
Nakiterpiosin targets tubulin and triggers mitotic catastrophe in human cancer cells
12
2010;9:3375-3385. Published OnlineFirst December 7, 2010. Mol Cancer Ther Jen-Hsuan Wei and Joachim Seemann Human Cancer Cells Nakiterpiosin Targets Tubulin and Triggers Mitotic Catastrophe in Updated version 10.1158/1535-7163.MCT-10-0305 doi: Access the most recent version of this article at: Cited Articles http://mct.aacrjournals.org/content/9/12/3375.full.html#ref-list-1 This article cites by 31 articles, 10 of which you can access for free at: E-mail alerts related to this article or journal. Sign up to receive free email-alerts Subscriptions Reprints and . [email protected] To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at Permissions . [email protected] To request permission to re-use all or part of this article, contact the AACR Publications Department at on October 18, 2013. © 2010 American Association for Cancer Research. mct.aacrjournals.org Downloaded from Published OnlineFirst December 7, 2010; DOI: 10.1158/1535-7163.MCT-10-0305 on October 18, 2013. © 2010 American Association for Cancer Research. mct.aacrjournals.org Downloaded from Published OnlineFirst December 7, 2010; DOI: 10.1158/1535-7163.MCT-10-0305 on October 18, 2013. © 2010 American Association for Cancer Research. mct.aacrjournals.org Downloaded from Published OnlineFirst December 7, 2010; DOI: 10.1158/1535-7163.MCT-10-0305 on October 18, 2013. © 2010 American Association for Cancer Research. mct.aacrjournals.org Downloaded from Published OnlineFirst December 7, 2010; DOI: 10.1158/1535-7163.MCT-10-0305 on October 18, 2013. © 2010 American Association for Cancer Research. mct.aacrjournals.org Downloaded from Published OnlineFirst December 7, 2010; DOI: 10.1158/1535-7163.MCT-10-0305 on October 18, 2013. © 2010 American Association for Cancer Research. mct.aacrjournals.org Downloaded from Published OnlineFirst December 7, 2010; DOI: 10.1158/1535-7163.MCT-10-0305 on October 18, 2013. © 2010 American Association for Cancer Research. mct.aacrjournals.org Downloaded from Published OnlineFirst December 7, 2010; DOI: 10.1158/1535-7163.MCT-10-0305 on October 18, 2013. © 2010 American Association for Cancer Research. mct.aacrjournals.org Downloaded from Published OnlineFirst December 7, 2010; DOI: 10.1158/1535-7163.MCT-10-0305 on October 18, 2013. © 2010 American Association for Cancer Research. mct.aacrjournals.org Downloaded from Published OnlineFirst December 7, 2010; DOI: 10.1158/1535-7163.MCT-10-0305 on October 18, 2013. © 2010 American Association for Cancer Research. mct.aacrjournals.org Downloaded from Published OnlineFirst December 7, 2010; DOI: 10.1158/1535-7163.MCT-10-0305 on October 18, 2013. © 2010 American Association for Cancer Research. mct.aacrjournals.org Downloaded from Published OnlineFirst December 7, 2010; DOI: 10.1158/1535-7163.MCT-10-0305 on October 18, 2013. © 2010 American Association for Cancer Research. mct.aacrjournals.org Downloaded from Published OnlineFirst December 7, 2010; DOI: 10.1158/1535-7163.MCT-10-0305
Transcript of Nakiterpiosin targets tubulin and triggers mitotic catastrophe in human cancer cells

2010;9:3375-3385. Published OnlineFirst December 7, 2010.Mol Cancer Ther Jen-Hsuan Wei and Joachim Seemann Human Cancer CellsNakiterpiosin Targets Tubulin and Triggers Mitotic Catastrophe in
Updated version
10.1158/1535-7163.MCT-10-0305doi:
Access the most recent version of this article at:
Cited Articles
http://mct.aacrjournals.org/content/9/12/3375.full.html#ref-list-1
This article cites by 31 articles, 10 of which you can access for free at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
To request permission to re-use all or part of this article, contact the AACR Publications Department at
on October 18, 2013. © 2010 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst December 7, 2010; DOI: 10.1158/1535-7163.MCT-10-0305
on October 18, 2013. © 2010 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst December 7, 2010; DOI: 10.1158/1535-7163.MCT-10-0305
on October 18, 2013. © 2010 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst December 7, 2010; DOI: 10.1158/1535-7163.MCT-10-0305
on October 18, 2013. © 2010 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst December 7, 2010; DOI: 10.1158/1535-7163.MCT-10-0305
on October 18, 2013. © 2010 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst December 7, 2010; DOI: 10.1158/1535-7163.MCT-10-0305
on October 18, 2013. © 2010 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst December 7, 2010; DOI: 10.1158/1535-7163.MCT-10-0305
on October 18, 2013. © 2010 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst December 7, 2010; DOI: 10.1158/1535-7163.MCT-10-0305
on October 18, 2013. © 2010 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst December 7, 2010; DOI: 10.1158/1535-7163.MCT-10-0305
on October 18, 2013. © 2010 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst December 7, 2010; DOI: 10.1158/1535-7163.MCT-10-0305
on October 18, 2013. © 2010 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst December 7, 2010; DOI: 10.1158/1535-7163.MCT-10-0305
on October 18, 2013. © 2010 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst December 7, 2010; DOI: 10.1158/1535-7163.MCT-10-0305
on October 18, 2013. © 2010 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst December 7, 2010; DOI: 10.1158/1535-7163.MCT-10-0305

Preclinical Development
Nakiterpiosin Targets Tubulin and Triggers MitoticCatastrophe in Human Cancer Cells
Jen-Hsuan Wei and Joachim Seemann
AbstractAgents that interfere with mitotic progression by perturbing microtubule dynamics are commonly used for
cancer chemotherapy. Here, we identify nakiterpiosin as a novel antimitotic drug that targets microtubules.
Nakiterpiosin induces mitotic arrest and triggers mitotic catastrophe in human cancer cells by impairing
bipolar spindle assembly. At higher concentration, it alters the interphase microtubule network and
suppresses microtubule dynamics. In the presence of nakiterpiosin, microtubules are no longer arranged
in a centrosomal array and centrosome-mediated microtubule regrowth after cold depolymerization is
inhibited. However, centrosome organization, the ultrastructure of Golgi stacks, and protein secretion are
not affected, suggesting that the drug has minimal toxicity toward other cellular functions. Nakiterpiosin
interacts directly with tubulin, inhibits microtubule polymerization in vitro, and decreases polymer mass in
cells. Furthermore, it enhances tubulin acetylation and reduces viability of paclitaxel-resistant cancer cells. In
conclusion, nakiterpiosin exerts antiproliferative activity by perturbing microtubule dynamics during mitosis
that activates the spindle assembly checkpoint and triggers cell death. These findings suggest the potential use
of nakiterpiosin as a chemotherapeutic agent. Mol Cancer Ther; 9(12); 3375–85. �2010 AACR.
Introduction
Cancer chemotherapy relies primarily on agents calledantimitotics that prevent or interferewithmitotic progres-sion (1). These compounds have provenwidely successfuland currently all such drugs in clinical use target micro-tubules. Microtubules are dynamic cytoskeletal filamentsessential for a wide variety of cellular processes such aschromosome segregation (2), organelle positioning (3),protein trafficking (4), and ciliogenesis (5). Composedof a- and b-tubulin heterodimers, microtubules alternatebetween phases of growth and shrinkage, a processtermed dynamic instability (6). In interphase, microtu-bules are normally arranged into a radial array emanatingfrom the centrosomes (7). On entry into mitosis, themicrotubule network is disassembled to allow assemblyof the mitotic spindle. At this transition, the dynamicinstability of microtubules is significantly increased (8),making it an excellent target for microtubule poisons tointerfere with mitosis. In contrast to most cells in thehuman body that are postmitotic, cancer cells have the
ability to proliferate and divide autonomously. By per-turbingmicrotubule dynamics during cell division,whichin turn induces mitotic arrest and cell death, highly pro-liferative cancer cells can be selectively eliminated.
Nakiterpiosin was initially isolated from a marinesponge as a potent cytotoxic compound against mouseleukemia P388 cells (9). Furthermore, it was recentlyshown that a synthetic form (for structure, see Fig. 1)caused loss of the primary cilium and thus reducedHedgehog (Hh) signaling in NIH3T3 mouse fibroblasts(10). Therefore, nakiterpiosin has been suggested to func-tion as an Hh antagonist that could be used againstHh-dependent tumors.
In this article, we identify nakiterpiosin as a novelmicrotubule-targeting agent that triggers mitotic cata-strophe in cancer cells. Surprisingly, we found that naki-terpiosin alters microtubule dynamics, which is anentirely different mechanism of action than previouslyreported (10). Nakiterpiosin induces mitotic arrest andcell death by impairing spindle assembly. In addition tomitotic abnormalities, nakiterpiosin also causes inter-phase defects including disintegration of the centrosomalmicrotubule array and suppression of microtubuledynamics. However, protein secretion is not affected.Nakiterpiosin interacts directly with tubulin, inhibitsmicrotubule polymerization, enhances tubulin acetyla-tion, and reduces the viability of paclitaxel-resistant can-cer cells. In summary, we report the first identification ofnakiterpiosin as a novel antimitotic agent and a detailedcharacterization of its effects on microtubules. Thesefindings highlight the potential of nakiterpiosin as achemotherapeutic agent.
Authors' Affiliation: Department of Cell Biology, University of TexasSouthwestern Medical Center, Dallas, Texas
Note: Supplementary material for this article is available at MolecularCancer Therapeutics Online (http://mct.aacrjournals.org/).
Corresponding Author: Joachim Seemann, Department of Cell Biology,University of Texas Southwestern Medical Center, 5323 Harry HinesBlvd, Dallas, TX 75390. Phone: 214-648-0317; Fax: 214-648-9093.E-mail: [email protected]
doi: 10.1158/1535-7163.MCT-10-0305
�2010 American Association for Cancer Research.
MolecularCancer
Therapeutics
www.aacrjournals.org 3375

Materials and Methods
Cell culture, antibodies, and reagentsHeLa cells were obtained from the American Type
Culture Collection and were grown in Dulbecco’s mod-ified Eagle’s medium containing antibiotics (Mediatech)and 10% of cosmic calf serum (CCS; HyClone). Cellsfrom early-passage (<10) stocks were used withoutfurther authentication. NCI-H1155 and HCC366 cellswere obtained in June 2010 from the laboratory ofMichael A. White and were DNA fingerprinted andconfirmed in March 2010 in the laboratory of John D.Minna (University of Texas Southwestern Medical Cen-ter, Dallas, TX). The cells were used within 2 months ofreceipt and were grown in RPMI 1640 (Invitrogen)containing antibiotics and 5% of CCS as previouslydescribed (11). The following primary antibodies wereused: mouse monoclonal antibodies against a-tubulin(TAT1, K. Gull), acetylated a-tubulin (6-11B-1, Sigma),g-tubulin (GTU-88, Sigma), BubR1 (Upstate Biotechnol-ogy), CD8 (OKT8, F. Watt), rabbit polyclonal antibodiesagainst a-tubulin (Thermo Fisher Scientific), EB1(Sigma), pericentrin (Covance and Abcam), g-tubulin(Axyll), GM130 (MLO7, M. Lowe), and human CRESTautoimmune serum (ImmunoVision). The following sec-ondary antibodies were used: anti-mouse and anti-rabbitconjugated toAlexaFluor 488, 555, 594, or 647 (Invitrogen),Cy2-conjugated anti-human, and horseradish peroxi-dase–conjugated anti-mouse and anti-rabbit antibodies(Pierce). Hoechst 33342 was from Invitrogen; nocodazoleand paclitaxel from Sigma. Synthetic nakiterpiosin wasprovided by Chuo Chen (10). All drugs were dissolved indimethyl sulfoxide (DMSO).
In vitro tubulin polymerization assayThe fluorescence-based in vitro tubulin polymerization
assay was performed according to the manufacturer’sinstructions (Cytoskeleton) at 37�Cwith 2mg/mL bovinebrain tubulin (>99% pure), 1 mmol/L GTP and theindicated drugs. Reactions were monitored by emissionat 450 nm.
Determination of microtubule polymer massHeLa cells treated with vehicle or drugs for 2 hours
were lysed at room temperature with 80 mmol/L PIPES(pH 6.8), 1 mmol/L MgCl2, 1 mmol/L EGTA, 0.5%
NP-40, and 2 mmol/L paclitaxel. Supernatant and pelletfractions were separated by centrifugation at 13,000 rpmfor 5 minutes at room temperature. Equal volumes wereimmunoblotted for a-tubulin. The ‘‘Gels’’ function ofImageJ was employed to quantify band intensity.
Microtubule regrowth assayHeLa cells were placed on ice for 30 minutes to depo-
lymerize microtubules, then incubated on ice for another30 minutes with vehicle or 25 mmol/L nakiterpiosin,warmed to 37�C to initiate microtubule regrowth, andfixed with methanol at the indicated time points.
ImmunofluorescenceImmunostaining was performed as described (12).
Images were acquired using LD Plan-Neofluar 40x/1.3DIC and Plan-Apochromat 63x/1.4 DIC objectives, anAxiovert 200M microscope (Zeiss), an Orca 285 camera(Hamamatsu), and the software Openlab 4.0.2 (Improvi-sion). To determine the spindle length, the ‘‘Measure’’function of ImageJ was employed and the shortest dis-tance between 2 g-tubulin foci was averaged. The statis-tical significance was determined by unpaired, 2-tailedStudent’s t test.
Live cell imagingVehicle- and drug-treated cells were imaged in parallel
using a motorized stage (M€arzh€auser) in CO2-indepen-dent medium containing 2 mmol/L GlutaMAX (Invitro-gen) and 10% CCS. Images were taken every 5 minuteswith an A-PLAN 20x/0.3 Ph1 objective, an Axiovert200M microscope (Zeiss), a Retiga 2000R camera (QIma-ging), and the software MetaMorph 7.1.3 (MolecularDevices).
Microinjection and CD8 transport assayA solution of 0.2 mg/mL pcDNA-CD8 and 2.5 mg/mL
of lysine-fixable Texas Red-dextran (Invitrogen) was co-injected into the nuclei as described (13). Cycloheximide(100 mg/mL) was included during the microinjectionperiod to prevent protein synthesis. Cycloheximidewas then washed out to allow expression and transportof CD8 and the cells were then incubated with theindicated drugs. After 90 minutes at 37�C, cells werefixed in 3.7% formaldehyde without permeabilizationand stained for CD8.
Figure 1. Chemical structures ofnakiterpiosin (NAK), nocodazole(NOC), and paclitaxel (PTX).
Wei and Seemann
Mol Cancer Ther; 9(12) December 2010 Molecular Cancer Therapeutics3376

Electron microscopyElectron microscopy (EM) analysis of cells grown on
glass cover slips was performed as described (14). Fornegative staining ofmicrotubules, the end products of thepolymerization reactions were fixed by diluting withequal volume of 2.5% glutaraldehyde in 100 mmol/Lsodium cacodylate, pH 7.4 (prewarmed to 37�C). Sampleswere absorbed on glow-discharged, formvar-coatedgrids, stained with 2% uranyl acetate and washed withwater (15). Images were captured using a USC1000 2kCCD camera (Gatan) and a Tecnai G2 Spirit EM (FEI).
Cell viability assayHeLa, NCI-H1155, and HCC366 cells were grown for
48 hours in the presence of vehicle or the indicated drugs.Cell viability was then determined by the relative amountof ATP present (normalized to vehicle-treated cells) usingthe CellTiter-Glo luminescent cell viability assay accord-ing to the manufacturer’s instructions (Promega).
Results
Nakiterpiosin arrests cells in mitosis and causesmitotic catastrophe
To determine the effects of nakiterpiosin, we moni-tored human cervical carcinoma HeLa cells treated withvarying concentrations of nakiterpiosin for 24 hoursusing phase-contrast, time-lapse microscopy. The major-ity of cells treated with 1 mmol/L or less nakiterpiosinenteredmitosis and progressed through cytokinesis (datanot shown). In contrast, cells treated with 5 mmol/L ormore also entered mitosis but became arrested, as indi-cated by a gradual accumulation of rounded-up mitoticcells (Fig. 2A, from�24:00 to 0:00). The mitotic arrest wasnot because of long-term imaging, as control cells treatedwith vehicle and imaged in parallel divided normally(Supplementary Fig. S1). In addition, we observed exten-sive membrane blebbing in some nakiterpiosin-treatedcells that died after prolonged mitotic arrest (Fig. 2A,
A
B C
D
Figure 2. Nakiterpiosin (NAK) arrests cells in mitosis and causes mitotic catastrophe by impairing spindle assembly. A, NAK causes mitotic arrest and celldeath. Still images from a representative phase-contrast, time-lapsemovie (n¼ 3) at the time points indicated in hh:mm. HeLa cells were imaged for 24 hours in10 mmol/L NAK (�24:00). The drug was washed out (0:00) and the cells were imaged in fresh medium without the drug for another 24 hours (þ24:00). Arrowsindicate cells undergoing mitotic catastrophe (mitotic cell death). The lower panels show 2 representative cells at higher magnification. Arrowheads indicate aNAK-treated cell that entered mitosis (�0:45), became arrested (0:00), and divided (þ1:55) after washout. Arrows point to a cell that underwent prolongedmitotic arrest (�6:30) and subsequent cell death (þ1:55). B–D, HeLa cells were treated for 2 hours with DMSOor 5 mmol/L NAK and labeled for DNA, for BubR1,and with CREST serum tomark kinetochores (B) or for a-tubulin, g-tubulin, and DNA (C and D). B, the spindle assembly checkpoint is activated in NAK-treatedcells, as indicated by BubR1 recruitment to kinetochores. Vehicle-treated cells in prophase and anaphase were located next to each other in the same field.Bar, 10 mm. C, NAK arrests cells in mitosis with aberrant spindles. Fifty-seven percent of the NAK-arrested cells assembled bipolar-like spindles withuncongressed chromosomes (type I), 34% formed multiple asters (type II), and 9% developed monopolar spindles (type III; 170 mitotic cells counted). Bar,5 mm. D, the spindle length was measured as the distance between 2 g-tubulin foci at metaphase spindle poles. The interpolar distance was significantlydecreased by 42%, from 11.5 � 0.2 mm in DMSO-treated cells to 6.7 � 0.1 mm in NAK-treated cells (mean � SEM; 25–27 spindles per condition; P < 0.0001).
Nakiterpiosin Targets Microtubules
www.aacrjournals.org Mol Cancer Ther; 9(12) December 2010 3377

arrows; Supplementary Fig. S1). This suggests that naki-terpiosin kills cells by inducing mitotic catastrophe, aform of cell death that occurs during mitosis (16).
We then investigated whether the mitotic block isreversible. After imaging the cells for 24 hours in naki-terpiosin, we removed the drug and imaged the cells foran additional 24 hours (Fig. 2A, from 0:00 to þ24:00). Onnakiterpiosin washout, the arrested cells divided andappeared healthy for the duration of the experiment(Fig. 2A, arrowheads). The reversibility implies thatnakiterpiosin interferes with mitotic progression throughnoncovalent binding to its cellular target and does notinvolve any chemical modification.
Nakiterpiosin impairs spindle assemblyand chromosome alignment
The live cell imaging data showed that nakiterpiosin-treated cells entered mitosis but were blocked from exit-ing, indicating that the spindle assembly checkpoint(SAC) could not be satisfied. To verify that nakiterpiosininduces SAC activation, we examined whether SAC pro-teins are recruited to the kinetochores. HeLa cells treatedfor 2 hours with vehicle or 5 mmol/L nakiterpiosin werestained for DNA, the SAC kinase BubR1 and with CRESTserum to mark kinetochores. In vehicle-treated cells,BubR1 was localized to kinetochores after mitotic entry(Fig. 2B, prophase) but was displaced on chromosomesegregation (Fig. 2B, anaphase). Kinetochore targeting ofBubR1wasclearlydetected innakiterpiosin-arrested cells,indicating that the SAC was indeed activated (Fig. 2B,NAK). To further confirm that the mitotic arrest is causedby defects in spindle assembly, we analyzed spindlemorphology of drug-treated cells. HeLa cells were cost-ained for a-tubulin to visualizemicrotubules, g-tubulin todetect the spindle poles, and DNA to label chromosomes.Nearly all vehicle-treated cells in metaphase displayednormal bipolar spindles with condensed chromosomesproperly aligned at themetaphase plate (Fig. 2C, normal).In contrast, nakiterpiosin-arrested cells exhibited a rangeof spindle abnormalities that can be categorized into3 major types (170 mitotic cells counted; Fig. 2C, NAK).Fifty-seven percent of mitotic cells assembled bipolar-likespindles but chromosome alignment was not as compactas in control cells (Fig. 2C, type I). In most cases, uncon-gressed chromosomes were found in close proximity tothe spindle poles (Fig. 2C, arrowheads). Thirty-four per-cent of mitotic cells formed multiple asters with chromo-somes appearing as an irregular-shaped DNA mass(Fig. 2C, type II). The remaining 9% of the mitotic cellsdevelopedmonopolar spindleswhere chromosomeswerearranged in a circular configuration (Fig. 2C, type III).
When we increased the concentration to 10 mmol/L,we observed a lower percentage of bipolar-like spin-dles. Instead, multiple asters became the dominantphenotype. Interestingly, these asters tended to clustertogether rather than spread throughout the cytoplasm(data not shown). At 25 mmol/L, the spindles severelydegenerated into small asters or punctate aggregates
and, in some cases, were entirely depolymerized (datanot shown). At this concentration, the chromosomesappeared less condensed.
The spindle length is decreased by nakiterpiosinWhile analyzing spindle abnormalities induced by
5 mmol/L nakiterpiosin, we noticed that bipolar-like spin-dles were much smaller. To evaluate the difference, wemeasured the spindle length in vehicle- and drug-treatedcells, as determined by the distance between 2 g-tubulinfoci atmetaphase spindlepoles.On treatmentwith5mmol/L nakiterpiosin, the interpolar distance was dramaticallydecreased by 42%, from 11.5 � 0.2 mm in control cells to6.7 � 0.1 mm in drug-treated cells (25–27 spindles percondition; P < 0.0001; Fig. 2D). These data, together withother observed spindle defects, suggest that nakiterpiosinblocks cells inmitosisby interferingwith spindle assembly.
Nakiterpiosin alters interphase microtubuleorganization
Because of the inhibitory effect on spindle assembly,weinvestigated whether nakiterpiosin alters the interphasemicrotubule network. HeLa cells were treated for 2 hourswith 5 mmol/L nakiterpiosin or, as controls, incubatedwith vehicle, 5 mg/mL nocodazole, 1 mmol/L paclitaxel orcooled on ice for 30 minutes (Fig. 3A). In vehicle-treatedcells, microtubules concentrated in the perinuclear regionand extended outwards to the cell periphery (Fig. 3A,DMSO). As expected, filaments were completely depoly-merized on nocodazole or cold treatment, as indicated bythehazy fluorescence stainingofa-tubulin throughout thecytoplasm (Fig. 3A, NOC and cold). In contrast, stabiliza-tion of microtubules by paclitaxel induced parallelbundles (Fig. 3A, PTX). Interestingly, although low con-centration of nakiterpiosin caused prominent mitoticarrest and aberrant mitotic phenotypes (Fig. 2), the inter-phase microtubule network seemed virtually unaffectedand resembled the characteristic centrosomal array incontrol cells (Fig. 3A, NAK 5 mmol/L).
Becausemicrotubules exhibit lessdynamic instability ininterphase than in mitosis (17), we reasoned that inter-phase microtubules might be less sensitive to nakiterpio-sin. To address this, we increased the concentration andexamined the organization of the interphase network.When treatedwith 25mmol/Lnakiterpiosin,microtubulesbecame extremely distorted, and most strikingly, nolonger originated from the centrosomes or fully extendedto the cell periphery (Fig. 3A, NAK 25 mmol/L, the cellborder is outlinedwithdash lines). Instead, they appearedshorter and twisted together.Moreover, the cell peripherywas often devoid of filaments, suggesting that microtu-bules were shortened and might not reach the cell cortexdue to partial depolymerization.
Nakiterpiosin displaces EB1 from microtubule tipsEB1 is a microtubule-associated protein that preferen-
tially binds to the plus end of growingmicrotubules and isoften used as a marker for microtubule dynamics (18). In
Wei and Seemann
Mol Cancer Ther; 9(12) December 2010 Molecular Cancer Therapeutics3378
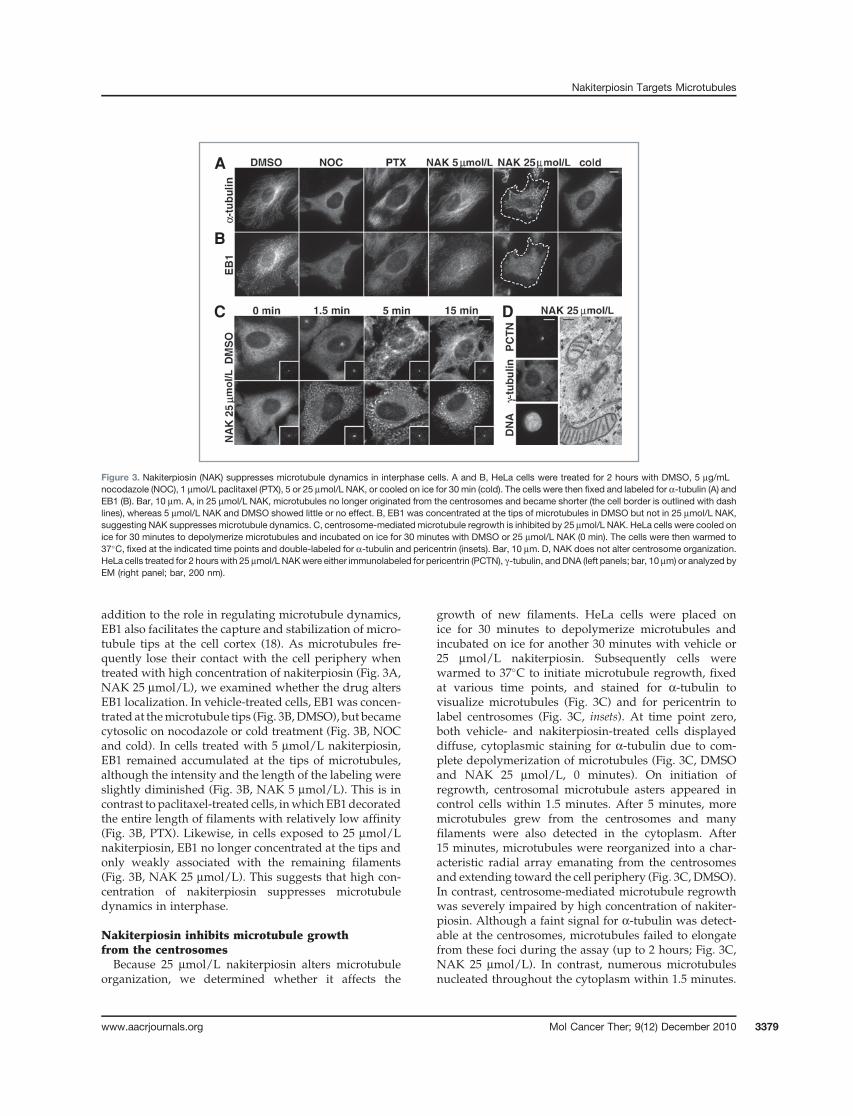
addition to the role in regulating microtubule dynamics,EB1 also facilitates the capture and stabilization of micro-tubule tips at the cell cortex (18). As microtubules fre-quently lose their contact with the cell periphery whentreated with high concentration of nakiterpiosin (Fig. 3A,NAK 25 mmol/L), we examined whether the drug altersEB1 localization. In vehicle-treated cells, EB1 was concen-trated at themicrotubule tips (Fig. 3B,DMSO), but becamecytosolic on nocodazole or cold treatment (Fig. 3B, NOCand cold). In cells treated with 5 mmol/L nakiterpiosin,EB1 remained accumulated at the tips of microtubules,although the intensity and the length of the labeling wereslightly diminished (Fig. 3B, NAK 5 mmol/L). This is incontrast to paclitaxel-treated cells, inwhichEB1decoratedthe entire length of filaments with relatively low affinity(Fig. 3B, PTX). Likewise, in cells exposed to 25 mmol/Lnakiterpiosin, EB1 no longer concentrated at the tips andonly weakly associated with the remaining filaments(Fig. 3B, NAK 25 mmol/L). This suggests that high con-centration of nakiterpiosin suppresses microtubuledynamics in interphase.
Nakiterpiosin inhibits microtubule growthfrom the centrosomesBecause 25 mmol/L nakiterpiosin alters microtubule
organization, we determined whether it affects the
growth of new filaments. HeLa cells were placed onice for 30 minutes to depolymerize microtubules andincubated on ice for another 30 minutes with vehicle or25 mmol/L nakiterpiosin. Subsequently cells werewarmed to 37�C to initiate microtubule regrowth, fixedat various time points, and stained for a-tubulin tovisualize microtubules (Fig. 3C) and for pericentrin tolabel centrosomes (Fig. 3C, insets). At time point zero,both vehicle- and nakiterpiosin-treated cells displayeddiffuse, cytoplasmic staining for a-tubulin due to com-plete depolymerization of microtubules (Fig. 3C, DMSOand NAK 25 mmol/L, 0 minutes). On initiation ofregrowth, centrosomal microtubule asters appeared incontrol cells within 1.5 minutes. After 5 minutes, moremicrotubules grew from the centrosomes and manyfilaments were also detected in the cytoplasm. After15 minutes, microtubules were reorganized into a char-acteristic radial array emanating from the centrosomesand extending toward the cell periphery (Fig. 3C, DMSO).In contrast, centrosome-mediated microtubule regrowthwas severely impaired by high concentration of nakiter-piosin. Although a faint signal for a-tubulin was detect-able at the centrosomes, microtubules failed to elongatefrom these foci during the assay (up to 2 hours; Fig. 3C,NAK 25 mmol/L). In contrast, numerous microtubulesnucleated throughout the cytoplasm within 1.5 minutes.
A
B
C D
Figure 3. Nakiterpiosin (NAK) suppresses microtubule dynamics in interphase cells. A and B, HeLa cells were treated for 2 hours with DMSO, 5 mg/mLnocodazole (NOC), 1 mmol/L paclitaxel (PTX), 5 or 25 mmol/L NAK, or cooled on ice for 30 min (cold). The cells were then fixed and labeled for a-tubulin (A) andEB1 (B). Bar, 10 mm. A, in 25 mmol/L NAK, microtubules no longer originated from the centrosomes and became shorter (the cell border is outlined with dashlines), whereas 5 mmol/L NAK and DMSO showed little or no effect. B, EB1 was concentrated at the tips of microtubules in DMSO but not in 25 mmol/L NAK,suggesting NAK suppresses microtubule dynamics. C, centrosome-mediated microtubule regrowth is inhibited by 25 mmol/L NAK. HeLa cells were cooled onice for 30 minutes to depolymerize microtubules and incubated on ice for 30 minutes with DMSO or 25 mmol/L NAK (0 min). The cells were then warmed to37�C, fixed at the indicated time points and double-labeled for a-tubulin and pericentrin (insets). Bar, 10 mm. D, NAK does not alter centrosome organization.HeLa cells treated for 2 hours with 25 mmol/L NAKwere either immunolabeled for pericentrin (PCTN), g-tubulin, and DNA (left panels; bar, 10 mm) or analyzed byEM (right panel; bar, 200 nm).
Nakiterpiosin Targets Microtubules
www.aacrjournals.org Mol Cancer Ther; 9(12) December 2010 3379

After 5 minutes until the end point of the assay, thecytoplasmic microtubules remained relatively shortand tangled and exhibited a very distinct pattern as smallstretches ofmicrotubules. In addition, somemicrotubulesformed around the nuclei, supporting the previous find-ing that the nuclear envelope can function as a micro-tubule-organizing center (19).
Nakiterpiosin does not alter centrosomeorganization
In principle, the inhibition of centrosomal microtu-bule regrowth is caused by either defective nucleationor suppressed elongation. Nucleation at the centro-somes requires g-tubulin and pericentrin (7), but naki-terpiosin did not affect the targeting of these 2 proteinsto the centrosomes [Fig. 3C (insets) and D (left panels)].Furthermore, EM analysis revealed that the centrosomein nakiterpiosin-treated cells maintained a normal orga-nization, as shown by a pair of centrioles arrangedperpendicularly to each other and made of 9 microtu-bule triplets surrounded by electron-dense matrix(Fig. 3D, right panel). We conclude that nakiterpiosindoes not impair the centrosome structure or centro-some-mediated microtubule nucleation, but interfereswith elongation of filaments.
Effects of nakiterpiosin on Golgi positioningand morphology
In addition to mitosis, microtubules are essential forinterphase functions such as intracellular trafficking andorganelle positioning. In particular, Golgi positioning inthe perinuclear region is crucial for cell polarity and cell
migration (20). Because nakiterpiosin influences themicrotubule network, we examined whether the struc-ture and function of the Golgi complex are affected. HeLacells treated for 2 hours with vehicle or the indicatedagents were stained for the Golgi-resident proteinGM130. In vehicle-treated cells, we observed the char-acteristic ribbon-like Golgi structure next to the nucleus(Fig. 4A, DMSO). Paclitaxel had little effect (Fig. 4A, PTX),but depolymerization of microtubules by nocodazolefragmented the Golgi into ministacks that were scatteredthroughout the cytoplasm (Fig. 4A, NOC). Nakiterpiosin(5 mmol/L), which interfered with spindle assembly, hadno effect on Golgi morphology (Fig. 4A, NAK 5 mmol/L).However, 25 mmol/L nakiterpiosin, which impaired theorganization of the centrosomal microtubule array,caused the Golgi ribbon to partially disperse, althoughthe stacks were more interconnected compared withnocodazole treatment (Fig. 4A, NAK 25 mmol/L). Wefurther analyzed the Golgi structure by EM. Cells treatedwith vehicle or nakiterpiosin showed virtually indistin-guishable Golgi stacks composed of closely apposedcisternae (Fig. 4B). Furthermore, we observed numerousbudding profiles at the rims of the cisternae (Fig. 4B),indicating that nakiterpiosin does not block vesiculartransport.
Protein trafficking is not affected by nakiterpiosinNext, we tested whether nakiterpiosin influences pro-
tein transport through theGolgi to the plasmamembrane.We microinjected a plasmid encoding the plasma mem-brane protein CD8 into HeLa cells that do not expressendogenous CD8. Cycloheximide was included during
A
B
C
Figure 4. Effects of nakiterpiosin(NAK) on Golgi morphology andprotein transport. HeLa cellstreated for 2 hours with DMSO orthe indicated drugs were eitherimmunolabeled for the Golgi-resident protein GM130 (A) oranalyzed by EM (B). A, the Golgiribbon is partially dispersed by25 mmol/L NAK but not by5 mmol/L NAK. Bar, 10 mm.B, the structure of Golgi stackscomposed of closely apposedcisternae is not altered by NAK.Bar, 500 nm. C, CD8 transportthrough the secretory pathway isnot affected by NAK. A plasmidencoding the plasma membraneprotein CD8 was microinjectedinto HeLa cells in the presence ofcycloheximide. The cells werereleased from the cycloheximideblock, incubated for 90 min withthe indicated agents at 37�C,fixed, and immunolabeled for CD8at the cell surface withoutpermeabilization. Bar, 10 mm.NOC, nocodazole; PTX, paclitaxel.
Wei and Seemann
Mol Cancer Ther; 9(12) December 2010 Molecular Cancer Therapeutics3380

the microinjection period to prevent protein synthesis.We then removed the cycloheximide and incubated thecells with the indicated drugs for 90 minutes to allowexpression and transport of CD8. The cells were thenfixed without permeabilization and CD8 transported tothe cell surface was detected using an antibody. Asshown in Fig. 4C, CD8 was transported to the plasmamembrane irrespective of the treatment (31–52 cells percondition). However, in nocodazole-treated cells, onlyabout one third of CD8 reached the cell surface, consistentwith a previous report, in which nocodazole causes adelay in transport at early time points (data not shown;21). Therefore, although nakiterpiosin affects the overallorganization of interphase microtubules at high concen-tration, it does not influence the transport of proteins tothe cell surface, which may minimize the toxicity tointerphase cells and enhance its specificity as an anti-mitotic drug.
Nakiterpiosin directly targets tubulin andsuppresses microtubule polymerization in vitroBecause nakiterpiosin interferes with microtubule
growth in cells, we tested whether it inhibits polymeriza-tion of highly purified tubulin in vitro (Fig. 5A). In DMSO,tubulin polymerization was complete within 5 minutesbut was completely blocked by nocodazole as expected.In the presence of 1 mmol/L nakiterpiosin, polymeriza-tion proceeded with an efficiency comparable to that ofDMSO. In contrast, 5 mmol/L and 10 mmol/L nakiterpio-sin strongly inhibited the reaction, and at 25 mmol/L, nopolymerization was observed (Fig. 5A).Next, we examined the end products of the polymer-
ization reactions by EM. At low magnification, longfilaments were clearly visible in DMSO, 1 and 5 mmol/L nakiterpiosin samples (Fig. 5B, left panels, DMSO andNAK 5 mmol/L). In addition, we also detected shortpolymers with few interspersed tubulin aggregates inthe 5 mmol/L sample (Fig. 5B, left panels, NAK 5 mmol/L).At 10 mmol/L, more tubulin aggregates and short poly-mers were seen (data not shown). In samples treated withnocodazole or 25 mmol/L nakiterpiosin, filaments werelargely absent and, presumably due to fixation, largeamounts of protein aggregates were observed instead(Fig. 5B, left panels, NAK 25 mmol/L and NOC). At highmagnification, most filament ends displayed a straightconformation, which is an indication of growing micro-tubules (Fig. 5B, right panels, DMSO). With increasingdrug concentrations, shrinking microtubules becamemore abundant, as indicated by the banana-peel structureat the ends (Fig. 5B, right panels, NAK 5 mmol/L). Takentogether, these data demonstrate that nakiterpiosindirectly binds to tubulin and suppresses microtubulepolymerization in a concentration-dependent manner.
Nakiterpiosin decreases microtubule polymermass in cellsBecause nakiterpiosin inhibits microtubule polymeri-
zation in vitro, we determined whether it influences
polymer mass in cells. To evaluate the ratio of tubulinin polymers, HeLa cells were treated with vehicle or theindicated agents for 2 hours and then lysed at roomtemperature in the presence of paclitaxel to stabilize allpolymers. Polymer and soluble forms were separatedand the amount of a-tubulin was analyzed by Westernblotting. In vehicle-treated cells, about two third of thetubulin was in polymers (8; Fig. 5C, DMSO). On noco-dazole treatment, most of the tubulin became soluble(Fig. 5C, NOC), whereas paclitaxel treatment shiftedthe equilibrium toward polymers (Fig. 5C, PTX). Con-sistent with our immunofluorescence data, 5 mmol/Lnakiterpiosin did not change the polymer mass(Fig. 5C, 5 mmol/L NAK). In contrast, 25 mmol/L naki-terpiosin induced partial depolymerization and morethan half of the tubulin was present in the soluble form(Fig. 5C, 25 mmol/L NAK). These results, together withour in vitro characterization, suggest that nakiterpiosinacts as a microtubule-depolymerizing agent both in vitroand in cells.
Nakiterpiosin enhances tubulin acetylationTubulin acetylation is thought to be an indication of
microtubule stabilization and it occurs most efficientlyaftermicrotubule assembly (22, 23). Todeterminewhethertubulin acetylationwas affectedbynakiterpiosin, theblotsfrom theprevious experimentswereprobed for acetylateda-tubulin. As expected, nocodazole depolymerizedmicrotubules and abolished acetylation (Fig. 5D, NOC).In contrast, the microtubule-stabilizing agent paclitaxelincreased tubulin acetylation by 7-fold (Fig. 5D, PTX).Surprisingly, although 5 mmol/L nakiterpiosin had littleeffect on microtubule organization or cellular polymermass (Figs. 3A and 5C, 5 mmol/L NAK), tubulin acetyla-tion was enhanced to a comparable extent as 1 mmol/Lpaclitaxel treatment (Fig. 5D, 5mmol/LNAK). In addition,although 25 mmol/L nakiterpiosin partially depolymer-ized microtubules in cells (Figs. 3A and 5C, 25 mmol/LNAK), the acetylation of the remaining filamentswas increased by 20-fold (Fig. 5D, 25 mmol/L NAK).These data demonstrate that, unlike nocodazole, whichinhibits both tubulin polymerization and acetylation,nakiterpiosin inhibits the formation of new microtubulesand simultaneously enhances the stability of existingfilaments.
Nakiterpiosin inhibits the viabilityof paclitaxel-resistant cancer cells
Paclitaxel stabilizes microtubules, which blocks micro-tubule reorganization in early mitosis, thereby killingfast-proliferating cancer cells. Despite the routine useof paclitaxel in the treatment of ovarian, breast and lungcancers, tumor cells often have intrinsic or develop resis-tance to paclitaxel treatment. Because nakiterpiosin alsostabilizes microtubules, we reason that nakiterpiosinmight be effective to eliminate cancer cells that respondpoorly to paclitaxel. We tested this possibility with NCI-H1155 and HCC366 cells, 2 human non-small cell lung
Nakiterpiosin Targets Microtubules
www.aacrjournals.org Mol Cancer Ther; 9(12) December 2010 3381

cancer cell lines (11) that show different degrees ofpaclitaxel resistance (Fig. 6A). After 48 hours treatmentwith vehicle or varying concentrations of paclitaxel, wemeasured the cellular ATP level of HeLa and the 2 cancer
cell lines to determine cell viability. After 48 hours in100 nmol/L and 1 mmol/L paclitaxel, respectively, 66% ofNCI-H1155 and 87% of HCC366 cells were still viable,whereas most HeLa cells died under these 2 conditions
A
C
D
B
Figure 5. Nakiterpiosin (NAK) inhibits microtubule polymerization in vitro and in cells and enhances tubulin acetylation. A, NAK inhibits microtubulepolymerization in vitro in a concentration-dependent manner. Microtubule polymerization from purified tubulin at 37�Cwith vehicle or the indicated drugs wasmonitored by an increase in fluorescence. B, the end products of the polymerization reactions were fixed, negatively stained, and analyzed by EM. At lowmagnification (left panels; bar, 1 mm), filamentous polymers were observed in 5 mmol/L NAK, although they appeared shorter than in DMSO. No polymers buttubulin aggregates were detected in 25 mmol/L NAK and nocodazole (NOC). At higher magnification (right panels; bar, 100 nm), most filament ends werestraight, indicating growing microtubules (DMSO). As NAK concentration increased, microtubule ends frequently showed banana peel structures, indicatingshrinking microtubules (NAK, 5 mmol/L). C and D, HeLa cells treated for 2 h with vehicle or the indicated agents were lysed at room temperature. Tubulin inpolymers was separated from soluble tubulin by centrifugation. Equal fractions of the supernatant (S) and the pellet (P) were analyzed by Western blotting fora-tubulin or acetylated a-tubulin and band intensity was quantified (n ¼ 3). C, the microtubule polymer mass in cells is decreased by 25 mmol/L NAK. D, NAKenhances tubulin acetylation. PTX, paclitaxel.
Wei and Seemann
Mol Cancer Ther; 9(12) December 2010 Molecular Cancer Therapeutics3382

(42% survival in 100 nmol/L and 9% survival in 1 mmol/Lpaclitaxel; Fig. 6A). We then compared the viability ofHeLa and NCI-H1155 cells treated with nakiterpiosin or100 nmol/L paclitaxel. Consistent with our results(Figs. 2A and 6A), HeLa cells were killed by both drugs(Fig. 6B). By contrast, although NCI-H1155 cells wererefractory to paclitaxel, their proliferation was signifi-cantly reduced from 66% in 100 nmol/L paclitaxel to 30%and 9% in 5 mmol/L and 25 mmol/L nakiterpiosin,respectively (Fig. 6B). A similar inhibition was alsoobserved in HCC366 cells treated with nakiterpiosin,which decreased cell viability from 86% in 1 mmol/Lpaclitaxel to 60% and 39% in 5 mmol/L and 25 mmol/Lnakiterpiosin, respectively (Fig. 6C). These data implythat nakiterpiosin acts through a different mechanismthan paclitaxel. In conclusion, our results demonstrate theeffectiveness of nakiterpiosin in killing cancer cells andthe potential to be applicable to a broad spectrum oftumors.
Discussion
Microtubule-targeting drugs have been used withremarkable success in cancer chemotherapy. However,despite their extensive use in treating various types ofcancers, several problems remain unsolved (24). Inparticular, patients often develop drug resistance dueto repeated exposure to the same chemical agent. Differ-ential expression of multiple tubulin isotypes may alsocompromise drug efficiency in certain tissues (25). Toovercome these limitations, tremendous efforts have beenmade to develop agents that either bind to or act differ-ently on microtubules. In addition, combination therapywith 2 or more antimicrotubule drugs improves drugefficiency and minimizes toxicity (24). Thus, new micro-tubule-targeting compounds can significantly impactcancer treatment. In the current study, we identify naki-terpiosin as a novel antimitotic agent that suppresses
microtubule dynamics and triggers cell death in cancercells (Figs. 2A, 6B, and 6C).
Nakiterpiosin was originally extracted from a marinesponge that grows on corals and causes the destruction oflarge areas of coral reefs (9). It was shown to have potentcytotoxicity against mouse leukemia cells (9), but thecellular target and mechanism of action were unknown.Our results demonstrate that nakiterpiosin targets micro-tubules and induces cell death by interfering with mitoticprogression. At 1 mmol/L or less, nakiterpiosin has littleor no effect on cell division when observed by time-lapsemicroscopy (Fig. 2A) and only marginal effects on cellcycle progression when analyzed by FACS (10). How-ever, cells that enter mitosis in the presence of 5 mmol/Lor greater concentrations of nakiterpiosin form aberrantspindles (Fig. 2C) and become arrested (Fig. 2A) due toactivation of the SAC (Fig. 2B). On prolonged arrest, cellsundergo mitotic catastrophe, a form of cell death thatoccurs during mitosis (16). The mitotic inhibition is fullyreversible and arrested cells divide on drug removal(Fig. 2A), suggesting that nakiterpiosin does not perma-nently damage cells. Protein secretion and viability ofinterphase cells are also not impaired at this concentra-tion (Fig. 3C). Altogether, our results show that nakiter-piosin induces mitotic cell death by perturbing spindledynamics with little effect on interphase cells.
In vitro microtubule polymerization from purifiedtubulin is completely abolished by a high concentrationof nakiterpiosin (Fig. 5A), demonstrating that nakiterpio-sin directly binds to tubulin. This is consistent with thefinding that, in interphase cells treated at the same con-centration, the amount of tubulin incorporated into poly-mers is markedly reduced (Fig. 5C). Moreover, theorganization of the microtubule network is altered underthis condition (Fig. 3A). Unlike nocodazole-treated cells,in which the microtubules are completely depolymer-ized, filaments are still present in nakiterpiosin-treatedcells, although the microtubules appear to be tangled and
A B C
Figure 6. Nakiterpiosin (NAK) reduces the viability of paclitaxel (PTX)-resistant cancer cells. HeLa, NCI-H1155, and HCC366 cells were treated for 48 h withDMSO or the indicated drugs. Cell viability was then determined by the relative amount of ATP present (normalized to DMSO-treated cells) using the CellTiter-Glo assay. A, NCI-H1155 and HCC366 cells show different levels of PTX resistance (n ¼ 3). B, NAK reduces cell viability of NCI-H1155 cells (n ¼ 4). C, NAKreduces cell viability of HCC366 cells (n ¼ 5).
Nakiterpiosin Targets Microtubules
www.aacrjournals.org Mol Cancer Ther; 9(12) December 2010 3383

shorter. Strikingly, these filaments no longer originatefrom the centrosomes.
In addition to reducing cellular polymer mass, naki-terpiosin negatively regulates microtubule dynamics, asshown by the loss of EB1 from the microtubule tips(Fig. 3B). EB1 is a microtubule plus end-tracking proteinthat preferentially binds to the tip of growing filaments(26). Reduced EB1 end labeling suggests that microtu-bules become less dynamic. Another line of evidence thatnakiterpiosin perturbs microtubule dynamics comesfrom the dramatic increase in tubulin acetylation(Fig. 5D). Tubulin acetylation is thought to be an indica-tion of microtubule stabilization and it occurs most effi-ciently after microtubule assembly (22, 23). In contrast tonocodazole treatment, microtubules are still present andbecome highly acetylated in nakiterpiosin-treated cells(Figs. 3A, 5C, and 5D), despite the fact that it interfereswith microtubule polymerization in vitro and in cells(Fig. 5A–C). Thus, the increase in tubulin acetylation innakiterpiosin-treated cells is presumably an outcome ofsuppressed microtubule dynamics. In the regrowthassay, nakiterpiosin strongly inhibits polymerization ofnew filaments from the centrosomes (Fig. 3C), but thecentrosomes appear unaltered when analyzed by EMand immunofluorescence (Fig. 3D). In addition, althoughthe Golgi could function as a nucleation site for micro-tubules (27, 28), filament formation is suppressed inthe perinuclear area where the Golgi is localized(Fig. 3C). These findings, together with enhanced tubulinacetylation, suggest that nakiterpiosin simultaneouslystabilizes existing filaments and prevents formation ofnew microtubules.
In addition to the unique properties described earlier,nakiterpiosin has a very different chemical structureand mechanism of action compared with other micro-tubule-targeting drugs such as paclitaxel (Figs. 1 and 6)(10). Paclitaxel perturbs mitotic progression by stabiliz-ing microtubules, which increases tubulin acetylation
(Fig. 5D; ref. 23). Several studies have provided evi-dence that tubulin acetylation is linked to drug resis-tance (23, 29, 30). In particular, acquisition of paclitaxelresistance in tumors is one of the most challengingobstacles to successful cancer chemotherapy. Cancercells can develop paclitaxel resistance by acquiringmutations in b-tubulin (31). Intriguingly, paclitaxel-resistant cells have reduced levels of acetylated tubulinand less stable filaments (31). Because nakiterpiosinstimulates tubulin acetylation more effectively thanpaclitaxel and interferes with microtubule dynamicsdifferently, it could be used as a chemotherapeuticagent against paclitaxel-resistant tumors. Indeed, ourdata show that nakiterpiosin reduces the viability of 2paclitaxel-resistant cancer cell lines (Fig. 6). Further-more, nakiterpiosin could be used in combination withother microtubule-targeting drugs to improve efficiencyor minimize toxicity. In summary, here we report thecellular target and biological effects of nakiterpiosin andsuggest its potential use in cancer chemotherapy.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Chuo Chen for the generous gift of nakiterpiosin; Christo-pher Gilpin and Tom Januszewski for EM assistance; Michael White forNCI-H1155 and HCC366 cell lines; Martin Lowe and Keith Gull forantibodies; Dorothy Mundy, Chih-Wen Chu, and Katie Zhang for criticalreading of the manuscript.
Grant Support
This work was supported by grants from the American Heart Association(8065090F) and the NIH (GM096070) to J. Seemann
Received 03/27/2010; revised 09/29/2010; accepted 10/01/2010;published OnlineFirst 12/07/2010.
References1. Gascoigne KE, Taylor SS. How do anti-mitotic drugs kill cancer cells?
J Cell Sci 2009;122:2579–85.2. Walczak CE, Heald R. Mechanisms of mitotic spindle assembly and
function. Int Rev Cytol 2008;265:111–58.3. Bornens M. Organelle positioning and cell polarity. Nat Rev Mol Cell
Biol 2008;9:874–86.4. Lane J, Allan V. Microtubule-based membrane movement. Biochim
Biophys Acta 1998;1376:27–55.5. Berbari NF, O’Connor AK, Haycraft CJ, Yoder BK. The primary cilium
as a complex signaling center. Curr Biol 2009;19:R526–35.6. Mitchison T, Kirschner M. Dynamic instability of microtubule growth.
Nature 1984;312:237–42.7. Luders J, Stearns T. Microtubule-organizing centres: a re-evaluation.
Nat Rev Mol Cell Biol 2007;8:161–7.8. Zhai Y, Kronebusch PJ, Simon PM, Borisy GG. Microtubule dynamics
at the G2/M transition: abrupt breakdown of cytoplasmic microtu-bules at nuclear envelope breakdown and implications for spindlemorphogenesis. J Cell Biol 1996;135:201–14.
9. Teruya T, Nakagawa S, Koyama T, Arimoto H, Kita M, Uemura D.Nakiterpiosin and nakiterpiosinone, novel cytotoxic C-nor-D-homo-
steroids from the Okinawan sponge Terpios hoshinotaTetrahedron2004;60:6989–93.
10. Gao S, Wang Q, Huang LJ, Lum L, Chen C. Chemical and biologicalstudies of nakiterpiosin and nakiterpiosinone. J Am Chem Soc2010;132:371–83.
11. Whitehurst AW, Bodemann BO, Cardenas J, Ferguson D, Girard L,Peyton M, et al. Synthetic lethal screen identification of chemosensi-tizer loci in cancer cells. Nature 2007;446:815–9.
12. Wei JH, Seemann J. The mitotic spindle mediates inheritance of theGolgi ribbon structure. J Cell Biol 2009;184:391–7.
13. Wang Y, Wei JH, Bisel B, Tang D, Seemann J. Golgi cisternalunstacking stimulates COPI vesicle budding and protein transport.PLoS ONE 2008;3:e1647.
14. Wei JH, Seemann J. Induction of asymmetrical cell division to analyzespindle-dependent organelle partitioning using correlative micro-scopy techniques. Nat Protoc 2009;4:1653–62.
15. Sandoval IV, Weber K. Different tubulin polymers are produced bymicrotubule-associated proteins MAP2 and tau in the presence ofguanosine 5’-(alpha, beta-methylene)triphosphate. J Biol Chem1980;255:8952–4.
Wei and Seemann
Mol Cancer Ther; 9(12) December 2010 Molecular Cancer Therapeutics3384

16. Castedo M, Perfettini JL, Roumier T, Andreau K, Medema R, KroemerG. Cell death bymitotic catastrophe: amolecular definition. Oncogene2004;23:2825–37.
17. Jordan MA, Wilson L. Microtubules as a target for anticancer drugs.Nat Rev Cancer 2004;4:253–65.
18. Lansbergen G, Akhmanova A. Microtubule plus end: a hub of cellularactivities. Traffic 2006;7:499–507.
19. Tassin AM, Maro B, Bornens M. Fate of microtubule-organizingcenters during myogenesis in vitro. J Cell Biol 1985;100:35–46.
20. Bisel B, Wang Y, Wei JH, Xiang Y, Tang D, Miron-Mendoza M, et al.ERK regulates Golgi and centrosome orientation towards the leadingedge through GRASP65. J Cell Biol 2008;182:837–43.
21. Cole NB, Sciaky N, Marotta A, Song J, Lippincott-Schwartz J. Golgidispersal during microtubule disruption: regeneration of Golgi stacks atperipheralendoplasmic reticulumexit sites.MolBiolCell 1996;7:631–50.
22. Matsuyama A, Shimazu T, Sumida Y, Saito A, Yoshimatsu Y,Seigneurin-Berny D, et al. In vivo destabilization of dynamic micro-tubules by HDAC6-mediated deacetylation. The EMBO journal2002;21:6820–31.
23. Piperno G, LeDizet M, Chang XJ. Microtubules containing acetylatedalpha-tubulin in mammalian cells in culture. J Cell Biol 1987;104:289–302.
24. Kavallaris M. Microtubules and resistance to tubulin-binding agents.Nat Rev Cancer 2010;10:194–204.
25. Luduena RF. Multiple forms of tubulin: different gene products andcovalent modifications. Int Rev Cytol 1998;178:207–75.
26. Komarova Y, De Groot CO, Grigoriev I, Gouveia SM, Munteanu EL,Schober JM, et al. Mammalian end binding proteins control persis-tent microtubule growth. J Cell Biol 2009;184:691–706.
27. Rivero S, Cardenas J, Bornens M, Rios RM. Microtubule nucleation atthe cis-side of the Golgi apparatus requires AKAP450 and GM130.The EMBO journal 2009;28:1016–28.
28. Efimov A, Kharitonov A, Efimova N, Loncarek J, Miller PM, AndreyevaN, et al. Asymmetric CLASP-dependent nucleation of noncentroso-mal microtubules at the trans-Golgi network. Dev Cell 2007;12:917–30.
29. Sangrajrang S, Denoulet P, Laing NM, Tatoud R, Millot G, Calvo F,et al. Association of estramustine resistance in human prostaticcarcinoma cells withmodified patterns of tubulin expression. BiochemPharmacol 1998;55:325–31.
30. Geyp M, Ireland CM, Pittman SM. Increased tubulin acetyla-tion accompanies reversion to stable ploidy in vincristine-resistant CCRF-CEM cells. Cancer Genet Cytogenet 1996;87:116–22.
31. Hari M, Loganzo F, Annable T, Tan X, Musto S, Morilla DB, et al.Paclitaxel-resistant cells have a mutation in the paclitaxel-bindingregion of beta-tubulin (Asp26Glu) and less stable microtubules. MolCancer Ther 2006;5:270–8.
Nakiterpiosin Targets Microtubules
www.aacrjournals.org Mol Cancer Ther; 9(12) December 2010 3385




















