Signs of cardiac autonomic imbalance and proarrhythmic remodeling in FTO deficient mice
Upload
independentCategory
view
5download
0
OSTEOCLASTOGENESIS IN CHILDREN WITH 21-HYDROXYLASE DEFICIENCY ON LONG
TERM GLUCOCORTICOID THERAPY: THE ROLE OF RANKL/OPG IMBALANCE
Maria Felicia Faienza1, Giacomina Brunetti2, Silvia Colucci2, Laura Piacente1, Maria Ciccarelli1, Lucia
Giordani1, Giovanni Carlo Del Vecchio1, Massimo D’Amore3, Livia Albanese3, Luciano Cavallo1, Maria
Grano2
1 Department of Biomedicine of Developmental Age, University of Bari, Bari, Italy; 2 Department of Human Anatomy and Histology, University of Bari, Bari, Italy; 3 Department of Internal Medicine and Public Medicine, Section of Rheumatology, University of Bari, Bari,
Italy.
Abbreviated title: OSTEOCLASTOGENESIS IN 21-OHD CHILDREN
Key words: osteoclastogenesis, 21-hydroxylase deficiency, RANKL, OPG, glucocorticoids
Corresponding author
Maria Felicia Faienza, MD, PhD
Department of Biomedicine of Developmental Age, University of Bari, Bari, Italy
Piazza G. Cesare, 11
70100 Bari, Italy
Phone +39805593075
Fax +39805592287
e-mail: [email protected]
For reprint requests:
Maria Felicia Faienza
Department of Biomedicine of Developmental Age, University of Bari, Bari, Italy
Piazza G. Cesare, 11
70100 Bari, Italy
The present study highlights, for the first time, that spontaneous osteoclastogenesis occurs in patients
affected by 21-OHD on long term GC treatment
J Clin Endocrin Metab. First published ahead of print April 28, 2009 as doi:10.1210/jc.2008-2446
Copyright (C) 2009 by The Endocrine Society

DISCLOSURE STATEMENT: The authors have nothing to disclosure
Word count: 3600
Number of figures: 5
Number of tables: 1

ABSTRACT
Context. Children with 21-hydroxylase deficiency (21-OHD) need chronic glucocorticoid (cGC) therapy to
replace congenital deficit of cortisol synthesis. cGC therapy is the most frequent and severe form of drug-
induced osteoporosis and different mechanisms have been proposed to explain its pathogenesis.
Objective. We investigated the osteoclastogenic potential of peripheral blood mononuclear cells (PBMCs)
from 18 children with 21-OHD on cGC therapy and 25 controls who never received GCs. We also evaluated
the presence of circulating osteoclast precursors (OCPs) and the role of T cells in osteoclast (OC) formation.
Results. Spontaneous osteoclastogenesis, without adding macrophage-colony stimulating factor (M-CSF)
and receptor activator of NF-kB ligand (RANKL), and significantly higher OCs resorption activity occurred
in 21-OHD patients. Conversely, MCSF and RANKL were essential to trigger and sustain osteoclastogenesis
in controls. Futhermore, in 21-OHD patients we identified a significant percentage of CD11b-CD51/CD61
and CD51/61-RANK positive cells, which are OCPs strongly committed. Additionally, we demonstrated a
T-cell-dependent osteoclastogenesis from 21-OHD patients’ PBMCs. T-cells from patients expressed high
levels of RANKL and low levels of osteoprotegerin (OPG) respect to controls. Moreover, 21-OHD patients
had higher sRANKL and lower OPG serum levels compared with controls, thus sRANKL/OPG ratio was
significantly higher in patients than in controls.
Conclusions. The present study showed for the first time a high osteoclastogenic potential of PBMCs from
21-OHD patients on cGC therapy. This spontaneous osteoclastogenesis seems to be supported both by the
presence of circulating OCPs and by factors released by T cells.

INTRODUCTION
21-hydroxylase deficiency (21-OHD) is the most common cause of congenital adrenal hyperplasia (CAH),
resulting from deletions or mutations of the P450 21-hydroxylase gene (CYP21) (1). This disorder is
characterized by accumulation of the precursors immediately proximal to the 21-hydroxylation step along the
pathway of cortisol synthesis, that are shunted into the androgen pathway. Three forms of 21-OHD can be
distinguished by means of clinical, hormonal, and molecular genetic criteria: the classical salt-wasting (SW),
classical simple-virilizing (SV) and nonclassical forms (NC). Children with 21-OHD need chronic
glucocorticoid (GC) therapy as soon as they are diagnosed with the disease, both to replace congenital deficit
in cortisol synthesis and to reduce androgen secretion by adrenal cortex (2). GC-induced osteoporosis (GIO)
represents the most common cause of drug-induced osteoporosis and different mechanisms have been
proposed to explain its pathogenesis (3,4). Previous reports on 21-OHD patients showed increased (5),
decreased (6-13) or normal bone mineral density (BMD) (14-18).
Bone remodelling is an ever-occurring event characterized by sequential tethering of the activities of
osteoclasts (OCs), the bone resorbing cells, and osteoblasts (OBs), the bone forming cells. In fact, the
majority of acquired, systemic diseases of the skeleton reflect imbalance in OC and OB activity in the
remodelling process.
OCs are members of the monocyte-macrophage family, derived from the fusion of marrow-derived
mononuclear phagocyte, the OCs precursors (OCPs), which circulate in peripheral blood (PB) (19). These
cells differentiate under the influence of two cytokines, namely macrophage-colony stimulating factor (M-
CSF) and receptor activator of NF-kB ligand (RANK-L) (20). RANKL expressed on OBs and stromal cells
as a membrane-bound protein and cleaved into a soluble molecule (sRANKL) by metalloproteinase (20)
promotes differentiation and fusion of OCPs and activates mature OCs to reabsorb bone by binding to its
specific receptor RANK (20). Osteoprotegerin (OPG), a soluble decoy receptor secreted by OBs and bone
marrow stromal cells, competes with RANK in binding to RANKL, preventing its osteoclastogenic effect
(20). RANKL and OPG are also secreted by activated T cells, which are a crucial paracrine link between
bone metabolism and the immune system (21). In fact, the critical role of T cells as regulators of bone
turnover is emerging in different pathological conditions (22, 23). However, data regarding the 21-OHD

patients systemic circulating cell populations (such as T lymphocytes and/or cells of the monocytes-
macrophage lineage) are at present not available.
Thus, since OCs can be generated from peripheral blood mononuclear cells (PBMCs) in the presence of M-
CSF and RANKL, the aim of this paper is to highlight the osteoclastogenic potential of unstimulated and
unfractionated PBMCs from 21-OHD children on long-term GC therapy, and to evaluate the presence of
circulating OCPs and the role of T cell in OC formation.
MATHERIALS AND METHODS
Subjects
The samples included PB from 18 Caucasian patients (9 females) with 21-OHD, aged 3.0 to 16.0 yr.
Diagnosis was made on the basis of clinical evidence, basal serum concentrations and peaks of 17α-hydroxy
progesterone (17α-OHP) after Adrenocorticotrophin (ACTH) test. Molecular analysis of the CYP21 gene
was performed in all patients and in their parents. Subjects with risk factors for reduced bone mass were
excluded from the study. The main characteristics of the patients are summarized in Table 1. Eight patients
were diagnosed at birth because of severe renal salt loss or virilization of external genitalia. The patients with
the NC form had been diagnosed within the first sixteen years of life because of precocious pubarche,
advanced bone age or hirsutism. All patients had been treated from the time of diagnosis (7.6 ± 4.3 yr).
Patients with the SW form received GCs and mineralcorticoid (9-alpha-fludrocortisone: 0.1-0.2 mg/day)
therapy since early infancy, while the patients with the SV form were treated with GCs alone. Treatment at
the time of the study consisted of hydrocortisone, expressed as dose per body surface per day (mg/m2/d),
given twice or three times daily. The four patients with SW form received a total hydrocortisone dose of 20-
25 mg/m2/d. The patients with SV and NC form were treated with 10-15 mg/m2/d of hydrocortisone. The
mean dose of GCs calculated over the 5 years preceding the investigation was 17.53 ± 4.49 mg/m2/d.
We studied 15 controls (patients’ siblings) aged 3.0 to 16.0 yr (12 ± 4.7 years) who resulted heterozygous for
CYP21 gene mutations and never received steroid therapy (subgroup A), and 10 healthy volunteers aged
18.0-30 yr, recruited from the same geographic area (subgroup B). Informed consent was obtained from all
subjects or from their parents.

Hormonal control Serum concentrations of 17α−OHP, Δ4-Androstenedione (Δ4-A) and testosterone (T)
from the patients’ records in the preceding 5 yr were measured by RIA (Sorin Diagnostic, Saluggia, Italy).
All parameters were also evaluated in the control group.
Bone mineral measurements BMD of 21-OHD patients and 10 volunteers was measured at the proximal
femur and lumbar spine (L2-L4), using Dual energy X-Ray absorptiometry (ACN Unigamma X- Ray Plus),
and converted to standard deviation scores (Z-scores) in relation to age and sex-matched normal population.
Biochemical measurements Serum levels of PTH, bone-specific alkaline phosphatase (BALP), calcium and
phosphate were measured in the patients and controls. Intact PTH was measured by RIA (Nichols Institute
Diagnostics, San Juan Capistrano, CA). BALP was measured in serum using a commercial immunoassay
(Dade Behring Inc, Newark, DE, USA).
Flow cytometry
Aliquots of 1 × 105 whole blood cells were incubated with fluorescein (FITC)-conjugated anti-CD11b
(Beckman Coulter, Inc. Fullerton, CA, USA), phycoerythrin (PE)-conjugated anti-CD51/CD61 (BD
Pharmingen, San Diego, CA, USA), and related isotype controls. The expression of RANK by CD51/61+
subsets of PBMCs was assessed by indirect flow cytometry by using rabbit anti-RANK (Santa Cruz
Biotecnology, Santa Cruz, CA, USA) as primary antibody and goat anti-rabbit IgG-FITC as secondary
antibody (AlexaFluor 488, Invitrogen, Carlsbad, CA). Appropriate controls were used to determine optimal
voltage settings and electronic subtraction for the spectral fluorescence overlap correction. Samples were
analyzed in a COULTER® EPICS® XL-MCL™ Flow Cytometer and elaborated by CellQuest software
(Beckman Coulter, Inc.). At least 10,000 positive events were collected for each sample. Data represent a
percentage of positive cells, determined by subtracting the percentage value of the appropriate isotype
controls from each sample.
Cell cultures

OCs were obtained from unfractionated PBMCs of 21-OHD patients and controls as well as from T-cell–
depleted PBMCs of the patients. PBMCs were isolated by centrifugation over Histopaque 1077 density
gradient (Sigma Chemical, St Louis, MO), diluted at 1 x 106 cells/ml in α-Minimal Essential Medium (α-
MEM; Invitrogen, UK) and supplemented with 10% fetal bovine serum, 100 IU/ml penicillin, and 100 μg/ml
streptomycin (Gibco Limited, Uxbridge, United Kingdom). To obtain fully differentiated human OCs, the
PBMCs were then cultured for about 15 days in the presence or absence of 25 ng/ml rh-MCSF and 30 ng/ml
RANKL (R&D Systems, Minneapolis, MN). In some experiments, PBMCs were cultured in the presence of
increasing concentrations of RANKL (30–100 ng/ml), or RANK-Fc (20-100 ng/ml, R&D Systems,
Minneapolis, MN) or anti-TNF-� antibody (0.05-8 �g/ml, R&D Systems, Minneapolis, MN). At the end of
the culture period, mature OCs were identified as tartrate-resistant acid phosphatase–positive (TRAP+)
multinucleated cells (Sigma Aldrich, Milan, Italy) containing three or more nuclei. Their resorbing activity
was demonstrated by plating the cells on Millennium multiwell slides (Millennium Biologix, Kingston, ON,
Canada). To visualize the pits formed by the OCs, the cells were removed by adding NaOCl to each well.
The photomicrographs of Figures 1 and 3 were obtained using a Nikon Ellipse E400 microscope equipped
with Nikon Plan Fluor 10x/0.30 dicl. The microscope was connected with a Nikon digital camera DxM
1200; the acquisition software was Lucia G version 4.61 (build 0.64) for Nikon Italy. The T-cell depletion
was performed by using anti-CD2 antibody (Ab)–coated immunomagnetic Dynabeads (Dynal, Lake Success,
NY). Briefly, CD2+ cells were captured from the PB buffy coats, incubating 2 x 107 beads/ml with 5 x 106
cells/ml for 30 minutes at 4°C on an apparatus which allows both gentle tilting and rotation. The T-cell–
depleted 21-OHD PBMCs were cultured with or without exogenous cytokines, in the same conditions of the
unfractionated cultures.
RNA isolation and RT–PCR amplification
The mature OCs were characterized by reverse transcriptase–polymerase chain reaction (RT-PCR) for the
expression of specific markers such as β3-integrin, cathepsin K, matrix metalloproteinase-9 (MMP-9),
calcitonin receptor (CTR), and c-fos. RNA was also extracted from freshly prepared T cells from PBMCs of
21-OHD patients and controls to evaluate the expression of TNF-α, RANKL and OPG.

Western blot analysis
Proteins from OCs, developed in PBMC cultures of 21-OHD patients and controls, were solubilized with
lysis buffer [50 mM Tris (tris(hydroxymethyl)aminomethane)–HCl (pH 8), 150 mM NaCl, 5 mM
ethylenediaminetetraacetic acid, 1% NP40, and 1 mM phenylmethyl sulfonyl fluoride]. Protein determination
was performed by BCA (bicinchoninic acid) Protein assay Reagent Kit (Pierce Biotechnology, Rockford,
MN). Cell proteins (50 μg) were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis
(SDS-PAGE) and subsequently transferred to nitrocellulose membranes (Hybond; Amersham Pharmacia,
London, United Kingdom). The blots were probed overnight at 4°C with mouse anti–β-actin (Santa Cruz
Biotechnology, Santa Cruz, CA), anti-RANKL monoclonal Ab (R&D Systems), rabbit anti-OPG polyclonal
Ab (Santa Cruz Biotechnology). After incubation with appropriate peroxidase-conjugated secondary Ab,
specific reactions were revealed with the electrochemiluminescence detection kit and visualized on
Hyperfilm (Amersham Pharmacia, Buckinghamshire, United Kingdom).
ELISA assay
The amount of RANKL and OPG were detected in the sera and in the media of unfractionated and T-cell–
depleted PBMC cultures from the patients as well as from the controls by enzyme-linked immunosorbent
assay (ELISA, Biomedica, GmbH, Wien, Austria), according to the manufacturer's instructions. The samples
were diluted to the concentrations within the standard curve range. The absorption was determined with an
ELISA reader at 450 nm (550 Microplate Reader; Bio-Rad), and the results were expressed as mean ± SE.
Statistical analysis
Statistical analyses were performed by Student t test. Correlations between osteoclastogenic potential,
expressed as number of OCs, and mean dose of glucocorticoid, duration of treatment, androgen levels and
age were analyzed with Spearman correlation test. The effect of the type of 21-OHD and sex on
osteoclastogenic potential was evaluated by ANOVA.
Statistical Package for the Social Sciences (spssx/pc) software (SPSS, Chicago, IL) was used.
The results were considered statistically significant for P values less than 0.05

RESULTS
Spontaneous osteoclastogenesis in unstimulated cultures of unfractionated PBMCs from 21-OHD
patients
Numerous large TRAP+ OCs were identified in the unstimulated PBMC cultures from 21-OHD patients (OC
average number/well = 62 ± 5; Fig. 1A), whereas smaller and fewer OCs appeared in the unstimulated
PBMC cultures from controls (OC average number/well = 15 ± 2; Fig.1B). Moreover, the addition of M-
CSF and RANKL to PBMC cultures from patients significantly increased the OC number (OC average
number/well = 73 ± 5; p< 0.02; Fig.1C), compared with the parallel unstimulated cultures (Fig. 1A). No
differences were found in the osteoclastogenic potential evaluated separately in the two subgroups of
controls (OC average number/well = 62 ± 4; Fig. 1D) showing that cytokines were essential to trigger and
sustain the PBMCs’ osteoclastogenesis in both of them. For all patients, the mature OCs phenotype was
assessed by studying the expression of specific markers, such as �3-integrin, cathepsin K, MMP-9, CTR, and
c-Fos by RT-PCR. All these markers were expressed by spontaneous OCs originated from 21-OHD
patients’ samples, and the levels were similar to those observed in control OCs generated in the presence of
M-CSF/RANKL (Fig. 1E-F). Moreover, the resorbing activity of the OCs obtained from 21-OHD patients
unstimulated PBMC cultures was also demonstrated (Fig. 1G), resulting significantly higher than that
detected in the unstimulated PBMC cultures from controls (Fig. 1H). The results prompted us to investigate
if the spontaneous OCs formation could be dependent on an increased number of circulating OCPs or on the
presence of osteoclastogenic factors in the PBMC cultures from patients.
The number of circulating osteoclast precursors is higher in 21-OHD patients
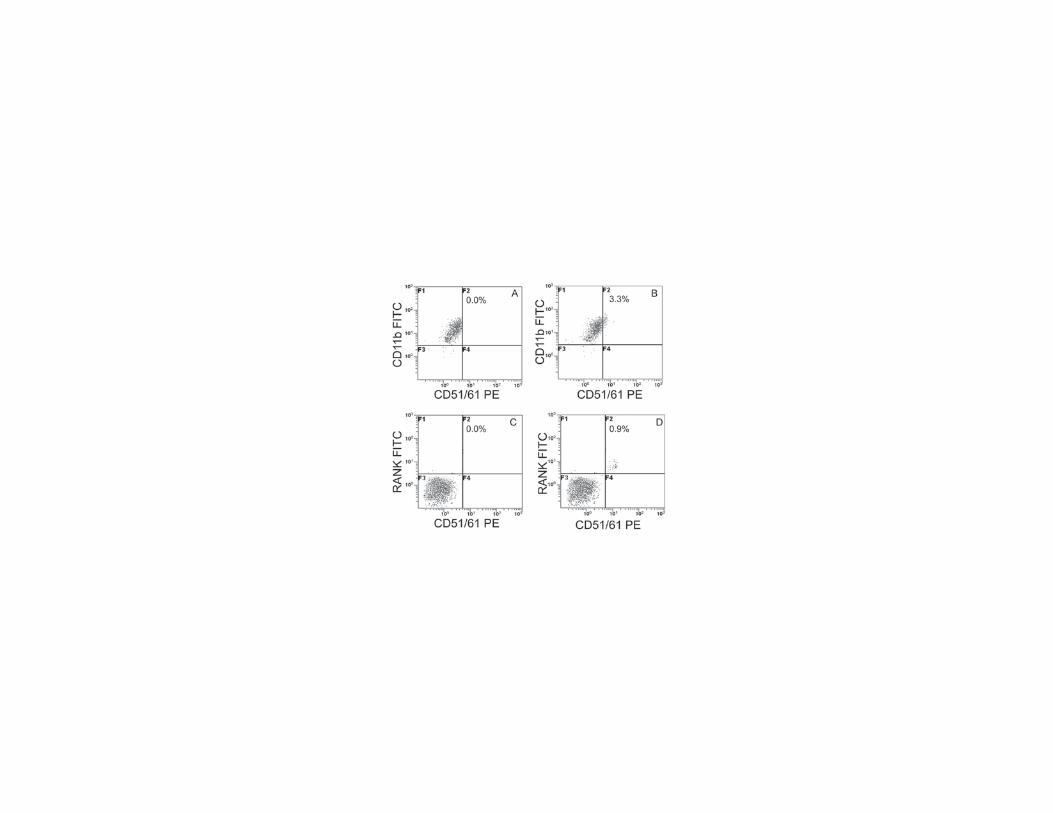
Freshly isolated PBMCs from 21-OHD patients and controls were stained and the expression of surface
markers of circulating OCPs, such as CD11b, CD51/CD61 and RANK was analyzed by flow cytometry.
There were no differences in the percentage of PBMCs expressing CD11b in 21-OHD patients with respect
to controls (data not shown). Moreover a percentage (3.3 ± 1.2%) of CD11b-CD51/CD61 positive cells,
which can be considered OCPs strongly committed (Fig. 2B),was identified in 21-OHD patients, but not in
controls (p<0.001; Fig. 2A). Also CD11b-CD51/CD61 positive cells coexpressing RANK (0.9 ± 0.05%; Fig.
2D), another marker of OCPs, were found.

T cells mediate osteoclastogenesis in 21-OHD patients
To assess the potential regulatory role of T cells in osteoclastogenesis, we studied unstimulated cultures of T
cell-depleted PBMCs from 21-OHD patients which resulted in the development of very few OCs (OC
average number/well = 7 ± 5; Fig. 3A). By contrast, the addition of M-CSF and RANKL to these cultures
induced the formation of numerous large TRAP+ OCs (OC average number/well = 55 ± 5; Fig. 3B), similar
to those observed in the presence of T cells.
Cytokine expression by fresh T cells and culture media
In order to understand the mechanism used by T cells to support OC formation, we analyzed the expression
of the factors possibly involved in osteoclastogenesis. In 21-OHD patients, fresh T cells purified from
PBMCs, overexpressed RANKL at mRNA and protein levels, while no expression was found in the T cells
from the controls (Fig. 4A). Additionally, we found that mRNA and protein levels of OPG resulted lower
than controls (Fig. 4A).
Our findings prompted us to evaluate the concentration of RANKL and OPG, by ELISA assay, in the media
derived from unfractionated as well as T cell-depleted PBMCs of both 21-OHD patients and controls. Higher
levels of RANKL were found in the media from unfractionated PBMC cultures of 21-OHD patients [SW
(1.05 ± 0.16 pmol/L), SV (1.01 ± 0.12 pmol/L), NC (1.10 ± 0.10 pmol/L)] vs controls (0.86 ± 0.01 pml/L),
p<0.005; these cytokines were undetectable in the media from T cell-depleted 21-OHD patients PBMCs and
controls. No significant differences in the OPG levels were found in the media derived from 21-OHD
patients unfractionated PBMC cultures, compared to controls. Thus, in the culture media collected from 21-
OHD patients PBMCs there was a RANKL/OPG ratio in favour of RANKL, compared to controls (Fig. 4B).
These data supported the spontaneous OCs formation observed in PBMC cultures from 21-OHD patients and
pointed to RANKL as the major cytokine involved in the spontaneous osteoclastogenesis from PBMCs of
21-OHD patients. However, the experiments performed in the presence of RANK-Fc, indicate RANKL as
the major cytokine involved in the spontaneous osteoclastogenesis from PBMCs of 21-OHD patients (Fig.
4C), even if the OC formation was not completely inhibited.
RANKL and OPG serum levels

21-OHD patients had elevated serum levels of sRANKL [SW (1.02 ± 0.20 pmol/L), SV (0.93 ± 0.20
pmol/L), NC (1.04 ± 0.16 pmol/L)], compared with controls (0.54 ± 0.07 pmol/L), p<0.01 (Fig. 5A), but
OPG serum levels were lower than in controls [SW (4.54 ± 0.90 pmol/L), SV (5.17 ± 0.42 pmol/L), NC (4.42
± 0.32 pmol/L), controls (7.07 ± 0.55 pmol/L), p<0.01; Fig. 5B]. The ratio of sRANKL/OPG was also
significantly higher in 21-OHD patients than in the controls (p < 0.005; Fig. 5C).
Bone mineral measurements and biochemical markers
Lumbar spine and proximal femur BMD Z- scores of 21-OHD patients were within the normal range (Z-
scores > -0.2) according to World Health Organization (WHO) criteria for osteopenia and osteoporosis (24).
However, a slight, but significant, reduction in bone mass was detected in 21-OHD patients (p<0.05).
Serum levels of BALP, total calcium and phosphate of 21-OHD patients did not differ significantly from
those of healthy subjects.
Correlation between osteoclastogenic potential and glucocorticoid dose, duration of treatment,
androgen levels, type of 21-OHD, age and sex of patients
No significant correlation was found between osteoclastogenic potential and glucocorticoid dose (Rho= 0.03;
p= 0.89), duration of treatment (Rho=0.13; p=0.58), androgen levels (Rho=0.12; p=0.63) and age (Rho= -
0.01; p=0.96) respectively.
Type of 21-OHD and sex of patients had no effect on osteoclastogenic potential (p= 0.78).
DISCUSSION
The present study highlights that spontaneous osteoclastogenesis occurs in patients with 21-OHD on long
term GC treatment. The OCs obtained from these patients are fully differentiated, mature and active in
resorption of bone matrix. This spontaneous osteoclastogenesis seems to be supported both by the presence
of circulating OCPs and by factors released by T cells.
Osteoporosis is a well-known side effect of long-term GC treatment (3,4). It is generally accepted that GCs
decrease bone formation (25) and increase bone resorption in vitro (26) as well as in vivo (27), the
combination of which leads to extremely rapid bone loss. The decrease in bone formation has been attributed

to GC effects on osteoblastogenesis (28), osteocyte apoptosis (28) and to alteration in skeletal growth factors
such as insulin-like growth factor-1 and transforming growth factor-β (4), while increase in bone resorption
has been linked to GC effects on OC formation and survival (29,30).
21-OHD patients receiving GCs at the time of diagnosis in order to replace congenital deficit in cortisol
synthesis, are therefore at risk of a greater incidence of low bone mass, although studies on BMD reported
discordant results. In this study we found a slight reduction in bone mass in 21-OHD patients, with normal
serum levels of 17α-OHP, Δ4-A, T, BALP, calcium, phosphate and PTH. In parallel, spontaneous formation
of numerous, mature, multinucleated and bone resorbing OCs in unstimulated and unfractionated PBMC
culture derived from 21-OHD patients was demonstrated, which was not evident in the same type of cultures
derived from controls. This spontaneous osteoclastogenesis seems to correlate directly with the higher
number of circulating OCPs present in 21-OHD patients compared with controls, and demonstrates, for the
first time, that OCPs are markedly increased in the circulation of 21-OHD patients.
No significant differences between osteoclastogenic potential and mean glucocorticoid dose, duration of
treatment, type of 21-OHD, serum androgen levels, age and sex of patients have been found.
We also evaluated OCPs from a child newly diagnosed with 21-OHD. Notably, this patient did not show
circulating OCPs, suggesting a link between GC therapy and the presence of circulating OCPs. However,
this important observation is limited by the lack of statistical significance, and it needs to be extended to
other subjects.
Data about an expanded pool of OCPs in specific pathologies were published on Paget disease, multiple
myeloma, psoriatic arthritis, and bone metastasis of cancer (31-34). Furthermore, a regulatory role of bone
turnover played by T cells in physiological and pathological conditions is emerging from the recent literature
(35,36). Thus, we also focused our attention on T cells as possible activators of osteoclastogenesis in vitro.
Evidences supporting a T-cell regulation of osteoclastogenesis came from 21-OHD patients T-cell–depleted
PBMC cultures, in which the addition of exogenous M-CSF and RANKL was necessary for OC formation,
suggesting that T cells alone could provide the above mentioned cytokines in our system and demonstrating,
for the first time, T-cell–dependent osteoclastogenesis from human 21-OHD PBMCs. These findings
prompted us to evaluate the cytokines possibly involved in OC formation produced by T-cells. In particular,
we analyzed (at both mRNA and protein levels), RANKL and OPG, and we showed that RANKL was

overexpressed, while OPG downregulated in T-cells from 21-OHD patients compared to controls.
Furthermore, high RANKL levels were measured in media from unfractionated PBMC culture of 21-OHD
patients, leading to a RANKL/OPG ratio higher in these patients than in controls. Additionally, we analyzed,
in the sera from 21-OHD patients and controls, RANKL and OPG levels, as well as their relative ratio: we
noticed that concurrently RANKL was significantly elevated, while OPG decreased in these patients
compared to controls and therefore RANKL/OPG ratio was higher in the sera of 21-OHD patients.
The key roles of RANKL and OPG in osteoclastogenesis regulation have been clearly identified: OC activity
is likely to depend, at least partly, on the relative balance of RANKL and OPG. Moreover, the RANKL/OPG
ratio in serum, rather than the individual protein concentrations, has been suggested to be the critical factor in
determining osteoclastic activation at bone level, with higher serum RANKL/OPG ratios being a marker for
up-regulation of osteoclastogenesis (37). Alteration in RANKL/OPG axis has been demonstrated in several
bone loss associated diseases (38). In particular, Hofbauer et al, reported that dexamethasone inhibits OPG
mRNA expression, and increases RANKL mRNA expression in human osteoblastic lineage cells (37).
Furthermore, the findings of Kobayashi et al, demonstrating that dexamethasone stimulation induces
RANKL expression in human T lymphoblastic cell line Jurkat (39), support our results. However, in our
system RANKL is not the only factor that produces spontaneous osteoclastogenesis. In fact, in the
unfractionated and unstimulated PBMC culture from 21-OHD patients, the presence of RANK-Fc does not
completely inhibit osteoclastogenesis. Additionally, in unstimulated T-depleted cultures, few OCs develop
spontaneously, maybe because OCPs receive a priming by GC stimulation in vivo, that commits these cells
towards complete differentiation. On the other hand, RANKL involvement in chronic GC therapy was also
supported by the very recent findings demonstrating that Denosumab, a fully human anti-RANKL antibody,
prevents bone loss in a murine model of GCs-induced osteoporosis (40). Thus, spontaneous
osteoclastogenesis, high levels of OCPs and high RANK/OPG ratio in 21-OHD patients could explain the
slight reduction in the BMD of these subjects.
In conclusion, the present study showed, for the first time, a high osteoclastogenic potential of PBMCs from
21-OHD patients. This spontaneous osteoclastogenesis seems to be supported by both the presence of
circulating OCPs and by factors released by T cells. In particular, we found the RANKL/OPG imbalance

both in T cells and sera from these patients. Thus, monitoring periodically OCPs, RANKL and OPG levels,
in these patients, could be useful to control bone metabolism, and eventually adjust GC doses.

REFERENCES
1.White PC, Speiser PW 2000 Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocr Rev
21:245-291
2. Joint LWPES/ESPE CAH Working Group 2002 Consensus statement on 21-hydroxylase deficiency from
the Lawson Wilkins Pediatric Endocrine Society and the European Society for Paediatric Endocrinology. J
Clin Endocrinol Metab 87:4048–4053
3. Mazziotti G, Angeli A, Bilezikian JP, Canalis E, Giustina A 2006 Glucocorticoid-induced osteoporosis: an
update. Trends Endocrinol Metab 17:144-149
4. Canalis E, Mazziotti G, Giustina A, Bilezikian JP 2007 Glucocorticoid-induced osteoporosis:
pathophysiology and therapy. Osteoporos Int 18:1319-1328
5. Speiser PW, New MI 1993 Increased bone mineral density in congenital adrenal hyperplasia. Pediatr Res
33:S81
6. Cameron FJ, Kaimakci B, Byrt EA, Ebeling PR, Warne GL, Wark JD 1995 Bone mineral density and
body composition in congenital adrenal hyperplasia. J Clin Endocrinol Metab 80:2238-2243
7. Jaaskelainen J, Voutilainen R 1996 Bone mineral density in relation to glucocorticoid substitution therapy
in adult patients with 21-hydroxylase deficiency. Clin Endocrinol 45:707-713
8. Girgis R, Winter JSD 1997 The effects of glucocorticoid replacement therapy on growth, bone mineral
density and bone turnover markers in children with congenital adrenal hyperplasia. J Clin Endocrinol Metab
82:3926-3929
9. Hagenfeldt K, Ritzen EM, Ringertz H, Helleday J, Carlstrom K 2000 Bone mass and body composition of
adult women with congenital virilizing 21-hydroxylase deficiency after glucocorticoid treatment since
infancy. Eur J Endocrinol 143:667-671
10. Paganini C, Radetti G, Livieri C, Braga V, Migliavacca D, Adami S 2000 Height, bone mineral density
and bone markers in congenital adrenal hyperplasia. Horm Res 54:164-168
11. de Almeida Freire PO, de Lemos-Marini SH, Maciel-Guerra AT, Morcillo AM, Matias Baptista MT, de
Mello MP, Guerra G Jr 2003 Classical congenital adrenal hyperplasia due to 21-hydroxylase deficiency: a
cross selectional study of factors involved in bone mineral density. J Bone Miner Metab 21:396-401

12. King JA, Wisniewski AB, Bankowski BJ, Carson KA, Zacur HA, Migeon CJ 2006 Long term
glucocorticoid replacement and bone mineral density in adult women with classical congenital adrenal
hyperplasia. J Clin Endocrinol Metab 91:865-869
13. Sciannamblo M, Russo G, Cuccato D, Chiumello G, Mora S 2006 Reduced bone mineral density and
increased bone metabolism rate in young adult patients with 21-hydroxylase. J Clin Endocrinol Metab
91:4453-4458
14. Guo CY, Weetman AP, Eastell R 1996 Bone turnover and bone mineral density in patients with
congenital adrenal hyperplasia. Clin Endocrinol 45:535-541
15. Mora S, Saggion F, Russo G, Weber G, Bellini A, Prinster C, Chiumello G 1996 Bone density in young
patients with congenital adrenal hyperplasia. Bone 18:337-340
16. Gussinyè M, Carrascosa A, Potau N, Enrubia M, Vicens-Calvet E, Ibanez L, Yeste D 1997 Bone mineral
density in prepubertal and in adolescent and young adult patients with the salt-wasting form of congenital
adrenal hyperplasia. Pediatrics 100:671-674
17. Stikkelbroeck NMML, Oyen WJG, van der Wilt GJ, Hermus ARMM, Otten BJ 2003 Normal bone
mineral density and lean body mass, but increased fat mass, in young adult patients with congenital adrenal
hyperplasia. J Clin Endocrinol Metab 88:1036-1042
18. Christiansen P, Moolgard C, Muller J 2004 Normal bone mineral content in young adults with congenital
adrenal hyperplasia due to 21-hydroxylase deficiency. Horm Res 61:133-136
19. Massey HM, Flanagan AM 1999 Human osteoclasts derive from CD14-positive monocytes. Br J
Haematol 106:167–170
20. Boyle WJ, Simonet WS, Lacey DL 2003 Osteoclast differentiation and activation. Nature 423:337-342
21. Theill LE, Boyle WJ, Penninger JM 2002 RANK-L and RANK: T cells, bone loss, and mammalian
evolution. Annu Rev Immunol 20:795–823
22. Kong YY, Feige U, Sarosi I, Bolon B, Tafuri A, Morony S, Capparelli C, Li J, Elliott R, McCabe S,
Wong T, Campagnuolo G, Moran E, Bogoch ER, Van G, Nguyen LT, Ohashi PS, Lacey DL, Fish E, Boyle
WJ, Penninger JM 1999 Activated T cells regulate bone loss and joint destruction in adjuvant arthritis
through osteoprotegerin ligand. Nature 402:304-309

23. Colucci S, Brunetti G, Rizzi R, Zonno A, Mori G, Colaianni G, Del Prete D, Faccio R, Liso A, Capalbo
S, Liso V, Zallone A, Grano M 2004 T cells support osteoclastogenesis in an in vitro model derived from
human multiple myeloma bone disease: the role of the OPG/TRAIL interaction. Blood 104:3722-3730
24. Kanis JA, Glu¨er CC, for the Committee of Scientific Advisors, International Osteoporosis Foundation*
2000 An Update on the Diagnosis and Assessment of Osteoporosis with Densitometry. Osteoporos Int
11:192–202
25. Reid IR 1997 Glucocorticoid osteoporosis—mechanisms and management. Eur J Endocrinol 137:209–
217
26. Conaway HH, Grogorie D, Lerner UH 1996 Stimulation of neonatal mouse calvarial bone resorption by
the glucocorticoids hydrocortisone and dexamethasone. J Bone Miner Res 11:1419–1429
27. Dempster D 1998 Bone histomorphometry in glucocorticoid-induced osteoporosis. J Bone Miner Res
13:137–141
28. Weinstein RS, Jilka RL, Parfitt AM, Manolagas SC 1998 Inhibition of osteoblastogenesis and promotion
of apoptosis of osteoblasts and osteocytes by glucocorticoids. Potential mechanism of their deleterious
effects on bone. J Clin Invest 102:274–282
29. Sivagurunathan S, Muir MM, Brennan TC, Seale JP, Mason RS 2005 Influence of glucocorticoids on
human osteoclast generation and activity. J Bone Miner Res 20:390-398
30. Hofbauer LC, Gori F, Riggs BL, Lacey DL, Dunstan CR, Spelsberg TC, Khosla S 1999 Stimulation of
osteoprotegerin ligand and inhibition of osteoprotegerin production by glucocorticoids in human osteoblastic
lineage cells: potential paracrine mechanisms of glucocorticoid-induced osteoporosis. Endocrinology
140:4382–4389
31. Demulder A, Takahashi S, Singer FR, Hosking DJ, Roodman GD 1993 Abnormalities in osteoclast
precursors and marrow accessory cells in Paget's disease. Endocrinology 133:1978–1982
32. Gregoretti MG, Bergui L, Aragno M, Cremona O, Marchisio PC, Caligaris-Cappio F 1995 Osteoclast
precursors circulate in the peripheral blood of patients with aggressive multiple myeloma. Leukemia 9:1392–
1397
33. Ritchlin CT, Haas-Smith SA, Li P, Hicks DG, Schwarz EM 2003 Mechanisms of TNF-alpha- and
RANKL-mediated osteoclastogenesis and bone resorption in psoriatic arthritis. J Clin Invest 111:821–831

34. Roato I, Grano M, Brunetti G, Colucci S, Mussa A, Bertetto O, Ferracini R 2005 Mechanisms of
spontaneous osteoclastogenesis in cancer with bone involvement. FASEB J 19:228-230
35. Nosaka K, Miyamoto T, Sakai T, Mitsuya H, Suda T, Matsuoka M 2002 Mechanism of hypercalcemia
in adult T-cell leukemia: overexpression of receptor activator of nuclear factor kappaB ligand on adult T-cell
leukemia cells. Blood 99:634–640
36. Giuliani N, Colla S, Sala R, Moroni M, Lazzaretti M, La Monica S, Bonomini S, Hojden M, Sammarelli
G, Barille S, Bataille R, Rizzoli D 2002 Human myeloma cells stimulate the receptor activator of nuclear
factor-kappa B ligand (RANKL) in T lymphocytes: a potential role in multiple myeloma bone disease. Blood
100:4615–4621
37. Hofbauer LC, Khosla S, Dunstan CR, Lacey DL, Boyle WJ, Riggs BL 2000 The roles of osteoprotegerin
and osteoprotegerin ligand in the paracrine regulation of bone resorption. J Bone Miner Res 15:2-12
38. Bostanci N, Ilgenli T, Emingil G, Afacan B, Han B, Töz H, Atilla G, Hughes FJ, Belibasakis GN 2007
Gingival crevicular fluid levels of RANKL and OPG in periodontal diseases: implications of their relative
ratio. J Clin Periodontol 34:370-376
39. Kobayashi A, Hirano F, Makino I 2005 The inhibitory effect of bisphosphonates on glucocorticoid-
induced RANKL expression in human cells. Scand J Rheumatol 34:480-484
40. Hofbauer LC, Zeitz U, Shoppet M, Scalicky M, Stolina M, Kostenuik PJ, Erben RG 2007 RANK ligang
inhibition by Denosumab prevents cortical bone loss in a murine nodel of glucocorticoid-induced
osteoporosis. 29th ASBMR J Bone Miner Res 22(S1)

FIGURE LEGENDS.
Figure 1. OCs generated from human 21-OHD PBMCs
OCs were obtained from unfractionated PBMCs of 21-OHD patients and controls. Numerous and large-sized
OCs developed in the unstimulated cultures from 21-OHD patients (A), whereas rare and small-sized OCs
were observed in the cultures from controls (B). A significant increase in OC formation was observed in 21-
OHD patient PBMCs by exogenous M-CSF and RANKL (C), whereas these cytokines were essential to
trigger the OC formation in controls (D). Multinucleated (>3 nuclei per cell) and TRAP+ cells were
identified as OCs. The arrows point to the OCs (magnification 200 X). The mRNA levels of CTR, �3
integrin, cathepsin K, c-fos, and MMP-9 in TRAP+ and multinucleated cells from PBMCs and SFMCs of
21-OHD patients were detected by RT-PCR (E). The housekeeping gene GAPDH was used as control gene.
The intensity of the bands obtained by RT-PCR was quantified by densitometry (histograms) and each gene
was normalized to GAPDH (F). Numerous and large resorption areas were observed in the MM bone disease
samples (G) respect to the few and small pits detected in the controls (H). The percentage of mineral surface
resorbed by the OCs was quantified with an image analyzer. The data reported in graph corresponded to the
mean ± SE (I).
Figure 2. OCPs expressing CD51/CD61 circulate in peripheral blood of 21-OHD patients
Flow cytometry analyses of circulating osteoclast precursors. The figures show representative dot plots of
CD11b and CD51/CD61 (A, B), CD51/61 and RANK (C, D) staining from 21-OHD patients, and controls.
OCPs express CD51/61 and RANK only in patients.
Figure 3. T cells mediate osteoclastogenesis in human 21-OHD PBMCs
Osteoclastogenesis occurred in T cell-depleted 21-OHD PBMCs cultured in the absence (A) or in the
presence (B) of M-CSF and RANKL. Small-sized OCs developed in the absence of M-CSF and RANKL
(A), whereas exogenous cytokines triggered the formation of large-sized OCs (B). Multinucleated and
TRAP+ cells were identified as OCs. The arrows point to the OCs (magnification 200 X).

Figure 4. Cytokine expression by fresh T cells and culture media
(A) Fresh T cells purified from PBMCs of 21-OHD patients as well as from controls were analyzed for
RANKL and OPG expression, respectively, by RT-PCR and western-blotting. The intensity of the bands
obtained by RT-PCR was quantified by densitometry (histograms) and normalized to GAPDH. (B) In the
culture media collected from PBMCs of patients with SW, SV and NC forms of 21-OHD, there was a
RANKL/OPG ratio in favour of RANKL respect to the controls (p<0.001). (C) The inhibition of RANKL by
RANK-Fc prevented in a dose-dependent manner the OC formation in unstimulated and unfractionated
PBMC cultures from 21-OHD patients.
Figure 5. sRANKL and OPG serum levels in 21-OHD patients
Patients with SW, SV and NC forms of 21-OHD had elevated serum levels of sRANKL, compared with
controls (A), but serum levels of OPG, were lower than in controls (B). The ratio of sRANKL/OPG was also
significantly higher in patients with SW, SV and NC forms of 21-OHD than in controls (C).

Table 1. Main clinical and hormonal characteristics of 18 patients with 21-OHD
Patients
no. Sex Age,y Weight
(kg) Height (cm)
BMI (Kg/m2)
Clinical form
17αααα-OHP * (ng/ml)
ΔΔΔΔ4-A* (ng/ml)
Testosterone* (ng/ml)
1 M 15 65 165 24 SW 40 2.4 4 2 M 16 68 168 24 SW 50 3 4.5 3 M 14 53.4 152.6 23.2 SW 50 7 5.1 4 M 15 59.5 173.3 19.8 NC 50 4.5 4.4 5 M 11 38.2 149.5 17 NC 50 6 3.4 6 M 16 70 170 24.2 NC 21.7 1.2 2.7 7 M 13 50 153.2 21.3 NC 5.6 0.6 3.3 8 M 16 70.5 169.3 24.6 NC 10 1.2 3.5 9 M 5 22 98 22 NC 10 2.5 3.2 10 F 14 60.5 155 25.2 SW 40 2.3 0.30 11 F 16 49.3 158.3 19.7 NC 40 2.2 0.35 12 F 16 48.2 148.2 22 SV 50 3.7 0.51 13 F 14 58.6 158.7 23.4 NC 21 2.4 0.34 14 F 9 37.6 135.5 20.5 NC 14.9 2.4 0.42 15 F 8 38 137 20 SV 50 4 0.4 16 F 3 11.7 82 17.5 SV 39 <0.1 <0.1 17 F 16 62.9 147.9 28.8 NC 5 2.3 0.45 18 F 16 52 155.1 21.6 SV 20 2.4 0.34
* mean value of the preceding 5 years




Copyright © 2022 FDOKUMEN