L�1 TiO2 P-25, (K) 1 gL�1 TiO2 P-25+
400 mgL�1 H2O2, (m) 1 g L�1 TiO2 (A) and (.) 1 g L�1 TiO2 (A)+
400 mgL�1 H2O2.
WA T E R R E S E A R C H 4 2 ( 2 0 0 8 ) 3 8 6 – 3 9 4392
addition of H2O2. After 2 h of illumination, it was possible to
completely remove chlorine from the molecule of Chloram-
phenicol in the presence of 1 g L�1 TiO2 P-25, as well as by the
synergetic effect of 1 g L�1 TiO2 P-25 and 400 mg L�1 H2O2,
while in the presence of TiO2 (A) only 28% of the expected
value of Cl� has been released.
Moreover, the organic nitrogen conversion to the respective
inorganic ions ðNO�3 ; NH�4 Þ is presented in Fig. 8. Again TiO2
P-25 appears to have better photocatalytic properties than
TiO2 (A) and after 3 h of illumination, the organic nitrogen has
been completely released in the form of NO�3 and NH�4 ions.
In photocatalytic processes involving nitrogen-containing
organic compounds, the final oxidation state of nitrogen or
the NHþ4 =NO�3 ratio, when mineralization of the organic
carbon has occurred, may depend on several factors, the
most significant being the nature and the concentration of
the initial organic compound, the concentration of molecular
oxygen, the pH of the solution, the loading and nature of the
catalyst, as well as the intensity of the irradiation source (Low
et al., 1991).
In the present case, the organic nitrogen conversion to the
respective inorganic ions under the given experimental
conditions, as presented in Fig. 8, is a very efficient process.
After 3 h of illumination in the presence of TiO2 P-25 a 93%
conversion has been achieved, while the respective one in the
presence of TiO2 (A) reaches the lower value of 60%. The
addition of H2O2 enhances slightly the conversion of organic
nitrogen to inorganic anions. In the presence of TiO2 P-25, we
measure a 39% conversion of the organic nitrogen to nitrate,
which corresponds to a 90% conversion of the nitrogroup in
the Chloramphenicol molecule to nitrate. According to the
literature, the nitrogroup in nitroaromatics is a light leaving
group, which can easily be eliminated, favoring the electro-
philic substitution of the dOH radicals at the para position in
the aromatic ring. This group ðNO�2 Þ can then be further
oxidized to NO�3 ions. Taking into account that the amount of
organic nitrogen converted to ammonium ions is more than
the expected one from the aliphatic amid group in the
molecule, we could hypothetize that except the oxidative
mechanism, Chloramphenicol could also participate to a
reductive degradation pathway. Generally, nitrate does not
represent the total nitrogen available from the nitrophenols.
Maurino et al. (1997) reported that the quantity of nitrate ions
reached approximately 70–80% of the total initial organic
nitrogen, whilst Augugliaro et al. (1991) found 60–70% of
nitrate at the end of the photodegradation runs. The
remaining percentages of inorganic nitrogen were usually
due to the formation of ammonium ions (Dieckmann and
Gray, 1996). Maurino et al. (1997) have reported that the direct
reduction of the nitrogroup in nitrophenols can be an
important degradation pathway, even in oxygenated solu-
tions. This means that oxygen and nitrophenol compete for
electrons as confirmed also by the presence of 4-aminophenol
during the initial steps of degradation of 4-nitrophenol
(Maurino et al., 1997; Di Paola et al., 2003). The reaction path
leading to ammonia must involve the nitrogroup before it
forms nitrate ions. Mahdavi et al. (1993) have shown that the
nitrogroup can be reduced to an amine group on the ring and
then is released into the system as ammonia.
On the other hand, the nitrogen mineralization in the
presence of TiO2 (A) is a slower process in comparison to the
TiO2 P-25 one, with a 60% conversion after 4 h of illumination.
This is indicative of the presence of various aliphatic
nitrogen-containing reaction products and is also in accor-
dance with the lower organic carbon reduction in the same
time, leading to the conclusion that longer illumination times
are needed for the complete conversion to inorganic end
products.
3.5. Inhibition of the antimicrobial activity ofChloramphenicol
The results concerning the inhibition of the drug activity of
Chloramphenicol before and after the photocatalytic treatment

ARTICLE IN PRESS
0 20 40 60 80 1000
20
40
60
80
100
drug
act
ivity
inhi
bitio
n (%
)
illumination time (min)
Fig. 9 – Antimicrobial susceptibility test. The initial
antibiotic activity of 50 lg mL�1 Chloramphenicol (100%)
was reduced to 40%, 20% and 0% after 30, 60 and 90 min
photocatalytic treatment, respectively.
WAT E R R E S E A R C H 4 2 ( 2 0 0 8 ) 3 8 6 – 3 9 4 393
in the presence of 1 g L�1 TiO2 P-25 for given irradiation times
are presented in Fig. 9.
The drug activity of either the untreated solution contain-
ing 50 mg L�1 of Chloramphenicol or the photocatalytically
treated solution at different time points was evaluated in
comparison to the activity of standards containing known
amounts of the antibiotic. As shown in Fig. 9 after 30 min of
the treatment, only 40% of the initial antibiotic activity was
detectable according to the Kirby–Bauer disk diffusion test.
Further illumination of the TiO2/Chloramphenicol disper-
sion for 90 min causes a 100% reduction of the initial
antibiotic activity.
4. Conclusions
In the present work the photocatalytic oxidative degradation
of Chloramphenicol, an antibiotic drug, has been demon-
strated. It was observed that, with TiO2 P-25 and ZnO as
photocatalysts, quantitative degradation of the organic mo-
lecule occurs after 4 h. During this time in the presence of
TiO2 P-25, the dechlorination of the substrate is complete,
while the nitrogen in the organic compound is recovered in
the form of nitrate and ammonium ions. The activation
energies for both photocatalysts calculated according to
Arrhenius equation shows that temperature influences
slightly the photodegradation process. The fast reduction of
the drug activity shows that heterogeneous photocatalysis
could be employed as a powerful tool for the deactivation of
this class of materials. The use of a catalyst such as TiO2, and
the possibility of activating it with UV-A or solar light,
combined with the simple technology required for this
method, can offer economically reasonable and practical
solutions to processing of wastewater containing pharma-
ceutically active compounds such as analgestics, antibiotics,
steroid and hormones.
R E F E R E N C E S
Ajit, K., Sarmah, A.K., Meyer, M.T., Boxall, A.B.A., 2006. A globalperspective on the use, sales, exposure pathways, occurrence,fate and effects of veterinary antibiotics (VAs) in the environ-ment. Chemosphere 65 (5), 725–759.
Andreozzi, R., Raffaele, M., Nicklas, P., 2003. Pharmaceuticals inSTP effluents and their solar photodegradation in aquaticenvironment. Chemosphere 50 (10), 1319–1330.
Augugliaro, V., Palmisano, L., Schiavello, M., Sclafani, A., March-ese, L., Martra, G., Miano, F., 1991. Photocatalytic degradationon nitrophenols in aqueous titanium dioxide dispersion. Appl.Catal. 69 (2), 323–340.
Bauer, A.W., Kirby, W.M., Sherris, J.C., Turck, M., 1966. Antibioticsusceptibility testing by a standardized single disk method.Tech Bull Regist Med Technol 36 (3), 49–52.
Blake, D., 2001. Bibliographic work on the heterogeneous photo-catalytic removal of hazardous compounds from water andair. National Renewable Energy Laboratory, Technical Report,NREL/TP-510-31319.
Blanco, J., Malato, S., 2003. Solar Detoxification. UNESCO Publishing.Braun, A.M., Maurette, M., Oliveros, E., 1991. Photochemical
Technology. Wiley, UK, p. 80.Chen, D., Ray, A., 1998. Photodegradation kinetics of 4-Nitrophe-
nol in TiO2 suspension. Water Res. 32 (11), 3223–3234.Council Regulation (EEC) No. 2377/90-laying down a Community
procedure for establishment of maximum residue limits ofveterinary medicinal products in foodstuffs of animal origin,Off. J. Eur. Commun. L224, 1990.
Cunningham, J., Al-Sayyed, G., Srijaranai, S., 1994. Adsorption ofmodel pollutants onto TiO2 particles in relation to photo-remedation of contaminated water. In: Helz, G., Zepp, R.,Crosby, D. (Eds.), Aquatic and Surface Photochemistry. CRCPress, Boca Raton, FL, pp. 317–348 (Chapter 22).
Dieckmann, M.S., Gray, K.A., 1996. A comparison of the degrada-tion of 4-nitrophenol via direct and sensitized photocatalysisin TiO2 slurries. Water Res. 30 (5), 1169–1183.
Dionysiou, D.D., Suidan, M.T., Baudin, I., Laine, J.M., 2004. Effect ofhydrogen peroxide on the destruction of organic contami-nants-synergism and inhibition in a continuous-modephotocatalytic reactor. Appl. Catal. B—Environ. 50 (4), 259–269.
Di Paola, A., Augugliaro, V., Palmisano, L., Pantaleo, G., Savinov, E.,2003. Heterogeneous photocatalytic degradation of nitrophe-nols. J. Photochem. Photobiol. A: Chem. 155 (1–3), 207–214.
Evgenidou, E., Fytianos, K., Poulios, I., 2005. Semiconductor-sensitized photo-degradation of dichlorvos in water usingTiO2 and ZnO as catalysts. Appl. Catal. B: Environ. 59, 83–91.
Fujishima, A., Rao, T.N., Tryk, D.A., 2000. Titanium dioxidephotocatalysis. J. Photochem. Photobiol. C: Photochem. Rev. 1(1), 1–21.
Halling-Sorensen, B., Nielsen, S.N., Lanzky, P.F., Ingerslev, F.,Lutzhoft, H.C.H., Jorgensen, S.E., 1998. Occurrence, fate andeffects of pharmaceutical substances in the environment. Areview. Chemosphere 36 (2), 357–394.
Heberer, T., 2002. Occurrence, fate, and removal of pharmaceu-tical residues in the aquatic environment: a review of recentresearch data. Toxicol. Lett. 131 (1–2), 5–17.
Herrmann, J.-M., 1999. Heterogeneous photocatalysis: fundamen-tals and applications to the removal of various types ofaqueous pollutants. Catal. Tod. 53 (1), 115–129.
Hoffman, M.R., Martin, S., Choi, W., Bahnemann, D., 1995.Environmental applications of semiconductor photocatalysis.Chem. Rev. 95 (1), 69–96.
Hurum, D.C., Agrios, A.G., Gray, K.A., Rajh, T., Thurnauer, M.C.,2003. Explaining the enhanced photocatalytic activity ofDegussa P25 mixed-phase TiO2 using EPR. J. Phys. Chem. B 107(19), 4545–4549.

ARTICLE IN PRESS
WA T E R R E S E A R C H 4 2 ( 2 0 0 8 ) 3 8 6 – 3 9 4394
Kolpin, D.W., Furlong, E.T., Meyer, M.T., Thurman, E.M., Zaugg, S.D.,Barber, L.B., Buxton, H.T., 2002. Pharmaceuticals, hormones,and other organic wastewater contaminants in US streams,1999–2000: a national reconnaissance. Environ. Sci. Technol.36 (6), 1202–1211.
Low, G.K.C., McEvoy, S.R., Matthews, R.W., 1991. Formation ofnitrate and ammonium-ions in titanium-dioxide mediatedphotocatalytic degradation of organic-compounds containingnitrogen-atoms. Environ. Sci. Technol. 25 (3), 460–467.
Mahdavi, F., Bruton, T.C., Li, Y.Z., 1993. Photoinduced reduction ofnitro-compounds on semiconductor particles. J. Org. Chem. 58(3), 744–746.
Maurino, V., Minero, C., Pelizzetti, E., Piccinini, P., Serpone, N.,Hidaka, H., 1997. The fate of organic nitrogen under photo-catalytic conditions: degradation of nitrophenols and amino-phenols on irradiated TiO2. J. Photochem. Photobiol. A: Chem.109 (2), 171–176.
Mills, A., Le Hunte, S., 1997. An overview of semiconductorphotocatalysis. J. Photochem. Photobiol. A: Chem. 108 (1), 1–35.
Muneer, M., Bahnemann, D., 2002. Semiconductor-mediatedphotocatalyzed degradation of two selected pesticide deriva-tives, terbacil and 2,4,5-tribromoimidazole, in aqueous sus-pension. Appl. Catal. B: Environ. 36 (2), 95–111.
Parra, S., Olivero, J., Pulgarin, C., 2002. Relationships betweenphysicochemical properties and photoreactivity of four bior-ecalcitrant phenylurea herbicides in aqueous TiO2 suspension.Appl. Catal. B: Environ. 36 (1), 75–85.
Pleskov, Y., Gurevich, Y., 1986. Semiconductor Photoelectrochem-istry. Consultants Bureau, New York, 223pp.
Ravina, M., Campanella, L., Kiwi, K., 2002. Accelerated miner-alization of the drug Diclofenac via Fenton reactions in aconcentric photo-reactor. Water Res. 36 (14), 3553–3560.
Sauer, T., Neto, G.C., Jose, H.J., Moreira, R., 2002. Kinetics ofphotocatalytic degradation of reactive dyes in a TiO2 slurryreactor. J. Photochem. Photobiol. A—Chem. 149 (1–3), 147–154.
Sioi, M., Bolosis, A., Kostopoulou, E., Poulios, I., 2006. Photo-catalytic treatment of colored wastewater from medicallaboratories: photocatalytic oxidation of hematoxylin. J.Photochem. Photobiol. A: Chem. 184 (1–2), 18–25.
Wolfrum, E.J., Ollis, D.F., 1994. Hydrogen peroxide in heteroge-neous photocatalysis. In: Helz, G., Zepp, R., Crosby, D. (Eds.),Aquatic and Surface Photochemistry. Lewis Publishers, CRCPress, UK, p. 261 Chapter 32.
Woodward, K.N., 2004. In: Watson, D.H. (Ed.), Pesticide, Veterinaryand Other Residues in Food. Woodward Publisher Limited,Cambridge, p. 176 (Chapter 8).




















