
The kinase activity of the Ser/Thr kinase BUB1 promotes TGF-β signaling

MOLECULAR AND CELLULAR BIOLOGY, Sept. 2011, p. 3515–3530 Vol. 31, No. 170270-7306/11/$12.00 doi:10.1128/MCB.05424-11Copyright © 2011, American Society for Microbiology. All Rights Reserved.
Nuclear Extracellular Signal-Regulated Kinase 1 and 2 TranslocationIs Mediated by Casein Kinase 2 and Accelerated
by Autophosphorylation�
Alexander Plotnikov,1 Dana Chuderland,1 Yael Karamansha,2 Oded Livnah,2 and Rony Seger1*Department of Biological Regulation, The Weizmann Institute of Science, Rehovot 76100, Israel,1 and Department of Biological
Chemistry and The Wolfson Centre for Applied Structural Biology, The Hebrew University, Jerusalem 91904, Israel2
Received 30 March 2011/Returned for modification 19 April 2011/Accepted 24 June 2011
The extracellular signal-regulated kinases (ERK) 1 and 2 (ERK1/2) are members of the mitogen-activatedprotein kinase [MAPK] family. Upon stimulation, these kinases translocate from the cytoplasm to the nucleus,where they induce physiological processes such as proliferation and differentiation. The mechanism of trans-location of this kinase involves phosphorylation of two Ser residues within a nuclear translocation signal(NTS), which allows binding to importin7 and a subsequent penetration via nuclear pores. Here we show thatthe phosphorylation of both Ser residues is mediated mainly by casein kinase 2 (CK2) and that active ERK mayassist in the phosphorylation of the N-terminal Ser. We also demonstrate that the phosphorylation is depen-dent on the release of ERK from cytoplasmic anchoring proteins. Crystal structure of the phosphomimeticERK revealed that the NTS phosphorylation creates an acidic patch in ERK. Our model is that in resting cellsERK is bound to cytoplasmic anchors, which prevent its NTS phosphorylation. Upon stimulation, phosphor-ylation of the ERK TEY domain releases ERK and allows phosphorylation of its NTS by CK2 and active ERKto generate a negatively charged patch in ERK, binding to importin 7 and nuclear translocation. These resultsprovide an important role of CK2 in regulating nuclear ERK activities.
Extracellular signal-regulated kinases (ERK) 1 and 2(ERK1/2) are central signaling proteins that mediate a varietyof vital cellular processes, including proliferation, survival, andeven apoptosis (1, 4, 15, 29, 50). In order to execute theirfunctions, ERK molecules activate a large number of regula-tory proteins, which are localized either in the cytoplasm orwithin various organelles, including mainly the nucleus (52).Indeed, the number of nuclear targets and downstream effec-tors of ERK, including a variety of transcription factors (17), iswell over 100. These direct and indirect targets participate inthe regulation of transcription as well as chromatin remodel-ing, and therefore they play a central role in mediating essen-tially all stimulated cellular processes (29). Moreover, becausethese ERK-induced nuclear activities are such central signalingprocesses, their dysregulation often leads to severe pathologi-cal processes, including oncogenic transformation, neurode-generative diseases, and developmental diseases (15). In orderto transmit their nuclear signals, ERK molecules that are lo-calized in the cytoplasm of quiescent cells rapidly translocateinto the nucleus upon stimulation. Although many details onERK in the nucleus were already provided, the mechanisms ofits translocation are not fully worked out yet.
The nucleus is separated from the cytoplasm by a doublemembrane envelope (25). Nuclear shuttling of proteins occursthrough a specialized nuclear pore complex (NPC), which en-sures high selectivity of molecules for nuclear import/export,thus supporting proper cytoplasmic/nuclear molecular balance.
The majority of proteins enter the nucleus by an active trans-port mechanism, based on a nuclear localization signal (NLS)in the shuttling proteins that binds to importin-� (Imp�) andimportin-� (Imp�), which act as shuttling transport factorsthrough the NPCs (13). However, a significant number of sig-naling proteins, which rapidly translocate into the nucleusupon cell stimulation, do not contain an NLS and thereforemust utilize distinct mechanisms for their translocation. In aprevious study, our group identified a novel NLS-independentmechanism of stimulated nuclear translocation of signalingproteins, including ERK, SMAD3, and MEK1 (6). This mech-anism involves a novel nuclear translocation signal (NTS),which contains either Ser or Thr residues that are phosphor-ylated upon stimulation to allow binding to importin 7 (Imp7)and thereby nuclear shuttling via the NPCs. The NTS of ERKcontains the sequence Ser-Pro-Ser (SPS) within its kinase in-sertion domain (KID), which undergoes stimulus-dependentphosphorylation by as-yet-unknown kinases.
Casein kinase 2 (CK2), formerly known as casein kinase II,is a ubiquitous protein Ser/Thr kinase that plays a central rolein the regulation of a variety of cellular processes (14, 28). Thiskinase acts a tetramer containing two catalytic (� and/or ��)and two regulatory (�) subunits (20). It is a constitutively activekinase whose minimal consensus phosphorylation site is Ser-X-X-Glu/Asp, but additional Glu or Asp residues in some ofthe surrounding residues often allow the phosphorylation ofmore than 300 substrates identified to date (22). Upon phos-phorylation, these substrates regulate a variety of processes,including proliferation, transformation, apoptosis, senescence,and also malignant cell transformation (8, 41). In the presentpaper, we show that CK2 is the main kinase that phosphoryl-ates both Ser residues within the ERK NTS. The phosphory-lation of one of the N-terminal Ser residues is facilitated by
* Corresponding author. Mailing address: Department of BiologicalRegulation, Weizmann Institute of Science, Rehovot 76100, Israel.Phone: 972-8-9343922. Fax: 972-8-9344186. E-mail: [email protected].
� Published ahead of print on 5 July 2011.
3515

active ERK transautophosphorylation. We also demonstratethat the phosphorylation is dependent on ERK release fromhindering cytoplasmic anchors. The crystal structure showedthat CK2 phosphorylation creates an acidic patch importantfor the translocation. These results provide an important roleof CK2 in regulating ERK activity in the nucleus as well asproliferation and oncogenic transformation.
MATERIALS AND METHODS
Reagents and antibodies. Tetradecanoyl phorbol acetate (TPA), epidermalgrowth factor (EGF), the CK2 inhibitor TBB, and transforming growth factor(TGF)–4�,6-diamidino-2-phenylindole (DAPI) were obtained from Sigma (St.Louis, MO). Chelating Sepharose-coupled nickel-nitrilotriacetic acid agarosebeads and a Resource-15Q anion-exchange column were from Amersham (Eng-land). Secondary antibody (Ab) conjugates were from Jackson ImmunoResearch(West Grove, PA). Anti-histone H1, antitubulin, antihemagglutinin (anti-HA),anti-Elk-1, anti-RSK-1, anti-CK2�, and anti-MEK1 Abs were acquired fromSanta Cruz Biotechnology (CA); anti-pElk-1 (2B1, Ser383) and anti-pRSK(Ser381) Abs were from Cell Signaling Technology (Beverly, MA); and anti-green fluorescent protein (anti-GFP) Abs were from Roche Diagnostics GmbH(Mannheim, Germany). Anti-Imp7 Ab was from ABNova (Taipei, Taiwan). Absto doubly TEY-phosphorylated ERK (pTEY-ERK) and general ERK (gERK)were from Sigma (Rehovot, Israel), who also produced and purified the poly-clonal and monoclonal anti-phospho-SPS-ERK Abs.
DNA constructs and mutations. GFP-ERK2 was prepared in pEGFP-C1 vec-tor (Clontech, Mountain View, CA). Human MEK1 was ligated into HindIII andApaI sites downstream of the red fluorescent protein (RFP) gene of theDsRed1-N1 vector (Clontech), and glutathione S-transferase (GST)-ERK2 con-structs were prepared in the pGEX vector. All mutations described were per-formed with a site-directed mutagenesis kit (Stratagene, La Jolla, CA) andconfirmed by sequencing. C-terminally tagged wild-type HA-CK2 in pRc/CMVvector was a gift from D. W. Litchfield (University of Western Ontario, London,ON, Canada). The 6His-MEK construct was produced as described previously(3). All small interfering RNAs (siRNAs) were from Dharmacon (Lafayette,CO).
Cell culture and transfection. HeLa, MDA-MB-231, MCF-7, and HEK-293Tcells were grown in Dulbecco’s modified Eagle’s medium (DMEM), and CHOcells were grown in F12-DMEM, all supplemented with 10% fetal bovine serum.Transfection used either polyethylenimine or Lipofectamine 2000 (Invitrogen,Carlsbad, CA). siRNAs were transfected using Oligofectamine (Dharmacon). Inorder to reach equal transfection efficiencies, transfected cells were distributedamong the necessary number of plates 24 h after transfection.
Immunofluorescence microscopy. Cells were fixed in 3% paraformaldehyde inphosphate-buffered saline (PBS) for 20 min and then permeabilized with 0.2%Triton X-100 in PBS-bovine serum albumin (BSA) (2%) for 20 min at roomtemperature. The fixed cells were sequentially incubated with appropriated Abs(diluted in PBS-BSA, 1:200) for 1 h, followed by either Cy-2- or rhodamine-conjugated secondary Abs and DAPI (diluted in PBS-BSA, 1:200) for 1 h. Slideswere analyzed and photographed by a fluorescence microscope (�600 magnifi-cation; Nikon, Japan).
ImageStream analysis. Nuclear translocation of ERKs was examined by theImageStream system (Amnis Corp., Seattle, WA) using the IDEAS image anal-ysis program. About 1 million cells per sample were rinsed in cold PBS and gentlyscripted into 1.5-ml tubes. Cells were spun down by centrifugation, fixed in 3%paraformaldehyde (20 min), and then permeabilized with 0.1% Triton X-100 inPBS-BSA (2%) (20 min, 23°C). For endogenous ERK staining, fixed cells weresequentially incubated with anti-ERK Abs (1:200 in PBS-BSA; 1 h, 4°C) followedby incubation with Cy-2-conjugated secondary Abs (1:200) and propidium iodide(PI) (1 �g/ml, for nuclear staining) in PBS containing 100 �g/ml of RNase and0.1 mM EDTA (1 h, 40°C). The cells transfected with GFP-containing plasmidswere incubated with secondary Abs and PI. The green ERK1/2 image and the rednucleus image were first compensated into separate channels, and then an over-lay of green and red (creating yellow) was quantified. Additional informationregarding the ImageStream technique can be found at the Internet site of thecompany (Amnis Corp.).
Cell extraction, cell fractionation, Western blotting, and coimmunoprecepita-tion. (i) Preparation of cellular extracts for Western blotting. Cells were rinsedtwice with ice-cold PBS and scraped into radioimmunoprecipitation assay buffer(RIPA) (137 mM NaCl, 20 mM Tris [pH 7.4], 10% glycerol, 1% Triton X-100,0.5% deoxycholate, 0.1% SDS, 2 mM EDTA, 1 mM phenylmethylsulfonyl fluo-
ride [PMSF], and 20 �M leupeptin). The extracts were centrifuged (20,000 � g,15 min, 4°C), and the supernatants were further analyzed by Western blotting.The blots were developed with horseradish peroxidase-conjugated anti-mouse oranti-rabbit Abs.
(ii) Cellular fractionation. Cells were rinsed twice with ice-cold PBS, sus-pended in ice-cold hypotonic buffer (10 mM HEPES [pH 7.9], 1.5 mM MgCl2, 10mM KCl, 0.2 mM PMSF, 0.5 mM dithiothreitol [DTT], and 0.5% Nonidet P-40),incubated (10 min, 4°C), disrupted by repeated aspiration through a 20-gaugeneedle, and centrifuged (12,000 � g, 5 min). The supernatant containing thecytosolic fraction was boiled in sample buffer. The pellet was suspended inextraction buffer (420 mM NaCl, 50 mM �-glycerophosphate, 0.5 mM Na3VO4,1.5 mM MgCl2, 0.2 mM EDTA, 1 mM DTT, 25% glycerol), incubated on ice for30 min, sonicated (50 W, 2 times for 7 s), and centrifuged (15,000 � g, 30 min).The supernatant containing the nuclear fraction was subjected to Western blot-ting with the appropriate Abs.
(iii) Coimmunoprecipitation. Cells were rinsed twice with ice-cold PBS andscraped into buffer H (50 mM �-glycerophosphate [pH 7.3], 1.5 mM EGTA, 1mM EDTA, 1 mM dithiothreitol, 0.1 mM sodium vanadate, 1 mM benzamidine,10 �g/ml aprotinin, 10 �g/ml leupeptin, 2 �g/ml pepstatin). The extracts weresonicated (50 W, 2 times for 7 s), and centrifuged (15,000 � g, 15 min, 4°C).Cellular extracts were incubated overnight at 4°C with the appropriate Abspreconjugated to A/G beads (1 h, 23°C). Subsequently, the beads were washedthree times with coimmunoprecipitation washing buffer (20 mM HEPES [pH7.4], 2 mM MgCl2, 2 mM EGTA, 150 mM NaCl, and 0.1% Triton) and once withPBS and were subjected to Western blot analysis.
Luciferase reporter assay. The following plasmids (from the ForchheimerPlasmid Collection of the Weizmann Institute of Science, Rehovot, Israel) wereused for the experiment: FR-Luc (reporter plasmid), pFA2-Elk1 (fusion trans-activator plasmid), pFC2-dbd (negative control), and pFC-MEK1 (positive con-trol). The plasmids were transfected into HEK-293T cells with Lipofectamine2000 according to the manufacturer’s protocol. At 36 h after transfection, cellswere starved overnight, pretreated with TBB (10 �M, 2 h) or left untreated as acontrol, and then stimulated with EGF (50 ng/ml, 6 h) or left untreated as acontrol. The assay was performed using the dual-luciferase reporter assay system(Promega) in accordance with the manufacturer’s protocol. The luminescencewas detected using a Viktor2 multilabel counter (Perkin Elmer).
In vitro kinase assay. The substrates used for the in vitro kinase assay werewild-type or mutated GST-ERK2, 6His-MEK1, and dephosphorylated �-casein(the last two from Sigma, St. Louis, MO). The substrates were mixed with coldATP (0.5 mM), [�-32P]ATP (4,000 cpm/pmol), and CK2 or active ERK2 (250units each; New England BioLabs, MA) or with GST-ERK2 (ERK), activeGST-ERK2 (Act-ERK), or kinase-dead GST-KA-ERK2 (KA-ERK) producedin our lab, in appropriate kinase buffers (supplied by the company; 1� CK2buffer contained 20 mM Tris-HCl [pH 7.5], 50 mM KCl, and 10 mM MgCl2; 1�ERK2 buffer contained 50 mM Tris-HCl [pH 7.5], 10 mM MgCl2, 2 mM DTT,and 1 mM EGTA, 0.01% Brij 35; both were used in a total volume of 20 �l). Themixture was shaken at 900 rpm (30 min, 30°C), mixed with 4� SDS, and boiledfor 5 min. Proteins were separated by 12 to 15% SDS-PAGE, followed byWestern blotting. The loading of �-casein was assayed by protein staining withReagent Blue (Pierce, IL).
Expression and purification of ERK2 mutants for crystallization purposes.The SPE-ERK2 proteins were expressed in Escherichia coli strain Rosetta asN-terminally hexahistidine-tagged proteins. The bacteria were grown, lysed, andthen purified with chelating Sepharose-coupled nickel-nitrilotriacetic acid aga-rose beads. Protein elution was obtained with a linear gradient of imidazole from10 mM to 250 mM, using 50 mM Tris-HCl (pH 8.0), 0.3 M NaCl, and 250 mMimidazole buffer. The fraction that contained ERK2 protein was then dialyzedagainst 50 mM Tris-HCl (pH 8.0), 0.1 M NaCl, and 1 mM EDTA for 4 h at 4°Cand then transferred to dialysis buffer containing Tris-HCl (pH 8.0), 0.1 M NaCl,1 mM DTT, and 5% glycerol for 16 to 18 h. The protein solution was thensubjected to a second purification step on a 23-ml Resource 15Q anion-exchangecolumn equilibrated with 50 mM Tris-HCl (pH 8.0), 0.1 M NaCl, 5% glycerol,and 1 mM DTT. The protein was eluted using a linear gradient of NaCl with thebuffer as follows: 50 mM Tris-HCl (pH 8.0), 1 M NaCl, 5% glycerol, and 1 mMDTT. Purified protein was concentrated using Vivaspin (VivaScience) up to 6 to12 mg/ml, as determined by A280. The purified protein was divided into aliquotsand stored at �80°C.
Crystallization, data collection, and solution of the SPE-ERK2 mutant. TheSPE-ERK2 proteins were expressed in E. coli (data not shown). Crystals ofSPE-ERK2 protein were obtained by the vapor diffusion sitting drop techniqueby mixing equal amounts of protein solution (2.3 to 2.5 mg/ml) and reservoirsolution in 61 mM ammonium sulfate, 100 mM Bis-Tris (pH 7.0), 12 to 15%(vol/vol) polyethylene glycol (PEG) 3350. Streak seeding (42) using wild-type
3516 PLOTNIKOV ET AL. MOL. CELL. BIOL.

(WT)-ERK2 crystals was conducted in order to initiate crystallization. Crystalsappeared within a day of incubation at 20°C and reached their final size severaldays later. For data collection, crystals were transferred into cryoprotectantsolution containing the crystallization solution and 15% glycerol. Crystallo-graphic data were collected from a single crystal at 100 K using an OxfordCryostream cryosystem cooling device on an ADSC Quantum 315R charge-coupled-device (CCD) detector with an oscillation range of 1.0° at beam lineID23-1 at the European Synchrotron Radiation Facility (ESRF), Grenoble,France. The crystals belonged to the monoclinic space group P21 with one ERK2molecule in the asymmetric unit (data not shown). Data were integrated, re-duced, and scaled using the HKL2000 suite (27). The structures were solved viamolecular replacement methods using Molrep (45) implemented in the CCP4isuite (30) with the ERK2 structure (1ERK) as the search model after removingall solvent molecules. Following molecular replacement, the models were refinedusing rigid-body and then restrained options in REFMAC5 (23). Solvent mole-cules were added utilizing ARP/WARP (19). The models were fitted into elec-tron-density maps using the graphics program Coot (9).
RESULTS
CK2 is important for nuclear translocation of ERK. The twophosphorylated Ser residues of the ERK NTS lie within con-sensus phosphorylation sites of several protein kinases. Sincethe C-terminal Ser seems to fall within a CK2 site, we under-took to examine the role of the latter kinase in the nucleartranslocation of ERK. For this purpose, we first pretreatedCHO cells with CK2 inhibitors, stimulated the cells with TPAfor 15 min, and followed the nuclear translocation of endoge-nous ERK by staining with an anti-ERK antibody (Ab). Asexpected, most ERK molecules were localized in the cytoplasmof resting cells and translocated into the nucleus after TPAstimulation (Fig. 1A). However, pretreatment with the CK2inhibitors TBB (Fig. 1A) and DMAT (data not shown) pre-vented the stimulated nuclear translocation, without any sig-nificant effect on the localization of ERK in resting cells. Sim-ilar effects of CK2 inhibitors were observed with MCF7 (Fig.1A), MBA-MB-231, and HeLa cells (data not shown). Theeffect of the CK2 inhibitors was not due to modulation of theactivating TEY phosphorylation, as judged by Western blottingwith anti-pTEY Ab (Fig. 1B). Since pharmacological inhibitorsare often not fully specific, we ascertained the CK2 effect byspecifically knocking down its expression. For this purpose, weused siRNA of CSNK2A1 (� subunit of CK2), which reducedthe expression of CK2� (but not CK2��) in HeLa and (unex-pectedly) CHO cells (Fig. 1C). As with the pharmacologicalinhibitors, knockdown of CK2� resulted in prevention of TPA-induced nuclear translocation of ERK (Fig. 1C).
In order to quantify our results, we used ImageStream An-alyzer, which simultaneously detects the localization of a flu-orescent dye in many individual cells, thus obtaining statisti-cally significant data. For this purpose, we treated HeLa cellswith the CK2 inhibitor TBB, stimulated the cells with TPA for15 min, and subjected them to the machine. Pictures of thesorted cells (see a representative cell in Fig. 1D, left), as well asquantification of the nuclear fluorescence of the cells (Fig. 1D,right), revealed that in the great majority of resting cells theERK molecules were localized in the cytoplasm. Upon stimu-lation, most ERK staining was shifted to the nucleus, whileTBB pretreatment prevented TPA-stimulated ERK transloca-tion. Finally, another quantitative measure of nuclear translo-cation of ERK was the subcellular fractionation of the treatedcells. Similar to the results in Fig. 1D, this method also dem-onstrated that TBB significantly prevents nuclear accumula-
tion of ERK upon stimulation (Fig. 1E). Taken together, ourresults strongly suggest that CK2 participates in the regulationof nuclear ERK translocation upon stimulation.
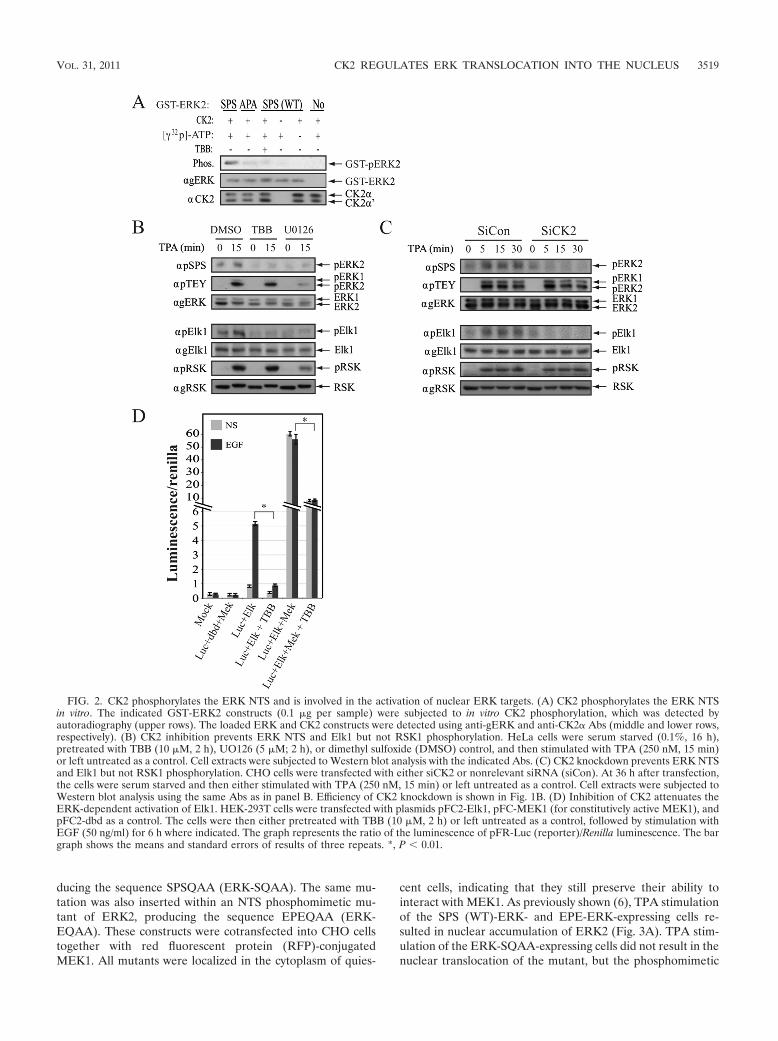
CK2 phosphorylates the ERK NTS and is involved in theactivation of nuclear ERK targets. The finding that CK2 in-hibitors prevent the nuclear translocation of ERK suggestedthat CK2 may act as an NTS kinase. In order to verify thispoint, we examined the ability of CK2 to phosphorylate puri-fied GST-ERK2 in vitro. As expected, CK2 phosphorylatedWT ERK2 but not ERK2 mutated in the Ser residues withinthe NTS (ERK-APA; Fig. 2A); this indicates that the Serresidues within the NTS are the only CK2 sites within ERK2.This phosphorylation was abolished by TBB, signifying that thephosphorylation is indeed mediated by CK2 and not caused byautophosphorylation. We then examined the in vivo effect ofCK2 inhibition or knockdown on NTS phosphorylation usinganti-pSPS Ab, which primarily recognizes the doubly phos-phorylated NTS. We found that TBB as well as siCK2 from twodifferent sources (Fig. 2B and C and data not shown) results inthe prevention of NTS phosphorylation in HeLa and CHO celllines. Since this effect is not dependent on changes in TEYphosphorylation, at any time after stimulation (Fig. 1B and2C), it is likely that CK2 is not involved in the upstreamactivation of the ERK cascade but rather directly phosphoryl-ates the Ser residues in the NTS. We also found that MEKinhibition had an effect similar to that of CK2 inhibition, anda possible reason for it will be explained below. In order tofurther verify the role of the CK2 effect, we postulated that theimpaired nuclear translocation of active ERK should lead to adecrease in the phosphorylation of nuclear ERK targets, with-out much change in the phosphorylation of cytoplasmic sub-strates. Indeed, pretreatment of cells with CK2 inhibitor orknockdown of CK2� resulted in a significant decrease in thephosphorylation and activation of the transcription factorElk1, which is a well-known nuclear target of ERK (Fig. 2A, B,and D). On the other hand, the phosphorylation of a cytoplas-mic ERK target (RSK) was reduced by MEK inhibition but notsignificantly affected by the inhibition of CK2 activity. We alsoexamined the effect of CK2 inhibition on cell viability, which isknown to be regulated by the ERK cascade, and found thatTBB indeed inhibits it in nontransfected cells as well as in cellsoverexpressing constitutively active MEK1. Interestingly, over-expression of the ERK2-EPE construct rescued the TBB effect(data not shown), indicating that although CK2 affects cellproliferation by inhibiting various cellular processes, its effectin our system might be mediated, at least in part, by the nuclearactivity of ERK. These results clearly indicate that NTS phos-phorylation is executed primarily by CK2, acting downstreamof TEY phosphorylation, and therefore does not affect theactivation of ERK. Thus, CK2 affects only the nuclear targetsof ERK, without much influence on cytoplasmic activity.
The most important requirement for substrate recognitionby CK2 within its consensus site is the presence of acidic aminoacid 3 amino acids C-terminal to the phosphorylated site (po-sition �3), while acidic amino acid at position �2 may con-tribute to the recognition as well (18, 36). In order to furtherverify the importance of CK2 in NTS phosphorylation, weconstructed GFP-conjugated mutants of ERK2, in which theGlu and Asp residues at positions of 248 and 249 (�2 and �3from the phosphorylated Ser246) were changed to Ala, pro-
VOL. 31, 2011 CK2 REGULATES ERK TRANSLOCATION INTO THE NUCLEUS 3517
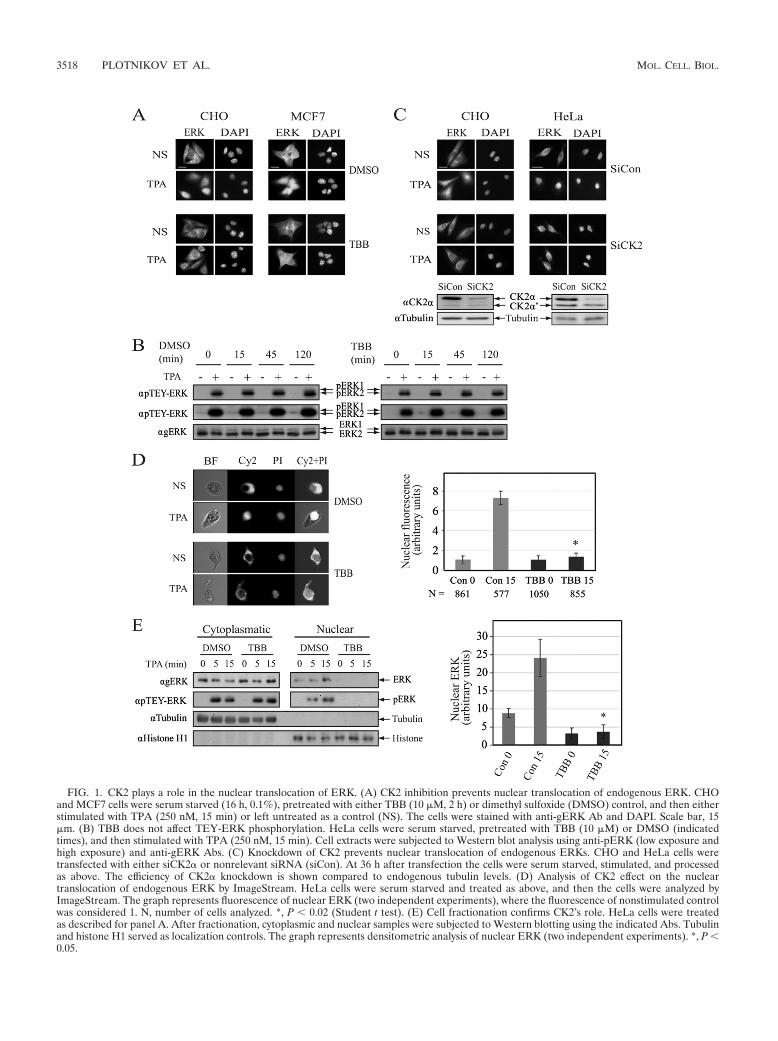
FIG. 1. CK2 plays a role in the nuclear translocation of ERK. (A) CK2 inhibition prevents nuclear translocation of endogenous ERK. CHOand MCF7 cells were serum starved (16 h, 0.1%), pretreated with either TBB (10 �M, 2 h) or dimethyl sulfoxide (DMSO) control, and then eitherstimulated with TPA (250 nM, 15 min) or left untreated as a control (NS). The cells were stained with anti-gERK Ab and DAPI. Scale bar, 15�m. (B) TBB does not affect TEY-ERK phosphorylation. HeLa cells were serum starved, pretreated with TBB (10 �M) or DMSO (indicatedtimes), and then stimulated with TPA (250 nM, 15 min). Cell extracts were subjected to Western blot analysis using anti-pERK (low exposure andhigh exposure) and anti-gERK Abs. (C) Knockdown of CK2 prevents nuclear translocation of endogenous ERKs. CHO and HeLa cells weretransfected with either siCK2� or nonrelevant siRNA (siCon). At 36 h after transfection the cells were serum starved, stimulated, and processedas above. The efficiency of CK2� knockdown is shown compared to endogenous tubulin levels. (D) Analysis of CK2 effect on the nucleartranslocation of endogenous ERK by ImageStream. HeLa cells were serum starved and treated as above, and then the cells were analyzed byImageStream. The graph represents fluorescence of nuclear ERK (two independent experiments), where the fluorescence of nonstimulated controlwas considered 1. N, number of cells analyzed. *, P 0.02 (Student t test). (E) Cell fractionation confirms CK2’s role. HeLa cells were treatedas described for panel A. After fractionation, cytoplasmic and nuclear samples were subjected to Western blotting using the indicated Abs. Tubulinand histone H1 served as localization controls. The graph represents densitometric analysis of nuclear ERK (two independent experiments). *, P 0.05.
3518 PLOTNIKOV ET AL. MOL. CELL. BIOL.
ducing the sequence SPSQAA (ERK-SQAA). The same mu-tation was also inserted within an NTS phosphomimetic mu-tant of ERK2, producing the sequence EPEQAA (ERK-EQAA). These constructs were cotransfected into CHO cellstogether with red fluorescent protein (RFP)-conjugatedMEK1. All mutants were localized in the cytoplasm of quies-
cent cells, indicating that they still preserve their ability tointeract with MEK1. As previously shown (6), TPA stimulationof the SPS (WT)-ERK- and EPE-ERK-expressing cells re-sulted in nuclear accumulation of ERK2 (Fig. 3A). TPA stim-ulation of the ERK-SQAA-expressing cells did not result in thenuclear translocation of the mutant, but the phosphomimetic
FIG. 2. CK2 phosphorylates the ERK NTS and is involved in the activation of nuclear ERK targets. (A) CK2 phosphorylates the ERK NTSin vitro. The indicated GST-ERK2 constructs (0.1 �g per sample) were subjected to in vitro CK2 phosphorylation, which was detected byautoradiography (upper rows). The loaded ERK and CK2 constructs were detected using anti-gERK and anti-CK2� Abs (middle and lower rows,respectively). (B) CK2 inhibition prevents ERK NTS and Elk1 but not RSK1 phosphorylation. HeLa cells were serum starved (0.1%, 16 h),pretreated with TBB (10 �M, 2 h), UO126 (5 �M; 2 h), or dimethyl sulfoxide (DMSO) control, and then stimulated with TPA (250 nM, 15 min)or left untreated as a control. Cell extracts were subjected to Western blot analysis with the indicated Abs. (C) CK2 knockdown prevents ERK NTSand Elk1 but not RSK1 phosphorylation. CHO cells were transfected with either siCK2 or nonrelevant siRNA (siCon). At 36 h after transfection,the cells were serum starved and then either stimulated with TPA (250 nM, 15 min) or left untreated as a control. Cell extracts were subjected toWestern blot analysis using the same Abs as in panel B. Efficiency of CK2 knockdown is shown in Fig. 1B. (D) Inhibition of CK2 attenuates theERK-dependent activation of Elk1. HEK-293T cells were transfected with plasmids pFC2-Elk1, pFC-MEK1 (for constitutively active MEK1), andpFC2-dbd as a control. The cells were then either pretreated with TBB (10 �M, 2 h) or left untreated as a control, followed by stimulation withEGF (50 ng/ml) for 6 h where indicated. The graph represents the ratio of the luminescence of pFR-Luc (reporter)/Renilla luminescence. The bargraph shows the means and standard errors of results of three repeats. *, P 0.01.
VOL. 31, 2011 CK2 REGULATES ERK TRANSLOCATION INTO THE NUCLEUS 3519
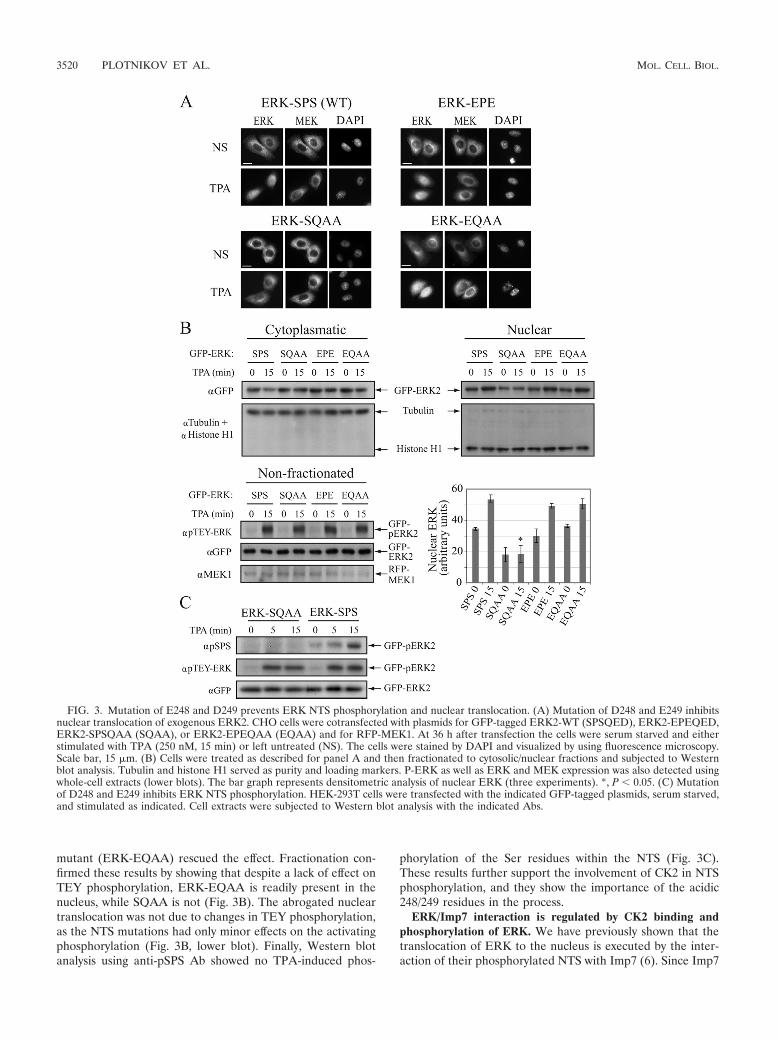
mutant (ERK-EQAA) rescued the effect. Fractionation con-firmed these results by showing that despite a lack of effect onTEY phosphorylation, ERK-EQAA is readily present in thenucleus, while SQAA is not (Fig. 3B). The abrogated nucleartranslocation was not due to changes in TEY phosphorylation,as the NTS mutations had only minor effects on the activatingphosphorylation (Fig. 3B, lower blot). Finally, Western blotanalysis using anti-pSPS Ab showed no TPA-induced phos-
phorylation of the Ser residues within the NTS (Fig. 3C).These results further support the involvement of CK2 in NTSphosphorylation, and they show the importance of the acidic248/249 residues in the process.
ERK/Imp7 interaction is regulated by CK2 binding andphosphorylation of ERK. We have previously shown that thetranslocation of ERK to the nucleus is executed by the inter-action of their phosphorylated NTS with Imp7 (6). Since Imp7
FIG. 3. Mutation of E248 and D249 prevents ERK NTS phosphorylation and nuclear translocation. (A) Mutation of D248 and E249 inhibitsnuclear translocation of exogenous ERK2. CHO cells were cotransfected with plasmids for GFP-tagged ERK2-WT (SPSQED), ERK2-EPEQED,ERK2-SPSQAA (SQAA), or ERK2-EPEQAA (EQAA) and for RFP-MEK1. At 36 h after transfection the cells were serum starved and eitherstimulated with TPA (250 nM, 15 min) or left untreated (NS). The cells were stained by DAPI and visualized by using fluorescence microscopy.Scale bar, 15 �m. (B) Cells were treated as described for panel A and then fractionated to cytosolic/nuclear fractions and subjected to Westernblot analysis. Tubulin and histone H1 served as purity and loading markers. P-ERK as well as ERK and MEK expression was also detected usingwhole-cell extracts (lower blots). The bar graph represents densitometric analysis of nuclear ERK (three experiments). *, P 0.05. (C) Mutationof D248 and E249 inhibits ERK NTS phosphorylation. HEK-293T cells were transfected with the indicated GFP-tagged plasmids, serum starved,and stimulated as indicated. Cell extracts were subjected to Western blot analysis with the indicated Abs.
3520 PLOTNIKOV ET AL. MOL. CELL. BIOL.

recognizes only NTS-phosphorylated ERK, we examinedwhether CK2 inhibition affects the endogenous ERK/Imp7interaction in stimulated cells. As expected, stimulation ofHeLa cells with TPA resulted in an enhanced interaction ofERK with Imp7 (Fig. 4A). Inhibition of CK2 phosphorylationby TBB as well as prevention of NTS phosphorylation due toSQAA mutation abolished this interaction (Fig. 4A and B).These results clearly support the involvement of CK2-medi-ated NTS phosphorylation in the interaction with Imp7 andthereby in the nuclear translocation of ERK. We then enter-tained the possibility that the phosphorylation of NTS by CK2is dependent on its physical association with ERK. For thispurpose we overexpressed GFP-ERK2 and HA-CK2 in HEK-293T cells, which were starved and then stimulated with TPA.The extracts of these cells were then used for coimmunopre-cipitation with anti-GFP Ab, which revealed that the two over-expressed proteins interact with each other after cell stimula-tion but not in resting cells (Fig. 4C). Therefore, it wasimportant to show that the interaction occurs between theendogenous proteins as well. To this end, coimmunoprecipita-tion revealed that as for the overexpressed proteins, there wasvery little interaction in resting cells, but TPA treatment stim-ulated this interaction mainly upon 5 min of stimulation (Fig.
4D). Therefore, it is likely that shortly after stimulation, ERKinteracts with and is phosphorylated by CK2. The phosphory-lation then allows ERK binding to Imp7, and this promotes thenuclear translocation of ERK.
Ser246 phosphorylation by CK2 is sufficient for full ERKtranslocation, while Ser244 phosphorylation accelerates it.Our previous study using APE and EPA mutants of the ERK2-NTS suggested that phosphorylation of Ser246 is more impor-tant for ERK nuclear translocation than that of Ser244 (6). Inorder to further study the role of the distinct residues, wemutated NTS within GFP-ERK2 to APS, SPA, and APA.These constructs were cotransfected into CHO cells togetherwith the RFP-conjugated MEK1 for cytoplasmic anchoring(11, 34). After serum starvation, the cells were treated withTPA and the nuclear translocation of the various constructswas monitored. As expected, all overexpressed-ERK2 con-structs were localized in the cytoplasm, but after stimulationthe distinct constructs translocated into the nucleus with dif-ferent kinetics (Fig. 5A). Thus, ERK-SPS (WT) translocationwas fastest, and this construct was found in the nuclei of 65%of the cells already 6 min after stimulation. ERK-APS trans-located slightly more slowly and reached this level 12 min afterstimulation. The translocation of the ERK-SPA was much
FIG. 4. ERK binding to Imp7 and CK2. (A) Imp7 interaction with ERK depends on CK2 phosphorylation of the ERK NTS. HeLa cells wereserum starved and treated with TPA (250 nM) for the indicated times. ERK was immunoprecipitated using anti-gERK Abs or normal rabbit serum(NRS), which served as an irrelevant Ab control. Coimmunoprecipitated Imp7 was detected by Western blot analysis using anti-Imp7 Abs (upperpanel). ERK activation and Imp7 amount were determined using the indicated Abs. (B) Mutation of E248 and D249 prevents the interaction ofERK with Imp7. HEK-293T cells were transfected with the GFP-tagged plasmids of ERK2-WT (SPS) and ERK2-SPSQAA, serum starved, andstimulated with TPA (250 nM) for the indicated times. ERK constructs were immunoprecipitated using either anti-GFP Ab or NRS control, andcoimmunoprecipitated Imp7 was detected by Western blot analysis with anti-Imp Abs. ERK activation and Imp7 amount were detected using theindicated Abs. (C) Ectopically expressed CK2 interacts with overexpressed ERK2 in a stimulus-dependent manner. HEK-293T cells weretransfected with plasmids for HA-tagged CK2� or GFP-ERK2 or with both plasmids. At 36 h after transfection, the cells were serum starved andthen stimulated with TPA (250 nM) for the indicated times. ERK2 was immunoprecipitated using anti-GFP Ab, and the coimmunoprecipitatedCK2� was detected by Western blot analysis using anti-HA Ab. The amount of expressed proteins was detected with anti-HA and antitubulin Abs.(D) Stimulus-dependent interaction of endogenous CK2 and ERK. HeLa cells were serum starved and then stimulated with TPA (250 nM) forthe indicated times. Endogenous ERKs were immunoprecipitated using either anti-gERK Ab or NRS control, and coimmunoprecipitated CK2�was detected by Western blot analysis with anti-CK2� Ab. ERK activation and CK2 loading were detected using the indicated Abs.
VOL. 31, 2011 CK2 REGULATES ERK TRANSLOCATION INTO THE NUCLEUS 3521
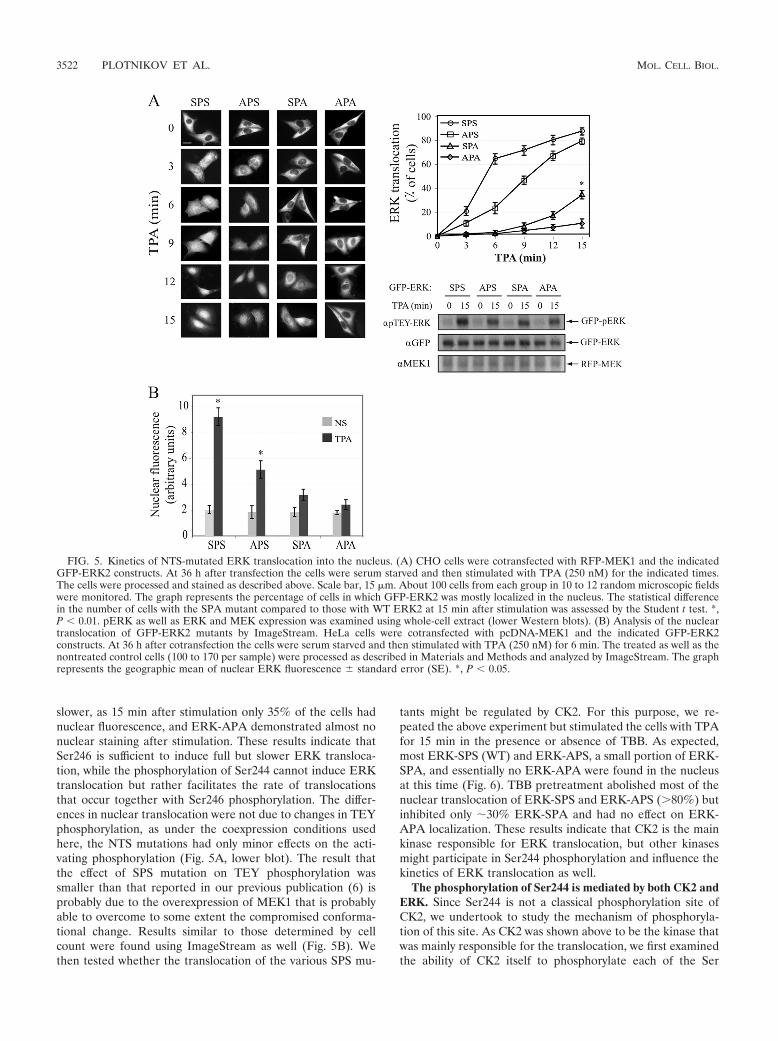
slower, as 15 min after stimulation only 35% of the cells hadnuclear fluorescence, and ERK-APA demonstrated almost nonuclear staining after stimulation. These results indicate thatSer246 is sufficient to induce full but slower ERK transloca-tion, while the phosphorylation of Ser244 cannot induce ERKtranslocation but rather facilitates the rate of translocationsthat occur together with Ser246 phosphorylation. The differ-ences in nuclear translocation were not due to changes in TEYphosphorylation, as under the coexpression conditions usedhere, the NTS mutations had only minor effects on the acti-vating phosphorylation (Fig. 5A, lower blot). The result thatthe effect of SPS mutation on TEY phosphorylation wassmaller than that reported in our previous publication (6) isprobably due to the overexpression of MEK1 that is probablyable to overcome to some extent the compromised conforma-tional change. Results similar to those determined by cellcount were found using ImageStream as well (Fig. 5B). Wethen tested whether the translocation of the various SPS mu-
tants might be regulated by CK2. For this purpose, we re-peated the above experiment but stimulated the cells with TPAfor 15 min in the presence or absence of TBB. As expected,most ERK-SPS (WT) and ERK-APS, a small portion of ERK-SPA, and essentially no ERK-APA were found in the nucleusat this time (Fig. 6). TBB pretreatment abolished most of thenuclear translocation of ERK-SPS and ERK-APS (80%) butinhibited only �30% ERK-SPA and had no effect on ERK-APA localization. These results indicate that CK2 is the mainkinase responsible for ERK translocation, but other kinasesmight participate in Ser244 phosphorylation and influence thekinetics of ERK translocation as well.
The phosphorylation of Ser244 is mediated by both CK2 andERK. Since Ser244 is not a classical phosphorylation site ofCK2, we undertook to study the mechanism of phosphoryla-tion of this site. As CK2 was shown above to be the kinase thatwas mainly responsible for the translocation, we first examinedthe ability of CK2 itself to phosphorylate each of the Ser
FIG. 5. Kinetics of NTS-mutated ERK translocation into the nucleus. (A) CHO cells were cotransfected with RFP-MEK1 and the indicatedGFP-ERK2 constructs. At 36 h after transfection the cells were serum starved and then stimulated with TPA (250 nM) for the indicated times.The cells were processed and stained as described above. Scale bar, 15 �m. About 100 cells from each group in 10 to 12 random microscopic fieldswere monitored. The graph represents the percentage of cells in which GFP-ERK2 was mostly localized in the nucleus. The statistical differencein the number of cells with the SPA mutant compared to those with WT ERK2 at 15 min after stimulation was assessed by the Student t test. *,P 0.01. pERK as well as ERK and MEK expression was examined using whole-cell extract (lower Western blots). (B) Analysis of the nucleartranslocation of GFP-ERK2 mutants by ImageStream. HeLa cells were cotransfected with pcDNA-MEK1 and the indicated GFP-ERK2constructs. At 36 h after cotransfection the cells were serum starved and then stimulated with TPA (250 nM) for 6 min. The treated as well as thenontreated control cells (100 to 170 per sample) were processed as described in Materials and Methods and analyzed by ImageStream. The graphrepresents the geographic mean of nuclear ERK fluorescence � standard error (SE). *, P 0.05.
3522 PLOTNIKOV ET AL. MOL. CELL. BIOL.
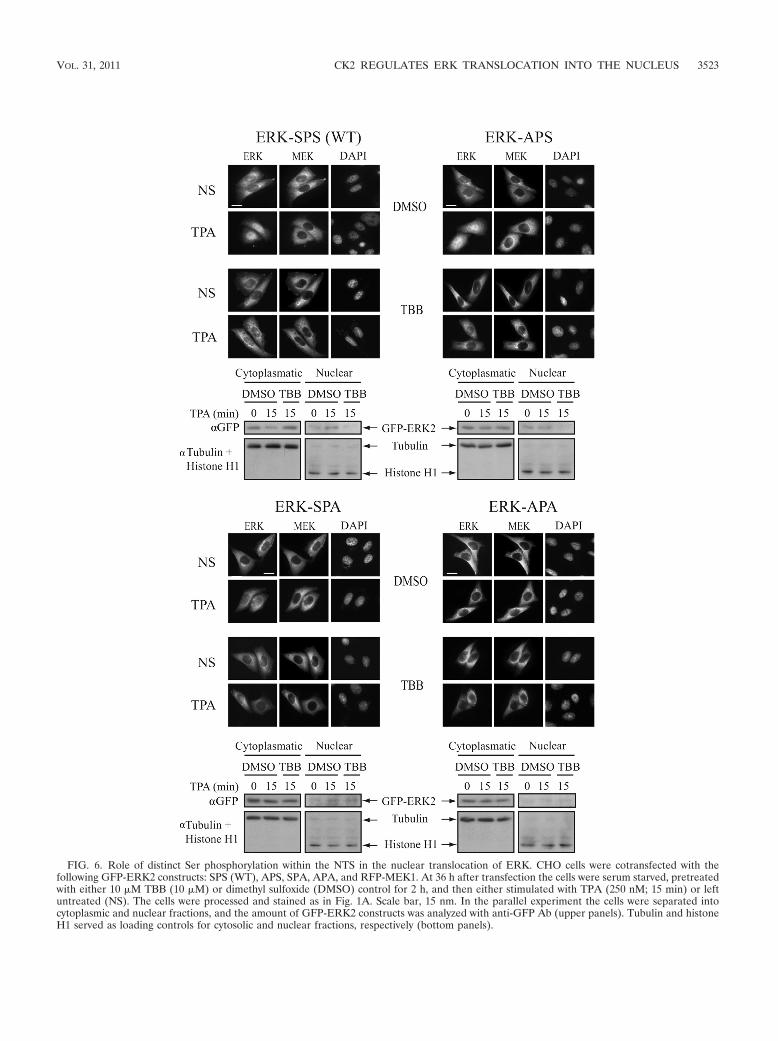
FIG. 6. Role of distinct Ser phosphorylation within the NTS in the nuclear translocation of ERK. CHO cells were cotransfected with thefollowing GFP-ERK2 constructs: SPS (WT), APS, SPA, APA, and RFP-MEK1. At 36 h after transfection the cells were serum starved, pretreatedwith either 10 �M TBB (10 �M) or dimethyl sulfoxide (DMSO) control for 2 h, and then either stimulated with TPA (250 nM; 15 min) or leftuntreated (NS). The cells were processed and stained as in Fig. 1A. Scale bar, 15 nm. In the parallel experiment the cells were separated intocytoplasmic and nuclear fractions, and the amount of GFP-ERK2 constructs was analyzed with anti-GFP Ab (upper panels). Tubulin and histoneH1 served as loading controls for cytosolic and nuclear fractions, respectively (bottom panels).
VOL. 31, 2011 CK2 REGULATES ERK TRANSLOCATION INTO THE NUCLEUS 3523
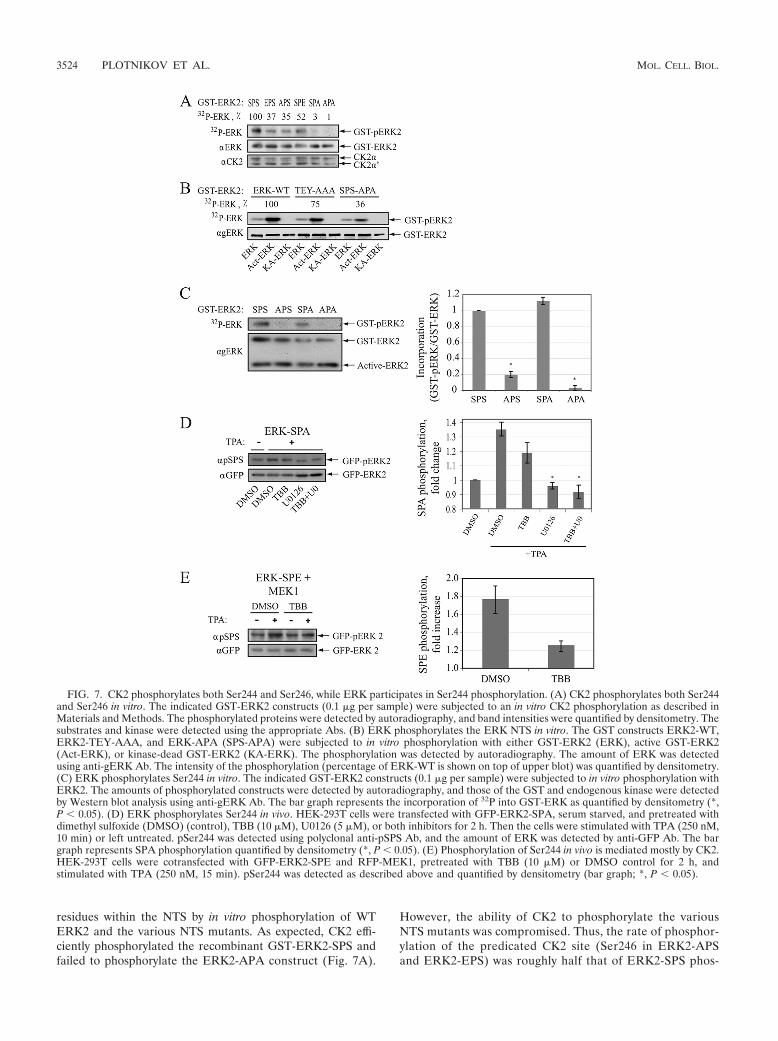
residues within the NTS by in vitro phosphorylation of WTERK2 and the various NTS mutants. As expected, CK2 effi-ciently phosphorylated the recombinant GST-ERK2-SPS andfailed to phosphorylate the ERK2-APA construct (Fig. 7A).
However, the ability of CK2 to phosphorylate the variousNTS mutants was compromised. Thus, the rate of phosphor-ylation of the predicated CK2 site (Ser246 in ERK2-APSand ERK2-EPS) was roughly half that of ERK2-SPS phos-
FIG. 7. CK2 phosphorylates both Ser244 and Ser246, while ERK participates in Ser244 phosphorylation. (A) CK2 phosphorylates both Ser244and Ser246 in vitro. The indicated GST-ERK2 constructs (0.1 �g per sample) were subjected to an in vitro CK2 phosphorylation as described inMaterials and Methods. The phosphorylated proteins were detected by autoradiography, and band intensities were quantified by densitometry. Thesubstrates and kinase were detected using the appropriate Abs. (B) ERK phosphorylates the ERK NTS in vitro. The GST constructs ERK2-WT,ERK2-TEY-AAA, and ERK-APA (SPS-APA) were subjected to in vitro phosphorylation with either GST-ERK2 (ERK), active GST-ERK2(Act-ERK), or kinase-dead GST-ERK2 (KA-ERK). The phosphorylation was detected by autoradiography. The amount of ERK was detectedusing anti-gERK Ab. The intensity of the phosphorylation (percentage of ERK-WT is shown on top of upper blot) was quantified by densitometry.(C) ERK phosphorylates Ser244 in vitro. The indicated GST-ERK2 constructs (0.1 �g per sample) were subjected to in vitro phosphorylation withERK2. The amounts of phosphorylated constructs were detected by autoradiography, and those of the GST and endogenous kinase were detectedby Western blot analysis using anti-gERK Ab. The bar graph represents the incorporation of 32P into GST-ERK as quantified by densitometry (*,P 0.05). (D) ERK phosphorylates Ser244 in vivo. HEK-293T cells were transfected with GFP-ERK2-SPA, serum starved, and pretreated withdimethyl sulfoxide (DMSO) (control), TBB (10 �M), U0126 (5 �M), or both inhibitors for 2 h. Then the cells were stimulated with TPA (250 nM,10 min) or left untreated. pSer244 was detected using polyclonal anti-pSPS Ab, and the amount of ERK was detected by anti-GFP Ab. The bargraph represents SPA phosphorylation quantified by densitometry (*, P 0.05). (E) Phosphorylation of Ser244 in vivo is mediated mostly by CK2.HEK-293T cells were cotransfected with GFP-ERK2-SPE and RFP-MEK1, pretreated with TBB (10 �M) or DMSO control for 2 h, andstimulated with TPA (250 nM, 15 min). pSer244 was detected as described above and quantified by densitometry (bar graph; *, P 0.05).
3524 PLOTNIKOV ET AL. MOL. CELL. BIOL.

phorylation by CK2. The reason for this reduced phosphor-ylation of Ser246 in APS or EPS is not clear, but it might bedue to changes in ERK conformation. On the other hand,Ser244 was not phosphorylated at all when Ser246 was mu-tated to Ala (ERK2-SPA), but the phosphorylation was res-cued when Ser246 was mutated to Glu (ERK2-SPE). Theseresults indicate that both Ser residues within the NTS can bephosphorylated by CK2, but Ser244 can be phosphorylatedonly after Ser246 phosphorylation, supporting the ability ofCK2 to phosphorylate Ser residues when position �2 isprimed by phosphorylation (36).
The ability of CK2 to phosphorylate both Ser244 and thepredicted Ser246 might indicate that this kinase is sufficientto induce full phosphorylation of the NTS. However, sincethe results described above (Fig. 6) showed that the trans-location of ERK2-SPA is only partially inhibited by TBB, weundertook to examine the possibility that this site is phos-phorylated by other kinases as well. Since this site lies withinan ERK consensus phosphorylation site and ERK is acti-vated upon stimulation, we entertained the possibility thatSer244 is partially phosphorylated by this kinase as well. Forthis purpose, we first examined the ability of active ERK tophosphorylate the NTS in vitro, using immunoprecipitatedWT GFP-ERK2, inactive ERK2 (TEY-AAA), and ERK2 inwhich the two Ser residues within the NTS were replacedwith Ala (SPS-APA). These three constructs were subjectedto in vitro phosphorylation by either GST-ERK2, which hasvery low activity, active GST-ERK2 (Act-ERK), or inactiveGST-ERK2 (ATP binding site mutation; KA-ERK). 32P in-corporation into GFP-ERK2 was detected mainly when thisprotein was incubated with Act-ERK2. The phosphorylationof TEY-AAA was reduced by �25%, while that of SPS-APAwas reduced by �80% (Fig. 7B). Next, we analyzed theability of active ERK2 to phosphorylate GST-ERK2 and itsNTS mutants. As expected, active ERK2 phosphorylatedboth ERK2-SPS and ERK2-SPA (Fig. 7C) but not ERK2-APS or ERK2-APA. These findings suggest that whileSer246 is phosphorylated mainly by CK2, Ser244 phosphor-ylation may be mediated by active ERK as well.
The ability of ERK to phosphorylate Ser244 in vitro raisedthe question as whether these kinases can also phosphory-late this site in vivo. To answer this question we transfectedthe GFP-ERK2-SPA construct into HEK-293T cells, whichwere then serum starved and pretreated with TBB, U0126,or both. Then, the cells were stimulated with TPA, andSer244 phosphorylation of the construct was detected usingspecial anti-pSPS Ab that recognizes monophosphorylatedas well as dually phosphorylated NTS. This experiment re-vealed that the phosphorylation of ERK2-SPA was signifi-cantly increased upon TPA stimulation (Fig. 7D). TBB hadno significant effect on Ser244 phosphorylation, whileU0126 or the combination of TBB and U0126 abolished it.Since the ERK2-SPA construct cannot be phosphorylated byCK2, these results indicate that Ser244 is indeed phosphor-ylated by ERK upon stimulation in the examined cells. Toexplore the relative roles of CK2 and ERK in the phosphor-ylation of Ser244, we then transfected the cells with anERK-SPE mutant that can be phosphorylated by both ki-nases, pretreated them with TBB, and stimulated the cellswith TPA. As expected, NTS phosphorylation of this con-
struct was elevated after stimulation, and this elevation wasinhibited by TBB (�70%) (Fig. 7E). Since TBB inhibitedmore than 95% of the CK2 activity (data not shown), thesmall degree of elevation of phosphorylation is likely medi-ated by active ERK. Indeed U0126 added to the TBB abol-ished the residual phosphorylation, indicating that Ser244phosphorylation is mediated mainly by CK2 but also byactive ERK.
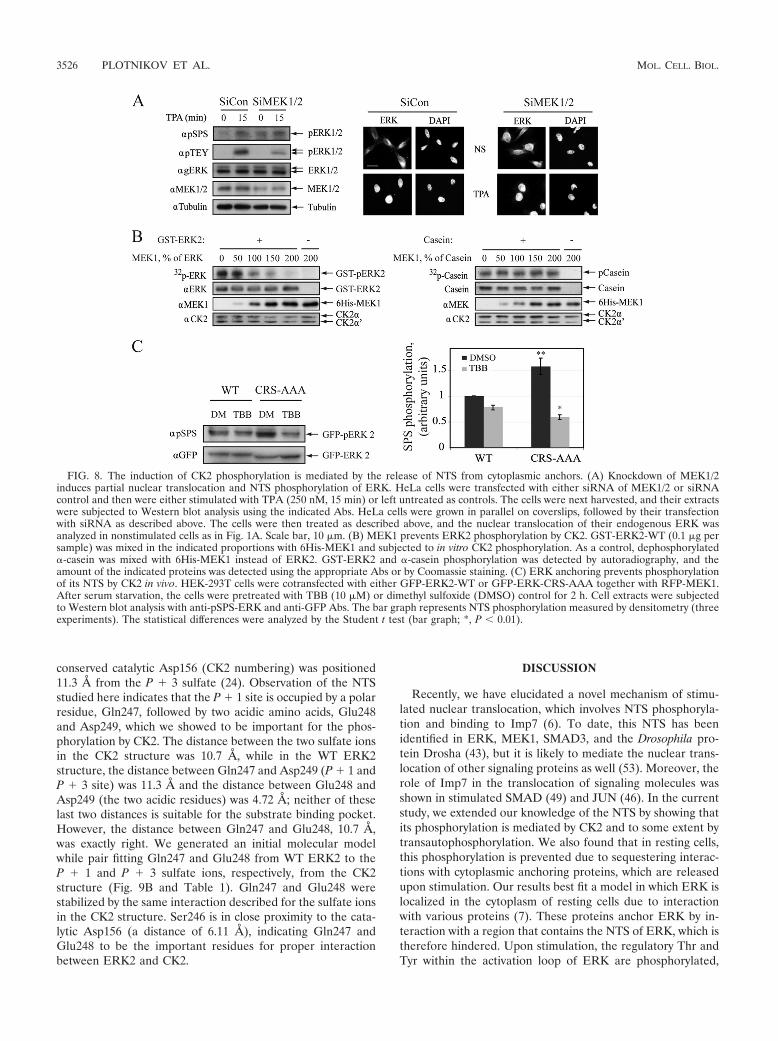
The induction of CK2 phosphorylation is mediated by therelease of hindered NTS from cytoplasmic anchors. Since CK2is a constitutively active kinase whose activity is not changedupon stimulation, the reason for the significant increase in theCK2-mediated phosphorylation upon stimulation was notclear. One possibility was that the CK2 phosphorylation site ishindered in resting cells and is exposed only after stimulation.Indeed, in resting cells, ERK interacts with various anchoringproteins, and the interaction with many of them is reversedupon stimulation and TEY phosphorylation (47). Therefore,we undertook to study whether MEK1, which serves as one ofthe cytoplasmic anchors, prevents the phosphorylation of NTSby CK2. First we confirmed that MEK1 and -2 serve as anchorproteins, and we found that knocking down these kinases in-deed causes nuclear translocation of about 20 to 25% of theERK molecules (Fig. 8A). In addition, MEK1/2 knockdownelevated NTS phosphorylation in quiescent but not stimulatedcells. These results indicate that endogenous MEK1 and -2 areresponsible for the anchoring of a portion of ERK molecules,which upon release from MEK1/2 are phosphorylated on theirNTS by CK2. To further establish this finding, we examined theability of CK2 to phosphorylate GST-ERK2 in the presence ofincreasing concentrations of His-MEK1. We found that theNTS phosphorylation by CK2 was decreased upon increasingHA-MEK1 concentration (Fig. 8B). This effect was not due toinhibition of CK2 activity, because the latter had no effect onthe phosphorylation of casein in a similar experiment. Thus, itis likely that MEK1 hinders the NTS and protects it from CK2phosphorylation. Finally, we used CRS-AAA, which does notinteract with most of the anchoring proteins (34). When thisconstruct was transfected together with MEK1, its basal CK2-dependent NTS phosphorylation was much higher than that ofthe WT (Fig. 8C), and this phosphorylation was prevented byTBB. These data clearly suggest that the NTS is hindered byanchoring proteins and is released upon stimulation to allow itsphosphorylation by CK2.
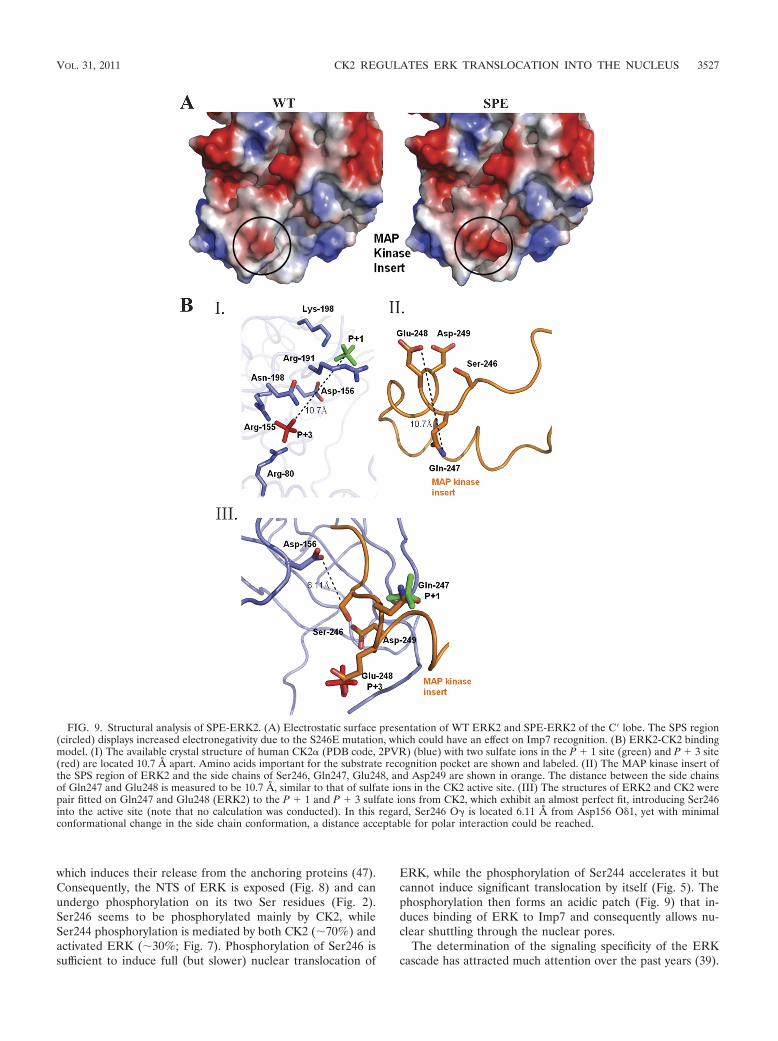
The SPE mutant exhibits an increased electronegative patchon the protein surface. Structural comparison of ERK2-SPEand ERK2-SPS (WT) revealed no significant difference in theirinterlobe orientations. When we observed the Ser246 area inthe WT structure, we found that two negatively charged aminoacids, Glu248 and Asp249, were located on the protein surface.In the SPE mutant structure, Glu246 is in close proximity tothese amino acids, making this region more electronegative(Fig. 9A and Table 1). Recently, the mechanism enabling sub-strate recognition in CK2 was discovered (24). The crystalstructure of human CK2 (PDB code, 2PVR) was solved, and itexhibited sulfate ions in the substrate-binding pocket mimick-ing the P � 1 and P � 3 acidic residues. This structure reveledthat the first P � 1 sulfate is stabilized by interaction withamino acids Arg191 and Lys198, while the P � 3 sulfate isstabilized by interactions with Arg155, Asn189, and Arg80. The
VOL. 31, 2011 CK2 REGULATES ERK TRANSLOCATION INTO THE NUCLEUS 3525
conserved catalytic Asp156 (CK2 numbering) was positioned11.3 Å from the P � 3 sulfate (24). Observation of the NTSstudied here indicates that the P � 1 site is occupied by a polarresidue, Gln247, followed by two acidic amino acids, Glu248and Asp249, which we showed to be important for the phos-phorylation by CK2. The distance between the two sulfate ionsin the CK2 structure was 10.7 Å, while in the WT ERK2structure, the distance between Gln247 and Asp249 (P � 1 andP � 3 site) was 11.3 Å and the distance between Glu248 andAsp249 (the two acidic residues) was 4.72 Å; neither of theselast two distances is suitable for the substrate binding pocket.However, the distance between Gln247 and Glu248, 10.7 Å,was exactly right. We generated an initial molecular modelwhile pair fitting Gln247 and Glu248 from WT ERK2 to theP � 1 and P � 3 sulfate ions, respectively, from the CK2structure (Fig. 9B and Table 1). Gln247 and Glu248 werestabilized by the same interaction described for the sulfate ionsin the CK2 structure. Ser246 is in close proximity to the cata-lytic Asp156 (a distance of 6.11 Å), indicating Gln247 andGlu248 to be the important residues for proper interactionbetween ERK2 and CK2.
DISCUSSION
Recently, we have elucidated a novel mechanism of stimu-lated nuclear translocation, which involves NTS phosphoryla-tion and binding to Imp7 (6). To date, this NTS has beenidentified in ERK, MEK1, SMAD3, and the Drosophila pro-tein Drosha (43), but it is likely to mediate the nuclear trans-location of other signaling proteins as well (53). Moreover, therole of Imp7 in the translocation of signaling molecules wasshown in stimulated SMAD (49) and JUN (46). In the currentstudy, we extended our knowledge of the NTS by showing thatits phosphorylation is mediated by CK2 and to some extent bytransautophosphorylation. We also found that in resting cells,this phosphorylation is prevented due to sequestering interac-tions with cytoplasmic anchoring proteins, which are releasedupon stimulation. Our results best fit a model in which ERK islocalized in the cytoplasm of resting cells due to interactionwith various proteins (7). These proteins anchor ERK by in-teraction with a region that contains the NTS of ERK, which istherefore hindered. Upon stimulation, the regulatory Thr andTyr within the activation loop of ERK are phosphorylated,
FIG. 8. The induction of CK2 phosphorylation is mediated by the release of NTS from cytoplasmic anchors. (A) Knockdown of MEK1/2induces partial nuclear translocation and NTS phosphorylation of ERK. HeLa cells were transfected with either siRNA of MEK1/2 or siRNAcontrol and then were either stimulated with TPA (250 nM, 15 min) or left untreated as controls. The cells were next harvested, and their extractswere subjected to Western blot analysis using the indicated Abs. HeLa cells were grown in parallel on coverslips, followed by their transfectionwith siRNA as described above. The cells were then treated as described above, and the nuclear translocation of their endogenous ERK wasanalyzed in nonstimulated cells as in Fig. 1A. Scale bar, 10 �m. (B) MEK1 prevents ERK2 phosphorylation by CK2. GST-ERK2-WT (0.1 �g persample) was mixed in the indicated proportions with 6His-MEK1 and subjected to in vitro CK2 phosphorylation. As a control, dephosphorylated�-casein was mixed with 6His-MEK1 instead of ERK2. GST-ERK2 and �-casein phosphorylation was detected by autoradiography, and theamount of the indicated proteins was detected using the appropriate Abs or by Coomassie staining. (C) ERK anchoring prevents phosphorylationof its NTS by CK2 in vivo. HEK-293T cells were cotransfected with either GFP-ERK2-WT or GFP-ERK-CRS-AAA together with RFP-MEK1.After serum starvation, the cells were pretreated with TBB (10 �M) or dimethyl sulfoxide (DMSO) control for 2 h. Cell extracts were subjectedto Western blot analysis with anti-pSPS-ERK and anti-GFP Abs. The bar graph represents NTS phosphorylation measured by densitometry (threeexperiments). The statistical differences were analyzed by the Student t test (bar graph; *, P 0.01).
3526 PLOTNIKOV ET AL. MOL. CELL. BIOL.
which induces their release from the anchoring proteins (47).Consequently, the NTS of ERK is exposed (Fig. 8) and canundergo phosphorylation on its two Ser residues (Fig. 2).Ser246 seems to be phosphorylated mainly by CK2, whileSer244 phosphorylation is mediated by both CK2 (�70%) andactivated ERK (�30%; Fig. 7). Phosphorylation of Ser246 issufficient to induce full (but slower) nuclear translocation of
ERK, while the phosphorylation of Ser244 accelerates it butcannot induce significant translocation by itself (Fig. 5). Thephosphorylation then forms an acidic patch (Fig. 9) that in-duces binding of ERK to Imp7 and consequently allows nu-clear shuttling through the nuclear pores.
The determination of the signaling specificity of the ERKcascade has attracted much attention over the past years (39).
FIG. 9. Structural analysis of SPE-ERK2. (A) Electrostatic surface presentation of WT ERK2 and SPE-ERK2 of the C� lobe. The SPS region(circled) displays increased electronegativity due to the S246E mutation, which could have an effect on Imp7 recognition. (B) ERK2-CK2 bindingmodel. (I) The available crystal structure of human CK2� (PDB code, 2PVR) (blue) with two sulfate ions in the P � 1 site (green) and P � 3 site(red) are located 10.7 Å apart. Amino acids important for the substrate recognition pocket are shown and labeled. (II) The MAP kinase insert ofthe SPS region of ERK2 and the side chains of Ser246, Gln247, Glu248, and Asp249 are shown in orange. The distance between the side chainsof Gln247 and Glu248 is measured to be 10.7 Å, similar to that of sulfate ions in the CK2 active site. (III) The structures of ERK2 and CK2 werepair fitted on Gln247 and Glu248 (ERK2) to the P � 1 and P � 3 sulfate ions from CK2, which exhibit an almost perfect fit, introducing Ser246into the active site (note that no calculation was conducted). In this regard, Ser246 O� is located 6.11 Å from Asp156 O 1, yet with minimalconformational change in the side chain conformation, a distance acceptable for polar interaction could be reached.
VOL. 31, 2011 CK2 REGULATES ERK TRANSLOCATION INTO THE NUCLEUS 3527

One of the main parameters that control the ERK selectivity isthe subcellular distribution of this kinase as well as its up-stream regulators (4). In this sense it has previously beenshown that the nuclear activity of ERK is important for theactivation of transcription factors necessary for relevant geneexpression (29). Other roles of the nuclear activities, such aschromatin remodeling, direct induction, or suppression oftranscription and regulation of the cell cycle, have been dem-onstrated as well. Therefore, nuclear translocation of ERK isessential for the induction of many ERK-dependent processes,and indeed, specific abrogation of ERK nuclear translocationblocks the progression of proliferation and oncogenic transfor-mation (2, 10, 44). It should be noted, however, that the nu-clear activity is probably not sufficient to induce all ERK-dependent processes, as their activity in the cytoplasm (5) andmitochondria (12) as well as ERK1c activity in the Golgi ap-paratus (37, 38) is also necessary to induce proliferation andsurvival. Therefore, ERK translocation seems to be regulatedby a variety of methods, such as cytoplasmic anchoring byinteraction with specific proteins (7, 16), changes in NPC’snumber in different cell types (40), interaction with Imp7 (21),and phosphorylation by CK2 (the study presented here). All ofthese mechanisms are likely to play important roles in govern-ing the specificity and efficiency of ERK signaling in health anddisease.
ERK plays a key role in regulating cell cycle progressionupon extracellular stimulation, which enhances proliferationand may lead to oncogenic transformation (31). Since CK2 is
the main kinase that phosphorylates the NTS of ERK, it wasreasonable to assume that it participates in the regulation ofthese processes as well. Indeed, it was clearly shown that CK2participates in the regulation and progression of several stagesof the cell cycle (41). This was verified in several systems,including mainly mammalian cells, in which reduction of CK2expression or activity inhibits G0/G1, G1/S, and G2/M transi-tions (28). Indeed, addition of CK2 inhibitors or knockingdown CK2 in our cellular system attenuated cell proliferation.Although much information on the involvement of CK2 inproliferation is already available, the regulatory mechanismscoordinating its numerous functions are not clear enough. Inthis sense it is possible that our identification of ERK-CK2cooperation might be one of the ways by which CK2 exerts itsfunction on cell proliferation. In addition to the role of CK2 inproliferation, CK2 was also shown to participate in the induc-tion of oncogenic transformation and tumor maintenance (8).However, studies in experimental transgenic mice models sug-gested that CK2 itself is not an oncogene but cooperates withoncogenes as well as protumorigenic and signaling molecules,thus increasing their oncogenic potential (35, 48). Some stud-ies indicate that CK2 may cooperate with the ERK cascade inthe promotion of tumorigenicity. For example, it was shownthat the CK2�� catalytic subunit synergized with H-Ras inBALB/c 3T3 fibroblast transformation (26). In addition, CK2promoted a transformed phenotype and survival through Her-2/neu signaling in NF639 breast cancer cells (33). Although themechanisms suggested in the above studies did not includeERK, the well-known involvement of the latter in oncogenictransformation may suggest that at least part of the effectmight involve the CK2-regulated nuclear accumulation ofERK described here.
The involvement of CK2 in the phosphorylation of both Serresidues in the NTS has another implication in our understand-ing of both passive and active translocation of ERK (51). SinceCK2 is a constitutively active kinase, it may phosphorylate theNTS whenever this region is not hindered. Although in restingcells most of the ERK molecules are attracted to anchoringproteins that hinder the SPS, some of them attached throughdistinct sites (32) that probably do not cover the NTS. Thesefree NTS regions may be phosphorylated by CK2 at any time,and this therefore may explain the relatively high basal NTSphosphorylation in resting cells, which results in a relativelysmall induction in this phosphorylation. In addition, this phe-nomenon may explain the free nuclear shuttle of overex-pressed proteins that are mostly free of anchoring interaction(34), leaving their NTS accessible for constitutive phosphory-lation by CK2. Another interesting issue that resulted from ourstudy is related to the consensus phosphorylation site of CK2.This site was traditionally thought to include an acidic aminoacid, 3 residues C-terminal to the phosphorylated Ser/Thr (po-sition �3), while other acidic residues in the vicinity of theSer/Thr were thought to accelerate the rate of phosphorylation(18, 36). Here we show that aside from the canonical Glu atposition �3, a phosphorylated amino acid or acidic residue atposition �2 may dictate the phosphorylation by CK2 (Ser244).Therefore, these results may expand the knowledge on CK2phosphorylation in various unknown substrates.
In summary, we found that the ERK NTS is phosphorylatedby CK2, demonstrating for the first time the cross talk between
TABLE 1. Data collection and refinement statistics for SPE-ERK2
Parameter Result for SPE-ERK2
ESRF beamline.........................................ID23-1Wavelength (Å) ........................................0.98Space group...............................................P21Unit cell parameters (Å) .........................a � 48.8; b � 70.0; c � 60.1;
� � 109.0°Resolution range
(last resolution shell) .......................50.0–1.70 (1.73–1.70)No. of unique reflections.........................40,797Redundancy...............................................3.6Rsym(I)a (%) ............................................5.8 (70.3)Completeness (%) ....................................97.1 (96.0)I/� ...............................................................36.2 (1.7)No. of protein atoms................................2,834No. of solvent atoms ................................161R factor......................................................18.8R-freeb........................................................24.5
Average B factor (Å2)Protein....................................................36.2Solvent....................................................40.8
RMSD from idealityBond length (Å) ...................................0.017Bond angle (°).......................................1.7
Ramachandran plot (PROCHECK)Favored (%)..........................................95.6Allowed (%)..........................................4.1Generously allowed (%)......................0Disallowed (%) .....................................0
a Rsym(I) � ��I��I��/�I.b Test set consists of 5% for all data.
3528 PLOTNIKOV ET AL. MOL. CELL. BIOL.

them. Unexpectedly, CK2 phosphorylates not only Ser246within its consensus site but, after initial Ser246 phosphoryla-tion, also Ser244. Ser244 is phosphorylated by activated ERKas well. We further found that Ser246 phosphorylation is suf-ficient to induce slow nuclear translocation, while pSer244cannot induce this translocation by itself but may acceleratethe pSer246 effect. Binding of inactive ERK to anchoring pro-teins (e.g., MEK1) hinders the NTS, and upon stimulation, theNTS is released to allow their phosphorylation by CK2 andERK. Finally, we crystallized the phosphomimetic mutants ofERK2 and found that they form a strong electronegative patchin the KID of ERK2, which was shown by mutations to par-ticipate in the interaction of ERK with Imp7. Overall, ourresults provide a new insight into three distinct signaling prob-lems. First, we shed light on the mechanism of ERK translo-cation into the nucleus. Second, we provide new understandingof CK2 activity by (i) demonstrating cooperation of CK2 withERK in regulating nuclear activities and (ii) identifying anunexpected phosphorylation site of CK2 (Ser244). Finally, weprovide new data regarding the binding of Imp7 to its cargoproteins.
ACKNOWLEDGMENTS
We thank Tamar Hanoch and Eldar Zehorai for their help in per-forming various experiments, David Litchfield (University of WesternOntario, London, Ontario, Canada) for the CK2 constructs, and thestaff of ESRF, Grenoble, France, for their help in maintaining andupgrading the facility.
This work was supported by the ISF research center of excellencenumber 180/09 awarded to R.S. and O.L. R.S. is an Incumbent of theYale S. Lewine and Ella Miller Lewine Professorial Chair for CancerResearch.
REFERENCES
1. Bodart, J. F. 2010. Extracellular-regulated kinase-mitogen-activated proteinkinase cascade: unsolved issues. J. Cell. Biochem. 109:850–857.
2. Brunet, A., et al. 1999. Nuclear translocation of p42/p44 mitogen-activatedprotein kinase is required for growth factor-induced gene expression and cellcycle entry. EMBO J. 18:664–674.
3. Burgermeister, E., et al. 2007. Interaction with MEK causes nuclear exportand downregulation of peroxisome proliferator-activated receptor gamma.Mol. Cell. Biol. 27:803–817.
4. Calvo, F., L. Agudo-Ibanez, and P. Crespo. 2010. The Ras-ERK pathway:understanding site-specific signaling provides hope of new anti-tumor ther-apies. Bioessays 32:412–421.
5. Casar, B., A. Pinto, and P. Crespo. 2008. Essential role of ERK dimers in theactivation of cytoplasmic but not nuclear substrates by ERK-scaffold com-plexes. Mol. Cell 31:708–721.
6. Chuderland, D., A. Konson, and R. Seger. 2008. Identification and charac-terization of a general nuclear translocation signal in signaling proteins. Mol.Cell 31:850–861.
7. Chuderland, D., and R. Seger. 2005. Protein-protein interactions in theregulation of the extracellular signal-regulated kinase. Mol. Biotechnol. 29:57–74.
8. Duncan, J. S., and D. W. Litchfield. 2008. Too much of a good thing: the roleof protein kinase CK2 in tumorigenesis and prospects for therapeutic inhi-bition of CK2. Biochim. Biophys. Acta 1784:33–47.
9. Emsley, P., and K. Cowtan. 2004. Coot: model-building tools for moleculargraphics. Acta Crystallogr. D Biol. Crystallogr. 60:2126–2132.
10. Formstecher, E., et al. 2001. PEA-15 mediates cytoplasmic sequestration ofERK MAP kinase. Dev. Cell 1:239–250.
11. Fukuda, M., Y. Gotoh, and E. Nishida. 1997. Interaction of MAP kinase withMAP kinase kinase: its possible role in the control of nucleocytoplasmictransport of MAP kinase. EMBO J. 16:1901–1908.
12. Galli, S., et al. 2009. A new paradigm for MAPK: structural interactions ofhERK1 with mitochondria in HeLa cells. PLoS One 4:e7541.
13. Gorlich, D. 1997. Nuclear protein import. Curr. Opin. Cell Biol. 9:412–419.14. Hessenauer, A., C. C. Schneider, C. Gotz, and M. Montenarh. 2011. CK2
inhibition induces apoptosis via the ER stress response. Cell. Signal. 23:145–151.
15. Kim, E. K., and E. J. Choi. 2010. Pathological roles of MAPK signalingpathways in human diseases. Biochim. Biophys. Acta 1802:396–405.
16. Kolch, W. 2005. Coordinating ERK/MAPK signalling through scaffolds andinhibitors. Nat. Rev. Mol. Cell Biol. 6:827–837.
17. Kolch, W., and A. Pitt. 2010. Functional proteomics to dissect tyrosine kinasesignalling pathways in cancer. Nat. Rev. Cancer 10:618–629.
18. Kuenzel, E. A., J. A. Mulligan, J. Sommercorn, and E. G. Krebs. 1987.Substrate specificity determinants for casein kinase II as deduced fromstudies with synthetic peptides. J. Biol. Chem. 262:9136–9140.
19. Lamzin, V. S., and K. S. Wilson. 1993. Automated refinement of proteinmodels. Acta Crystallogr. D Biol. Crystallogr. 49:129–147.
20. Litchfield, D. W. 2003. Protein kinase CK2: structure, regulation and role incellular decisions of life and death. Biochem. J. 369:1–15.
21. Lorenzen, J. A., et al. 2001. Nuclear import of activated D-ERK by DIM-7,an importin family member encoded by the gene moleskin. Development128:1403–1414.
22. Meggio, F., and L. A. Pinna. 2003. One-thousand-and-one substrates ofprotein kinase CK2? FASEB J. 17:349–368.
23. Murshudov, G. N., A. A. Vagin, and E. J. Dodson. 1997. Refinement ofmacromolecular structures by the maximum-likelihood method. Acta Crys-tallogr. D Biol. Crystallogr. 53:240–255.
24. Niefind, K., C. W. Yde, I. Ermakova, and O. G. Issinger. 2007. Evolved to beactive: sulfate ions define substrate recognition sites of CK2alpha and em-phasise its exceptional role within the CMGC family of eukaryotic proteinkinases. J. Mol. Biol. 370:427–438.
25. Nigg, E. A. 1997. Nucleocytoplasmic transport: signals, mechanisms andregulation. Nature 386:779–787.
26. Orlandini, M., et al. 1998. Protein kinase CK2alpha� is induced by serum asa delayed early gene and cooperates with Ha-ras in fibroblast transformation.J. Biol. Chem. 273:21291–21297.
27. Otwinowski, Z., and W. Minor. 1997. Processing of X-ray diffraction datacollected in oscillation mode. Methods Enzymol. 276:307–326.
28. Pinna, L. A., and J. E. Allende. 2009. Protein kinase CK2 in health anddisease: CK2: an ugly duckling in the kinome pond. Cell. Mol. Life Sci.66:1795–1799.
29. Plotnikov, A., E. Zehorai, S. Procaccia, and R. Seger. The MAPK cascades:signaling components, nuclear roles and mechanisms of nuclear transloca-tion. Biochim. Biophys. Acta, in press.
30. Potterton, E., P. Briggs, M. Turkenburg, and E. Dodson. 2003. A graphicaluser interface to the CCP4 program suite. Acta Crystallogr. D 59:1131–1137.
31. Raman, M., W. Chen, and M. H. Cobb. 2007. Differential regulation andproperties of MAPKs. Oncogene 26:3100–3112.
32. Reszka, A. A., J. C. Bulinski, E. G. Krebs, and E. H. Fischer. 1997. Mitogen-activated protein kinase/extracellular signal-regulated kinase 2 regulates cy-toskeletal organization and chemotaxis via catalytic and microtubule-specificinteractions. Mol. Biol. Cell 8:1219–1232.
33. Romieu-Mourez, R., E. Landesman-Bollag, D. C. Seldin, and G. E. Sonen-shein. 2002. Protein kinase CK2 promotes aberrant activation of nuclearfactor-kappaB, transformed phenotype, and survival of breast cancer cells.Cancer Res. 62:6770–6778.
34. Rubinfeld, H., T. Hanoch, and R. Seger. 1999. Identification of a cytoplas-mic-retention sequence in ERK2. J. Biol. Chem. 274:30349–30352.
35. Ruzzene, M., and L. A. Pinna. 2010. Addiction to protein kinase CK2: acommon denominator of diverse cancer cells? Biochim. Biophys. Acta 1804:499–504.
36. Schwartz, D., and S. P. Gygi. 2005. An iterative statistical approach to theidentification of protein phosphorylation motifs from large-scale data sets.Nat. Biotechnol. 23:1391–1398.
37. Shaul, Y. D., G. Gibor, A. Plotnikov, and R. Seger. 2009. Specific phosphor-ylation and activation of ERK1c by MEK1b: a unique route in the ERKcascade. Genes Dev. 23:1779–1790.
38. Shaul, Y. D., and R. Seger. 2006. ERK1c regulates Golgi fragmentationduring mitosis. J. Cell Biol. 172:885–897.
39. Shaul, Y. D., and R. Seger. 2007. The MEK/ERK cascade: from signalingspecificity to diverse functions. Biochim. Biophys. Acta 1773:1213–1226.
40. Smith, E. R., et al. 2010. Nuclear entry of activated MAPK is restricted inprimary ovarian and mammary epithelial cells. PLoS One 5:e9295.
41. St-Denis, N. A., and D. W. Litchfield. 2009. Protein kinase CK2 in health anddisease: from birth to death: the role of protein kinase CK2 in the regulationof cell proliferation and survival. Cell. Mol. Life Sci. 66:1817–1829.
42. Stura, E. A., and I. A. Wilson. 1991. The streak seeding technique in proteincrystallization. J. Cryst. Growth 110:270–282.
43. Tang, X., Y. Zhang, L. Tucker, and B. Ramratnam. 2010. Phosphorylation ofthe RNase III enzyme Drosha at serine300 or serine302 is required for itsnuclear localization. Nucleic Acids Res. 38:6610–6619.
44. Tresini, M., A. Lorenzini, C. Torres, and V. J. Cristofalo. 2007. Modulationof replicative senescence of diploid human cells by nuclear ERK signaling.J. Biol. Chem. 282:4136–4151.
45. Vagin, A. A., and M. N. Isupov. 2001. Spherically averaged phased transla-tion function and its application to the search for molecules and fragmentsin electron-density maps. Acta Crystallogr. D Biol. Crystallogr. 57:1451–1456.
VOL. 31, 2011 CK2 REGULATES ERK TRANSLOCATION INTO THE NUCLEUS 3529

46. Waldmann, I., S. Walde, and R. H. Kehlenbach. 2007. Nuclear import ofc-Jun is mediated by multiple transport receptors. J. Biol. Chem. 282:27685–27692.
47. Wolf, I., et al. 2001. Involvement of the activation loop of ERK in thedetachment from cytosolic anchoring. J. Biol. Chem. 276:24490–24497.
48. Xu, X., E. Landesman-Bollag, P. L. Channavajhala, and D. C. Seldin.1999. Murine protein kinase CK2: gene and oncogene. Mol. Cell.Biochem. 191:65–74.
49. Yao, X., X. Chen, C. Cottonham, and L. Xu. 2008. Preferential utilizationof Imp7/8 in nuclear import of Smads. J. Biol. Chem. 283:22867–22874.
50. Yao, Z., and R. Seger. 2009. The ERK signaling cascade—views from dif-ferent subcellular compartments. Biofactors 35:407–416.
51. Yazicioglu, M. N., et al. 2007. Mutations in ERK2 binding sites affect nuclearentry. J. Biol. Chem. 282:28759–28767.
52. Yoon, S., and R. Seger. 2006. The extracellular signal-regulated kinase:multiple substrates regulate diverse cellular functions. Growth Factors 24:21–44.
53. Zehorai, E., Z. Yao, A. Plotnikov, and R. Seger. 2010. The subcellularlocalization of MEK and ERK—a novel nuclear translocation signal (NTS)paves a way to the nucleus. Mol. Cell. Endocrinol. 314:213–220.
3530 PLOTNIKOV ET AL. MOL. CELL. BIOL.
Copyright © 2022 FDOKUMEN




















