
Nitric oxide deficiency determines global chromatin changes in Duchenne muscular dystrophy
Upload
independentCategory
view
7download
0
SchottNewbury-Ecob, Vinh Tran, Ian Young, Jean-Noel Trochu, Hervé Le Marec and Jean-JacquesFrançoise Le Bouffant, Claire Toquet, Estelle Roy, Lesley McGregor, Sally Ann Lynch, Ruth
Florence Kyndt, Jean-Pierre Gueffet, Vincent Probst, Philippe Jaafar, Antoine Legendre,Dystrophy
Mutations in the Gene Encoding Filamin A as a Cause for Familial Cardiac Valvular
Print ISSN: 0009-7322. Online ISSN: 1524-4539 Copyright © 2006 American Heart Association, Inc. All rights reserved.
is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Circulation doi: 10.1161/CIRCULATIONAHA.106.622621
2007;115:40-49; originally published online December 26, 2006;Circulation.
http://circ.ahajournals.org/content/115/1/40World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://circ.ahajournals.org/content/suppl/2006/12/15/CIRCULATIONAHA.106.622621.DC1.htmlData Supplement (unedited) at:
http://circ.ahajournals.org//subscriptions/
is online at: Circulation Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer this process is available in the
click Request Permissions in the middle column of the Web page under Services. Further information aboutOffice. Once the online version of the published article for which permission is being requested is located,
can be obtained via RightsLink, a service of the Copyright Clearance Center, not the EditorialCirculationin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on August 25, 2014http://circ.ahajournals.org/Downloaded from by guest on August 25, 2014http://circ.ahajournals.org/Downloaded from

Mutations in the Gene Encoding Filamin A as a Cause forFamilial Cardiac Valvular Dystrophy
Florence Kyndt, PharmD, PhD; Jean-Pierre Gueffet, MD; Vincent Probst, MD, PhD; Philippe Jaafar, MD;Antoine Legendre, MD; Françoise Le Bouffant, PhD; Claire Toquet, MD; Estelle Roy, BS;
Lesley McGregor, MD; Sally Ann Lynch, MD; Ruth Newbury-Ecob, MD; Vinh Tran, PhD;Ian Young, MD, PhD; Jean-Noel Trochu, MD, PhD; Hervé Le Marec, MD, PhD; Jean-Jacques Schott, PhD
Background—Myxomatous dystrophy of the cardiac valves affects �3% of the population and remains one of the mostcommon indications for valvular surgery. Familial inheritance has been demonstrated with autosomal and X-linkedtransmission, but no specific molecular abnormalities have been documented in isolated nonsyndromic forms. We haveinvestigated the genetic causes of X-linked myxomatous valvular dystrophy (XMVD) previously mapped tochromosome Xq28.
Methods and Results—A familial and genealogical survey led us to expand the size of a large, previously identified familyaffected by XMVD and to refine the XMVD locus to a 2.5-Mb region. A standard positional cloning approach identifieda P637Q mutation in the filamin A (FLNA) gene in all affected members. Two other missense mutations (G288R andV711D) and a 1944-bp genomic deletion coding for exons 16 to 19 in the FLNA gene were identified in 3 additional,smaller, unrelated families affected by valvular dystrophy, which demonstrates the responsibility of FLNA as a causeof XMVD. Among carriers of FLNA mutation, the penetrance of the disease was complete in men and incomplete inwomen. Female carriers could be mildly affected, and the severity of the disease was highly variable among mutationcarriers.
Conclusions—Our data demonstrate that FLNA is the first gene known to cause isolated nonsyndromic MVD. This is thefirst step to understanding the pathophysiological mechanisms of the disease and to defining pathways that may lead tovalvular dystrophy. Screening for FLNA mutations could be important for families affected by XMVD to provideadequate follow-up and genetic counseling. (Circulation. 2007;115:40-49.)
Key Words: genetics � mitral valve � regurgitation � valves
Myxomatous dystrophy of the cardiac valves is a heteroge-neous group of disorders that includes syndromic dis-
eases, such as Marfan syndrome, and isolated valvular diseases.Mitral valve prolapse, the most common form of this disease, ispresumed to affect �2% to 3% of the population and remainsone of the most common indications for valvular surgery.1
Editorial p 2Clinical Perspective p 49
Typical histopathologic features of myxomatous valvulardystrophy include fragmentation of collagenous bundleswithin the valve fibrosa and accumulation of proteoglycans,which produces excessive valve tissue that leads to billowing
of the valve leaflets, with or without prolapse and regurgita-tion. The causes of these histological changes remain un-known, however.
The diagnosis of myxomatous valvular dystrophy is pre-dominantly determined by echocardiography. There is noeffective treatment to prevent the progressive alteration of thevalves, and follow-up is limited to prevention of complica-tions, which include endocarditis and periodic evaluation ofthe severity of the valvular defects until surgery is required.This situation is largely due to the limited understanding ofthe pathophysiology of the valvular defects.2
Familial inheritance has been demonstrated, and a familialstudy is recommended when a case of mitral valve prolapse is
Received February 22, 2006; accepted September 5, 2006.From INSERM, U533, /l’Institut du thorax/ (F.K., J.-P.G., V.P., P.J., A.L., F.L.B., C.T., E.R., J.-N.T., H.L.M., J.-J.S.) Nantes, France; Universite de
Nantes, Faculte de medecine, Centre Hospitalier Universitaire de Nantes, Clinique Cardiologique (F.K., J.-P.G., V.P., P.J., A.L., F.L.B., C.T., E.R.,J.-N.T., H.L.M., J.-J.S.) Nantes, France; New Guy’s House (L.M.), Guy’s Hospital, London, UK; the National Centre for Medical Genetics (S.A.L.), OurLady’s Hospital for Sick Children, Dublin, Ireland; the Department of Clinical Genetics (R.N.-E.), Royal Hospital for Sick Children, Bristol, UK; theUnite Biotechnologie, Biocatalyse, Bioregulation (V.T.), Centre National de Recherche Scientifique, Unite Mixte de Recherche 6204, Faculte desSciences et Techniques, Nantes, France; and the Department of Clinical Genetics (I.Y.), Leicester Royal Infirmary, Leicester, UK.
The online-only Data Supplement, consisting of a table and a figure, is available at http://circ.ahajournals.org/cgi/content/full/CIRCULATIONAHA.106.622621/DC1.
Correspondence to Hervé Le Marec, MD, PhD, l’Institut du Thorax, Service de Cardiologie du CHU de Nantes, CHU de Nantes, Hôpital G and RLaennec, Bd Jacques Monod, 44093 Nantes Cedex, France. E-mail [email protected]
© 2007 American Heart Association, Inc.
Circulation is available at http://www.circulationaha.org DOI: 10.1161/CIRCULATIONAHA.106.622621
40
Genetics
by guest on August 25, 2014http://circ.ahajournals.org/Downloaded from

identified.3 Autosomal dominant transmission is the usualinheritance, with reduced penetrance and variable expressiv-ity.4 Three loci have been mapped to chromosomes 16p11–p12,5 11p15.46 and 13q31–32,7 but the underlying geneticdefects are not currently known. An X-linked recessive formwas originally described by Monteleone and Fagan8 in 1969and by Newbury-Ecob et al9 in 1993.
We previously clinically and genetically characterized alarge family (family 1a) affected by X-linked myxomatousvalvular dystrophy (XMVD) (OMIM 314400) and usedlinkage analysis to map the gene to an 8-cM interval onchromosome Xq28.10,11 A large familial and genealogicalsurvey led us to refine the locus of the XMVD gene, and astandard positional cloning approach identified a P637Qmutation in the filamin A (FLNA) gene in all affectedmembers of this family. Screening of additional, smaller,unrelated families with severe forms of valvular dystrophyallowed us to find 3 other mutations in this gene. The presentstudy demonstrates that FLNA is the first gene known to beresponsible for isolated nonsyndromic valvulopathy.
MethodsClinical EvaluationThe present study was conducted according to French guidelines forgenetic research and approved by the ethics committee of NantesUniversity Hospital.
Written informed consent was obtained from all participants.Clinical investigation included a review of medical history and aphysical examination, with particular attention given to the cardio-vascular system and any connective tissue diseases. The phenotypicassignment of family members was based on echocardiographicexamination. Family members were followed up with regular echo-cardiography, and the present data are those recorded during theirlast examination, with the exception of patients who underwentvalvular surgery. Transthoracic echocardiograms were recordedaccording to the criteria of the American Society of Echocardiogra-phy3 with a Sequoia C256 (Acuson Inc., Mountain View, Calif)equipped with a multifrequency probe (3.5 to 2.0 MHz). Measure-ments of mitral valves were performed on parasternal, long-axis,2-dimensional images without second harmonic.12 The length ofeach leaflet was determined immediately before valve closure. Thethickness of the free edge of the mitral leaflets was measured on aselected diastolic frame that clearly separated the mitral leaflets andchordae. Mitral valve prolapse was considered to exist when2-dimensional recordings in the parasternal long-axis view showedprotrusion of mitral leaflets into the left atrium, crossing the linebetween the annular hinge points, and when the coaptation point ofthe leaflets remained at or above the mitral annular plane duringsystole. Mitral regurgitation was estimated by using standard meth-ods, which include the proximal isovelocity surface area (PISA)analysis. Aortic regurgitation was considered to exist if an abnormaldiastolic flow that originated from aortic cusps was identified in theleft ventricular outflow tract. Tricuspid valve images were recordedin 4-chamber apical views, and the pulmonary valve was analyzed inthe high, left, parasternal, short-axis view.
Patients were defined as affected if the thickness of the free edgeof either or both mitral leaflets was �4 mm, with or without mitralvalve prolapse and mitral regurgitation on the echocardiogramsbecause of the inconsistency of mitral valve prolapse and mitralregurgitation in affected individuals. Because aortic valve dystrophyis difficult to assess by transthoracic echocardiography, we chose toquantify aortic regurgitation. Patients were also considered as af-fected in cases of mild to severe aortic regurgitation.
Genetic AnalysisFor molecular studies, DNA was isolated from peripheral bloodlymphocytes or paraffin-embedded valve tissue with standardmethods.
Linkage Analysis, Refined Mapping, and HaplotypeConstructionsFamily 1 was already linked to chromosome Xq28. To narrow thecandidate region, we selected 8 microsatellite markers (DXS998,DXS8091, DXS8069, DXS8061, DXS15, DXS1073, F8, andDXS1108) from the candidate interval on Xq28 for use in linkageanalysis in the expanded family. F8 is a dinucleotide marker locatedwithin factor VIII gene intron 13.13 We carried out 2-point linkageanalysis with the FASTLINK program in the easyLINKAGE soft-ware package (version 4.0; Medical University Clinic at the Univer-sity of Wurzburg, Wurzburg, Germany).14 For linkage calculations,we assumed X-linked inheritance with a disease-allele frequency of0.0001 and phenocopies at 2%. Penetrance was set at 100% for malefamily members and 70% for female family members. All familymembers were included in the analysis. Patients were defined asaffected if the thickness of the mitral leaflets was �4 mm and if mildto severe aortic regurgitation was present. Subjects with a mitralleaflet thickness �2 mm without aortic regurgitation were defined asunaffected, and subjects with a 2- to 4-mm thickness of mitralleaflets were considered phenotypically undetermined. To detectmore recombination events and refine the locus, 11 microsatellitemarkers (DXS8103, DXS1684, DXS10052, DXS10053, DXS10054,Afm308yh1, DXS10051, DXS10049, Afm082xa5, GABRA3, andDXS10047) were genotyped in the recombinant individuals todetermine the centromeric boundary.15 Two intragenic microsatellitemarkers, GAB3 and F8, were used to determine the telomericboundary.
Analysis of Candidate Genes and FLNAMutation AnalysisCandidate genes in the Xq28 interval were screened for mutations bysequence analysis in 1 affected patient of family 1a. Coding exonsand short flanking intronic sequences were amplified by polymerasechain reaction (PCR), and excess primer was removed from theamplified fragments with the use of exoSAP (Amersham Bio-sciences, Piscataway, NJ) and sequenced with a dye-terminatorcycle-sequencing system (ABI PRISM 377, Perkin-Elmer AppliedBiosystems, Foster City, Calif). The coding sequence of FLNA(exons 2 to 48)16 was amplified from genomic DNA with the use ofprimers as previously described.15
Variants of FLNA identified by sequence analysis were confirmedby restriction-enzyme digestion with HaeIII (P637Q) and Sau96I(G288R): The required exons were amplified by PCR, digested withrestriction enzyme, and size fractionated on an 8% acrylamide gel. Inthe 2 cases, the variant abolished 1 restriction site. The V711Dmutation was confirmed by derived cleaved amplified polymorphicsequencing.17 To assess the frequency of the mutations, we usedsequence analysis, restriction-enzyme digestion, or derived cleavedamplified polymorphic sequencing with DNA from family membersand from 500 control chromosomes of European, African, or Asianorigin.
Model BuildingTo study the structural consequences of the identified mutations, weconstructed a 3-dimensional model of human filamin repeats with thecrystal coordinates of Dictyostelium filamin deposited in ProteinData Bank (identification: 1WLH).18 The automated homologymodel construction was performed by the protein structure modelingprogram INSIGHTII (Accelrys, Cambridge, UK).
The authors had full access to and take full responsibility for theintegrity of the data. All authors have read and agree to themanuscript as written.
ResultsClinical EvaluationFamily 1a (Figure 1A) is a previously reported white Frenchfamily10,11 that is affected with XMVD and currently consists
Kyndt et al Filamin A in Myxomatous Valvular Dystrophy 41
by guest on August 25, 2014http://circ.ahajournals.org/Downloaded from
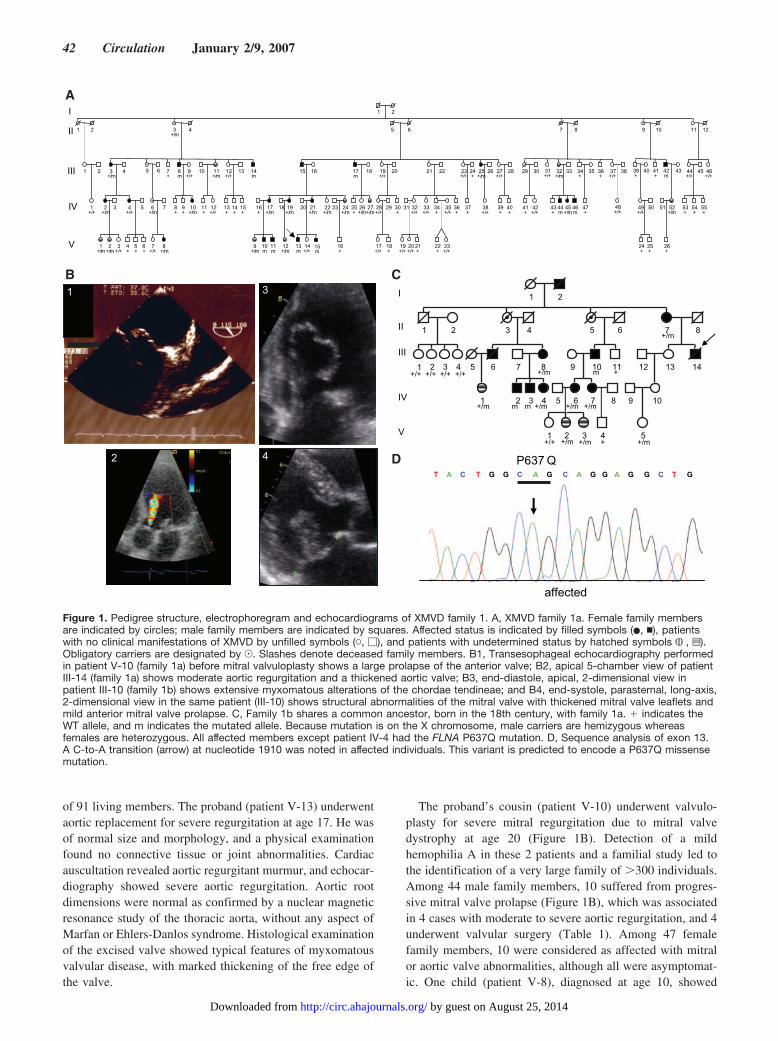
of 91 living members. The proband (patient V-13) underwentaortic replacement for severe regurgitation at age 17. He wasof normal size and morphology, and a physical examinationfound no connective tissue or joint abnormalities. Cardiacauscultation revealed aortic regurgitant murmur, and echocar-diography showed severe aortic regurgitation. Aortic rootdimensions were normal as confirmed by a nuclear magneticresonance study of the thoracic aorta, without any aspect ofMarfan or Ehlers-Danlos syndrome. Histological examinationof the excised valve showed typical features of myxomatousvalvular disease, with marked thickening of the free edge ofthe valve.
The proband’s cousin (patient V-10) underwent valvulo-plasty for severe mitral regurgitation due to mitral valvedystrophy at age 20 (Figure 1B). Detection of a mildhemophilia A in these 2 patients and a familial study led tothe identification of a very large family of �300 individuals.Among 44 male family members, 10 suffered from progres-sive mitral valve prolapse (Figure 1B), which was associatedin 4 cases with moderate to severe aortic regurgitation, and 4underwent valvular surgery (Table 1). Among 47 femalefamily members, 10 were considered as affected with mitralor aortic valve abnormalities, although all were asymptomat-ic. One child (patient V-8), diagnosed at age 10, showed
affected
P637 QT A C T G G C A G C A G G A G G C T G
3
17+/m
16+
21+/m
2019+/m
22+/m
24+/m
23
1615 17m
21 27+/+
19+/+
18
26+/m
25+
27+/m
20
28+/+
30+
29
22
32+/+
33+/+
34+
36+
28
39+
40+
9+/m
10m
11m
13m
15m
14+/+
16+
5 6 7 8
32+/m
33
46m
3+/m
4
8m
9+/+
1+/m
2+/m
3+/+
4+
5+
6+
2+/m
3 4+/+
5
3+/m
4
6+/m
5 6 7+
8+
9+
10+/m
12+/+
11+
11+/m
10
13+
14+
15+
12+/+
13 14m
41+
42+/+
29 30 31+/+
43+
44m
45+/m
47+
34+
35 36+
48+/+
37+/+
38
9 10
24+
25+
49+/+
50 52+/m
11 12
53+
54+
55+
44+/+
45 46+/+
17+/+
18+
31
19+/+
20+/+
21+
35+/+
22+
23+/+
1 2
23+/+
24+
26
37+
38+/+
18 51
26+
12+/m
25+/m
1 2
1 2
1+/+
7
7+/+
8+/m
39+
41+
42m
40 43
I
II
III
IV
V
21
1 2 4 5 6 7+/m
8
1+/+
2+/+
3+/+
4+/+
65 8+/m
10m
11+
13 14
1+/m
102m
3m
4+/m
6+/m
7+/m
5+/m
7 9 12
9
3
5 8
3+/m
2+/m
1+/+
4+
I
II
III
IV
V
42
1
A
B C
D
Figure 1. Pedigree structure, electrophoregram and echocardiograms of XMVD family 1. A, XMVD family 1a. Female family membersare indicated by circles; male family members are indicated by squares. Affected status is indicated by filled symbols (�, �), patientswith no clinical manifestations of XMVD by unfilled symbols (�, �), and patients with undetermined status by hatched symbols , z).Obligatory carriers are designated by J. Slashes denote deceased family members. B1, Transesophageal echocardiography performedin patient V-10 (family 1a) before mitral valvuloplasty shows a large prolapse of the anterior valve; B2, apical 5-chamber view of patientIII-14 (family 1a) shows moderate aortic regurgitation and a thickened aortic valve; B3, end-diastole, apical, 2-dimensional view inpatient III-10 (family 1b) shows extensive myxomatous alterations of the chordae tendineae; and B4, end-systole, parasternal, long-axis,2-dimensional view in the same patient (III-10) shows structural abnormalities of the mitral valve with thickened mitral valve leaflets andmild anterior mitral valve prolapse. C, Family 1b shares a common ancestor, born in the 18th century, with family 1a. � indicates theWT allele, and m indicates the mutated allele. Because mutation is on the X chromosome, male carriers are hemizygous whereasfemales are heterozygous. All affected members except patient IV-4 had the FLNA P637Q mutation. D, Sequence analysis of exon 13.A C-to-A transition (arrow) at nucleotide 1910 was noted in affected individuals. This variant is predicted to encode a P637Q missensemutation.
42 Circulation January 2/9, 2007
by guest on August 25, 2014http://circ.ahajournals.org/Downloaded from
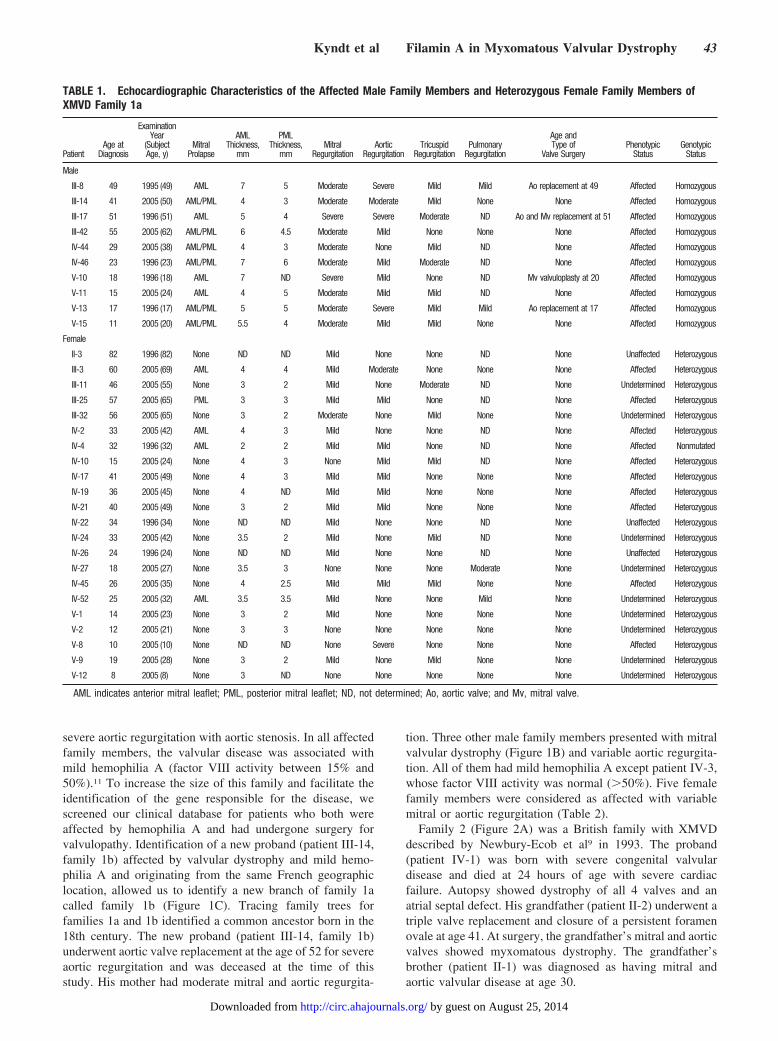
severe aortic regurgitation with aortic stenosis. In all affectedfamily members, the valvular disease was associated withmild hemophilia A (factor VIII activity between 15% and50%).11 To increase the size of this family and facilitate theidentification of the gene responsible for the disease, wescreened our clinical database for patients who both wereaffected by hemophilia A and had undergone surgery forvalvulopathy. Identification of a new proband (patient III-14,family 1b) affected by valvular dystrophy and mild hemo-philia A and originating from the same French geographiclocation, allowed us to identify a new branch of family 1acalled family 1b (Figure 1C). Tracing family trees forfamilies 1a and 1b identified a common ancestor born in the18th century. The new proband (patient III-14, family 1b)underwent aortic valve replacement at the age of 52 for severeaortic regurgitation and was deceased at the time of thisstudy. His mother had moderate mitral and aortic regurgita-
tion. Three other male family members presented with mitralvalvular dystrophy (Figure 1B) and variable aortic regurgita-tion. All of them had mild hemophilia A except patient IV-3,whose factor VIII activity was normal (�50%). Five femalefamily members were considered as affected with variablemitral or aortic regurgitation (Table 2).
Family 2 (Figure 2A) was a British family with XMVDdescribed by Newbury-Ecob et al9 in 1993. The proband(patient IV-1) was born with severe congenital valvulardisease and died at 24 hours of age with severe cardiacfailure. Autopsy showed dystrophy of all 4 valves and anatrial septal defect. His grandfather (patient II-2) underwent atriple valve replacement and closure of a persistent foramenovale at age 41. At surgery, the grandfather’s mitral and aorticvalves showed myxomatous dystrophy. The grandfather’sbrother (patient II-1) was diagnosed as having mitral andaortic valvular disease at age 30.
TABLE 1. Echocardiographic Characteristics of the Affected Male Family Members and Heterozygous Female Family Members ofXMVD Family 1a
PatientAge at
Diagnosis
ExaminationYear
(SubjectAge, y)
MitralProlapse
AMLThickness,
mm
PMLThickness,
mmMitral
RegurgitationAortic
RegurgitationTricuspid
RegurgitationPulmonary
Regurgitation
Age andType of
Valve SurgeryPhenotypic
StatusGenotypic
Status
Male
III-8 49 1995 (49) AML 7 5 Moderate Severe Mild Mild Ao replacement at 49 Affected Homozygous
III-14 41 2005 (50) AML/PML 4 3 Moderate Moderate Mild None None Affected Homozygous
III-17 51 1996 (51) AML 5 4 Severe Severe Moderate ND Ao and Mv replacement at 51 Affected Homozygous
III-42 55 2005 (62) AML/PML 6 4.5 Moderate Mild None None None Affected Homozygous
IV-44 29 2005 (38) AML/PML 4 3 Moderate None Mild ND None Affected Homozygous
IV-46 23 1996 (23) AML/PML 7 6 Moderate Mild Moderate ND None Affected Homozygous
V-10 18 1996 (18) AML 7 ND Severe Mild None ND Mv valvuloplasty at 20 Affected Homozygous
V-11 15 2005 (24) AML 4 5 Moderate Mild Mild ND None Affected Homozygous
V-13 17 1996 (17) AML/PML 5 5 Moderate Severe Mild Mild Ao replacement at 17 Affected Homozygous
V-15 11 2005 (20) AML/PML 5.5 4 Moderate Mild Mild None None Affected Homozygous
Female
II-3 82 1996 (82) None ND ND Mild None None ND None Unaffected Heterozygous
III-3 60 2005 (69) AML 4 4 Mild Moderate None None None Affected Heterozygous
III-11 46 2005 (55) None 3 2 Mild None Moderate ND None Undetermined Heterozygous
III-25 57 2005 (65) PML 3 3 Mild Mild None ND None Affected Heterozygous
III-32 56 2005 (65) None 3 2 Moderate None Mild None None Undetermined Heterozygous
IV-2 33 2005 (42) AML 4 3 Mild None None ND None Affected Heterozygous
IV-4 32 1996 (32) AML 2 2 Mild Mild None ND None Affected Nonmutated
IV-10 15 2005 (24) None 4 3 None Mild Mild ND None Affected Heterozygous
IV-17 41 2005 (49) None 4 3 Mild Mild None None None Affected Heterozygous
IV-19 36 2005 (45) None 4 ND Mild Mild None None None Affected Heterozygous
IV-21 40 2005 (49) None 3 2 Mild Mild None None None Affected Heterozygous
IV-22 34 1996 (34) None ND ND Mild None None ND None Unaffected Heterozygous
IV-24 33 2005 (42) None 3.5 2 Mild None Mild ND None Undetermined Heterozygous
IV-26 24 1996 (24) None ND ND Mild None None ND None Unaffected Heterozygous
IV-27 18 2005 (27) None 3.5 3 None None None Moderate None Undetermined Heterozygous
IV-45 26 2005 (35) None 4 2.5 Mild Mild Mild None None Affected Heterozygous
IV-52 25 2005 (32) AML 3.5 3.5 Mild None None Mild None Undetermined Heterozygous
V-1 14 2005 (23) None 3 2 Mild None None None None Undetermined Heterozygous
V-2 12 2005 (21) None 3 3 None None None None None Undetermined Heterozygous
V-8 10 2005 (10) None ND ND None Severe None None None Affected Heterozygous
V-9 19 2005 (28) None 3 2 Mild None Mild None None Undetermined Heterozygous
V-12 8 2005 (8) None 3 ND None None None None None Undetermined Heterozygous
AML indicates anterior mitral leaflet; PML, posterior mitral leaflet; ND, not determined; Ao, aortic valve; and Mv, mitral valve.
Kyndt et al Filamin A in Myxomatous Valvular Dystrophy 43
by guest on August 25, 2014http://circ.ahajournals.org/Downloaded from
A male infant was the first child of healthy black Africanparents (family 3) (Figure 2B). He was diagnosed antenatallywith abnormally thick cardiac valves by ultrasound and fetalechocardiography and was born at 38 weeks in good health.Postnatal echocardiography confirmed moderate tricuspidincompetence, trivial mitral and pulmonary incompetence,and mild aortic incompetence. All valves were thickened anddystrophic. At 4 months of age, his growth and developmen-tal assessment were within normal limits and he showed nosigns of cardiac failure. An echocardiogram showed excellentventricular function. The mitral valve remained dystrophicwithout evidence of regurgitation and with only very mildaortic regurgitation. His mother was examined clinically andshowed no evidence of cardiac involvement.
Family 4 (Figure 2C) was of Hong Kong Chinese origin.The 2 boys, 12 and 4 years old, had both mitral and aorticdystrophy. A heart murmur was identified in patient II-1 atage 4 months on a routine check. Subsequent echocardiogra-phy revealed he had polyvalvular disease with myxomatousthickening of the mitral tricuspid and aortic valves. He hadsignificant mitral and tricuspid regurgitation with mild aorticregurgitation. Patient II-2 was identified because of hisbrother’s history. He was shown to have polyvalvular diseasewith mitral incompetence and stenosis, tricuspid regurgita-tion, and mild aortic regurgitation. Their mother had anessentially normal echocardiogram at age 38 with mild aorticand pulmonary incompetence.
All 4 families presented no clinically apparent extracar-diac abnormalities, no dysmorphic features, and no epilep-tic seizures.
Linkage Analysis and Refined Mapping to Narrowthe LocusA significant linkage (Zmax�8.6 at ��0 for DXS1108) wasobtained for family 1 (Data Supplement Table). To refine theXMVD locus, additional markers were genotyped. Recombi-nation at DXS10049 for unaffected male patient IV-54 of
family 1a (Data Supplement Figure) set the centromericboundary for the locus. Analysis of markers from members offamily 1b revealed the same disease haplotype, with theexception of patient IV-3. This person, affected by valvulardystrophy but with normal coagulation factor activity, har-bored recombination at microsatellite marker GAB3 (DataSupplement Figure), which set the telomeric boundary for thelinked region and showed that valvulopathy and hemophiliawere transmitted as independent traits. Therefore, the diseasegene was located between DXS10049 and GAB3, spanned 2.5Mb, and excluded the factor VIII gene.
Mutation IdentificationAfter exclusion of candidate genes in the disease interval, eg,BGN, G4.5, ZNF185, CETN2, DUSP9, STK23, SSR4,RENBP, MGC29729, and several predicted genes, we exam-ined FLNA with a direct sequencing approach. The gene iscomposed of 48 exons that span �26 Kb immediately distalto the emerin gene. Mutational analysis of all coding regionsof FLNA in affected members of family 1 (families 1a and 1bcombined) revealed that all have a C-to-A transition atnucleotide 1910 in exon 13 (NM_001456: c.1910C�A),which predicts a missense mutation–substitution of a proline(P) for a glutamine (Q) at amino acid 637 (P637Q) (Figure1D). In family 1, a total of 13 affected male family membersand 30 female family members are mutation carriers. Oneclinically affected woman (patient IV-4) did not inherit themutation and appeared to be a phenocopy. A G-to-A transi-tion at nucleotide 862 in exon 5 (NM_001456: c.862G�A),which predicts a missense mutation of a glycine (G) for anarginine (R) (G288R), was found in the affected members offamily 2 (Figure 2A). A T-to-A transition at nucleotide 2132in exon 14 (NM_001456: c.2132T�A), which predicts amissense mutation of a valine (V) for an aspartic acid (D) atamino acid 711 (V711D), was found in the affected memberof family 3 (Figure 2B). These sequence variations were notfound in unaffected relatives of either family or in 500 control
TABLE 2. Echocardiographic Characteristics of the Affected Male Family Members and Heterozygous Female Family Members ofXMVD Family 1b
PatientAge at
Diagnosis
ExaminationYear (Subject
Age, y)Mitral
Prolapse
AMLThickness,
mm
PMLThickness,
mmMitral
RegurgitationAortic
RegurgitationTricuspid
RegurgitationPulmonary
Regurgitation
Age andType of
Valve SurgeryPhenotypic
StatusGenotypic
Status
Male
III-10 49 2005 (55) AML/PML 5 5 Moderate Moderate Mild Mild None Affected Homozygous
III-14 50 1982 (50) None ND ND Moderate Severe None None Ao replacement at 52 Affected Unknown
IV-2 24 2005 (30) None 5 4 Moderate Mild Mild Mild None Affected Homozygous
IV-3 21 2005 (27) AML/PML 5 5 Moderate Moderate Mild None None Affected Homozygous
Female
II-7 86 1992 (86) ND ND ND Moderate Moderate ND ND None Affected Heterozygous
III-8 50 2005 (56) None 3 3 Moderate Moderate None None None Affected Heterozygous
IV-1 29 2005 (34) None 3 3 Mild None Mild None None Undetermined Heterozygous
IV-4 18 2005 (24) None 4.5 4 Moderate None None None None Affected Heterozygous
IV-6 28 2005 (34) None 4 4 Mild Mild Mild ND None Affected Heterozygous
IV-7 26 2005 (32) None 3 3 None Mild Mild Mild None Affected Heterozygous
V-2 6 2005 (6) None ND ND None None Mild None None Undetermined Heterozygous
V-3 7 2005 (7) None 3 3 Mild None ND None None Undetermined Heterozygous
V-5 23 2000 (23) None ND ND Mild None None ND None Unaffected Heterozygous
Abbreviations as in Table 1.
44 Circulation January 2/9, 2007
by guest on August 25, 2014http://circ.ahajournals.org/Downloaded from
chromosomes of white or African origin. The mutations wereconfirmed by testing for loss of a HaeIII (P637Q) and Sau96I(G288R) restriction sites and derived cleaved amplifiedpolymorphic sequence (V711D), respectively (data notshown).
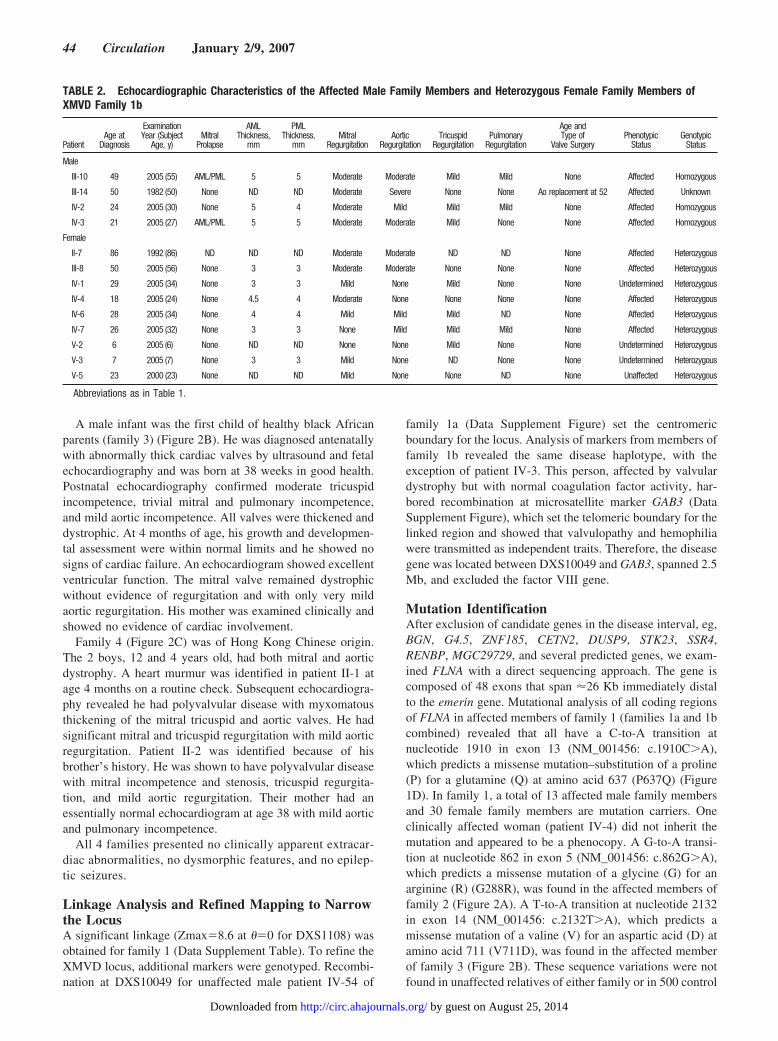
Finally, we identified a 1944-bp genomic deletion codingfor exons 16 to 19, which corresponds to the 546-bp codingsequence (NT_025965.13:g.942144_944086del1944insTG)that predicts an in-frame deletion of 182 residues (from V761to Q943) in 2 boys (family 4) diagnosed with plurivalvulardystrophy. The deletion was visualized by PCR analysis andsequencing reaction from intron 15 to 19 to show thataffected individuals had a smaller PCR product that corre-sponds to a deletion of 1944 bp (Figure 2C). This deletionwas not found in 200 control chromosomes, which included100 Asian chromosomes.
In addition to the mutations identified in FLNA, otherknown single-nucleotide polymorphisms were identified inthe patients. These single-nucleotide polymorphisms oc-curred at frequencies comparable to those listed in thepublic National Center for Biotechnology Information(NCBI) database, some of them resulting in amino acidsubstitutions. No additional disease-associated variantswere observed. Screening of 3 other families with potential
X-linked valvular dystrophy did not find any mutation inFLNA.
Genotype-to-Phenotype Relations in Family 1Genotype-to-Phenotype Relations in MaleFamily MembersIn family 1, 13 male subjects were carriers of the P637Qmutation (Table 1). All male carriers had mitral valvulopathy,leading to mitral valve replacement or mitral valvuloplasty in2 cases. Age for mitral surgery ranged from 20 to 51 years.All but 1 male carrier had mitral valve prolapse, found on theanterior valve in 4 patients (Figure 1B) and on both valves in8 patients. None had an isolated posterior mitral valveprolapse. The thickness of the mitral valve was 5.3�1.1 mmfor the anterior valve and 4.4�0.9 mm for the posterior valve.All but 1 male carrier also had aortic valve regurgitation. Thisregurgitation was considered mild in 6 cases, moderate in 3cases, and severe in 3 cases that led to aortic valve replace-ment. There was no abnormality of the aortic root, and allaortic valves were tricuspid. Age for aortic valve surgeryranged from 17 to 52 years. Mild to moderate tricuspid valveregurgitation was found in 11 male carriers, and mild pulmo-nary regurgitation was found in 4 male carriers. However,there was no surgery of the tricuspid or pulmonary valves inthe family.
3216bp
1272bp
C
A B
I
II
+/m
m m
intron 15 intron 19 exon 20
1944bpdeletion
ex16 ex17 ex18 ex19
161bp 91bp 170bp124bp1
1
2
2
1 2
1 2 3+/+
1+
2+/m
3
1m
I
II
III
IV
C C C G T G C C T A C G/A G G C C A G G T G A G G G A
G288R
Ind III-2
I
II
+/m
m
1 2
V711DG T C C A A G A C C A G G T A G
affected
1
affected
Figure 2. Pedigree structures and electrophoregrams of three other XMVD families. A, Pedigree structure of family 2 shows that all affectedmembers had the mutation G288R (m) in FLNA, and sequence analysis of exon 5 shows a G-to-A transition (arrow) at nucleotide 862 inaffected individuals. This variant is predicted to encode a G288R missense mutation in FLNA. B, Pedigree structure of family 3 and sequenceanalysis of exon 14 show a T-to-A transition at nucleotide 2132 that predicts a V711D missense mutation in affected individual. C, Pedigreestructure of family 4. The deletion was visualized by PCR analysis from intron 15 to intron 19 of the FLNA genomic sequence. The father(patient I-1) produced 1 fragment of 3216 bp, the heterozygous mother (patient I-2) produced 2 fragments of 3216 bp and 1272 bp, and the2 affected boys (patients II-1 and II-2) produced 1 fragment of 1272 bp, which corresponds to the deletion of 1944 bp.
Kyndt et al Filamin A in Myxomatous Valvular Dystrophy 45
by guest on August 25, 2014http://circ.ahajournals.org/Downloaded from

Genotype-to-Phenotype Relations in FemaleFamily MembersNone of the 30 heterozygous women was symptomatic, andnone underwent valvular surgery. Among female carriers ofthe P637Q mutation, 14 were considered affected, 12 wereconsidered undetermined because of minor valve disease, and4 were considered unaffected. Mitral valve prolapse wasfound in 4 cases; these involved the anterior valve in 3 casesand the posterior valve in 1 case. Among heterozygouswomen, 19 had mild mitral regurgitation and 4 had moderatemitral regurgitation. Mitral valve thickness was 3.3�0.5 mmfor the anterior valve and 2.8�0.7 mm for the posterior valve.The anterior mitral valve (P�0.001) and the posterior mitralvalve (P�0.001) were thicker in male than in female familymembers. Eight heterozygous women had mild aortic valveregurgitation, 3 had moderate aortic valve regurgitation, and1 had severe aortic valve regurgitation. Nine heterozygouswomen had mild tricuspid valve regurgitation, and 1 hadmoderate tricuspid valve regurgitation. Two heterozygouswomen had mild pulmonary valve regurgitation, and 1 hadmoderate pulmonary valve regurgitation.
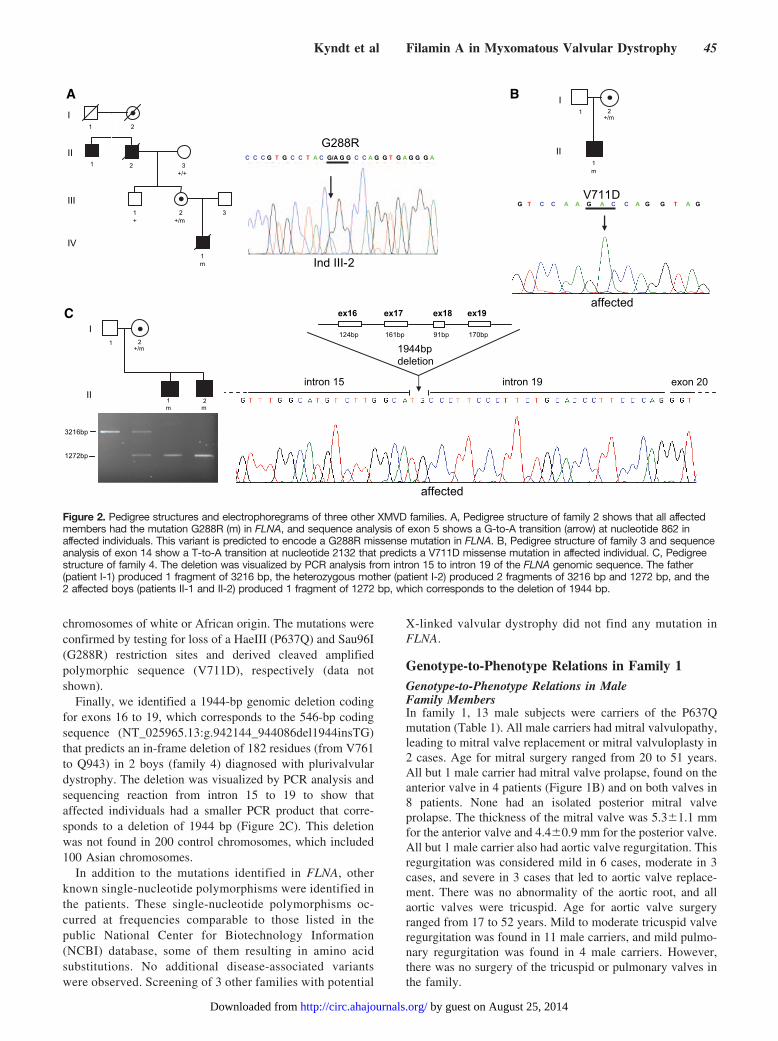
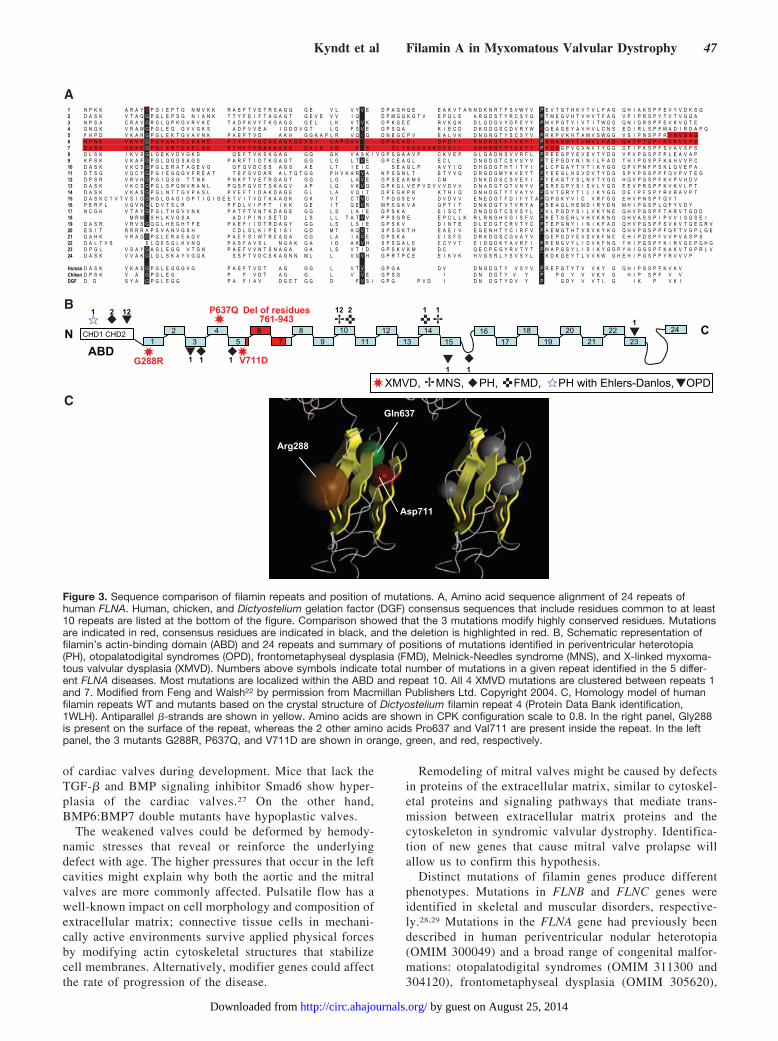
Structural Aspects of FLNA Mutations in XMVDSequence comparisons revealed that all 3 missense mutationsmodify highly conserved residues and affect the repeatconsensus sequence, which includes residues that are com-mon to at least 10 repeats and are highly conserved across awide range of vertebrate filamins, as compared with consen-sus sequences derived from the repeating backbones ofchicken filamin and Dictyostelium gelation factor (Figure3A).18–21
The G288R, P637Q, and V711D mutations are locatedwithin the first, the fourth, and the fifth repeat, respectively,and the truncated protein corresponds to a smaller protein thatlacks repeats 5 to 7 (Figure 3B).
The predicted 3-dimensional model of human filamin repeatsshows remarkable similarity to Dictyostelium gelation factor inthe overall 3-dimensional fold. The complete structure of filaminrepeat consists of 7 antiparallel �-strands arranged in 2 �-sheetsof 3 and 4 �-strands (Figure 3C). Modeling the 3-dimensionalstructure of the filamin repeat showed that the Gly288 residue islocated on the external face of the repeat, whereas the Pro637and Val711 residues are internal. For all mutants, the replace-ment of a nonpolar residue with a polar residue (basic, un-charged, and acidic for G288R, P637Q, and V711D, respec-tively) could account for an increase in polarity. It can beexpected, therefore, that these mutations would cause a signifi-cant change in the structural conformation of the �-strands.
All 3 substitutions, located in the same region andpredicted to impact the antiparallel �-strands organizationof the protein, could result in impaired partner binding.
DiscussionIn this study, we describe the identification of FLNA as thefirst gene responsible for a nonsyndromic valvulardystrophy.
The identification of this genetic defect transmitted witha X-linked recessive pattern was facilitated by a genealog-ical and geographic approach to the disease. Screening of
our hospital files coupled with genealogical analysis ofpatients affected with myxomatous valvular dystrophy ledto the identification of 2 kindreds from the same geo-graphic location who have inherited the same mutation inthe FLNA gene from a common ancestor born in the 18thcentury.
We have several lines of evidence to show that theP637Q mutation in the FLNA gene is the gene responsiblefor the valvular defect in family 1. Linkage analysis thatincludes all male and female patients gave a significantpositive lod score, and all affected males are carriers of themutation. Furthermore, screening of additional unrelatedfamilies affected by valvular dystrophy potentially trans-mitted with an X-linked recessive pattern identified 3 othermutations (G288R, V711D, and a 182–amino acid dele-tion) in the FLNA gene. None of these mutations was foundin 500 control chromosomes. All missense mutations arehighly conserved across filamin repeats and species.
Within the families with X-linked valvular dystrophy,the disease was inherited with complete penetrance in malefamily members and incomplete penetrance in femalefamily members, with variable degrees of expression,consistent with different X-inactivation patterns. Among13 male carriers of the P637Q mutation, 12 had typicalmitral valve prolapse characterized by thicker leafletsassociated with mild to severe aortic regurgitation, and 11also had tricuspid regurgitation. Among 30 female carriers,14 were considered affected with aortic or mitral valveabnormalities. Female mutation carriers had thicker mitralleaflets than normal women but were less severely affectedthan male patients (no valvular surgery).
Age for valvular surgery ranged from 17 to 52 years infamily 1. Age at diagnosis was also highly variable. Withinfamily 1, male family members were diagnosed betweenage 11 and 55 years. Within family 2, 2 patients (II-1 andII-2) were diagnosed with progressive polyvalvular dys-trophy between age 25 and 30 years, and 1 patient (IV-1)was diagnosed at birth. In family 3, polyvalvular disorderwas diagnosed antenatally, and it was diagnosed at age 4and 12 years in family 4.
Filamin A is a ubiquitous phosphoprotein that X-linksactin filaments and links the actin cytoskeleton to theplasma membrane by interacting with both actin andmembrane proteins such as �-integrins.23,24 Filamin con-sists of an actin-binding domain at the N-terminus and 24homologous repeats that correspond to the rod backbone ofthe protein.16,19 Each repeat consists of 7 antiparallel�-strands that are arranged in 2 �-sheets. Filamins exist invivo as dimers mediated by interactions betweenC-terminal sequences.
In addition to its role as a structural component of thecytoskeleton, filamin A has also been implicated in regu-lating many cellular signaling pathways. Filamin A maycontribute to the development of myxomatous changes ofthe cardiac valves by regulation of transforming growthfactor-� (TGF-�) signaling through its interaction withSmads activated by TGF-� receptors.25,26 Defective sig-naling cascades that involve members of the TGF-�superfamily have been described in impaired remodeling
46 Circulation January 2/9, 2007
by guest on August 25, 2014http://circ.ahajournals.org/Downloaded from
of cardiac valves during development. Mice that lack theTGF-� and BMP signaling inhibitor Smad6 show hyper-plasia of the cardiac valves.27 On the other hand,BMP6:BMP7 double mutants have hypoplastic valves.
The weakened valves could be deformed by hemody-namic stresses that reveal or reinforce the underlyingdefect with age. The higher pressures that occur in the leftcavities might explain why both the aortic and the mitralvalves are more commonly affected. Pulsatile flow has awell-known impact on cell morphology and composition ofextracellular matrix; connective tissue cells in mechani-cally active environments survive applied physical forcesby modifying actin cytoskeletal structures that stabilizecell membranes. Alternatively, modifier genes could affectthe rate of progression of the disease.
Remodeling of mitral valves might be caused by defectsin proteins of the extracellular matrix, similar to cytoskel-etal proteins and signaling pathways that mediate trans-mission between extracellular matrix proteins and thecytoskeleton in syndromic valvular dystrophy. Identifica-tion of new genes that cause mitral valve prolapse willallow us to confirm this hypothesis.
Distinct mutations of filamin genes produce differentphenotypes. Mutations in FLNB and FLNC genes wereidentified in skeletal and muscular disorders, respective-ly.28,29 Mutations in the FLNA gene had previously beendescribed in human periventricular nodular heterotopia(OMIM 300049) and a broad range of congenital malfor-mations: otopalatodigital syndromes (OMIM 311300 and304120), frontometaphyseal dysplasia (OMIM 305620),
1 N P K K A R A Y G P G I E P T G N M V K K R A E F T V E T R S A G Q G E V L V Y V E D P A G H Q E E A K V T A N N D K N R T F S V W Y V P E V T G T H K V T V L F A G Q H I A K S P F E V Y V D K S Q2 D A S K V T A Q G P G L E P S G N I A N K T T Y F E I F T A G A G T G E V E V V I Q D P M G Q K G T V E P Q L E A R G D S T Y R C S Y Q P T M E G V H T V H V T F A G V P I P R S P Y T V T V G Q A3 N P S A C R A V G R G L Q P K G V R V K E T A D F K V Y T K G A G S G E L L K V T V K G P K G E E R V K Q K D L G D G V Y G F E Y Y P M V P G T Y I V T I T W G G Q N I G R S P F E V K V G T E4 G N Q K V R A W G P G L E G G V V G K S A D F V V E A I G D D V G T L G F S V E G P S Q A K I E C D D K G D G S C D V R Y W P Q E A G E Y A V H V L C N S E D I R L S P F M A D I R D A P Q5 F H P D V K A R G P G L E K T G V A V N K P A E F T V D A K H G G K A P L R V Q V Q D N E G C P V E A L V K D N G N G T Y S C S Y V P R K P V K H T A M V S W G G V S I P N S P F R V N V G A G6 H P N K V K V Y G P G V A K T G L K A H E P T Y F T V D C A E A G K G D V S I C A P G V V G P A E A D I D F D I I R N D N D T F T V K Y T P R G A G S Y T I M V L F A D Q A T P T S P I R V K V E P S7 D A S K V K A E G P G L S R T G V E L G K P T H F T V N A K A A G K G K L D V Q F S G L T K G D A V R D V D I I D H H D N T Y T V K Y T P V Q Q G P V G V N V T Y G G D P I P K S P F S V A V S P S8 D L S K I K V S G L G E K V D V G K D Q E F T V K S K G A G G Q G K V A S K I V G P S G A A V P C K V E P G L G A D N S V V R F L P R E E G P Y E V E V T Y D G V P V P G S P F P L E A V A P9 K P S K V K A F G P G L Q G G S A G S P A R F T I D T K G A G T G G L G L T V E G P C E A Q L E C L D N G D G T C S V S Y V P T E P G D Y N I N I L F A D T H I P G S P F K A H V V P C10 D A S K V K C S G P G L E R A T A G E V G Q F Q V D C S S A G S A E L T I E I C S E A G L P A V Y I Q D H G D G T H T I T Y I P L C P G A Y T V T I K Y G G Q P V P N F P S K L Q V E P A11 D T S G V Q C Y G P G I E G Q G V F R E A T T E F S V D A R A L T Q T G G P H V K A R V A N P S G N L T E T Y V Q D R G D G M Y K V E Y T P Y E E G L H S V D V T Y D G S P V P S S P P F Q V P V T E G12 D P S R V R V H G P G I Q S G T T N K P N K F T V E T R G A G T G G L G L A V E G P S E A K M S C M D N K D G S C S V E Y I P Y E A G T Y S L N V T Y G G H Q V P G S P F K V P V H D V13 D A S K V K C S G P G L S P G M V R A N L P Q S F Q V D T S K A G V A P L Q V K V Q G P K G L V E P V D V V V D V V D N A D G T Q T V N Y V P S R E G P Y S I S V L Y G D E E V P R S P F K V K V L P T14 D A S K V K A S G P G L N T T G V P A S L P V E F T I D A K D A G E G L L A V Q I T D P E G K P K K T H I Q D N H D G T Y T V A Y V P D V T G R Y T I L I K Y G G D E I P F S P Y R V R A V P T15 D A S K C T V T V S I G G H G L G A G I G P T I Q I G E E T V I T V D T K A A G K G K V T C T V C T P D G S E V D V D V V E N E D G T F D I F Y T A P Q P G K Y V I C V R F G G E H V P N S P F Q V T16 P E R P L V G V N G L D V T S L R P F D L V I P F T I K K G E I T G E V R M P S G K V A Q P T I T D N K D G T V T V R Y A P S E A G L H E M D I R Y D N M H I P G S P L Q F Y V D Y17 N C G H V T A Y G P G L T H G V V N K P A T F T V N T K D A G E G G L S L A I E G P S K A E I S C T D N Q D G T C S V S Y L P V L P G D Y S I L V K Y N E Q H V P G S P F T A R V T G D D18 M R M S H L K V G S A A D I P I N I S E T D L S L L T A T V V P P S G R E E P C L L K R L R N G H V G I S F V P K E T G E H L V H V K K N G Q H V A S S P I P V V I S Q S E I19 D A S R V R V S G Q G L H E G H T F E P A E F I I D T R D A G Y G G L S L S I E G P S K V D I N T E D L E D G T C R V T Y C P T E P G N Y I I N I K F A D Q H V P G S P F S V K V T G E G R V20 E S I T R R R R A P S V A N V G S H C D L S L K I P E I S I Q D M T A Q V T S P S G K T H E A E I V E G E N H T Y C I R F V P A E M G T H T V S V K Y K G Q H V P G S P P F Q F T V G P L G E21 G A H K V R A G G P G L E R A E A G V P A E F S I W T R E A G A G G L A I A V E G P S K A E I S F E D R K D G S C G V A Y V V Q E P G D Y E V S V K F N E E H I P D S P F V V P V A S P S22 D A L T V S S L Q E S G L K V N Q P A S F A V S L N G A K G A I D A K V H S P S G A L E E C Y V T E I D Q D K Y A V R F I P R E N G V Y L I D V K F N G T H I P G S P F K I R V G E P G H G23 D P G L V S A Y G A G L E G G V T G N P A E F V V N T S N A G A G A L S V T I D G P S K V K M D C Q E C P E G Y R V T Y T P M A P G S Y L I S I K Y G G P Y H I G G S P F K A K V T G P R L V24 D A S K V V A K G L G L S K A Y V G Q K S S F T V D C S K A G N N M L L V G V H G P R T P C E E I K V K H V G S R L Y S V S Y L L K D K G E Y T L V V K W G H E H I P G S P P Y R V V V P
Human D A S K V K A S G P G L E G G G V G P A E F T V D T A G G G L V T V G P G A D V D N G D G T Y V S Y V P R E P G T Y T V V K Y G Q H I P G S P F K V K VChiken D P S K V A G P G L E G P F V D T A G G L V V E G P S G I D N D G T Y V Y P P G Y V V K Y G H I P S P F V VDGF D D S Y A G P G L E G G P A F I A V D G E T G G D F V S I G P G P V D I D N D G T Y D V Y P G D Y V V T L G I K P V K I
N CCHD1 CHD2
ABD1 3
425
6 89
1011
1213
1415
1617
1819
2021
2223
24
G288R V711D
Del of residues761-943
P637Q
XMVD, MNS, PH, FMD, PH with Ehlers-Danlos, OPD
1 2 12
1 1 11
1
1
2 11
12
7
Arg288
Gln637
Asp711
A
B
C
�
�
� �
�
�
�� �
�
�
�
�
�
�
�
�
Figure 3. Sequence comparison of filamin repeats and position of mutations. A, Amino acid sequence alignment of 24 repeats ofhuman FLNA. Human, chicken, and Dictyostelium gelation factor (DGF) consensus sequences that include residues common to at least10 repeats are listed at the bottom of the figure. Comparison showed that the 3 mutations modify highly conserved residues. Mutationsare indicated in red, consensus residues are indicated in black, and the deletion is highlighted in red. B, Schematic representation offilamin’s actin-binding domain (ABD) and 24 repeats and summary of positions of mutations identified in periventricular heterotopia(PH), otopalatodigital syndromes (OPD), frontometaphyseal dysplasia (FMD), Melnick-Needles syndrome (MNS), and X-linked myxoma-tous valvular dysplasia (XMVD). Numbers above symbols indicate total number of mutations in a given repeat identified in the 5 differ-ent FLNA diseases. Most mutations are localized within the ABD and repeat 10. All 4 XMVD mutations are clustered between repeats 1and 7. Modified from Feng and Walsh22 by permission from Macmillan Publishers Ltd. Copyright 2004. C, Homology model of humanfilamin repeats WT and mutants based on the crystal structure of Dictyostelium filamin repeat 4 (Protein Data Bank identification,1WLH). Antiparallel �-strands are shown in yellow. Amino acids are shown in CPK configuration scale to 0.8. In the right panel, Gly288is present on the surface of the repeat, whereas the 2 other amino acids Pro637 and Val711 are present inside the repeat. In the leftpanel, the 3 mutants G288R, P637Q, and V711D are shown in orange, green, and red, respectively.
Kyndt et al Filamin A in Myxomatous Valvular Dystrophy 47
by guest on August 25, 2014http://circ.ahajournals.org/Downloaded from

and Melnick-Needles syndrome (OMIM 309350).30 Val-vular disease was reported in frontometaphyseal dyspla-sia31 and periventricular nodular heterotopia with Ehlers-Danlos syndrome.32 No signs of human periventricularnodular heterotopia, otopalatodigital syndromes, fronto-metaphyseal dysplasia, or Melnick-Needles or Ehlers-Danlos syndromes were found in these families. Becauseno extracardiac abnormalities were found, the valvulardystrophy reported in the present article is an isolateddisease and not part of a syndrome.
The mechanisms by which different mutations in FLNAlead to variable expression of disease remain unknown. Alarge number of mutations identified in periventricularheterotopia are nonsense and splicing mutations, and allmutations identified in the other disorders are missensemutations that cluster in small regions of the protein withno clear genotype–phenotype correlation. The differentphenotypes may highlight distinct interactions of filaminwith protein partners, and so far �30 binding partners havebeen found for FLNA. Among �30 mutations identified inFLNA, most are localized in the actin-binding domain andthe repeat 10 (Figure 3B), which suggests a functionalimportance of these regions of the protein.22 The 4 XMVDmutations are located within the same region of the protein(repeats 1 to 7) and could alter binding affinity for proteinpartners not yet identified. Screening of additional pro-bands for FLNA mutations is required to determine howparticular mutations are associated with differentphenotypes.
In conclusion, our data have demonstrated that FLNA isthe first gene to cause isolated nonsyndromic myxomatousvalvular dystrophy. This is the first step to understand theunderlying mechanisms of the disease and to definepathways that may lead to valvular dystrophy. In thefuture, the identification of the proteins involved in thiscascade could provide therapeutic targets for the medicalmanagement of this disease. Screening of FLNA mutationscould be important to provide adequate follow-up andgenetic counseling for families affected by XMVD.
AcknowledgmentsWe thank Professor Jean-Baptiste Michel and Dr Marie-PauleJacob (INSERM U460, Hôpital Bichat-Claude Bernard, France)for fruitful discussion. We also thank Dr Stéphane Bézieau(LEPA, France) for sharing control DNA. We thank FrançoiseGros for technical assistance. We are grateful to the familymembers for their active participation in the study and toChristine Fruchet and Régine Valéro for their essential contribu-tion to the recruitment of families.
Sources of FundingThis work was supported by grants from Projet Hospitalier deRecherche Clinique (PHRC 20-05), Fondation de France, Asso-ciation Française contre les Myopathies, and Groupementd’Intérêt Scientifique-Institut de la longévité et du vieillissement(GIS).
DisclosuresNone.
References1. Levy D, Savage D. Prevalence and clinical features of mitral valve
prolapse. Am Heart J. 1987;113:1281–1290.2. Towbin JA. Toward an understanding of the cause of mitral valve
prolapse. Am J Hum Genet. 1999;65:1238–1241.3. Cheitlin MD, Alpert JS, Armstrong WF, Aurigemma GP, Beller GA,
Bierman FZ, Davidson TW, Davis JL, Douglas PS, Gillam LD.ACC/AHA guidelines for the clinical Application of Echocardiography: areport of the American College of Cardiology/American Heart Asso-ciation Task Force on Practice Guidelines (committee on clinical appli-cation of Echocardiography), developed in collaboration with theAmerican Society of Echocardiography. Circulation. 1997;95:1686–1744.
4. Devereux RB, Brown WT, Kramer-Fox R, Sachs I. Inheritance of mitralvalve prolapse: effect of age and sex on gene expression. Ann Intern Med.1982;97:826–832.
5. Disse S, Abergel E, Berrebi A, Houot A-M, Le Heuzey JY, Diebold B,Guize L, Carpentier A, Corvol P, Jeunemaitre X. Mapping of a first locusfor autosomal dominant myxomatous mitral-valve prolapse to chro-mosome 16p11.2-p12.1. Am J Hum Genet. 1999;65:1242–1251.
6. Freed LA, Acierno JS, Dai D, Leyne M, Marshall JE, Nesta F, LevineRA, Slaugenhaupt SA. A locus for autosomal dominant mitral valveprolapse on chromosome 11p15.4. Am J Hum Genet. 2003;72:1551–1559.
7. Nesta F, Leyne M, Yosefy C, Simpson C, Dai D, Marshall JE, Hung J,Slaugenhaupt SA, Levine RA. New locus for autosomal dominant mitralvalve prolapse on chromosome 13: clinical insights from genetic studies.Circulation. 2005;112:2022–2230.
8. Monteleone PL, Fagan LF. Possible X-linked congenital heart disease.Circulation. 1969;39:611–614.
9. Newbury-Ecob RA, Zuccollo JM, Rutter N, Young ID. Sex-linked val-vular dysplasia. J Med Genet. 1993;30:873–874.
10. Kyndt F, Schott JJ, Trochu JN, Baranger F, Herbert O, Scott V,Fressinaud E, David A, Moisan JP, Bouhour JB, Le Marec H, BenichouB. Mapping of X-linked myxomatous valvular dystrophy to chromosomeXq28. Am J Hum Genet. 1998;62:627–632.
11. Trochu JN, Kyndt F, Schott JJ, Gueffet JP, Probst V, Benichou B, LeMarec H. Clinical characteristics of a familial inherited myxomatousvalvular dystrophy mapped to Xq28. J Am Coll Cardiol. 2000;35:1890–1897.
12. Weissman NJ, Pini R, Roman MJ, Kramer-Fox R, Andersen HS,Devereux RB. In vivo mitral valve morphology and motion in mitralvalve prolapse. Am J Cardiol. 1994;73:1080–1088.
13. Lalloz MR, Schwaab R, McVey JH, Michaelides K, Tuddenham EG.Haemophilia A diagnosis by simultaneous analysis of two variable dinu-cleotide tandem repeats within the factor VIII gene. Br J Haematol.1994;86:804–809.
14. Lindner TH, Hoffmann K. EasyLINKAGE: a PERL script for easy andautomated two-/multi-point linkage analyses. Bioinformatics. 2005;21:405–407.
15. Minassian BA, Aiyar R, Alic S, Banwell B, Villanova M, Fardeau M,Mandell JW, Juel VC, Rafii M, Auranen M, Kalimo H. Narrowing in onthe causative defect of an intriguing X-linked myopathy with excessiveautophagy. Neurology. 2002;59:596–601.
16. Patrosso C, Repetto M, Villa A, Milanesi L, Frattini A, Faranda S,Mancini M, Maestrini E, Toniolo D, Vezzoni P. The exon-intron orga-nization of the human X-linked gene (FLN1) encoding actin-bindingprotein 280. Genomics. 1994;21:71–76.
17. Neff MM, Turk E, Kalishman M. Web-based primer design for singlenucleotide polymorphism analysis. Trends Genet. 2002;18:613–615.
18. Popowicz GM, Muller R, Noegel AA, Schleicher M, Huber R, Holak TA.Molecular structure of the rod domain of Dictyostelium filamin. J MolBiol. 2004;342:1637–1646.
19. Gorlin JB, Yamin R, Egan S, Stewart M, Stossel TP, Kwiatkowski DJ,Hartwig JH. Human endothelial actin-binding protein (ABP-280, non-muscle filamin): a molecular leaf spring. J Cell Biol. 1990;111:1089–1105.
20. Barry CP, Xie J, Lemmon V, Young AP. Molecular characterization of amulti-promoter gene encoding a chicken filamin protein. J Biol Chem.1993;268:25577–25586.
21. Van der Flier A, Sonnenberg A. Structural and functional aspects offilamins. Biochim Biophys Acta. 2001;1538:99–117.
22. Feng Y, Walsh CA. The many faces of filamin: a versatile molecularscaffold for cell motility and signalling. Nat Cell Biol. 2004;6:1034–1038.
48 Circulation January 2/9, 2007
by guest on August 25, 2014http://circ.ahajournals.org/Downloaded from

23. Cunningham CC, Gorlin JB, Kwiatkowski DJ, Hartwig JH, Janmey PA,Byers HR, Stossel TP. Actin-binding protein requirement for corticalstablity and efficient locomotion. Science. 1992;255:325–327.
24. Cunningham CC. Actin polymerization and intracellular solvent flow incell surface blebbing. J Cell Biol. 1995;129:1589–1599.
25. Sasaki A, Masuda Y, Ohta Y, Ikeda K, Watanabe K. Filamin associateswith Smads and regulates transforming growth factor-� signalling. J BiolChem. 2001;276:17871–17877.
26. Derynck R, Zhang YE. Smad-dependent and smad-independent pathwaysin TGF-� family signalling. Nature. 2003;425:577–584.
27. Glavin KM, Donovan MJ, Lynch CA, Meyer RI, Paul RJ, Lorenz V,Fairchild-Huntress V, Dixon KL, Dunmore JH, Gimbrone MA, Falb D,Huszar D. A role of smad6 in development and homeostasis of thecardiovascular system. Nat Genet. 2000;24:171–174.
28. Vorgerd M, van der Ven PF, Bruchertseifer V, Lowe T, Kley RA,Schroder R, Lochmuller H, Himmel M, Koehler K, Furst DO, Huebner A.A mutation in the dimerization domain of filamin C causes a novel typeof autosomal dominant myofibrillar myopathy. Am J Hum Genet. 2005;77:297–304.
29. Krakow D, Robertson SP, King LM, Morgan T, Sebald ET, Bertolotto C,Wachsmann-Hogiu S, Acuna D, Shapiro SS, Takafuta T, Aftimos S, Kim
CA, Firth H, Steiner CE, Cormier-Daire V, Superti-Furga A, Bonafe L,Graham JM Jr, Grix A, Bacino CA, Allanson J, Bialer MG, Lachman RS,Rimoin DL, Cohn DH. Mutations in the gene encoding filamin B disruptvertebral segmentation, joint formation and skeletogenesis. Nat Genet.2004;36:405–410.
30. Robertson SP, Twigg SRF, Sutherland-Smith AJ, Biancalana V, GorlinRJ, Horn D, Kenwrick SJ, Kim CA, Morava E, Newbury-Ecob R,Orstavik KH, Quarrell OWJ, Schwartz CE, Shears DJ, Suri M,Kendrick-Jones J; The OPD-spectrum Disorders Clinical CollaborativeGroup & Wilkie O.M.A. Localized mutations in the gene encoding thecytoskeletal protein filamin A cause diverse malformations in humans.Nat Genet. 2003;33:487–491.
31. Park JM, Contreras EA, Garcia RR. Mitral valve prolapse in a patientwith frontometaphyseal dysplasia. Clin Pediatr. 1986;25:469–471.
32. Sheen VL, Jansen A, Chen MH, Parrini E, Morgan T, Ravenscroft R, GaneshV, Underwood T, Wiley J, Leventer R, Vaid RR, Ruiz DE, Hutchins GM,Menasha J, Willner J, Geng Y, Gripp KW, Nicholson L, Berry-Kravis E,Bodell A, Apse K, Hill RS, Dubeau F, Andermann F, Barkovich J,Andermann E, Shugart YY, Thomas P, Viri M, Veggiotti P, Robertson S,Guerrini R, Walsh CA. Filamin A mutations cause periventricular heter-otopia with Ehlers-Danlos syndrome. Neurology. 2005;64:254–262.
CLINICAL PERSPECTIVEFamilial inheritance has been demonstrated for mitral valve prolapse, the most common form of valvular dystrophy, anda familial study is recommended when a case is identified. Autosomal dominant transmission is the usual inheritance, withreduced penetrance and variable expressivity, and 3 loci have been mapped on chromosomes 11, 13, and 16. An X-linkedmyxomatous valvular dystrophy is a less frequent form of the disease and we have previously mapped it to Xq28, but theunderlying genetic defect was not previously known. Clinically affected patients are predominantly male with either mitralor aortic valve defects, whereas female patients present with minor forms of valvular dystrophy. The present studydemonstrates that a defect in the filamin A (FLNA) gene is responsible for X-linked valvulopathy. These findings couldhave important clinical implications. For example, the identification of the genetic basis for nonsyndromic valvulardystrophy offers a potential molecular diagnostic tool to detect patients at risk. Early identification of presymptomaticpatients might allow a better clinical follow-up to prevent complication of the disease.
Kyndt et al Filamin A in Myxomatous Valvular Dystrophy 49
by guest on August 25, 2014http://circ.ahajournals.org/Downloaded from
Copyright © 2022 FDOKUMEN




















