
Periodic Trends in Adsorption and Activation Energies for Heterometallic Diffusion on (100)...
7
Periodic Trends in Adsorption and Activation Energies for Heterometallic Diffusion on (100) Transition Metal Surfaces Handan Yildirim, Subramanian K.R.S. Sankaranarayanan,* and Jeffrey P. Greeley* Center for Nanoscale Materials, Argonne National Laboratory, Argonne, Illinois 60439, United States * S Supporting Information ABSTRACT: A first-principles analysis of trends in metal-on-metal hopping diffusion for 64 admetal/substrate systems is presented. Focusing on the (100) facets of various transition metal substrates, we demonstrate that the calculated hopping diffusion barriers may be interpreted in terms of the cohesive energies of the admetals and substrates, as well as the lattice constants of the substrates. We further show that general linear relationships exist between the diffusion barriers and the corresponding adsorption energies on each transition metal substrate. The slopes in these Brønsted-Evans- Polanyi relationships are related to the degree of resemblance between the initial states and the transition states for hopping diffusion, and the slopes are found to depend sensitively on the nature of the transition metal substrate. Substrates with higher cohesive energies and smaller lattice constants generally exhibit smaller slopes and, therefore, a closer correspondence between the transition states and the initial states. These relationships, in addition to providing fundamental insights into trends in diffusion across different transition metal surfaces, give a powerful and convenient means of predicting diffusional kinetics from purely thermodynamic quantities. The results may ultimately provide a useful input to kinetic Monte Carlo (kMC)-type simulations, enabling efficient and accurate studies of heteroepitaxial metal-on-metal growth. I. INTRODUCTION Metal-on-metal surface diffusion is central to both the basic physics of crystal and thin film growth and to a variety of technologically important fields, including catalysis, micro- electronics, and corrosion. In spite of these diverse and significant applications, however, fundamental knowledge of the kinetics and dynamics of diffusing metal adspecies is far from complete, and nearly all atomistic studies of these phenomena have focused on the diffusion of specific metals across specific substrates. 1-6 While such studies, involving both experimen- tal 7,8 and computational techniques, have identified many important principles of surface diffusive processes, a more general understanding of atomic-scale trends in surface diffusion across different admetals and substrates is lacking. The development of such a generalized understanding, in turn, could be of significant benefit in controlling the structure of alloys during growth or dealloying processes and in designing bimetallic materials for desired applications. Theoretical surface science studies have emerged in recent years as a powerful tool to elucidate the kinetics, dynamics, and atomistic details of the mechanisms governing surface processes. Such studies, based primarily on periodic Density Functional Theory (DFT) calculations, have found extensive uses in a variety of applications. 9-15 These calculations have been shown, for example, to be useful for obtaining linear energy scaling relationships for a variety of molecular adsorbates on transition metal surfaces. 16 Additional linear relationships are of the Brønsted-Evans-Polanyi (BEP) type, 17-19 which describes the correlations between the kinetics of elementary surface processes and the corresponding thermodynamics. 20,21 For complex catalytic reactions, these relationships have permitted the description of fundamental reactivity trends across diverse catalyst surfaces using just a few independent parameters, or descriptors. 22-27 To a significantly lesser extent, simplified forms of these general classes of linear correlations have been used to describe metal adatom diffusion on transition metal substrates. 28-31 In particular, adatom diffusion on corrugated surfaces such as (100) has often been studied, 28-31 and some correlations between the adsorbate binding strengths and the bulk bond energies have been suggested for self-diffusion processes 32 wherein the admetal and the substrate have the same elemental identity (these systems are also referred to with the term “homodiffusion” in the remainder of this article). A linear relation between the hopping barriers over step edges and the (111) terrace adsorption energies has also been reported for a few heterodiffusive processes, wherein the elemental identities of the admetal and substrate are different. 33 Similarly, a relatively recent first- principles study, using related ideas, has reported that BEP-type correlations exist for the diffusion of several atomic and molecular adsorbates (C, N, NO) on close-packed transition metal surfaces. 34 In spite of the advances described above, there are relatively few general principles that have been identified for describing atomistic details of heterodiffusion of metal adatoms on metal Received: September 7, 2012 Published: September 29, 2012 Article pubs.acs.org/JPCC © 2012 American Chemical Society 22469 dx.doi.org/10.1021/jp3089275 | J. Phys. Chem. C 2012, 116, 22469-22475
-
Upload
independent -
Category
Documents
-
view
4 -
download
0
Transcript of Periodic Trends in Adsorption and Activation Energies for Heterometallic Diffusion on (100)...

Periodic Trends in Adsorption and Activation Energies forHeterometallic Diffusion on (100) Transition Metal SurfacesHandan Yildirim, Subramanian K.R.S. Sankaranarayanan,* and Jeffrey P. Greeley*
Center for Nanoscale Materials, Argonne National Laboratory, Argonne, Illinois 60439, United States
*S Supporting Information
ABSTRACT: A first-principles analysis of trends in metal-on-metal hopping diffusionfor 64 admetal/substrate systems is presented. Focusing on the (100) facets of varioustransition metal substrates, we demonstrate that the calculated hopping diffusion barriersmay be interpreted in terms of the cohesive energies of the admetals and substrates, aswell as the lattice constants of the substrates. We further show that general linearrelationships exist between the diffusion barriers and the corresponding adsorptionenergies on each transition metal substrate. The slopes in these Brønsted−Evans−Polanyi relationships are related to the degree of resemblance between the initial statesand the transition states for hopping diffusion, and the slopes are found to dependsensitively on the nature of the transition metal substrate. Substrates with highercohesive energies and smaller lattice constants generally exhibit smaller slopes and,therefore, a closer correspondence between the transition states and the initial states.These relationships, in addition to providing fundamental insights into trends in diffusion across different transition metalsurfaces, give a powerful and convenient means of predicting diffusional kinetics from purely thermodynamic quantities. Theresults may ultimately provide a useful input to kinetic Monte Carlo (kMC)-type simulations, enabling efficient and accuratestudies of heteroepitaxial metal-on-metal growth.
I. INTRODUCTION
Metal-on-metal surface diffusion is central to both the basicphysics of crystal and thin film growth and to a variety oftechnologically important fields, including catalysis, micro-electronics, and corrosion. In spite of these diverse andsignificant applications, however, fundamental knowledge of thekinetics and dynamics of diffusing metal adspecies is far fromcomplete, and nearly all atomistic studies of these phenomenahave focused on the diffusion of specific metals across specificsubstrates.1−6 While such studies, involving both experimen-tal7,8 and computational techniques, have identified manyimportant principles of surface diffusive processes, a moregeneral understanding of atomic-scale trends in surfacediffusion across different admetals and substrates is lacking.The development of such a generalized understanding, in turn,could be of significant benefit in controlling the structure ofalloys during growth or dealloying processes and in designingbimetallic materials for desired applications.Theoretical surface science studies have emerged in recent
years as a powerful tool to elucidate the kinetics, dynamics, andatomistic details of the mechanisms governing surfaceprocesses. Such studies, based primarily on periodic DensityFunctional Theory (DFT) calculations, have found extensiveuses in a variety of applications.9−15 These calculations havebeen shown, for example, to be useful for obtaining linearenergy scaling relationships for a variety of molecularadsorbates on transition metal surfaces.16 Additional linearrelationships are of the Brønsted−Evans−Polanyi (BEP)type,17−19 which describes the correlations between the kinetics
of elementary surface processes and the correspondingthermodynamics.20,21 For complex catalytic reactions, theserelationships have permitted the description of fundamentalreactivity trends across diverse catalyst surfaces using just a fewindependent parameters, or descriptors.22−27 To a significantlylesser extent, simplified forms of these general classes of linearcorrelations have been used to describe metal adatom diffusionon transition metal substrates.28−31 In particular, adatomdiffusion on corrugated surfaces such as (100) has often beenstudied,28−31 and some correlations between the adsorbatebinding strengths and the bulk bond energies have beensuggested for self-diffusion processes32 wherein the admetal andthe substrate have the same elemental identity (these systemsare also referred to with the term “homodiffusion” in theremainder of this article). A linear relation between the hoppingbarriers over step edges and the (111) terrace adsorptionenergies has also been reported for a few heterodiffusiveprocesses, wherein the elemental identities of the admetal andsubstrate are different.33 Similarly, a relatively recent first-principles study, using related ideas, has reported that BEP-typecorrelations exist for the diffusion of several atomic andmolecular adsorbates (C, N, NO) on close-packed transitionmetal surfaces.34
In spite of the advances described above, there are relativelyfew general principles that have been identified for describingatomistic details of heterodiffusion of metal adatoms on metal
Received: September 7, 2012Published: September 29, 2012
Article
pubs.acs.org/JPCC
© 2012 American Chemical Society 22469 dx.doi.org/10.1021/jp3089275 | J. Phys. Chem. C 2012, 116, 22469−22475
substrates. This lack of generality, in turn, is largely due to theabsence of a comprehensive, trends-based, first-principlesdatabase of energetic information for diffusion of differentclasses of transition metal adspecies on metal substrates. Suchresults would be highly useful in identifying periodic trends inadsorption and activation energies for hetero metal-on-metaldiffusion and in developing correlations between thethermodynamics and kinetics of diffusion for use in longertime scale kinetic Monte Carlo (kMC)-based simulations.35−43
In this paper, we report on first-principles, periodic DFTcalculations of the adsorption energies and activation barriersfor hopping diffusion of the metal adatoms Cu, Ag, Au, Pd, Pt,Rh, Ru, and Ir on the terraces of the unreconstructed (100)surfaces of Cu, Ag, Au, Pd, Pt, Rh, Ni, and Ir, for 64 systems intotal (we do not explicitly consider diffusion via exchangemechanisms although exchange processes are known, in somecases, to lead to surface alloying on (100) surfaces44). Weconfirm that the energetics of self-diffusion for homodiffusionsystems are closely related to the bulk cohesive energies of thestudied metals, and we demonstrate that more general classes ofcorrelations, in the form of BEP relationships, exist between thekinetics and thermodynamics of metal diffusion in many heterosystems. We discuss how the slopes of these relationships arerelated to the degree of correspondence between the transitionstates and the stable initial states on various metals, and wefurther analyze how knowledge of substrate lattice constantsand the cohesive energies of the adsorbates and substrates canbe used to obtain a qualitative understanding of the presentedtrends. Finally, we briefly mention how these results could beextended to provide the necessary input for predicting thenature of growth modes associated with each system.45,46
II. THEORETICAL METHODSFirst-principles calculations have been carried out within theperiodic Density Functional Theory framework, as embodied inthe Vienna ab initio Simulation Package (VASP).47−50 ThePerdew−Wang 91 (PW91) gradient-corrected potential is usedto represent the electron exchange-correlation functional in thegeneralized gradient approximation (GGA).51,52 We choose theGGA approximation over the local density approximation(LDA) as it gives activation barriers that better matchexperimental results.53 For all the calculations, a kinetic-energycutoff of 400 eV is used for the wave functions to obtainconverged results. Brillouin zones are sampled using 14 × 14 ×14 Monkhorst−Pack k-point meshes for determining the bulklattice constants and 5 × 5 × 1 k-point meshes for the surfacecalculations.54 The optimization of the total energies isachieved via the conjugated-gradient (CG) algorithm55 withthe force criterion on each atom set for the convergence to bearound 0.02 eV/Å. Spin-polarized calculations are performedfor calculations involving Ni and are also employed to obtainthe energies of the isolated metal atoms in the gas phase. Thebulk lattice constants obtained with these sets of parameters are3.52, 3.64, 3.84, 3.88, 3.96, 3.99, 4.16, and 4.18 Å for Ni, Cu,Rh, Ir, Pd, Pt, Ag, and Au, respectively.For all the calculations, the (100) surfaces are constructed
using a super cell with a vacuum region of 15 Å between thesurfaces. For all the slabs considered, five layers of a 5 × 5surface unit cell with an adatom adsorbed on only one side ofthe slab are used to model the adsorbate−substrate systems.The structure (surface with adsorbed atom) is optimized usingthe conjugate-gradient method, and during the optimization wefixed the bottom layer of atoms (the results are unchanged if
the bottom two layers are held fixed). The (100) surfacecontains three adsorption sites: top, 4-fold, and bridge. To testthe energetically most stable adsorption sites for the adatoms,we calculated the adsorption energies at each site for selectedadsorbate−substrate pairs (Au, Ag, Pd, and Pt adatoms onCu(100) and Au and Pt adatoms on Ag(100), Au(100),Pd(100), Pt(100), and Ir(100)). Our results suggest that the 4-fold site (FF) is the energetically most stable adsorption site,while the bridge is the next most stable and the top site theleast stable, and we therefore focus on FF calculations in whatfollows.The activation barriers for the adatom diffusion via hopping
mechanism on (100) are calculated using either the dragmethod (1D scan of the potential energy surface) or thenudged elastic band method56 (see also discussion in the text).For the drag method, the potential energy surface is scannedwith about 20 intermediate points between the two FF sites toobtain the highest energy transition state configuration. TheNEB calculations are performed using five images, which isfound to be sufficient for such a symmetric diffusion pathway.
III. RESULTS AND DISCUSSIONSIII.1. Diffusion Barriers and Binding Energies. We begin
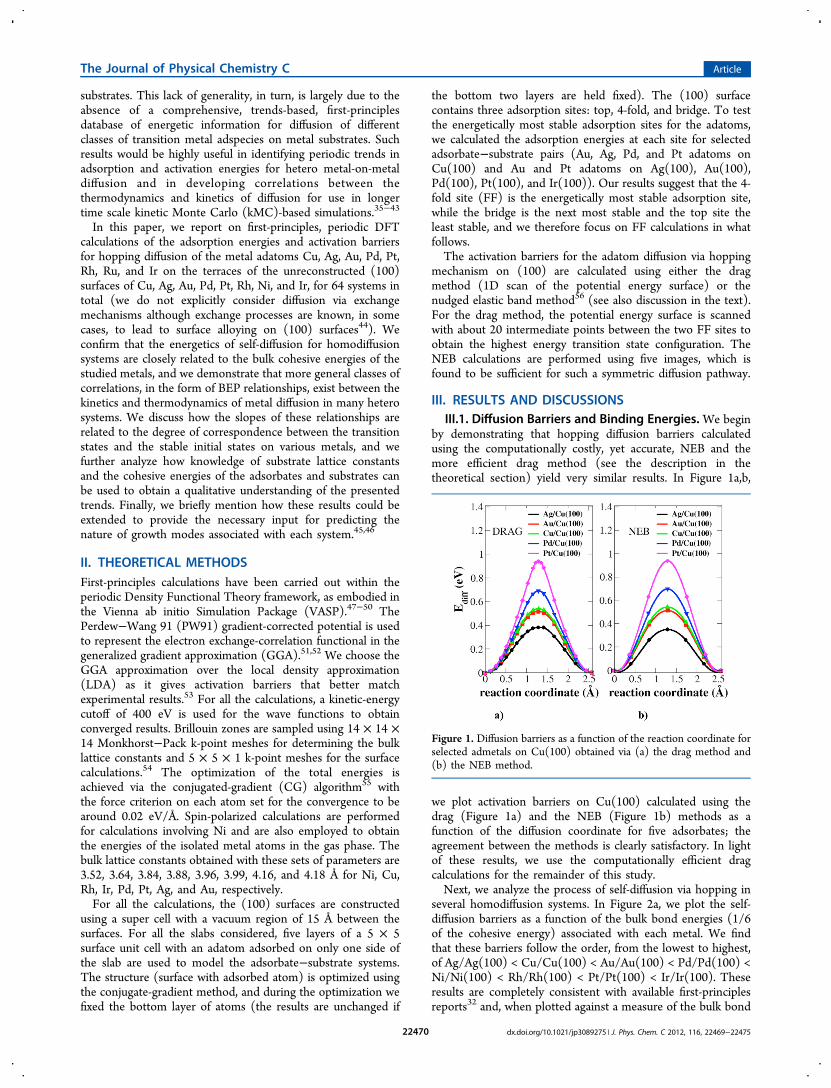
by demonstrating that hopping diffusion barriers calculatedusing the computationally costly, yet accurate, NEB and themore efficient drag method (see the description in thetheoretical section) yield very similar results. In Figure 1a,b,
we plot activation barriers on Cu(100) calculated using thedrag (Figure 1a) and the NEB (Figure 1b) methods as afunction of the diffusion coordinate for five adsorbates; theagreement between the methods is clearly satisfactory. In lightof these results, we use the computationally efficient dragcalculations for the remainder of this study.Next, we analyze the process of self-diffusion via hopping in
several homodiffusion systems. In Figure 2a, we plot the self-diffusion barriers as a function of the bulk bond energies (1/6of the cohesive energy) associated with each metal. We findthat these barriers follow the order, from the lowest to highest,of Ag/Ag(100) < Cu/Cu(100) < Au/Au(100) < Pd/Pd(100) <Ni/Ni(100) < Rh/Rh(100) < Pt/Pt(100) < Ir/Ir(100). Theseresults are completely consistent with available first-principlesreports32 and, when plotted against a measure of the bulk bond
Figure 1. Diffusion barriers as a function of the reaction coordinate forselected admetals on Cu(100) obtained via (a) the drag method and(b) the NEB method.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp3089275 | J. Phys. Chem. C 2012, 116, 22469−2247522470

energy obtained from experimental cohesive energies,57 clearlyconfirm that homodiffusion is related to the metals’ bulkcohesive energies, with the least cohesive metals having thelowest diffusion barriers. To further analyze the relationshipbetween the kinetics and thermodynamics of surface diffusionin these systems, we extend this analysis by plotting thediffusion barriers against one-sixth of the adsorption energies atthe FF (EFF
bind), and diffusional transition state, TS (ETSbind,
found at bridge positions), sites (the factor of 1/6, in thesecases, is for a consistent comparison with the cohesive energyanalysis). A linear relation is also observed, demonstrating thatsurface thermodynamic properties can also be used tounderstand trends in admetal diffusion barriers.To our knowledge, generalized analyses of surface diffusional
properties, discussed above for homodiffusion systems, havenot previously been investigated for a wide variety of heterosystems from first principles. The elucidation of such patterns,in turn, would provide useful atomistic insights into simulationsof dealloying, electrodeposition of heterometallic films, and thesurface stability of metallic alloys, among other processes.58−61
As a first step in the determination of these trends, we plot, inFigure 2b, the calculated diffusion barriers as a function of theadsorbate (admetal) identities in order of increasing cohesiveenergy of the metal adsorbate. While we discuss these trendsprimarily in terms of lattice constants and cohesive energies, wenote that analysis of other electronic and energetic propertiescould also provide insights into diffusional properties (seeSupporting Information for an example of an alternativedescription). For a given substrate element, the barriers exhibita generally monotonic increase with increasing cohesive energyof the admetal, with Ag adspecies generally having the lowest
diffusion barriers and Ir the highest. Although a broadrelationship between the barrier and the cohesive energy canbe seen when the adsorbate/substrate pairs are considered inaggregate, the clearest trends are observed for the diffusion on agiven substrate, when the geometric and electronic/energeticproperties of the substrate do not vary. For all of theadsorbates, the largest diffusion barriers are in general foundon Au(100) substrates, while the smallest barriers are onNi(100) substrates. Au has the largest lattice constant of all thesystems studied, and it is one of the least cohesive metals. Nihas the smallest lattice constant, and it is quite cohesive incomparison to Au. Although the trends are not completelyregular for metals between these extremes (see also discussionbelow), these observations suggest that, for heteroepitaxialsystems, the cohesive energy and lattice constants are usefulparameters for describing qualitative trends in diffusionalproperties.When considering the inverse set of trends to that described
above, corresponding to diffusion of a single admetal elementacross different substrates, trends in diffusional propertiesbecome less well-defined. For example, a plot of diffusionbarrier against the substrate element (plot not shown) for givenadmetals, with the substrate elements listed in order ofincreasing cohesive energy, does not show a clearly definedmonotonic trend; this result is due to the fact that, whenmoving from one substrate element to the next, there aresubstantial changes in both the substrate’s electronic andenergetic properties, as well as in the substrate lattice constant.Nevertheless, a limited number of trends governing thediffusion of given adsorbate species across multiple substratemetals can be deduced from the results in Figure 2b. Mostsignificantly, adsorbates with higher cohesive energies, such asPt, Rh, Ru, and Ir, show a broader range of diffusion barriersacross different substrates (the vertical spread in Figure 2bincreases accordinglynote that the point for Ir/Au(100) isomitted since a certain amount of surface alloying was observedin this case). This increased sensitivity of diffusion barrier to thesubstrate element is likely due to the fact that more cohesivemetals will naturally interact more strongly with thesurrounding metal atoms, leading to a greater sensitivity tothe properties of the substrate.In addition to kinetic quantities such as diffusion barriers,
trend-based analyses may also be performed for thermody-namic quantities such as admetal adsorption energies. In Figure2c, we plot the adsorption energies at the FF site as a functionof the elemental identities of the admetal adsorbates, in order ofincreasing cohesive energy of the adsorbate metals (a largeradsorption energy, in this figure, denotes a stronger admetal−substrate bond). As with the diffusion barriers, there is anapproximately monotonic increase in the strength of adsorptionas the cohesive energy of the adsorbate atoms increases whilekeeping the identity of the substrate unchanged; a smallnumber of admetals, howevermost notably Ptdeviate fromthis trend. Additionally, we note that there is a general trend forless cohesive substrates, such as Ag(100), to exhibit weakeradsorption of all adsorbates as compared to substrates withhigher cohesive energies, such as Ir(100). This trend is moreclearly consistent in the case of the FF adsorption energies thanin the case of the diffusion barriers, as will be further discussedbelow. Finally, as is the case for the diffusion barriers, thereseems to be a variable range of adsorption energies for a givenadmetal on different metal substrates, with the largest rangeexhibited by the most cohesive admetals.
Figure 2. (a) Self-diffusion barriers (homodiffusion systems) as afunction of one-sixth of the bulk cohesive energies and of the surfaceadsorption energies at the 4-fold (FF) and diffusional transition stateadsorption sites, (b) diffusion barriers for all of the admetal/substratepairs as a function of the adsorbate identities, listed in order ofincreasing adsorbate cohesive energy, and (c) adsorption energies atthe FF site for all the pairs as a function of adsorbate identities. A morepositive adsorption energy indicates a more strongly bound adatom.(b) and (c) exclude the value for Ir/Au(100), as Ir penetrates into thesubstrate and hence cannot be considered to have the same diffusionmechanism. The cohesive energies of the metals in (a), (b), and (c)are 2.94, 3.48, 3.84, 3.90, 4.44, 5.76, 5.82, 6.74, and 6.96 eV for Ag, Cu,Au, Pd, Ni, Rh, Pt, Ru, and Ir, respectively (from reference 57).
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp3089275 | J. Phys. Chem. C 2012, 116, 22469−2247522471

Figure 3 shows an alternative presentation of the kinetic andthermodynamic diffusion data. In Figure 3a, the hoppingdiffusion barriers are summarized in a periodic grid, with theelements listed in order of increasing cohesive energy from leftto right and from top to bottom. The shading of each boxreflects the magnitude of the diffusion barrier of eachadsorbate−substrate pair, with the darkest-colored boxescorresponding to the highest diffusion barriers. Figure 3bshows the FF adsorption energy data in a similar periodicarrangement. Consistent with our discussion of Figures 2b and2c above, a general increase in diffusion barriers and adsorptionstrength is clearly seen when moving across the rows,corresponding to changing the admetal with a given substrate.For the diffusion barriers, moving down the columns,corresponding to changing the substrate element for a fixedadmetal, does not yield such well-defined trends; as discussedabove, this effect is likely due to the simultaneous changing ofelectronic (cohesive energy) and geometrical (lattice constant)effects. The fact that the diffusion barriers do not show a cleartrend when changing the substrate, while the qualitative trend ismore clearly established in the case of the FF site adsorptionenergies, is likely due to the fact that the scale of the diffusionbarriers, which are effectively differences between the energiesof the diffusional transition states and the FF adsorptionenergies, is much smaller than the corresponding scale of theadsorption energies (see also discussion below).III.2. Correlation between Diffusion Barriers and
Binding Energies. The similarities between the trends,described above, for heteroatom diffusion barriers andadsorption energies suggest that monotonic relationships mayexist between these two quantities across different admetal/substrate pairs, especially for diffusion of atoms across a givensubstrate metal. Demonstrating the existence of such BEPrelationships, in turn, would not only extend a valuablefundamental concept from catalytic surface science to surface
diffusive processes but also provide an important practical toolfor rapidly and accurately estimating diffusion barriers fromthermodynamic data alone. In Figure 4a, we plot the diffusion
barriers as a function of the adsorption energies at the FF sitesfor each considered adsorbate on each metal substrate (total of64 systems). The overall correlation, considering all 64admetal/substrate pairs together, is only qualitative in nature;while a general trend is apparent, the scatter is considerable.However, when admetal diffusion and adsorption are
Figure 3. Periodic plots constructed for the (a) diffusion barriers and (b) admetal adsorption energies at FF sites. Corresponding numerical values ofthe diffusion and adsorption energies are given in (c) and (d). The figures exclude the value for Ir/Au(100), as Ir significantly perturbs the structureof the Au(100) surface.
Figure 4. (a) Linear correlations obtained for the admetal diffusionbarriers as a function of the adsorbate adsorption energies at 4-foldsites, (b) correlations between the adsorption energies at the transitionstates (ETSbind) and those at the most stable adsorption sites (EFFbind),and (c) linear fits for the BEP-type relationships for admetal diffusionon each metal substrate.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp3089275 | J. Phys. Chem. C 2012, 116, 22469−2247522472

considered only on a given substrate, the correlations becomemuch more quantitative, with standard errors of 0.04, 0.05,0.03, 0.02, 0.01, 0.02, 0.02, and 0.02 eV for diffusion on Ag, Au,Cu, Ir, Ni, Pd, Pt, and Rh(100), respectively; these errors arewell within the standard uncertainties of DFT calculations. Thefact that better correlations exist for given metal substrates isnot surprising in light of the discussions above; for givensubstrates, both the geometrical and the energetic/electronicproperties of the substrate are fixed, and all remaining variationsare due only to changes in the nature of the admetal. Theexistence of these correlations, in turn, provides a powerful toolfor combining the accuracy of DFT calculations withaccelerated strategies for simulating metal-on-metal growth ordealloying processes; with only the computationally inexpensivedeterminations of adsorption energies, diffusional propertiescan be determined with great efficiency.The slopes of the correlations on Ag(100) and Au(100) are
the largest (dimensionless values of 0.33 and 0.36, respectively),followed by Pt(100) and Pd(100), with slopes of 0.23 and 0.26,by Cu(100) and Ni(100), with values of 0.24 and 0.20, andfinally by Ir(100) and Rh(100), with slopes of 0.15 and 0.16.These slopes, in turn, provide insights into the degree ofcorrespondence between the transition and initial states ofdiffusion on different substrate metals. Since the hoppingdiffusion barrier is the difference in adsorption energiesbetween the diffusing adatom at the transition state and themost stable FF adsorption sites (Figure 1), a smaller slope inthese relationships implies a closer relationship between theenergy at the transition state and that at the initial (FF) state. Aslope of zero would suggest that there is a constant diffusionbarrier, which in turn indicates that there is merely a constantenergy offset between the FF and transition states for alladatoms on a given metal substrate; such a constant, or near-constant, offset has generally been interpreted, in the BEPliterature, as evidence that the transition states resemble, ineither a geometric or an energetic sense, the correspondingthermodynamically stable states.62,63
As with our analysis in Figures 2 and 3 above, we discuss thelinear correlations in Figure 4a in terms of two properties, thesubstrate lattice constant (a geometric property) and thesubstrate cohesive energy (an energetic/electronic property).These properties provide an important qualitative under-standing of the BEP behavior, which is in line with otherlargely qualitative interpretations that have been given for theexistence of BEP relationships in the heterogeneous catalysisliterature62,63 (we again note, however, that related properties,such as the metal substrate electronic d-band centers anddensities of states, could have similar explanatory power). Thecorrelations with the largest slopes are generally associated withthe metal substrates that have the largest lattice constants andsmaller cohesive energies (Au(100) and Ag(100), Table 1). Alarger slope also implies, on average, that there is a weakermagnitude of binding of the admetal species at FF sites on thesubstrates. In contrast, smaller slopes are often associated withthose substrate metals with smaller lattice constants (Ir and Rhhave the third smallest lattice constants among all the systems)
and higher cohesive energies; these smaller slopes also indicatea higher average strength of binding of the admetals to thesubstrates. Pt and Pd have both correlation slopes and latticeconstants/cohesive energies in between these extremes. Theseassociations suggest that substrates with smaller latticeconstants or larger cohesive energies exhibit more energeticsimilarities between the diffusional transition states and thecorresponding FF adsorption sites. Both factors could beintuitively thought to promote closer interactions with thesubstrates, in a geometric and an energetic sense, respectively,thus leading to the closer correspondence.While consideration of the substrate lattice constants and
cohesive energies provides useful understandings of numerouscharacteristics of the diffusion/adsorption correlations on anumber of transition metal substrates, neither of theseparameters provides a universal description. Diffusion on theCu(100) substrate illustrates this point. If we consider only theCu lattice constant, we would expect the slope of thecorrelation to be lower than the slopes of the correspondingcorrelations for Ir and Rh substrates, according to thearguments presented above. An analysis based solely oncohesive energies, on the other hand, would suggest a slopeclose to that of the Au or Ag substrates. Figure 4a shows,however, that neither prediction corresponds perfectly to thecalculated results; indeed, the slope is somewhat higher thanwhat we would expect based on lattice constant trends andsomewhat lower than the expectation based on cohesiveenergies. This different behavior likely results from a couplingof the lattice constant and cohesive effects; the result may, inturn, be related to the unusual combination of Cu’s small latticeconstant and its low cohesive energy (Table 1). Ni is anotherinteresting case, with the smallest lattice constant of any of thesubstrates but an intermediate value of the cohesive energy;again, the calculated slope of the BEP relationship does notcorrespond perfectly to the qualitative arguments made basedon either lattice constant or cohesive energy. We note that thisresult is not surprising, given that a full explanation of linearityin BEP relationships remains elusive even for systems that havebeen extensively studied, such as the reactions of organicspecies on metal surfaces.16,22,23,34
An alternative formulation of BEP relationships involves anexplicit correlation of adsorption energies of transition stateswith adsorption energies of the stable initial states; thisformulation more directly illuminates the fundamental linksbetween transition state and initial state characteristics. InFigure 4b, such a relationship is plotted for the hoppingdiffusion calculations. The results confirm that a linearrelationship between the adsorption energies at the FF andthe TS sites exists, even considering the broad range ofadsorption energies (from 2.0 to 7 eV) corresponding to the 64systems in this analysis. The reason that a reasonable linearrelationship can be obtained in this case for all heterodiffusionalsystems, whereas a similar relationship does not exist whenplotting diffusion barriers against adsorption energies (Figure4a), is simply related to the different choice of dependentvariable in Figures 4a and 4b. The larger range of the transition
Table 1. Calculated Lattice Constants and Experimental Cohesive Energies (from reference 57) for Main Group TransitionMetals
property Ni Cu Rh Ir Pd Pt Ag Au
lattice constant (Å) 3.52 3.64 3.84 3.88 3.96 3.99 4.16 4.18cohesive energy (eV) 4.44 3.48 5.76 6.96 3.90 5.82 2.94 3.84
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp3089275 | J. Phys. Chem. C 2012, 116, 22469−2247522473

state adsorption energies, compared to the corresponding rangeof diffusion barriers, enhances the linearity of the relationships.We note that this enhancement is also likely related to the moreobvious relationship between adsorption energy and cohesiveenergy, described in Figure 2c, compared to the correspondingrelationship for diffusion barriers and cohesive energy (Figure2b). In spite of the overall linearity of the generalized BEPrelationship in Figure 4b, it is possible to obtain even betterrelationships by generating separate correlations for each metalsubstrate (Figure 4c). The ordering of the slopes of thesecorrelations is reversed compared to the ordering in Figure 4b,but the interpretation of the results in terms of thecorrespondence between the transition states and the initialstates, described earlier, remains unchanged.
IV. CONCLUSIONSWe have used periodic density functional theory calculations toscreen the diffusion and adsorption energetics for hetero metal-on-metal diffusion via a hopping mechanism on the (100)surfaces of 64 admetal/metal substrate systems. The resultsdemonstrate that relationships between hopping diffusionbarriers and metal cohesive energies, previously establishedfor homodiffusion systems, can be generalized to heterometallicsystems, and the resulting relationships can be described byconsidering the cohesive energies of the admetals andsubstrates, as well as the lattice constants of the substrates. Inaddition, there is a linear relationship between the adsorptionenergies and the diffusion barriers when the adsorption isconsidered on a given substrate. The slopes of these linearrelationships indicate the degree of similarity between thediffusional transition states and initial states, and the slopes areat least partly determined by the cohesive energies and latticeconstants of the metal substrates. The relationships providefundamental insights into the links between the diffusionaltransition state and the corresponding initial state properties,and they yield a powerful and convenient means of predictingdiffusional kinetics from purely thermodynamic quantities.Similar studies could be carried out to describe hopping andexchange diffusional processes on other surface geometries, andtaken together, these results may ultimately provide a veryuseful input to KMC-type simulations, enabling studies ondynamics and time evolution of the growth processes for theseadsorbate−substrate heterometallic pairs.
■ ASSOCIATED CONTENT*S Supporting InformationA simple computational decomposition procedure thatseparates the contribution of lattice constant changes fromchanges in the electronic/alloying properties of the systems isshown. This material is available free of charge via the Internetat http://pubs.acs.org.
■ AUTHOR INFORMATIONCorresponding Author*E-mail: [email protected]; [email protected] authors declare no competing financial interest.
■ ACKNOWLEDGMENTSA DOE Early Career Award for J.G., together with use of theCenter for Nanoscale Materials, was supported by the U.S.Department of Energy, Office of Science, and Office of Basic
Energy Sciences, under Contract No. DE-AC02-06CH11357.The authors also acknowledge the use of the computationalfacilities provided by CNM-ANL (Carbon Cluster) and FusionClusters.
■ REFERENCES(1) Brune, H. Microscopic View of Epitaxial Metal Growth:Nucleation and Aggregation. Surf. Sci. Rep. 1998, 31, 121−229.(2) Antczak, G.; Ehrlich, G. Jump Processes in Surface Diffusion.Surf. Sci. Rep. 2007, 62, 39−61.(3) Antczak, G.; Ehrlich, G. Long Jumps in Diffusion of Iridium onW(110). Phys. Rev. B 2005, 71, 115422.(4) Ratsch, C.; Ruggerone, P.; Scheffler, M. Density-functional Theoryof Surface Diffusion and Epitaxial Growth of Metals; Plenum Press: NewYork, 1997.(5) Ruggerone, P.; Kley, A.; Scheffler, M. Microscopic Aspects ofHomoepitaxial Growth. Prog. Surf. Sci. 1997, 54, 331−340.(6) Kurpick, U.; Kara, A.; Rahman, T. S. Role of Lattice Vibrations inAdatom Diffusion. Phys. Rev. Lett. 1997, 78, 1086−1089.(7) Kellog, G. L. Field Ion Microscope Studies of Single-atom SurfaceDiffusion and Cluster Nucleation on Metal Surfaces. Surf. Sci. Rep.1994, 21, 1−88.(8) Antczak, G.; Ehrlich, G. Surface Diffusion: Metals, Metal Atoms,and Clusters; Cambridge University Press: New York, 2010.(9) Nørskov, J. K.; Abild-Pedersen, F. Catalysis: Bond Control inSurface Reactions. Nature 2009, 461, 1223−1225.(10) Greeley, J.; Stephens, I. E. L.; Bondarenko, A. S.; Johansson, T.P.; Hansen, H. A.; Jaramillo, T. F.; Rossmeisl, J.; Chorkendorff, I.;Nørskov, J. K. Alloys of Platinum and Early Transition Metals asOxygen Reduction Electrocatalysts. Nat. Chem. 2009, 1, 552−556.(11) Jiang, T.; Mowbray, D. J.; Dobrin, S.; Falsig, H.; Hvolbæk, B.;Bligaard, T.; Nørskov, J. K. Trends in CO Oxidation Rates for MetalNanoparticles and Close-Packed, Stepped, and Kinked Surfaces. J.Phys. Chem. C 2009, 113, 10548−10553.(12) Freund, H. J.; Meijer, G.; Scheffler, M.; Schlogl, R.; Wolf, M.CO Oxidation as a Prototypical Reaction for HeterogeneousProcesses. Angew. Chem., Int. Ed. 2011, 50, 10064−10094.(13) Reuter, K.; Frenkel, D.; Scheffler, M. The Steady State ofHeterogeneous Catalysis, Studied by First-Principles StatisticalMechanics. Phys. Rev. Lett. 2004, 93, 116105.(14) Reuter, K.; Scheffler, M. First-principles Kinetic Monte CarloSimulations for Heterogeneous Catalysis: Application to the COOxidation at RuO2(110). Phys. Rev. B 2006, 73, 045433.(15) Rogal, J.; Reuter, K.; Scheffler, M. First-Principles StatisticalMechanics Study of the Stability of a Sub-Nanometer Thin SurfaceOxide in Reactive Environments: CO Oxidation at Pd(100). Phys. Rev.Lett. 2007, 98, 046101.(16) Nørskov, J. K.; Bligaard, T.; Rossmeisl, J.; Christensen, C. H.Towards the Computational Design of Solid Catalysts. Nat. Chem.2009, 1, 37−46.(17) Wang, S. G.; Temel, B.; Shen, J. A.; Jones, G.; Grabow, L. C.;Studt, F.; Bligaard, T.; Abild-Pedersen, F.; Christensen, C. H.;Nørskov, J. K. Universal Bronsted-Evans-Polanyi Relations for C-C,C-O, C-N, N-O, N-N, and O-O Dissociation Reactions. Catal. Lett.2011, 141, 370−373.(18) Michaelides, A.; Liu, Z. -P.; Zhang, C. J.; Alavi, A.; King, D. A.;Hu, P. Identification of General Linear Relationships betweenActivation Energies and Enthalpy Changes for Dissociation Reactionsat Surfaces. J. Am. Chem. Soc. 2003, 125, 3704−3705.(19) Logadottir, A.; Rod, T. H.; Nørskov, J. K.; Hammer, B.;Jacobsen, C. J. H. The Brønsted−Evans−Polanyi Relation and theVolcano Plot for Ammonia Synthesis over Transition Metal Catalysts.J. Catal. 2001, 197, 229−231.(20) Greeley, J.; Mavrikakis, M. Alloy Catalysts Designed from FirstPrinciples. Nat. Mater. 2004, 3, 810−815.(21) Xu, Y.; Ruban, A. V.; Mavrikakis, M. Adsorption andDissociation of O2 on Pt−Co and Pt−Fe Alloys. J. Am. Chem. Soc.2004, 126, 4717−4725.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp3089275 | J. Phys. Chem. C 2012, 116, 22469−2247522474

(22) Bligaard, T.; Nørskov, J. K.; Dahl, S.; Matthiesen, J.;Christensen, C. H.; Sehested, J. The Brønsted-Evans-Polanyi Relationand the Volcano Curve in Heterogeneous Catalysis. J. Catal. 2004,224, 206−217.(23) Liu, B; Greeley, J. Decomposition Pathways of Glycerol via C−H, O−H, and C−C Bond Scission on Pt(111): A Density FunctionalTheory Study. J. Phys. Chem. C 2011, 115, 19702−19709.(24) Studt, F.; Abild-Pedersen, F.; Bligaard, T.; Sorensen, R. Z.;Christensen, C. H.; Nørskov, J. K. Identification of Non-PreciousMetal Alloy Catalysts for Selective Hydrogenation of Acetylene. Science2008, 320, 1320−1322.(25) Andersson, M. P.; Bligaard, T.; Kustov, A.; Larsen, K. E.;Greeley, J.; Johannessen, T.; Christensen, C. H.; Nørskov, J. K.Toward Computational Screening in Heterogeneous Catalysis: Pareto-optimal Methanation Catalysts. J. Catal. 2006, 239, 501−506.(26) Ferrin, P.; Simonetti, D.; Kandoi, S.; Kunkes, E.; Dumesic, J. A.;Nørskov, J. K.; Mavrikakis, M. Modeling Ethanol Decomposition onTransition Metals: A Combined Application of Scaling and Brønsted−Evans−Polanyi Relations. J. Am. Chem. Soc. 2009, 131, 5809−5815.(27) Liu, B.; Greeley, J. Density Functional Theory Study ofSelectivity Considerations for C−C Versus C−O Bond Scission inGlycerol Decomposition on Pt(111). Top. Catal. 2012, 55, 280−289.(28) Yu, B. D.; Scheffler, M. Anisotropy of Growth of the Close-Packed Surfaces of Silver. Phys. Rev. Lett. 1996, 77, 1095−1098.(29) Feibelman, P. J. Rebonding Effects in Separation and Surface-diffusion Barrier Energies of an Adatom Pair. Phys. Rev. Lett. 1987, 58,2766−2769.(30) Kellogg, G. L.; Feibelman, P. J. Surface Self-diffusion on Pt(001)by an Atomic Exchange Mechanism. Phys. Rev. Lett. 1990, 64, 3143−3146.(31) Michaelides, A.; Scheffler, M. An Introduction to the Theory ofMetal Surfaces; Wiley-VCH: New York, 2010.(32) Feibelman, P. J. Scaling of Hopping Self-diffusion Barriers onfcc(100) Surfaces with Bulk Bond Energies. Surf. Sci. 1999, 423, 169−174.(33) Mo, Y.; Zhu, W.; Kaxiras, E.; Zhang, Z. Electronic Nature ofStep-Edge Barriers against Adatom Descent on Transition-MetalSurfaces. Phys. Rev. Lett. 2008, 101, 216101.(34) Udaykumar, N. A.; Greeley, J.; Mavrikakis, M. A Simple Rule ofThumb for Diffusion on Transition-Metal Surfaces. Angew. Chem., Int.Ed. 2006, 45, 7046−7049.(35) Voter, A. F. Classically Exact Overlayer Dynamics: Diffusion ofRhodium Clusters on Rh(100). Phys. Rev. B 1986, 34, 6819−6829.(36) Fichthorn, K. A.; Weinberg, W. H. Theoretical Foundations ofDynamical Monte Carlo Simulations. J. Chem. Phys. 1991, 95, 1090−1096.(37) Blue, J. L.; Beichl, I.; Sullivan, F. Faster Monte CarloSimulations. Phys. Rev. E 1995, 51, R867−R868.(38) Henkelman, G.; Jonsson, H. Long Time Scale Kinetic MonteCarlo Simulations without Lattice Approximation and PredefinedEvent Table. J. Chem. Phys. 2001, 115, 9657−9666.(39) Kara, A.; Trushin, O.; Yildirim, H.; Rahman, T. S. Off-latticeSelf-learning Kinetic Monte Carlo: Application to 2D ClusterDiffusion on the fcc(111) surface. J. Phys.: Condens. Matter 2009, 21,084213.(40) Beland, L. K.; Brommer, P.; El-Mellouhi, F.; Joly, J-F; Mousseau,N. Kinetic Activation-Relaxation Technique. Phys. Rev. E 2011, 84,046704.(41) Kleiner, K.; Comas-Vives, A.; Naderian, M.; Mueller, J. E.;Fantauzzi, D.; Mesgar, M.; Keith, J. A.; Anton, J.; Jacob, T. MultiscaleModeling of Au-Island Ripening on Au(100). Adv. Phys. Chem. 2011,2011, 252591.(42) Soisson, F.; Becquart, C. S.; Castin, N.; Domain, C.; Malerba, L.;Vincent, E. Atomistic Kinetic Monte Carlo Studies of MicrochemicalEvolutions Driven by Diffusion Processes under Irradiation. J. Nucl.Mater. 2010, 406, 55−67.(43) Ratsch, C.; Zangwill, A.; Smilauer, P.; Vvedensky, D. D.Saturation and Scaling of Epitaxial Island Densities. Phys. Rev. Lett.1994, 72, 3194−3197.
(44) Zaum, C.; Rieger, M.; Reuter, K.; Morgenstern, K. AnomalousScaling in Heteroepitaxial Island Dynamics on Ag(100). Phys. Rev. Lett.2011, 107, 046101.(45) Ehrlich, G.; Hudda, F. G. Atomic View of Surface Self-Diffusion:Tungsten on Tungsten. J. Chem. Phys. 1966, 44, 1039−1049.(46) Schwoebel, R. L.; Shipsey, E. J. Step Motion on Crystal Surfaces.J. Appl. Phys. 1966, 37, 3682−3686.(47) Kresse, G.; Hafner, J. Ab initio Molecular Dynamics for LiquidMetals. Phys. Rev. B 1993, 47, 558−561.(48) Kresse, G.; Hafner, J. Ab Initio Molecular Dynamics Simulationof the Liquid-Metal−Amorphous-Semiconductor Transition inGermanium. Phys. Rev. B 1994, 49, 14251−14269.(49) Kresse, G.; Furthmuller, J. Efficiency of Ab-initio Total EnergyCalculations for Metals and Semiconductors Using a Plane-wave BasisSet. Comput. Mater. Sci. 1996, 6, 15−50.(50) Kresse, G.; Furthmuller, J. Efficient Iterative Schemes for Abinitio Total-energy Calculations Using a Plane-wave Basis Set. Phys.Rev. B 1996, 54, 11169−11186.(51) Perdew, J. P.; Wang, Y. Accurate and Simple AnalyticRepresentation of the Electron-gas Correlation Energy. Phys. Rev. B1992, 45, 13244−13249.(52) Perdew, J. P.; Chevary, J. A.; Vosko, S. H.; Jackson, K. A.;Pederson, M. R.; Singh, D. J.; Fiolhais, C. Atoms, Molecules, olids, andSurfaces: Applications of the Generalized Gradient Approximation forExchange and Correlation. Phys. Rev. B 1992, 46, 6671−6687.(53) Perdew, J. P.; Zunger, A. Self-interaction Correction to Density-functional Approximations for Many-electron Systems. Phys. Rev. B1981, 23, 5048−5079.(54) Monkhorst, H. J.; Pack, J. P. Special Points for Brillouin-zoneIntegrations. Phys. Rev. B 1976, 13, 5188−5192.(55) Press, W. H.; Teukolsky, S. A.; Vetterling, W. T.; Flannery, B. P.Numerical Recipes in Fotran; Cambridge University Press: Cambridge,1992.(56) Jonsson, H.; Mills, G.; Jacobsen, K. W. Nudged Elastic BandMethod for Finding Minimum Energy Paths of Transitions, in Classicaland Quantum Dynamics in Condensed Phase Simulations; WorldScientific: River Edge, NJ, 1998.(57) Kittel, C. Introduction to Solid State Physics; Wiley: New York,1997.(58) Stamenkovic, V. R.; Mun, B. S.; Mayrhofer, K. J. J.; Ross, P. N.;Markovic, N. M. Effect of Surface Composition on ElectronicStructure, Stability, and Electrocatalytic Properties of Pt-TransitionMetal Alloys: Pt-Skin versus Pt-Skeleton Surfaces. J. Am. Chem. Soc.2006, 128, 8813−8819.(59) Vitos, L.; Ropo, M.; Kokko, K.; Punkkinen, M. P. J.; Kollar, J.;Johansson, B. Exceptional Surface Stability in Late Transition MetalAlloys Driven by Lattice Strain. Phys. Rev. B 2008, 77, 121401.(60) Strasser, P.; Koh, S.; Greeley, J. Voltammetric SurfaceDealloying of Pt Bimetallic Nanoparticles: and Experimental andDFT Computational Analysis. Phys. Chem. Chem. Phys. 2008, 10,3670−3683.(61) Bicelli, L. P.; Bozzini, B.; Mele, B.; D’Urzo, L. A Review ofNanostructural Aspects of Metal Electrodeposition. Int. J. Electrochem.Sci. 2008, 3, 356−408.(62) Alcala, R.; Dumesic, J. A.; Mavrikakis, M. DFT Studies forCleavage of C-C and C-O Bonds in Surface Species Derived fromEthanol on Pt(111). J. Catal. 2003, 218, 178−190.(63) Nørskov, J. K.; Bligaard, T.; Logadottir, A.; Bahn, S.; Hansen, L.B.; Bollinger, M.; Bengaard, H.; Hammer, B.; Sljivancanin, Z.;Mavrikakis, M.; et al. Universality in Heterogeneous Catalysis. J.Catal. - Priority Commun. 2002, 209, 275−278.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp3089275 | J. Phys. Chem. C 2012, 116, 22469−2247522475




















