
Photocatalytic reactions over TiO2 supported on porcelain spheres
16
1 PHOTOCATALYTIC REACTIONS OVER TiO 2 SUPPORTED ON PORCELAIN SPHERES GABRIELA PIPERATA, JORGE M. MEICHTRY AND MARTA I. LITTER ∗ Unidad de Actividad Química, Centro Atómico Constituyentes, Comisión Nacional de Energía Atómica, Buenos Aires, Argentina Av. Gral. Paz 1499, 1650 San Martín, Prov. de Buenos Aires, Argentina 541167727016 541167727886 [email protected] Abstract Results of immobilisation of TiO 2 on low-cost porcelain beads and the resulting photocatalytic activities of the samples are presented. A very simple technique for immobilisation by immersion in a TiO 2 suspension, previously acidified to pH 2.5, followed by calcination at 450 o C, was used. After a vigorous washing with a water jet, no detachment of the catalyst was observed at the end of the fixation process. Reasons for the stability of the material are discussed. The crystallographic nature of P-25 was not affected after attachment to the porcelain surface. The beads were submitted for 100 h to turbulent regimes corresponding to Re numbers of 100 and 400 in a recirculating system. The effect of the recirculation flow was carefully examined to evaluate the degree of detachment of the catalyst from the support and to assess how the treatment influences on the activity of the supported catalyst. For this, photocatalytic tests were performed on two model compounds, 4-chlorophenol and potassium dichromate in the presence of ethylenediaminetetraacetic acid, and results were compared with those of untreated beads. A good activity remained in the beads even after washing for more than 100 hours in a turbulent regime (both Re 100 and 400). The activity of particles detached from the support by ultrasonication was also compared with that of the pure precursor. Key words: supported TiO 2 , heterogeneous photocatalysis, porcelain beads Introduction Heterogeneous photocatalysis (HP) for disinfection and detoxification of effluents is an emergent technology already applied in industrialised countries [1, 2 and references therein]. By irradiation of a semiconductor with UV light (below 400 ∗ Corresponding author
Transcript of Photocatalytic reactions over TiO2 supported on porcelain spheres

1
PHOTOCATALYTIC REACTIONS OVER TiO2 SUPPORTED ON PORCELAIN SPHERES
GABRIELA PIPERATA, JORGE M. MEICHTRY AND MARTA I. LITTER∗
Unidad de Actividad Química, Centro Atómico Constituyentes, Comisión Nacional de Energía Atómica, Buenos Aires, Argentina
Av. Gral. Paz 1499, 1650 San Martín, Prov. de Buenos Aires, Argentina 541167727016 541167727886
Abstract
Results of immobilisation of TiO2 on low-cost porcelain beads and the resulting photocatalytic
activities of the samples are presented. A very simple technique for immobilisation by immersion
in a TiO2 suspension, previously acidified to pH 2.5, followed by calcination at 450 oC, was used.
After a vigorous washing with a water jet, no detachment of the catalyst was observed at the end of
the fixation process. Reasons for the stability of the material are discussed. The crystallographic
nature of P-25 was not affected after attachment to the porcelain surface.
The beads were submitted for 100 h to turbulent regimes corresponding to Re numbers of 100 and
400 in a recirculating system. The effect of the recirculation flow was carefully examined to
evaluate the degree of detachment of the catalyst from the support and to assess how the treatment
influences on the activity of the supported catalyst. For this, photocatalytic tests were performed
on two model compounds, 4-chlorophenol and potassium dichromate in the presence of
ethylenediaminetetraacetic acid, and results were compared with those of untreated beads. A good
activity remained in the beads even after washing for more than 100 hours in a turbulent regime
(both Re 100 and 400). The activity of particles detached from the support by ultrasonication was
also compared with that of the pure precursor.
Key words: supported TiO2, heterogeneous photocatalysis, porcelain beads
Introduction
Heterogeneous photocatalysis (HP) for disinfection and detoxification of effluents
is an emergent technology already applied in industrialised countries [1, 2 and
references therein]. By irradiation of a semiconductor with UV light (below 400
∗ Corresponding author

2
nm), electron-hole pairs are created that can react with acceptors and donors
present in the suspension. Active species such as HO• radicals are formed,
provoking mineralization of organic pollutants [3]. Even toxic ionic metals can be
transformed in less noxious species [4].
So far, the most used photocatalyst is TiO2, especially in its commercial Degussa
P-25 powdered form. One of the most important drawbacks to achieve the
successful implementation of HP is that the photocatalyst must be separated from
the system after the treatment, and the cost of this separation stage may even
invalidate economically the technology [5, 6]. To solve this problem, several
attempts to immobilise TiO2 have been performed. For the immobilisation, two
techniques can be envisaged [6-9]: i) “in situ” synthesis of the oxide on the
support by a sol-gel, CVD technique or others; ii) direct impregnation of a
commercial or previously prepared TiO2 powder on the surface of the support.
The second one is the easiest and most economical procedure. The fixation is
achieved starting from a TiO2 suspension in an adequate dispersant followed by
sinterization, in some cases with the addition of ligand agents [10-12]. The
process by which the particles are fixed is not totally clear. Probably, electrostatic
interactions between particles and charge surfaces are involved, although covalent
interactions cannot be discarded when the adherence is extremely strong (for
example, with glass). There is no agreement on which method produces films with
better photocatalytic activity, and this depends generally on the nature and
experimental conditions of the system [6].
There is an important variety of materials that can be used to support TiO2. The
most studied are the ones based on SiO2, owing to their low cost and easy
availability [6]. The success of these coatings is based on the large adherence
between TiO2 and glass, attributed to the occurrence of some sintering in the
calcination process [13]. The thermal treatment is limited by the glass softening
temperature that, in the case of borosilicate, is next to 500 ºC. To overcome this
limitation, more resistant and then more expensive glasses must be used. Other
support materials include metals, ceramics, zeolites, plastics, cements, red bricks
and inorganic fibres [5, 7, 14-19].
The first prototypes of photoreactors with supported TiO2 were constructed by
simply covering the internal wall of glass tubes with powdered TiO2 [20-21]. To
have more surface area, beads, rings or glass fibres inside the tubes have been

3
filled [5]. These materials have been successfully used in commercial devices for
air purification [22]. Conversely to gas systems, the catalyst has been found
mechanically unstable in aqueous systems, generally submitted to strong abrasion
and turbulent regimes, which cause removal of the TiO2 coating with the time of
use. The geometrical shape of the support is a very important parameter for
fixation: whereas the fixation to a flat or cylindrical surface yields rather good
permanent adhesion, glass spherical shapes produce poor results, and TiO2 is
rapidly detached.
As it is known, the surface area as well as the nature of the TiO2 phase strongly
influences photocatalytic activity. Changes in the electronic and crystalline
structure of TiO2 due to interaction with the support may result in alteration of the
efficiency. The attachment of TiO2 to a surface decreases considerably the
available surface area and the amount of light reaching the particles. Also, mass
transfer limitations due to a poorer access or contact of the pollutant with the
catalyst are usually invoked. As a general behaviour, supported TiO2 is always
less active than suspended TiO2.
In this work, we discuss results of immobilisation of TiO2 on low-cost small
porcelain spheres where TiO2 has been impregnated by a simple technique. The
degree of detachment of the catalyst from the surface and the photocatalytic
activity on model compounds as 4-chlorophenol (4-CP) and potassium dichromate
(Cr(VI)) have been evaluated.
Experimental section
Materials and equipment
As support for TiO2, commercial porcelain beads according to 40685 DIN
Standard, Group 100 (Del Morro S.A., Argentina), 6 mm diameter were used.
Basically, this porcelain is a silicoaluminate containing vestiges of potassium.
TiO2 Degussa P-25 was a commercial sample, used as provided. K2Cr2O7 (Carlo
Erba), 4-CP (Fluka), Na2EDTA (Mallinkrodt) and all other chemicals were of
analytical reagent grade. Doubly distilled water was used for the preparation of all
solutions and for final washings. Diluted HClO4 was used for pH adjustments.
X-ray diffraction (XRD) patterns were obtained at room temperature with a
Philips PW-3710 diffractometer, using CuKα radiation.

4
Spectrophotometric measurements were performed with a Hewlett-Packard diode
array UV-visible spectrophotometer, model HP 8453 A.
Ultrasonications were performed with a Cleanson (25 KHz), ultrasonicator, model
CS-1109.
Impregnation procedure
Beads were impregnated with TiO2 according to the following procedure. Spheres
were introduced in a 0.1 M HNO3 solution for 2 h, then washed four times with
water and dried at 100 ºC. This step was followed by immersion in a 1M KOH
solution for 48 h, washing four times with water and drying again at 100 ºC. A
10% w/v TiO2 suspension, previously adjusted at pH 2.5 with HClO4, was
ultrasonicated for 30 min and the spheres were immersed in the suspension for 15
min. The excess of suspension was drained and the material was dried at 100 ºC
and calcined at 450 ºC for 2 h at a 3.3 ºC min-1 heating rate. At the end, the
spheres were rinsed for 15-30 min with a vigorous water jet to eliminate loose
particles, and dried at 100 ºC. The impregnation procedure was repeated six times.
Evaluation of the abrasion resistance
The mechanical stability of TiO2 on the porcelain spheres was tested by washing
with bidistilled water for more than 100 hours. For this sake, a recirculation set-up
was used, consisting of a 30-mm diameter, 140-mm length glass tube, in which
230 spheres were arranged as a fixed bed. From a reservoir, 500 mL of distilled
water was continuously recirculated by means of a peristaltic pump. Two
recirculation rates, 0.6 L min-1 and 2.4 L min-1, corresponding to Re 100 and Re
400 regimes, were tested. The Reynolds number for spherical beads can be
calculated according to [23]:
Re = Dp ρ v0 µ-1 (1)
where:
Dp: beads diameter
ρ: fluid density
v0: flow rate in an empty bed
µ: fluid viscosity

5
For this geometry and configuration, a turbulent flow is defined for Re > 40.
Therefore, both recirculation rates correspond to turbulent regimes.
Periodically, samples of washings were taken and analysed
spectrophotometrically against pure water to evaluate the bleeding of TiO2 by
changes in the UV region due to the scattering caused by TiO2 particles. After
around 100 h of recirculation, a 45-mL sample was filtered through a 0.2 µm
Sartorius filter membrane and the membrane was analysed by XRD for a possible
solid residue.
Hereafter in this work, beads will be named “uncoated” (without TiO2),
“untreated” (with TiO2 but not submitted to the recirculation treatment), “Re 100”
and “Re 400”, respectively.
To estimate the amount of TiO2 attached to the beads, 21 of them were
ultrasonicated (25 KHz) for 30 min in 10 mL of bidistilled water. Ultrasonication
at this frequency did not provoke any deterioration of the beads. The beads were
withdrawn from the suspension, rinsed and dried at 100 ºC, and the mass of TiO2
was calculated from differences in weight before and after the detachment. This
procedure was repeated three times and the results averaged. The amount of TiO2
deposited on untreated beads was 2.1 mg TiO2/g spheres (≈ 0.8 mg TiO2/cm2).
For “Re 100”, 1.7 mg TiO2/g spheres was calculated (≈ 0.7 mg TiO2/cm2) and for
Re 400, 0.6 mg TiO2/g spheres (≈ 0.3 mg TiO2/cm2). The suspensions obtained
from the ultrasonication procedure were used for photocatalytic experiments, as
described later.
Photocatalytic tests
Photocatalytic irradiations were carried out in a 30-mL cylindrical cell, open to
air, irradiated from the top, using high-pressure xenon arc lamps (Osram XBO,
150 or 450 W). The set-up was thermostatted at 25 ºC. The cell was provided with
a 300-nm cut off filter, avoiding wavelengths lower than 300 nm. The IR fraction
of the incident light was removed by a 50-mm cylindrical water filter.
The solution containing the model pollutant (4-CP or Cr(VI)) at a fixed
concentration and pH, together with TiO2, either supported or in suspension, was
introduced in the cell. In the case of supported TiO2, a layer of 21 beads was
evenly arranged on the bottom of the cell. When suspended TiO2 was used, the
suspension was first ultrasonicated for 15 min and magnetically stirred during the

6
run. Prior to irradiation, solutions or suspensions were kept in the dark for 30 min,
to assure substrate-surface equilibrium. No significant differences in
concentrations were observed after this dark period. Once the irradiation was
initiated, periodical 500 µL samples were taken and diluted to 5 mL for analysis.
Suspensions were filtered through a 0.45 µm MSF filter before analysis. At least,
duplicated runs were carried out for each condition, averaging the results. Photon
flows per unit volume were determined with ferrioxalate [24], using the same
volume of actinometric solution as those used in the experiments to keep the same
conditions.
For 4-chlorophenol, 25 mL (corresponding to a 2.8-cm height of the solution in
the cell) of a 0.2 mM solution at initial pH 3 were irradiated. 4-CP degradation
was followed by the decrease of the peak at 225 nm. During the photocatalytic
reaction, some intermediates (phenol, hydroquinone, among others) can be
formed. Under our conditions, the amount of them is very low and their
absorption coefficient at 225 nm is lower than that of 4-CP [25, 26]. This validates
our measurement methodology.
For chromium (VI) experiments, 20 mL (corresponding to a 2.5-cm height of the
solution in the cell) of 0.4 mM K2Cr2O7, together with 0.9 mM Na2EDTA as a
sacrificial synergetic agent [4], was used. Initial pH was adjusted to 2. Cr(VI)
measurement was performed spectrophotometrically at 249 nm [27].
The concentration of TiO2 in the different experiments is indicated in the legends
of the figures. For the case of supported TiO2, the values correspond to equivalent
concentrations in suspension, calculated from the estimated amount of TiO2 in the
beads.
Results and discussion
Preparation of the impregnated beads
A very simple procedure was used to impregnate the porcelain beads with TiO2. A
pretreatment with HNO3 solution (pH 1) for 2 h, allowed to eliminate any
previous impurity of the surface to improve further treatments. As the surface of
beads was very smooth, an alkaline treatment on the porcelain by immersion in
KOH solution was done. A 10% w/v TiO2 suspension, adjusted at pH 2.5 with
HClO4, was ultrasonicated to break possible aggregates. The supports were

7
introduced then in this suspension and kept for a time as short as 15 min. At this
pH value and TiO2 concentration, the deposition of the catalyst is facilitated by
electrostatic attraction. Use of another mineral acid than HClO4 could have left
anions on the surface that would have reduced the photocatalytic activity of the
semiconductor [12]. Calcination at 450 ºC for 2 h at a controlled temperature
ramp was performed. In this way, beads were resistant to crack. Finally, the
material was rinsed vigorously with water to eliminate loose particles. Previous
experiments in our laboratory had shown that the activity of the beads were higher
when two TiO2 layers were applied compared to only one impregnation. In this
work, we decided to apply 6 layers, but this number should be still optimised.
The impregnation treatment did not affect the crystallographic composition of the
TiO2 coated onto the spheres, as it will be discussed later.
Evaluation of the abrasion resistance under different recirculation regimes
The adherence of TiO2 to porcelain spheres was tested by washing with bidistilled
water in two vigorous turbulent regimes for more than 100 hours. After this
treatment, washings were analysed for the presence of TiO2 particles,
spectrophotometrically as well as by DRX after filtration. Concerning “Re 100”,
no TiO2 was observed in any case in the washings or as a solid residue, but
actually, some detachment was detected by difference of weight before and after
treatment in the turbulent regime (2.1 against 1.7 mg TiO2/g spheres). In the case
of “Re 400”, a much lower amount of TiO2 remained after the treatment (0.6 mg
TiO2/g spheres) and a very weak but visible peak of anatase was detected by DRX
as a solid residue in the membrane used for filtration.
It was verified that neither the turbulent recirculation nor the impregnation
procedure had affected the crystallographic composition of the coated TiO2. For
this sake, Re 100 beads were submitted to ultrasonication for 30 min and a DRX
spectrum of the detached TiO2 powder was obtained. Comparison of this spectrum
with that of pure P-25 (not shown), indicated that both samples presented a similar
anatase:rutile ratio (74% anatase), calculated from the intensity of the main
diffraction peaks ((101) anatase and (110) rutile), according to ref. [28].

8
On the other hand, it was proven that on this type of beads, made of porcelain,
TiO2 presented a higher adherence than that on Pyrex glass spheres, tested
previously by us in preliminary experiments.
Photocatalytic experiments
To evaluate the activity of the coated beads, two photocatalytic model systems,
tested previously in our and other laboratories were used: a) degradation of 4-CP;
b) reduction of Cr(VI) in the presence of excess of ethylenediaminetetraacetic acid
(EDTA). No attempts to compare the results of the different experiments after
normalization by TiO2 mass have been made.
Figure 1 shows 4-CP degradation with different TiO2-containing beads at pH 3.
Untreated beads as well as beads submitted to both Re regimes are compared,
together with TiO2 in suspension at the same concentration as in the untreated
beads (0.7 g l-1). As expected, immobilized TiO2 presented lower efficiency than
that of the suspended catalyst at the same concentration. As it can be seen,
although the activity is decreased after the turbulent recirculation treatment, it is
worthwhile to remark that the treatment was prolonged for more than 100 h. The
activity loss was higher with the highest Re number. Of course, the amount of
TiO2 in the reaction volume was also lower due to the previous detachment.
However, both Re 100 and Re 400 beads show still some efficiency.
⟨Figure 1⟩ As the photocatalytic degradation of 4-CP with immobilised TiO2 was found a
slow system to compare the activities of the samples, another very well known
and faster photocatalytic reaction, reduction of Cr(VI) in the presence of EDTA at
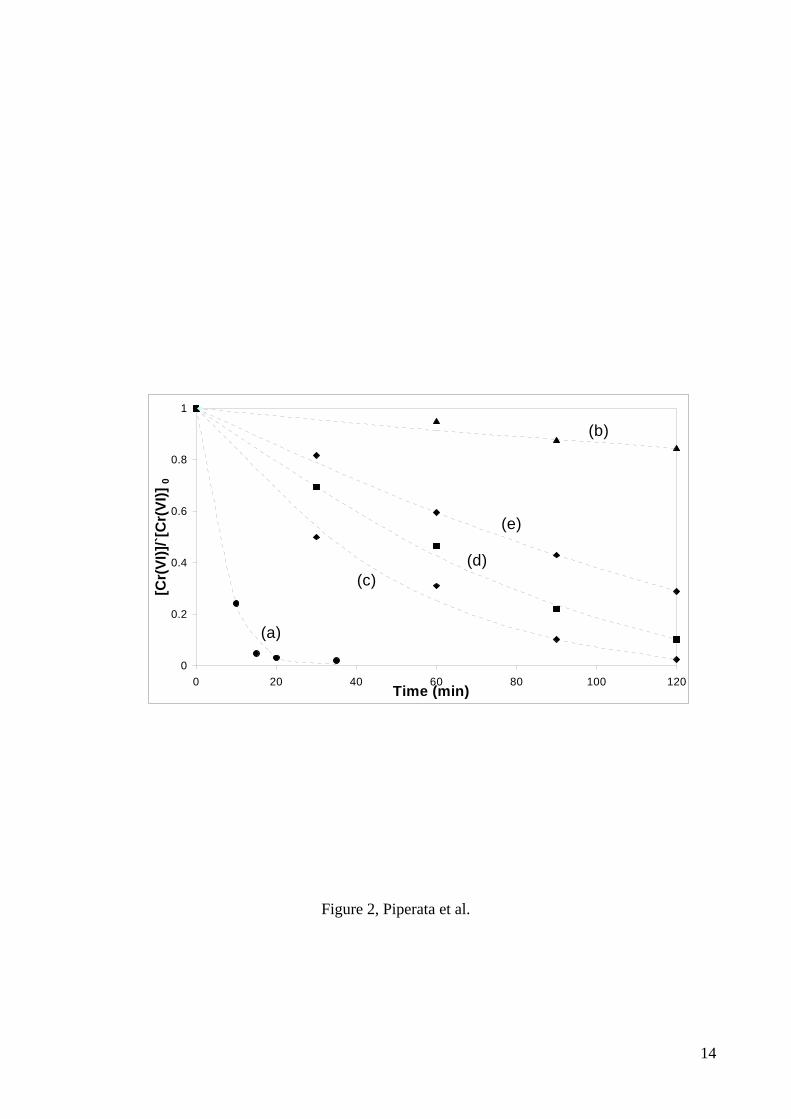
pH 2 [4], was tested. Results are shown in Figure 2. In the absence of TiO2, a
minor photochemical reaction between Cr(VI) and EDTA takes place. The results
were similar to those obtained for 4-CP, Cr(VI) reduction efficiency decreasing in
immobilised TiO2 compared to that in suspension and in “Re 100” and “Re 400”
with respect to “untreated”.
⟨Figure 2⟩ Activities of “Re 100” and a suspension prepared from TiO2 detached from “Re
100” by ultrasonication are compared in Figure 3, together with a P-25 suspension
of the same concentration. As expected, due to a lower surface area, mass transfer

9
limitations and a lower amount of light reaching the beads only from the top, the
efficiency was lower for the immobilised catalyst. It must be taken into account
also that suspensions were magnetically stirred in contrast with experiments done
with beads. TiO2 detached from “Re 100” shows a lower activity compared with
that of the precursor suspension. This lower activity can be explained by the
presence of some impurities coming from the support [17].
⟨Figure 3⟩ Same experiments were performed using “Re 400” and results are presented in
Figure 4. As in the previous case, fixed TiO2 presents a lower activity than TiO2
in suspension. It is remarkable that here the activity of detached TiO2 is almost
identical to the activity of P-25. We think that a more profound washing than in
“Re 100” has removed impurities, increasing the activity.
⟨Figure 4⟩
Conclusions
Our results indicate that a very good adherence of TiO2 is obtained onto this
porcelain beads compared with Pyrex glass spheres. This aspect is important as
this is one of the main drawbacks when spherical geometries are used to fix the
catalyst by impregnation. We have then obtained a cost effective material by a
very simple technique, in comparison with sol-gel or other preparative synthesis
of TiO2 on the support.
We have noticed that the adherence of TiO2 to the support is affected by a
turbulent regime in a recirculation system. However, Reynolds number
corresponding to 100 would be still suitable for application in a fixed bed reactor
without significant detachment of the catalyst. Further experiments are needed to
determine the optimal Re number that guarantees a turbulent regime without TiO2
detachment.
Although immobilised TiO2 on porcelain beads does not exhibit a very good
photocatalytic activity for Cr(VI) reduction and 4-CP degradation under the
conditions explored in this paper compared with TiO2 in suspension, the new
material would able to be applied in a fixed bed in which, with an optimum flow
rate, geometry and other parameters, a better global performance of the system
would be obtained.

10
Further studies are underway to optimise the fixation time and the number of
TiO2 layers to be applied on the support.
Acknowledgements
We thank G. Leyva for taking DRX spectra. Work performed as part of Comisión
Nacional de Energía Atómica CNEA P5-PID-36-4 Program, Agencia Nacional de
Promoción de la Ciencia y la Tecnología, PICT98-13-03672 ANPCYT project
and Consejo Nacional de Investigaciones Científicas y Técnicas, PIP662/98
CONICET project. G.P. thanks CNEA-CONICET for a fellowship to perform this
work. M.I.L. is a member of CONICET.

11
References
[1] Hoffmann MR, Martin ST, Choi W, Bahnemann DW (1995) Chem. Rev. 95: 69.
[2] Domènech X, Jardim W, Litter M (2001) Tecnologías avanzadas de oxidación para la eliminación de
contaminantes. In: Blesa MA (ed). Eliminación de contaminantes por fotocatálisis heterogénea. Texto
colectivo elaborado por la Red CYTED VIII-G. Digital Grafic, La Plata, pp 3-26. Available in:
http://www.cnea.gov.ar/xxi/ambiental/cyted.asp.
[3] Bahnemann D (1999) Photocatalytic detoxification of polluted waters. In: P. Boule (ed). Handbook of
environmental photochemistry. Springer Verlag Berlin, p. 285.
[4] Litter MI (1999) Appl. Catal. B: Environ. 23: 89.
[5] Candal RJ, Rodríguez J, Colón G, Gelover S, Vigil Santos E, Jiménez González A, Blesa MA (2001)
Materiales para fotocatálisis y electrofotocatálisis. In: Blesa MA (ed). Eliminación de contaminantes por
fotocatálisis heterogénea. Texto colectivo elaborado por la Red CYTED VIII-G. Digital Grafic, La Plata, pp
143-166. Available in: http://www.cnea.gov.ar/xxi/ambiental/cyted.asp.
[6] Pozzo RL, Baltanás MA, Cassano AE (1997) Catal. Today 39: 219.
[7] Anandan S, Yoon M (2003) J. Photochem. Photobiol. C: Photochem. Rev. 4: 5.
[8] Hu C, Yizhong W, Hongxiao T (2001) Appl. Catal. B: Environ. 30: 277.
[9] Monneyron P, Manero M-H, Foussard J-N, Benoit-Marquisse F, Maurette M-T (2003) Chemical
Engineering Science 58: 971.
[10] Barbé C, Arendse F, Comte P, Jirousek M, Lenzman F, Shklover V, Grätzel M (1997) J. Am. Ceram.
Soc. 80: 3157.
[11] Burnside SD, Shklover V, Barbé C, Comte P, Arendse F, Brooks K, Grätzel M (1998) Chem Mater. 10:
2419.
[12] Haarstrick A, Kut OM, Heinzle E (1996) Environ. Sci. Technol, 30: 817.
[13] Zeltner WA, Hill CG Jr., Anderson MA (1993) Chemtech 21.
[14] Candal RJ, Zeltner WA, Anderson MA (1999) J. Environ. Eng. 125: 906.
[15] Candal RJ, Zeltner WA., Anderson MA (1998) J. Adv. Oxid. Tech. 3: 270.
[16] Candal RJ, Zeltner WA, Anderson MA (1999) J. Environ. Eng. 125: 906.
[17] Fernández A, Lassaletta G, Jiménez VM, Justo A, González-Elipe AR, Herrmann JM, Tahiri H, Ait-
Ichou Y (1995) App. Catal. B: Environ. 7: 49.
[18] Anpo M, Takeuchi M (2003) Journal of Catalysis 216: 505.
[19] Rachel A, Subrahmanyam M, Boule P (2002) Appl. Catal. B: Environ. 37: 301.
[20] Matthews R (1987) J Phys. Chem. 91:3328.
[21] Sabate J, Anderson MA, Kikkawa H, Edwards M, Hill CG Jr. (1991) J. Catal. 127: 167.
[22] Zorn ME, Tompkins DT, Zeltner WA, Anderson MA (2000) Environ. Sci. Technol. 34: 5206.
[23] Perry JH (1979) Manual del Ingeniero Químico, 3ª. Edición, Mc Graw Hill, México.
[24] Hatchard CG, Parker CA (1956) Proc. Roy. Soc. A 235: 518.
[25] Theurich J, Lindner M, Bahnemann DW (1996) Langmuir 12:6368.
[26] Hodak J, Quinteros C, Litter MI, San Román (1996) J. Chem. Soc. Faraday Trans. 92:5081.
[27] Wei C, German S, Basak SR, Rajeshwar K (1993) J. Electrochem. Soc. 140: L60.
[28] Criado JM, Real C (1983) J. Chem. Soc. Faraday Trans. 1: 2765.

12
Legends to the figures
Figure 1: 4-CP degradation with immobilised and suspended TiO2. Conditions: [4-CP] = 0.2 mM;
pH0 3; T = 25oC; open to air; P0 = 12 µeinstein s-1 l-1; λ > 300 nm. a) “untreated”, [TiO2] = 0.7 g l-
1; b) P-25 suspension, [TiO2] = 0.7 g l-1 ; c) “Re 100”, [TiO2] = 0.5 g l-1; c) “Re 400”, [TiO2] =
0.15 g l-1.
Figure 2: Cr(VI) reduction with suspended TiO2 and beads submitted to different treatments.
Conditions: [Cr(VI)] = 0.2 mM; [EDTA] = 0.9 mM; pH0 2; T = 25oC; open to air; P0 = 18
µeinstein s-1 l-1; λ > 300 nm. a) P-25 suspension, [TiO2] = 0.9 g l-1; b) “uncoated”, [TiO2] = 0; c)
“untreated”, [TiO2] = 0.9 g l-1; d) “Re 100”, [TiO2] = 0.6 g l-1; e) “Re 400”, [TiO2] = 0.2 g l-
1.Figure 3: Cr(VI) reduction efficiencies of immobilised and suspended TiO2. The equivalent
concentration of TiO2 in all cases is 0.6 g l-1. Conditions: [Cr(VI)] = 0.2 mM; [EDTA] = 0.9 mM;
pH0 2; T = 25oC; open to air; P0 = 3.6 µeinstein s-1 l-1; λ > 300 nm. a) “Re 100”; b) TiO2 detached
from “Re 100” after ultrasonication; c) P-25 suspension.
Figure 4: Cr(VI) reduction efficiencies of immobilised and suspended TiO2. The equivalent
concentration of TiO2 in all cases is 0.2 g l-1. Conditions: [Cr(VI)] = 0.2 mM; [EDTA] = 0.9 mM;
pH0 2; T = 25 oC; open to air; P0 = 3.6 µeinstein s-1 l-1; λ > 300 nm. a) “Re 400”; b) TiO2 detached
from “Re 400” after ultrasonication; c) P-25 suspension

0
0.2
0.4
0.6
0.8
1
0 1 2 3Time (h)
[4-C
P]/[4
-CP]
0
4
(c)
(b)
(a)
(d)
Figure 1, Piperata et al.
13
0
0.2
0.4
0.6
0.8
1
0 20 40 60 80 100Time (min)
[Cr(
VI)]/
`[Cr(
VI)]
0
120
(b)
(d)(c)
(a)
(e)
Figure 2, Piperata et al.
14

0
0.2
0.4
0.6
0.8
1
0 20 40 60 80 100 120Time (min)
[Cr(
VI)]/
[Cr(
VI)] 0
(b)(c)
(a)
Figure 3, Piperata et al.
15

0
0.2
0.4
0.6
0.8
1
0 20 40 60 80 100 120Time (min)
[Cr(
VI)]/
[Cr(
VI)]
0
(b)
(a)
(c)
Figure 4, Piperata et al.References
16




















