Neutrophil-associated activation markers in healthy smokers relates to a fall in DL COand to...
11
Neutrophil-associated activation markers in healthy smokers relates to a fall in DL CO and to emphysematous changes on high resolution CT A. EKBERG-JANSSON*, B. ANDERSSON { , B. BAKE*, M. BOIJSEN { , I. ENANDEN } , A. ROSENGREN } , B.-E, SKOOGH*, U. TYLE ´ N { ,P.VENGE k AND C.-G. LO ¨ FDAHL** Departments of *Pulmonary Medicine, { Immunology, { Radiology and } Section of Preventive Cardiology, Sahlgrensaka University Hospital, Go ¨teborg; } Pharmacia and Upjohn Diagnostics AB, Uppsala; k Department of Clinical Chemistry, University Hospital, Uppsala; **Department of Respiratory Medicine, University Hospital, Lund, Sweden Smoking is a risk factor for developing chronic obstructive pulmonary disease (COPD), but there are no good indicators for early identification of subjects who will develop symptomatic COPD. The aim of this study was to investigate inflammatory mechanisms related to changes in lung function and emphysematous changes on high resolution computed tomography (HRCT) in ‘healthy’ smokers. Subjects were 60-year-old men from a population study. Bronchoscopy was performed in 30 smokers and 18 who had never smoked. Blood tests, lung function measurements and HRCT were carried out in 58 and 34 subjects, respectively. In comparison with never-smokers, smokers had higher levels of myeloperoxidase (MPO), human neutrophil lipocalin (HNL), eosinophil cationic protein (ECP) and lysozyme in blood, higher levels of MPO, interleukin-8 (IL- 8) and HNL in bronchial lavage (BL), and of IL-8, HNL and interleukin-1b (IL-1b) in bronchoalveolar lavage (BAL). Smokers also had lower levels of Clara cell protein 16 (CC-16) in blood. HNL in BL and BAL showed strong correlations to other inflammatory markers (MPO, IL-8, IL-1b). The variations in MPO in BL were explained by variations in HNL (R 2 =0?69), while these variations in BAL were explained by variations in HNL and IL-1b (R 2 =0?76). DL CO was the lung function variable most closely related to MPO and IL-8 in BL and BAL and to IL-1b in BAL. In a multiple regression analysis, MPO, IL-1b, IL-8 and CC-16 in BL and MPO in BAL contributed to the explanation of variations in DL CO to 41% and 22%, respectively, independent of smoking habits. In smokers with emphysematous lesions on HRCT, HNL in BAL correlated to emphysema score (r s =0?71). We conclude that ‘healthy’ smoking men with a near normal FEV 1 show signs of inflammation in the lower airways that are related to a decrease in DL CO and to emphysematous lesions on HRCT. This inflammation seems to be the result of both monocyte/macrophage and neutrophil activation. Key words: smoking; inflammation; chronic obstructive pulmonary disease; bronchoalveolar lavage; cytokines; computed tomography; lung function. RESPIR.MED. (2001) 95, 363–373 # 2001 HARCOURT PUBLISHERS LTD Introduction Smoking-induced chronic obstructive pulmonary disease (COPD) is common in the industrialized part of the world and causes high morbidity and increasing mortality (1). About 50% of all smokers develop chronic bronchitis and at least 15% develop COPD (2). By the time the COPD patient seeks medical attention, the lung function is often seriously deteriorated and the subject already severely handicapped. At present, there are no good indicators to enable early identification of subjects who will later develop symptomatic COPD, except for the knowledge that smoking is a risk factor. The slope of phase III N 2 test and DL CO have been advocated to show early airway changes in smokers (3–5). There are indications that histologic changes in the small airways are related to lung function test thought to reflect small airways (6). Smokers with chronic bronchitis have increased numbers of neutrophils in fluids from the lower airways, and also of various neutrophil associated Received 25 January 2001 and accepted 26 January 2001. Correspondence should be addressed to: Ann Ekberg-Jansson. Department of Allergology and Pulmonary Medicine, Sahlgrenska University Hospital, 413 45 Go¨teborg, Sweden. Fax: (46) 31 82 49 04; E-mail: [email protected] RESPIRATORY MEDICINE (2001) 95, 363–373 doi:10.1053/rmed.2001.1050, available online at http://www.idealibrary.com on 0954-6111/01/050363+11 $35?00/0 # 2001 HARCOURT PUBLISHERS LTD
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of Neutrophil-associated activation markers in healthy smokers relates to a fall in DL COand to...

Neutrophil-associated activation markers in healthysmokers relates to a fall in DLCO andto emphysematous changes on high resolution CT
A. EKBERG-JANSSON*, B. ANDERSSON{, B. BAKE*, M. BOIJSEN
{, I. ENANDEN}, A. ROSENGREN
},B.-E, SKOOGH*, U. TYLEN
{, P. VENGEk
AND C.-G. LOFDAHL**
Departments of *Pulmonary Medicine, {Immunology, {Radiology and }Section of Preventive Cardiology,
Sahlgrensaka University Hospital, Goteborg; }Pharmacia and Upjohn Diagnostics AB, Uppsala; kDepartment ofClinical Chemistry, University Hospital, Uppsala; **Department of Respiratory Medicine, University Hospital,
Lund, Sweden
Smoking is a risk factor for developing chronic obstructive pulmonary disease (COPD), but there are no goodindicators for early identification of subjects who will develop symptomatic COPD. The aim of this study was to
investigate inflammatory mechanisms related to changes in lung function and emphysematous changes on highresolution computed tomography (HRCT) in ‘healthy’ smokers. Subjects were 60-year-old men from a populationstudy. Bronchoscopy was performed in 30 smokers and 18 who had never smoked. Blood tests, lung function
measurements and HRCT were carried out in 58 and 34 subjects, respectively.In comparison with never-smokers, smokers had higher levels of myeloperoxidase (MPO), human neutrophil
lipocalin (HNL), eosinophil cationic protein (ECP) and lysozyme in blood, higher levels of MPO, interleukin-8 (IL-
8) and HNL in bronchial lavage (BL), and of IL-8, HNL and interleukin-1b (IL-1b) in bronchoalveolar lavage(BAL). Smokers also had lower levels of Clara cell protein 16 (CC-16) in blood. HNL in BL and BAL showedstrong correlations to other inflammatory markers (MPO, IL-8, IL-1b). The variations in MPO in BL wereexplained by variations in HNL (R2=0?69), while these variations in BAL were explained by variations in HNL
and IL-1b (R2=0?76). DLCO was the lung function variable most closely related to MPO and IL-8 in BL and BALand to IL-1b in BAL. In a multiple regression analysis, MPO, IL-1b, IL-8 and CC-16 in BL and MPO in BALcontributed to the explanation of variations in DLCO to 41% and 22%, respectively, independent of smoking
habits. In smokers with emphysematous lesions on HRCT, HNL in BAL correlated to emphysema score (rs=0?71).We conclude that ‘healthy’ smoking men with a near normal FEV1 show signs of inflammation in the lower
airways that are related to a decrease in DLCO and to emphysematous lesions on HRCT. This inflammation seems
to be the result of both monocyte/macrophage and neutrophil activation.
Key words: smoking; inflammation; chronic obstructive pulmonary disease; bronchoalveolar lavage; cytokines;
computed tomography; lung function.
RESPIR. MED. (2001) 95, 363–373 # 2001 HARCOURT PUBLISHERS LTD
RESPIRATORY MEDICINE (2001) 95, 363–373doi:10.1053/rmed.2001.1050, available online at http://www.idealibrary.com on
Introduction
Smoking-induced chronic obstructive pulmonary disease(COPD) is common in the industrialized part of the world
and causes high morbidity and increasing mortality (1).About 50% of all smokers develop chronic bronchitis andat least 15% develop COPD (2). By the time the COPD
Received 25 January 2001 and accepted 26 January 2001.
Correspondence should be addressed to: Ann Ekberg-Jansson.
Department of Allergology and Pulmonary Medicine, Sahlgrenska
University Hospital, 413 45 Goteborg, Sweden. Fax: (46) 31 82 49
04; E-mail: [email protected]
0954-6111/01/050363+11 $35?00/0
patient seeks medical attention, the lung function is often
seriously deteriorated and the subject already severelyhandicapped. At present, there are no good indicators toenable early identification of subjects who will later develop
symptomatic COPD, except for the knowledge thatsmoking is a risk factor.The slope of phase III N2 test and DLCO have been
advocated to show early airway changes in smokers (3–5).
There are indications that histologic changes in the smallairways are related to lung function test thought to reflectsmall airways (6). Smokers with chronic bronchitis have
increased numbers of neutrophils in fluids from the lowerairways, and also of various neutrophil associated
# 2001 HARCOURT PUBLISHERS LTD

364 A. EKBERG-JANSSON ET AL.
activation markers such as MPO and IL-8 (7–11). However,in the early phase of COPD, the influence of cellular
inflammation and soluble inflammatory markers has notbeen thoroughly studied. Knowledge on the relationbetween the inflammatory markers and lung function
impairment in the early phase of disease is also poor.In this study we recruited both smoking and never-
smoking subjects from a population study of 60-year-old
men who had never sought medical attention for airwaydisease (‘healthy’). Among the smokers, we expected to findsubjects with slightly impaired lung function considered tobe mild COPD or a preclinical phase of the development of
COPD as described by Cherniack (12). Many of thesesubjects were shown in an earlier study to have emphyse-matous changes on high resolution computed tomography
(HRCT) (13). The aim was to study inflammatorymechanisms related to changes in lung function i.e. forcedexpiratory volume in 1 sec (FEV1), the slope of phase III
N2-test (N2 test) and DLCO, and to emphysematouschanges on HRCT in healthy smokers.For this purpose we measured markers of neutrophils,
monocytes/macrophages and Clara cells in bronchial fluidsand in blood. Human neutrophil lipocalin (HNL) wasmeasured as a specific marker of neutrophils (14), whilemeasurement of myeloperoxidase (MPO) was considered to
reflect both neutrophils and monocytes/macrophages(15,16). Lysozyme measurements in blood served as amarker of monocytes/macrophages and in bronchial fluids
as a marker of the secretory activity of glands (17).Interleukin 1b (IL-1b) served as a marker of macrophageactivity in bronchial fluids (18) together with the measure-
ment of interleukin-8 (IL-8) (19), although both thesecytokines may originate from other sources as well. Finally,we measured the Clara cell protein (CC-16) in blood and
bronchial fluids as an indicator of the activity of the thesecells (20).
Materials and methods
SUBJECTS
The subjects were recruited from the population study ‘men
born in 1933 in Goteborg’, a random half of all men born in1933 and residing in Goteborg in 1983 (n=1016) (21). In1993, 879 men of the original cohort who were still alive
and living in Goteborg were recruited for a secondexamination. Of the 602 participating men, 532 wereevaluated by spirometry. Of these men, 112 were smokers,
198 were never-smokers and 222 were former smokers whohad not smoked for 1 month. All smokers and a randomsample of 60 lifelong never-smokers were invited toundergo a lung examination.
Fifty-eight smokers and 34 never-smokers consented tofurther investigations and, of these, 30 smoking and 18never-smoking men accepted to undergo a bronchoscopy.
Smoking habits were designated as 0=never-smokers,1=1–515 cigarettes day71, 2=�15 cigarettes day71.Thirty-three of 58 (57%) were current heavy smokers
(�15 cigarettes day71) and 25/58 (43%) were current light
smokers (515 cigarettes day71). The median number ofpack-years was 35 (9–79) years.
Among the subjects originally included in the study,subjects were excluded if they (i) had any airway disease forwhich they had sought medical attention (n=11), (ii) had a
history of congestive heart failure or unstable anginapectoris (n=4), or (iii) had any other severe disease(n=5). Subjects would also have been excluded if they
had (iv) scoliosis or other diseases with deformation ofthorax, (v) any kind of infection during the 4 weekspreceding the examination, or (vi) if they were on treatmentwith corticosteroids, N-acetylcysteine (NAC) or acetylsa-
licylic acid (ASA) less than 4 weeks prior to blood tests orbronchoscopy. No subject was excluded according to iv–vi.ASA treatment was allowed in subjects with prior heart
infarction (n=8). Four smokers had quit smoking beforethey came to the follow-up and were therefore excluded.The rest of the drop-outs were not willing to take part in the
study for various reasons (n=56). The drop-out frequencywas equally distributed in smokers and never-smokers.The study was approved by the local Ethics Commitee at
Sahlgrenska University Hospital, Goteborg, Sweden.
FIBREOPTIC BRONCHOSCOPY ANDSAMPLE COLLECTIONS
The subjects were examined in the supine position.Bronchoscopies were performed by one experiencedbronchoscopist between 08?00 and 10?00 hours. Premedica-
tion was given with 5 mg diazepam orally and 0?5 mlmorphine–scopolamin intra-muscularly. In case of kidneyor gall bladder disease petidine 75 mg intra-muscularly and
atropine 0?5 mg subcutaneously were given. Additionaldiazepam (2?5–5?0 mg) intravenousely could also be given.All persons were given nebulized terbutalin 0?25 mg dose71
263 before bronchoscopy. Local anaesthesia was giveninitially with 1% tetracaine spray in the mouth andlaryngeal tract. Additional anaesthesia for the lowerrespiratory tract was given via the bronchoscope (Olym-
pus). Oxygen saturation was measured during the broncho-scopy, and the subjects were given supplemental oxygen(2–3 lmin71) via a nasal catheter.
Bronchoalveolar lavage was made as a fractionated smallvolume lavage 10 mlþ3650 ml of sterile and endotoxin-free phosphate buffered saline (PBS). The first 10 ml were
processed separately as bronchial lavage (BL). Theremainder, the bronchoalveolar lavage (BAL), were pooledinto a sterile siliconized bottle and transportated on ice
immediately to the laboratory. At the laboratory the totalvolume of the lavage was measured and centrifuged at 250 gand 48C for 10 min. The supernatant was centrifuged asecond time at 10 000 g and frozen at 7708C for later
analysis of soluble inflammatory markers.The total cell number was determined using a haemo-
cytometer. Cell viability was estimated using tryptan blue
exclusion. Calculation of cell differentials was done oncytocentrifuged preparations (Cytospin 2; Shandon South-ern Products LTD, Runcorn, U.K.) stained with May–
Grunwald–Giemsa counting 1000 cells.

INFLAMMATORY MARKERS AND EMPHYSEMATOUS CHANGES IN SMOKERS 365
Blood samples for analysis of lymphocyte counts werealso taken.
ANALYSIS OF INFLAMMATORY MARKERS
Human neutrophil lipocalin (HNL) was determined in
serum and BL and BAL fluid using a solid phase double-ligand fluoroimmunoassay (FEIA) (22). Briefly, HNL instandard or sample was allowed to bind to an anti-HNL
mouse monoclonal antibody (mAb) coupled to Immuno-CAP (Pharmacia & Upjohn, Diagnostic AB, Uppsala,Sweden). After washing, b-galatosidase-labelled anti-HNL
mab was added and allowed to bind to the HNL. After afinal wash, the amount of HNL was determined byfluorescence after adding 4-methylumbelliferyl-b-D-galac-tosidase as enzyme substrate. HNL, purified from neutro-
phil granulocytes, was used as standard ranging from20 mg l71 to 600 mg l71. The intra-assay and inter-assaycoefficients of variation were less than 10%, and the
detection limit was 3 mg l71.Clara cell protein 16 (CC-16) was determined in serum
and BL and BAL fluid using a solid phase double ligand
fluoroimmunoassay (FEIA) (23). Briefly, CC-16 in standardor sample was allowed to bind to an anti-CC-16 mousemAb coupled to ImmunoCAP (Pharmacia & Upjohn).
After washing, b-galactosidase-labelled anti-CC-16 mAbwas added and allowed to bind to the CC-16. After a finalwash, the amount of CC-16 was determined by fluorescenceafter adding 4-methylumberiferyl-b-D-galactosidase as en-
zyme substrate. CC-16, purified from human urine, wasused as standard ranging from 1 mg l71 to 100 mg l71. Theintra-assay and inter-assay coefficients of variation were less
than 10%, and the detection limit was less than 1 mg l71.Interleukin-8 (IL-8) was determined in serum and
bronchial lavage (BL) and BAL fluid using a solid phase
fluorescent enzyme immuno assay (24). Briefly, the BALfluid was incubated in micro-titre wells coated with a mousemonoclonal antibody against IL-8. After washing, a goatanti-IL-8 b-galactosidase conjugate was added and the
plates were incubated. The amount of IL-8 was measuredby adding 4-methylumbelliferyl-b-D-galactosidase as en-zyme substrate. Recombinant IL-8 was used as standard,
ranging from 25 ng l71 to 1600 ng l71. The intra-assay andinter-assay coefficients of variation were less than 10%, andthe detection limit was 5 ng l71.
Lysozyme was titrated in serum and BL and BAL fluidby incubating standard solution or samples with Sephadex-bound anti-lysozyme for 2 h before an addition of 125I-
labelled lysozyme (25). Incubation was then continued for20 h during rotation end-over-end. After washing fourtimes, the radioactivity of the pellet was determined in awell-type scintillation counter. Lysozyme purified from
human blood was used as standard ranging from 3 mg l71
to 900 mg l71. The intra-assay and inter-assay coefficients ofvariation were less than 10%, and the detection limit was
less than 3 mg l71.MPO and interleukin-1b (IL-1b; ultra-sensitive kit), were
determined in serum and BL and BAL (except for IL-1bwhich was determined only in BL and BAL) by commer-
cially available kits (Pharmacia & Upjohn Diagnostics ABand Biosource International, Camarillo, CA, U.S.A.),
according to the instructions of the manufacturer.
LUNG FUNCTION TESTS
In all subjects lung volumes, spirometry, single breath N2
test and diffusion capacity test were determined. Lungvolumes were obtained by a flow displacement body
pletysmograph (Body Box 2800; SensorMedics Co., Biltho-ven, The Netherlands). FEV1 and vital capacity (VC) wereobtained on a water-sealed regularly calibrated bellspirometer. The slope of phase III was obtained by the
single breath N2 method (26). Carbon monoxide transfer(DLCO) was assessed by the single breath method withstandard equipment (SensorMedics 2200; SensorMedics
Co.) We used the European Respiratory Society referencevalues (27) for lung volumes and spirometry, and for thesingle breath N2 test we used data from Sixt et al. (28). For
DLCO and carbon monoxide transfer factor (DLCO/VA) weused the reference values according to Salorinne (29).
HIGH RESOLUTION COMPUTEDTOMOGRAPHY (HRCT)
The examination was performed with a Picker PQ 2000.The subjects were examined in a supine position and in full
inspiration. The entire thorax was scanned with a slicethickness of 1?5 mm and 3 cm inter-slice distance. Exposuredata were 130 kV and 200 mA. The images were recon-
structed with the sharp algorithm of Picker and six imageswere copied to each 14@617@ film. Window width was set at1400 HU and the level at 7400HU.
The images were analysed from the hard copies by twoexperienced chest radiologists independently, withoutknowledge of whether the subject was a smoker. The
radiologists analysed each slice in each lung. The diagnosisof emphysematous changes was based on findings of areasof low attenuation and/or presence of stretched, narrowedvessels. The degree of emphysema was scored as 0=no
emphysema, 1=1–25% of the parenchymal area wasinvolved, 2=26–50% involvement, 3=51–75% involve-ment and 4=more than 75%. The mean scores for the slices
obtained were calculated. In this way, a score of total lunginvolvement could be calculated and the individual obser-ver’s evaluations compared. The kappa value was 0?78.
All subjects were examined by HRCT. The technicalquality was insufficient in one smoker and two never-smokers so they were excluded. As demonstrated in an
earlier paper emphysematous changes were shown in 25/57smokers (44%) and 1/32 (3%) never-smokers. In subjectswith emphysematous changes the mean values of theemphysema score ranged from 0?09 to 1?63 (13).
STATISTICAL METHODS
Group data are presented with their mean and range. For
lung function variables mean and standard deviation (SD)
366 A. EKBERG-JANSSON ET AL.
are presented. The Mann–Whitney U-test was used for thecomparison between smokers and never-smokers and for
the comparison of inflammatory markers between subjectswith and without HRCT lesions. The w2 test and Fischer’sexact test were used for the comparison between categories.
The non-parametric Spearman rank correlation coefficient(rs) was used for correlation analyses between lung functionand inflammatory markers as well as between emphysema
score and inflammatory markers. A multi-variate stepwisebackward regression analysis was used. All tests were two-sided, and P-values lower than 0?05 were regarded assignificant. We used Stat View 4?51 and Statistica1 for
Windows (Stat Soft) as the statistical packages.
Results
INFLAMMATORY CELLS RELATED TOSMOKING
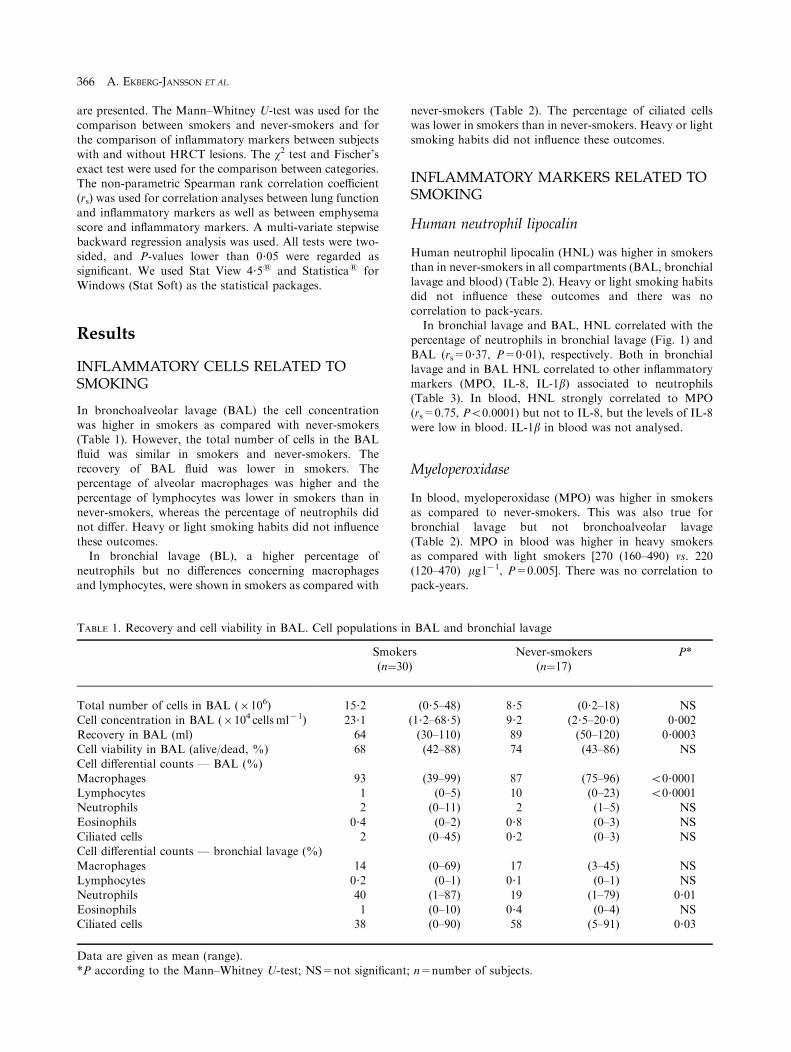
In bronchoalveolar lavage (BAL) the cell concentrationwas higher in smokers as compared with never-smokers(Table 1). However, the total number of cells in the BAL
fluid was similar in smokers and never-smokers. Therecovery of BAL fluid was lower in smokers. Thepercentage of alveolar macrophages was higher and the
percentage of lymphocytes was lower in smokers than innever-smokers, whereas the percentage of neutrophils didnot differ. Heavy or light smoking habits did not influence
these outcomes.In bronchial lavage (BL), a higher percentage of
neutrophils but no differences concerning macrophages
and lymphocytes, were shown in smokers as compared with
TABLE 1. Recovery and cell viability in BAL. Cell populations i
Smok(n¼3
Total number of cells in BAL (6106) 15?2
Cell concentration in BAL (6104 cellsml71) 23?1Recovery in BAL (ml) 64Cell viability in BAL (alive/dead, %) 68
Cell differential counts — BAL (%)Macrophages 93Lymphocytes 1Neutrophils 2
Eosinophils 0?4Ciliated cells 2Cell differential counts — bronchial lavage (%)
Macrophages 14Lymphocytes 0?2Neutrophils 40
Eosinophils 1Ciliated cells 38
Data are given as mean (range).*P according to the Mann–Whitney U-test; NS=not significan
never-smokers (Table 2). The percentage of ciliated cellswas lower in smokers than in never-smokers. Heavy or light
smoking habits did not influence these outcomes.
INFLAMMATORY MARKERS RELATED TOSMOKING
Human neutrophil lipocalin
Human neutrophil lipocalin (HNL) was higher in smokers
than in never-smokers in all compartments (BAL, bronchiallavage and blood) (Table 2). Heavy or light smoking habitsdid not influence these outcomes and there was no
correlation to pack-years.In bronchial lavage and BAL, HNL correlated with the
percentage of neutrophils in bronchial lavage (Fig. 1) and
BAL (rs=0?37, P=0?01), respectively. Both in bronchiallavage and in BAL HNL correlated to other inflammatorymarkers (MPO, IL-8, IL-1b) associated to neutrophils(Table 3). In blood, HNL strongly correlated to MPO
(rs=0.75, P50.0001) but not to IL-8, but the levels of IL-8were low in blood. IL-1b in blood was not analysed.
Myeloperoxidase
In blood, myeloperoxidase (MPO) was higher in smokersas compared to never-smokers. This was also true forbronchial lavage but not bronchoalveolar lavage
(Table 2). MPO in blood was higher in heavy smokersas compared with light smokers [270 (160–490) vs. 220(120–470) mg l71, P=0.005]. There was no correlation to
pack-years.
n BAL and bronchial lavage
ers0)
Never-smokers(n¼17)
P*
(0?5–48) 8?5 (0?2–18) NS
(1?2–68?5) 9?2 (2?5–20?0) 0?002(30–110) 89 (50–120) 0?0003(42–88) 74 (43–86) NS
(39–99) 87 (75–96) 50?0001(0–5) 10 (0–23) 50?0001(0–11) 2 (1–5) NS
(0–2) 0?8 (0–3) NS(0–45) 0?2 (0–3) NS
(0–69) 17 (3–45) NS(0–1) 0?1 (0–1) NS(1–87) 19 (1–79) 0?01
(0–10) 0?4 (0–4) NS(0–90) 58 (5–91) 0?03
t; n=number of subjects.
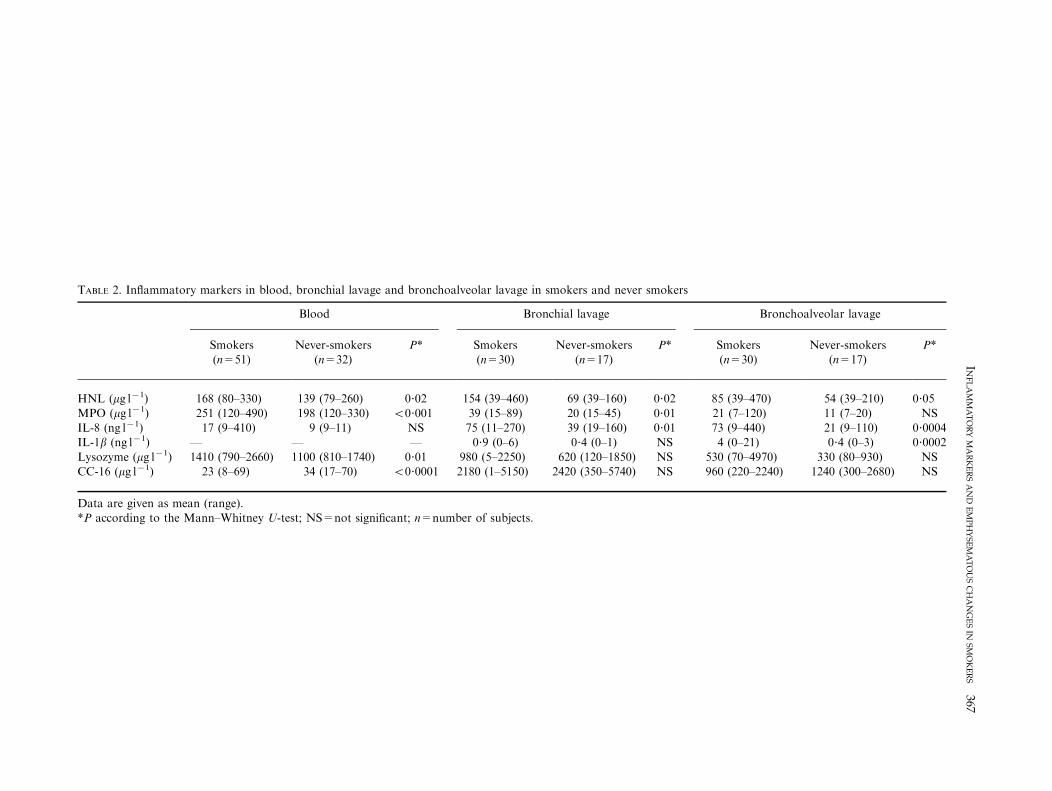
TABLE 2. Inflammatory markers in blood, bronchial lavage and bronchoalveolar lavage in smokers and never smokers
Blood Bronchial lavage Bronchoalveolar lavage
Smokers(n=51)
Never-smokers(n=32)
P* Smokers(n=30)
Never-smokers(n=17)
P* Smokers(n=30)
Never-smokers(n=17)
P*
HNL (mg l71) 168 (80–330) 139 (79–260) 0?02 154 (39–460) 69 (39–160) 0?02 85 (39–470) 54 (39–210) 0?05
MPO (mg l71) 251 (120–490) 198 (120–330) 50?001 39 (15–89) 20 (15–45) 0?01 21 (7–120) 11 (7–20) NSIL-8 (ng l71) 17 (9–410) 9 (9–11) NS 75 (11–270) 39 (19–160) 0?01 73 (9–440) 21 (9–110) 0?0004IL-1b (ng l71) — — — 0?9 (0–6) 0?4 (0–1) NS 4 (0–21) 0?4 (0–3) 0?0002
Lysozyme (mg l71) 1410 (790–2660) 1100 (810–1740) 0?01 980 (5–2250) 620 (120–1850) NS 530 (70–4970) 330 (80–930) NSCC-16 (mg l71) 23 (8–69) 34 (17–70) 50?0001 2180 (1–5150) 2420 (350–5740) NS 960 (220–2240) 1240 (300–2680) NS
Data are given as mean (range).*P according to the Mann–Whitney U-test; NS=not significant; n=number of subjects.
INF
LA
MM
AT
OR
YM
AR
KE
RS
AN
DE
MP
HY
SE
MA
TO
US
CH
AN
GE
SIN
SM
OK
ER
S367
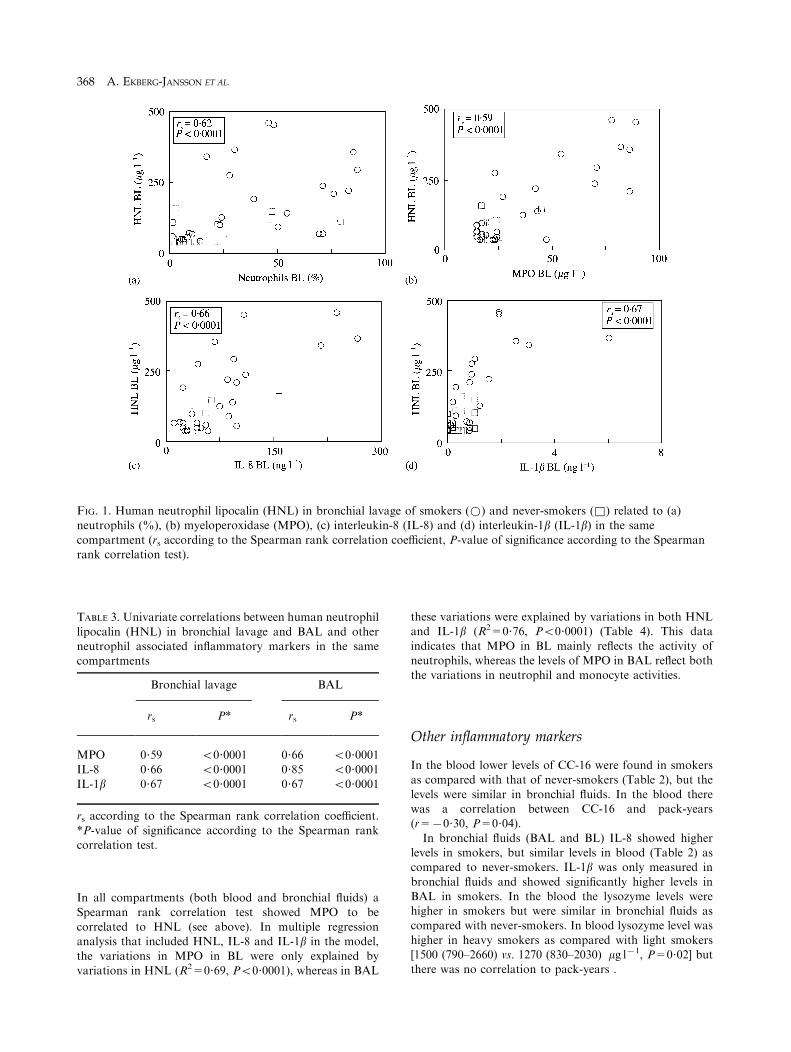
TABLE 3. Univariate correlations between human neutrophil
lipocalin (HNL) in bronchial lavage and BAL and otherneutrophil associated inflammatory markers in the samecompartments
Bronchial lavage BAL
rs P* rs P*
MPO 0?59 50?0001 0?66 50?0001IL-8 0?66 50?0001 0?85 50?0001
IL-1b 0?67 50?0001 0?67 50?0001
rs according to the Spearman rank correlation coefficient.
*P-value of significance according to the Spearman rankcorrelation test.
FIG. 1. Human neutrophil lipocalin (HNL) in bronchial lavage of smokers (*) and never-smokers (&) related to (a)neutrophils (%), (b) myeloperoxidase (MPO), (c) interleukin-8 (IL-8) and (d) interleukin-1b (IL-1b) in the same
compartment (rs according to the Spearman rank correlation coefficient, P-value of significance according to the Spearmanrank correlation test).
368 A. EKBERG-JANSSON ET AL.
In all compartments (both blood and bronchial fluids) a
Spearman rank correlation test showed MPO to becorrelated to HNL (see above). In multiple regressionanalysis that included HNL, IL-8 and IL-1b in the model,
the variations in MPO in BL were only explained byvariations in HNL (R2=0?69, P50?0001), whereas in BAL
these variations were explained by variations in both HNLand IL-1b (R2=0?76, P50?0001) (Table 4). This data
indicates that MPO in BL mainly reflects the activity ofneutrophils, whereas the levels of MPO in BAL reflect boththe variations in neutrophil and monocyte activities.
Other inflammatory markers
In the blood lower levels of CC-16 were found in smokersas compared with that of never-smokers (Table 2), but the
levels were similar in bronchial fluids. In the blood therewas a correlation between CC-16 and pack-years(r=70?30, P=0?04).
In bronchial fluids (BAL and BL) IL-8 showed higherlevels in smokers, but similar levels in blood (Table 2) ascompared to never-smokers. IL-1b was only measured in
bronchial fluids and showed significantly higher levels inBAL in smokers. In the blood the lysozyme levels werehigher in smokers but were similar in bronchial fluids ascompared with never-smokers. In blood lysozyme level was
higher in heavy smokers as compared with light smokers[1500 (790–2660) vs. 1270 (830–2030) mg l71, P=0?02] butthere was no correlation to pack-years .

TABLE 4. Stepwise multiple regression analysis (backwards) in smokers and never-smokers with MPO as the dependentvariable and HNL, IL-8 and IL-1b in bronchial lavage (BL) as the independent variables (n=47)
(a) MPO in bronchial lavage
R=0?83 R2=0?69 Adjusted R2=0?69 P50?0001 SE of estimate: 12?8
B SE of B P-level
Intercept 11?6 2?7 0?000HNL BL 0?16 0?02 50?0001
(b) MPO in BAL
R=0?87 R2=0?76 Adjusted R2=0?75 P50?0001 SE of estimate: 11?1B SE of B P-level
Intercept 3?1 2?4 0?000
HNL BAL 0?07 0?03 0?02II-1b BAL 3?4 0?49 50?0001
R=regression coefficient; B=the raw regression coefficient.
INFLAMMATORY MARKERS AND EMPHYSEMATOUS CHANGES IN SMOKERS 369
LUNG FUNCTION TESTS
Lung function measurements were impaired in smokers
seen in FEV1, VC, DLCO, carbon monoxide transfer factor(DLCO/VA ) and N2 test, as compared with never-smokers(Table 5).The influence of the degree of current smoking was only
significant between heavy and light smokers in total lungcapacity (TLC) [101% (SD 13%) vs. 93% (SD 12%),P=0?02] and residual volume (RV) [124% (SD 30%) vs.
104% (SD 30%), P=0?01]. The influence of pack-years i.e.when the smokers were divided into two groups under andabove the median pack-years (35), was only significant for
TLC [101% (SD 12%) vs. 94% (SD 13%), P=0?04].
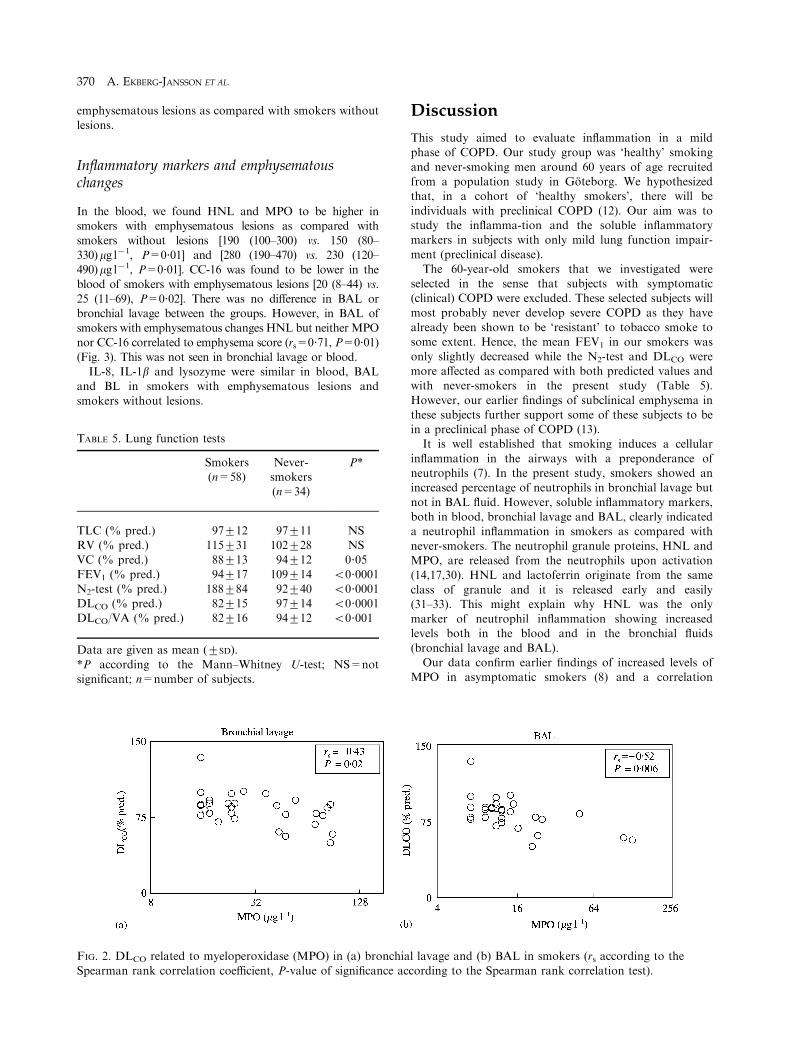
INFLAMMATORY MARKERS RELATED TOLUNG FUNCTION TESTS
Among smokers the Spearman rank correlation test showedcorrelations between MPO in bronchial fluids and DLCO
(Fig. 2) but not between MPO and FEV1 or the results of
the N2 test. DLCO showed a negative correlation to IL-1bin BAL (rs=70?48, P=0?007) and to IL-8 in BL andBAL (rs=70?37, P=0?03 and rs=70?42, P=0?02,respectively).
Multiple regression analysis that included all markers inthe model and smoking habits showed that MPO, IL-1b,IL-8 and CC-16 in BL as independent variables contributed
to the explanation of 41% (P50?0003) of the variations inDLCO (Table 6). The contribution of IL-8 and CC-16 wassmall, however. A similar estimation with the results in
BAL showed that MPO as the only independent variable
explained 22% of the variations in DLCO (P50?0009).When MPO was removed from the analysis, it was seen thatHNL in BL and HNL and IL-1b in BAL to a certain extent
replaced MPO as an independent explanatory variable.None of the blood variables offered any explanation to
the variations in DLCO, either by means of an univariate
analysis or in a multiple regression analysis when smokehabit was included in the model.
INFLAMMATORY CELLS AND MARKERSRELATED TO EMPHYSEMATOUSCHANGES
Inflammatory cells and emphysematous changes
In BAL, the cell concentration and the total number ofcells in the BAL fluid were higher in smokers withemphysematous lesions as compared with smokers with-
out lesions [31?0 (7?7–68?6) vs. 16?9 (1?2–43?6)6104
cellsml71, P=0?02] and [20?0 (4?5–48?0) vs. 10?8(0?5–25?0)6106 cells, P=0?05]. The number of alveolarmacrophages in BAL was also higher in smokers
with emphysematous lesions as compared with smokerswithout lesions [29?5 (7?5–65?1) vs. 16?0 (0?5–40?6)6104
cellsml71, P=0?03]. There was no difference concer-
ning cell recovery, cell viability, lymphocytes, neutro-phils, eosinophils and ciliated cells in BAL between thegroups.
In bronchial lavage, there was no significant differenceconcerning percentage of macrophages, neutrophils, lym-phocytes, eosinophils and ciliated cells in smokers with
370 A. EKBERG-JANSSON ET AL.
emphysematous lesions as compared with smokers withoutlesions.
Inflammatory markers and emphysematouschanges
In the blood, we found HNL and MPO to be higher insmokers with emphysematous lesions as compared withsmokers without lesions [190 (100–300) vs. 150 (80–
330)mg l71, P=0?01] and [280 (190–470) vs. 230 (120–490)mg l71, P=0?01]. CC-16 was found to be lower in theblood of smokers with emphysematous lesions [20 (8–44) vs.
25 (11–69), P=0?02]. There was no difference in BAL orbronchial lavage between the groups. However, in BAL ofsmokers with emphysematous changes HNL but neither MPOnor CC-16 correlated to emphysema score (rs=0?71, P=0?01)
(Fig. 3). This was not seen in bronchial lavage or blood.IL-8, IL-1b and lysozyme were similar in blood, BAL
and BL in smokers with emphysematous lesions and
smokers without lesions.
FIG. 2. DLCO related to myeloperoxidase (MPO) in (a) bronchi
Spearman rank correlation coefficient, P-value of significance a
TABLE 5. Lung function tests
Smokers(n=58)
Never-smokers(n=34)
P*
TLC (% pred.) 97+12 97+11 NSRV (% pred.) 115+31 102+28 NSVC (% pred.) 88+13 94+12 0?05
FEV1 (% pred.) 94+17 109+14 50?0001N2-test (% pred.) 188+84 92+40 50?0001DLCO (% pred.) 82+15 97+14 50?0001DLCO/VA (% pred.) 82+16 94+12 50?001
Data are given as mean (+SD).
*P according to the Mann–Whitney U-test; NS=notsignificant; n=number of subjects.
Discussion
This study aimed to evaluate inflammation in a mildphase of COPD. Our study group was ‘healthy’ smoking
and never-smoking men around 60 years of age recruitedfrom a population study in Goteborg. We hypothesizedthat, in a cohort of ‘healthy smokers’, there will be
individuals with preclinical COPD (12). Our aim was tostudy the inflamma-tion and the soluble inflammatorymarkers in subjects with only mild lung function impair-
ment (preclinical disease).The 60-year-old smokers that we investigated were
selected in the sense that subjects with symptomatic(clinical) COPD were excluded. These selected subjects will
most probably never develop severe COPD as they havealready been shown to be ‘resistant’ to tobacco smoke tosome extent. Hence, the mean FEV1 in our smokers was
only slightly decreased while the N2-test and DLCO weremore affected as compared with both predicted values andwith never-smokers in the present study (Table 5).
However, our earlier findings of subclinical emphysema inthese subjects further support some of these subjects to bein a preclinical phase of COPD (13).It is well established that smoking induces a cellular
inflammation in the airways with a preponderance ofneutrophils (7). In the present study, smokers showed anincreased percentage of neutrophils in bronchial lavage but
not in BAL fluid. However, soluble inflammatory markers,both in blood, bronchial lavage and BAL, clearly indicateda neutrophil inflammation in smokers as compared with
never-smokers. The neutrophil granule proteins, HNL andMPO, are released from the neutrophils upon activation(14,17,30). HNL and lactoferrin originate from the same
class of granule and it is released early and easily(31–33). This might explain why HNL was the onlymarker of neutrophil inflammation showing increasedlevels both in the blood and in the bronchial fluids
(bronchial lavage and BAL).Our data confirm earlier findings of increased levels of
MPO in asymptomatic smokers (8) and a correlation
al lavage and (b) BAL in smokers (rs according to the
ccording to the Spearman rank correlation test).
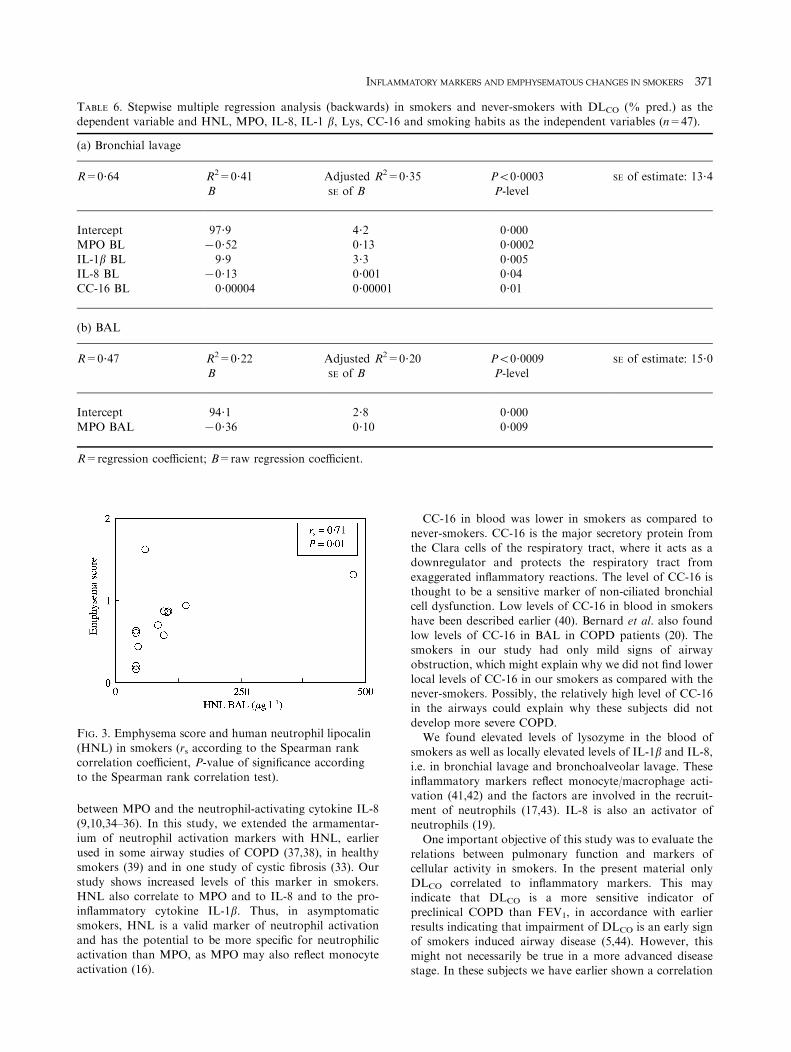
TABLE 6. Stepwise multiple regression analysis (backwards) in smokers and never-smokers with DLCO (% pred.) as thedependent variable and HNL, MPO, IL-8, IL-1 b, Lys, CC-16 and smoking habits as the independent variables (n=47).
(a) Bronchial lavage
R=0?64 R2=0?41 Adjusted R2=0?35 P50?0003 SE of estimate: 13?4
B SE of B P-level
Intercept 97?9 4?2 0?000MPO BL 70?52 0?13 0?0002
IL-1b BL 9?9 3?3 0?005IL-8 BL 70?13 0?001 0?04CC-16 BL 0?00004 0?00001 0?01
(b) BAL
R=0?47 R2=0?22 Adjusted R2=0?20 P50?0009 SE of estimate: 15?0B SE of B P-level
Intercept 94?1 2?8 0?000
MPO BAL 70?36 0?10 0?009
R=regression coefficient; B=raw regression coefficient.
FIG. 3. Emphysema score and human neutrophil lipocalin
(HNL) in smokers (rs according to the Spearman rankcorrelation coefficient, P-value of significance accordingto the Spearman rank correlation test).
INFLAMMATORY MARKERS AND EMPHYSEMATOUS CHANGES IN SMOKERS 371
between MPO and the neutrophil-activating cytokine IL-8(9,10,34–36). In this study, we extended the armamentar-
ium of neutrophil activation markers with HNL, earlierused in some airway studies of COPD (37,38), in healthysmokers (39) and in one study of cystic fibrosis (33). Our
study shows increased levels of this marker in smokers.HNL also correlate to MPO and to IL-8 and to the pro-inflammatory cytokine IL-1b. Thus, in asymptomaticsmokers, HNL is a valid marker of neutrophil activation
and has the potential to be more specific for neutrophilicactivation than MPO, as MPO may also reflect monocyteactivation (16).
CC-16 in blood was lower in smokers as compared to
never-smokers. CC-16 is the major secretory protein fromthe Clara cells of the respiratory tract, where it acts as adownregulator and protects the respiratory tract from
exaggerated inflammatory reactions. The level of CC-16 isthought to be a sensitive marker of non-ciliated bronchialcell dysfunction. Low levels of CC-16 in blood in smokers
have been described earlier (40). Bernard et al. also foundlow levels of CC-16 in BAL in COPD patients (20). Thesmokers in our study had only mild signs of airway
obstruction, which might explain why we did not find lowerlocal levels of CC-16 in our smokers as compared with thenever-smokers. Possibly, the relatively high level of CC-16in the airways could explain why these subjects did not
develop more severe COPD.We found elevated levels of lysozyme in the blood of
smokers as well as locally elevated levels of IL-1b and IL-8,
i.e. in bronchial lavage and bronchoalveolar lavage. Theseinflammatory markers reflect monocyte/macrophage acti-vation (41,42) and the factors are involved in the recruit-
ment of neutrophils (17,43). IL-8 is also an activator ofneutrophils (19).One important objective of this study was to evaluate the
relations between pulmonary function and markers ofcellular activity in smokers. In the present material onlyDLCO correlated to inflammatory markers. This mayindicate that DLCO is a more sensitive indicator of
preclinical COPD than FEV1, in accordance with earlierresults indicating that impairment of DLCO is an early signof smokers induced airway disease (5,44). However, this
might not necessarily be true in a more advanced diseasestage. In these subjects we have earlier shown a correlation

372 A. EKBERG-JANSSON ET AL.
between CD8+ lymphocytes in central bronchial biopsiesand FEV1 (45). This may indicate that FEV1 in a preclinical
disease stage mainly reflects more central airway events andto a lesser extent more peripheral events. This last statementgets further support by a recent paper con-cerning the same
population where we have shown that men with mildrespiratory symptoms had lower FEV1, while no associationcould be seen between symptoms and DLCO (46).
In the multiple regression analyses, MPO in both BLand BAL was the most important contributing variableto explain the variation in DLCO, even when smokinghabits were taken into account. The finding that HNL,
especially in BL, replaces MPO in the multiple regressionanalysis indicates that neutrophils are important in thedevelopment of impaired DLCO. The finding in BAL that
IL-1b also replaces MPO in the multiple regression analysisalso suggests that monocytes (together with neutrophils)are involved in the development of decreased DLCO in
the more peripheral segments of the airways. This conceptis also strengthened by the correlations seen in multipleregression analyses in this study between MPO and
HNL on the BL level and HNL together with IL-1b onthe BAL level.This study also extends its focus from lung function
variables to emphysematous changes seen on HRCT. There
seem to be parallel changes in HRCT and DLCO in thispreclinical phase of COPD, as well as cellular changes withincreased percentages of neutrophils and monocytes/
macrophages in the peripheral airway segments studied byBAL fluid. Furthermore, the most specific neutrophilactivation marker, HNL, correlates to the extent of
emphysema in the smokers. This indicates that, within thesmoking population, the neutrophilic activation is stronglyrelated to the degree of emphysema. There is also a relation
between neutrophil activation markers and a fall in DLCO
studied with multiple regression analysis, where smokinghabits was one of the independent variables.In conclusion, this study shows that smokers without
known airway disease do show several signs of airwayinflammation and lung function deterioration. Markers ofneutrophil and monocyte/macrophage activation correlate
to lung function deterioration and early emphysematouschanges, independently of smoking habits, which indicatesthat this could be a primary event in smoking induced
airway inflammation. These activation mechanisms arepossibly different in smokers who develop COPD and insmokers who do not develop COPD. The peripheralinflammatory events seem to be best reflected physiologi-
cally in DLCO measurements.
Acknowledgements
This study was supported by grants from the SwedishHeart–Lung Foundation, the Swedish Medical ResearchCouncil and Goteborg University, Sweden.
References
1. Lofdahl CG. Cost development of obstructive airway
disease in Sweden. Eur Respir Rev 1996; 6: 113–115.
2. Fletcher CM, Peto R, Tinker CM, Speizer FE. TheNatural History of Cronic Bronchitis and Emphysema.
Oxford: Oxford University Press, 1976; 47–91.3. Oxhoj H, Bake B, Wilhelmsen L. Ability of spirometry,
flow-volume curves and the nitrogen closing volume test
to detect smokers. Scand J Respir Dis 1977; 58: 80–96.4. Olofsson J, Bake B, Svardsudd K, Skoogh BE. The
single breath N2-test predicts the rate of decline in
FEV1. Eur J Respir Dis 1986; 69: 46–56.5. Knudson RJ, Kaltenborn WT, Burrows B. The effects
of cigarette smoking and smoking cessation on thecarbon monoxide diffusing capacity of the lung in
asymptomatic subjects. Am Rev Respir Dis 1989; 140:645–651.
6. Cosio MD, Ghezzo H, Hogg JC, et al. The relations
between structural changes in small airways andpulmonary function tests. N Engl J Med 1978; 298:
1277–1281.
7. Thompson A, Daughton D, Robbins R, Ghafouri M,Oehlerking M, Rennard S. Intraluminal airway inflam-mation in chronic bronchitis. Characterization and
correlation with clinical parameters. Am Rev Respir Dis1989; 140: 1527–1537.
8. Linden M, Rasmussen JB, Piitulainen E, et al. Airwayinflammation in smokers with nonobstructive and
obstructive chronic bronchitis. Am Rev Respir Dis1993; 148: 1226–1232.
9. Riise G, Ahlstedt S, Larsson S, et al. Bronchial
inflammation in chronic bronchitis assessed by mea-surement of cell products in bronchial lavage fluid.Thorax 1995; 50: 360–365.
10. Nocker RE, Schoonbrood DF, van de Graaf EA, et al.Interleukin-8 in airway inflammation in patients withasthma and chronic obstructive pulmonary disease. Int
Arch Allergy Immunol 1996; 109: 183–191.11. Pesci A, Balbi B, Majori M, et al. Inflammatory cells
and mediators in bronchial lavage of patients withchronic obstructive pulmonary disease. Eur Respir J
1998; 12: 380–386.12. Cherniack NS. Preclinical COPD: description, recogni-
tion and therapy. Chronic Obstructive Pulmonary Dis-
ease. 1st ed. Philadelphia: W.B. Saunders Company,1991; 420–427.
13. Tylen U, Boijsen M, Ekberg-Jansson A, Bake B,
Lofdahl CG. Emphysematous lesions and lung func-tion in healthy smokers 60 years of age. Respir Med2000; 94: 38–43.
14. Seveus L, Amin K, Peterson CGB, Roomans GM,
Venge P. Human neutrophil lipocalin (HNL) is aspecific granule constituent of the neutrophil granulo-cyte. Studies in bronchial and lung parenchymal tissue
and peripheral blood cells. Histochem Cell Biol 1997;107: 423–432.
15. Venge P, Bergstrand H, Hakansson L. Neutrophils and
eosinophils. In: Kelley WN, Harris ED, Ruddy S,Sledge CB, eds. Textbook of Rheumatology. 4th ed.Philadelphia: W.B. Saunders Company, 1993; 269–285.
16. Nichols BA, Bainton DF, Farquhar MG. Differentia-tion of monocytes; origin, nature, and fate of theirazurophil granules. J Cell Biol 1971; 50: 498–515.

INFLAMMATORY MARKERS AND EMPHYSEMATOUS CHANGES IN SMOKERS 373
17. Venge P. The monitoring of inflammation by specificcellular markers. Scand J Clin Lab Invest 1994; 54:
47–54.18. Oppenheim JJ, Kovacs EJ, Matsushinma K, Durum
SK. There is more than one interleukin-1. Immunol
Today 1986; 7: 45–48.19. Kunkel SL, Standiford T, Kasahara K, Strieter RM.
IL-8: the major neutrophil chemotactic factor in the
lung. Exp Lung Res 1991; 17: 17–23.20. Bernard A, Marchandise FX, Depelchin S, Lauwerys R,
Sibille Y. Clara cell protein in serum and bronchoal-veolar lavage. Eur Respir J 1992; 5: 1231–1238.
21. Rosengren A, Orth-Gomer K, Wedel H, Wilhelmsen L.Stressful life events, social support, and mortality inmen born in 1933. BMJ 1993; 307: 1102–1105.
22. Xu SY, Petersson C, Carlson M, Venge P. Thedevelopment of an assay for human neutrophillipocalin (HNL)-to be used as a specific marker of
neutrophil activity in vivo and vitro. J Immunol Methods1994; 171: 245–252.
23. Bernard A, Lauwerys R, Noel A, Vandeleene B,
Lambert A. Determination of protein 1 in normaland pathological urine. Clin Chem Acta 1991; 201:
213–246.24. Lindley I, Aschauer H, Seifert JM, et al. Synthesis and
expression in Escherichia coli of the gene encodingmonocyte- derived neutrophil-activating factor: biolo-gical equivalence between natural and recombinant
neutrophil-activating factor. Proc Natl Acad Sci USA1988; 85: 9199–9203.
25. Venge P, Hallgren R, Stalenheim G, Olsson I. Effects of
serum and cations on the selective release of granularproteins from human neutrophils during phagocytosis.Scand J Haematol 1979; 22: 317–326.
26. Oxhoj H, Bake B. Measurement of closing volume withthe single breath nitrogen method. Scand J Respir Dis1974; 55: 320–331.
27. Quanjer PH, Tammeling GJ, Cotes JE, Pedersen OF,
Peslin R, Yerault JC. Lung volumes and forcedventilatory flows. Official statment of the EuropeanRespiratory Society. Eur Respir J 1993; 6: 5–40.
28. Sixt R, Bake B, Oxhoj H. The single-breath N2-test andspirometry in healthy non-smoking males. Eur J RespirDis 1984; 65: 296–304.
29. Salorinne Y. Single-breath pulmonary diffusing capa-city. Reference values and application in connectivetissue diseases avd in various lung diseases. Scand JResp Dis 1976; (Suppl. 96): 9–28, 80–81.
30. Behera D, Dash S, Sen S. Neutrophil count andmyeloperoxidase activity in Indian bidi smokers.Respiration 1994; 61: 269–273.
31. Jensen EJ, Pedersen B, Schmidt E, Venge P, Dahl R.Serum eosinophilic cationic protein and lactoferrinrelated to smoking history and lung function. Eur
Respir J 1994; 7: 927–933.32. Xu SY, Carlson M, Engstrom A, Garcia R, Peterson
CG, B, Venge P. Purification and characterization of a
human neutrophil lipocalin (HNL) from the secondarygranules of human neutrophils. Scand J Clin Lab Invest1994; 54: 365–376.
33. Eichler I, Nilsson M, Rath R, Enander I, Venge P,Koller DY. Human neutrophil lipocalin, a highly
specific marker for acute exacerbation in cystic fibrosis.Eur Respir J 1999; 14: 1145–1149.
34. Chanez P, Enander I, Jones I, Godard P, Bousquet J.
Interleukin 8 in the bronchoalveolar lavage of asth-matics and chronic bronchitis patients. Int Arch AllergyImmunol 1996; 111: 83–88.
35. Yamamoto C, Yoneda T, Yoshikava M, et al. Airwayinflammation in COPD assessed by sputum levels ofinterleukin-8. Chest 1997; 112: 505–510.
36. Hill AT, Bayley D, Stockley RA. The interrelationship
of sputum inflammatory markers in patients withchronic bronchitis. Am J Respir Crit Care Med 1999;160: 893–898.
37. Keatings VM, Barnes PJ. Granulocyte activation markersin induced sputum: comparison between chronic ob-structive pulmonary disease, asthma, and normal sub-
jects. Am J Respir Crit Care Med 1997; 155: 449–453.38. Keatings VM, Jatakanon A, Worsdell YM, Barnes PJ.
Effects of inhaled and oral glucocorticoids on inflam-
matory indices in asthma and COPD. Am J Respir CritCare Med 1997; 155: 542–548.
39. Betsuyaku T, Nishimura M, Takeyabu K, et al.Neutrophil granule proteins in bronchoalveolar lavage
fluid from subjects with subclinical emphysema. Am JRespir Crit Care Med 1999; 159: 1985–1991.
40. Bernard A, Roels H, Buchet JP, Lauwerys R. Serum
Clara cell protein: an indicator of bronchial celldysfunction caused by tobacco smoking. Environ Res1994; 66: 96–104.
41. Janson RW, Hance KR, King TE. Human alveolarmacrophages produce predominantly the 35-kD pro-forms of interleukin-1a and interleukin-1b when
stimulated with lipopolysaccachrides. Am J Respir CritCare Med 1995; 151: 1613–1620.
42. McCrea KA, Ensor JE, Nall K, Bleecker ER, HasdayJD. Altered cytokine regulation in the lungs of cigarette
smokers. Am J Respir Crit Care Med 1994; 150: 696–703.43. Pober JS, Gimbrone MAJ, Lapierre LA, et al.
Overlapping patterns of activation of human endo-
thelial cells by interleukin-1, tumor necrosis factor,and immune interferon. J Immunol 1986; 137:
1893–1896.
44. Welle I, Eide GE, Bakke PS, Gulsvik A. The single-breath transfer factor for carbon monoxide andrespiratory symptoms in a Norwegian communitysample. Eur Respir J 1999; 14: 1320–1325.
45. Ekberg-Jansson A, Andersson B, Arva E, Nilsson O,Lofdahl CG. The expression of lymphocyte surfaceantigens in bronchial biopsies, bronchoalveolar
lavage cells and blood cells in healthy smoking andnever-smoking men, 60 years old. Respir Med 2000; 94:264–272.
46. Ekberg-Jansson A, Bake B, Andersson B, Skoogh BE,Lofdahl CG. Respiratory symptoms relate to physio-logic changes and inflammatory markers reflecting
central but not peripheral airways. A study in 60-year-old ‘healthy’ smokers and never-smokers. RespirMed 2001, 95: 40–47.