
Versican and CD44 in in vitro valvular interstitial cell injury and repair
9
Original Article Versican and CD44 in in vitro valvular interstitial cell injury and repair Jon M. Carthy, Seti Boroomand, Bruce M. McManus ⁎ UBC James Hogg Research Centre, Institute for Heart + Lung Health, Department of Pathology and Laboratory Medicine, University of British Columbia, Vancouver, BC, Canada V6Z 1Y6 Received 14 January 2011; accepted 14 March 2011 Abstract Background: Versican is one of the key components of the extracellular matrix (ECM) that is expressed during injury, inflammatory, and repair processes. The current study evaluated the relationship between versican and the membrane receptor CD44 during in vitro valvular interstitial cell (VIC) injury and repair. Methods: Subconfluent, confluent, and wounded cultures of human VICs were fixed and immunostained to detect versican and the membrane receptor CD44. To examine the relationship between versican and CD44, a blocking antibody to CD44 was added to cultured VICs, and in vitro wound repair along with pericellular versican organization and stress fiber formation were examined. Results: Immunohistochemistry demonstrated that versican is prominent intracellularly, as well as extracellularly, in actively proliferating VICs. In contrast, versican was only localized to fibrils in the extracellular space in between cells in confluent (quiescent) cultures. Following wounding, versican expression was up-regulated, and it was secreted as ECM at the trailing edge of migrating cells. The staining for CD44 was similarly localized to the trailing edge of migrating VICs in wounded cultures. Treatment of VICs with a CD44-blocking antibody disrupted the organization of versican in the pericellular matrix and inhibited stress fiber formation in these cells. Functionally, blocking CD44 significantly inhibited VIC-mediated contraction of type I collagen gels (35.7%±0.7% vs. 23.3%±1.4% of initial gel area, Pb.01). Conclusions: Versican is a key component of the provisional wound repair ECM that is expressed following injury to VICs. The receptor CD44 plays an important role in organizing the provisional ECM. Summary: Our data suggests VICs synthesize and secrete versican following injury. These cells also up-regulate CD44, a receptor that binds versican. Blocking CD44 disrupted the organization of versican and inhibited stress fiber formation. Functionally, blocking CD44 inhibited cell-mediated contraction of a collagen matrix. Collectively, these data suggest versican expression and organization are important to valve cell injury and repair. © 2012 Elsevier Inc. All rights reserved. Keywords: Versican; CD44; Valvular interstitial cells; Injury; Wound repair 1. Introduction Valvular heart disease is a major contributor to the overall burden of cardiovascular disease [1]. Pathological analysis of most diseased heart valves suggests a response to injury process is involved in the development of valvular heart disease, generally characterized by an accumulation of valvular interstitial cells (VICs) and extracellular matrix (ECM), along with various intensities of inflammation, neovascularization, and fibrosis, ultimately leading to valve sclerosis, possible calcification and dysfunction [2,3]. Being the predominant cell type found in the interstitium of heart valves, VICs are likely critical regulators of this repair process. In response to valve injury, VICs become activated and consistently display features of myofibroblasts, includ- ing increased contraction, stress fiber formation, and smooth muscle α-actin expression [4,5]. As myofibroblasts regulate wound repair in many organs [6], a better understanding of how VICs respond to injury will help us to gain insight into the pathobiology of valvular heart disease. The ECM is now widely recognized as more than simply a support scaffold and is emerging as one of the key regulators of cell phenotypes. This function is evidenced by the significant remodeling of tissues during injury, Cardiovascular Pathology 21 (2012) 74 – 82 ⁎ Corresponding author. The James Hogg iCAPTURE Centre for Cardiovascular and Pulmonary Research, Institute for Heart + Lung Health, Room 166, Burrard Building, 1081 Burrard St, BC, Canada V6Z 1Y6. Tel.: +1 604 806 8586; fax: +1 604 806 9274. E-mail address: [email protected] (B.M. McManus). 1054-8807/11/$ – see front matter © 2012 Elsevier Inc. All rights reserved. doi:10.1016/j.carpath.2011.03.003
Transcript of Versican and CD44 in in vitro valvular interstitial cell injury and repair

Cardiovascular Pathology 21 (2012) 74–82
Original Article
Versican and CD44 in in vitro valvular interstitial cell injury and repair
Jon M. Carthy, Seti Boroomand, Bruce M. McManus⁎
UBC James Hogg Research Centre, Institute for Heart + Lung Health, Department of Pathology and Laboratory Medicine, University of British Columbia,Vancouver, BC, Canada V6Z 1Y6
Received 14 January 2011; accepted 14 March 2011
Abstract
Background: Versican is one of the key components of the extracellular matrix (ECM) that is expressed during injury, inflammatory, andrepair processes. The current study evaluated the relationship between versican and the membrane receptor CD44 during in vitro valvularinterstitial cell (VIC) injury and repair. Methods: Subconfluent, confluent, and wounded cultures of human VICs were fixed andimmunostained to detect versican and the membrane receptor CD44. To examine the relationship between versican and CD44, a blockingantibody to CD44 was added to cultured VICs, and in vitro wound repair along with pericellular versican organization and stress fiberformation were examined. Results: Immunohistochemistry demonstrated that versican is prominent intracellularly, as well as extracellularly,in actively proliferating VICs. In contrast, versican was only localized to fibrils in the extracellular space in between cells in confluent(quiescent) cultures. Following wounding, versican expression was up-regulated, and it was secreted as ECM at the trailing edge of migratingcells. The staining for CD44 was similarly localized to the trailing edge of migrating VICs in wounded cultures. Treatment of VICs with aCD44-blocking antibody disrupted the organization of versican in the pericellular matrix and inhibited stress fiber formation in these cells.Functionally, blocking CD44 significantly inhibited VIC-mediated contraction of type I collagen gels (35.7%±0.7% vs. 23.3%±1.4% ofinitial gel area, Pb.01). Conclusions: Versican is a key component of the provisional wound repair ECM that is expressed following injury toVICs. The receptor CD44 plays an important role in organizing the provisional ECM. Summary: Our data suggests VICs synthesize andsecrete versican following injury. These cells also up-regulate CD44, a receptor that binds versican. Blocking CD44 disrupted theorganization of versican and inhibited stress fiber formation. Functionally, blocking CD44 inhibited cell-mediated contraction of a collagenmatrix. Collectively, these data suggest versican expression and organization are important to valve cell injury and repair. © 2012 ElsevierInc. All rights reserved.
Keywords: Versican; CD44; Valvular interstitial cells; Injury; Wound repair
1. Introduction
Valvular heart disease is a major contributor to the overallburden of cardiovascular disease [1]. Pathological analysis ofmost diseased heart valves suggests a response to injuryprocess is involved in the development of valvular heartdisease, generally characterized by an accumulation ofvalvular interstitial cells (VICs) and extracellular matrix(ECM), along with various intensities of inflammation,
⁎ Corresponding author. The James Hogg iCAPTURE Centre forCardiovascular and Pulmonary Research, Institute for Heart + Lung Health,Room 166, Burrard Building, 1081 Burrard St, BC, Canada V6Z 1Y6.Tel.: +1 604 806 8586; fax: +1 604 806 9274.
E-mail address: [email protected] (B.M. McManus).
1054-8807/11/$ – see front matter © 2012 Elsevier Inc. All rights reserved.doi:10.1016/j.carpath.2011.03.003
neovascularization, and fibrosis, ultimately leading to valvesclerosis, possible calcification and dysfunction [2,3]. Beingthe predominant cell type found in the interstitium of heartvalves, VICs are likely critical regulators of this repairprocess. In response to valve injury, VICs become activatedand consistently display features of myofibroblasts, includ-ing increased contraction, stress fiber formation, and smoothmuscle α-actin expression [4,5]. As myofibroblasts regulatewound repair in many organs [6], a better understanding ofhow VICs respond to injury will help us to gain insight intothe pathobiology of valvular heart disease.
The ECM is now widely recognized as more than simplya support scaffold and is emerging as one of the keyregulators of cell phenotypes. This function is evidenced bythe significant remodeling of tissues during injury,

75J.M. Carthy et al. / Cardiovascular Pathology 21 (2012) 74–82
inflammation, and repair processes [7,8]. The proteoglycanversican is one of the key ECM components up-regulatedafter injury in a number of tissue and cell types [9–14], suchas in diseased myxomatous heart valves [15,16]. Versican isa large multidomain chondroitin sulfate proteoglycan thatbinds hyaluronic acid at its N-terminus [17] and binds avariety of other matrix proteins at its C-terminus, includingfibrillin-1 [18], fibulin-1 and fibulin-2 [19,20], tenascin-R[21], type I collagen [22], and fibronectin [22]. Theseterminal domains are separated by a central glycosamino-glycan (GAG) attachment region consisting of two exonsnamed GAG-α and GAG-β. As a result of differentialsplicing to these central domains, the versican gene codes forfour variants (namely V0–V3) that differ in their length andnumber of attached GAG side chains [23,24]. Becauseversican connects hyaluronan at the cell surface to othermatrix proteins at its C-terminus, it is believed to play animportant role in the assembly and organization of the ECM.In addition, this versatile proteoglycan modifies a number ofcell processes that are thought to contribute to repair,including cell adhesion [25,26], migration [27–29], prolif-eration [27,30], and apoptosis [30,31]. Therefore, it is likelythat versican is a key regulator of cell phenotype duringnormal or aberrant wound healing.
Despite the apparent importance of versican to injury andrepair processes, the role of this versatile proteoglycan inheart valve repair has not been studied. In this study, wesought to determine whether versican is expressed bycultured VICs after injury. We used a scratch assay tomechanically injure confluent monolayers of human VICsand then observed repair over a period of 48 h. Our resultssuggest that human VICs synthesize and secrete versicaninto the ECM following injury. These cells also up-regulatethe expression of CD44, a cell membrane receptor involvedin cell adhesion and migration that binds versican andhyaluronan [32,33]. When a neutralizing antibody was usedto block CD44, wound repair in our model wassignificantly impaired. Further, blocking CD44 disruptedthe organization of versican in the pericellular matrix andinhibited stress fiber formation in VICs. Collectively, thesedata suggest versican expression and organization isimportant to VIC injury and repair events, and that CD44may serve as a link between the actin cytoskeleton and theprovisional wound repair ECM.
2. Methods
2.1. Cell culture
This study was approved by the University of BritishColumbia/Providence Health Care Research Ethics Boardand conforms with the principles outlined in the Declarationof Helsinki for use of human tissues or subjects. Human aorticvalve leaflets were collected by the Cardiovascular Registry,St. Paul's Hospital, Vancouver, British Columbia, Canada,
from the explanted heart at the time of cardiac transplantationfrom a 47–year-old male patient with no history of heartvalve disease. VICs were isolated from noncalcified valveleaflets by enzymatic digestion and phenotyped by immuno-fluorescence using a panel of antibodies against cytoskeletalcomponents. Valve interstitial cells were cultured in MCDB131 (Gibco) containing 15% fetal bovine serum and 100U/mlpenicillin/streptomycin. Cells were maintained in a humid-ified incubator at 37°C with 5% CO2. Cells were used forexperiments between passages 2 and 4. Subconfluent culturesthat had been grown for 3–4 days and confluent culturesgrown for 11 days were fixed in their nonwounded states with3.7% formaldehyde for 20 min before immunohistochemis-try. The in vitro wound model is described below. Forantibody treatments, cells were trypsinized and incubated for45 min at 4°C with 20 μg/ml of the CD44-blocking antibody(BD Pharmingen, clone IM7, product number: 553130) andthen seeded onto glass coverslips and cultured for 24 h priorto fixation and immunohistochemistry. All experiments wererepeated a minimum of three independent times.
2.2. In vitro scratch injury model
The in vitro scratch injury model was performedaccording to the protocol described previously [34]. Briefly,VICs were seeded on glass coverslips that had been placed insix-well culture dishes and grown to confluence (approxi-mately 11 days). A sterile P200 pipette tip was used to makea linear wound across the confluent monolayers. Cells weresubsequently fixed at the indicated time points with3.7% formaldehyde.
2.3. Immunohistochemistry
Fixed cells were permeabilized with 0.1% Triton X-100for 20 min, blocked for 30 min with 1% bovine serumalbumin (BSA) in phosphate-buffered saline, and incubatedovernight at 4°C with the versican (US Biologicals, productnumber: L1350) or CD44 (Santa Cruz Biotechnology,product number: sc-9960) primary antibodies at a concen-tration of 1:100 in 1% BSA. Following primary antibodies,cells were incubated with antimouse Alexa-fluor488 conju-gated secondary antibody (Invitrogen) at a concentration of1:200 in 1% BSA for 1 h at room temperature in the dark.Cells were then stained for 20 min with phalloidinconjugated to Alexa-fluor594 (Invitrogen) to visualizeF-actin and coverslipped with VectaShield mountingmedium containing 4′-6-diamidino-2-phenylindole (VectorLabs). Cells were imaged using a Leica AOBS SP2 confocalmicroscope as we have previously described [35–37].
2.4. Collagen gel contraction assay
Twelve-well culture dishes were coated with 1% BSAand incubated for 1 h at 37°C to create a nonstick surface thatprevents gels from attaching to the dishes. Cells were

76 J.M. Carthy et al. / Cardiovascular Pathology 21 (2012) 74–82
incubated at 4°C in the presence or absence of CD44-blocking antibody (20 μg/ml) and then seeded into a 0.5-mg/mltype I collagen solution (BD Biosciences, product number:354236) in growthmedia at a concentration of 2.5×104 cells/ml.The collagen/cell suspension was then vortexed, and 1 ml/well was added to the BSA-coated dishes, and the solutionwas allowed to polymerize for 45 min at 37°C. Fresh growthmedia was then added to the solidified collagen gels andplates were returned to the incubator. Collagen gelcontraction was monitored over a period of 24 h, and thesurface area of contracted gels was measured using Image-Pro Plus software (Media Cybernetics, Bethesda, MD). Thecollagen gels were then fixed and immunostained asdescribed above.
2.5. Statistics
Results are represented as the mean±S.D. Significantdifferences in treatment groups were determined using theunpaired Student's t test. For all analyses, Pb.05 wasconsidered statistically significant.
3. Results
3.1. Phenotypic analysis of VICs
A panel of antibodies to cytoskeletal elements was used tophenotype the VICs used in this study by immunohisto-
Fig. 1. Cultured VICs display a myofibroblast-like phenotype. Confocalimages of VICs immunostained for (A) smooth muscle α-actin, (B) desmin,(C) vimentin, and (D) smooth muscle myosin heavy chain. These cells hadpositive immunostaining for smooth muscle α-actin, desmin, and vimentin(green) but were negative for smooth muscle myosin heavy chain (scalebar=47.62 μm).
chemistry. The valve cells demonstrated positive staining forsmooth muscle α-actin, vimentin and desmin, but werenegative for smooth muscle myosin heavy chain (Fig. 1),suggesting these cells display a myofibroblast-like pheno-type in culture.
3.2. Versican immunostaining in VICs
Immunohistochemistry demonstrated that cultured VICsproduce an abundance of versican. In subconfluent culturesof VICs, versican was clearly observed intracellularly in theperinuclear region, as well as along the cell membrane, andwas deposited in the ECM (Fig. 2A,B). In contrast, confluentmonolayers of VICs displayed little detectable intracellularversican. In these cultures, versican was localized to theextracellular space between cells where it was organizedinto fibrils that aligned in parallel to the F-actin stressfibers (Fig. 2C,D). This staining pattern suggests thatversican is involved in VIC proliferation and migration andis down-regulated in quiescent (confluent) cells. A negativecontrol experiment, in which no primary antibody wasadded, demonstrated the specificity of the versican antibody(data not shown).
Wounding of the confluent monolayers led to themigration of elongated VICs moving outward from thewound edge, as has previously been described [2,38]. Therewas a loss of versican reactivity at the wound edge 4 h afterinjury, but migrating VICs clearly showed cytoplasmic
Fig. 2. Versican expression in cardiac VICs. Confocal images of subconfluentVICs immunostained for (A) versican and (B) Alexa-fluor594-labeledphalloidin (to identify F-actin stress fibers). Versican was detected in thecytoplasm (arrow), along the cell membrane and in cell protrusions(arrowhead), and was deposited in the ECM (empty arrow). (C) In confluentcultures, versican was present in the extracellular space between cells with noapparent intracellular staining and was organized into fibrils (arrows) thataligned in parallel with the F-actin stress fibers (D) (scale bar=47.62 μm).

Fig. 3. Versican expression during injury and repair in cardiac VICs. Confocal images of cells at the wound edge immunostained to detect versican at (A) 4, (B)24, and (C) 48 h after wounding. (A) At 4 h, cells had started migrating into the wound (w) and synthesized de novo versican that was present in the cytoplasm inthe perinuclear region (arrows). (B) Twenty-four hours after wounding, versican was apparent along the cell membrane of migrating cells and was beginning tobe deposited into the ECM (arrows). (C) Forty-eight hours after wounding, versican appeared predominantly in the ECM where it was organized into fibrils(arrows). Collectively, these results suggest versican is synthesized and secreted into the ECM following injury to VICs (scale bar=37.50 μm).
77J.M. Carthy et al. / Cardiovascular Pathology 21 (2012) 74–82
staining for versican in the perinuclear region (Fig. 3A).Twenty-four hours after wounding, the migrating VICsshowed increased versican expression along the cellmembrane and in the ECM (Fig. 3B). The newly synthesizedversican was deposited at the trailing edge of migrating cells,suggesting the cells leave behind a trail of versican as theymove into the wound. Forty-eight hours after injury, as cellsbegin to close the wound, versican was predominantlydetected in the ECM where it was organized into small thinfibrils (Fig. 3C). These versican fibrils were not as dense asthose seen in confluent cultures of VICs, nor did they displaythe same degree of organization.
Fig. 4. Expression of CD44 in cardiac VICs. Confocal images ofsubconfluent VICs immunostained for (A) CD44 and (B) F-actin showingimmunostaining for CD44 predominantly along the cell membrane (arrows)in nonconfluent cultures. (C) In confluent cultures of valve cells, expressionof CD44 was minimal with no clearly observable pattern. Actin staining isshown in (D) (scale bar=37.50 μm).
3.3. CD44 immunostaining in VICs
We next studied the expression pattern of CD44, a cellmembrane receptor that binds versican and is involved in cellmigration [32,39]. In subconfluent cultures of VICs, CD44was localized exclusively to the cell membrane, andexpression was often stronger on one side of the cell ascompared with the other in a polarized fashion (Fig. 4A,B).Interestingly, we observed little immunostaining for CD44 inconfluent cultures of VICs (Fig. 4C,D), suggesting thisprotein, like versican, is involved in the proliferation andmigration of VICs, but is down-regulated in quiescent cells.
Early after injury (at 4 h), the cells had started migratinginto the wound, yet the CD44 expression remained scarcelydetectable, similar to what was seen in the confluentmonolayers (Fig. 5A,B). Twenty-four hours after wounding,however, CD44 was detected intracellularly and along thecell membrane in migrating cells (Fig. 5C,D). Forty-eighthours after wounding, CD44 immunoreactivity was in-creased and was clearly seen along the cell membrane,predominantly localized to the trailing edge of migratingcells (Fig. 5E,F).
3.4. Blocking CD44 disrupts pericellular matrixorganization and stress fiber formation in VICs
To examine the relationship between versican and CD44in VICs, we used a blocking antibody to CD44 and observedany changes in cell phenotype. In the absence of CD44-blocking antibody, immunohistochemistry demonstratedthat VICs formed a highly organized versican pericellularmatrix (Fig. 6A) and were characterized by prominentintracellular stress fibers (Fig. 6B). In contrast, in thepresence of CD44-blocking antibody, the pericellularversican organization was clearly disrupted and versicantended to clump as deposits under the cells (Fig. 6C).Likewise, the actin cytoskeleton appeared disordered withlittle stress fiber formation following treatment with CD44-

Fig. 5. Expression of CD44 during injury and repair in cardiac VICs. Confocal images of cells at the wound edge immunostained to detect (A, C, E) CD44 and(B, D, F) F-actin at (A, B) 4 h, (C, D) 24 h, and (E, F) 48 h after wounding. (A, B) At 4 h, cells had started migrating into the wound (w) and demonstrated verylittle immunostaining for CD44. (C, D) Twenty-four hours after wounding, cells up-regulated the expression of CD44 that localized to the cell membrane ofmigrating cells (arrows). (E, F) Forty-eight hours after wounding, migrating cells demonstrated the highest expression of CD44 (arrows), localizedpredominantly along the cell membrane in trailing cell processes (scale bar=75.00 μm).
Fig. 6. Blocking CD44 inhibits pericellular matrix organization and stress fiberformation in VICs. Confocal images of cells incubated in the (A, B) absence or(C, D) presence of a CD44-blocking antibody and immunostained for (A, C)versican and (B, D) F-actin. In the absence of CD44-blocking antibody, thecells displayed a versican pericellular matrix that was highly organized anddeposited into the ECM as fibrils (A, arrows). These cells clearly demonstratedthe formation of well-organized F-actin stress fibers (B, arrows). In thepresence of CD44-blocking antibody, there was a lack of pericellular matrixorganization and the versican tended to clump as deposits under the cell(C, arrows). These cells displayed a poorly organized actin cytoskeleton withlittle stress fiber formation (D, arrows) (scale bar=23.81 μm).
78 J.M. Carthy et al. / Cardiovascular Pathology 21 (2012) 74–82
blocking antibody (Fig. 6D), highlighting a potential role forCD44 as a link between the actin cytoskeleton and versicanin the ECM.
3.5. Blocking CD44 inhibits VIC-mediated contraction oftype I collagen gels
To investigate the functional effect of blocking CD44, wefirst performed a scratch wound assay in the presence orabsence of CD44-neutralizing antibody. Interestingly, wedid not detect any difference in the wound closure rate seenin cells treated with CD44 antibody when compared withcontrol cells (PN.05, data not shown). As CD44 blockingdisrupted stress fiber formation in VICs, we next performed acollagen gel contraction assay, a commonly employed vitromodel that can be used to measure the contractile propertiesof a population of cells. Valve interstitial cells were seededinto type I collagen gels in the presence or absence of CD44-blocking antibody, and gel contraction was observed over aperiod of 24 h by measuring the surface area of contractedgels. Representative images and the quantified surface areaof contracted gels are shown in Fig. 7. Blocking CD44significantly inhibited VIC-mediated contraction of the typeI collagen gels (35.7%±0.7% vs. 23.3%±1.4% of initial gelarea, Pb.01).
Immunohistochemistry on VICs cultured in collagen gelsshowed that in the absence of CD44-blocking antibody,VICs appeared elongated, interconnected, and had clearlyvisible stress fibers (Fig. 8A–D). These cells also displayedstrong immunoreactivity for versican in the pericellular
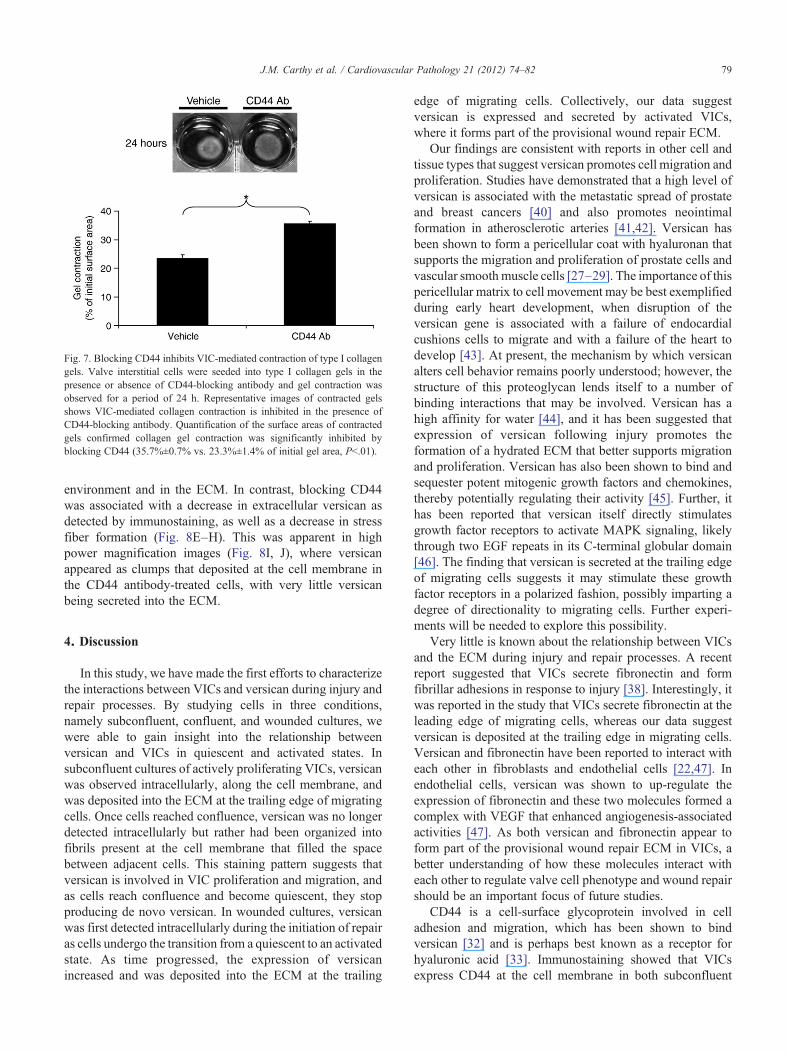
Fig. 7. Blocking CD44 inhibits VIC-mediated contraction of type I collagengels. Valve interstitial cells were seeded into type I collagen gels in thepresence or absence of CD44-blocking antibody and gel contraction wasobserved for a period of 24 h. Representative images of contracted gelsshows VIC-mediated collagen contraction is inhibited in the presence ofCD44-blocking antibody. Quantification of the surface areas of contractedgels confirmed collagen gel contraction was significantly inhibited byblocking CD44 (35.7%±0.7% vs. 23.3%±1.4% of initial gel area, Pb.01).
79J.M. Carthy et al. / Cardiovascular Pathology 21 (2012) 74–82
environment and in the ECM. In contrast, blocking CD44was associated with a decrease in extracellular versican asdetected by immunostaining, as well as a decrease in stressfiber formation (Fig. 8E–H). This was apparent in highpower magnification images (Fig. 8I, J), where versicanappeared as clumps that deposited at the cell membrane inthe CD44 antibody-treated cells, with very little versicanbeing secreted into the ECM.
4. Discussion
In this study, we have made the first efforts to characterizethe interactions between VICs and versican during injury andrepair processes. By studying cells in three conditions,namely subconfluent, confluent, and wounded cultures, wewere able to gain insight into the relationship betweenversican and VICs in quiescent and activated states. Insubconfluent cultures of actively proliferating VICs, versicanwas observed intracellularly, along the cell membrane, andwas deposited into the ECM at the trailing edge of migratingcells. Once cells reached confluence, versican was no longerdetected intracellularly but rather had been organized intofibrils present at the cell membrane that filled the spacebetween adjacent cells. This staining pattern suggests thatversican is involved in VIC proliferation and migration, andas cells reach confluence and become quiescent, they stopproducing de novo versican. In wounded cultures, versicanwas first detected intracellularly during the initiation of repairas cells undergo the transition from a quiescent to an activatedstate. As time progressed, the expression of versicanincreased and was deposited into the ECM at the trailing
edge of migrating cells. Collectively, our data suggestversican is expressed and secreted by activated VICs,where it forms part of the provisional wound repair ECM.
Our findings are consistent with reports in other cell andtissue types that suggest versican promotes cell migration andproliferation. Studies have demonstrated that a high level ofversican is associated with the metastatic spread of prostateand breast cancers [40] and also promotes neointimalformation in atherosclerotic arteries [41,42]. Versican hasbeen shown to form a pericellular coat with hyaluronan thatsupports the migration and proliferation of prostate cells andvascular smoothmuscle cells [27–29]. The importance of thispericellular matrix to cell movement may be best exemplifiedduring early heart development, when disruption of theversican gene is associated with a failure of endocardialcushions cells to migrate and with a failure of the heart todevelop [43]. At present, the mechanism by which versicanalters cell behavior remains poorly understood; however, thestructure of this proteoglycan lends itself to a number ofbinding interactions that may be involved. Versican has ahigh affinity for water [44], and it has been suggested thatexpression of versican following injury promotes theformation of a hydrated ECM that better supports migrationand proliferation. Versican has also been shown to bind andsequester potent mitogenic growth factors and chemokines,thereby potentially regulating their activity [45]. Further, ithas been reported that versican itself directly stimulatesgrowth factor receptors to activate MAPK signaling, likelythrough two EGF repeats in its C-terminal globular domain[46]. The finding that versican is secreted at the trailing edgeof migrating cells suggests it may stimulate these growthfactor receptors in a polarized fashion, possibly imparting adegree of directionality to migrating cells. Further experi-ments will be needed to explore this possibility.
Very little is known about the relationship between VICsand the ECM during injury and repair processes. A recentreport suggested that VICs secrete fibronectin and formfibrillar adhesions in response to injury [38]. Interestingly, itwas reported in the study that VICs secrete fibronectin at theleading edge of migrating cells, whereas our data suggestversican is deposited at the trailing edge in migrating cells.Versican and fibronectin have been reported to interact witheach other in fibroblasts and endothelial cells [22,47]. Inendothelial cells, versican was shown to up-regulate theexpression of fibronectin and these two molecules formed acomplex with VEGF that enhanced angiogenesis-associatedactivities [47]. As both versican and fibronectin appear toform part of the provisional wound repair ECM in VICs, abetter understanding of how these molecules interact witheach other to regulate valve cell phenotype and wound repairshould be an important focus of future studies.
CD44 is a cell-surface glycoprotein involved in celladhesion and migration, which has been shown to bindversican [32] and is perhaps best known as a receptor forhyaluronic acid [33]. Immunostaining showed that VICsexpress CD44 at the cell membrane in both subconfluent

Fig. 8. Blocking CD44 inhibits stress fiber formation and versican deposition into the ECM in VICs cultured in collagen gels. Immunohistochemistry wasperformed on VICs cultured in collagen gels in the (A–D) absence or (E–H) presence of CD44-blocking antibody. Cells were stained for (A, E) nuclei, (B, F)F-actin, and (C, G) versican. Overlay images (D, H) show the cells to be elongated and interconnected in the absence of CD44-blocking antibody, with clearlyvisible F-actin stress fibers and versican deposition around the cell membrane and in the ECM (D, arrows). In contrast, cells treated with CD44-blocking antibodylacked clear stress fibers, did not appear interconnected and had less versican present in the ECM (H, arrows). (I, J) High power confocal images highlight theabsence of extracellular versican in cells treated with CD44-blocking antibody as compared with control cells. In the CD44 antibody-treated cells, versicantended to clump around the cell membrane (J, arrows) instead of being secreted into the ECM as seen in control cells (I, arrows). The antibody treated cells alsolacked clear stress fibers as compared with control cells (scale bars=47.62 μm in a and 23.81 μm in I).
80 J.M. Carthy et al. / Cardiovascular Pathology 21 (2012) 74–82
cultures and following wounding, when the cells are in anactivated state. In contrast, we did not detect any CD44expression in confluent cultures of quiescent VICs. Theexpression pattern for CD44 mirrored that of versican in thewounded cultures, where CD44 was up-regulated afterinjury and localized to the trailing edge of migrating cells.Interestingly, we did not observe any difference in the rateof wound closure in VICs treated with a CD44-blockingantibody. There are conflicting reports in the literatureabout the role of CD44 in cell migration, and a recent reportshowed that CD44 knockout fibroblasts actually had ahigher rate of migration, but the migration was directionlessand disorganized in these cells and resulted in less efficientwound closure [48]. The polarized expression pattern seenfor CD44 in VICs supports the idea that CD44 mayinfluence the direction of migration. Thus, it seemsplausible that CD44 expression results in a slower rate of
migration, but promotes a more coordinated and efficientwound closure response.
The similar localization of CD44 and versican observedin activated VICs suggests CD44 may be involved inorganizing the provisional wound repair matrix secreted bythese cells. Consistent with this idea, cellular expression ofCD44 has been shown to be required for the formation of ahyaluronan/versican pericellular sheath during migration ofprostate cancer cells [29]. The cytoplasmic tail of CD44 isanchored to cytoskeletal elements via ankyrin [49] andtherefore may provide a link between the actin cytoskeletonand versican in the pericellular matrix, and could haveimportant implications to VIC function. Indeed, our studyhas demonstrated that blocking CD44 inhibited theorganization of versican in the pericellular matrix of VICsand was associated with a decreased ability of VICs to formstress fibers. Functionally, the change in cell structure

81J.M. Carthy et al. / Cardiovascular Pathology 21 (2012) 74–82
induced by blocking CD44 was associated with a decreasedcontractile ability of VICs when seeded into type I collagengels. These results support findings from studies in chronicinflammatory diseases, where CD44 has been suggested toplay a role in tissue remodeling and fibrosis [48], andsuggest valve myofibroblasts require CD44 to remodelextracellular matrices.
There are a number of limitations to the current study,most notably, the in vitro nature of the experiments that areclearly an overly simplified model of human disease. Thatbeing said, the in vitro scratch wound assay and collagengel contraction assay have been used extensively in thepast to gain valuable insight into cell–matrix interactionsduring injury and repair events. In addition, the data werecollected using cells from a single patient; however, thephenotype of these cells is consistent with the phenotypeof VICs isolated from other nondiseased humans or animalmodels. Further, we do not present data to show a directlink or physical interaction between versican and CD44 inVICs. Both of these proteoglycans are highly interactive,and while it has been demonstrated previously that theybind each other [32], it is quite possible that our datasimply reflect CD44 acting on a different component of theECM to which versican may be bound, such ashyaluronan. While this possibility cannot be ruled out, atthe very least, our data highlight a novel role for CD44 inregulating the organization of the ECM, which is reflectedin our studies by the altered organization of versican uponblocking CD44.
In summary, we have shown that activated VICs secreteversican into the ECM following wounding where it formspart of the provisional wound repair ECM. Our immunohis-tochemistry results also suggest valve cells up-regulate theexpression of CD44 following injury, a membrane receptorthat binds versican. The similar localization of versican andCD44 at the trailing edge of migrating valve cells suggeststhis receptor is involved in organizing the provisionalversican-rich ECM. Blocking antibodies to CD44 confirmedthis idea, highlighting CD44 as an important linker proteinbetween the actin cytoskeleton and the ECM. In functionalstudies, these changes were associated with decreasedcontraction of collagen gels by VICs treated with CD44-blocking antibody. As VICs are the predominant cell typefound in heart valves, a better understanding of how theyrespond to injury will help us to gain insight into valvularwound repair and the pathological complications that arisefrom abnormalities in this process.
Acknowledgments
This work was supported by a grant-in-aid from the Heartand Stroke Foundation of British Columbia and Yukon(to B.M.M.) and graduate fellowships from the University ofBritish Columbia (J.M.C) and the Canadian Institutes ofHealth Research (S.B). The authors would like to thank Dr.David Marchant for his critical reading of the manuscript.
References
[1] Lloyd-Jones D, Adams R, Carnethon M, De Simone G, Ferguson TB,Flegal K, Ford E, Furie K, Go A, Greenlund K, Haase N, Hailpern S,Ho M, Howard V, Kissela B, Kittner S, Lackland D, Lisabeth L,Marelli A, McDermott M, Meigs J, Mozaffarian D, Nichol G,O'Donnell C, Roger V, Rosamond W, Sacco R, Sorlie P, Stafford R,Steinberger J, Thom T,Wasserthiel-Smoller S, Wong N,Wylie-Rosett J,Hong Y. Heart disease and stroke statistics–2009 update: a reportfrom the American Heart Association Statistics Committee andStroke Statistics Subcommittee. Circulation 2009;119(3):e21–181.
[2] Durbin AD, Gotlieb AI. Advances towards understanding heart valveresponse to injury. Cardiovasc Pathol 2002;11(2):69–77.
[3] Liu AC, Joag VR, Gotlieb AI. The emerging role of valve interstitialcell phenotypes in regulating heart valve pathobiology. Am J Pathol2007;171(5):1407–18.
[4] Liu AC, Gotlieb AI. Transforming growth factor-beta regulates in vitroheart valve repair by activated valve interstitial cells. Am J Pathol2008;173(5):1275–85.
[5] Rabkin-Aikawa E, Farber M, Aikawa M, Schoen FJ. Dynamic andreversible changes of interstitial cell phenotype during remodeling ofcardiac valves. J Heart Valve Dis 2004;13(5):841–7.
[6] Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML,Gabbiani G. The myofibroblast: one function, multiple origins. Am JPathol 2007;170(6):1807–16.
[7] Daley WP, Peters SB, Larsen M. Extracellular matrix dynamics indevelopment and regenerative medicine. J Cell Sci 2008;121(Pt 3):255–64.
[8] Shaw TJ, Martin P. Wound repair at a glance. J Cell Sci 2009;122(Pt18):3209–13.
[9] Geary RL, Nikkari ST, Wagner WD, Williams JK, Adams MR,Dean RH. Wound healing: a paradigm for lumen narrowing afterarterial reconstruction. J Vasc Surg 1998;27(1):96–106 discussion106–108.
[10] Wang J, Dodd C, Shankowsky HA, Scott PG, Tredget EE. Deepdermal fibroblasts contribute to hypertrophic scarring. Lab Invest2008;88(12):1278–90.
[11] Provenzano PP, Alejandro-Osorio AL, Valhmu WB, Jensen KT,Vanderby R. Intrinsic fibroblast-mediated remodeling of damagedcollagenous matrices in vivo. Matrix Biol 2005;23(8):543–55.
[12] Farb A, Kolodgie FD, Hwang JY, Burke AP, Tefera K, Weber DK,Wight TN, Virmani R. Extracellular matrix changes in stented humancoronary arteries. Circulation 2004;110(8):940–7.
[13] Wang JF, Olson ME, Reno CR, Kulyk W, Wright JB, Hart DA.Molecular and cell biology of skin wound healing in a pig model.Connect Tissue Res 2000;41(3):195–211.
[14] Toeda K, Nakamura K, Hirohata S, Hatipoglu OF, Demircan K,Yamawaki H, Ogawa H, Kusachi S, Shiratori Y, Ninomiya Y.Versican is induced in infiltrating monocytes in myocardial infarction.Mol Cell Biochem 2005;280(1–2):47–56.
[15] Stephens EH, Chu CK, Grande-Allen KJ. Valve proteoglycan contentand glycosaminoglycan fine structure are unique to microstructure,mechanical load and age: relevance to an age-specific tissue-engineered heart valve. Acta Biomater 2008;4(5):1148–60.
[16] Gupta V, Barzilla JE, Mendez JS, Stephens EH, Lee EL, Collard CD,Laucirica R, Weigel PH, Grande-Allen KJ. Abundance and location ofproteoglycans and hyaluronan within normal and myxomatous mitralvalves. Cardiovasc Pathol 2009;18(4):191–7.
[17] LeBaron RG, Zimmermann DR, Ruoslahti E. Hyaluronate bindingproperties of versican. J Biol Chem 1992;267(14):10003–10.
[18] Isogai Z, Aspberg A, Keene DR, Ono RN, Reinhardt DP, Sakai LY.Versican interacts with fibrillin-1 and links extracellular microfibrilsto other connective tissue networks. J Biol Chem 2002;277(6):4565–72.
[19] Aspberg A, Adam S, Kostka G, Timpl R, Heinegard D. Fibulin-1 is aligand for the C-type lectin domains of aggrecan and versican. J BiolChem 1999;274(29):20444–9.

82 J.M. Carthy et al. / Cardiovascular Pathology 21 (2012) 74–82
[20] Olin AI, Morgelin M, Sasaki T, Timpl R, Heinegard D, Aspberg A.The proteoglycans aggrecan and Versican form networks with fibulin-2 through their lectin domain binding. J Biol Chem 2001;276(2):1253–61.
[21] Aspberg A, Binkert C, Ruoslahti E. The versican C-type lectin domainrecognizes the adhesion protein tenascin-R. Proc Natl Acad Sci U S A1995;92(23):10590–4.
[22] Yamagata M, Yamada KM, Yoneda M, Suzuki S, Kimata K.Chondroitin sulfate proteoglycan (PG-M-like proteoglycan) is in-volved in the binding of hyaluronic acid to cellular fibronectin. J BiolChem 1986;261(29):13526–35.
[23] Dours-Zimmermann MT, Zimmermann DR. A novel glycosamino-glycan attachment domain identified in two alternative splice variantsof human versican. J Biol Chem 1994;269(52):32992–8.
[24] Zako M, Shinomura T, Ujita M, Ito K, Kimata K. Expression of PG-M(V3), an alternatively spliced form of PG-M without a chondroitinsulfate attachment in region in mouse and human tissues. J Biol Chem1995;270(8):3914–8.
[25] Yamagata M, Kimata K. Repression of a malignant cell–substratumadhesion phenotype by inhibiting the production of the anti-adhesiveproteoglycan, PG-M/versican. J Cell Sci 1994;107(Pt 9):2581–90.
[26] Yamagata M, Saga S, Kato M, Bernfield M, Kimata K. Selectivedistributions of proteoglycans and their ligands in pericellular matrix ofcultured fibroblasts. Implications for their roles in cell–substratumadhesion. J Cell Sci 1993;106(Pt 1):55–65.
[27] Evanko SP, Angello JC, Wight TN. Formation of hyaluronan- andversican-rich pericellular matrix is required for proliferation andmigration of vascular smooth muscle cells. Arterioscler Thromb VascBiol 1999;19(4):1004–13.
[28] Huang R, Merrilees MJ, Braun K, Beaumont B, Lemire J, Clowes AW,Hinek A, Wight TN. Inhibition of versican synthesis by antisense alterssmooth muscle cell phenotype and induces elastic fiber formation invitro and in neointima after vessel injury. Circ Res 2006;98(3):370–7.
[29] Ricciardelli C, Russell DL, Ween MP, Mayne K, Suwiwat S, Byers S,Marshall VR, Tilley WD, Horsfall DJ. Formation of hyaluronan- andversican-rich pericellular matrix by prostate cancer cells promotes cellmotility. J Biol Chem 2007;282(14):10814–25.
[30] ShengW, Wang G, Wang Y, Liang J, Wen J, Zheng PS, Wu Y, Lee V,Slingerland J, Dumont D, Yang BB. The roles of versican V1 and V2isoforms in cell proliferation and apoptosis. Mol Biol Cell 2005;16(3):1330–40.
[31] Wu Y, Wu J, Lee DY, Yee A, Cao L, Zhang Y, Kiani C, Yang BB.Versican protects cells from oxidative stress-induced apoptosis. MatrixBiol 2005;24(1):3–13.
[32] KawashimaH, HiroseM,Hirose J, NagakuboD, Plaas AH,MiyasakaM.Binding of a large chondroitin sulfate/dermatan sulfate proteoglycan,versican, to L-selectin, P-selectin, and CD44. J Biol Chem 2000;275(45):35448–56.
[33] Aruffo A, Stamenkovic I, Melnick M, Underhill CB, Seed B. CD44 isthe principal cell surface receptor for hyaluronate. Cell 1990;61(7):1303–13.
[34] Liang CC, Park AY, Guan JL. In vitro scratch assay: a convenient andinexpensive method for analysis of cell migration in vitro. Nat Protoc2007;2(2):329–33.
[35] Abraham T, Carthy J, McManus B. Collagen matrix remodeling in 3-dimensional cellular space resolved using second harmonic generation andmultiphoton excitation fluorescence. J Struct Biol 2009;169(1):36–44.
[36] Marchant D, Sall A, Si X, Abraham T, Wu W, Luo Z, Petersen T,Hegele RG, McManus BM. ERK MAP kinase-activated Arf6trafficking directs coxsackievirus type B3 into an unproductivecompartment during virus host-cell entry. J Gen Virol 2009;90(Pt 4):854–62.
[37] Rezai N, Corbel SY, Dabiri D, Kerjner A, Rossi FM, McManus BM,Podor TJ. Bone marrow-derived recipient cells in murine transplantedhearts: potential roles and the effect of immunosuppression. Lab Invest2005;85(8):982–91.
[38] Fayet C, Bendeck MP, Gotlieb AI. Cardiac valve interstitial cellssecrete fibronectin and form fibrillar adhesions in response to injury.Cardiovasc Pathol 2007;16(4):203–11.
[39] Marhaba R, Zoller M. CD44 in cancer progression: adhesion,migration and growth regulation. J Mol Histol 2004;35(3):211–31.
[40] Ricciardelli C, Sakko AJ, Ween MP, Russell DL, Horsfall DJ. Thebiological role and regulation of versican levels in cancer. CancerMetastasis Rev 2009;28(1–2):233–45.
[41] Lin H, Wilson JE, Roberts CR, Horley KJ, Winters GL, Costanzo MR,McManus BM. Biglycan, decorin, and versican protein expressionpatterns in coronary arteriopathy of human cardiac allograft:distinctness as compared to native atherosclerosis. J Heart LungTransplant 1996;15(12):1233–47.
[42] Evanko SP, Raines EW, Ross R, Gold LI, Wight TN. Proteoglycandistribution in lesions of atherosclerosis depends on lesion severity,structural characteristics, and the proximity of platelet-derived growthfactor and transforming growth factor-beta. Am J Pathol 1998;152(2):533–46.
[43] Camenisch TD, Spicer AP, Brehm-Gibson T, Biesterfeldt J, AugustineML, Calabro A, Kubalak S, Klewer SE, McDonald JA. Disruption ofhyaluronan synthase-2 abrogates normal cardiac morphogenesis andhyaluronan-mediated transformation of epithelium to mesenchyme.J Clin Invest 2000;106(3):349–60.
[44] Wight TN, Merrilees MJ. Proteoglycans in atherosclerosis andrestenosis: key roles for versican. Circ Res 2004;94(9):1158–67.
[45] Hirose J, Kawashima H, Yoshie O, Tashiro K, Miyasaka M. Versicaninteracts with chemokines and modulates cellular responses. J BiolChem 2001;276(7):5228–34.
[46] Xiang YY, Dong H, Wan Y, Li J, Yee A, Yang BB, Lu WY. VersicanG3 domain regulates neurite growth and synaptic transmission ofhippocampal neurons by activation of epidermal growth factorreceptor. J Biol Chem 2006;281(28):19358–68.
[47] Zheng PS, Wen J, Ang LC, Sheng W, Viloria-Petit A, Wang Y, Wu Y,Kerbel RS, Yang BB. Versican/PG-M G3 domain promotes tumorgrowth and angiogenesis. FASEB J 2004;18(6):754–6.
[48] Acharya PS, Majumdar S, Jacob M, Hayden J, Mrass P, Weninger W,Assoian RK, Pure E. Fibroblast migration is mediated by CD44-dependent TGF beta activation. J Cell Sci 2008;121(Pt 9):1393–402.
[49] Lokeshwar VB, Bourguignon LY. Post-translational protein modifi-cation and expression of ankyrin-binding site(s) in GP85 (Pgp-1/CD44) and its biosynthetic precursors during T-lymphoma mem-brane biosynthesis. J Biol Chem 1991;266(27):17983–9.




















