
Myc down-regulation induces apoptosis in M14 melanoma cells by increasing p27kip1 levels
12
Myc down-regulation induces apoptosis in M14 melanoma cells by increasing p27 kip1 levels Igea D’Agnano* ,1 , Alessandra Valentini 1 , Cristina Fornari 1 , Barbara Bucci 1,4 , Giuseppe Starace 2 , Armando Felsani 1 and Gennaro Citro 3 1 CNR, Istituto di Tecnologie Biomediche, V.le Marx 43, 00137 Roma, Italy; 2 CNR, Istituto di Medicina Sperimentale, V.le Marx 43, 00137 Roma, Italy; 3 Istituto Regina Elena, Via delle Messi D’Oro 156, 00158, Roma, Italy In recent years, increasing evidence indicated the importance of a deregulated c-myc gene in the melanoma pathogenesis. We have previously demonstrated that treatment of melanoma cells with c-myc antisense oligodeoxynucleotides can inhibit cell proliferation and activate apoptosis. To gain insight into the mechanisms activated by Myc down-regulation, we have now developed an experimental model that allows modulating Myc protein expression in melanoma cells. This was achieved by originating stable melanoma cell clones expressing ecdysone-inducible c-myc antisense RNA. We show that the induction of c-myc antisense RNA in M14 melanoma cells leads to an inhibition of cell proliferation characterized by accumulation of cells in the G 1 phase of the cell cycle (up to 80%) and activation of apoptosis (50%). These data are associated with an increase of p27 kip1 levels and a significant reduction of the cdk2- associated kinase activity. In addition, we show that an ectopic overexpression of p27 kip1 in this experimental model can enhance the apoptotic rate. Our results indicate that down-regulation of Myc protein induces a G 1 arrest and activates apoptosis by increasing p27 kip1 content in melanoma cells, that are known to be defective for the p16-cyclinD/cdk4-pRb G 1 checkpoint. This is particularly relevant for identifying new therapeutic strategies based on the re-establishment of the apoptotic pathways in cancer cells. Oncogene (2001) 20, 2814 – 2825. Keywords: c-myc; melanoma; p27; apoptosis Introduction The c-myc gene encodes a nuclear phosphoprotein with extremely short half-life, which has been widely demonstrated to play a central role in the proliferation and dierentiation processes of normal cells. Following mitogenic stimulation, Myc is rapidly induced in quiescent cells thus participating in the cascade of events that result in mitogenesis (Ryan and Birnie, 1996; Dang, 1999; Schmidt, 1999). Myc protein expression remains elevated in proliferating cells and c-myc mRNA levels are stable throughout the phases of the cell cycle, suggesting that c-myc is required for continuous cell proliferation. The deregulation of the c-myc gene has been shown to be involved in the establishment of many types of human malignancy (Dang, 1999), however, c-myc deregulation is insu- cient to cause neoplasm by itself. The oncogenic potential of c-myc could simply be ascribed to its implication as a direct regulator of the cell cycle machinery, c-myc being poised at the intersection between cell growth and the cell cycle control. Consistent with the hypothesis that the loss of coordination between cell growth and DNA synthesis destabilizes cell proliferation, alterations in cell growth control can cause apoptosis in Myc-overexpressing cells. Indeed, Evan et al. (1992) demonstrated that removal of serum from growth media of Myc-over- expressing Rat1 fibroblasts induces dramatic apoptosis. Similarly, activation-induced apoptosis through the CD3-T cell receptor in T-cell hybridomas was blocked by antisense oligodeoxynucleotides targeting c-myc mRNA (Shi et al., 1992). This could simply reflect the loss of coordination of growth processes in a cell that cannot limit its DNA synthesis. On the other hand, data are reported in literature which demonstrate that a down-regulation of the Myc protein by using c-myc antisense oligodeoxynucleotides induces apopto- sis in BV173 leukemia cells (Skorski et al., 1995) as well as in HL60 cells (Kimura et al., 1995). In addition engagement of the B-cell receptor of WEHI 231 immature lymphoma cells led to a drop in Myc protein levels which resulted in the induction of apoptosis (Ezhevsky et al., 1996; Wu et al., 1999; Fischer et al., 1994). Furthermore, we have previously demonstrated that Myc down-regulation caused a significant inhibi- tion of cell proliferation and induced apoptosis in human melanoma experimental models (Leonetti et al., 1996; Citro et al., 1998). Although deregulation of melanoma cell growth has been widely associated with mutations in the p16-cyclin D/cdk4-pRb pathway (Bartkova et al., 1996; Maelandsmo et al., 1996), an Oncogene (2001) 20, 2814 – 2825 ª 2001 Nature Publishing Group All rights reserved 0950 – 9232/00 $15.00 www.nature.com/onc *Correspondence: I D’Agnano, 4 Current address: Centro Ricerca ‘San Pietro’ Fatebenefratelli (FBN) Hospital, Via Cassia 600, 00189 Roma, Italy Received 3 July 2000; revised 7 February 2001; accepted 13 February 2001
Transcript of Myc down-regulation induces apoptosis in M14 melanoma cells by increasing p27kip1 levels

Myc down-regulation induces apoptosis in M14 melanoma cells byincreasing p27kip1 levels
Igea D'Agnano*,1, Alessandra Valentini1, Cristina Fornari1, Barbara Bucci1,4, Giuseppe Starace2,Armando Felsani1 and Gennaro Citro3
1CNR, Istituto di Tecnologie Biomediche, V.le Marx 43, 00137 Roma, Italy; 2CNR, Istituto di Medicina Sperimentale, V.le Marx43, 00137 Roma, Italy; 3Istituto Regina Elena, Via delle Messi D'Oro 156, 00158, Roma, Italy
In recent years, increasing evidence indicated theimportance of a deregulated c-myc gene in the melanomapathogenesis. We have previously demonstrated thattreatment of melanoma cells with c-myc antisenseoligodeoxynucleotides can inhibit cell proliferation andactivate apoptosis. To gain insight into the mechanismsactivated by Myc down-regulation, we have nowdeveloped an experimental model that allows modulatingMyc protein expression in melanoma cells. This wasachieved by originating stable melanoma cell clonesexpressing ecdysone-inducible c-myc antisense RNA. Weshow that the induction of c-myc antisense RNA in M14melanoma cells leads to an inhibition of cell proliferationcharacterized by accumulation of cells in the G1 phase ofthe cell cycle (up to 80%) and activation of apoptosis(50%). These data are associated with an increase ofp27kip1 levels and a signi®cant reduction of the cdk2-associated kinase activity. In addition, we show that anectopic overexpression of p27kip1 in this experimentalmodel can enhance the apoptotic rate. Our resultsindicate that down-regulation of Myc protein induces aG1 arrest and activates apoptosis by increasing p27kip1
content in melanoma cells, that are known to be defectivefor the p16-cyclinD/cdk4-pRb G1 checkpoint. This isparticularly relevant for identifying new therapeuticstrategies based on the re-establishment of the apoptoticpathways in cancer cells. Oncogene (2001) 20, 2814 ±2825.
Keywords: c-myc; melanoma; p27; apoptosis
Introduction
The c-myc gene encodes a nuclear phosphoprotein withextremely short half-life, which has been widelydemonstrated to play a central role in the proliferationand di�erentiation processes of normal cells. Following
mitogenic stimulation, Myc is rapidly induced inquiescent cells thus participating in the cascade ofevents that result in mitogenesis (Ryan and Birnie,1996; Dang, 1999; Schmidt, 1999). Myc proteinexpression remains elevated in proliferating cells andc-myc mRNA levels are stable throughout the phasesof the cell cycle, suggesting that c-myc is required forcontinuous cell proliferation. The deregulation of thec-myc gene has been shown to be involved in theestablishment of many types of human malignancy(Dang, 1999), however, c-myc deregulation is insu�-cient to cause neoplasm by itself. The oncogenicpotential of c-myc could simply be ascribed to itsimplication as a direct regulator of the cell cyclemachinery, c-myc being poised at the intersectionbetween cell growth and the cell cycle control.Consistent with the hypothesis that the loss ofcoordination between cell growth and DNA synthesisdestabilizes cell proliferation, alterations in cell growthcontrol can cause apoptosis in Myc-overexpressingcells. Indeed, Evan et al. (1992) demonstrated thatremoval of serum from growth media of Myc-over-expressing Rat1 ®broblasts induces dramatic apoptosis.Similarly, activation-induced apoptosis through theCD3-T cell receptor in T-cell hybridomas was blockedby antisense oligodeoxynucleotides targeting c-mycmRNA (Shi et al., 1992). This could simply re¯ectthe loss of coordination of growth processes in a cellthat cannot limit its DNA synthesis. On the otherhand, data are reported in literature which demonstratethat a down-regulation of the Myc protein by usingc-myc antisense oligodeoxynucleotides induces apopto-sis in BV173 leukemia cells (Skorski et al., 1995) aswell as in HL60 cells (Kimura et al., 1995). In additionengagement of the B-cell receptor of WEHI 231immature lymphoma cells led to a drop in Myc proteinlevels which resulted in the induction of apoptosis(Ezhevsky et al., 1996; Wu et al., 1999; Fischer et al.,1994). Furthermore, we have previously demonstratedthat Myc down-regulation caused a signi®cant inhibi-tion of cell proliferation and induced apoptosis inhuman melanoma experimental models (Leonetti et al.,1996; Citro et al., 1998). Although deregulation ofmelanoma cell growth has been widely associated withmutations in the p16-cyclin D/cdk4-pRb pathway(Bartkova et al., 1996; Maelandsmo et al., 1996), an
Oncogene (2001) 20, 2814 ± 2825ã 2001 Nature Publishing Group All rights reserved 0950 ± 9232/00 $15.00
www.nature.com/onc
*Correspondence: I D'Agnano,4Current address: Centro Ricerca `San Pietro' Fatebenefratelli (FBN)Hospital, Via Cassia 600, 00189 Roma, ItalyReceived 3 July 2000; revised 7 February 2001; accepted 13February 2001

increasing number of recent reports showed that thec-myc gene also plays a role in the melanomapathogenesis (Castellano and Parmiani, 1999; Schramlet al., 1999; Schlagbauer-Wadl et al., 1999; Ross andWilson, 1998).
To gain insight into the molecular events regulatedby c-myc in human melanoma cells, we developed asuitable experimental model that allows modulation ofMyc protein synthesis; this was achieved by originatingstable cell clones expressing inducible c-myc antisensemRNA. This avoided using modi®ed oligodeoxynu-cleotides to inhibit gene expression. In fact, it is wellknown that modi®ed oligomers, besides sequence-speci®c e�ects, could also have non-antisense activitywhich could make the understanding of the underlyinggenetic mechanism di�cult (Stein, 1996, 1998; Crooke,1996; Krieg and Stein, 1995).
Furthermore, since it has been reported in theliterature that over-expression of the cyclin-dependentkinase inhibitor p27kip1 is able to promote apoptosis inseveral mammalian tumor cell lines, including breast,lung, colon, cervical carcinoma and melanoma (Ka-tayose et al., 1997; Wang et al., 1997), as well as in aline of B-cell lymphoma after treatment with anti-IgM(Ezhevsky et al., 1996; Wu et al., 1999; Fischer et al.,1994), the e�ects of c-myc inhibition on p27kip1 proteinexpression have also been investigated. Data hereinreported demonstrate, for the ®rst time in melanomacells, that the inhibitory e�ect on M14 melanoma cellproliferation caused by Myc protein down-regulationcan activate apoptosis by increasing the levels ofp27kip1.
Results
Expression of the p16, cyclin D1 and cdk4 in M14melanoma cells
Since deregulation of melanoma cell growth has beenmainly associated with alterations in the p16-cyclin D1/cdk4-pRb pathway, we have ®rst characterized theM14 melanoma cell line for the expression of cyclinD1, cdk4 and p16 (Figure 1). Western blot analysisrevealed that p16 is down-regulated in M14 cells ascompared with normal peripheral lymphocytes, used ascontrol cells. Indeed, to visualize detectable levels ofp16 in M14 cells, fourfold more protein amount, withrespect to the normal lymphocytes, had to be loadedon the acrylamide gel. Cyclin D1 as well as cdk4 werefound overexpressed in M14 cells as compared to thenormal cells, the increase of cyclin D1 and cdk4 levelsbeing 2.4-fold and sixfold, respectively.
Inducible expression of c-myc antisense RNA inhibits cellproliferation in M14 cells
In order to investigate the molecular pathwaysregulated by c-myc involved in the activation ofapoptosis in M14 melanoma cells, we developed asuitable tumor experimental model in which the
expression of the Myc protein can be modulated. Toreduce the levels of Myc protein expression weestablished a M14 cell line stably expressing ahormone-inducible c-myc antisense (AS) RNA. M14cells were co-transfected with either the pINDc-mycAS constructs or the pINDneo empty vector, togetherwith the pVgRXR plasmid, encoding for the ecdysonereceptor and carrying the zeocyn resistance. G418/zeocyn-resistant clones were isolated, under hormone-free conditions, after a selection period of 3 weeks.These clones were treated with 20 mM of hormone(ponasterone A) and scored for hormone inducibleinhibition of cell proliferation. Ten di�erent pINDc-myc AS clones displayed inhibition of cell prolifera-tion, already evident after 24 h of induction with thehormone, ranging from 30 ± 60% (data not shown).The control clones exposed to the same ponasteroneconcentration did not show any signi®cant inhibitionof the proliferation rate. Therefore, for furtherexperiments the pINDc-myc AS clone showing thehighest inhibition of cell proliferation upon hormone-exposure, was chosen. As control we used one of thepINDneo clones. To determine the optimal scheduleof hormone treatment pINDc-myc AS cells wereexposed to di�erent concentrations of ponasterone A(5, 10 and 20 mM). After addition of 20 mM ponaster-one an inhibition of cell proliferation of 38, 42 and40% was observed in pINDc-myc AS cells at 24, 48and 72 h, respectively, as compared with the controlcells transfected with the empty vector (pINDneo;Figure 2a). The pINDc-myc AS clone continuouslyexposed to 5 or 10 mM ponasterone did not elicitsigni®cant inhibition of cell growth up to 72 h afterinduction. The cell proliferation rate of control cells
Figure 1 Representative Western blot analysis demonstratingthe abundance of the cyclin D1, cdk4 and p16 G1-checkpointregulators in M14 melanoma cells compared to normal peripherallymphocytes. HSP 72/73 was used as control for loading sameamounts of proteins. The experiment was repeated three timesshowing similar results
Oncogene
p27kip1-mediated apoptosis in melanoma cellsI D'Agnano et al
2815
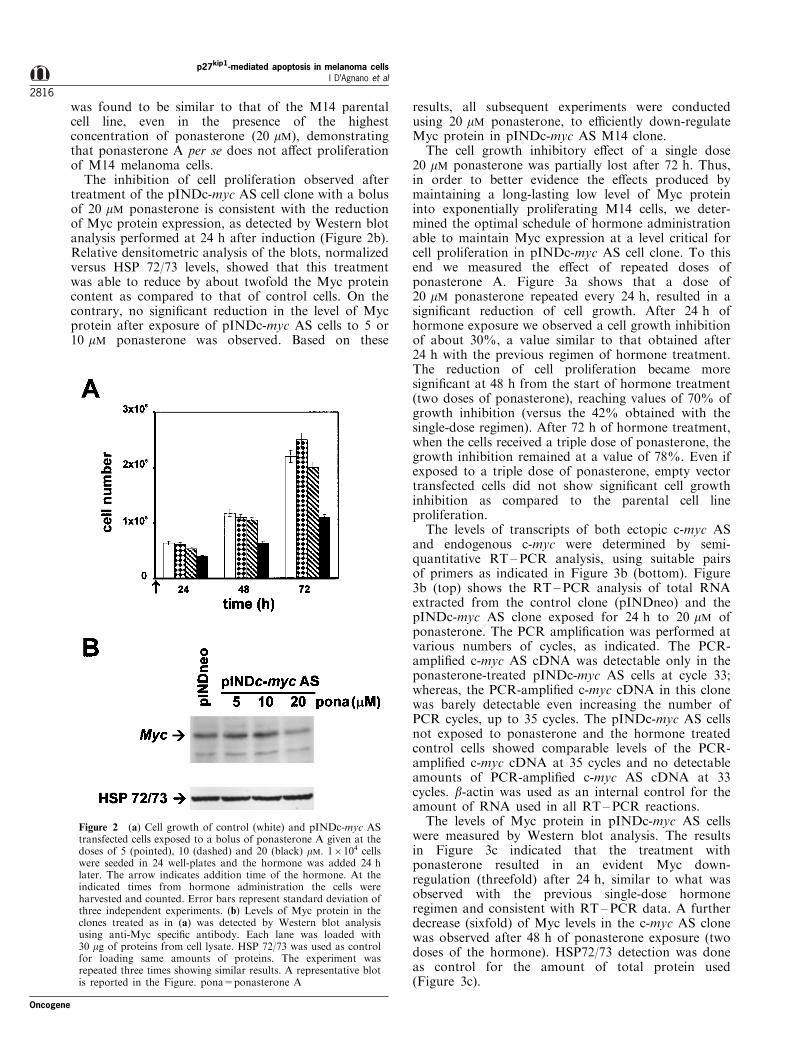
was found to be similar to that of the M14 parentalcell line, even in the presence of the highestconcentration of ponasterone (20 mM), demonstratingthat ponasterone A per se does not a�ect proliferationof M14 melanoma cells.
The inhibition of cell proliferation observed aftertreatment of the pINDc-myc AS cell clone with a bolusof 20 mM ponasterone is consistent with the reductionof Myc protein expression, as detected by Western blotanalysis performed at 24 h after induction (Figure 2b).Relative densitometric analysis of the blots, normalizedversus HSP 72/73 levels, showed that this treatmentwas able to reduce by about twofold the Myc proteincontent as compared to that of control cells. On thecontrary, no signi®cant reduction in the level of Mycprotein after exposure of pINDc-myc AS cells to 5 or10 mM ponasterone was observed. Based on these
results, all subsequent experiments were conductedusing 20 mM ponasterone, to e�ciently down-regulateMyc protein in pINDc-myc AS M14 clone.
The cell growth inhibitory e�ect of a single dose20 mM ponasterone was partially lost after 72 h. Thus,in order to better evidence the e�ects produced bymaintaining a long-lasting low level of Myc proteininto exponentially proliferating M14 cells, we deter-mined the optimal schedule of hormone administrationable to maintain Myc expression at a level critical forcell proliferation in pINDc-myc AS cell clone. To thisend we measured the e�ect of repeated doses ofponasterone A. Figure 3a shows that a dose of20 mM ponasterone repeated every 24 h, resulted in asigni®cant reduction of cell growth. After 24 h ofhormone exposure we observed a cell growth inhibitionof about 30%, a value similar to that obtained after24 h with the previous regimen of hormone treatment.The reduction of cell proliferation became moresigni®cant at 48 h from the start of hormone treatment(two doses of ponasterone), reaching values of 70% ofgrowth inhibition (versus the 42% obtained with thesingle-dose regimen). After 72 h of hormone treatment,when the cells received a triple dose of ponasterone, thegrowth inhibition remained at a value of 78%. Even ifexposed to a triple dose of ponasterone, empty vectortransfected cells did not show signi®cant cell growthinhibition as compared to the parental cell lineproliferation.
The levels of transcripts of both ectopic c-myc ASand endogenous c-myc were determined by semi-quantitative RT±PCR analysis, using suitable pairsof primers as indicated in Figure 3b (bottom). Figure3b (top) shows the RT±PCR analysis of total RNAextracted from the control clone (pINDneo) and thepINDc-myc AS clone exposed for 24 h to 20 mM ofponasterone. The PCR ampli®cation was performed atvarious numbers of cycles, as indicated. The PCR-ampli®ed c-myc AS cDNA was detectable only in theponasterone-treated pINDc-myc AS cells at cycle 33;whereas, the PCR-ampli®ed c-myc cDNA in this clonewas barely detectable even increasing the number ofPCR cycles, up to 35 cycles. The pINDc-myc AS cellsnot exposed to ponasterone and the hormone treatedcontrol cells showed comparable levels of the PCR-ampli®ed c-myc cDNA at 35 cycles and no detectableamounts of PCR-ampli®ed c-myc AS cDNA at 33cycles. b-actin was used as an internal control for theamount of RNA used in all RT ±PCR reactions.
The levels of Myc protein in pINDc-myc AS cellswere measured by Western blot analysis. The resultsin Figure 3c indicated that the treatment withponasterone resulted in an evident Myc down-regulation (threefold) after 24 h, similar to what wasobserved with the previous single-dose hormoneregimen and consistent with RT ±PCR data. A furtherdecrease (sixfold) of Myc levels in the c-myc AS clonewas observed after 48 h of ponasterone exposure (twodoses of the hormone). HSP72/73 detection was doneas control for the amount of total protein used(Figure 3c).
Figure 2 (a) Cell growth of control (white) and pINDc-myc AStransfected cells exposed to a bolus of ponasterone A given at thedoses of 5 (pointed), 10 (dashed) and 20 (black) mM. 16104 cellswere seeded in 24 well-plates and the hormone was added 24 hlater. The arrow indicates addition time of the hormone. At theindicated times from hormone administration the cells wereharvested and counted. Error bars represent standard deviation ofthree independent experiments. (b) Levels of Myc protein in theclones treated as in (a) was detected by Western blot analysisusing anti-Myc speci®c antibody. Each lane was loaded with30 mg of proteins from cell lysate. HSP 72/73 was used as controlfor loading same amounts of proteins. The experiment wasrepeated three times showing similar results. A representative blotis reported in the Figure. pona=ponasterone A
p27kip1-mediated apoptosis in melanoma cellsI D'Agnano et al
2816
Oncogene

The low proliferation rate of M14 pINDc-myc AS cells isdue to the prevention of G1?S transition
In order to determine which phase of the cell cycle wasa�ected by the decreased Myc protein expression wehave analysed the cell cycle distributions in pINDc-mycAS cells, exposed to repeated doses of ponasterone A,by BrdUrd incorporation and ¯ow cytometry. Thisanalysis, performed at di�erent times after c-myc ASinduction, revealed, as expected, an increasing accu-mulation of the hormone-treated pINDc-myc AS cellsin the G1 cell cycle phase as the time proceeded (Figure4a), pINDc-myc AS cells accumulated in the G1 phaseof the cell cycle (52% compared to 29% of controlcells) as early as 24 h from hormone exposure; after72 h of hormone treatment more than 80% of the cellswere blocked in G1, as compared to the 43% of controlcells. The arrest in the G1 phase was associated to aconcomitant depletion of cells from both the S and G2/M cell cycle compartments. The block in the G1 phasewas also con®rmed in cells continuously exposed toBrdUrd for 24 h, in order to label the entire cyclingcell population. As shown in Figure 4b after thecontinuous 24 h BrdUrd labeling of the hormone-induced pINDc-myc AS cells, performed 48 h after
hormone administration (two doses of ponasterone), aremarkable fraction of the cell population (about 50%)did not show BrdUrd positivity (right panel), while allthe control cells were labeled by BrdUrd (left panel).Indeed, within the continuous 24 h labeling time thecontrol cells have completed their DNA replication,mitosis and cell division, as evident from the presenceof BrdUrd-positive cells at G1 DNA content. Instead,the presence of a great proportion of unlabeled G1 cellsin the induced pINDc-myc AS clone, indicates that afraction of the cell population did not proceed throughthe G1?S transition during the same time interval.
The prevention of G1?S transition, following inhibitionof c-myc expression, correlates with an increase of p27kip1
level, a decrease of cdk2 kinase activity and a decrease ofpRb phosphorylation
Since Myc is known to act as an upstream regulator ofcdks activity, we investigated whether down-regulationof Myc protein levels could a�ect the expression of themain molecules that control cell cycle entry andprogression. The expression levels of cyclin E, cyclinA, p27kip1, p21cip1 and cdk2 as well as of the cdc25Aphosphatase were determined in pINDc-myc AS and
Figure 3 (a) Cell growth of pINDc-myc AS (black) and control (white) clones exposed to repeated 20 mM doses of ponasterone Agiven every 24 h starting after 24 h from cell seeding. Error bars represent standard deviation of three independent experiments. (b)Representative RT±PCR analysis of ectopic c-myc AS and endogenous c-myc in control and pINDc-myc AS transfected cellstreated as in (a). The results are referred to the 24 h time of exposure to the hormone. The scheme at the bottom of this panel showsthe relevant portions of pINDc-myc AS construct and c-myc coding sequence, the two pairs of PCR primers used to measure therespective transcripts and the length of the ampli®cation products. c-myc primers: Hmyc01 and Hmyc02, black arrows; this pair ofprimers, internal to the c-myc coding sequence, can also recognize the AS transcript. c-myc-AS primers: Hmyc03 and BGHrev, whitearrows; this pair of primers, spanning the boundary between the c-myc AS sequence and the plasmid polyadenylation sequence, isspeci®c for the AS transcript. 5XE/GRE=ecdysone/glucocorticoid response elements; PDHSP=heat shock minimal promoter; BGHpA=bovine growth hormone polyadenylation sequence; pona=ponasterone A. (c) Western blot analysis of the Myc protein levelsin the control (lanes 1) and pINDc-myc AS (lanes 2) clones treated with ponasterone A as in (a). HSP 72/73 was used as control forloading same amounts of proteins
Oncogene
p27kip1-mediated apoptosis in melanoma cellsI D'Agnano et al
2817

control cells, as compared to those of the M14 parentalcell line (Figure 5a). The Western blot analysis,performed after 48 h from hormone exposure, revealeda decrease of both cyclin E (1.5-fold) and cyclin A(threefold) expression, consistent with the accumula-tion of cells in the G1 phase. The cdk2 content was alsodecreased by about threefold in M14 pINDc-myc AScells exposed to the hormone. In addition, a more than10-fold reduction was observed in p21cip1 protein levels.Cdc25A levels were slightly decreased after c-myc ASinduction (1.1-fold). By contrast, an increase in thelevels of p27kip1 of more than threefold was detected.The expression levels of cyclin E, cyclin A, cdk2,p21cip1, p27kip1 and cdc25A in empty vector-transfectedcells appeared quite similar to those of the nontransfected parental cells. Consistent with the Westernblot data cdk2-associated kinase activity, evaluated inpINDc-myc AS cells after 48 h from hormoneexposure, was found decreased by about 70%, ascompared to those of control cells (Figure 5b).
In addition, since one of the major target of cdk2kinase activity is pRb, we have also analysed the statusof pRb in pINDc-myc AS cells induced with thehormone. Western blot analysis of pRb demonstratesthat Myc down-regulation led to an increased amountof hypophosphorylated pRb (Figure 5c, top).
It is known that the degradation of p27kip1 isdependent on the phosphorylation at the threonine187, then we decided to test whether the accumulationof p27kip1 protein could be due to a decrease of itsdegradation analysing the phosphorylation status ofp27kip1. p27kip1 was immunoprecipitated and thenanalysed by Western blot using an antibody recogniz-ing the phosphothreonine at position 187. Figure 5c,bottom, shows more than threefold decrease in theamount of phospho-p27kip1 in pINDc-myc AS cellstreated with the hormone, suggesting that Myc proteinregulates the pathway of phosphorylation of p27kip1.
To investigate whether the overexpression of p27kip1
in the M14 pINDc-myc AS cells treated with thehormone could also be ascribed to an accumulation ofp27kip1 mRNA, Northern blot analysis of total RNAwas performed. The analysis showed that the level ofp27kip1 mRNA did not signi®cantly change followingc-myc AS induction (Figure 5d).
The increased levels of p27kip1 expression correlate withactivation of programmed cell death
In order to determine the contribution of apoptosis tothe decrease of cell number observed in pINDc-myc AScells after down-regulation of Myc protein expression,
Figure 4 Cell cycle analysis following down-regulation of Myc protein. (a) control (^) and pINDc-myc AS (&) cells, treated as inFigure 2, were labeled with a BrdUrd pulse of 30 min at the indicated times and analysed by biparametric ¯ow cytometry. Errorbars represent standard deviation from three separate experiments. (b) Representative biparametric ¯ow cytometric dot plots of cellscontinuously labeled with BrdUrd for 24 h, starting 48 h from hormone treatment with two doses 20 mM of ponasterone A. Control(a) and pINDc-myc AS (b). The window in (a and b) represents BrdUrd negative G1 cells
p27kip1-mediated apoptosis in melanoma cellsI D'Agnano et al
2818
Oncogene

DNA fragmentation analysis was performed on thesame samples previously examined for cell prolifera-tion. Two di�erent methods of analysis by ¯owcytometry (Propidium Iodide-staining of the DNAand TUNEL assay) were used on each sample.TUNEL assay was used to exclude necrosis that couldnot be accurately discriminated using PI-staining.Figure 6a shows the percentages of apoptotic cells inpINDc-myc AS and control cultures exposed to thehormone, as determined by ¯ow cytometry, either inthe sub-G1 region of the ¯ow cytometric DNA contentdistributions or as FITC-dUTP associated positivity.The results obtained with the two methods of analysiswere superimposable, indicating that no necrosis hadoccurred after treatment of the cells with the hormone.The hormone-induced down-regulation of Myc proteinactivates apoptotic cell death only in the pINDc-mycAS clone as compared to the vector transfected cells.Apoptosis was already evident after 48 h of hormoneexposure, the apoptotic cell percentages being 20 and17% as calculated by PI-staining and TUNEL assay,respectively. The inhibition of cell proliferationobtained at 72 h of hormone exposure resulted in amarked increase of programmed cell death, withapoptotic cell percentages of 41 and 48% calculatedwith the two methods of analysis, respectively. Only2 ± 3% of the control cell population showed evidenceof apoptotic cell death. Morphological analysis of thecell cytospins further con®rmed the presence ofapoptosis in the pINDc-myc AS clone (data notshown).
Since p27kip1 overexpression has been demonstratedto activate apoptosis in di�erent tumor types, wesought to determine the e�ect of an ectopic p27kip1
expression in M14 melanoma cells. To better evidencethe role of p27kip1 in the activation of apoptoticprogram in M14 melanoma cells we also transfectedthe pINDc-myc AS clone with two di�erent p27kip1
mutants that have been demonstrated to fail inblocking the cells in G1 phase (Vlach et al., 1997).The p27wt, p27ck7 and p27k7 expression vectors weretransiently co-transfected with the GFP-spectrin ex-pression plasmid in pINDc-myc AS cells and the DNAcontent pro®le of transfected cells was analysed bymeans of two-color ¯ow cytometry (Figure 6b). In fact,gating out GFP-positive cells and analysing the cellcycle distribution of this population 48 h aftertransfection, we evidenced 40% of apoptosis in thepINDc-myc AS cells overexpressing p27wt (panels c andc'). By contrast, apoptosis was undetectable in GFP-positive cells transfected with the p27ck7 mutant (panelsd and d') and with the p27k7 mutant (panels e and e')as well as with the empty vector (panels b and b'). The
Figure 5 E�ects induced by the down-regulation of Myc proteinon the main cell cycle-regulating molecules. pINDc-myc AS andcontrol cells were exposed to 20 mM of ponasterone A, given every24 h as in Figure 3. Forty-eight hours after hormone addition,cells from the two clones were assayed for cellular proteinexpressions, H1 kinase activities and p27kip1 mRNA expression,and compared to those of the parental untransfected M14 cells.At the same times of analysis phosphorylation status of the pRband p27kip1 proteins was also studied. (a) Representativeimmunoblot analyses of the indicated proteins in M14 parentalcells, control and pINDc-myc AS transfected cells. At the time ofanalysis cells were subcon¯uent and pINDc-myc AS cells werearrested in G1 as shown in Figure 4. HSP 72/73 was used ascontrol for loading same amounts of proteins. (b) H1 kinaseactivity in immunoprecipitates of cyclin A and cdk2 from parental(gray), control (white) and pINDc-myc AS (black) cells. Eachmeasurement was from equal amounts of total cellular proteinand the activity in pINDc-myc AS was normalized to that incontrol cells (assumed as 100%). Error bars indicate standarddeviation of three independent experiments. (c) Representativeimmunoblots analysis of pRb and phospho-p27kip1 proteins.Lysates were prepared from cells treated as described above andanalysed for pRb expression. Concerning p27kip1 phosphorylation,extracts were immunoprecipitated with an anti-human p27kip1
antibody. Immunocomplexes were analysed by Western immuno-blot for phospho-p27kip1. (d) Representative Northern blotanalysis of p27kip1 mRNA in M14 parental cells, control and
pINDc-myc AS transfected cells. Equal loading and integrity ofthe RNA was veri®ed by ethidium bromide staining shown below(rRNA). Experiments in (a), (c) and (d) were repeated three timesshowing similar results
Oncogene
p27kip1-mediated apoptosis in melanoma cellsI D'Agnano et al
2819

overexpression of the p27kip1 proteins in transientlytransfected cells was veri®ed by sorting the cells on thebasis of GFP expression. Western blot analysis of thesorted cells con®rmed a threefold increase of the p27kip1
protein expression, as evaluated by densitometricanalysis of the blots, normalized to the HSP 72/73intracellular levels (data not shown).
In addition, to exclude the possibility that theapoptotic program could be activated by the G1 blockproduced by p27kip1, we have studied the e�ect of anectopic expression of a dominant negative cdk2 mutant(CDK2 DN) in pINDc-myc AS M14 cell clone. Cellswere co-transfected with GFP-spectrin expressionplasmid and pCMV/CDK2 DN or pCMV emptyvector and the DNA content pro®le of transfectedcells was analysed as described above. Indeed, theinactivation of the cdk2 kinase was not able to induce
apoptosis even though about 70% of the cells wereaccumulated in the G1 phase (Figure 6c). The fractionof the cells in the sub-G1 region was less than 10%. Inaddition, the analysis of the expression of the p27kip1
protein following inactivation of the cdk2 kinasedemonstrated that no increase in p27kip1 level occurred(Figure 6d), strongly supporting the hypothesis thatp27kip1 per se could promote apoptosis.
Discussion
The c-myc proto-oncogene has been shown to play arole in melanoma cell proliferation. In fact, in previousreports we have shown that inhibition of Myc proteinexpression leads to apoptosis in several melanoma celllines (Leonetti et al., 1996). However, the Myc-
Figure 6 (a) Percentages of apoptotic cells in pINDc-myc AS and control cells treated with ponasterone A. Apoptosis percentagescalculated at the indicated times by Propidium Iodide-staining (black) and TUNEL assay (gray) are reported for the pINDc-myc ASclone. Only apoptosis percentages calculated by PI-staining are shown for control cells (white). Arrows indicate hormone additions.(b) E�ect of ectopic p27wt, p27ck7 mutant and p27k7 mutant on apoptosis in pINDc-myc AS cells. These cells were co-transfectedwith p27wt, p27ck7, p27k7 or pBabe empty vector together with the GFP-spectrin plasmid. Untransfected cells are shown in (a) and(a') and used as negative control for setting the gating region. The GFP positive cells (upper region of the cytograms) were analysedfor DNA content distribution by ¯ow cytometry. On the top (b ± e) are the representative GFP-spectrin ¯uorescence cytograms andon the bottom (b' ± e') are the DNA content histograms of GFP positive p27wt (c and c'), p27ck7 mutant (d and d') , p27k7 mutant(e and e') and empty vector (b and b') transfected cells. The analysis of apoptosis, detected in the sub-G1 region, has been conducted48 h following transient transfection. The cell cycle distribution of the transfected p27wt, p27ck7, p27k7 and empty vector cells,respectively, are: G1=70.0, S=18.0, G2/M=12.0; G1=38.0, S=31.0, G2/M=30.0; G1=36.0, S=34.0, G2/M=30.0; G1=41.5,S=38.4, G2/M=20.1. (c) CDK2 DN on apoptosis in pINDc-myc AS cells. The cells were co-transfected with pCMV/CDK2 DN orpCMV empty vector together with the GFP-spectrin plasmid and analysed as in (b). The cell cycle distribution of the transfectedCDK2 DN and empty vector cells, respectively, are: G1=42.0, S=30.0, G2/M=28; G1=68.0, S=8.0, G2/M=24.0. (d) CDK2 DNon p27kip1 expression in pINDc-myc AS cells. Representative immunoblot analysis of p27kip1 in pCMV/CDK2 DN or pCMV emptyvector transfected cells. The experiment was repeated two times with similar results
p27kip1-mediated apoptosis in melanoma cellsI D'Agnano et al
2820
Oncogene

controlled pathways involved in the observed apoptosisare still obscure. In the present study we developed amelanoma model in which reduction of endogenousc-myc expression can be ectopically driven in ahormone-inducible way, providing a useful tool tostudy the molecular events controlled by c-myc whichcould be responsible for the activation of programmedcell death. Data herein reported provide, for the ®rsttime in melanoma cells, experimental evidence that`switching o�' the synthesis of Myc protein, the levelsof the kinase cyclin-dependent inhibitor p27kip1 increase,preventing G1?S transition and promoting apoptosis.
The 20 mM dose of the hormone/ponasterone givenevery 24 h induces the transcription of antisense c-mycRNA and is needed to reduce Myc expression at a levelthat is critical for cell proliferation. Other authors haveshown that cell proliferation was inhibited by inducingthe expression of c-myc antisense mRNA in a SmallCell Lung Carcinoma cell line, even though no Mycdown-regulation and cell cycle perturbations weredetected (van Waardenburg et al., 1997). Conversely,our data demonstrate that the induction of c-myc AStranscripts in M14 cells causes a reduction of Myclevels and an inhibition of proliferation of exponen-tially growing cells. This strongly favors the hypothesisthat the observed inhibition of cell proliferation isreally to be ascribed to the down-regulation of Mycprotein. We have previously demonstrated that anti-sense phosphorothioate oligodeoxynucleotides (ODNs)targeted to the c-myc mRNA can inhibit cellproliferation in melanoma cells both in vitro and invivo (Leonetti et al., 1996) and that Myc proteinreduction also makes the melanoma cells more sensitiveto cis-platin treatment (Citro et al., 1998). In thesestudies it has been reported that the decreased level ofMyc protein obtained by ODNs treatment accumulatedthe cells in the S phase of the cell cycle. Data hereinreported demonstrate that the inhibition of cellproliferation, following Myc protein down-regulation,is due to an arrest of the cells in the G1 phase. Theseapparently contrasting results could be explainedconsidering that: (1) To avoid enzymatic degradationODNs must be modi®ed in their chemical compositionand this could a�ect the ODNs binding a�nity to thetarget mRNA (Lesnikowski et al., 1990). (2) Modi®edODNs can also react with other cellular targets (suchas enzymes or cellular structural proteins (Burgess etal., 1995; Stein, 1998; Krieg and Stein, 1995). (3) To bee�ective against melanoma cells modi®ed ODNs mustbe administered at high doses. Thus, the use ofmodi®ed ODNs, employed at high doses, can render`signi®cant' the non-sequence speci®c interactions ofODNs, partially masking the sequence speci®c e�ectsof the antisense ODNs and interfering in the evaluationof the molecular mechanisms studied. An experimentalmodel based on the inhibition of gene function induceddirectly inside the cells appears to be more appropriateto investigate the role of the cell cycle-related Mycprotein in the induced-apoptosis. Our model allowed usto directly link the down-regulation of Myc proteinwith the observed e�ects on proliferation, thus
avoiding interference possibly caused by non-antisensemechanism.
The Myc down-regulation-mediated G1 block is alsostrongly supported by the decreased expression ofcyclins E and A, which are cell cycle regulators at theG1?S transition and at the S-progression/S?G2
transition, respectively. These observations are inagreement with results from other authors, whodemonstrated that a deregulation of c-myc is associatedto an increase of cyclin E expression (Jansen-Durr etal., 1993; Daksis et al., 1994; Hanson et al., 1994;Hoang et al., 1994). However, even though evidenceshave been provided that c-myc is able to directlytransactivate the expression of cyclin E, the mechanisminvolved remains unclear (Leone et al., 1997; Perez-Roger et al., 1997). In addition, the observed decreasein cyclin A levels could be ascribed to a reducedactivation of the cyclin A promoter by the cyclin E/cdk2 kinase. The cyclin E-cdk2 complex activity isneutralized by p27kip1 that increased its levels bythreefold following Myc down-regulation. In fact, asigni®cant reduction of cdk2 kinase activity was alsofound. These data are consistent with those fromothers who demonstrated that an increase of the Myclevels is related to the increase of cdk2-associatedkinase activity (Vlach et al., 1996) or that in c-myc-nullcells the activities of all cyclin-cdk complexes arereduced (Mateyak et al., 1999).
The increased level of p27kip1 observed in our modelis not regulated by a transcriptional mechanism sincethe amount of p27kip1 RNA did not change in cells withinhibited Myc expression. We hypothesize a regulationof this CKI due to reduced p27kip1 degradation. It hasbeen recently proposed that c-myc drives the synthesisof a putative p27kip1-sequestering protein, which rendersthe hyperphosphorylated p27kip1 available for ubiquiti-nation (Obaya et al., 1999). Accordingly, it is likelythat the down-regulation of Myc protein could preventthe synthesis of this putative protein making p27kip1
unavailable for the following ubiquitination. In fact, asrecently demonstrated in a study by O'Hagan et al.(2000) a Myc-target gene (Cul1), which encodes acritical component of the ubiquitin ligase SCFskip2, hasbeen identi®ed. p27kip1 is a known target of the SCFskip2
complex and the regulation of p27kip1 polyubiquitina-tion by this complex is dependent on the priorphosphorylation of p27kip1 on threonine 187. It is likelythat in the Myc down-regulated cells, both theinhibition of p27 phosphorylation and the down-regulation of Cul1, contribute to the increase of p27stabilization.
In addition, since p27kip1 is a substrate of the cyclinE/cdk2 phosphorylating activity (Shea� et al., 1997), itis also possible that the reduction of cyclin E levelsdecreases p27kip1 phosphorylation, thus contributing torender the CKI not available for proteasome degrada-tion. Indeed, we found that the amount of phosphory-lated p27kip1 decreases of about threefold following Mycdown-regulation, strongly supporting the hypothesisthat the accumulation of p27kip1 is to be ascribed to areduced degradation of the protein. On the other hand,
Oncogene
p27kip1-mediated apoptosis in melanoma cellsI D'Agnano et al
2821

the decrease of the activity of cyclin E/cdk2 complex isalso demonstrated by the increase of the amount ofhypophosphorylated fraction of pRb.
The increase of p27kip1 was found to be associated toa very strong decrease (10-fold) of the other CKIp21cip1. Similar results were reported by Mateyak et al.(1999), who demonstrated that in c-myc null cellsoverexpression of p27kip1 was accompanied by a quitecomplete disappearance of p21cip1.
Our results also show that down-regulating Mycprotein, melanoma cells underwent apoptotic celldeath. The role of c-myc in the apoptotic processeswas de®nitively established in the early 1990s byCleveland, Evan and their colleagues, who haveextensively demonstrated that c-myc is able to activateapoptosis (Evan et al., 1992). Indeed, cells where c-mycis enforced undergo apoptosis following growth factorwithdrawal. More recently Evan himself and Little-wood have proposed the appealing idea that c-mycdoes not act as a death e�ector per se but insteadsensitizes the cells to a variety of apoptotic triggers,thus supporting the hypothesis that c-myc has intrinsicfunctions related to cell death (Evan and Littlewood,1998).
In this study we clearly demonstrate that onestimulus which could activate apoptosis in melanomacells is the down-regulation of Myc protein. It is likelythat the down-regulation of Myc protein by itself couldmimic the growth factor withdrawal in a cell type inwhich the proliferative stimulus is assured by theinactivation of the p16-cyclinD/cdk4-pRb pathway(Bartkova et al., 1996; Maelandsmo et al., 1996).Therefore, the cells committed to proliferate are poisedin unfavorable conditions by diminishing Myc levelsand therefore undergo apoptosis. We hypothesize thatthis apoptosis is mediated by the increase of p27kip1. Tosupport this hypothesis we have investigated the e�ectsof an ectopic overexpression of p27wt and two p27mutants (ck7 and k7) that fail to block the cells in theG1 phase (Vlach et al., 1997). The results hereinreported show that the p27wt activates the apoptoticprogram, while the two mutants do not. In addition,we have also investigated whether the block in the G1
cell cycle phase caused by p27wt could act per se asdeath e�ector. We show that an ectopic expression of aCDK2 DN (Van den Heuvel and Harlow, 1993) wasable to arrest in G1 phase about 70% of the cellpopulation but no signi®cant apoptosis was evidencedand did not increase the p27kip1 levels. This stronglysupports the idea that p27kip1 is directly responsible forthe promotion of apoptosis in the melanoma cell type,in accordance with data reported in the literature(Katayose et al., 1997; Wang et al., 1997).
In conclusion, data herein reported evidenced, in themelanoma tumor type in which the p16-cyclinD/cdk4-pRb G1 checkpoint is defective, an alternative pathwayof growth- and death-regulation mediated by theincrease of p27kip1 levels. This is particularly relevantin view of the identi®cation of new therapeuticstrategies based on the re-establishment of theapoptotic pathways in cancer cells, thus providing
important information for improving melanomapatients management.
Materials and methods
Plasmid preparation
pINDc-myc AS was generated by subcloning from pBJ3O-Myc MER (kind gift from Bruno Amati, DNAx Institute,San Francisco, USA) the complete c-myc cDNA fragmentinto BamHI site of pIND downstream of the ecdysoneresponse element (Invitrogen, San Diego, CA, USA) whichcontains the neomycin (G418) resistance gene. pVgRXR,which encodes for a heterodimer of the ecdysone receptor,was purchased by Invitrogen and contains the Sh ble genethat permits stable selection in mammalian cells in thepresence of zeocyn. Bruno Amati (Vlach et al., 1996) kindlyprovided the puromycin resistant gene vectors pBabe-puro(pBP), pBP/p27kip1wt, p27kip1 CANA/NADA mutant (ck7),p27kip1 NADA mutant (k7) and the pCMV/CDK2 DN (Vanden Heuvel and Harlow, 1993). The pCMVEGFP-spectrinexpression vector was a kind gift from Andrew J Beavis(Kalejta et al., 1997).
Cell culture and DNA transfections
M14 human melanoma cell line was cultured in RPMI-1640medium supplemented with fetal calf serum (10%), gentamy-cin (0,1%) and L-glutamine (1%) at 378C in a 5% CO2/95%air atmosphere in a humidi®ed incubator. Generation ofstable c-myc AS inducible expression cell clones was achievedemploying the Ecdysone-Inducible Expression System (In-vitrogen, San Diego, CA, USA). Brie¯y, this system is basedon the molting induction system found in Drosophila andmodi®ed for inducible expression in mammalian cells. Thesystem uses the steroid hormone ecdysone analog, Ponaster-one A, to activate the expression of the gene of interest via aheterodimeric nuclear receptor. Two mg of double-strandedDNA of both pINDc-myc AS and pVgRXR plasmids weremixed in Optimem (GIBCO, BRL) with 10 ml of lipofecta-mine reagent (GIBCO, BRL) and incubated, for 5 h, with16105 M14 cells the day after seeding, according to themanufacturer's instructions. RPMI fresh medium supplemen-ted with 10% FCS was added and after 48 h culturing, thetransfected cells were maintained for 3 weeks in the presenceof both G418 (GIBCO, BRL) and zeocyn (Invitrogen, SanDiego, CA, USA). G418/Zeocyn resistant clones wereisolated and selected from pINDc-myc AS as well as fromthe empty vector (pINDneo) transfected cells. For transienttransfections of pBP/p27kip1 the pINDc-myc AS M14 cellclone was co-transfected with 8 mg of pBP/p27kip1 or p27kip1
CANA/NADA mutant (ck7) or p27kip1 NADA mutant (k7)or pCMV/CDK2 DN (7/7) and 2 mg of pCMVEGFP-spectrin plasmids, mixed in Optimem containing 25 mllipofectamine reagent and incubated, for 5 h, with 36105
cells (100 mm dishes) the day after seeding, according to themanufacturer's instructions. Afterwards, the transfected cellswere maintained in fresh growing medium for an additionaltime of 48 h and then were trypsinized for ¯ow cytometricapoptosis detection.
RNA extraction, Northern blot and semi-quantitative RT ±PCR
Total RNA was prepared by guanidium thiocyanate methodof extraction. Northern blots were made using standard
p27kip1-mediated apoptosis in melanoma cellsI D'Agnano et al
2822
Oncogene

procedures. For semi-quantitative RT ±PCR, 1 mg of totalRNA from each sample were reverse-transcribed in a 20 mlvolume reaction using 50 pmoles of random examers with200 units M-MLV reverse transcriptase (GIBCO, BRL),according to the manufacturer's instructions. PCR wascarried out in 100 ml volume containing 0.25 mM of eachdNTP, 20 pmoles/ml of each oligonucleotide primer, 2 mM
MgCl2, 200 mM (NH4)2SO4, 750 mM tris-HCl pH 9.0, 0.1%Tween 20, using 1 ± 2 ml of RT reaction mixture and 2.5 unitsof Thermoprime Plus DNA polymerase (Advanced Bio-technologies LTD). The mixtures were subjected to di�erentPCR cycles, as indicated, including the ®rst denaturationcycle at 948C for 4 min; denaturation at 948C for 30 s;annealing for 30 s and extension at 728C for 1 min.Ampli®cation products (20 ml) were analysed by electro-phoresis on a 1.5% Agarose gel stained with ethidiumbromide. Primers and annealing temperatures used were asfollows: c-myc (Hmyc01 ATTCTCTGCTCTCCTCGA;Hmyc02 TCTTGGCAGCAGGATAGT, TA=578C); c-mycAS (Hmyc03 CTCCTCGTCGCAGTAGAA; BGHrev TA-GAAGGCACAGTCGAGG, TA=578C); b-actin (senseGCGCGGCGTAGCCCCCGTCAG; antisense CGCGGC-AGGAAGCCAGGCCCC, TA=578C).
Apoptosis detection
Apoptosis was analysed in pINDc-myc AS and controlstable clones by ¯ow cytometry using both Propidium Iodide(PI)-staining and TUNEL assay. Cells induced withponasterone A were harvested, washed once in PBS andeither stained with PI (50 mg/ml, Sigma Chemical Co.) inPBS containing RNase A (75 Ku/ml, Sigma Chemical, Co.)or processed for TUNEL assay as previously described(D'Agnano et al., 1998). The analysis of apoptosis inpINDc-myc AS cells transiently transfected with expressionvectors containing p27wt, p27ck7, p27k7 was performed by¯ow cytometry after PI-staining. Cells transfected asdescribed above were trypsinized after 48 h culturing infresh medium and either analysed for DNA content orsorted for Western blot analysis. For DNA content analysisthe transfected cells were ®xed in 80% ethanol and stainedwith PI (50 mg/ml, Sigma Chemical Co.) in PBS containingRNase A (75 KU/ml, Sigma Chemical, Co.). Two-color ¯owcytometry was performed, simultaneously measuring GFP-spectrin (green channel) and PI (red channel) ¯uorescence,using a FACScan ¯ow cytometer (Becton Dickinson,Sunnyvale, CA, USA). The gates to analyse DNA contentdistribution of both GFP-spectrin/pBP/p27wt, p27ck7, p27k7
and pCMV/CDK2 DN transfected cells were established bymeasuring background levels of empty vectors transfectedcell ¯uorescence. For Western blot analysis, transfected cellswere sorted on the basis of the GFP-spectrin positivity,using an EPICS 541 ¯ow cytometer (Coulter ElectronicsInc), and then analysed for Western blot as describedsuccessively.
Cell cycle analysis
Cell cycle analysis of pINDc-myc AS and control stableclones was performed after bromodeoxyuridine (BrdUrd,Sigma, St. Louis, USA) incorporation. Both BrdUrd pulse-labeling and continuous-labeling experiments were carriedout. Pulse-labeling experiments were performed at 24, 48 and72 h from hormone induction, by adding 10 mM BrdUrd tothe medium during the last 30 min before analysis. ForBrdUrd continuous-labeling experiments the stable cell cloneswere continuously exposed for 24 h to 10 mM BrdUrd at 48 h
of hormone induction. At the indicated times the cells wereharvested, washed once in PBS, ®xed in 70% ethanol andstored at 48C until analysis. Then cell suspensions were rinsedwith cold PBS and incubated with 4 N HCl for 20 min atroom temperature to partially denature DNA. Cells werewashed twice with borax-borate bu�er (pH=9.1) toneutralize the acid pH and once with PBS. The sampleswere incubated with mouse monoclonal antibody anti-bromodeoxyuridine (Boehringer Mannheim, Monza, Italy)diluted 1 : 50 in complete medium containing 0.5% Tween-20(Calbiochem, San Diego, CA, USA) at 48C for 1 h. Afterwashing twice in PBS, cells were exposed to FITC-Conjugated F(ab') rabbit anti-mouse (Dako, SA, Glostrup,Denmark) at dilution 1 : 20 in PBS at 48C for 1 h. Finally, thecells were washed twice with PBS, stained with a solutioncontaining 50 mg/ml PI and 75 KU/ml RNase in PBS for30 min at room temperature. The samples that had not beenincubated with monoclonal antibody anti-bromodeoxyuridinewere used as negative controls. Twenty thousand events/sample were acquired using a FACScan cyto¯uorimeter(Becton Dickinson, Sunnyvale, CA, USA). The analysis wasperformed using a CellQuest software package (BectonDickinson).
Western blot analysis
Lymphocytes and M14 cells were solubilized in lysis bu�er(0.01 M Tris-HCl [pH 7.5], 0.144 M NaCl, 0.5% Nonidet P-40, 0.5% sodium dodecyl sulfate [SDS], 0.1% aprotinin,10 mg/ml leupeptin, and 2 mM phenylmethylsulfonyl ¯uoride)and treated by 10 s of sonication. The protein content in thedi�erent samples was quanti®ed using the BCA protein assay(Pierce Chemical Co., Rockford, IL, USA). Thirty-micro-gram aliquots of protein were subjected to 10% SDS-polyacrylamide gel electrophoresis. The resolved proteinswere blotted to a nitrocellulose membrane by semidry electrictransfer, and the membranes were blocked in TBS bu�er(20 mM tris Base, 137 mM sodium chloride, 1 M hydrochloricacid, pH 7.6) containing 0.2% blotting grade blocker non fatmilk (Biorad, Hercules, CA, USA) for 1 h. Blots were thenincubated with the primary antibodies: anti-human cyclin D1(clone HD11, Calbiochem, Cambridge, UK), anti-humancdk4 (clone H-22, Santa Cruz Biotechnology, Santa Cruz,CA, USA) and anti-human p16 (clone N-20, Santa CruzBiotechnology, Santa Cruz, CA, USA) MoAbs.Cells (16106) obtained from the di�erent experiments were
stored frozen at 7808C and processed as above described.Blots were then incubated with the primary antibodies: anti-human Myc Mab (clone 9E10, Santa Cruz Biotechnology,Santa Cruz, CA, USA), anti-human cyclin A (clone BF683,Calbiochem, Cambridge, UK) and cyclin E (C-19, SantaCruz Biotechnology, Santa Cruz, CA, USA) Mabs, anti-human p21cip1 (clone DF10, Calbiochem, Cambridge, UK)and p27kip1 (UPSTATE Biotechnology, Lake Placid, NY,USA) Mabs, anti-human cdk2 Mab (Calbiochem, Cam-bridge, UK), anti-human cdc25A (N-15 Santa Cruz Biotech-nology, Santa Cruz, CA, USA), anti-human pRB (clone G3-245, Pharmingen) and anti-human HSP 72/73 Mab (Ab-1,Oncogene Science Inc.). Peroxidase labeled anti-mouse andanti-rabbit antibodies (Amersham Life Science, ArlingtonHeights, IL, USA) were used as secondary antibodies. Theimmunoblots were processed for enhanced chemilumines-cence detection (Amersham Life Science). The relativeamount of transferred proteins in a given sample wasquanti®ed by scanning X-ray ®lms and estimating the relativearbitrary density units, normalized to the correspective HSP72/73 content in each sample.
Oncogene
p27kip1-mediated apoptosis in melanoma cellsI D'Agnano et al
2823

In vitro kinase assay
Cells were lysed by addition of ice-cold lysis bu�er (50 mM
HEPES, 250 mM NaCl, 0.1% Tween-20, 1 mM EDTA,2.5 mM EGTA, 10% Glycerolo, 10 mM b-glycerol-phospha-to, 1 mM DTT) containing 1 mM phenylmethylsulfonyl¯uoride, 10 mg/ml pepstatin, 25 mg/ml leupeptin, 25 mg/mlaprotinin, 50 mM sodium ¯uoride, 1 mM sodium orthovana-date. The lysates were sonicated, centrifuged at 13 000 r.p.m.for 30 min at 48C and total proteins were quanti®ed using theBCA protein assay (Pierce Chemical Co.). Cyclin A and cdk-2 immunoprecipitations were performed using 200 mg ofproteins from cell lysate. Anti-human cdk2 (Ab-1 Calbio-chem) and cyclin A (clone, BF683, Calbiochem, Cambridge,UK) Mabs were used. The immunocomplexes were bound toprotein A-agarose, centrifuged and then washed four timeswith lysis bu�er and twice with kinase bu�er preparative(50 mM HEPES, 1 mM DTT). Fifty ml kinase bu�er (50 mM
HEPES, 10 mM MgCl2, 1 mM DTT, 2.5 mM EGTA, 10 mM
b-glycerol-phosphato, 0.1 mM sodium orthovanadate, 1 mM
sodium ¯uoride) was added together with 2 mg histone H1and 50 mM ATP, 10 mCi [g-32P]ATP to the cyclin A- or cdk2-immunocomplexes, and the reaction was incubated at 308Cfor 30 min. The reaction was stopped by the addition of 25 ml46 Laemmli sample bu�er and boiling for 5 min. Thereaction products were analysed on a 10% acrylamide gel,stained with Coomassie blue (to control for the same
amounts of histone H1 protein added), dried and exposedto Kodak X-AR ®lm.
Immunoprecipitation
Protein extraction was done as described above and 1 mMokadaic acid was added at the lysis bu�er. Immunoprecipita-tion was performed using an anti-human p27kip1 (UpstateBiotechnology, Lake Placid, NY, USA) and the immuno-complexes were bound to protein A-agarose, centrifuged andthen washed four times with lysis bu�er. The reactionproducts were analysed on a 12% acrylamide gel, theresolved proteins were blotted to a nitrocellulose membraneby semidry electric transfer, and the membranes wereprocessed as described above using a rabbit anti-phospho-p27kip1 antibody (Zymed Laboratories, San Francisco, CA,USA).
AcknowledgementsWe thank Maurizia Caruso for critically reading themanuscript; and Alessandra Rinaldi for technical assis-tance. This work was supported by grants from CNR,Target Project on Biotechnology (to I D'Agnano) andProgetto Strategico `Oncologia' MURST-CNR (to AFelsani).
References
Bartkova J, Lukas J, Guldberg P, Alsner J, Kirkin AF,Zeuthen J and Bartek J. (1996). Cancer Res., 56, 5475 ±5483.
Burgess TL, Fisher EF, Ross SL, Bready JV, Qian YX,Bayewitch LA, Cohen AM, Herrera CJ, Hu SS andKramer TB. (1995). Proc. Natl. Acad. Sci. USA, 92, 4051 ±4055.
Castellano M and Parmiani G. (1999). Melanoma Res., 9,421 ± 432.
Citro G, D'Agnano I, Leonetti C, Perini R, Bucci B, Zon G,Calabretta B and Zupi G. (1998). Cancer Res., 58, 283 ±289.
Crooke ST. (1996). Antisense Nucleic Acid Drug Dev., 6,145 ± 147.
D'Agnano I, Antonelli A, Bucci B, Marcucci L, Petrinelli P,Ambra R, Zupi G and Elli R. (1998). Environ. Mol.Mutagen., 32, 56 ± 63.
Daksis JI, Lu RY, Facchini LM, Marhin WW and Penn LJ.(1994). Oncogene, 9, 3635 ± 3645.
Dang CV. (1999). Mol. Cell Biol., 19, 1 ± 11.Evan G and Littlewood T. (1998). Science, 281, 1317 ± 1322.Evan GI, Wyllie AH, Gilbert CS, Littlewood TD, Land H,
Brooks M, Waters CM, Penn LZ and Hancock DC.(1992). Cell, 69, 119 ± 128.
Ezhevsky SA, Toyoshima H, Hunter T and Scott DW.(1996). Mol. Biol. Cell, 7, 553 ± 564.
Fischer G, Kent SC, Joseph L, Green DR and Scott DW.(1994). J. Exp. Med., 179, 221 ± 228.
Hanson KD, Shichiri M, Follansbee MR and Sedivy JM.(1994). Mol. Cell Biol., 14, 5748 ± 5755.
Hoang AT, Cohen KJ, Barrett JF, Bergstrom DA and DangCV. (1994). Proc. Natl. Acad. Sci. USA, 91, 6875 ± 6879.
Jansen-Durr P, Meichle A, Steiner P, Pagano M, Finke K,Botz J, Wessbecher J, Draetta G and Eilers M. (1993).Proc. Natl. Acad. Sci. USA, 90, 3685 ± 3689.
Kalejta RF, Shenk T and Beavis AJ. (1997). Cytometry, 29,286 ± 291.
Katayose Y, Kim M, Rakkar AN, Li Z, Cowan KH and SethP. (1997). Cancer Res., 57, 5441 ± 5445.
Kimura S, Maekawa T, Hirakawa K, Murakami A and AbeT. (1995). Cancer Res., 55, 1379 ± 1384.
Krieg AM and Stein CA. (1995). Antisense Res. Dev., 5, 241.Leone G, DeGregori J, Sears R, Jakoi L and Nevins JR.
(1997). Nature, 387, 422 ± 426.Leonetti C, D'Agnano I, Lozupone F, Valentini A, Geiser T,
Zon G, Calabretta B, Citro GC and Zupi G. (1996). J.Natl. Cancer Inst., 88, 419 ± 429.
Lesnikowski ZJ, Jaworska M and Stec WJ. (1990). NucleicAcids Res., 18, 2109 ± 2115.
Maelandsmo GM, Florenes VA, Hovig E, Oyjord T,Engebraaten O, Holm R, Borresen AL and Fodstad O.(1996). Br. J. Cancer, 73, 909 ± 916.
Mateyak MK, Obaya AJ and Sedivy JM. (1999). Mol. CellBiol., 19, 4672 ± 4683.
O'Hagan RC, Ohh M, David G, de Alboran IM, Alt FW,Kaelin Jr WG and DePinho RA. (2000). Genes Dev., 14,2185 ± 2191.
Obaya AJ, Mateyak MK and Sedivy JM. (1999). Oncogene,18, 2934 ± 2941.
Perez-Roger I, Solomon DL, Sewing A and Land H. (1997).Oncogene, 14, 2373 ± 2381.
Ross DA and Wilson GD. (1998). Br. J. Surg., 85, 46 ± 51.Ryan KM and Birnie GD. (1996). Biochem. J., 314, 713 ±
721.Schlagbauer-Wadl H, Gri�oen M, van Elsas A, Schrier PI,
Pustelnik T, Eichler HG, Wol� K, Pehamberger H andJansen B. (1999). J. Invest. Dermatol., 112, 332 ± 336.
Schmidt EV. (1999). Oncogene, 18, 2988 ± 2996.
p27kip1-mediated apoptosis in melanoma cellsI D'Agnano et al
2824
Oncogene

Schraml P, Kononen J, Bubendorf L, Moch H, Bissig H,Nocito A, Mihatsch MJ, Kallioniemi OP and Sauter G.(1999). Clin. Cancer Res., 5, 1966 ± 1975.
Shea� RJ, Groudine M, Gordon M, Roberts JM andClurman BE. (1997). Genes Dev., 11, 1464 ± 1478.
Shi Y, Glynn JM, Guilbert LJ, Cotter TG, Bissonnette RPand Green DR. (1992). Science, 257, 212 ± 214.
Skorski T, Nieborowska-Skorska M, Campbell K, IozzoRV, Zon G, Darzynkiewicz Z and Calabretta B. (1995). J.Exp. Med., 182, 1645 ± 1653.
Stein CA. (1996). J. Natl. Cancer Inst., 88, 391 ± 393.Stein CA. (1998). Antisense Nucleic Acid Drug Dev., 8, 129 ±
132.
Van den Heuvel S and Harlow E. (1993). Science, 262, 2050 ±2054.
van Waardenburg RC, Meijer C, Burger H, Nooter K, deVries EG, Mulder NH and De Jong S. (1997). Int. J.Cancer, 73, 544 ± 550.
Vlach J, Hennecke S, Alevizopoulos K, Conti D and AmatiB. (1996). EMBO J., 15, 6595 ± 6604.
Vlach J, Hennecke S and Amati B. (1997). EMBO J., 16,5334 ± 5344.
Wang X, Gorospe M, Huang Y and Holbrook NJ. (1997).Oncogene, 15, 2991 ± 2997.
Wu M, Bellas RE, Shen J, Yang W and Sonenshein GE.(1999). J. Immunol., 163, 6530 ± 6535.
Oncogene
p27kip1-mediated apoptosis in melanoma cellsI D'Agnano et al
2825




















