
Mutations in MFSD8/CLN7 are a frequent cause of variant-late infantile neuronal ceroid...
11
DOI: 10.1002/humu.20975 MUTATION IN BRIEF © 2009 WILEY-LISS, INC. HUMAN MUTATION Mutation in Brief #1052, 30:E530-E540, (2008) Online HUMAN MUTATION Received 28 August 2008; accepted revised manuscript 17 December 2008. Mutations in MFSD8/CLN7 Are a Frequent Cause of Variant-Late Infantile Neuronal Ceroid Lipofuscinosis Chiara Aiello 1 , Alessandra Terracciano 1 , Alessandro Simonati 2 , Giancarlo Discepoli 3 , Natalia Cannelli 1 , Dianela Claps 1 , Yanick J. Crow 4 , Marzia Bianchi 1 , Claudia Kitzmuller 5 , Daniela Longo 1 , Antonietta Tavoni 2 , Emilio Franzoni 6 , Alessandra Tessa 1 , Edwige Veneselli 7 , Renata Boldrini 1 , Mirella Filocamo 7 , Ruth E. Williams 8 , Enrico S. Bertini 1 , Roberta Biancheri 7 , Rosalba Carrozzo 1 , Sara E. Mole 5, 9 , and Filippo M. Santorelli 1* 1 Molecular Medicine, Neurology, Radiology, and Pathology, IRCCS-Bambino Gesù Hospital, Rome; 2 Department Neurological and Visual Sciences-Neurology, University of Verona Medical School, Verona; 3Neuropediatrics and Medical Genetics, Az. Ospedaliera Salesi, Ancona, Italy; 4 Leeds Institute of Molecular Medicine, St James's University Hospital, Leeds; 5 MRC Laboratory for Molecular Cell Biology, University College London, United Kingdom; 6 Child Neuropsychiatry, University of Bologna, Italy; 7 Child Neuropsichiatry and Tissue Bank Service, IRCCS-G.Gaslini Institute, Genova, Italy; 8 Department of Paediatric Neurology, Evelina Childrens Hospital, London; 9 Molecular Medicine Unit, UCL Institute of Child Health, and Department of Genetics, Evolution and Environment, University College London, United Kingdom *Correspondence to F.M. Santorelli, MD; Molecular Medicine, Piazza S. Onofrio, 4 - 00165 Rome, Italy Tel +390668592105 Fax +390668592024 E-mail: [email protected] Communicated by David S. Rosenblatt ABSTRACT: The neuronal ceroid lipofuscinoses (NCL) are a group of genetically heterogeneous neurodegenerative disorders. The recent identification of the MFSD8/CLN7 gene in a variant-late infantile form of NCL (v-LINCL) in affected children from Turkey prompted us to examine the relative frequency of variants in this gene in Italian patients with v-LINCL. We identified nine children harboring 11 different mutations in MFSD8/CLN7. Ten mutations were novel and included three nonsense (p.Arg35Stop, p.Glu381Stop, p.Arg482Stop), four missense (p.Met1Thr, p.Gly52Arg, p.Thr294Lys, p.Pro447Leu), two splice site mutations (c.863+3_4insT, c.863+1G>C), and a 17-bp deletion predicting a frameshift and premature protein truncation (c.627_643del17/p.Met209IlefsX3). The clinical phenotype, which was similar to that of the Turkish v-LINCL cases, was not influenced by type and location of the mutation nor the length of the predicted residual gene product. As well as identifying novel variants in MFSD8/CLN7, this study contributes to a better molecular characterization of Italian NCL cases, and will facilitate medical genetic counseling in such families. The existence of a subset of v-LINCL cases without mutations in any of the known NCL genes suggests further genetic heterogeneity. © 2009 Wiley- Liss, Inc. KEY WORDS: MFSD8, CLN7, neuronal ceroid lipofuscinosis, NCL, v-LINCL INTRODUCTION The neuronal ceroid lipofuscinoses (NCL) are a heterogeneous group of pediatric neurodegenerative disorders (Mole et al., 2005; Haltia, 2006; Williams et al., 2006) sharing the clinical symptoms of macular-cerebral OFFICIAL JOURNAL www.hgvs.org
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Mutations in MFSD8/CLN7 are a frequent cause of variant-late infantile neuronal ceroid...

DOI: 10.1002/humu.20975
MUTATION IN BRIEF
© 2009 WILEY-LISS, INC.
HUMAN MUTATION Mutation in Brief #1052, 30:E530-E540, (2008) Online
HUMAN MUTATION
Received 28 August 2008; accepted revised manuscript 17 December 2008.
Mutations in MFSD8/CLN7 Are a Frequent Cause of Variant-Late Infantile Neuronal Ceroid Lipofuscinosis
Chiara Aiello1, Alessandra Terracciano1, Alessandro Simonati2, Giancarlo Discepoli3, Natalia Cannelli1, Dianela Claps1, Yanick J. Crow4, Marzia Bianchi1, Claudia Kitzmuller5, Daniela Longo1, Antonietta Tavoni2, Emilio Franzoni6, Alessandra Tessa1, Edwige Veneselli7, Renata Boldrini1, Mirella Filocamo7, Ruth E. Williams8, Enrico S. Bertini1, Roberta Biancheri7, Rosalba Carrozzo1, Sara E. Mole5, 9, and Filippo M. Santorelli1*
1Molecular Medicine, Neurology, Radiology, and Pathology, IRCCS-Bambino Gesù Hospital, Rome; 2Department Neurological and Visual Sciences-Neurology, University of Verona Medical School, Verona; 3Neuropediatrics and Medical Genetics, Az. Ospedaliera Salesi, Ancona, Italy; 4Leeds Institute of Molecular Medicine, St James's University Hospital, Leeds; 5MRC Laboratory for Molecular Cell Biology, University College London, United Kingdom; 6Child Neuropsychiatry, University of Bologna, Italy; 7Child Neuropsichiatry and Tissue Bank Service, IRCCS-G.Gaslini Institute, Genova, Italy;8Department of Paediatric Neurology, Evelina Childrens Hospital, London; 9Molecular Medicine Unit, UCL Institute of Child Health, and Department of Genetics, Evolution and Environment, University College London, United Kingdom
*Correspondence to F.M. Santorelli, MD; Molecular Medicine, Piazza S. Onofrio, 4 - 00165 Rome, Italy Tel +390668592105 Fax +390668592024 E-mail: [email protected] Communicated by David S. Rosenblatt
ABSTRACT: The neuronal ceroid lipofuscinoses (NCL) are a group of genetically heterogeneous neurodegenerative disorders. The recent identification of the MFSD8/CLN7 gene in a variant-late infantile form of NCL (v-LINCL) in affected children from Turkey prompted us to examine the relative frequency of variants in this gene in Italian patients with v-LINCL. We identified nine children harboring 11 different mutations in MFSD8/CLN7. Ten mutations were novel and included three nonsense (p.Arg35Stop, p.Glu381Stop, p.Arg482Stop), four missense (p.Met1Thr, p.Gly52Arg, p.Thr294Lys, p.Pro447Leu), two splice site mutations (c.863+3_4insT, c.863+1G>C), and a 17-bp deletion predicting a frameshift and premature protein truncation (c.627_643del17/p.Met209IlefsX3). The clinical phenotype, which was similar to that of the Turkish v-LINCL cases, was not influenced by type and location of the mutation nor the length of the predicted residual gene product. As well as identifying novel variants in MFSD8/CLN7, this study contributes to a better molecular characterization of Italian NCL cases, and will facilitate medical genetic counseling in such families. The existence of a subset of v-LINCL cases without mutations in any of the known NCL genes suggests further genetic heterogeneity. © 2009 Wiley-Liss, Inc.
KEY WORDS: MFSD8, CLN7, neuronal ceroid lipofuscinosis, NCL, v-LINCL
INTRODUCTION
The neuronal ceroid lipofuscinoses (NCL) are a heterogeneous group of pediatric neurodegenerative disorders (Mole et al., 2005; Haltia, 2006; Williams et al., 2006) sharing the clinical symptoms of macular-cerebral
OFFICIAL JOURNAL
www.hgvs.org

E531 Further mutations in CLN7
degeneration, drug-resistant epilepsy, progressive motor and cognitive deterioration, and premature death (Mitchinson et al., 2004; Zhong 2004). All are characterized by intracellular storage of autofluorescent material. Knowledge about NCL has expanded in recent years with the identification of underlying genetic defects in ten different human forms (Siintola et al., 2006) caused by mutations in eight genes (CLN1, CLN2, CLN3, CLN5, CLN6, CLN7, CLN8, CLN10/CTSD). The causal genes in two forms, CLN4 and CLN9, have yet to be identified (Mole et al., 2005), and the genetic defect remains unknown in ~10-15% of cases.
The CLN7 form was first described in variant-late infantile NCL (v-LINCL) children from Turkey (Mitchell et al., 2001; Topçu et al., 2004), and six different mutations were identified in a new gene, MFSD8/CLN7 (MIM# 611124; Siintola et al., 2007), in five Turkish and one Indian kindred. Recently, a new c.362A>G variant has been reported in an Egyptian family (Stogmann et al., 2008).
We previously demonstrated that Italian and Turkish v-LINCL patients share many clinical features, suggesting the presence of similar NCL forms in the Mediterranean basin (Cannelli et al., 2006). We therefore analyzed the MFSD8/CLN7 gene in Italian v-LINCL patients.
MATERIALS AND METHODS
Patients
Twenty-three patients, 12 boys and 11 girls, aged 2-16 years were studied if they met clinical and ultrastructural criteria for NCL as reported previously (Cannelli et al., 2006). All the patients had been referred to our centers in the past few years with a possible diagnosis of NCL and had already been tested biochemically for the activities of PPT1 and TPP-I and genetically for mutations in the CLN1-2-3 and CLN5-6-8 genes (Simonati and Rizzuto, 2000; Tessa et al., 2000; Bonsignore et al., 2006; Cannelli et al. 2007). Mutations in the CTSD gene associated with both congenital and v-LINCL forms (Siintola et al., 2006) were also ruled out by sequencing/enzyme analysis prior to this study (unpublished). With the exception of patient 4, who lived in south-east France, all were of known Italian origin. Patient 2 was independently screened for mutations in CLN7 in a separate parallel study, as patient 397Pa (unpublished).
Genetic studies All the studies were performed with parental consent following the guidelines of our Institutional Ethic
Committees. The MFSD8/CLN7gene was analyzed in peripheral blood DNA by PCR amplification and direct sequencing using BigDye 3.1 chemistry (Applied Biosystems, Foster City, CA, USA) and intronic oligonucleotide primers designed to flank coding exons of the MFSD8/CLN7 (Hs.480701) gene on chromosome 4 (Table 1).
Table 1. Oligonucleotide primers designed to amplify the coding exons of the MFSD8/CLN7 gene.
Exon Primer Forward / Reverse (5’-3’) 2 AGCTCTCCTGGGCTCTCAGT / GACGCCCTTTCTCCTACGTT 3 CTGTTGACACTTAAGATCCACATGTAA / CACAATAAAGATAATTTCAGAGGCAAT 4 GCCACGGTTCAGAAAATTGTA / GCACAATAAAGCATATGGTCACA 5 TTCTTGGATAACAATTCTTAAAGCAC / CATGAGCAAAGGCAAAATGA 6 TGTGTGATCATAAAGAGGAAGCA / CCATCTGTTGAGAATGCATGA 7 CCATCTGTTGAGAATGCATGA / TCTTCCAACAATTACAAAGCTCA 8 CAATGCCGATAATAAATTTAAACAA / ATCTTCCCCAATCACCCACT 9 GGTAATGTAATTTTTCTCCTATTTCC / CTACATTCAAGTTACAAGCAACAAAA
10 AAAGGCATGCACATTCTTGA / TCCTTTGCATTGTTAGCTCTG 11-12 GGAGTCCTGGTTATTTTTAGTGGA / AAATCCCTCAAATCAGTCTGTG
13 CCCCAATATCAATCTGCTTGT / TGTAGTCTGATTCTTGGAGACTGG Nomenclature of mutations followed the guidelines of the Human Genome Variation Society
(http://www.hgvs.org/mutnomen) and refers to the cDNA sequence (GenBank reference sequence version NM_152778.1) with the A of the translation initiation codon as +1. Synonymous, missense and splice site variations were systematically evaluated for modifications of exonic splicing enhancers (www.rulai.cshl.edu/cgi-

E532 Aiello et al.
bin/tools/ESE/esefinder.cgi) or consensus splicing sequences in order to determine the splice site score (rulai.cshl.edu/new_alt_exon_db2/HTML/score.html and www.fruitfly.org /seq_tools/splice.html), and the likelihood of a deleterious effect on structure or function (PolyPhen, www.genetics.bwh.harvard.edu/pph/, and SIFT, www.blocks.fhcrc.org/sift/SIFT.html). Multiple alignments with MFSD8/CLN7orthologs were performed using ClustalW (www.ebi.ac.uk/clustalw/) to evaluate the degree of conservation of missense variants. The predicted secondary structure of the wild-type and mutant proteins was performed using the PSIPRED software (McGuffin et al., 2000; Aydin et al., 2006).
The c.154G>A mutation was ruled out in control alleles by high resolution melting (HRM)-PCR (Rotor-Gene 6000, Corbett, Australia), and the presence of the remaining missense mutations in the normal population was tested by mutation specific PCR-restriction fragment length polymorphism (PCR-RFLP) analysis in 500 Italian control chromosomes. For this, the relevant amplicon was amplified adopting ad hoc designed PCR conditions and mismatched oligonucleotide primers, and digested with NlaIII (c.2T>C), DraI (c.881C>A), and HaeIII (c.1340C>T) (New England BioLabs Inc., Beverly, MA). Briefly, a 262-bp fragment is normally cleaved with NlaIII into fragments sized 239- and 23-bp, but the presence of the c.2T>C mutation removes the single site of cleavage. The c.881C>A mutation introduces an additional DraI site of cleavage in a 216-bp amplicon, with control sequences being cleaved into fragments sized 176- and 40-bp, and the presence of the mutation producing fragments sized 153-, 40-, and 23-bp. Finally, a 351-bp amplicon is normally cleaved with the endonuclease HaeIII into fragments sized 255-, and 96-bp but the c.1406C>T mutation removes the site of cleavage generating an uncut fragment. Fragments were resolved on a Metaphor 2%-agarose 1% gel and stained with ethidium bromide.
Only first-degree relatives in five families (cases #2, 3, and 6-8) could be genotyped a posteriori in peripheral blood DNA with fluorescently-labeled microsatellite markers surrounding the CLN7 locus as reported (Siintola et al., 2007). Haplotypes were constructed manually assuming a minimal number of recombination.
Other methods Total RNA was purified from cultured skin fibroblasts obtained from three children (patients 3, 7 and 9) and
five age-matched normal controls using TriReagent (Sigma-Aldrich, St.Louis, MO) and was reversely transcribed using the 1st Strand cDNA Synthesis Kit (Roche, Hamburg, Germany) according to the manufacturer's random primer protocol. The consequences of the c.863+3insT mutation on splicing in patient 3 were examined by RT-PCR using primers from exons 6 and 12. The determination of the expression level of the MFSD8/CLN7 mRNA in patients 3, 7, and 9 was analyzed by quantitative real-time PCR (qPCR) in a MicroAmp® optical 96-well plate using an inventoried TaqMan®-MBP probe (Hs00380724_m1, Applied Biosystems, Foster City, CA) and the automated ABI Prism 7500 detector system (Applied Biosystems). The reaction mixture (25 μl) contained template, 1X TaqMan Universal Master mix (Applied Biosystems), 200 nM NCL probe, and 900 nM each primer. To correct for differences in the amount of starting first-stranded cDNAs, the human GAPDH/mRNA (TaqMan®-MBP assay ID Hs99999905_m1) was amplified in parallel as a reference endogenous control. The relative quantification of the MFSD8-CLN7/mRNAs was performed according to the comparative method (2—ΔΔCt) (Livak and Schmittgen, 2001; Applied Biosystems User Bulletin 2 P/N 4303859). The mean result of three independent experiments run in triplicate was compared with that of normal controls whose expression was arbitrarily attributed the value of 1.
Further mutations in CLN7 E533
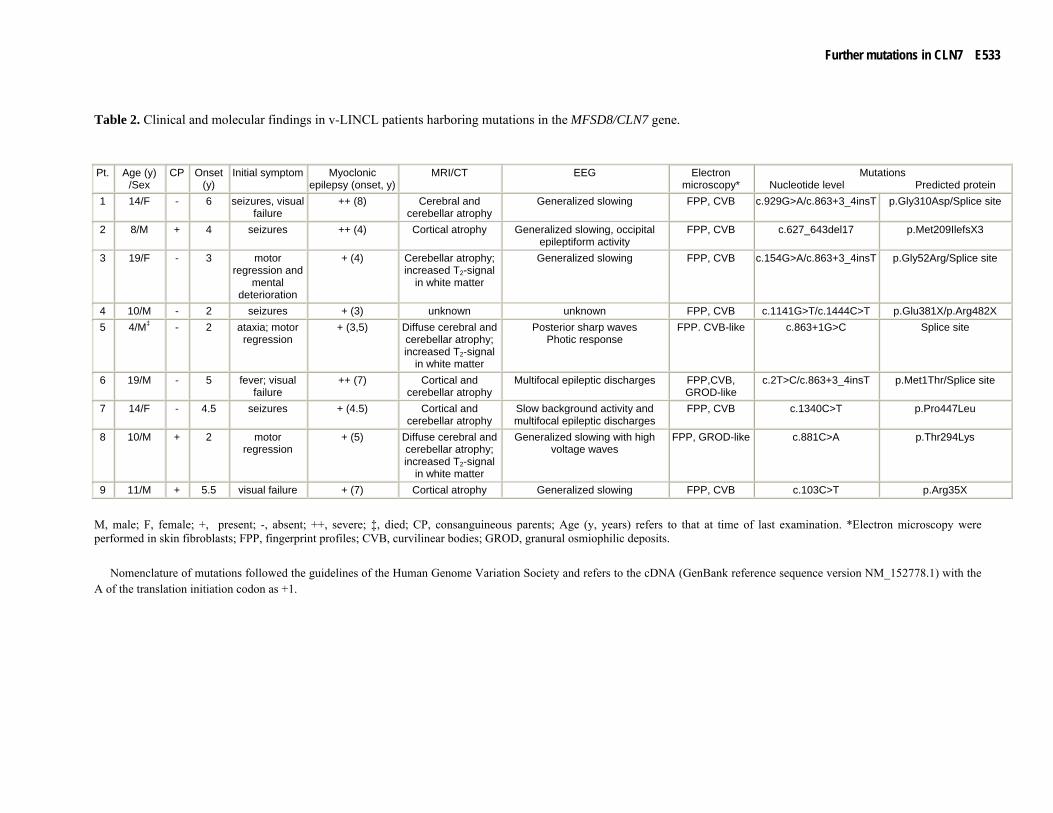
Table 2. Clinical and molecular findings in v-LINCL patients harboring mutations in the MFSD8/CLN7 gene.
M, male; F, female; +, present; -, absent; ++, severe; ‡, died; CP, consanguineous parents; Age (y, years) refers to that at time of last examination. *Electron microscopy were performed in skin fibroblasts; FPP, fingerprint profiles; CVB, curvilinear bodies; GROD, granural osmiophilic deposits.
Nomenclature of mutations followed the guidelines of the Human Genome Variation Society and refers to the cDNA (GenBank reference sequence version NM_152778.1) with the A of the translation initiation codon as +1.
Pt. Age (y) /Sex
CP Onset (y)
Initial symptom Myoclonic epilepsy (onset, y)
MRI/CT EEG Electron microscopy*
Mutations Nucleotide level Predicted protein
1 14/F - 6 seizures, visual failure
++ (8) Cerebral and cerebellar atrophy
Generalized slowing FPP, CVB c.929G>A/c.863+3_4insT p.Gly310Asp/Splice site
2 8/M + 4 seizures ++ (4) Cortical atrophy Generalized slowing, occipital epileptiform activity
FPP, CVB c.627_643del17 p.Met209IlefsX3
3 19/F - 3 motor regression and
mental deterioration
+ (4) Cerebellar atrophy; increased T2-signal
in white matter
Generalized slowing FPP, CVB c.154G>A/c.863+3_4insT p.Gly52Arg/Splice site
4 10/M - 2 seizures + (3) unknown unknown FPP, CVB c.1141G>T/c.1444C>T p.Glu381X/p.Arg482X 5 4/M‡ - 2 ataxia; motor
regression + (3,5) Diffuse cerebral and
cerebellar atrophy; increased T2-signal
in white matter
Posterior sharp waves Photic response
FPP. CVB-like c.863+1G>C Splice site
6 19/M - 5 fever; visual failure
++ (7) Cortical and cerebellar atrophy
Multifocal epileptic discharges FPP,CVB, GROD-like
c.2T>C/c.863+3_4insT p.Met1Thr/Splice site
7 14/F - 4.5 seizures + (4.5) Cortical and cerebellar atrophy
Slow background activity and multifocal epileptic discharges
FPP, CVB c.1340C>T p.Pro447Leu
8 10/M + 2 motor regression
+ (5) Diffuse cerebral and cerebellar atrophy; increased T2-signal
in white matter
Generalized slowing with high voltage waves
FPP, GROD-like c.881C>A p.Thr294Lys
9 11/M + 5.5 visual failure + (7) Cortical atrophy Generalized slowing FPP, CVB c.103C>T p.Arg35X

E534 Aiello et al.
RESULTS
Nine of 23 v-LINCL patients (39%) with genetically undefined disease showed pathogenic mutations in the MFSD8/CLN7 gene. Table 2 summarizes their associated major clinical, neurorimaging, neurophysiological, and ultrastructural features. Patients were characterized by early psychomotor regression and seizures. Mental regression, personality disorders, and speech impairment developed with disease progression and were documented in 7/9 cases within 3-4 years after onset. Unsteady gait and inability to walk unaided were reported in four patients within two years after onset. All this fits well with a clinical diagnosis of NCL. Generalized slowing at EEG in many children, with photic responses in one case, were recorded, resembling what is found in classical late-infantile NCL with mutations in CLN2 (Haltia, 2006).A total of 11 different sequence changes were detected in the coding regions or consensus splice sites; 10 of these are novel mutations whereas one has been previously reported (Siintola et al., 2007). We also identified two new heterozygous variants (c.334C>T/p.Leu112Leu; c.1089A>G/p.Lys363Lys) which were present in 3% and 5% of control alleles respectively.
In three children (patients 1, 3, 6) we detected the heterozygous c.863+3_4insT splice site mutation at the exon-intron 9 junction. The mutation is predicted to affect the correct splicing of the flanking exons. When tested in cultured skin fibroblasts from patient 3, we observed, in addition to the wild-type, extra transcripts lacking exons 8 and 9 or exon 8 only (Figure 1), probably the results of different expression or mRNA stability in that tissue. We cannot exclude that additional, less stable transcripts could be generated in other tissues as well. An additional mutation (c.863+1G>C) was homozygous in patient 5 and might have similar consequences on splicing, although we did not test this in cultured cells.
Figure 1. Scheme of the effects on cDNA from cultured skin fibroblasts of the c.863+3_4insT mutation detected in three children with v-LINCL. CD, patient 3; MW, 100-bp DNA molecular weight marker.
Five missense mutations were identified in this study. Patient 1 was heterozygous for c.929G>A/p.Gly310Asp
in exon 10, already reported in a Turkish child with v-LINCL (Siintola et al., 2007). Patient 3 was heterozygous for c.154G>A/p.Gly52Arg in exon 3 and patient 6 was heterozygous for c.2T>C/p.Met1Thr in exon 2. These three cases, originating from different regions in Italy, harbored the same c.863+3_4insT on the second disease allele but parents were unaware of any relationship. Finally, patients 7 and 8 were homozygous for c.1340C>T/p.Pro447Leu in exon 12 and c.881C>A/p.Thr294Lys in exon 10, respectively. These novel mutations were identified at the heterozygous state in parents and available relatives (whenever this was possible), were not detected in a large set of ethnically-matched control chromosomes, and they affected amino acids were conserved across vertebrates (Figure 2).

Further mutations in CLN7 E535
Figure 2. Predicted amino acid sequence alignment and relative conservation of MFSD8 among human (Homo sapiens), mouse (Mus musculus), frog (Xenopus laevis), and zebrafish (Danio rerio). Sites of the four novel missense (green), three nonsense variants (cyan) and the 17-bp deletion leading to a frameshift (yellow) identified in this study are indicated with colored shading. Amino acid sequences were aligned using Clustal W.
We also identified three mutations predicted to result in premature protein truncation. Patient 2 was homozygous for a 17-bp deletion (c.627_643del17) in exon 7 which predicts a frameshift and a premature translation termination with a protein likely to be cropped by 40% (p.Met209IlefsX3). Patient 4 was compound heterozygous for two nonsense mutations, c.1141G>T/p.Glu381Stop in exon 12 and c.1444C>T/p.Arg482Stop in exon 13. These mutations are predicted to truncate the protein by 137 and 36 residues respectively. Patient 9 was homozygous for c.103C>T/p.Arg35Stop, which is predicted to severely reduce the size of any mutant protein produced.
Segregation of mutations could be verified in 11 healthy relatives, and none of the control chromosomes harbored the changes detected in this study. Homozygosity was observed at flanking markers (D4S2975, D4S2567E, D4S1027, D4S587, D4S1002, D4S3304, and D4S2938) in the MFSD8/CLN7 region in patient 7, but not in the unaffected parents and younger sister (Figure 3). Analysis of flanking haplotypes in patients 1, 3, and 6 showed that the c.863+3_4insT mutation occurred on an almost identical genetic background. In particular, the flanking haplotype was identical in cases 3 and 6 (110-167-244- MFSD8/CLN7-132), suggesting a shared ancestor. The remaining mutations occurred on different genetic backgrounds.
MFSD8-CLN7 mRNA in patients 3, 7, and 9 was reduced when compared to controls (64%, 30%, and 40% of normal mRNA levels, respectively), as expected.

E536 Aiello et al.
Figure 3. Haplotype reconstruction in five Italian kindred harboring mutations in the MFSD8/CLN7 gene. M (capital letter or small letters) indicatets the mutations identified in the patients and their presence in relatives. +, wild-type sequence in MFSD8/CLN7.
DISCUSSION
Identification of the MFSD8/CLN7 gene represents the latest molecular determinant in NCL. As with all new disease etiologies, it is important to establish their frequency and clinical spectrum. This is crucial in disorders such as NCL where an almost homogeneous clinical picture in some affected families is associated with great genetic heterogeneity. To start this process, we sequenced the MFSD8/CLN7 gene in 23 patients with v-LINCL without mutations in any of the known NCL genes. We identified 11 mutations in MFSD8/CLN7, 10 of which were novel, in nine children. Figure 4 shows the location of new mutations with regard to predicted transmembrane domains of MFSD8/CLN7. The first conclusion suggested by our data is that mutations in MFSD8/CLN7 are a

Further mutations in CLN7 E537
significant cprior molecascertained ito that for CInterestinglyaffected v-Lthis family m
Figure 4. Loregard to tran2007. Circle s
The secopresentationwere usuallyonce at schopatient (caseInitial brainneurodegenematter mighprogressive 7). Similar tmaterial in osmiophilic that cytosomdeficiency (phenotype opredicted remutations se
cause of NCL, cular genetic in our centers
CLN6 (unpubliy, patient 8, wLINCL cases inmay also be of
ocation of reportnsmembrane domsymbols, missen
ond conclusionn in our patienty the presentinool, but at timee 1). Most of pn MRI scan eration involviht be due to ecerebral and ce
to patients withMFSD8/CLN
deposits (GROmes are more(Simonati andoriginally obsersidual gene preverely reduce
at least of the vconfirmation. to date, mutati
ished), and mowho had v-LINn Roma Gypsy Roma extracti
ted (black symbmains. MFSD8
nse mutations; sq
n of this studts and the genng symptom (Tes were presentpatients are sti
showed cereing cortical streither gray maerebellar atroph mutations in N7 cases wasOD), and variae frequently hd Rizzuto, 200rved in Turkis
roduct or the pMFSD8 functi
v-LINCL formWhen consid
ions in MFSD8ore common thNCL, harbored
families (Dvoion.
bols) and novel is predicted to
quare symbols, s
dy is that thenetic findings. Table 2). Retint at onset. Spasll alive in theibellar atrophyructures and, i
atter loss or anhies typically oCLN5 (Cannel
s ultrastructurants of rectilineheterogenous in00; Cannelli esh v-LINCL caposition of the ion.
m, as we found dering the wh8/CLN7 accounhan variations d the homozygoràkovà and Mo
mutations (redcontain 12 tranplice site; diamo
ere appeared tOnset occurrednal degeneratiosticity, dystonir teens and cany, but subseqin three case,n absolute incrobserved are shlli et al., 2007)rally polymorpear profiles wern NCL varianet al., 2007). ases, and appeamutated amino
them in 39% ohole group of nt for about 14in CLN1 (9%)
gous p.Thr294Lole, personal c
symbols) in thensmembrane domond, nonsense; s
to be little cod with a medion and visual ic posturing, ann barely walk quent scans swhite matter. rease in volumhown over a 6-) and CLN8 (Cphic: fingerprre observed. Tnts unrelated
Overall, the ared not to be o acid residue.
of this series of 65 Italian v4% of cases, a ), CLN8 (6%), Lys, recently dcommunication
e MFSD8/CLN7 mains, accordingtar, deletion
orrelation betwan age of 4 yefailure typicalnd tremor werewith support,
showed a proDiffuse hypei
me (Worgall e-year period in
Cannelli et al., 2rint profiles his is consistento primary lyclinical picturinfluenced by
. This may be
f children lackv-LINCL patiefrequency simand CLN5 (3%
detected in man), suggesting t
7 gene product wg to Siintola et
ween the cliniears, and seizully occurred lae observed in oor are bedridd
ogression of intensity of whet al., 2007). Tn Figure 5 (pati2006), the stora(FPPs), granunt with the not
ysosomal enzyre resembles the length of because all th
king ents
milar %). any that
with al.,
ical ures ater, one
den. the
hite The ient age ular tion yme the the
hese
E538 Aiello et al.
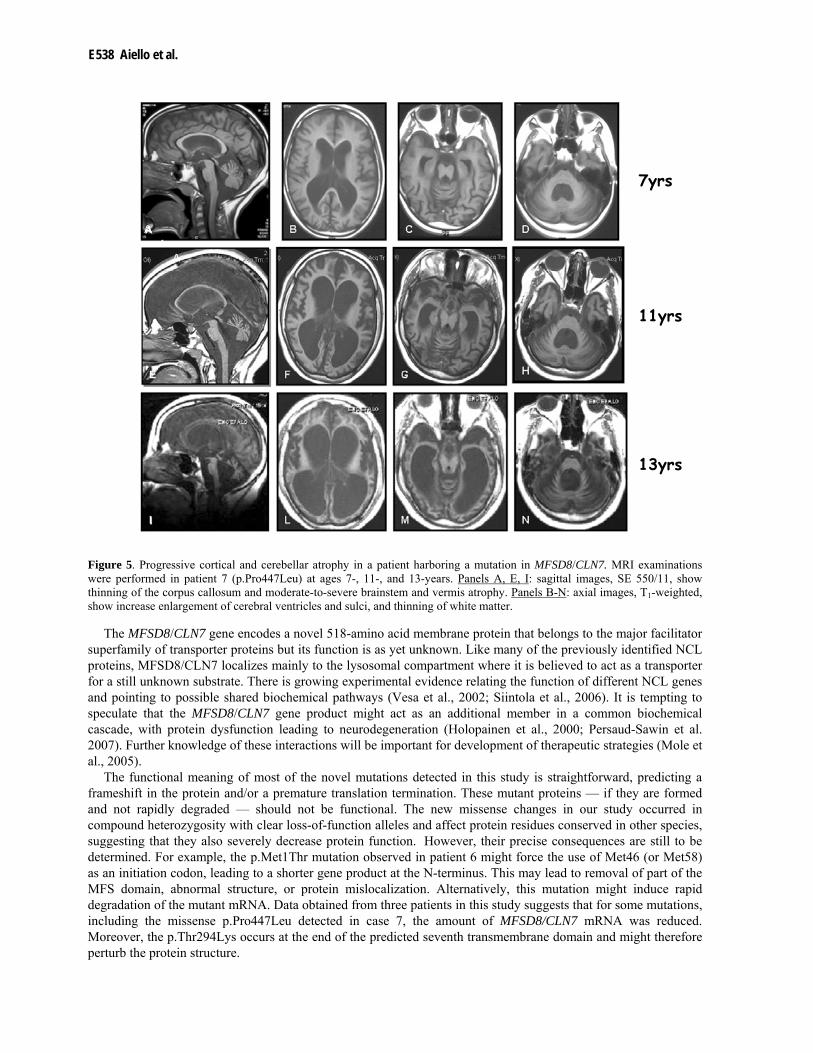
Figure 5. Progressive cortical and cerebellar atrophy in a patient harboring a mutation in MFSD8/CLN7. MRI examinations were performed in patient 7 (p.Pro447Leu) at ages 7-, 11-, and 13-years. Panels A, E, I: sagittal images, SE 550/11, show thinning of the corpus callosum and moderate-to-severe brainstem and vermis atrophy. Panels B-N: axial images, T1-weighted, show increase enlargement of cerebral ventricles and sulci, and thinning of white matter.
The MFSD8/CLN7 gene encodes a novel 518-amino acid membrane protein that belongs to the major facilitator superfamily of transporter proteins but its function is as yet unknown. Like many of the previously identified NCL proteins, MFSD8/CLN7 localizes mainly to the lysosomal compartment where it is believed to act as a transporter for a still unknown substrate. There is growing experimental evidence relating the function of different NCL genes and pointing to possible shared biochemical pathways (Vesa et al., 2002; Siintola et al., 2006). It is tempting to speculate that the MFSD8/CLN7 gene product might act as an additional member in a common biochemical cascade, with protein dysfunction leading to neurodegeneration (Holopainen et al., 2000; Persaud-Sawin et al. 2007). Further knowledge of these interactions will be important for development of therapeutic strategies (Mole et al., 2005).
The functional meaning of most of the novel mutations detected in this study is straightforward, predicting a frameshift in the protein and/or a premature translation termination. These mutant proteins — if they are formed and not rapidly degraded — should not be functional. The new missense changes in our study occurred in compound heterozygosity with clear loss-of-function alleles and affect protein residues conserved in other species, suggesting that they also severely decrease protein function. However, their precise consequences are still to be determined. For example, the p.Met1Thr mutation observed in patient 6 might force the use of Met46 (or Met58) as an initiation codon, leading to a shorter gene product at the N-terminus. This may lead to removal of part of the MFS domain, abnormal structure, or protein mislocalization. Alternatively, this mutation might induce rapid degradation of the mutant mRNA. Data obtained from three patients in this study suggests that for some mutations, including the missense p.Pro447Leu detected in case 7, the amount of MFSD8/CLN7 mRNA was reduced. Moreover, the p.Thr294Lys occurs at the end of the predicted seventh transmembrane domain and might therefore perturb the protein structure.
7yrs
11yrs
13yrs

Further mutations in CLN7 E539
Our final conclusion supports further genetic heterogeneity in v-LINCL. In our study, 14 v-LINCL patients did not harbor mutations in any of the known NCL genes, including MFSD8/CLN7. Most patients presented with a progressive myoclonic epilepsy and cytosomes of mixed ultrastructural features (FPPs, curvilinear (CVBs) and, more rarely, GROD-like) on skin biopsy. Although the present study did not consider unconventional mutations such as rearrangements or mutations in as yet unidentified regulatory sequences, it seems reasonable to expect the existence of further NCL genes in these cases. In this regard, a recent whole genome scanning (Siintola et al., 2007) of similar patients from Turkey proposed the existence of at least three additional genes which will be pertinent candidates in these Italian cases.
To summarize, we showed that mutations in MFSD8/CLN7 are a significant cause of v-LINCL in Italian children. Our data will facilitate medical genetic counseling and promote more accurate prenatal diagnosis. The clinical picture observed in our patients suggests that cases of late-infantile or early-juvenile onset blindness and myoclonic epilepsy require appropriate morphological and genetic testing. Taking our findings together with information on previous Turkish kindreds (Siintola et al., 2007), particular consideration should be given to mutation screening of the MFSD8/CLN7 gene in v-LINCL patients originating from southern Europe.
ACKNOWLEDGMENTS
The Authors thank Mrs C.J. Wrenn for critically reading and editing the manuscript, Mr Keith Parker who helped with DNA extraction from important blood samples, the families and their physicians. C.A. is a fellow of the Bambino Gesù-Roma Tre University PhD program. This work was supported in part by grants from the Italian Ministry of Health (Ricerca Corrente e Finalizzata) to EB, Fondazione CARIVR-2005 (to AS), and the Wellcome Trust (054606) and the Batten Disease and Research Association (to SM). The contribution of the RNGC (Rare NCL Gene Consortium) is also acknowledged.
REFERENCES
Aydin Z, Altunbasak Y, Borodovsky M. 2006. Protein secondary structure prediction for a single-sequence using hidden semi-Markov models. BMC Bioinformatics 7: 178.
Bonsignore M, Tessa A, Di Rosa G, Piemonte F, Dionisi-Vici C, Simonati A, Calamoneri F, Tortorella G, Santorelli FM. 2006. Novel CLN1 mutation in two Italian sibs with late infantile neuronal ceroid lipofuscinosis. Eur J Paediatr Neurol 10: 154-6.
Cannelli N, Cassandrini D, Bertini E, Striano P, Fusco L, Gaggero R, Specchio N, Biancheri R, Vigevano F, Bruno C, Simonati A, Zara F, Santorelli FM. 2006. Novel mutations in CLN8 in Italian variant late infantile neuronal ceroid lipofuscinosis: Another genetic hit in the Mediterranean. Neurogenetics 7: 111-7.
Cannelli N, Nardocci N, Cassandrini D, Morbin M, Aiello C, Bugiani M, Criscuolo L, Zara F, Striano P, Granata T, Bertini E, Simonati A, Santorelli FM. 2007. Revelation of a novel CLN5 mutation in early juvenile neuronal ceroid lipofuscinosis. Neuropediatrics 38: 46-9.
Haltia M. 2006. The neuronal ceroid-lipofuscinoses: from past to present. Biochim Biophys Acta 1762: 850-6.
Holopainen JM, Saarikoski J, Kinnunen PK, Jarvela II. 2001. Elevated lysosomal pH in neuronal ceroid lipofuscinoses (NCLs). Eur. J. Biochem 268: 5851-6.
Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2–ΔΔCT method. Methods 25:402–8.
McGuffin LJ, Bryson K, Jones DT. 2000. The PSIPRED protein structure prediction server Bioinformatics 16: 404-5.
Mitchell WA, Wheeler RB, Sharp JD, Bate SL, Gardiner RM, Ranta US, Lonka L, Williams RE, Lehesjoki AE, Mole SE. 2001. Turkish variant late infantile neuronal ceroid lipofuscinosis (CLN7) may be allelic to CLN8. Eur J Paediatr Neurol 5, Suppl A:21-7.
Mitchison HM, Lim MJ, Cooper JD. 2004. Selectivity and types of cell death in the neuronal ceroid lipofuscinoses. Brain Pathol 14: 86-96
Mole SE, Williams RE, Goebel HH. 2005. Correlations between genotype, ultrastructural morphology and clinical phenotype in the neuronal ceroid lipofuscinoses. Neurogenetics 6:107-126.

E540 Aiello et al.
Persaud-Sawin DA, Mousallem T, Wang C, Zucker A, Kominami E, Boustany RM. 2007. Neuronal ceroid lipofuscinosis: a common pathway? Pediatr Res 61:146-52.
Ranta S, Topçu M, Tegelberg S, Tan H, Ustübütün A, Saatci I, Dufke A, Enders H, Pohl K, Alembik Y, Mitchell WA, Mole SE, Lehesjoki AE. 2004. Variant late infantile neuronal ceroid lipofuscinosis in a subset of Turkish patients is allelic to Northern epilepsy. Hum Mutat 23: 300-5.
Siintola E, Lehesjoki AE, Mole SE. 2006. Molecular genetics of the NCLs - status and perspectives. Biochim Biophys Acta 1762: 857-64.
Siintola E, Topçu M, Aula N, Lohi H, Minassian BA, Paterson AD, Liu XQ, Wilson C, Lahtinen U, Anttonen AK, Lehesjoki AE. 2007. The novel neuronal ceroid lipofuscinosis gene MFSD8 encodes a putative lysosomal transporter. Am J Hum Genet 81: 136-46.
Simonati A, Rizzuto N. 2000. Neuronal ceroid lipofuscinoses: pathological features of bioptic specimens from 28 patients. Neurol Sci 21: S63-70.
Stogmann E, El Tawil S, Wagenstaller J, Gaber A, Edris S, Abdelhady A, Assem-Hilger E, Leutmezer F, Bonelli S, Baumgartner C, Zimprich F, Strom TM, Zimprich A. 2008. A novel mutation in the MFSD8 gene in late infantile neuronal ceroid lipofuscinosis. Neurogenetics. Oct 11 [Epub ahead of print].
Tessa A, Simonati A, Tavoni A, Bertini E, Santorelli FM. 2000. A novel nonsense mutation (Q509X) in three Italian Late infantile neuronal ceroid-lipofuscinosis children. Hum Mut (on line) 15:577.
Topçu M, Tan H, Yalnizoğlu D, Usubütün A, Saatçi I, Aynaci M, Anlar B, Topaloğlu H, Turanli G, Köse G, Aysun S. 2004. Evaluation of 36 patients from Turkey with neuronal ceroid lipofuscinosis: clinical, neurophysiological, neuroradiological and histopathologic studies. Turk J Pediatr 46: 1-10.
Vesa J, Chin MH, Oelgeschläger K, Isosomppi J, DellAngelica EC, Jalanko A, Peltonen L. 2002. Neuronal Ceroid Lipofuscinoses are connected at molecular level: interaction of CLN5 protein with CLN2 and CLN3. Mol Biol Cell 13:2410-20., Isosomppi
Williams RE, Aberg L, Autti T, Goebel HH, Kohlschütter A, Lönnqvist T. 2006. Diagnosis of the neuronal ceroid lipofuscinoses: an update. Biochim Biophys Acta 1762: 865-72.
Worgall S, Kekatpure MV, Heier L, Ballon D, Dyke JP, Shungu D, Mao X, Kosofsky B, Kaplitt MG, Souweidane MM, Sondhi D, Hackett NR, Hollmann C, Crystal RG. 2007. Neurological deterioration in late infantile neuronal ceroid lipofuscinoses Neurology 69:521-35
Zhong N. 2000. Neuronal ceroid lipofuscinoses and possible pathogenic mechanism. Mol Genet Metab 71: 195-206.




















