
Nuclear localization of Vpr is crucial for the efficient replication of HIV-1 in primary CD4+ T...
13
Nuclear localization of Vpr is crucial for the efficient replication of HIV-1 in primary CD4 + T cells Sayuki Iijima a,b , Yuko Nitahara-Kasahara a , Kiyonori Kimata a , Wen Zhong Zhuang a , Masakazu Kamata a , Maya Isogai c , Masanao Miwa b , Yasuko Tsunetsugu-Yokota c , Yoko Aida a, * a Retrovirus Research Unit, RIKEN, Wako, Saitama 351-0198, Japan b Institute of Medical Sciences, University of Tsukuba, Tsukuba, Ibaraki 305-8575, Japan c Department of Immunology, National Institute of Infectious Disease, Toyama, Shinjuku-ku, Tokyo 162-8640, Japan Received 22 March 2004; accepted 17 June 2004 Available online 8 August 2004 Abstract The human immunodeficiency virus type 1 (HIV-1) accessory protein Vpr appears to make a substantial contribution to the replication of HIV-1 in established Tcell lines when HIV-1 is present at very low multiplicities of infection. However, the role of Vpr in viral replication in primary CD4 + T cells remains to be clarified. In this study, we generated a panel of viruses that encoded mutant forms of Vpr that lacked either the ability to accumulate in the nucleus and induce G 2 arrest or the ability to induce apoptosis, which has been shown to occur independently of G 2 arrest of the cell cycle. We demonstrate here that the nuclear localization of Vpr and consequent G 2 arrest but not the induction of apoptosis by Vpr are important for viral replication in primary CD4 + T cells at both high and low multiplicities of infection. Viruses that encoded mutant forms of Vpr that failed to be imported into the nucleus in the presence of cytoplasmic extracts from primary CD4 + T cells in an in vitro nuclear import assay replicated at drastically reduced rates. Thus, Vpr might be a key regulator of the viral nuclear import process during infection in primary CD4 + T cells. By contrast, a mutant form of Vpr that exhibited diffuse cytosolic staining exclusively in an immunofluorescence assay of HeLa cells and was not imported into nucleus by the cytosol from HeLa cells was effectively imported into the nucleus by cytosol from primary CD4 + T cells. This Vpr mutant virus replicated well in primary CD4 + T cells, indicating that cellular factors in primary CD4 + T cells are indispensable for the accumulation of Vpr in the nucleus and, thus, for viral replication. Our results suggest that the nuclear import of Vpr might be a good target in efforts to block the early stages of replication of HIV-1. D 2004 Elsevier Inc. All rights reserved. Keywords: Vpr; Nuclear localization; HIV-1; Primary CD4 + T cell; G 2 arrest; Apoptosis Introduction The accessory gene vpr of human immunodeficiency virus type 1 (HIV-1) encodes a 15-kDa nuclear protein of 96 amino acids that is strongly conserved in primate lentivi- ruses, all of which include a Vpr-like protein (Emerman, 1996; Planelles et al., 1996). Thus, it is likely that Vpr plays an important role in the life cycle of such viruses. Vpr has many biological functions, being involved in incorporation of virions (Cohen et al., 1990a; Connor et al., 1995; Mahalingam et al., 1995a; Schuler et al., 1999; Tung et al., 1997); nuclear localization (Gallay et al., 1996; Heinzinger et al., 1994; Jenkins et al., 1998; Kamata and Aida, 2000; Lu et al., 1993; Popov et al., 1998a; Vodicka et al., 1998); induction of cell cycle arrest at the G 2 /M phase (He et al., 1995; Jowett et al., 1995; Re et al., 1995) or at the G 1 phase (Nishizawa et al., 1999, 2000a, 2000b); weak activation of 0042-6822/$ - see front matter D 2004 Elsevier Inc. All rights reserved. doi:10.1016/j.virol.2004.06.024 * Corresponding author. Mailing address: Retrovirus Research Unit, RIKEN, 2-1 Hirosawa, Wako, Saitama 351-0198, Japan. Fax: +81 48 462 4399. E-mail address: [email protected] (Y. Aida). Virology 327 (2004) 249– 261 www.elsevier.com/locate/yviro
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Nuclear localization of Vpr is crucial for the efficient replication of HIV-1 in primary CD4+ T...

www.elsevier.com/locate/yviro
Virology 327 (20
Nuclear localization of Vpr is crucial for the efficient replication
of HIV-1 in primary CD4+ T cells
Sayuki Iijimaa,b, Yuko Nitahara-Kasaharaa, Kiyonori Kimataa, Wen Zhong Zhuanga,
Masakazu Kamataa, Maya Isogaic, Masanao Miwab, Yasuko Tsunetsugu-Yokotac, Yoko Aidaa,*
aRetrovirus Research Unit, RIKEN, Wako, Saitama 351-0198, JapanbInstitute of Medical Sciences, University of Tsukuba, Tsukuba, Ibaraki 305-8575, Japan
cDepartment of Immunology, National Institute of Infectious Disease, Toyama, Shinjuku-ku, Tokyo 162-8640, Japan
Received 22 March 2004; accepted 17 June 2004
Available online 8 August 2004
Abstract
The human immunodeficiency virus type 1 (HIV-1) accessory protein Vpr appears to make a substantial contribution to the replication of
HIV-1 in established T cell lines when HIV-1 is present at very low multiplicities of infection. However, the role of Vpr in viral replication
in primary CD4+ T cells remains to be clarified. In this study, we generated a panel of viruses that encoded mutant forms of Vpr that lacked
either the ability to accumulate in the nucleus and induce G2 arrest or the ability to induce apoptosis, which has been shown to occur
independently of G2 arrest of the cell cycle. We demonstrate here that the nuclear localization of Vpr and consequent G2 arrest but not the
induction of apoptosis by Vpr are important for viral replication in primary CD4+ T cells at both high and low multiplicities of infection.
Viruses that encoded mutant forms of Vpr that failed to be imported into the nucleus in the presence of cytoplasmic extracts from primary
CD4+ T cells in an in vitro nuclear import assay replicated at drastically reduced rates. Thus, Vpr might be a key regulator of the viral
nuclear import process during infection in primary CD4+ T cells. By contrast, a mutant form of Vpr that exhibited diffuse cytosolic staining
exclusively in an immunofluorescence assay of HeLa cells and was not imported into nucleus by the cytosol from HeLa cells was
effectively imported into the nucleus by cytosol from primary CD4+ T cells. This Vpr mutant virus replicated well in primary CD4+ T cells,
indicating that cellular factors in primary CD4+ T cells are indispensable for the accumulation of Vpr in the nucleus and, thus, for viral
replication. Our results suggest that the nuclear import of Vpr might be a good target in efforts to block the early stages of replication of
HIV-1.
D 2004 Elsevier Inc. All rights reserved.
Keywords: Vpr; Nuclear localization; HIV-1; Primary CD4+ T cell; G2 arrest; Apoptosis
Introduction
The accessory gene vpr of human immunodeficiency
virus type 1 (HIV-1) encodes a 15-kDa nuclear protein of 96
amino acids that is strongly conserved in primate lentivi-
ruses, all of which include a Vpr-like protein (Emerman,
0042-6822/$ - see front matter D 2004 Elsevier Inc. All rights reserved.
doi:10.1016/j.virol.2004.06.024
* Corresponding author. Mailing address: Retrovirus Research Unit,
RIKEN, 2-1 Hirosawa, Wako, Saitama 351-0198, Japan. Fax: +81 48 462
4399.
E-mail address: [email protected] (Y. Aida).
1996; Planelles et al., 1996). Thus, it is likely that Vpr plays
an important role in the life cycle of such viruses. Vpr has
many biological functions, being involved in incorporation
of virions (Cohen et al., 1990a; Connor et al., 1995;
Mahalingam et al., 1995a; Schuler et al., 1999; Tung et al.,
1997); nuclear localization (Gallay et al., 1996; Heinzinger
et al., 1994; Jenkins et al., 1998; Kamata and Aida, 2000;
Lu et al., 1993; Popov et al., 1998a; Vodicka et al., 1998);
induction of cell cycle arrest at the G2/M phase (He et al.,
1995; Jowett et al., 1995; Re et al., 1995) or at the G1 phase
(Nishizawa et al., 1999, 2000a, 2000b); weak activation of
04) 249–261

S. Iijima et al. / Virology 327 (2004) 249–261250
several viral promoters, including those of HIV-1 (Cohen et
al., 1990b); induction of the terminal differentiation of
certain types of cells (Le Rouzic et al., 2002); and
association with a variety of several cellular factors
(Agostini et al., 1996; Bouhamdan et al., 1996; Mahalingam
et al., 1998; Refaeli et al., 1995; Sawaya et al., 1998; Wang
et al., 1995; Withers-Ward et al., 1997; Zhao et al., 1994).
The various biological activities of Vpr appear to be
correlated with specific structural features of the protein
(Di Marzio et al., 1995; Mahalingam et al., 1995b, 1995c,
1995d, 1997; Piller et al., 1996; Wang et al., 1996; Yao et
al., 1995; Zhao et al., 1996). Analysis of the three-
dimensional structure of Vpr by nuclear magnetic resonance
(NMR) revealed three well-defined a-helices, namely,
amino acids 17–33, 38–50, and 56–77, which are sur-
rounded by flexible amino- and carboxy-terminal domains
(Morellet et al., 2003). Mutational analysis has suggested
the importance of the first of the three a-helical domains in
nuclear localization and in the expression, stability, and
incorporation into virions of Vpr (Mahalingam et al., 1995a,
1995c; Yao et al., 1995). The second domain also appears to
be critical for the incorporation of Vpr into the virions and
for the oligomerization feature of Vpr (Singh et al., 2000),
while the third domain contains a determinant that is
involved in nuclear localization and, in particular, in the
translocation to the nucleus of the preintegration complex
(PIC) in nondividing cells (Kamata and Aida, 2000; Nie et
al., 1998). The carboxy-terminal 20-aa arginine-rich tail,
which contains a cryptic nuclear localization signal (NLS),
makes a major contribution to the nuclear localization of
Vpr (Lu et al., 1993; Mahalingam et al., 1997; Yao et al.,
1995; Zhao et al., 1996, 1998). Truncation of amino acid
substitutions in this region results in failure to induce cell
cycle arrest (Di Marzio et al., 1995; Mahalingam et al.,
1997; Zhao et al., 1998). Thus, it appears that the carboxy-
terminal region of Vpr is also required for the induction of
cell cycle arrest.
It appears that Vpr is not absolutely required for viral
replication because deletion of Vpr is not lethal to viral
replication in vitro. However, it was recently reported that
Vpr is present in significant amounts in the serum of HIV-1-
infected patients and that it activates viral expression in
latently infected cell lines and resting peripheral blood
mononuclear cells (PBMC) (Levy et al., 1993, 1994). These
observations suggest that Vpr is very much involved in the
life cycle of HIV-1 and controls both the replication and
pathogenesis of this virus. Moreover, the fact that many
copies of Vpr are packaged in progeny virions suggests that
the protein might play a role early in the infectious process
(Cohen et al., 1990a). Experimental support for an early
functional role for Vpr comes from observations of a
contribution by Vpr to the nuclear import of proviral DNA
in nondividing cells, such as macrophages (Heinzinger et
al., 1994). In addition, Vpr synthesized de novo plays an
additional role in the replication of HIV-1 because Vpr that
is supplied in trans does not fully complement the
replication of Vpr-negative viruses in infected macrophages
(Connor et al., 1995). It is possible that Vpr might
upregulate the expression of viral genes.
By contrast to its well-documented role in nondividing
cells, the role of Vpr in the replication of HIV-1 in dividing
cells remains to be fully characterized. Vpr is not essential
for the replication of HIV-1 in vitro in proliferating T cells
and stimulated PBMC (Balliet et al., 1994; Cohen et al.,
1990a, 1990b; Connor et al., 1995; Hattori et al., 1990;
Ogawa et al., 1989; Rogel et al., 1995). However, Vpr
contributes substantially to the replication of HIV-1 in these
cells because viral replication is increased in dividing cells
when the multiplicity of infection is very low (Balliet et al.,
1994; Connor et al., 1995; Ogawa et al., 1989). Previous
evidence indicates that expression of the viral genome is
maximal during the G2 phase of the cell cycle and that Vpr
increases the production of virions by progression of the cell
cycle beyond the point when the LTR is most active (Goh et
al., 1998). These observations suggest a novel mechanism
for maximizing the production of virions in the face of the
rapid killing of infected target cells. Thus, the G2 arrest
induced by Vpr might enhance the production of HIV-1.
However, the role of Vpr in the replication of HIV-1 both in
dividing cells and in nondividing cells remains to be fully
clarified.
In this study, we attempted to confirm the roles of
various phenomena induced by Vpr in the replication of
HIV-1 in primary CD4+ T cells. CD4+ monocytes exist in
PBMC fraction (3–10%, depending on the donor), which
differentiate into macrophages during cultivation. Further-
more, macrophages can be infected with an X4-type HIV,
although the replication of this T-cell line-adapted HIV is
much less efficiently in macrophages compared to that in
activated T cells. Therefore, to characterize the biological
function of Vpr in T cells, we consider that it is crucial to
purify CD4+ T cells from PBMC. First, we developed a
panel of expression vectors that encoded Vpr proteins with
site-directed mutations in each of the putative domains,
namely, in the three a-helical domains and in the arginine-
rich carboxy-terminal domain, we produced mutants of Vpr
that lacked either the ability to localize to the nucleus and to
induce G2 arrest or the ability to induce apoptosis, which
occurs independently of G2 arrest (Nishizawa et al., 1999,
2000a, 2000b). Furthermore, to explore the roles, if any, of
Vpr-mediated G2 arrest, nuclear localization, and apoptosis
in viral replication, we monitored the replication of viruses
that encoded the various mutant forms of Vpr during
infection of primary CD4+ T cells by HIV-1. Our results
indicate that the nuclear localization of Vpr, as well as G2
arrest, is important for viral replication in primary CD4+ T
cells. We also found that Vpr mutant viruses with impaired
nuclear import of Vpr in vitro in the presence of
cytoplasmic extracts of primary CD4+ T cells exhibited
drastically reduced levels of viral replication, while mutants
that retained importability replicated well in primary CD4+
T cells.

S. Iijima et al. / Virology 327 (2004) 249–261 251
Results
Construction of plasmids and expression of substitution
mutants of Vpr
The Vpr protein does not include a canonical nuclear
localization signal (NLS). However, it appears to interact
with PIC and to play a critical role in nuclear transport of
the PIC (Bukrinsky and Haffar, 1998). Moreover, Vpr
interrupts the cell cycle at G2 by inhibiting the activation of
Cdc2 kinase, which is required for entry into the M phase
of the cell cycle (He et al., 1995; Jowett et al., 1995; Re et
al., 1995). Furthermore, a recent report indicates that Vpr
enhances virus production by delaying cells at the G2 phase
of the cell cycle, when the LTR is most active (Goh et al.,
1998). By contrast, the induction of apoptosis by Vpr
occurs independently of G2 arrest (Nishizawa et al., 1999,
2000a, 2000b). Interestingly, the various biological activ-
ities of Vpr are correlated with specific structural features
or domains of the protein as noted in the introduction and
shown in Fig. 1A.
Therefore, to identify the domains of Vpr that are
involved in nuclear localization, G2 arrest, and the apoptosis
that occurs independently of G2 arrest, we generated a panel
of expression vectors derived from pME18Neo that encoded
Vpr and variants with site-directed mutagenesis in each
putative domain of Vpr as follows. (i) We replaced His and
Ile at positions 45 and 46 within the second a-helical
domain by Trp and Ala, respectively (HI4546WA); (ii) we
replaced the bulky nonpolar Leu at position 67 within the
third a-helical domain by smaller Ala to introduce a
Fig. 1. Construction and expression of mutant forms of Vpr. (A) Plasmids
that included cDNAs for the mutant forms of Vpr were generated by site-
directed mutagenesis. The diagram shows schematic representations of the
variations of proteins and putative domains. (B) Western blotting of wild-
type and Vpr mutant proteins; cells were co-transfected with pME18Neo
that encoded Flag-tagged wild-type Vpr, L67A, R8788EA, L67P,
aLAL67P, HI4546WA, or the control plasmid pME18Neo-Flag, together
with pSV-h-galactosidase. Transfected cells were harvested and lysed 24 h
after transfection. Lysates with equal h-galactosidase activity were
subjected to Western blotting analysis with the Flag-specific MAb M2.
significant change of hydrophobicity (L67A); (iii) we
replaced Arg at positions 87 and 88 within the arginine-
rich domain by Glu and Ala (R8788EA); (iv) we replaced
Leu at position 67 within the third a-helical domain by Pro
(L67P) to disrupt helical structure (L67P); and (v) we
replaced four bulky nonpolar Leu at positions 20, 22, 23,
and 26 within the first a-helical domain and the Leu at
position 67 within the third a-helical domain by Ala and Pro
(aLAL67P) (Fig. 1A). We transfected HeLa cells with these
expression vectors, which included, for monitoring the
expression of wild-type and mutant proteins, cDNA for an
amino terminal Flag tag that encoded the sequence NH2-
Met-Asp-Tyr-Lys-Asp-Asp-Asp-Asp-Lys (Fig. 1A). West-
ern blotting analysis with MAb M2, which recognizes the
Flag-tag, indicated that each mutant form of Vpr was
expressed at detectable levels in the corresponding trans-
fected cells 24 h after transfection (Fig. 1B). Interestingly,
the aLAL67P mutant exhibited migration slightly slower
than that of wild-type Vpr. This difference in electrophoretic
migration may have resulted from the altered conformation
of mutant Vpr proteins relative to the wild-type polypeptide
or changes in the hydrophobic face (aLA) of the first helical
domain (Mahalingam et al., 1997).
Analysis of G2 arrest, apoptosis, and nuclear localization of
substitution mutants of Vpr
To determine the effects of substitutions within the
putative domains of Vpr on G2 arrest, we transfected HeLa
cells with pME18Neo that encoded wild-type Vpr, L67A,
R8788EA, HI4546WA, or the control pME18Neo-Flag.
Forty-eight hours after transfection, we stained cells with the
Flag-specific MAb M2 and then with Alexa 488-conjugated
goat antibodies against mouse IgG for detection of cells that
expressed Vpr. Then we stained cells with PI for analysis of
DNA content (Fig. 2). Flow cytometric analysis revealed
that, in the case of cells transfected with the expression
vector that encoded wild-type Vpr, there was a dramatic
increase in the proportion of cells in the G2 phase of the cell
cycle (approximately 21.5% and 68.3% were at the G1 and
the G2/M phase, respectively, and the G2/M:G1 ratio was
2.31) compared with cells transfected with the control
vector pME18Neo-Flag (approximately 68.5% and 8.4%
were at the G1 and G2 phase, respectively, and the G2/M:G1
ratio was 0.13). Similarly, approximately 69.9% and 65.8%
of HeLa cells that expressed the mutant proteins L67A and
HI4546WA, respectively, were arrested at the G2 phase. By
contrast, cells that expressed the mutant protein R8788EA
had a reduced ability to induce G2 arrest (approximately
46.2% of cells were in G2 phase) as compared to the wild-
type Vpr.
The Vpr protein can inhibit cell proliferation by arresting
the cell cycle at the G2 phase and then it can induce
apoptosis after G2 arrest (Stewart et al., 1997). On the other
hand, we previously found that expression of endogenous
Vpr appears also to cause apoptosis independently of its role
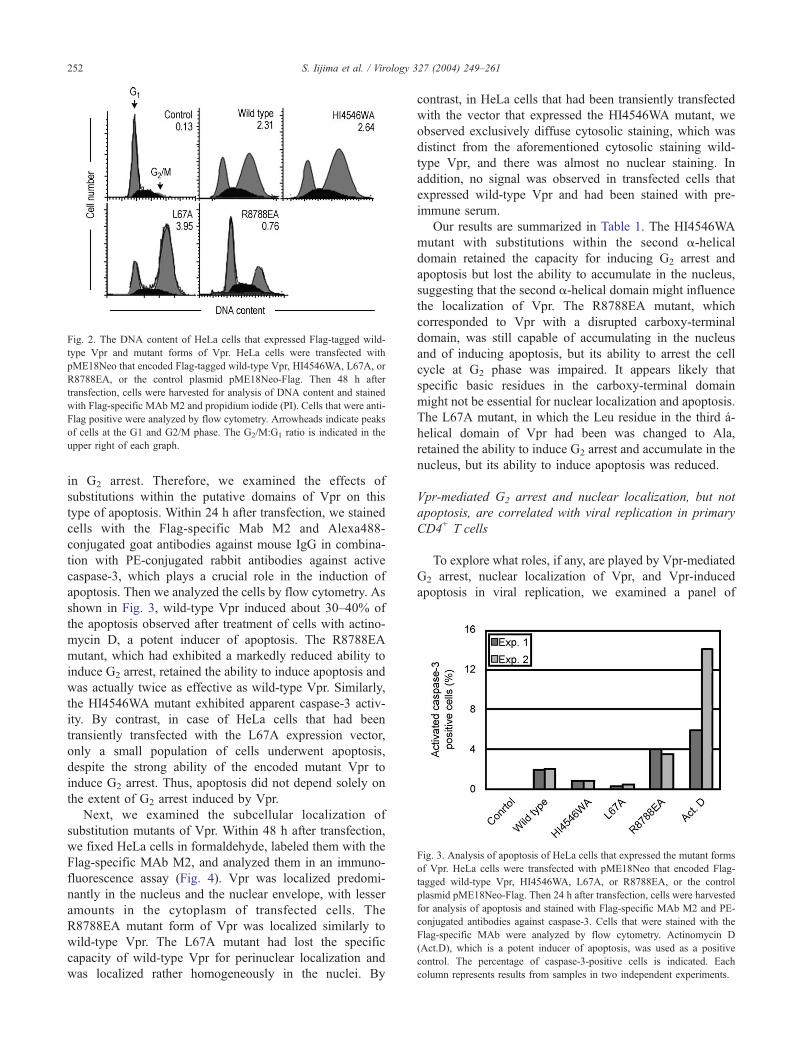
Fig. 2. The DNA content of HeLa cells that expressed Flag-tagged wild-
type Vpr and mutant forms of Vpr. HeLa cells were transfected with
pME18Neo that encoded Flag-tagged wild-type Vpr, HI4546WA, L67A, or
R8788EA, or the control plasmid pME18Neo-Flag. Then 48 h after
transfection, cells were harvested for analysis of DNA content and stained
with Flag-specific MAb M2 and propidium iodide (PI). Cells that were anti-
Flag positive were analyzed by flow cytometry. Arrowheads indicate peaks
of cells at the G1 and G2/M phase. The G2/M:G1 ratio is indicated in the
upper right of each graph.
Fig. 3. Analysis of apoptosis of HeLa cells that expressed the mutant forms
of Vpr. HeLa cells were transfected with pME18Neo that encoded Flag-
tagged wild-type Vpr, HI4546WA, L67A, or R8788EA, or the control
plasmid pME18Neo-Flag. Then 24 h after transfection, cells were harvested
for analysis of apoptosis and stained with Flag-specific MAb M2 and PE-
conjugated antibodies against caspase-3. Cells that were stained with the
Flag-specific MAb were analyzed by flow cytometry. Actinomycin D
(Act.D), which is a potent inducer of apoptosis, was used as a positive
control. The percentage of caspase-3-positive cells is indicated. Each
column represents results from samples in two independent experiments.
S. Iijima et al. / Virology 327 (2004) 249–261252
in G2 arrest. Therefore, we examined the effects of
substitutions within the putative domains of Vpr on this
type of apoptosis. Within 24 h after transfection, we stained
cells with the Flag-specific Mab M2 and Alexa488-
conjugated goat antibodies against mouse IgG in combina-
tion with PE-conjugated rabbit antibodies against active
caspase-3, which plays a crucial role in the induction of
apoptosis. Then we analyzed the cells by flow cytometry. As
shown in Fig. 3, wild-type Vpr induced about 30–40% of
the apoptosis observed after treatment of cells with actino-
mycin D, a potent inducer of apoptosis. The R8788EA
mutant, which had exhibited a markedly reduced ability to
induce G2 arrest, retained the ability to induce apoptosis and
was actually twice as effective as wild-type Vpr. Similarly,
the HI4546WA mutant exhibited apparent caspase-3 activ-
ity. By contrast, in case of HeLa cells that had been
transiently transfected with the L67A expression vector,
only a small population of cells underwent apoptosis,
despite the strong ability of the encoded mutant Vpr to
induce G2 arrest. Thus, apoptosis did not depend solely on
the extent of G2 arrest induced by Vpr.
Next, we examined the subcellular localization of
substitution mutants of Vpr. Within 48 h after transfection,
we fixed HeLa cells in formaldehyde, labeled them with the
Flag-specific MAb M2, and analyzed them in an immuno-
fluorescence assay (Fig. 4). Vpr was localized predomi-
nantly in the nucleus and the nuclear envelope, with lesser
amounts in the cytoplasm of transfected cells. The
R8788EA mutant form of Vpr was localized similarly to
wild-type Vpr. The L67A mutant had lost the specific
capacity of wild-type Vpr for perinuclear localization and
was localized rather homogeneously in the nuclei. By
contrast, in HeLa cells that had been transiently transfected
with the vector that expressed the HI4546WA mutant, we
observed exclusively diffuse cytosolic staining, which was
distinct from the aforementioned cytosolic staining wild-
type Vpr, and there was almost no nuclear staining. In
addition, no signal was observed in transfected cells that
expressed wild-type Vpr and had been stained with pre-
immune serum.
Our results are summarized in Table 1. The HI4546WA
mutant with substitutions within the second a-helical
domain retained the capacity for inducing G2 arrest and
apoptosis but lost the ability to accumulate in the nucleus,
suggesting that the second a-helical domain might influence
the localization of Vpr. The R8788EA mutant, which
corresponded to Vpr with a disrupted carboxy-terminal
domain, was still capable of accumulating in the nucleus
and of inducing apoptosis, but its ability to arrest the cell
cycle at G2 phase was impaired. It appears likely that
specific basic residues in the carboxy-terminal domain
might not be essential for nuclear localization and apoptosis.
The L67A mutant, in which the Leu residue in the third a-
helical domain of Vpr had been was changed to Ala,
retained the ability to induce G2 arrest and accumulate in the
nucleus, but its ability to induce apoptosis was reduced.
Vpr-mediated G2 arrest and nuclear localization, but not
apoptosis, are correlated with viral replication in primary
CD4+ T cells
To explore what roles, if any, are played by Vpr-mediated
G2 arrest, nuclear localization of Vpr, and Vpr-induced
apoptosis in viral replication, we examined a panel of

Fig. 4. Subcellular localization of mutant forms of Vpr. HeLa cells were transfected with pME18Neo that encoded Flag-tagged wild-type Vpr, HI4546WA,
L67A, or R8788EA, or with the control plasmid pME18Neo-Flag. Then 24 h after transfection, cells were subjected to immunofluorescence staining with Flag-
specific MAb M2 and Alexa488-conjugated goat antibodies against mouse IgG and analyzed by confocal laser scanning microscopy. Scale bar, 20 Am.
S. Iijima et al. / Virology 327 (2004) 249–261 253
viruses that encoded Vpr and its mutant, derivatives such as
L67A, R8788EA, and HI4546WA. We compared the
replication of a virus coding for a wild-type Vpr and of
the a Vpr-negative ATG-mutant (DVpr) to that of viruses
that encoded substitution mutants of Vpr using activated
primary CD4+ T cells from three donors. At 3- to 4-day
intervals, we collected culture supernatants and evaluated
the kinetics of virus production as by quantitation of the p24
antigen by an ELISA. Typical kinetics of replication of the
various viruses in activated primary CD4+ T cells from one
donor (TM) are shown in Fig. 5. The virus that encoded
wild-type Vpr was apparently infectious at low (0.5 ng of
p24 antigen) and high (5 ng of p24 antigen) viral inputs, and
replication reached a maximum 21 days after infection and
then decreased. Some delay in the production of viruses was
observed in cultures after infection of cells with viruses that
encoded Vpr+ and DVpr. In the case of viruses that encoded
Table 1
Characterization of Vpr and mutants
Mutant or virus G2 arrest Apoptosis Localization
Vpr +++ +++ WTb
HI4546WA +++ ++ Cytoplasm
L67A +++ Fc Most at the nuclear envelope
R8788EA F +++ WT
DVprc NTd NT NT
a The viruses containing an amount of 0.5 ng (low) and 5 ng (high) of p24 antigb Subcellular pattern of Vpr expression detected by immunofluorescent staining:
nuclear envelope and a certain amount was present in the cytoplasm.c Effect on activity compared to that of wild-type Vpr, which was taken as 100%; +d NT, not tested.
HI4546WA and R8788EA, respectively, a form of Vpr that
is unable to localize in the nucleus and a form that has a
reduced ability to induce G2 arrest, the kinetics of HIV-1
replication were similar to those of the DVpr virus. The
growth of these mutated viruses was severely impaired at a
low viral input as compared to that at a high viral input. By
contrast, the L67A mutant, which retained its ability to
induce G2 arrest and to localize in the nucleus but had lost
its ability to induce apoptosis, showed an increased rate of
viral replication. This result was reproducible and was
observed also in primary CD4+ T cells from the two other
donors.
Our results suggest that the ability of the Vpr protein to
localize in the nucleus and its ability to induce G2 arrest, but
not its ability to induce apoptosis, are important for viral
replication in primary CD4+ T cells at both of high and low
viral inputs (Table 1).
Infectivitya
High Low
After 14 days After 21 days After 14 days After 21 days
+++ +++ +++ +++
+ ++ + +
+++ +++ +++ +++
+ ++ + +
+ +++ + +
en were used.
bwtQ indicated wild-type pattern with intense expression in the nucleus and
++++, N200%; +++, 100–80%; ++, 79–50%; +, 49–10%;F, 9–1%;�, b1%.

Fig. 5. Kinetics of p24 antigen production after infection with wild-type and
mutant forms of Vpr. Activated CD4+ T cells were infected with wild-type
virus or with viruses that encoded mutant forms of Vpr. Results are shown
for infections that corresponded to 5 ng (A) and 0.5 ng (B) p24 antigen.
Cells were maintained for 4 weeks, and virus production was monitored in
terms of levels of p24 in the culture supernatant, as determined by an
ELISA every at 3- to 4-day intervals.
S. Iijima et al. / Virology 327 (2004) 249–261254
Mutant viruses in which Vpr has lost its ability of nuclear
import are unable to proliferate in primary CD4+ T cells
In previous studies, Vpr appeared to contribute substan-
tially to the replication of HIV-1 in proliferating T cells.
Viral replication was enhanced in dividing cells when Vpr
was present under certain conditions, such as when the
multiplicity of infection was very low (Cohen et al., 1990b,
Yao et al., 1998). A previous report by Goh et al. (1998)
indicated that Vpr increases virus production by delaying
cells at the G2 phase of the cell cycle, when the LTR is most
active. In the present study, we found that the ability of Vpr
to accumulate in the nucleus, as well as its ability to induce
G2 arrest, might contribute to the efficient replication of
HIV-1 in primary CD4+ T cells, as shown in Table 1 and
Fig. 5. However, the extent to which Vpr contributes to the
nuclear import of proviral DNA during viral infection in
primary CD4+ T cells remains to be clarified.
To examine whether the nuclear import of Vpr is
promoted by a cytoplasmic extract from activated primary
CD4+ T cells, we performed an assay in vitro in which we
monitored the nuclear import of Vpr in digitonin-permea-
bilized HeLa cells (Fig. 6). As reported previously, the
region between residues 17 and 74 (N17C74), which is the
actual NLS of Vpr, appears to be indispensable for the
nuclear translocation of wild-type Vpr (Kamata and Aida,
2000). Indeed, using a chimeric protein that consisted of the
region between residues 17 and 74, designated N17C74,
fused at its amino-terminal end to GST and at its carboxy-
terminal end to GFP, we were able to confirm the subcellular
localization of the chimeric protein in a nuclear import assay
in vitro (Kamata et al., unpublished data). As shown in Figs.
6A and B, nuclear import of N17C74 was rescued upon
addition to the in vitro system of a cytoplasmic extract of
HeLa cells and of primary CD4+ T cells.
Next, in addition to the HI4546WA mutant, which had
lost the ability to localize in the nucleus in HeLa cells, as
described above, we selected two mutant forms of Vpr,
namely, L67P and aLAL67P that localize in the cytoplasm
of HeLa cells that had been transfected with expression
vectors that encoded these mutants as shown Figs. 1B and
6C. Moreover, these two mutants retained weak ability to
induce G2 arrest and apoptosis (data not shown). First, we
constructed plasmids for the expression L67PN17C74,
aLAL67PN17C74, and HI4546WAN17C74 as chimeric pro-
teins in which Vpr sequences were fused to GFP and GST,
and then equal amounts of recombinant protein used in a
nuclear import assay in vitro (Figs. 6A and B). We also
compared the replication of viruses that encoded for
HI4546WA, L67P, and aLAL67P in stimulated primary
CD4+ T cells from three donors (Fig. 6E). Interestingly,
nuclear import of HI4546WAN17C74 was rescued by
cytoplasmic extracts of primary CD4+ T cells but not of
HeLa cells, and the virus that encoded HI4546WA
replicated efficiently in activated primary CD4+ T cells.
These results indicate that a cellular factor, present in
stimulated primary CD4+ T cells but not in HeLa cells, can
support the nuclear entry of Vpr and, also, viral replication.
Indeed, in Fig. 6D, immunofluorescence staining revealed
the HI4546WA mutant yielded intense signals in the nuclear
in human lymphoid Jurkat. By contrast, the L67PN17C74 and
aLAL67PN17C74 proteins were unable to enter nucleus even
in the presence of cytoplasmic extracts of HeLa cells or of
primary CD4+ T cells. Moreover, the viruses encoding these
mutant proteins were unable to proliferate in primary CD4+
T cells (Fig. 6E).
As summarized in Table 2, our results indicate that the
viral phenotype with respect to the nuclear import of Vpr
closely correlates with infection by HIV-1 in primary CD4+
T cells.
Discussion
The result of the present study of mutant forms of Vpr
leads to four major conclusions. First, the results suggest the
importance of Vpr in the replication of HIV-1 in primary
CD4+ T cells at both low and high viral inputs. At low viral
input, in particular, the growth of virus that encoded the
Vpr-negative ATG mutant (DVpr) was considerably
impaired as compared to that of the wild-type virus. This
result supports previous observations that viral replication is
enhanced in proliferating T cells and primary PBMC when
Vpr is present at very low viral input (Balliet et al., 1994;
Connor et al., 1995; Ogawa et al., 1989). At high viral input
(5 ng), we found that the replication of DVpr virus was
significantly less effective than that of the wild-type virus in
primary CD4+ T cells during infection for 14 days. Second,
our results show that the phenotype with respect to nuclear
import of Vpr is closely correlated with HIV-1 infection in
primary CD4+ T cells. Moreover, viruses that encoded
mutant forms of Vpr that were unable to enter the nucleus
upon addition of a cytoplasmic extract of activated primary
CD4+ T cells were also unable to proliferate in stimulated

Fig. 6. Nuclear import and kinetics of viral replication. (A) Coomassie blue-stained gel of GST- and GFP-tagged N17C74 and substitution mutants of Vpr. (B)
In vitro nuclear import assay of the mutant forms of Vpr. Scale bar, 20 Am. (C and D) Subcellular localization of mutant forms of Vpr in CD4+ T cells and HeLa
cells (C) and Jurkat cells (D). Cells were transfected with pME18Neo that encoded Flag-tagged wild-type Vpr, L67P, aLAL67P, HI4546WA. Then 24 h after
transfection, cells were subjected to immunofluorescence staining with Flag-specific MAb M2 and Alexa488-conjugated goat antibodies against mouse IgG
and analyzed by confocal laser scanning microscopy. Scale bar, 20 Am. (E) Kinetics of viral replication. Activated CD4+ T cells were infected with viruses that
encoded wild-type or mutant Vpr equivalent to 0.5 ng of p24 antigen. The production of viruses was monitored in terms of levels of p24 in the culture
supernatant determined by ELISA at 3- or 4-day intervals for 3 weeks.
S. Iijima et al. / Virology 327 (2004) 249–261 255
primary CD4+ T cells. Thus, the nuclear import of Vpr
might be a good target in attempts to block early events in
the replication of HIV-1. Third, our data revealed that the
ability of Vpr to induce G2 arrest might be important for
viral replication in primary CD4+ T cells because replication
of the virus that encoded R8788EA, which had reduced
ability to induce G2 arrest, was clearly impaired. This
observation strongly supports the results of Cohen et al.
(1990b), Goh et al. (1998), and Yao et al. (1998), who
reported that Vpr appeared to make a substantial contribu-
tion to the replication of HIV-1 in proliferating T cells. They
found that viral replication was enhanced in dividing cells
when Vpr was present under certain conditions, such as
when the multiplicity of infection was very low. In
particular, Goh et al. (1998) indicated that the G2 arrest
induced by Vpr contributed to the enhanced replication of
HIV-1 in CD4+ T cells, suggesting that this phenomenon
might explain why Vpr is so strongly conserved in primate
lentiviruses. Fourth, our results demonstrate that any cellular
factors in stimulated primary CD4+ T cells are indispensable
for nuclear entry of Vpr into the nucleus and for viral
replication. Although the Vpr mutant HI4546WA exhibited
diffuse cytosolic staining exclusively in an immunofluor-
escence assay of HeLa cells, this mutant protein yielded
intense signals in the nucleus in Jurkat cells, its nuclear
import was effectively restored by cytoplasmic extract of
primary CD4+ T cells, and a virus that encoded this mutant
Vpr was able to infect primary CD4+ T cells. This

Table 2
Correlation between nuclear import of wild-type or mutant Vpr in vitro and infectivity
Virus Localization Nuclear import with extract from Infectivitya
HeLa cells Jurkat cells HeLa cells CD4+ T cells
Vpr Nucleus Nucleus ++b + +++
HI4546WA Cytoplasm Nucleus F ++ ++
L67Pc Nucleus b Cytoplasm NTd � � FaLAL67Pc Cytoplasm NT � � F
a The viruses containing an amount of 0.5 ng (low) of p24 antigen were used.b Extent of activity; �, negative; F, weakly positive; +, positive; ++, strongly positive.c The L67P and aLAL67P mutants exhibited week abilities to induce G2 arrest and apoptosis.d NT, not tested.
S. Iijima et al. / Virology 327 (2004) 249–261256
observation suggests that host proteins might play key roles
in the life cycle of HIV-1.
In the viral replication assay in primary CD4+ T cells,
the viruses that encoded the mutant forms of Vpr that had
lost the ability of nuclear import, namely, aLAL67P and
L67P exhibited significantly reduced rate of replication.
The extent of such reductions was greater than observed
for the DVpr virus. The structure of Vpr is characterized
by three a-helices, which are folded around a hydrophobic
core that consists of Leu, Ile, Val, and aromatic residues,
and this structure might be related to the interactions of
Vpr with different targets (Morellet et al., 2003). Several
cellular proteins have been reported to associate with Vpr,
such as a 41-kDa cytosolic protein that forms a complex
with the glucocorticoid receptor protein (Refaeli et al.,
1995; Zhao et al., 1994), an unidentified 180-kDa protein
(Refaeli et al., 1995), Sp1 (Wang et al., 1995), TFIIB
(Agostini et al., 1996), uracil DNA glycosylase (Bouham-
dan et al., 1996), HHR23A (Withers-Ward et al., 1997), p53
(Sawaya et al., 1998), and a human 34-kDa homolog of
mov34 (Mahalingam et al., 1998). It was also suggested
recently that Vpr binds importin-a, which is known as a
classical NLS (Agostini et al., 2000; Popov et al., 1998a,
1998b), in addition to phenylalanine-glycine repeat (FG-
repeat) nucleoporins, which are components of the nuclear
pore complex (Fouchier et al., 1998; Le Rouzic et al., 2002;
Popov et al., 1998a; Vodicka et al., 1998). A critical step in
the nuclear transport process is the recognition of the NLS
by the importin a/h complex followed by the docking of the
NLS-containing importin a/h complex to nucleoporins that
are residents of nuclear pores. These results suggest that,
because mutations at positions 20, 22, 23, and 26 within the
first a-helical domain or at position 67 within the third a-
helical domain, aLAL67P and L67P might lose the ability
to interact with the cellular nuclear transport machinery.
This interference might be exerted through steric hindrance
of the Vpr domain involved in interaction with the cellular
factor nuclear transport machinery. By contrast, Zhao et al.
(1994) and Zhang et al. (2001) identified a cellular protein
of 180 kDa (VprBP) that interacted with Vpr specifically in
co-immunoprecipitation assays and they obtained experi-
mental evidence that recombinant VprBP blocks the nuclear
import of Vpr. Thus, a human cytoplasmic protein VprBP
might help retain Vpr in the cytoplasm and might induce
changes in cellular functions as well as in the life cycle of
HIV-1. It is possible that aLAL67P and L67P are able to
interact with factors that block the nuclear transport of these
mutants and do not interact with wild-type Vpr because the
viruses that encoded these mutant proteins replicated
significantly less efficiently than DVpr virus. Therefore,
our results suggest that cellular factors carrying key roles in
nuclear import of Vpr might be a good target in efforts to
block the early phase of the HIV-1 life cycle. By contrast,
HI4546WA was found almost exclusively in the cytoplasm
in our immunofluorescence assay of HeLa cells but
imported at a high level in the nucleus upon addition of
cytosol from primary CD4+ T cells, but not from HeLa cells,
and the virus that encoded HI4546WA successfully infected
primary CD4+ T cells. This observation in fact indicates that
the nuclear entry of Vpr may be dependent on cell-type-
specific factors. Further studies are required to define the
cellular factors that interact with Vpr and are involved in the
targeting of the PIC to the nucleus.
Future therapies will likely target viral proteins other
than the reverse transcriptase, protease, and integrase, or
select host proteins that have key roles in the HIV life
cycles. Indeed, small chemicals already exist that block Tat
transactivation (Lind et al., 2002) and Rev-dependent
export of viral transcripts from the nucleus to the cytoplasm
(Chao et al., 2000). Moreover, as a proof of principle,
dominant-negative mutant Tat, Rev, and Gag proteins have
been shown to block viral replication. The present study
suggests that it might be possible to exploit our Vpr mutants
as dominant-negative inhibitors of infection by HIV-1 and
that it might also be possible to target Vpr in the treatment
of AIDS.
Materials and methods
Construction of plasmids and infectious molecular clones
Generation of expression vectors, based on pME18Neo,
which encoded Flag-tagged wild-type Vpr; the substitution

S. Iijima et al. / Virology 327 (2004) 249–261 257
mutants of Vpr, designated L67A, aLAL67P, and L67P; and
the control plasmid pME18Neo-Flag, was described pre-
viously (Kamata and Aida, 2000; Nishino et al., 1997;
Nishizawa et al., 1999, 2000a, 2000b). To generate the
substitution mutants designated R8788EA and HI4546WA,
we introduced site-specific mutations into pSK-Fvpr
(Tajima et al., 1998) by ExSite PCR-based site-directed
mutagenesis using a kit from Stratagene (La Jolla, CA) and
the following primers: 5V-AGGGCAGCAAGAAATG-
GAGCCAG-3Vand 5V-CTGTCGAGTAACGCCTATTC-
TGC-3Vfor R8788EA; and 5V-TATGAAACTTACGGGGA-TACTTGGG-3Vand 5V-CGCCCATTGTCCTAAGTTATGG-3Vfor HI4546WA. Each XhoI–NotI fragment, including
mutated vpr and Flag sequences, in pSK-Fvpr was excised
and subcloned into pME18Neo (Tajima et al., 1998).
To generate the infectious molecular clone HIV-1
pNL432 (Adachi et al., 1986), parent clones that encoded
for the Vpr-negative ATG-mutant (DVpr), and clones that
encoded substitution mutants of Vpr designated L67A,
HI4546WA, L67P, and aLA, we introduced site-specific
mutations into pUC19 included the NdeI–SalI (NdeI site at
5122 and SalI site at 5785) fragment of the infectious
molecular clone HIV-1 NF462 (Kawamura et al., 1994; a
kind gift from A. Adachi, Tokushima University, Japan) by
PCR with the following primers: 5V-GAACAAGCCCCAG-AAGACCAAGG-3V and 5V-ACCTGTCCTCTGTC-
AGTTTCCT-3Vfor DVpr; 5V-AAGCACTGTTTATCCATTT-CAGA-3V and 5V-GTTGCAGAATTCTTATTATGG-3VforL67A; 5V-TATGAAACTTACGGGGATACTTGGG-3V and
5V-CGCCCATTGTCCTAAGTTATGG-3V for HI4546WA;
5V-CAACCCCTGTTTATCCATTTC-3V and 5V-TTGCA-
GAATTCTTATTATGGCTTCC-3V for L67P; and 5V-CCGAGGAAGCCAAGAGTGAAGCTGTTAGA-3V and5V-CCGCCTCGGCTGTCCATTCATTGTATGGCTCC - 3Vfor aLA. Furthermore, to generate the pNL432 parent clone
that encoded for the substitution mutant aLAL67P, we
introduced a site-specific mutation into pUC19 that included
the construct for aLA, described above, by PCR with
primers 5V-CAACCCCTGTTTATCCATTTC-3Vand 5V-TTGCAGAATTCTTATTATGGCTTCC-3V. Each PflMI and
SalI fragment, including mutations, in pUC19 was excised
and subcloned into pSK that included the NdeI–SalI (NdeI
site at 5122 and SalI site at 5785) fragment of pNL432
(Adachi et al., 1986). To generate the pNL432 parent clone
that encoded the substitution mutant R8788EA, we intro-
duced site-specific mutations into the construct for aLA,
described above, with primers 5V-AGGGCAGCAA-
GAAATGGAGCCAG-3Vand 5V-CTGTCGAGTAACGCC-TATTCTGC-3V. The EcoRI–NdeI fragment, including
R8788EA, in pUC19 was excised and subcloned into pSK
which included the EcoRI–XhoI (EcoRI site at 5743 and
XhoI site at 8887) fragment of pNL432 (Adachi et al., 1986).
The EcoRI–XhoI fragment in pSK, in which included
R8788EA, was subcloned into pNL432.
For constructions of GST-N17C74-GFP, GST-
L67PN17C74-GFP, and GST-aLAL67PN17C74-GFP, fragments
encoding each sequence were obtained by PCR with 5V-GCGGATATCCGAATGGACACTAGAG-3Vand 5V-CGCGGATCCCCAATTCTGAAA-3Vusing pSK-Fvpr,
pSK-FL67P, and pSK-FaLAL67P (Kamata and Aida,
2000; Nishizawa et al., 1999) as templates, respectively.
For constructions of GST-HI4546WAN17C74-GFP, the frag-
ment of interest was obtained by PCRwith 5V-TATGAAACT-TACGGGGATACTTGGG-3Vand 5V-CGCCCATTGTCCTA-AGTTATGG-3V using pSK-FHI4546WA as templates. The
fragment was then subcloned into pGEX-6P3 at the BamH1
and Not1 sites.
All constructs described above were verified by nucleo-
tide sequencing with a BigDye Terminator Cycle Sequenc-
ing Kit and a Genetic Analyzer (ABI PRISM 310; PF
Applied Biosystems, Norwalk, Conn.).
Cell culture and transfection
Human cervical HeLa cells, African green monkey
COS-1 cells, and human T-lymphoid cell line Jurkat were
maintained in RPMI 1640 medium supplemented with 2–
10% heat-inactivated fetal calf serum (FCS), penicillin
(100 Ag/ml), and streptomycin (100 Ag/ml). For Western
blotting, immunofluorescence staining, and examinations
of apoptosis and the cell cycle, HeLa cells (1 � 107) were
transfected with 20 Ag of expression vector by electro-
poration in a 0.4-cm diameter cuvette using a gene pulsar
(Bio-Rad, Richmond, CA) operated at 300 V and 975 AF.COS-1 cells were used for generation of viral stocks.
Western blotting
Twenty-four hours after transfection, the expression of
Vpr was examined by Western blotting as described
previously (Nishizawa et al., 2000b).
Immunofluorescence assay
Twenty-four hours after transfection, HeLa cells growing
on coverslips were fixed in 1% formaldehyde-phosphate-
buffered saline (PBS) for 1 h at 4 8C, permeabilized for 10
min in PBS that contained 0.2% Triton X-100, and washed
twice with PBS. The cells were incubated for 1 h at 4 8C with
Flag-specific MAb M2 (Eastman Kodak, Rochester, N.Y.) or
normal mouse immunoglobulin G (IgG). After washing with
PBS, the cells were incubated for 45 min at 4 8C with
Alexa488-conjugated goat against mouse IgG (Molecular
Probes, Eugene, OR). Coverslips were washed with PBS and
mounted on glass slides. Immunofluorescence was visual-
ized with a confocal laser scanning microscope (LSM510;
Carl Zeiss, Jena, Germany).
Analysis of the cell cycle
Forty-eight hours after transfection, cells were harvested,
fixed in 1% formaldehyde and 70% ethanol, and then

S. Iijima et al. / Virology 327 (2004) 249–261258
incubated for 1 h with the Flag-specific MAb M2 (Eastman
Kodak). After washing with PBS, cells were incubated for 45
min with Alexa488-conjugated goat against mouse IgG
(Molecular Probes). Then cells were washed again with PBS,
incubated in PBS that contained propidium iodide (PI; 50 Ag/ml), RNaseA (50 Ag/ml), and FCS (2%, v/v) for 15 min at 37
8C. The fluorescence of 10000 cells was analyzed on a
FACScan system (Becton-Dickinson, Mountain View, CA)
with Cell Quest software (Becton-Dickinson). Data are
presented after gating to eliminate cells in which Alexa did
not emit strong fluorescence. Relative numbers of cells in the
G2/M phase were calculated with ModFit LT software
(Verity Software House, Topsham, ME).
Analysis of apoptosis
To analyze apoptosis, we used flow cytometry and two-
color immunofluorescence staining caspase-3 activity was
evaluated as follows. Twenty-four hours after transfection
of cells with 20 Ag of Vpr expression and mutant
expression plasmids, HeLa cells were harvested, fixed in
1% formaldehyde and 70% ethanol, and then incubated for
1 h with the anti-Flag-specific MAb M2 (Eastman Kodak).
After washing with PBS, cells were incubated for 45 min at
4 8C with Alexa488-conjugated goat antibodies against
mouse IgG (Molecular Probes). After cells had been
washed again with PBS, they were incubated for 1 h at 4
8C with phycoerythrin (PE)-conjugated rabbit antibodies
against active caspase-3 (BD Biosciences, Franklin Lakes,
NJ). The fluorescence of 10000 cells was analyzed on the
FACScan system (Becton-Dickinson). Data are presented
after gating to eliminate cells in which Alexa did not emit
strong fluorescence. Relative numbers of positive cells
were calculated as percentages.
Generation of virus stocks
We used two methods for the generation of virus
stocks, as follows. We performed transfections using
FuGENE 6 reagent (Roche, Indianapolis, IN) initially
according to the manufacture’s instructions and then, in
later experiments, under optimized conditions. Then the
DNA was diluted 1:10 (w/v) with serum-free Dulbecco’s
modified Eagle medium (GIBCO Laboratories, Grand
Island, N.Y.). And FuGENE 6 reagent (Roche) was
diluted 1:2 with 25 mM HEPES and incubated at room
temperature for 5 min. The mixtures were combined and
incubated together for a minimum of 15 min at room
temperature. The complex DNA was added to a culture of
cells in fresh medium, then COS-1 cells were incubated at
37 8C in an atmosphere of 5% CO2 in air for 4 h. Then
the medium was replaced by fresh medium and culture
was continued for 72 h. We introduced DNA by electro-
poration using COS-1 cells that had been washed once
with PBS and then resuspended at a concentration of 1 �107 cells/ml in 1� K-PBS-buffered media [10� K-PBS
buffer contained 308 mM NaCl, 1.207 mM KCl, 81 mM
Na2HPO4, 146 mM KH2PO42] plus 30 Ag/Al plasmid
DNA. Cells were transfected in a cuvette using the gene
pulsar (Bio-Rad), which was operated at 260 V and 970
AF. Then cells were cultured in fresh medium at 37 8C in
an atmosphere of 5% CO2 in air. Seventy-two hours after
introduction of DNA, culture supernatants were harvested
and filtered (pore size, 0.45 Am).
Titers of virus stocks were measured on the basis of the
amount of p24 antigen in each culture supernatants by an
enzyme-linked immunosorbent assay (ELISA) using a
combination of two antibodies, namely a p24-specific
MAb (Nu24) and peroxidase-labeled MAb10B5 (Tsunet-
sugu-Yokota et al., 1995).
Preparation of CD4+ T cells
We isolated PBMC on a Ficoll (lymphoceparll; IBL,
Hamburg, Germany) gradient from a healthy HIV-1-
seronegative donor. After PBMC had been collected, they
were washed twice, and then monocytes were eliminated
using a magnetic cell-separation system and microbeads
coated with a CD14-specific MAb (MACS system;
Militenyi Biotec, Bergisch Gladback, Germany). After
removal of monocytes, CD4+ T cells were isolated from
CD14� PBMC on a T-cell column, as described pre-
viously (Tsunetsugu-Yokota et al., 2003). Purified CD4+ T
cells (approximately 95–98% purity) were stimulated with
CD3-specific and CD28-specific MAbs in the presence of
IL-2 (20 U/ml; Genzyme, Cambridge, MA) and were
cultured at 37 8C in a humidified atmosphere of 5% CO2
in air.
Kinetics of virus production
CD4+ T cells were exposed to diluted virus stocks that
contained 0.5 or 5 ng of p24 antigen for 2 h at 37 8C.They were then washed three times and the infected cells
(5 � 105/well) were seeded in a 48-well tissue-culture
plate (Corning, Acton, MA). Cells were maintained in
RPMI 1640 that contained 10% FCS and IL2 (20 U/ml;
Genzyme). Culture supernatants were harvested at 3- or 4-
day intervals and viral production was monitored by
sequential quantitation of p24 antigen in cell-free super-
natants with an HIV-1 p24gag ELISA kit (RETRO TEC;
ZeptoMetrix, NY).
Expression and purification of Vpr and its derivatives
cDNAs encoding GST-N17C74-GFP, GST-L67PN17C74-
GFP, and GST-HI4546WA N17C74-GFP were cloned into
pGEX-6P3 (Amersham Biosciences, Uppsala, Sweden) at
the BamH1 and Not1 sites. The proteins were expressed in
Escherichia coli strains NovaBlue (Novagen). The cells
were cultured in 500 ml of Super Broth medium that
contained ampicillin at 37 8C with constant shaking (190

S. Iijima et al. / Virology 327 (2004) 249–261 259
rpm). When absorbance at 550 nm had reached 0.8, 0.1 mM
isopropyl-1-thio-h-d-galactopyranoside was added to the
medium at 16 8C. After overnight culture at 16 8C, cells werecollected by centrifugation, resuspended in sonication buffer
[50 mM Tris–HCl (pH 8.3), 500 mM NaCl, 1 mM DTT, 1
mM EDTA], and lysed by sonication. Each lysate was
centrifuged and GST-tagged proteins in the supernatant were
allowed to absorb to glutathione-Sepharose 4B (Amersham
Biosciences). The resin was washed with sonication buffer
and then proteins were eluted with 16 mM glutathione-
containing sonication buffer. The proteins were dialyzed
against transport buffer [20 mM HEPES-KOH (pH 7.3), 110
mM potassium acetate, 2 mM magnesium acetate, 5 mM
sodium acetate, 2 mM EGTA, 2 mM DTT] and then
concentrated in a Vivaspin (Sartorius AG, Goettingen,
Germany) centrifuge concentrator. The solution was applied
to a HiTrap Q FF column (Amersham Biosciences) and
eluted with gradient buffer [1 M NaCl in transport buffer].
Peak fractions containing individual proteins were pooled
and dialyzed against transport buffer. The purity of each
recombinant protein was checked by SDS-polyacrylamide
gel electrophoresis (SDS-PAGE) and preparations of pro-
teins were stored at �80 8C.
In vitro nuclear import assay
In vitro nuclear import assay was performed based on a
previously described method (Adam et al., 1990). Cyto-
plasmic extracts of HeLa cells and activated CD4+T cells
were prepared by lysis of cells in cold, hypotonic buffer
[10 mM Tris–HCl (pH 7.5), 2 mM MgCl2, 3 mM CaCl2,
0.3 M sucrose, 1 mM dithiothreitol, 1 Ag/ml leupeptin, 1
Ag/ml aprotinin, and 1 mM phenylmethylsulfonylfluoride].
HeLa cells were permeabilized by treatment with 35 Ag/ml
digitonin (Fluka, Buchs SG, Switzerland) in transport
buffer on ice for 5 min. Adjusted to 40 Al reaction solution
with transport buffer were contained 0.7 AM GST- and
GFP-fusion protein plus in the presence of cytosol extract.
Cells were incubated in this reaction mixture at 29 8C for
20 min and reactions were stopped by fixation of cells with
1% formaldehyde in PBS for 30 min on ice. Cells have
been analyzed by confocal laser-scanning microscopy
(Radiance 2100; Bio-Rad, Hemel Hempsted, UK).
Acknowledgments
We thank Dr. A. Adachi (Tokushima University,
Tokushima, Japan) for kindly providing HIV-1 pNL432
and pNF462. This study was supported in part by a grant
for AIDS Research from the Japan Health Sciences
Foundation; by a Health Sciences Research Grant from
the Ministry of Hearth, Labour and Welfare of Japan
(Research on HIV/AIDS 13110201 and 16150301), by a
grant from the Japanese Foundation for AIDS Prevention;
by a grant for AIDS Research from the Ministry and
Education, Science and Culture of Japan; and by a
President’s Special Research Grant from RIKEN.
References
Adachi, A., Gendelman, H.E., Koenig, S., Folks, T., Willey, R.,
Rabson, A., Martin, M.A., 1986. Production of acquired immuno-
deficiency syndrome-associated retrovirus in human and nonhuman
cells transfected with an infectious molecular clone. J. Virol. 59,
284–291.
Adam, S.A., Marr, R.S., Gerace, L., 1990. Nuclear protein import in
permeabilized mammalian cells requires soluble cytoplasmic factors.
J. Cell Biol. 111, 807–816.
Agostini, I., Navarro, J.M., Rey, F., Bouhamdan, M., Spire, B., Vigne, R.,
Sire, J., 1996. The human immunodeficiency virus type 1 Vpr
transactivator: cooperation with promoter-bound activator domains
and binding to TFIIB. J. Mol. Biol. 261, 599–606.
Agostini, I., Popov, S., Li, J., Dubrovsky, L., Hao, T., Bukrinsky, M., 2000.
Heat-shock protein 70 can replace viral protein R of HIV-1 during
nuclear import of the viral preintegration complex. Exp. Cell Res. 259,
398–403.
Balliet, J.W., Kolson, D.L., Eiger, G., Kim, F.M., McGann, K.A.,
Srinivasan, A., Collman, R., 1994. Distinct effects in primary macro-
phages and lymphocytes of the human immunodeficiency virus type 1
accessory genes vpr, vpu, and nef: mutational analysis of a primary
HIV-1 isolate. Virology 200, 623–631.
Bouhamdan, M., Benichou, S., Rey, F., Navarro, J.M., Agostini, I.,
Spire, B., Camonis, J., Slupphaug, G., Vigne, R., Benarous, R., Sire,
J., 1996. Human immunodeficiency virus type 1 Vpr protein binds
to the uracil DNA glycosylase DNA repair enzyme. J. Virol. 70,
697–704.
Bukrinsky, M.I., Haffar, O.K., 1998. HIV-1 nuclear import: matrix protein is
back on center stage, this time together with Vpr. Mol. Med. 4, 138–143.
Chao, S.H., Fujinaga, K., Marion, J.E., Taube, R., Sausville, E.A.,
Senderowicz, A.M., Peterlin, B.M., Price, D.H., 2000. Flavopiridol
inhibits P-TEFb and blocks HIV-1 replication. J. Biol. Chem. 275,
28345–28348.
Cohen, E.A., Dehni, G., Sodroski, J.G., Haseltine, W.A., 1990a. Human
immunodeficiency virus vpr product is a virion-associated regulatory
protein. J. Virol. 64, 3097–3099.
Cohen, E.A., Terwilliger, E.F., Jalinoos, Y., Proulx, J., Sodroski, J.G.,
Haseltine, W.A., 1990b. Identification of HIV-1 vpr product and
function. J. Acquired Immune Defic. Syndr. 3, 11–18.
Connor, R.I., Chen, B.K., Choe, S., Landau, N.R., 1995. Vpr is required for
efficient replication of human immunodeficiency virus type-1 in
mononuclear phagocytes. Virology 206, 935–944.
Di Marzio, P., Choe, S., Ebright, M., Knoblauch, R., Landau, N.R., 1995.
Mutational analysis of cell cycle arrest, nuclear localization and virion
packaging of human immunodeficiency virus type 1 Vpr. J. Virol. 69,
7909–7916.
Emerman, M.,1996.HIV-1,Vprandthecellcycle. Curr. Biol. 6, 1096–1103.
Fouchier, R.A., Meyer, B.E., Simon, J.H., Fischer, U., Albright, A.V.,
Gonzalez-Scarano, F., Malim, M.H., 1998. Interaction of the human
immunodeficiency virus type 1 Vpr protein with the nuclear pore
complex. J. Virol. 72, 6004–6013.
Gallay, P., Stitt, V., Mundy, C., Oettinger, M., Trono, D., 1996. Role of the
karyopherin pathway in human immunodeficiency virus type 1 nuclear
import. J. Virol. 70, 1027–1032.
Goh, W.C., Rogel, M.E., Kinsey, C.M., Michael, S.F., Fultz, P.N., Nowak,
M.A., Hahn, B.H., Emerman, M., 1998. HIV-1 Vpr increases viral
expression by manipulation of the cell cycle: a mechanism for selection
of Vpr in vivo. Nat. Med. 4, 65–71.
Hattori, N., Michaels, F., Fargnoli, K., Marcon, L., Gallo, R.C., Franchini,
G., 1990. The human immunodeficiency virus type 2 vpr gene is

S. Iijima et al. / Virology 327 (2004) 249–261260
essential for productive infection of human macrophages. Proc. Natl.
Acad. Sci. U.S.A. 87, 8080–8084.
He, J., Choe, S., Walker, R., Di Marzio, P., Morgan, D.O., Landau, N.R.,
1995. Human immunodeficiency virus type 1 viral protein R (Vpr)
arrests cells in the G2 phase of the cell cycle by inhibiting p34cdc2
activity. J. Virol. 69, 6705–6711.
Heinzinger, N.K., Bukinsky, M.I., Haggerty, S.A., Ragland, A.M.,
Kewalramani, V., Lee, M.A., Gendelman, H.E., Ratner, L., Stevenson,
M., Emerman, M., 1994. The Vpr protein of human immunodefi-
ciency virus type 1 influences nuclear localization of viral nucleic
acids in nondividing host cells. Proc. Natl. Acad. Sci. U.S.A. 91,
7311–7315.
Jenkins, Y., McEntee, M., Weis, K., Greene, W.C., 1998. Characterization
of HIV-1 Vpr nuclear import: analysis of signals and pathways. J. Cell
Biol. 143, 875–885.
Jowett, J.B., Planelles, V., Poon, B., Shah, N.P., Chen, M.L., Chen, I.S.,
1995. The human immunodeficiency virus type 1 vpr gene arrests
infected T cells in the G2 + M phase of the cell cycle. J. Virol. 69,
6304–6313.
Kamata, M., Aida, Y., 2000. Two putative alpha-helical domains of human
immunodeficiency virus type 1 Vpr mediate nuclear localization by at
least two mechanisms. J. Virol. 74, 7179–7186.
Kawamura, M., Ishizaki, T., Ishimoto, A., Shioda, T., Kitamura, T., Adachi,
A., 1994. Growth ability of human immunodeficiency virus type 1
auxiliary gene mutants in primary blood macrophage cultures. J. Gen.
Virol. 75, 2427–2431.
Le Rouzic, E., Mousnier, A., Rustum, C., Stutz, F., Hallberg, E.,
Dargemont, C., Benichou, S., 2002. Docking of HIV-1 Vpr to the
nuclear envelope is mediated by the interaction with the nucleoporin
hCG1. J. Biol. Chem. 277, 45091–45098.
Levy, D.N., Fernandes, L.S., Williams, W.V., Weiner, D.B., 1993. Induction
of cell differentiation by human immunodeficiency virus 1 vpr. Cell 72,
541–550.
Levy, D.N., Refaeli, Y., MacGregor, R.R., Weiner, D.B., 1994. Serum Vpr
regulates productive infection and latency of human immunodeficiency
virus type 1. Proc. Natl. Acad. Sci. U.S.A. 91, 10873–10877.
Lind, K.E., Du, Z., Fujinaga, K., Peterlin, B.M., James, T.L., 2002.
Structure-based computational database screening, in vitro assay, and
NMR assessment of compounds that target TAR RNA. Chem. Biol. 9,
185–193.
Lu, Y.L., Spearman, P., Ratner, L., 1993. Human immunodeficiency virus
type 1 viral protein R localization in infected cells and virions. J. Virol.
67, 6542–6550.
Mahalingam, S., Khan, S.A., Jabbar, M.A., Monken, C.E., Collman, R.G.,
Srinivasan, A., 1995a. Identification of residues in the N-terminal acidic
domain of HIV-1 Vpr essential for virion incorporation. Virology 207,
297–302.
Mahalingam, S., Khan, S.A., Murali, R., Jabbar, M.A., Monken, C.E.,
Collman, R.G., Srinivasan, A., 1995b. Mutagenesis of the putative
alpha-helical domain of the Vpr protein of human immunodeficiency
virus type 1: effect on stability and virion incorporation. Proc. Natl.
Acad. Sci. U.S.A. 92, 3794–3798.
Mahalingam, S., Collman, R.G., Patel, M., Monken, C.E., Srinivasan, A.,
1995c. Role of the conserved dipeptide Gly75 and Cys76 on HIV-1 Vpr
function. Virology 210, 495–500.
Mahalingam, S., Collman, R.G., Patel, M., Monken, C.E., Srinivasan, A.,
1995d. Functional analysis of HIV-1 Vpr: identification of determinants
essential for subcellular localization. Virology 212, 331–339.
Mahalingam, S., Ayyavoo, V., Patel, M., Kieber-Emmons, T., Weiner, D.B.,
1997. Nuclear import, virion incorporation, and cell cycle arrest/
differentiation are mediated by distinct functional domains of human
immunodeficiency virus type 1 Vpr. J. Virol. 71, 6339–6347.
Mahalingam, S., Ayyavoo, V., Patel, M., Kieber-Emmons, T., Kao, G.D.,
Muschel, R.J., Weiner, D.B., 1998. HIV-1 Vpr interacts with a human
34-kDa mov34 homologue, a cellular factor linked to the G2/M phase
transition of the mammalian cell cycle. Proc. Natl. Acad. Sci. U.S.A.
95, 3419–3424.
Morellet, N., Bouaziz, S., Petitjean, P., Roques, B.P., 2003. NMR
structure of the HIV-1 regulatory protein VPR. J. Mol. Biol. 327,
215–227.
Nie, Z., Bergeron, D., Subbramanian, R.A., Yao, X.J., Checroune, F.,
Rougeau, N., Cohen, E.A., 1998. The putative alpha helix 2 of human
immunodeficiency virus type 1 Vpr contains a determinant which is
responsible for the nuclear translocation of proviral DNA in growth-
arrested cells. J. Virol. 72, 4104–4115.
Nishino, Y., Myojin, T., Kamata, M., Aida, Y., 1997. Human immunode-
ficiency virus type 1 Vpr gene product prevents cell proliferation on
mouse NIH3T3 cells without the G2 arrest of the cell cycle. Biochem.
Biophys. Res. Commun. 232, 550–554.
Nishizawa, M., Myojin, T., Nishino, Y., Nakai, Y., Kamata, M., Aida, Y.,
1999. A carboxy-terminally truncated form of the Vpr protein of human
immunodeficiency virus type 1 retards cell proliferation independently
of G(2) arrest of the cell cycle. Virology 263, 313–322.
Nishizawa, M., Kamata, M., Katsumata, R., Aida, Y., 2000a. A carboxy-
terminally truncated form of the human immunodeficiency virus type 1
Vpr protein induces apoptosis via G(1) cell cycle arrest. J. Virol. 74,
6058–6067.
Nishizawa, M., Kamata, M., Myojin, T., Nakai, Y., Aida, Y., 2000b.
Induction of apoptosis by the Vpr protein of human immunodeficiency
virus type 1 occurs independently of G(2) arrest of the cell cycle.
Virology 276, 16–26.
Ogawa, K., Shibata, R., Kiyomasu, T., Higuchi, I., Kishida, Y., Ishimoto,
A., Adachi, A., 1989. Mutational analysis of the human immunodefi-
ciency virus vpr open reading frame. J. Virol. 63, 4110–4114.
Piller, S.C., Ewart, G.D., Premkumar, A., Cox, G.B., Gage, P.W., 1996.
Vpr protein of human immunodeficiency virus type 1 forms cation-
selective channels in planar lipid bilayers. Proc. Natl. Acad. Sci. U.S.A.
93, 111–115.
Planelles, V., Jowett, J.B., Li, Q.X., Xie, Y., Hahn, B., Chen, I.S., 1996.
Vpr-induced cell cycle arrest is conserved among primate lentiviruses.
J. Virol. 70, 2516–2524.
Popov, S., Rexach, M., Zybarth, G., Reiling, N., Lee, M.A., Ratner, L.,
Lane, C.M., Moore, M.S., Blobel, G., Bukrinsky, M., 1998a. Viral
protein R regulates nuclear import of the HIV-1 pre-integration
complex. EMBO J. 17, 909–917.
Popov, S., Rexach, M., Ratner, L., Blobel, G., Bukrinsky, M., 1998b. Viral
protein R regulates docking of the HIV-1 preintegration complex to the
nuclear pore complex. J. Biol. Chem. 273, 13347–13352.
Re, F., Braaten, D., Franke, E.K., Luban, J., 1995. Human immunodefi-
ciency virus type 1 Vpr arrests the cell cycle in G2 by inhibiting the
activation of p34cdc2-cyclin B. J. Virol. 69, 6859–6864.
Refaeli, Y., Levy, D.N., Weiner, D.B., 1995. The glucocorticoid receptor
type II complex is a target of the HIV-1 vpr gene product. Proc. Natl.
Acad. Sci. U.S.A. 92, 3621–3625.
Rogel, M.E., Wu, L.I., Emerman, M., 1995. The human immunodeficiency
virus type 1 vpr gene prevents cell proliferation during chronic
infection. J. Virol. 69, 882–888.
Sawaya, B.E., Khalili, K., Mercer, W.E., Denisova, L., Amini, S., 1998.
Cooperative actions of HIV-1 Vpr and p53 modulate viral gene
transcription. J. Biol. Chem. 273, 20052–20057.
Schuler, W., Wecker, K., de Rocquigny, H., Baudat, Y., Sire, J., Roques,
B.P., 1999. NMR structure of the (52–96) C-terminal domain of the
HIV-1 regulatory protein Vpr: molecular insights into its biological
functions. J. Mol. Biol. 285, 2105–2117.
Singh, S.P., Tomkowicz, B., Lai, D., Cartas, M., Mahalingam, S.,
Kalyanaraman, V.S., Murali, R., Srinivasan, A., 2000. Functional
role of residues corresponding to helical domain II (amino acids 35
to 46) of human immunodeficiency virus type 1 Vpr. J. Virol. 74,
10650–10657.
Stewart, S.A., Poon, B., Jowett, J.B., Chen, I.S., 1997. Human immuno-
deficiency virus type 1 Vpr induces apoptosis following cell cycle
arrest. J. Virol. 71, 5579–5592.
Tajima, S., Zhuang, W.Z., Kato, M.V., Okada, K., Ikawa, Y., Aida, Y.,
1998. Function and conformation of wild-type p53 protein are

S. Iijima et al. / Virology 327 (2004) 249–261 261
influenced by mutations in bovine leukemia virus-induced B-cell
lymphosarcoma. Virology 243, 735–746.
Tsunetsugu-Yokota, Y., Akagawa, K., Kimoto, H., Suzuki, K., Iwasaki, M.,
Yasuda, S., Hausser, G., Hultgren, C., Meyerhans, A., Takemori, T.,
1995.Monocyte-derived cultured dendritic cells are susceptible to human
immunodeficiency virus infection and transmit virus to resting T cells in
the process of nominal antigen presentation. J. Virol. 69, 4544–4547.
Tsunetsugu-Yokota, Y., Morikawa, Y., Isogai, M., Kawana-Tachikawa, A.,
Odawara, T., Nakamura, T., Grassi, F., Autran, B., Iwamoto, A., 2003.
Yeast-derived human immunodeficiency virus type 1 p55gag virus-like
particles activate dendritic cells (DCs) and induce perforin expression in
Gag-specific CD8+ T cells by cross-presentation of DCs. J. Virol. 77,
10250–10259.
Tung, H.Y., De Rocquigny, H., Zhao, L.J., Cayla, X., Roques, B.P., Ozon,
R., 1997. Direct activation of protein phosphatase-2A0 by HIV-1
encoded protein complex NCp7:vpr. FEBS Lett. 401, 197–201.
Vodicka, M.A., Koepp, D.M., Silver, P.A., Emerman, M., 1998. HIV-1 Vpr
interacts with the nuclear transport pathway to promote macrophage
infection. Genes Dev. 12, 175–185.
Wang, L., Mukherjee, S., Jia, F., Narayan, O., Zhao, L.J., 1995. Interaction
of virion protein Vpr of human immunodeficiency virus type 1 with
cellular transcription factor Sp1 and trans-activation of viral long
terminal repeat. J. Biol. Chem. 270, 25564–25569.
Wang, L., Mukherjee, S., Narayan, O., Zhao, L.J., 1996. Characterization of
a leucine-zipper-like domain in Vpr protein of human immunodefi-
ciency virus type 1. Gene 178, 7–13.
Withers-Ward, E.S., Jowett, J.B., Stewart, S.A., Xie, Y.M., Garfinkel, A.,
Shibagaki, Y., Chow, S.A., Shah, N., Hanaoka, F., Sawitz, D.G.,
Armstrong, R.W., Souza, L.M., Chen, I.S., 1997. Human immuno-
deficiency virus type 1 Vpr interacts with HHR23A, a cellular
protein implicated in nucleotide excision DNA repair. J. Virol. 71,
9732–9742.
Yao, X.J., Subbramanian, R.A., Rougeau, N., Boisvert, F., Bergeron, D.,
Cohen, E.A., 1995. Mutagenic analysis of human immunodeficiency
virus type 1 Vpr: role of a predicted N-terminal alpha-helical structure
in Vpr nuclear localization and virion incorporation. J. Virol. 69,
7032–7044.
Yao, X.J., Mouland, A.J., Subbramanian, R.A., Forget, J., Rougeau, N.,
Bergeron, D., Cohen, E.A., 1998. Vpr stimulates viral expression and
induces cell killing in human immunodeficiency virus type 1-infected
dividing Jurkat T cells. J. Virol. 72, 4686–4693.
Zhang, S., Feng, Y., Narayan, O., Zhao, L.J., 2001. Cytoplasmic
retention of HIV-1 regulatory protein Vpr by protein–protein
interaction with a novel human cytoplasmic protein VprBP. Gene
263, 131–140.
Zhao, L.J., Mukherjee, S., Narayan, O., 1994. Biochemical mechanism of
HIV-I Vpr function. Specific interaction with a cellular protein. J. Biol.
Chem. 269, 15577–15582.
Zhao, Y., Cao, J., O’Gorman, M.R., Yu, M., Yogev, R., 1996. Effect of
human immunodeficiency virus type 1 protein R (vpr) gene expression
on basic cellular function of fission yeast Schizosaccharomyces pombe.
J. Virol. 70, 5821–5826.
Zhao, Y., Yu, M., Chen, M., Elder, R.T., Yamamoto, A., Cao, J., 1998.
Pleiotropic effects of HIV-1 protein R (Vpr) on morphogenesis and cell
survival in fission yeast and antagonism by pentoxifylline. Virology
246, 266–276.




















