
Analysis of the Calcium-Dependent Regulation of Proline-Rich Tyrosine Kinase 2 by...
14
Analysis of the Calcium-Dependent Regulation of Proline-Rich Tyrosine Kinase 2 by Gonadotropin- Releasing Hormone Jianjun Xie, Krystal H. Allen, Amelia Marguet, Kathie A. Berghorn, Stuart P. Bliss, Amy M. Navratil, Jun Lin Guan, and Mark S. Roberson Department of Biomedical Sciences (J.X., K.H.A., A.M., K.A.B., S.P.B., A.M.N., M.S.R.), Cornell University, Ithaca, New York 14853; and Division of Molecular Medicine and Genetics (J.L.G.), University of Michigan, Ann Arbor, Michigan 48109 Calcium influx through L-type voltage-gated calcium channels (VGCC) is required for ERK activation in- duced by GnRH in pituitary gonadotropes. The cur- rent studies investigate VGCC-sensitive catalytic ac- tivities that may lie upstream of ERKs within the GnRH signaling network. Ion exchange fractionation of T3-1 cell lysates subjected to anti-phosphoty- rosine Western blot analysis revealed a nifedipine- sensitive activity that colocalized with proline-rich tyrosine kinase (Pyk) 2 immunoreactivity. Phosphor- ylated Pyk2 was present in T3-1 cells after GnRH agonist administration for a time course that lasted up to 4 h. Pyk2 phosphorylation was also evident in gonadotropes in vivo after administration of a bolus of GnRH. Knockdown of Pyk2 using specific small interfering RNAs revealed that Pyk2 contributed to modulation of GnRH-induced ERK but not c-Jun N- terminal kinase activation. Using pharmacological approaches, calmodulin (Cam) was also demon- strated to be required for the phosphorylation of Pyk2. Pyk2 was shown to bind specifically to a Cam agarose affinity column in a calcium-dependent manner, suggesting Cam and Pyk2 are capable of forming a complex. Specific mutation of a putative Cam binding motif within the catalytic domain of Pyk2 blocked association with Cam and uncoupled Pyk2’s ability to activate ERK-dependent gene tran- scription. Thus, GnRH induces Pyk2 tyrosine phos- phorylation dependent upon calcium flux within go- nadotropes. Furthermore, association of Pyk2 and Cam may be required to mediate the effects of cal- cium on Pyk2 phosphorylation and subsequent acti- vation of ERKs by GnRH. (Molecular Endocrinology 22: 2322–2335, 2008) H YPOTHALAMIC SYNTHESIS and secretion of GnRH and expression of the type I GnRH receptor (GnRHR) in pituitary gonadotropes are central to the regulation of the hypothalmo-pituitary-gonadal axis and are required for normal reproductive function in mam- mals. In the absence of GnRH stimulation to the anterior pituitary, hypogonadism results due to a lack of gona- dotropic stimulation to the gonads (1–3). The GnRHR is a member of a superfamily of G protein-coupled recep- tors (GPCR), classically identified by seven transmem- brane-spanning domains. However, unlike other GPCRs studied to date, the GnRHR is unique in that following transmembrane domain 7, this receptor does not have an extensive carboxyl-terminal tail extending into the cytosol of the gonadotrope. This observation and sub- sequent studies supported speculation that this unique tailless attribute of the GnRHR leads to slowed internal- ization and desensitization kinetics (4, 5). Slowed desen- sitization kinetics relative to tailed GPCRs (such as the type II GnRHR) may also contribute to the complex sig- naling network induced by GnRH in gonadotrope cell models. GnRH induces the activation of a number of different second messengers and intracellular catalytic activities, including phospholipase C, diacylglycerol, and inositol 1,4,5-trisphosphate, protein kinase C iso- zymes; release of intracellular calcium; and influx of ex- tracellular calcium leading to activation of multiple MAPKs. MAPK activities induced by GnRH include the ERKs 1 and 2, c-Jun N-terminal kinase (JNK), and the p38 MAPK (reviewed in Ref. 6). Key links between these pathways and activation of several genes necessary for the differentiated function of the gonadotrope have emerged over the past decade. These include regulation of the glycoprotein hormone -subunit gene via putative Ets factors and activating transcription factor-3 (7, 8), the LH subunit and MKP-2 genes via alterations in early growth response factor (Egr)-1 activity (9–11), the FSH subunit and the GnRHR genes via regulation by activator protein-1 activity, all potentially linked to ERK and/or JNK activity induced by GnRH (12–15). We have explored the regulation of MAPK pathways by GnRH in both the T3-1 cell model as well as in rat First Published Online July 17, 2008 Abbreviations: Cam, Calmodulin; CamK, Cam-dependent kinase; Egr, early growth response factor; FAK, focal adhe- sion kinase; GnRHa, GnRH agonist; GnRHR, GnRH receptor; GPCR, G protein-coupled receptor; HA, hemagglutinin; IQGAP1, IQ domain-containing GTPase activating protein; JNK, c-Jun N-terminal kinase; MEK1, MAP/ERK kinase; PMSF, phenylmethylsulfonyl fluoride; Pyk2, proline-rich ty- rosine kinase 2; VGCC, voltage-gated calcium channel. Molecular Endocrinology is published monthly by The Endocrine Society (http://www.endo-society.org), the foremost professional society serving the endocrine community. 0888-8809/08/$15.00/0 Molecular Endocrinology 22(10):2322–2335 Printed in U.S.A. Copyright © 2008 by The Endocrine Society doi: 10.1210/me.2008-0061 2322
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of Analysis of the Calcium-Dependent Regulation of Proline-Rich Tyrosine Kinase 2 by...

Analysis of the Calcium-Dependent Regulationof Proline-Rich Tyrosine Kinase 2 by Gonadotropin-Releasing Hormone
Jianjun Xie, Krystal H. Allen, Amelia Marguet, Kathie A. Berghorn, Stuart P. Bliss, Amy M. Navratil,Jun Lin Guan, and Mark S. Roberson
Department of Biomedical Sciences (J.X., K.H.A., A.M., K.A.B., S.P.B., A.M.N., M.S.R.), CornellUniversity, Ithaca, New York 14853; and Division of Molecular Medicine and Genetics (J.L.G.),University of Michigan, Ann Arbor, Michigan 48109
Calcium influx through L-type voltage-gated calciumchannels (VGCC) is required for ERK activation in-duced by GnRH in pituitary gonadotropes. The cur-rent studies investigate VGCC-sensitive catalytic ac-tivities that may lie upstream of ERKs within theGnRH signaling network. Ion exchange fractionationof �T3-1 cell lysates subjected to anti-phosphoty-rosine Western blot analysis revealed a nifedipine-sensitive activity that colocalized with proline-richtyrosine kinase (Pyk) 2 immunoreactivity. Phosphor-ylated Pyk2 was present in �T3-1 cells after GnRHagonist administration for a time course that lastedup to 4 h. Pyk2 phosphorylation was also evident ingonadotropes in vivo after administration of a bolusof GnRH. Knockdown of Pyk2 using specific smallinterfering RNAs revealed that Pyk2 contributed tomodulation of GnRH-induced ERK but not c-Jun N-
terminal kinase activation. Using pharmacologicalapproaches, calmodulin (Cam) was also demon-strated to be required for the phosphorylation ofPyk2. Pyk2 was shown to bind specifically to a Camagarose affinity column in a calcium-dependentmanner, suggesting Cam and Pyk2 are capable offorming a complex. Specific mutation of a putativeCam binding motif within the catalytic domain ofPyk2 blocked association with Cam and uncoupledPyk2’s ability to activate ERK-dependent gene tran-scription. Thus, GnRH induces Pyk2 tyrosine phos-phorylation dependent upon calcium flux within go-nadotropes. Furthermore, association of Pyk2 andCam may be required to mediate the effects of cal-cium on Pyk2 phosphorylation and subsequent acti-vation of ERKs by GnRH. (Molecular Endocrinology22: 2322–2335, 2008)
HYPOTHALAMIC SYNTHESIS and secretion ofGnRH and expression of the type I GnRH receptor
(GnRHR) in pituitary gonadotropes are central to theregulation of the hypothalmo-pituitary-gonadal axis andare required for normal reproductive function in mam-mals. In the absence of GnRH stimulation to the anteriorpituitary, hypogonadism results due to a lack of gona-dotropic stimulation to the gonads (1–3). The GnRHR isa member of a superfamily of G protein-coupled recep-tors (GPCR), classically identified by seven transmem-brane-spanning domains. However, unlike other GPCRsstudied to date, the GnRHR is unique in that followingtransmembrane domain 7, this receptor does not havean extensive carboxyl-terminal tail extending into thecytosol of the gonadotrope. This observation and sub-
sequent studies supported speculation that this uniquetailless attribute of the GnRHR leads to slowed internal-ization and desensitization kinetics (4, 5). Slowed desen-sitization kinetics relative to tailed GPCRs (such as thetype II GnRHR) may also contribute to the complex sig-naling network induced by GnRH in gonadotrope cellmodels. GnRH induces the activation of a number ofdifferent second messengers and intracellular catalyticactivities, including phospholipase C�, diacylglycerol,and inositol 1,4,5-trisphosphate, protein kinase C iso-zymes; release of intracellular calcium; and influx of ex-tracellular calcium leading to activation of multipleMAPKs. MAPK activities induced by GnRH include theERKs 1 and 2, c-Jun N-terminal kinase (JNK), and thep38 MAPK (reviewed in Ref. 6). Key links between thesepathways and activation of several genes necessary forthe differentiated function of the gonadotrope haveemerged over the past decade. These include regulationof the glycoprotein hormone �-subunit gene via putativeEts factors and activating transcription factor-3 (7, 8), theLH� subunit and MKP-2 genes via alterations in earlygrowth response factor (Egr)-1 activity (9–11), the FSH�subunit and the GnRHR genes via regulation by activatorprotein-1 activity, all potentially linked to ERK and/or JNKactivity induced by GnRH (12–15).
We have explored the regulation of MAPK pathwaysby GnRH in both the �T3-1 cell model as well as in rat
First Published Online July 17, 2008Abbreviations: Cam, Calmodulin; CamK, Cam-dependent
kinase; Egr, early growth response factor; FAK, focal adhe-sion kinase; GnRHa, GnRH agonist; GnRHR, GnRH receptor;GPCR, G protein-coupled receptor; HA, hemagglutinin;IQGAP1, IQ domain-containing GTPase activating protein;JNK, c-Jun N-terminal kinase; MEK1, MAP/ERK kinase;PMSF, phenylmethylsulfonyl fluoride; Pyk2, proline-rich ty-rosine kinase 2; VGCC, voltage-gated calcium channel.
Molecular Endocrinology is published monthly by TheEndocrine Society (http://www.endo-society.org), theforemost professional society serving the endocrinecommunity.
0888-8809/08/$15.00/0 Molecular Endocrinology 22(10):2322–2335Printed in U.S.A. Copyright © 2008 by The Endocrine Society
doi: 10.1210/me.2008-0061
2322

pituitary cells in primary culture (16, 17). These studieshave led to the hypothesis that calcium flux from bothintra- and extracellular stores are critical for the acti-vation of several components of this signaling net-work, particularly regarding activation of the ERK andJNK modules. Most notable is the specific require-ment for influx of extracellular calcium through L-typevoltage-gated calcium channels (VGCC) on activationof the ERK cascade. Dihydropyridine receptor antag-onists such as nifedipine or acute withdrawal of extra-cellular calcium were shown to specifically blockGnRH-induced ERK (but not JNK) activation in boththe �T3-1 cell gonadotrope model and in rat pituitariesin primary culture. The impact of calcium flux on theERK cascade may be due in part to the apparentassociation between the calcium-binding protein cal-modulin (Cam) and c-Raf kinase, an upstream activa-tor of MAP/ERK kinase (MEK1) and subsequentlyERKs (18). Taken together, these data suggested amodel whereby localized or spatially restricted cal-cium signals may be playing a fundamental role inregulation of MAPK pathways within the GnRH signal-ing network.
The central aim of the current studies was to identifypossible catalytic activities capable of mediating theeffects of calcium influx through VGCCs on the acti-vation of the ERK pathway by GnRH. These studiesrevealed that GnRHR stimulation results in calcium-dependent activation of proline-rich tyrosine kinase 2(Pyk2) in the homologous gonadotrope cell line �T3-1and in vivo in mouse pituitary gonadotropes. Specificknockdown of Pyk2 using small interfering RNAs (siR-NAs) resulted in specific loss of ERK but not JNK acti-vation. Pyk2 was determined to be a Cam-interactingprotein, and Cam association with Pyk2 appears to oc-cur via a specific Cam binding site within the catalyticdomain of Pyk2.
RESULTS
GnRH Treatment Induces TyrosinePhosphorylation of Pyk2 in �T3-1 Cells
Our previous studies demonstrated that inhibition ofL-type VGCCs or withdrawal of extracellular calciumimmediately before GnRH agonist (GnRHa) (buserelin)treatment specifically blocked GnRHa-induced ERKactivation in �T3-1 cells and rat pituitary cells in pri-mary culture (16, 17). We examined the possibility thatspecific upstream activators of the ERK cascade maybe revealed by examining global tyrosine phosphory-lation induced by GnRHa in the absence or presenceof VGCC blockade by nifedipine. �T3-1 cells wereserum starved, pretreated with control solution or ni-fedipine, and then treated with saline or GnRHa.Whole-cell lysates were prepared and fractionated onMono Q ion exchange chromatography. Column frac-tions were then subjected to Western blot analysisusing an anti-phosphotyrosine antibody (Fig. 1, A–C).
GnRHa induced a number of tyrosine-phosphorylatedproteins including ERK1 and -2. Nifedipine pretreat-ment blocked GnRHa-induced ERK phosphorylationas well as tyrosine phosphorylation of several otherproteins including a rather strong activity at approxi-mately 115–120 kDa in fractions 29–32. Because Pyk2activity has been shown to be regulated by calciumand the molecular mass of Pyk2 was consistent withour observations, blots were stripped and reprobedwith a pan-specific anti-Pyk2 monoclonal antibody(Fig. 1D). The nifedipine-sensitive tyrosine-phosphory-lated band at 115–120 kDa colocalized with Pyk2immunoreactivity.
To confirm Pyk2 tyrosine phosphorylation in MonoQ fractions 29–32, fractions were reexamined byWestern blot analysis using a phosphospecific anti-body recognizing Pyk2 phosphorylated at Y402. Gn-RHa treatment clearly induced increased Pyk2 Y402
immunoreactivity in these Mono Q fractions comparedwith similar fractions obtained from saline-treated cells(data not shown). GnRH induced Pyk2 Y402 phosphor-ylation over a 4-h time course (Fig. 1E), and this effectcould be mimicked by addition of a phorbol ester as adirect pharmacological activator of protein kinase C(data not shown).
GnRHa Induces Phosphorylation of MultipleTyrosine Residues on Pyk2
Structurally, Pyk2 includes a central kinase domain,and tyrosine residues at positions Y402, Y580, and Y881
are key sites of phosphorylation (19). Using a series ofphosphospecific antibodies in Western blotting stud-ies, GnRHa administration induced phosphorylation atall three tyrosine residues within Pyk2. Pretreatmentwith nifedipine abolished Y402 and Y580 phosphoryla-tion and greatly reduced phosphorylation at Y881 (datanot shown). Consistent with the effects of nifedipineon GnRHa-induced Pyk2 phosphorylation, isotonicsubstitution of magnesium for calcium ions in the cellculture media immediately before GnRHa administra-tion also blocked GnRHa-induced phosphorylation ofPyk2 at Y402 (data not shown). This same paradigmalso blocked GnRHa-induced ERK phosphorylation asshown previously (16).
GnRHa Treatment Induces Pyk2 Phosphorylationin Mouse Gonadotropes in Vivo
To confirm an in vivo correlate to our �T3-1 cell go-nadotrope cell model, we used a GnRH immunoneu-tralization paradigm to examine the effects of a GnRHanalog on Pyk2 phosphorylation in vivo in the contextof a physiological milieu (20). Mature female mice wereovariectomized and allowed a 14-d recovery period.Mice were then administered an antiserum againstendogenous GnRH. Three days later, mice receivedeither saline injection or 100 ng of a non-immunoreactiveanalog of GnRH (D-Ala6-GnRH), and mice were killed at45 and 90 min after injection of GnRHa. Trunk blood was
Xie et al. • Pyk2 Regulation by GnRH and Calmodulin Mol Endocrinol, October 2008, 22(10):2322–2335 2323

collected at the time of exsanguination and LH measuredin a pilot study after 60 min of D-Ala6-GnRH administra-tion. In this preliminary study, mean concentrations ofLH in serum at 1 h after saline or D-Ala6-GnRH admin-istration were 0.53 � 0.31 and 53.2 � 13.2 ng/ml,respectively. Double-label fluorescent immunocyto-chemistry studies indicated an increase in phospho-Pyk2 (Y881) immunoreactivity in individual gonado-tropes in pituitary sections from animals receiving 45and 90 min of D-Ala6-GnRH treatment compared withcontrols (Fig. 2). Phospho-Pyk2 Y881 was compart-mentalized primarily to the cytosol and colocalizedwith LH� immunoreactivity to identify gonadotropepopulations within the anterior pituitary. Preimmunerabbit serum was used as a negative control in lieu ofthe phospho-Pyk2 (Y881) antiserum. These studiesconfirm that Pyk2 becomes phosphorylated by Gn-RHa administration in vivo and supports the utility ofthe �T3-1 cell model in our studies.
Knockdown of Pyk2 Using siRNA Resultsin Modulation of GnRHa-InducedERK Phosphorylation
Thus far, our data suggest the possibility that Pyk2may potentially lie upstream of the ERK pathway ingonadotropes. In an effort to determine the functionalconsequence of Pyk2 in the GnRH signaling network,we designed a series of siRNAs against Pyk2. Fourstable cell lines were created that overexpressed ei-ther a control scrambled siRNA or three distinct siR-NAs for mouse Pyk2. All three Pyk2 siRNA cell lineshad reduced levels of Pyk2 protein; however, none ofthese siRNAs used alone produced knockdowngreater that 20–30% (data not shown). To increase theefficacy of the Pyk2 knockdown, a stable cell line wascreated using all three siRNAs as a mixture. The resultantcell line demonstrated Pyk2 knockdown of 52.0 � 6.0%over all studies (P � 0.05; Fig. 3). Using this cell line
A B
C D
E
Pyk2 p-Y402
GnRHa (h) 0 0.5 1 2 4
Pan Pyk2
20 21 22 23 24 25 26 27 28 29 30 31 32200
50
15
25
75
150100
50
15
25
75
200150100
MW(Kda)
50
15
25
75
200150100
50
15
25
75
200150100
IB: pY - Control IB: pY - GnRHa
IB: pY - nifedipine + GnRHa IB: Pyk2
Pyk2
ERKs
FPLC Fraction #
ERKs
Fig. 1. Detection of Nifedipine-Sensitive Tyrosine Phosphorylation Implicates Pyk2 in the GnRHa Signaling Network in �T3-1 Cells�T3-1 whole-cell lysates were fractionated by FPLC using an ion-exchange Mono Q column. Before fractionation, cells were
treated with control solution (A), GnRHa buserelin (10 nM, 15 min; B), or nifedipine (2 �M) followed by GnRHa (nifedipine � GnRHa;C). Mono Q fractions 20–32 were resolved by SDS-PAGE and subjected to phosphotyrosine immunoblot analysis (IB: pY; A–C).The molecular markers are listed to the left of each panel. Short arrowheads identify tyrosine phosphorylation of ERKs in B andC. In D, blots were stripped and reprobed with antibody against Pyk2. Longer arrows identify tyrosine phosphorylation andcolocalization with Pyk2 immunoreactivity. GnRHa treatment (100 nM) induced phosphorylation at Pyk2 Y402 as determined inwhole-cell �T3-1 lysates over a 4-h time course (E) using a phosphospecific antibody and Western blot analysis. A Pyk2 antibody(Pan Pyk2) was used to demonstrate equivalent lane loading in E. MW, Molecular weight.
2324 Mol Endocrinol, October 2008, 22(10):2322–2335 Xie et al. • Pyk2 Regulation by GnRH and Calmodulin

(along with the control siRNA cell line), we examined thephosphorylation state of ERK and JNK proteins afterGnRHa treatment. GnRHa-induced ERK phosphoryla-tion was blunted (P � 0.05) in the Pyk2 siRNA cell linecompared with the control siRNA cell line (Fig. 2). Forexample, using the control siRNA cell line, ERK phos-phorylation increased 10.4-fold at the 30-min time point.ERK phosphorylation induced by GnRHa in the Pyk2siRNA cell line was increased by 3-fold. In contrast,GnRHa-induced phosphorylation of JNK was not ap-preciably altered (P � 0.05) comparing these cell lines(Fig. 3). Importantly, control and Pyk2-specific siRNAsdid not affect expression levels of actin, ERK2, or JNK1over all of the experiments, suggesting the effects of thePyk2 siRNA mixture were specific. These results supportthe conclusion that specific disruption of Pyk2 proteinlevels in �T3-1 cells uncouples the ability of the GnRHRto effectively activate the ERK cascade.
These Pyk2 knockdown studies were further corrob-orated by transfection studies examining the ability ofGnRHa to induce known ERK- and JNK-dependent re-porter gene systems (Fig. 4A). The c-Fos gene promoter
has been shown to be ERK dependent within the GnRHsignaling network (10, 16, 21), and expression of thisreporter gene construct after GnRHa administration wasmarkedly reduced (P � 0.05) in the Pyk2 siRNA cell line(Fig. 4A). In contrast, the GnRHR promoter-luciferasereporter has been shown to be sensitive to JNK pathwayactivity and relatively insensitive to ERK activity in �T3-1cells (22). Consistent with these observations, GnRHa-induced (fold) activation of the GnRHR promoter-lucif-erase reporter was similar in all siRNA cell lines. A re-duction in basal expression of the GnRHR luciferasereporter was detected in the Pyk2 siRNA cell line com-pared with the control cell line (P � 0.05).
To provide additional corroboration to the Pyk2 siRNAdata, studies examined the ability of a dominant-nega-tive form of Pyk2 to interfere with GnRHa-induced ex-pression of a known ERK target, the transcription factorElk1 (Fig. 4B). GnRHa induced a robust activation of aGal4-Elk1 fusion protein that was nearly abolished withpretreatment using the MEK1 inhibitor PD98059 (P �0.05). Overexpression of a dominant-negative form ofPyk2 also reduced (P � 0.05) the ability of GnRH signal-ing to transactivate the Gal4-Elk1 fusion protein. Com-bined, these studies provide strong evidence that acti-
LH βphospho-
Pyk2 Merge
NRSdAla6-GnRH(t = 90 min)
Control
dAla6-GnRH(t = 90 min)
dAla6-GnRH(t = 45 min)
N NN
N NN
N NN
N NN
-
-
-
-
N NN
N NN
N NN
N NN
Fig. 2. GnRH Induces Pyk2 Phosphorylation in VivoMature ovariectomized mice received anti-GnRH anti-
serum (250 �l) ip to immunoneutralize endogenous GnRH.Three days later, immunoneutralized mice received eithersaline or an analog of GnRH (D-Ala6-GnRH; 100 ng ip) andwere killed 45 or 90 min later. Pituitaries were dissected out,fixed, sectioned, and examined for Pyk2 (pY881) phosphory-lation or LH� protein by double-labeled fluorescent immuno-cytochemistry. The first column depicts labeling for LH�(green), the second column of images phospho-Pyk2 (pY881;red), and the final column of images is a merge of the twoimages. The first row depicts results from cells collected frommice receiving saline for 60 min (control). The second andthird rows of images depict cells collected 45 and 90 min(respectively) after D-Ala6-GnRH administration. The fourthrow of images is immunocytochemical analysis using normalrabbit serum substituted for the phospho-Pyk2 antibody ontissues collected at 90 min after D-Ala6-GnRH treatment. Thefigure reflects a representative example from three or fourmice per treatment. n, Nucleus. Bar, 5 �m.
Cont siRNA Pyk2 siRNA mix
Pyk2
Actin
phospho-ERKs
GnRHa (mins) 0 15 30 60 0 15 30 60
ERK2
phospho-JNKs
JNK1
-
-
Fig. 3. Knockdown of Pyk2 Using siRNA Uncouples GnRHaStimulation with Activation of the ERK But Not JNK Pathway
Preliminary studies identified three separate Pyk2 siRNAsthat all had partial knockdown effects when transfected into�T3-1 cells. A stably transfected population of �T3-1 cellswas created using a mixture of all three Pyk2 siRNAs (Pyk2siRNA mix). A second stable cell line was transfected with acontrol siRNA. After selection, GnRHa (10 nM) was adminis-tered over a 60-min time course to both cell lines, and whole-cell lysates were prepared and subjected to Western blotanalysis using antibodies for Pan-Pyk2, actin, phospho-ERKs, ERK2, phospho-JNK, and JNK1. Actin, ERK2, andJNK1 immunoreactivity was used to confirm the specificity ofthe siRNAs used as well as lane loading controls.
Xie et al. • Pyk2 Regulation by GnRH and Calmodulin Mol Endocrinol, October 2008, 22(10):2322–2335 2325

vation of Pyk2 within the GnRH signaling network isnecessary for ERK pathway activation and subsequentERK-dependent gene promoter activity.
Inhibition of Cam But Not Cam-DependentKinase (CamK) II Blocks GnRHa-Induced Pyk2Phosphorylation in �T3-1 Cells
We have previously demonstrated that pharmaco-logical perturbation of Cam resulted in blockade in
GnRH-induced signaling to ERK (18). Because Pyk2appears to reside within the GnRH pathway leading toERK activation (Figs. 3 and 4) and Pyk2 is known to becalcium-dependent, it was logical to consider the pos-sibility that Cam may be playing a role as a calciumsensor mediating the effects of VGCC calcium onPyk2 activity and subsequently ERK phosphorylation.To assess the potential contribution of Cam in theregulation of Pyk2 phosphorylation by GnRHa, W7 (a
A
Luci
fera
se A
ctiv
ity(li
ght u
nits
/µg
prot
ein)
[ [
0
10000
20000
30000ControlGnRHa
c-Fos Luc
Fold 56 35 [ [
Con
t siR
NA
Pyk2
siR
NA
mix
0
1000
2000
3000GnRH-R Luc
13 16
Con
t siR
NA
Pyk2
siR
NA
mix
Luci
fera
se A
ctiv
ity(li
ght u
nits
/µg
prot
ein)
[ [
0
10000
20000
30000ControlGnRHa
c-Fos Luc
Fold 56 35 [ [
Con
t siR
NA
Pyk2
siR
NA
mix
0
1000
2000
3000GnRH-R Luc
13 16
Con
t siR
NA
Pyk2
siR
NA
mix
B
0
500
1000
1500
ControlGnRHa
[Py
k2 K
D
3.0[PD
9805
9
2.7[C
ontr
ol
32.3
1 2
HA
Gal4-Elk1 + 5xGal4-E1b Luc
Luc
ifera
se A
ctiv
ity(li
ght u
nits
/µg
prot
ein)
Fold
a
b
a
c
a
b
c
b
a
b
a a a
c
Fig. 4. Knockdown of Pyk2 or Overexpression of a Dominant-Negative Pyk2 in �T3-1 Cells Results in Inhibition of ERK- But NotJNK-Dependent Gene Transcription
A, c-Fos and GnRHR luciferase reporter genes (c-Fos-Luc and GnRHR-Luc, respectively; 1 �g/reporter) were transfected intostable cell lines expressing control (Cont siRNA) or Pyk2 (Pyk2 siRNA mix) siRNAs as described in Fig. 3. After transfection, cellsreceived control solution (saline) or GnRHa for 6 h. Transfected cells were then lysed, and luciferase activity was determined permicrogram total cell protein. Fold induction relative to saline-treated controls is shown. B, �T3-1 cells were transfected with anexpression vector for a Gal4 DNA binding domain-Elk1 fusion protein along with a luciferase reporter containing five copies ofthe Gal4 binding site cloned upstream of the E1b minimal promoter (Gal4-Elk1 � 5xGal4–E1b Luc; 500 ng of each plasmid). Sometransfected cells received dimethylsulfoxide or PD98059 (50 �M) 30 min before administration of either saline (control) or GnRHafor 6 h. Some transfections included a control expression vector (pKH3) or an expression vector for an HA-tagged dominant-negative kinase dead Pyk2 (Pyk2KD; 5 �g). Luciferase activity was assayed as described above. Fold induction relative to controlis shown. In addition, expression level of Pyk2KD was determined by Western blot analysis using an HA antibody (see inset: lane1, pKH3 control plasmid; lane 2, Pyk2KD).
2326 Mol Endocrinol, October 2008, 22(10):2322–2335 Xie et al. • Pyk2 Regulation by GnRH and Calmodulin

relatively nonspecific Cam inhibitor) was used to in-hibit Cam (Fig. 5A). Pretreatment of �T3-1 cells withW7 for 30 min greatly diminished (P � 0.05) Pyk2 Y402
phosphorylation induced by GnRHa. For example, 30min after GnRHa administration, ERK phosphorylationwas increased 3.0 � 1.0-fold in control treated cells;GnRHa administration induced a 1.3 � 0.27-fold in-crease in cells pretreated with W7. Similar results wereobtained using another Cam inhibitor, W13 (data notshown). In contrast, KN62 (a Cam inhibitor with greaterspecificity toward Cam-dependent protein kinases)did not disrupt GnRHa-induced Pyk2 phosphorylation(Fig. 5B).
The Catalytic Domain of Pyk2 Binds toCam Directly
To examine the possibility that Cam and Pyk2 mayinteract directly, �T3-1 cell lysates were prepared andapplied to a Cam agarose affinity column in the ab-sence or presence of calcium. The column waswashed extensively and Cam-binding proteins wereeluted by chelating calcium within the column. Analy-sis of the elution fractions revealed the presence ofIQ domain-containing GTPase activating protein(IQGAP1) as a positive control (Fig. 6). Notably, Pyk2was retained on the Cam affinity column in a calcium-dependent manner, whereas the related tyrosine ki- nase, Src, did not bind to this column, providing evi-
dence that Pyk2 interaction with the Cam affinitycolumn was specific and calcium dependent.
To determine whether the activity state of Pyk2 mayaffect Cam binding, we synthesized recombinant wild-type and mutant forms of full-length Pyk2 with muta-tions in either the kinase domain (catalytically inactive)or at residue Y402. 35S-labeled proteins were sub-jected to Cam agarose binding in the absence or pres-ence of calcium. As observed with endogenously ex-pressed Pyk2 derived from �T3-1 cells, recombinantPyk2 bound Cam agarose in a calcium-dependentmanner. All of the forms of Pyk2 tested bound the Camcolumn in a calcium-dependent manner, suggestingthat kinase activity or activation state related to phos-phorylation at Y402 does not determine the ability ofPyk2 to form complexes with Cam (data not shown).
We next investigated the possibility that a putativeCam binding site may be identifiable within the primaryamino acid sequence of Pyk2. This search revealedthe presence of a potential Cam binding site withinthe catalytic domain of Pyk2 that was conserved inmouse and human (Fig. 7A). Potential Cam bindingsites are generally characterized by IQ domains or thepresence of hydrophobic residues within amphiphilic�-helices where the hydrophobic residues are spacedin positions 1–10, 1–14, or 1–16 or some variant of thispositioning (as reviewed in Ref. 23). Within the Pyk2catalytic domain, putative Cam binding sites includeda 1–14, 1–8-14, and 1–10 motif (Fig. 7A). Making use ofweb-based protein structure prediction programs(ProteinPredict and nnPredict), the hydrophobic resi-dues at positions 1, 10, and 14 (positions 514, 523,
GnRHa (min) 0 15 30 0 15 30
W7Control
Pyk2 p-Y402
Pan Pyk2
A
B
GnRHa (min) 0 15 30 0 15 30 0 15 30
Control
Pyk2 p-Y402
Pan Pyk2
KN62(10 µM)
KN62(5 µM)
Fig. 5. GnRHa-Induced Pyk2 Phosphorylation Is Blocked byCam Inhibition
�T3-1 cells were pretreated with saline (control) or the Caminhibitor W7 (15 �M) for 30 min. Cells then received GnRHa for15 or 30 min, and whole-cell lysates were prepared. Westernblot analysis was used to determine the phosphorylation statusof Pyk2 (p-Y402). A, The Pan-Pyk2 antibody was used to deter-mine lane loading. B, A similar study was performed except cellswere pretreated with increasing doses of the CamK II-specificinhibitor KN62 (5 or 10 �M), and Western blots were used todetermine phosphorylation status of Pyk2 as described above.
CalmodulinColumn Elutions
1 2 3 4 1 2 3 420%
Inpu
t
+ Ca2+ - Ca2+
Pyk2
IQGAP
Src
Fig. 6. Pyk2 Binds to a Cam Agarose Affinity Column�T3-1 whole-cell lysates were prepared (see Materials and
Methods) and subjected to Cam agarose affinity chromatog-raphy in the presence or absence of Ca2� followed by columnelution with EGTA. Input lysates (20%; positive control forWestern blots) and fractions 1–4 were resolved on SDS-PAGE, and Western blot analysis was used to determine thepresence of IQGAP, Pyk2, and Src.
Xie et al. • Pyk2 Regulation by GnRH and Calmodulin Mol Endocrinol, October 2008, 22(10):2322–2335 2327

and 527 of mouse Pyk2) within the putative Cam bind-ing site aligned with predicted �-helices. Although thecrystal structure of the catalytic domain of Pyk2 hasnot been solved, the catalytic domain of a closelyrelated family member, focal adhesion kinase (FAK)has been crystallized and solved (24). Using this as amodel, the predicted positioning of �-helices withinthe catalytic domain of Pyk2 and FAK (using Protein-Predict and nnPredict) were consistent with the crystalstructure in the region of the putative Cam binding site(Fig. 7B).
We then determined whether the catalytic domain ofPyk2 alone was sufficient to bind Cam in a calcium-dependent manner. For these studies, �T3-1 cells
were transiently transfected with expression vectorsfor hemagglutinin (HA)-tagged full-length wild-typePyk2, a truncated form of Pyk2 (Pyk2411–696) that spe-cifically represented the catalytic domain and an ex-pression vector for HA-tagged ERK2 as a negativecontrol (Fig. 8A). Transfected cells were cultured for24 h, lysed, and subjected to the Cam agarose bindingassay described above. As predicted, full-length Pyk2clearly bound Cam as did the Pyk2 catalytic domain ina calcium-dependent fashion. HA-tagged ERK2 didnot bind the Cam agarose. Thus, the catalytic domainof Pyk2 alone was sufficient for Cam interactions.Based upon these studies, we prepared alanine sub-stitution mutations within the putative Cam binding
A1 411 1008696
NTD CAT PBD
.Catalytic domain Cam binding site
Mouse 514 NH3 LERNKNSLKVPTLV COOHHuman 514 NH3 LERNKNSLKVLTLV COOH
Mutant 514 NH3 AERNKNSAKALTLA COOH
(1-14) 1 14 (1-8-14) 1 8 14
(1-10) 1 10
.
A
--
.
-
B
Pyk2 454 VAVKTCKKDCTLDN-KEKFMSEAVIMKNLDHPHIVKLIGIIEEEPTWIIMELYPYGELGH 512VA+KTCK +CT D+ +EKF+ EA+ M+ DHPHIVKLIG+I E P WIIMEL GEL
FAK 451 VAIKTCK-NCTSDSVREKFLQEALTMRQFDHPHIVKLIGVITENPVWIIMELCTLGELRS 509
* * * *Pyk2 513 YLERNKNSLKVLTLVLYSLQICKAMAYLESINCVHRDIAVRNILVASPECVKLGDFGLSR 572
+L+ K SL + +L+LY+ Q+ A+AYLES VHRDIA RN+LV+S +CVKLGDFGLSR FAK 510 FLQVRKYSLDLASLILYAYQLSTALAYLESKRFVHRDIAARNVLVSSNDCVKLGDFGLSR 569
Pyk2 573 YIEDEDYYKASVTRLPIKWMSPESINFRRFTTASDVWMFAVCMWEILSFGKQPFFWLENK 632Y+ED YYKAS +LPIKWM+PESINFRRFT+ASDVWMF VCMWEIL G +PF ++N
FAK 570 YMEDSTYYKASKGKLPIKWMAPESINFRRFTSASDVWMFGVCMWEILMHGVKPFQGVKNN 629
Pyk2 633 DVIGVLEKGDRLPKPDLCPPVLYTLMTRCWDYDPSDRPRFTELVCSLSDV 682DVIG +E G+RLP P CPP LY+LMT+CW YDPS RPRFTEL LS +
FAK 630 DVIGRIENGERLPMPPNCPPTLYSLMTKCWAYDPSRRPRFTELKAQLSTI 679
Fig. 7. A Putative Cam Binding Site Is Present within the Catalytic Domain of Pyk2A, Schematic of mouse Pyk2 highlighting the amino-terminal domain (NTD), the catalytic domain (CAT), and the paxillin-binding
domain (PBD). Amino acid sequences for mouse and human Pyk2 were subjected to a web-based search engine to identifypotential Cam binding sites (Calmodulin Target Database). The 14 residues shown are about 93% conserved between mouse andhuman and match potential (1–14; [FILVW]xxxxxxxxxxxx[FILVW]) or (1–8-14; [FILVW]xxxxxx [FAILVW]xxxxx[FILVW]) or (1–10;[FILVW]xxxxxxxx[FILVW]) Cam binding sites. Within these putative sites, alanine was substituted for key hydrophobic residuesat positions 1, 8, 10, and 14 (Mutant). B, Alignment of the catalytic domains of human Pyk2 and FAK. This alignment revealed a64% identity within this region. The positioning of the FAK secondary structures [�-sheets (flat ribbon shape) and �-helices (curledribbon shape)] based upon crystallography studies (24) is shown. Asterisks identify the positioning of the hydrophobic residueswithin the putative Cam binding site. The residues shown in bold for Pyk2 and FAK indicate structural predictions using nnPredictand ProteinPredict (ca.expasy.org).
2328 Mol Endocrinol, October 2008, 22(10):2322–2335 Xie et al. • Pyk2 Regulation by GnRH and Calmodulin
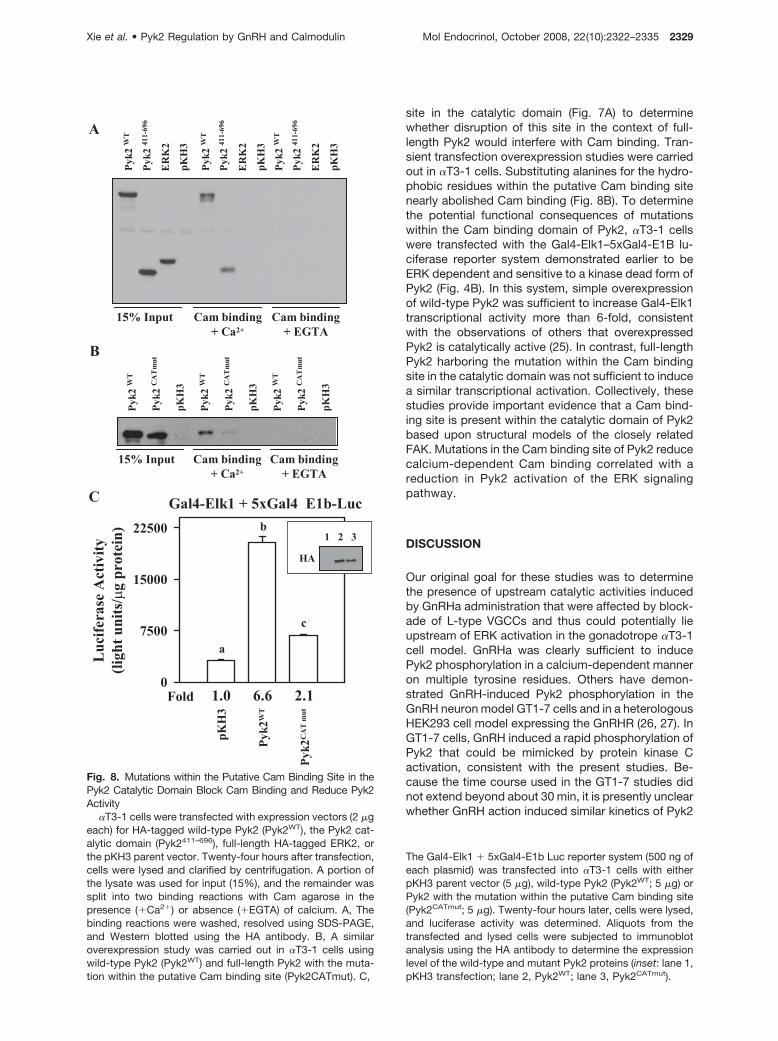
site in the catalytic domain (Fig. 7A) to determinewhether disruption of this site in the context of full-length Pyk2 would interfere with Cam binding. Tran-sient transfection overexpression studies were carriedout in �T3-1 cells. Substituting alanines for the hydro-phobic residues within the putative Cam binding sitenearly abolished Cam binding (Fig. 8B). To determinethe potential functional consequences of mutationswithin the Cam binding domain of Pyk2, �T3-1 cellswere transfected with the Gal4-Elk1–5xGal4-E1B lu-ciferase reporter system demonstrated earlier to beERK dependent and sensitive to a kinase dead form ofPyk2 (Fig. 4B). In this system, simple overexpressionof wild-type Pyk2 was sufficient to increase Gal4-Elk1transcriptional activity more than 6-fold, consistentwith the observations of others that overexpressedPyk2 is catalytically active (25). In contrast, full-lengthPyk2 harboring the mutation within the Cam bindingsite in the catalytic domain was not sufficient to inducea similar transcriptional activation. Collectively, thesestudies provide important evidence that a Cam bind-ing site is present within the catalytic domain of Pyk2based upon structural models of the closely relatedFAK. Mutations in the Cam binding site of Pyk2 reducecalcium-dependent Cam binding correlated with areduction in Pyk2 activation of the ERK signalingpathway.
DISCUSSION
Our original goal for these studies was to determinethe presence of upstream catalytic activities inducedby GnRHa administration that were affected by block-ade of L-type VGCCs and thus could potentially lieupstream of ERK activation in the gonadotrope �T3-1cell model. GnRHa was clearly sufficient to inducePyk2 phosphorylation in a calcium-dependent manneron multiple tyrosine residues. Others have demon-strated GnRH-induced Pyk2 phosphorylation in theGnRH neuron model GT1-7 cells and in a heterologousHEK293 cell model expressing the GnRHR (26, 27). InGT1-7 cells, GnRH induced a rapid phosphorylation ofPyk2 that could be mimicked by protein kinase Cactivation, consistent with the present studies. Be-cause the time course used in the GT1-7 studies didnot extend beyond about 30 min, it is presently unclearwhether GnRH action induced similar kinetics of Pyk2
A
B
Pyk2
WT
Pyk2
CA
Tm
ut
pKH
3
Pyk2
WT
Pyk2
CA
Tm
ut
pKH
3
Pyk2
WT
Pyk2
CA
Tm
ut
pKH
3
15% Input Cam binding+ EGTA
Cam binding+ Ca2+
Pyk2
WT
Pyk2
411-
696
ER
K2
Pyk2
WT
Pyk2
411-
696
ER
K2
Pyk2
WT
Pyk2
411-
696
ER
K2
15% Input Cam binding+ EGTA
Cam binding+ Ca2+
pKH
3
pKH
3
pKH
3
C
pKH
3
Pyk2
WT
Luc
ifera
se A
ctiv
ity(li
ght u
nits
/µg
prot
ein)
0
7500
15000
22500
Gal4-Elk1 + 5xGal4 E1b-Luc
Pyk2
CA
T m
ut
2.16.61.0Fold
1 2 3
HA
a
b
c
Fig. 8. Mutations within the Putative Cam Binding Site in thePyk2 Catalytic Domain Block Cam Binding and Reduce Pyk2Activity
�T3-1 cells were transfected with expression vectors (2 �geach) for HA-tagged wild-type Pyk2 (Pyk2WT), the Pyk2 cat-alytic domain (Pyk2411–696), full-length HA-tagged ERK2, orthe pKH3 parent vector. Twenty-four hours after transfection,cells were lysed and clarified by centrifugation. A portion ofthe lysate was used for input (15%), and the remainder wassplit into two binding reactions with Cam agarose in thepresence (�Ca2�) or absence (�EGTA) of calcium. A, Thebinding reactions were washed, resolved using SDS-PAGE,and Western blotted using the HA antibody. B, A similaroverexpression study was carried out in �T3-1 cells usingwild-type Pyk2 (Pyk2WT) and full-length Pyk2 with the muta-tion within the putative Cam binding site (Pyk2CATmut). C,
The Gal4-Elk1 � 5xGal4-E1b Luc reporter system (500 ng ofeach plasmid) was transfected into �T3-1 cells with eitherpKH3 parent vector (5 �g), wild-type Pyk2 (Pyk2WT; 5 �g) orPyk2 with the mutation within the putative Cam binding site(Pyk2CATmut; 5 �g). Twenty-four hours later, cells were lysed,and luciferase activity was determined. Aliquots from thetransfected and lysed cells were subjected to immunoblotanalysis using the HA antibody to determine the expressionlevel of the wild-type and mutant Pyk2 proteins (inset: lane 1,pKH3 transfection; lane 2, Pyk2WT; lane 3, Pyk2CATmut).
Xie et al. • Pyk2 Regulation by GnRH and Calmodulin Mol Endocrinol, October 2008, 22(10):2322–2335 2329

phosphorylation seen in the present studies. Pyk2 ac-tivity induced by GnRH was critical for ERK activationin GT1-7 cells; however, this mechanisms did notinvolve GnRHR-dependent shedding of epidermalgrowth factor isoforms (28). In the heterologousHEK293 cell model where GnRHR is overexpressed,treatment with GnRH induced a rapid phosphorylationof Pyk2 and translocation into the nuclear compart-ment; however, the role of nuclear Pyk2 is presentlynot clear. Interestingly, Pyk2 phosphorylation inHEK293 cells did not appear to contribute to ERKactivation by GnRH (27). In the present studies,GnRHa induced Pyk2 phosphorylation for an ex-tended time course (up to 4 h), and phosphorylation ofY881 was localized primarily to the cytosol in vivo inpituitary gonadotropes. Consistent with the GT1-7and HEK293 cell studies, GnRHa-induced Pyk2 phos-phorylation in the homologous �T3-1 cell line could bemimicked by the addition of phorbol ester via diacyl-glycerol-dependent protein kinase C isozymes. Mostrecently, Pamela Brown and her colleagues (29) pro-vided important new evidence that GnRH activation ofthe ERK pathway was indeed linked to phosphoryla-tion of Pyk2 in the �T3-1 and L�T2 gonadotrope cellmodels. The present studies extend these findings inseveral key areas. First, the present studies provideimportant corroboration of Pyk2 phosphorylation onmultiple specific tyrosine residues in the gonadotrope-derived cell line �T3-1. Our studies provide evidencefor the fidelity of these cell lines because Pyk2 appearsto be phosphorylated in vivo in response to GnRHadministration using our GnRH immunoneutralizationmouse model (Fig. 2). Our Pyk2 knockdown studiesprovide evidence for the role of Pyk2 upstream of theERK but not JNK modules with the GnRH signalingnetwork (Figs. 3 and 4). Lastly, our studies define acritical interface between the calcium-binding proteinCam and Pyk2 through a discrete interaction withinthe catalytic domain of Pyk2 (Figs. 6–8).
Our original prediction was that Pyk2 would bean important intermediate effectively linking GnRH-induced ERK activation with influx of extracellular cal-cium through L-type VGCCs consistent with earlierstudies (16, 17). A number of studies have implicatedPyk2 as a signaling intermediate upstream of ERKs ina diverse array of cell types. In the central nervoussystem, Pyk2 is widely expressed and thought to be afunctional link between calcium and MAPK signaling(30). In cardiac fibroblasts, angiotensin II induces acalcium/Cam-dependent increase in Pyk2 phosphor-ylation that is functionally linked to downstream acti-vation of the ras/ERK cascade (31). Our initial studiesexamining the effects of nifedipine on influx of calciumthrough VGCCs and Pyk2 phosphorylation induced byGnRHa were consistent with the hypothesis that Pyk2was potentially upstream of ERK activation in theGnRH signaling network.
The specific mechanism(s) by which calcium medi-ates Pyk2 activation is not currently clear. Activation ofPyk2 appears to involve a complex mechanism of
trans-acting autophosphorylation of Y402, followed byphosphorylation on additional tyrosine residues thatlead to full activation of Pyk2, capable of phosphory-lation of downstream substrates (32). In the presentstudies, GnRH-induced Pyk2 phosphorylation in�T3-1 cells clearly required mobilization of cell calciumand could be blocked by Cam inhibitors. We reasonedthat Cam may serve as a calcium sensor in this sys-tem, and our studies implicate a role for Cam in thiscapacity. Others have demonstrated a requirement forcalcium/Cam-dependent kinases in calcium-mediatedPyk2 phosphorylation. In studies using carbachol andangiotensin II as agonists, Pyk2 phosphorylation wasclearly calcium/Cam dependent in kidney cells (33).Similar observations were reported for KCl/depolariza-tion- and bradykinin-stimulated PC12 cells (34). In thelatter study, the signaling link between calcium influxand Pyk2 phosphorylation induced by membrane de-polarization was dependent upon CamK II, whereasthe effects of bradykinin appeared to be independentof CamK II but fully dependent on Cam. Other studiesexamining the effects of cerebral ischemia on Pyk2phosphorylation in rats (35) or the induction of Pyk2phosphorylation with ionomycin in vascular smoothmuscle cells (36) demonstrate sensitivity to or require-ment for CamKs to mediate Pyk2 phosphorylation.The role of GnRH-induced CamK II activity was re-cently demonstrated in L�T2 cells and in rat pituitarycells, demonstrating that inhibition of CamK II affectedthe regulation of gonadotropin subunit gene expres-sion (37, 38). Thus, although GnRH is clearly capableof activating CamK II, its role in the calcium/Cam-dependent phosphorylation of Pyk2 appears to benegligible based upon limited pharmacological studiespresented here. It will be interesting to determinewhether the trans-acting autophosphorylation of Pyk2recently reported (32) is sensitive to Cam disruption,suggesting a possible role for calcium and Cam in theinitial activation steps leading to Pyk2 catalytic activityvia aggregation of Pyk2.
Corroborating the pharmacology studies describedabove was the observation that Pyk2 could be re-tained on a Cam agarose column, and this associationwas dependent upon the presence of calcium, sug-gesting the formation of a putative Cam-Pyk2 com-plex. Another nonreceptor tyrosine kinase, Src, did notassociate with the Cam agarose column, whereas aknown Cam-interacting protein, IQGAP, was retainedon the column, contributing to the notion that Pyk2association with the Cam agarose affinity column wasindeed specific. Because Pyk2 and Src have beenshown to specifically interact associated with integrinstimulation (reviewed in Ref. 19) and in the context ofthe GnRH signaling network (29), it is interesting tonote a lack of Src binding to the Cam agarose columnin conjunction with Pyk2. It is certainly plausible thatSrc cannot bind Cam directly, and the conditions usedin the present studies were incompatible with Pyk2-Src association in the binding assays. Additional stud-
2330 Mol Endocrinol, October 2008, 22(10):2322–2335 Xie et al. • Pyk2 Regulation by GnRH and Calmodulin

ies will be necessary to determine the precise interac-tions between Pyk2, Cam, and potentially Src.
The solved crystal structure of the catalytic domainof FAK (24) was instrumental to our prediction of aspecific Cam binding interface within the catalytic do-main of Pyk2. Modeling studies provide the basis forthe prediction that key �-helices and hydrophobic res-idues adjacent to the putative catalytic cleft (predictedbased upon FAK structure) form the interface for Camassociation. Two separate structure prediction data-bases (ProteinPredict and nnPredict) accurately pre-dicted �-helices within the Pyk2 catalytic domain thatwere entirely consistent with the FAK crystal structure(Fig. 7B). Indeed, the catalytic domain alone was suf-ficient to bind Cam, suggesting that the folding of thisdomain was likely correct. The predicted Cam bindingsite within the catalytic domain of Pyk2 was charac-terized by either a 1–10 or 1–14 motif. Unique to Pyk2was that the �-helix containing the hydrophobic resi-due in position 1 in either motif was separated from thehydrophobic residue at position 10 or 14 by a nonhe-lical stretch of amino acids (Fig. 7B). Generally, the1–10 or 1–14 motif resides within an uninterrupted�-helix. Mutations within this putative Cam bindingsite were sufficient to block Cam-Pyk2 physical inter-actions and Pyk2 functional activity (Fig. 8). The po-tential caveat here is that alanine substitution muta-tions within the putative Cam binding site resulted inaberrant folding of full-length Pyk2 in our studies.However, this seems unlikely because modeling thePyk2 catalytic domain (based upon the FAK structure)with these alanine substitutions did not alter theprediction of �-helices within this domain (data notshown).
The substrates for Pyk2 catalytic activity within go-nadotropes are currently not known, limiting our un-derstanding of the functional consequence of Pyk2activation within the GnRH signaling network. Re-markable tyrosine phosphorylation of intracellular pro-teins is evident after GnRH administration to �T3-1cells, presumably some of which reflects substrates ofactivated Pyk2 (Fig. 1). Indirectly, Pyk2 substrateswould include any of the putative gene targets forERK activation. This would be consistent with the re-cent findings that Pyk2 perturbation reduced GnRH-induced LH� subunit gene expression in L�T2 cells(29). Consistent with this finding are studies from ourgroup suggesting that Cam activity is required forGnRH-induced up-regulation of the immediate-earlygene, Egr-1 (18), a required transcription factor sup-porting LH� subunit gene expression in vivo (9). Sev-eral potential direct substrates of Pyk2 have also beendescribed. In a CHO cell model, Pyk2 was shown todirectly tyrosine phosphorylate glycogen synthase ki-nase 3B, a serine threonine kinase known to play animportant role in many cellular functions (39). Metabo-tropic glutamate receptor 1 activity appears to bemodulated by Pyk2/Src activity in cortical neurons(40). The large-conductance, calcium-activated K(BKCa) channel is directly phosphorylated by Src fam-
ily members including Pyk2 functionally enhancingBKCa channel activity (41). And likely one of the bestcharacterized substrates of Pyk2, gelsolin directly in-teracts with Pyk2 where Pyk2 tyrosine phosphorylatesgelsolin, modulating its bioactivity in regulating theactin cytoskeleton in osteoclasts (42). Certainly, a lim-itation of the current studies is ascertaining whetherGnRH-induced tyrosine-phosphorylated proteins aredirect substrates of Pyk2, and the challenge for futurestudies will be to identify these tyrosine-phosphory-lated proteins by proteomic approaches.
MATERIALS AND METHODS
Reagents
Antibodies for phosphotyrosine (titer 1:2000), ERK2 (titer1:1000), JNK1 (titer 1:1000), IQGAP (titer 1:1000), actin (titer1:1000), and all horseradish peroxidase-coupled secondaryantibodies (titer 1:5000) were purchased from Santa CruzBiotechnology (Santa Cruz, CA) and were used according tothe manufacturer’s instructions. Antibody for pan-Pyk2 (titer1:1000) was purchased from BD Biosciences (Palo Alto, CA)and was used according to manufacturer’s instructions. Thephospho-Pyk2 antibodies (Y402, Y580, and Y881; titer 1:1000)and the Src antibody (titer 1:500) were purchased from Bio-source (Camarillo, CA) and were used according to the man-ufacturer’s instructions. The HA epitope tag antibody (YPY-DVPDYA; titer 1:1000) was purchased from BerkeleyAntibody Co. (Berkeley, CA) and used according to the man-ufacturer’s instructions. Antibodies against phosphorylatedforms of ERK and JNK (titer 1:1000) were obtained from CellSignaling Technologies (Danvers, MA) and were used ac-cording to the manufacturer’s instructions. The GnRHa buse-relin was purchased from Sigma Chemical Co. (St. Louis,MO) and was used at 10 nM for all studies. Nifedipine, W7,and KN62 were obtained from CalBiochem (San Diego, CA)and were used at 1, 15, and 10 �M, respectively. Camagarose was purchased from Stratagene (La Jolla, CA),and the matrix was prepared according to the manufac-turer’s instructions. Expression vector for ERK2 was gen-erously provided by Melanie Cobb (University of Texas,Southwestern, Dallas, TX).
Cell Culture and siRNA Stable Cell Selection
�T3-1 cells were generously provided by Dr. Pamela Mellon(University of California, San Diego) (43, 44). Cells were cul-tured in monolayers in the presence of DMEM containing10% fetal bovine serum and supplemented with penicillin andstreptomycin. Cells were maintained at 37 C in a 5% CO2,humidified atmosphere. For all studies, �T3-1 cells were splitwithin 2 d of experimentation and used as subconfluentcultures.
In some specific knockdown studies, control and Pyk2siRNAs were designed and cloned into the expression vectorpSuper-Retro-Neo using the same strategy described previ-ously (45). The control siRNA contained the following se-quence: 5�-TTCTCCGAACGTGTC ACGT-3�. Three mousePyk2-specific siRNA target sequences were identified as fol-lows: 5�-GAACATGGCTGATCTCATA-3�, 5�-CTACCT GGAA-CGAAATAAA-3�, and 5�-TTGAGGACG AAGACTATTA-3�.Each siRNA in pSuper-Retro-Neo was expressed as a hairpinlooped siRNA structure. Briefly, �T3-1 cells were plated at30–40% confluence in 100-mm dishes and were transfectedthe following day with pSuper-Retro-Neo-Pyk2 or controlsiRNAs using Fugene transfection reagent (Roche Applied
Xie et al. • Pyk2 Regulation by GnRH and Calmodulin Mol Endocrinol, October 2008, 22(10):2322–2335 2331

Science, Indianapolis, IN) following the manufacturer’s in-structions. After 18 h of transfection, stably transfected cellswere selected by geneticin treatment (Invitrogen, Carlsbad,CA) at 500 �g/ml in DMEM. After 3 wk of neomycin selection,the stable cell populations were ready for use in experiments.Originally, the three Pyk2 siRNAs were tested individually instable cell lines to identify the effectiveness of knockdown ofPyk2. Although all three Pyk2 siRNAs resulted in a modestlevel of knockdown at the protein level, the combination of allthree Pyk2 siRNA in the same stable cell population provedmost effective for Pyk2 knockdown.
Mono Q Chromatography
�T3-1 cell lysates were prepared and subjected to fraction-ation as described previously (46). Briefly, cells were serumstarved for 2 h and then pretreated with either control solutionor nifedipine for 30 min. Some cells then received GnRHa for15 min. Cells were then lysed with gentle agitation in a buffercontaining 0.5% Triton X-100. Lysates were cleared of debrisusing centrifugation and equal amounts of cellular protein(1–2 mg total protein; equal within an individual replicate)were loaded on a Mono Q column. Fractions were collectedand subjected to Western blot analysis outlined below. MonoQ fractionation procedures were completed on two separateoccasions with identical results.
Preparation of Whole-Cell Lysates and WesternBlot Analysis
For all Western blot studies, cells were serum starved for 2 hfollowed by treatment application. After treatments (for ex-ample buserelin time-course studies) within individual exper-iments, cells were lysed in a standard RIA immunoprecipita-tion (RIPA) buffer as described (10, 21). Lysates were clarifiedby centrifugation and denatured by boiling in an equal volumeof buffer containing 100 mM Tris (pH 6.8), 4% sodium dodecylsulfate (SDS), 20% glycerol, and 200 mM dithiothreitol (SDS-loading buffer). Samples were resolved on 10% polyacryl-amide gels (SDS-PAGE), transferred to polyvinylidene diflu-oride or nitrocellulose membranes, blocked in either 5% BSA(phosphotyrosine antibody) or nonfat dried milk in Tris-buff-ered saline (pH 7.5)/0.1% Tween 20. Primary antibodies wereadded at the appropriate dilution and incubated at 4 C over-night with constant agitation. Blots were washed in Tris-buffered saline (pH 7.5)/0.1% Tween 20 and then exposed tosecondary antibody for 1–2 h at room temperature andwashed again, and protein bands were visualized using en-hanced chemiluminescence reagents (PerkinElmer, Boston,MA). Lane loading was determined using the pan-specificantibodies to the corresponding phosphospecific antiseraused in individual studies. For all Western blot analyses,studies were carried out at least three times. ImageJ (http://rsb.info.nih.gov/ij/) was used to quantitate band intensity.Representative experiments are shown.
Animal Studies and Double-LabelingFluorescent Immunocytochemistry
All animal studies were completed in accordance with theCornell University Institutional Animal Care and Use Commit-tee. NSA (CF-1) female mice (Harlan, Indianapolis, IN) wereovariectomized and allowed 14 d to recover. All ovariecto-mized females were administered 0.25 ml ovine anti-GnRHserum ip and were maintained for 3 d to immunosuppressendogenous GnRH. On the fourth day, control animals (n � 3)received an ip injection of sterile saline (50 �l), whereastreated animals (n � 4 per group) received 100 ng (in 50 �lsaline) of a nonimmunoreactive analog of GnRH (D-Ala6-GnRH). GnRH analog-treated animals were then killed 45 and90 min after injection. Control animals were killed at approx-
imately 60 min after saline injection. Whole pituitaries weredissected free and fixed in 4% paraformaldehyde. Fixed tis-sues were sectioned at 7 �m. Endogenous peroxides wereblocked by incubating sections in methanol containing 0.6%H2O2. Slides were then rinsed one time with 70% ethanol andthree times over 15 min in 150 mM NaCl, 40 mM K2HPO4, 10mM KH2PO4 (pH 7.4) (KPBS). Next, tissue sections were putthrough an antigen retrieval method using trypsin (Sigma).Sections were incubated in a 0.1% trypsin solution contain-ing 0.1% CaCl2 and 20 mM Tris (pH 7.6). Sections then wererinsed with KPBS and incubated in primary antibody (anti-Pyk2 Y881) at 4 C overnight. The next morning, the tissuesections were rinsed with KPBS and then incubated for 30min at room temperature in biotinylated donkey antirabbit IgG(Vector Laboratories, Burlingame, CA) at a concentration of1:5000 in KPBS. Sections were rinsed and then incubated inan avidin-biotin complex solution (1.12 �l/ml KPBS, EliteABC kit; Vector) for 30 min at room temperature. After addi-tional rinsing, sections were incubated in streptavidin TexasRed (Vector) for 45 min at 37 C. Rinsing followed, and thenthe sections were incubated in the second primary antibody(LH� obtained from the National Hormone and Peptide Pro-gram, National Institute of Diabetes and Digestive and KidneyDiseases and Dr. A. F. Parlow, Torrance, CA) for 6 h at roomtemperature. Tissue was rinsed again and then incubated inCy2-donkey antirabbit IgG (Jackson ImmunoResearch Lab-oratories, West Grove, PA) for 45 min at 37 C. After rinsingwith KPBS, sections were dehydrated in graded ethanols,cleared in xylene, and coverslipped with Krystalon (Harleco,EM Science, Gibbstown, NJ).
In a pilot study to determine the effectiveness of the im-munosuppression model on LH secretion, control (n � 4) andGnRHa-treated (n � 4) animals were prepared as describedabove, and trunk blood was collected after exsanguination at60 min after ip injection of control saline or GnRHa. Serumwas collected and assayed for mouse LH using a specific LHRIA (Colorado State University Reproductive EndocrinologyLaboratory, Fort Collins, CO).
Transient Transfection and Reporter Gene Assay
For transient transfections, �T3-1 cells were plated at 60–70% confluence in 60-mm plates and were transiently trans-fected with a c-Fos promoter-luciferase fusion (500 ng) (46),a GnRHR promoter-luciferase fusion (15, 47, 48), or a Gal-Elk1 and 5xGal4-EIB luciferase reporter (500 ng of eachplasmid) (7) using lipofection reagents (either Fugene fromRoche Applied Science or Lipofectamine 2000 from Invitro-gen) following the manufacturer’s instructions. In specificexperiments, reporter systems were cotransfected with 5 �gempty parent vector (pKH3), wild-type Pyk2 (Pyk2WT), orcatalytically inactive Pyk2 (Pyk2KD) (25, 49). In other studies,site-directed mutagenesis was used to prepare a mutationwithin the putative Cam binding site in the catalytic domain infull-length Pyk2 (Pyk2cat-mut). The mutation was constructedby replacing the leucine residues with alanine as described inFig. 7A using a PCR-based strategy. To facilitate cloning intothe pKH3 vector, EcoR1 and Cla1 restriction sites were engi-neered to the forward and reverse primers, respectively. Themutation was verified by nucleotide sequence analysis. Eigh-teen to twenty hours after transfection, cells were collected forluciferase activity as previously described (45). All data are pre-sented as mean � SEM for luciferase activity standardized bytotal protein as measured by Bradford assay.
Cam Agarose Affinity Chromatography
�T3-1 cell lysates were prepared in a buffer containing 50 mM
Tris (pH 7.5), 1.0% Triton X-100, 5 mM EDTA, 250 mM NaCl,1 mM sodium vanadate, 25 mM �-glycerophosphate, 5 mM
benzamidine, and 0.2 mM phenylmethylsulfonyl fluoride(PMSF) (referred to as buffer A). This buffer was supple-
2332 Mol Endocrinol, October 2008, 22(10):2322–2335 Xie et al. • Pyk2 Regulation by GnRH and Calmodulin

mented with a protease inhibitor cocktail (Sigma; catalognumber P-8340). Lysates were subjected to two freeze-thawcycles (�80 C, then thawed on ice) and clarified by centrif-ugation. To prepare the Cam agarose, beads were sedi-mented by low-speed centrifugation, the storage buffer wasremoved, and beads were washed four times in a buffercontaining 20 mM Tris (pH 7.5), 4 mM MgCl2, 2 mM CaCl2, 10mM KCl, and 2 mM PMSF (buffer B �Ca2�), or in some cases,buffer B was supplemented with 10 mM EGTA and is referredto as buffer B �Ca2�. Cell lysates in buffer A were diluted10-fold in buffer B �Ca2� or in some cases buffer B �Ca2�.Diluted lysates were then mixed with washed Cam agarosebeads (in the presence or absence of calcium) for 3–4 h at 4C with constant mixing. The lysate/agarose solutions werethen loaded into a disposable column, and the retained Camagarose matrix was washed with 20 column volumes ofbuffer B �Ca2� or in some cases buffer B �Ca2�. Thecolumns were eluted with a buffer containing 20 mM Tris (pH7.5), 4 mM MgCl2, 10 mM EGTA, 10 mM KCl, and 1 mM PMSF.Fractions were collected in approximately 250-�l volumes.Equal volumes of fractions were used for Western blot anal-ysis. In some overexpression studies, control plasmid (pKH3)or HA-tagged wild-type and Pyk2 mutants (2 �g each) weretransiently transfected into �T3-1 cells as described above,and cell lysates were isolated and subjected to Cam agaroseaffinity chromatography. Chromatography experiments usingCam agarose were carried out at least three times with similarresults.
FAK Structural Modeling
Expasy (ca.expansy.org) was used to identify the domains forhuman FAK and Pyk2 based upon the sequences publishedin accession numbers Q05397 (FAK) and Q14289 (Pyk2). Thecatalytic domains of each protein were identified and alignedusing NCBI Blast. The FAK structure (24) was manipulatedusing Deep View/Swiss-PDB Viewer version 3.7. The FAKstructure was obtained from the Swiss Protein Databankwhere the ID number is 1MP8. Primary amino acid sequencedata for the catalytic domain of human FAK and Pyk2 weresubjected to two different secondary structure predictiontools (nnPredict and ProteinPredict). Predicted structures ofeach sequence were compared between the two search en-gines, and sequences common to both prediction tools aswell as consistency with the solved FAK structure were iden-tified. Similar analyses were carried out substituting criticalhydrophobic residues within the putative Cam binding site inthe Pyk2 catalytic domain with alanine residues to determinewhether predicted secondary structure would be altered.
Statistical Analysis
Data sets were subjected to an ANOVA, followed by individ-ual pairwise comparisons using the Tukey honestly signifi-cant difference test. P � 0.05 was deemed significant. Dataare reported as the treatment group means � SEM for repre-sentative studies. Studies were carried out at least threetimes on separate occasions.
Acknowledgments
We are grateful to Colin Clay, Pamela Mellon, MelanieCobb, and Terry Nett for helpful reagents. We are indebted toDr. Bill Horne at Cornell University for helpful discussions inthe development of this manuscript.
Received February 25, 2008. Accepted July 10, 2008.Address all correspondence and requests for reprints to:
Mark S. Roberson, Ph.D., T4-018 Veterinary Research Tower,
Department of Biomedical Sciences, Cornell University,Ithaca, New York 14853. E-mail: [email protected].
This work was supported by a grant from the NationalInstitutes of Child Health and Human Development (R01HD34722) to M.S.R.
Disclosure Statement: The authors have nothing to dis-close.
REFERENCES
1. Fink G, Sheward WJ, Charlton HM 1982 Priming effect ofluteinizing hormone releasing hormone in the hypogo-nadal mouse. J Endocrinol 94:283–287
2. Fink G, Sheward WJ, Plant TM 1984 The hypogonadalmouse pituitary contains bioactive LH. J Reprod Fertil70:277–280
3. Mason AJ, Pitts SL, Nikolics K, Szonyi E, Wilcox JN,Seeburg PH, Stewart TA 1986 The hypogonadal mouse:reproductive functions restored by gene therapy. Sci-ence 234:1372–1378
4. Hislop JN, Madziva MT, Everest HM, Harding T, Uney JB,Willars GB, Millar RP, Troskie BE, Davidson JS, McArdleCA 2000 Desensitization and internalization of humanand Xenopus gonadotropin-releasing hormone receptorsexpressed in �T4 pituitary cells using recombinant ade-novirus. Endocrinology 141:4564–4575
5. Vrecl M, Heding A, Hanyaloglu A, Taylor PL, Eidne KA 2000Internalization kinetics of the gonadotropin-releasing hor-mone (GnRH) receptor. Pflugers Arch 439:R19–R20
6. Ruf F, Fink MY, Sealfon SC 2003 Structure of the GnRHreceptor-stimulated signaling network: insights fromgenomics. Front Neuroendocrinol 24:181–199
7. Roberson MS, Misra-Press A, Laurance ME, Stork PJ,Maurer RA 1995 A role for mitogen-activated proteinkinase in mediating activation of the glycoprotein hor-mone �-subunit promoter by gonadotropin-releasinghormone. Mol Cell Biol 15:3531–3539
8. Xie J, Bliss SP, Nett TM, Ebersole BJ, Sealfon SC, RobersonMS 2005 Transcript profiling of immediate early genesreveals a unique role for activating transcription factor 3in mediating activation of the glycoprotein hormone�-subunit promoter by gonadotropin-releasing hormone.Mol Endocrinol 19:2624–2638
9. Lee SL, Sadovsky Y, Swirnoff AH, Polish JA, Goda P,Gavrilina G, Milbrandt J 1996 Luteinizing hormone defi-ciency and female infertility in mice lacking the transcrip-tion factor NGFI-A (Egr-1). Science 273:1219–1221
10. Zhang T, Wolfe MW, Roberson MS 2001 An early growthresponse protein (Egr) 1 cis-element is required for gona-dotropin-releasing hormone-induced mitogen-activatedprotein kinase phosphatase 2 gene expression. J BiolChem 276:45604–45613
11. Tremblay JJ, Drouin J 1999 Egr-1 is a downstream ef-fector of GnRH and synergizes by direct interaction withPtx1 and SF-1 to enhance luteinizing hormone � genetranscription. Mol Cell Biol 19:2567–2576
12. Coss D, Jacobs SB, Bender CE, Mellon PL 2004 A novelAP-1 site is critical for maximal induction of the follicle-stimulating hormone � gene by gonadotropin-releasinghormone. J Biol Chem 279:152–162
13. Strahl BD, Huang HJ, Sebastian J, Ghosh BR, MillerWL 1998 Transcriptional activation of the ovine follicle-stimulating hormone �-subunit gene by gonadotropin-releasing hormone: involvement of two activating protein-1-binding sites and protein kinase C. Endocrinology 139:4455–4465
14. Strahl BD, Huang HJ, Pedersen NR, Wu JC, Ghosh BR,Miller WL 1997 Two proximal activating protein-1-bindingsites are sufficient to stimulate transcription of the ovinefollicle-stimulating hormone-� gene. Endocrinology 138:2621–2631
Xie et al. • Pyk2 Regulation by GnRH and Calmodulin Mol Endocrinol, October 2008, 22(10):2322–2335 2333

15. Duval DL, Nelson SE, Clay CM 1997 The tripartite basalenhancer of the gonadotropin-releasing hormone (GnRH)receptor gene promoter regulates cell-specific expres-sion through a novel GnRH receptor activating se-quence. Mol Endocrinol 11:1814–1821
16. Mulvaney JM, Zhang T, Fewtrell C, Roberson MS 1999Calcium influx through L-type channels is required forselective activation of extracellular signal-regulated ki-nase by gonadotropin-releasing hormone. J Biol Chem274:29796–29804
17. Mulvaney JM, Roberson MS 2000 Divergent signalingpathways requiring discrete calcium signals mediateconcurrent activation of two mitogen-activated proteinkinases by gonadotropin-releasing hormone. J BiolChem 275:14182–14189
18. Roberson MS, Bliss SP, Xie J, Navratil AM, Farmerie TA,Wolfe MW, Clay CM 2005 Gonadotropin-releasing hor-mone induction of extracellular-signal regulated kinase isblocked by inhibition of calmodulin. Mol Endocrinol 19:2412–2423
19. Avraham H, Park SY, Schinkmann K, Avraham S 2000RAFTK/Pyk2-mediated cellular signalling. Cell Signal 12:123–133
20. Duval DL, Farris AR, Quirk CC, Nett TM, Hamernik DL,Clay CM 2000 Responsiveness of the ovine gonado-tropin-releasing hormone receptor gene to estradiol andgonadotropin-releasing hormone is not detectable invitro but is revealed in transgenic mice. Endocrinology141:1001–1010
21. Zhang T, Mulvaney JM, Roberson MS 2001 Activation ofmitogen-activated protein kinase phosphatase 2 by gona-dotropin-releasing hormone. Mol Cell Endocrinol 172:79–89
22. Ellsworth BS, White BR, Burns AT, Cherrington BD, OtisAM, Clay CM 2003 c-Jun N-terminal kinase activation ofactivator protein-1 underlies homologous regulation ofthe gonadotropin-releasing hormone receptor gene in�T3-1 cells. Endocrinology 144:839–849
23. O’Day DH, Myre MA 2004 Calmodulin-binding do-mains in Alzheimer’s disease proteins: extending thecalcium hypothesis. Biochem Biophys Res Commun320:1051–1054
24. Nowakowski J, Cronin CN, McRee DE, Knuth MW, NelsonCG, Pavletich NP, Rogers J, Sang BC, Scheibe DN,Swanson RV, Thompson DA 2002 Structures of the cancer-related Aurora-A, FAK, and EphA2 protein kinases fromnanovolume crystallography. Structure 10:1659–1667
25. Ueda H, Abbi S, Zheng C, Guan JL 2000 Suppression ofPyk2 kinase and cellular activities by FIP200. J Cell Biol149:423–430
26. Shah BH, Soh JW, Catt KJ 2003 Dependence of gona-dotropin-releasing hormone-induced neuronal MAPKsignaling on epidermal growth factor receptor transacti-vation. J Biol Chem 278:2866–2875
27. Farshori PQ, Shah BH, Arora KK, Martinez-Fuentes A,Catt KJ 2003 Activation and nuclear translocation ofPKC�, Pyk2 and ERK1/2 by gonadotropin releasing hor-mone in HEK293 cells. J Steroid Biochem Mol Biol 85:337–347
28. Shah BH, Farshori MP, Catt KJ 2004 Neuropeptide-induced transactivation of a neuronal epidermal growthfactor receptor is mediated by metalloprotease-depen-dent formation of heparin-binding epidermal growth fac-tor. J Biol Chem 279:414–420
29. Maudsley S, Naor Z, Bonfil D, Davidson L, Karali D,Pawson AJ, Larder R, Pope C, Nelson N, Millar RP,Brown P 2007 Proline-rich tyrosine kinase 2 mediatesgonadotropin-releasing hormone signaling to a specificextracellularly regulated kinase-sensitive transcriptionallocus in the luteinizing hormone �-subunit gene. MolEndocrinol 21:1216–1233
30. Lev S, Moreno H, Martinez R, Canoll P, Peles E, MusacchioJM, Plowman GD, Rudy B, Schlessinger J 1995 Protein
tyrosine kinase PYK2 involved in Ca2�-induced regulationof ion channel and MAP kinase functions. Nature 376:737–745
31. Murasawa S, Mori Y, Nozawa Y, Masaki H, Maruyama K,Tsutsumi Y, Moriguchi Y, Shibasaki Y, Tanaka Y,Iwasaka T, Inada M, Matsubara H 1998 Role of calcium-sensitive tyrosine kinase Pyk2/CAK�/RAFTK in angioten-sin II induced Ras/ERK signaling. Hypertension 32:668–675
32. Park SY, Avraham HK, Avraham S 2004 RAFTK/Pyk2activation is mediated by trans-acting autophosphoryla-tion in a Src-independent manner. J Biol Chem 279:33315–33322
33. Espiritu DJ, Bernardo AA, Robey RB, Arruda JA 2002 Acentral role for Pyk2-Src interaction in coupling diversestimuli to increased epithelial NBC activity. Am J PhysiolRenal Physiol 283:F663–F670
34. Zwick E, Wallasch C, Daub H, Ullrich A 1999 Distinctcalcium-dependent pathways of epidermal growth factorreceptor transactivation and PYK2 tyrosine phosphory-lation in PC12 cells. J Biol Chem 274:20989–20996
35. Guo J, Meng F, Fu X, Song B, Yan X, Zhang G 2004N-methyl-D-aspartate receptor and L-type voltage-gatedCa2� channel activation mediate proline-rich tyrosine ki-nase 2 phosphorylation during cerebral ischemia in rats.Neurosci Lett 355:177–180
36. Ginnan R, Singer HA 2002 CaM kinase II-dependentactivation of tyrosine kinases and ERK1/2 in vascularsmooth muscle. Am J Physiol Cell Physiol 282:C754–C761
37. Haisenleder DJ, Burger LL, Aylor KW, Dalkin AC, MarshallJC 2003 Gonadotropin-releasing hormone stimulation ofgonadotropin subunit transcription: evidence for the in-volvement of calcium/calmodulin-dependent kinase II (Ca/CAMK II) activation in rat pituitaries. Endocrinology 144:2768–2774
38. Haisenleder DJ, Ferris HA, Shupnik MA 2003 The calciumcomponent of gonadotropin-releasing hormone-stimulatedluteinizing hormone subunit gene transcription is mediatedby calcium/calmodulin-dependent protein kinase type II.Endocrinology 144:2409–2416
39. Hartigan JA, Xiong WC, Johnson GV 2001 Glycogensynthase kinase 3� is tyrosine phosphorylated by PYK2.Biochem Biophys Res Commun 284:485–489
40. Heidinger V, Manzerra P, Wang XQ, Strasser U, Yu SP,Choi DW, Behrens MM 2002 Metabotropic glutamatereceptor 1-induced upregulation of NMDA receptorcurrent: mediation through the Pyk2/Src-family kinasepathway in cortical neurons. J Neurosci 22:5452–5461
41. Ling S, Sheng JZ, Braun AP 2004 The calcium-dependentactivity of large conductance, calcium-activated K� chan-nels is enhanced by Pyk2 and Hck-induced tyrosine phos-phorylation. Am J Physiol Cell Physiol 287:C698–C706
42. Wang Q, Xie Y, Du QS, Wu XJ, Feng X, Mei L, McDonaldJM, Xiong WC 2003 Regulation of the formation of os-teoclastic actin rings by proline-rich tyrosine kinase 2interacting with gelsolin. J Cell Biol 160:565–575
43. Windle JJ, Weiner RI, Mellon PL 1990 Cell lines of thepituitary gonadotrope lineage derived by targeted onco-genesis in transgenic mice. Mol Endocrinol 4:597–603
44. Horn F, Bilezikjian LM, Perrin MH, Bosma MM, Windle JJ,Huber KS, Blount AL, Hille B, Vale W, Mellon PL 1991Intracellular responses to gonadotropin-releasing hor-mone in a clonal cell line of the gonadotrope lineage. MolEndocrinol 5:347–355
45. Xie J, Roberson MS 2008 3�,5�-Cyclic adenosine 5�-mono-phosphate response element-dependent transcriptionalregulation of the secretogranin II gene promoter dependson gonadotropin-releasing hormone-induced mitogen-activated protein kinase activation and the transactivatoractivating transcription factor 3. Endocrinology 149:783–792
2334 Mol Endocrinol, October 2008, 22(10):2322–2335 Xie et al. • Pyk2 Regulation by GnRH and Calmodulin

46. Roberson MS, Zhang T, Li HL, Mulvaney JM 1999 Acti-vation of the p38 mitogen-activated protein kinase path-way by gonadotropin-releasing hormone. Endocrinology140:1310–1318
47. Duval DL, Nelson SE, Clay CM 1997 A binding site forsteroidogenic factor-1 is part of a complex enhancerthat mediates expression of the murine gonadotropin-releasing hormone receptor gene. Biol Reprod 56:160–168
48. Cherrington BD, Farmerie TA, Lents CA, Cantlon JD,
Roberson MS, Clay CM 2005 Activin responsiveness ofthe murine gonadotropin-releasing hormone receptorgene is mediated by a composite enhancer containingspatially distinct regulatory elements. Mol Endocrinol 19:898–912
49. Zheng C, Xing Z, Bian ZC, Guo C, Akbay A, Warner L,Guan JL 1998 Differential regulation of Pyk2 and focaladhesion kinase (FAK). The C-terminal domain of FAKconfers response to cell adhesion. J Biol Chem 273:2384–2389
Molecular Endocrinology is published monthly by The Endocrine Society (http://www.endo-society.org), the foremostprofessional society serving the endocrine community.
Chemical Approaches to Nuclear Receptors and MetabolismApril 16-17, 2009
Bethesda, Maryland
The NIDDK-sponsored workshop ‘Chemical Approaches to Nuclear Receptors and Metabolism‘ will focusattention on the use of chemical biology approaches to address the role of ligands in the regulation of nuclearreceptor action.
Additional information may be found on the workshop website (http://www3.niddk.nih.gov/fund/other/chemi-calapproach/) or by contacting Ron Margolis (NIDDK) at 301-594-8819/[email protected].
Xie et al. • Pyk2 Regulation by GnRH and Calmodulin Mol Endocrinol, October 2008, 22(10):2322–2335 2335




















