
The management of congenital malpositions of eyelids, eyes ...
Upload
khangminh22Category
view
0download
0
ORIGINAL ARTICLE
Synaptic Changes in the Thalamocortical System of CathepsinDYDeficient Mice: A Model of Human Congenital Neuronal
Ceroid-Lipofuscinosis
Sanna Partanen, PhD, Aleksi Haapanen, BM, Catherine Kielar, MSc, Charles Pontikis, PhD,
Noreen Alexander, MSc, Teija Inkinen, MS, Paul Saftig, PhD, Thomas H. Gillingwater, PhD,
Jonathan D. Cooper, PhD, and Jaana Tyynela, PhD
AbstractCathepsin D (CTSD; EC 3.4.23.5) is a lysosomal aspartic
protease, the deficiency of which causes early-onset and particu-
larly aggressive forms of neuronal ceroid-lipofuscinosis in infants,
sheep, and mice. Cathepsin D deficiencies are characterized by
severe neurodegeneration, but the molecular mechanisms behind
the neuronal death remain poorly understood. In this study, we have
systematically mapped the distribution of neuropathologic changes
in CTSD-deficient mouse brains by stereologic, immunologic, and
electron microscopic methods. We report highly accentuated
neuropathologic changes within the ventral posterior nucleus
(ventral posteromedial [VPM]/ventral posterolateral [VPL]) of
thalamus and in neuronal laminae IV and VI of the somatosensory
cortex (S1BF), which receive and send information to the thalamic
VPM/VPL. These changes included pronounced astrocytosis and
microglial activation that begin in the VPM/VPL thalamic nucleus
of CTSD-deficient mice and are associated with reduced neuronal
number and redistribution of presynaptic markers. In addition, loss
of synapses, axonal pathology, and aggregation of synaptophysin
and synaptobrevin were observed in the VPM/VPL. These synaptic
alterations are accompanied by changes in the amount of
synaptophysin/synaptobrevin heterodimer, which regulates forma-
tion of the SNARE complex at the synapse. Taken together, these
data reveal the somatosensory thalamocortical circuitry as a
particular focus of pathologic changes and provide the first
evidence for synaptic alterations at the molecular and ultrastructural
levels in CTSD deficiency.
Key Words: Cathepsin D, Electron microscopy, Lysosome, Neuro-
degeneration, Stereology, Synapse, Thalamus.
INTRODUCTIONCathepsin D (CTSD; EC 3.4.23.5) is a lysosomal
aspartic protease that, like many other lysosomal enzymes, issynthesized as an inactive precursor and proteolyticallyprocessed to yield the mature protein (1, 2). In addition toits enzymatic function, CTSD has a complex, but poorlycharacterized, role in both cell death and proliferation, and ithas been suggested to function in a novel lysosome-associated apoptosis-like cell death pathway (3, 4).
Mutations leading to complete inactivation of CTSDunderlie congenital neuronal ceroid-lipofuscinosis (NCL) inhuman infants and lambs (5, 6). These are fatal neuro-degenerative disorders characterized by extreme brainatrophy and death within the first days of life (5, 6). Incontrast, CTSD-deficient (CTSDj/j) mice are normal atbirth but develop severe neurologic signs, including tremor,epileptic seizures, and motor problems, beginning at the ageof 2 weeks (7). Subsequently, these mice also developatrophic changes in their lymphoid system and smallintestine; they die prematurely at the age of 26 T 2 days(8). Cathepsin D-deficient mice exhibit the main neuro-pathologic characteristics of NCLs, including neuronaldeposition of autofluorescent storage material with granularor fingerprint-type ultrastructure (7, 9, 10), and immunohis-tologic evidence for the accumulation of subunit c of themitochondrial adenosine triphosphate synthase (7). In thebrains of CTSDj/j mice, neuronal loss is accompanied bypronounced microglial activation (11). Activated microgliaare capable of producing nitric oxide, which has beensuggested to play an important role in the neuronal apoptosisand intestinal necrosis in CTSDj/j mice (11). However,inhibition of nitric oxide synthesis provided only a modestimprovement in the life span of these mice (11). Theinvolvement of autophagy in neuronal degeneration and
J Neuropathol Exp Neurol � Volume 67, Number 1, January 200816
J Neuropathol Exp NeurolCopyright � 2007 by the American Association of Neuropathologists, Inc.
Vol. 67, No. 1January 2008
pp. 16Y29
From the Institute of Biomedicine/Biochemistry and Neuroscience ResearchProgram (SP, AH, TI, JT), University of Helsinki, Finland; PediatricStorage Disorders Laboratory (CK, CP, NA, JDC), the James BlackCentre, Institute of Psychiatry, King’s College London, UK; BiochemicalInstitute (PS), Christian-Albrechts-University Kiel, Kiel, Germany; andCentre for Integrative Physiology & Centre for Neuroscience Research(THG), University of Edinburgh, Edinburgh, UK.
Send correspondence and reprint requests to: Jaana Tyynela, Institute ofBiomedicine/Biochemistry, POB 63, 00014 University of Helsinki,Finland; E-mail: [email protected]
The authors Partanen and Haapanen contributed equally to this work.This study was financially supported by grants from the Academy of Finland
(214343; JT), National Institutes of Health (NS41930; JDC), EuropeanCommission (LSHM-CT-2003-503051; JT, JDC), The Batten DiseaseSupport and Research Association (JDC, CK, THG), The Natalie Fund(JDC), The Batten Disease Family Association (JDC), Biotechnologyand Biological Sciences Research Council (THG), and Scottish HospitalEndowments Research Trust (THG).
Sanna Partanen is a member of the Finnish Graduate School of Neuro-science.
Copyright © 2007 by the American Association of Neuropathologists, Inc. Unauthorized reproduction of this article is prohibited.
Dow
nloaded from https://academ
ic.oup.com/jnen/article/67/1/16/2916840 by guest on 27 July 2022

storage deposition has also been implicated in CTSDj/j
mice (7), but despite active research, the molecular mech-anisms involved in the neurodegeneration caused by CTSDdeficiency remain obscure, and more detailed informationabout the events in different brain areas is needed to gaininsights into disease pathogenesis.
To gain a better understanding of the cellular andmolecular mechanisms involved in the pathogenesis ofCTSD deficiency, we have systematically mapped neuro-pathologic alterations in the brains of CTSDj/j mice using acombination of unbiased stereology, immunologic techni-ques, and electron microscopy. We report a series oflocalized neurodegenerative and reactive events within thesomatosensory thalamocortical system of CTSDj/j miceand provide the first evidence for synaptic abnormalities inthe NCLs at both the molecular and ultrastructural levels.
MATERIALS AND METHODS
AnimalsCathepsin DYdeficient mice produced by Saftig et al
(8) were maintained on a mixed C57BL6J strain backgroundin the animal facility of the Helsinki University, Biomedi-cum, where food and water were available ad libitum, andlight-dark cycle was 12-12 hours. The study protocol wasapproved by the ethical committee of Helsinki University.
AntibodiesThe following mouse monoclonal antibodies were
used for immunohistochemical and immunofluorescencestaining of paraffin sections (IHP) or cryosections (IHC)and Western blotting (WB): >-Synaptobrevin (StressgenBiotechnologies, Victoria, Canada, 1:1000 for IHP and WB;Chemicon International, Temecula, CA, 1:500 for IHC),>-syntaxin (Stressgen Biotechnologies, 1:2000 for IHP and1:1500 for WB; Chemicon International, 1:250 for IHC),>-synaptophysin (Stressgen Biotechnologies, 1:200 for IHP,1:750 for WB, and, 1:1000 for IHC), and >-synaptosomalYassociated protein of 25 kd (synaptosomal-associated proteinof 25 kd [SNAP25]; BD Transduction Laboratories, Lexington,KY; 1:1500 for IHP, 1:1000 for WB and IHC) or themicroglial markers F4/80 (monoclonal rat anti-mouse F4/80;Serotec, Oxford, UK; 1:100 for IHC) and CD68 (clone PG-M1; DAKO, Cambridge, UK; 1:100 for IHC and IHP).Polyclonal antisera against the calcium-binding protein par-valbumin (PV) (polyclonal rabbit anti-PV; Swant, Bellinzona,Switzerland; 1:5000) and the astrocytic marker glial fibrillaryacidic protein (GFAP; DAKO, Cambridge, UK; 1:5000) wereused for IHC. Polyclonal antisera against the lysosome-associated membrane protein 1 (Lamp-1; Sigma-Aldrich,Steinheim, Germany; 1:500) and the autophagosome markerlight chain 3 of neuronal microtubule-associated protein 1A/B(LC3; a kind gift of professor Takashi Ueno, JuntendoUniversity Medical School, Tokyo, Japan; 1:100) were usedfor immunofluorescence.
Histologic ProcessingFor light microscopy analysis of cryopreserved sam-
ples, brains were dissected from CTSDj/j mice and age-
matched control+/+ littermates at postnatal day (P) 16, P20,and P23 T 1 (n = 5Y6 for P23 T 1, n = 2 for P16, and n = 3for P20). The brains were fixed for 24 hours in a solutioncontaining 4% paraformaldehyde in PBS (pH 7.4), cryopro-tected at 4-C in Tris-buffered saline (TBS; 50 mmol/L Tris,pH 7.6) containing 30% sucrose and 0.05% NaN3. Brainswere subsequently processed and sectioned as describedpreviously (12) with serial 40-Km frozen coronal andsagittal sections stored at j40-C in cryoprotectant solution(TBS/30% ethylene glycol/15% sucrose/0.05% sodiumazide) in a 96-well plate well before histologic andimmunohistologic processing of the free-floating sections.For paraffin-embedded samples, brains from 23 T 1-day-oldCTSDj/j and CTSD+/+ mice (n = 4) were fixed in 4%neutral-buffered formaldehyde and subsequently processed,embedded in paraffin, and cut into 4-Km-thick sections.
Nissl StainingEvery fifth section was mounted onto gelatin-chrome
alum-coated Superfrost microscope slides (VWR, Dorset,UK), air-dried overnight, and incubated for 45 minutes at60-C in a solution of 0.05% Cresyl fast violet and 0.05%acetic acid (VWR). Stained sections were then rinsed indistilled water and differentiated through alcohol seriesbefore clearing in xylene (VWR) and coverslipping with[p-xylene-bis(pyridinium bromine)] (VWR).
Immunohistochemical andImmunofluorescence Stainings
Every sixth 40-Km free-floating cryosection wasstained for markers of astrocytosis (GFAP and F4/80;n = 5), synapses (synaptobrevin [Syb], synaptophysin [Syp],SNAP25, and syntaxin [Syx]; n = 6), or PV expressed inGABAergic neurons in the reticular thalamic nucleus (n = 5).Sections were first incubated in 1% H2O2 in TBS for 30 (glialmarkers) and 15 minutes (synaptic markers), rinsed in TBS,and blocked with 15% normal serum in TBS for 40 minutes.Sections were incubated with primary antibody diluted inTBS containing 0.3% Triton X-100 and 10% normal serumovernight at 4-C. After washing, sections were incubated inbiotinylated secondary antisera (Vector Laboratories, Peter-borough, UK; 1:2000) in TBS containing 0.3% Triton X-100containing 10% normal serum for 2 hours at 4-C. Afterwashing, sections were incubated with an avidin-biotin-peroxidase complex in TBS (Vectastain Elite ABC kit;Vector Laboratories) for 2 hours. Sections were then rinsedin TBS, and immunoreactivity was visualized by incubationin 0.05% 3,3¶-diaminobenzidine HCl (Sigma, Dorset, UK)and 0.001% H2O2 in TBS for 6 to 20 minutes (a range of timethat represented saturation for this reaction). Sections werethen transferred to excess ice-cold TBS and rinsed severaltimes, mounted, air-dried, cleared in xylene, and coverslippedwith [p-xylene-bis(pyridinium bromine)] (VWR). To obtainreproducible and quantitative results, particular care wastaken to fully saturate the color reaction and to treat allsections stained for the same antigen as 1 batch.
Paraffin-embedded CTSDj/j and control mouse brainsections were immunohistochemically stained for synapticmarkers (Syb, Syp, SNAP25, and Syx). Sections were
J Neuropathol Exp Neurol � Volume 67, Number 1, January 2008 Thalamus and Synapses in CTSD Deficiency
� 2007 American Association of Neuropathologists, Inc. 17
Copyright © 2007 by the American Association of Neuropathologists, Inc. Unauthorized reproduction of this article is prohibited.
Dow
nloaded from https://academ
ic.oup.com/jnen/article/67/1/16/2916840 by guest on 27 July 2022

dewaxed in xylene, antigen retrieval was performed bymicrowaving in citric acid buffer, and endogenous peroxidaseactivity was blocked by incubating the sections in methanolcontaining 1.6% H2O2 at room temperature for 30 minutes.Before mounting, the sections were counterstained withhematoxylin.
For double labeling immunofluorescence studies, thetissue sections were treated with the primary antibodies afterblocking as above, and the primary antibodies were detectedusing species-specific fluorescent secondary antibody con-jugates (Alexa Fluor 488 and Alexa Fluor 568; MolecularProbes Inc., Eugene, OR). The double stainings were visu-alized using Leica TCS SP2 AOBS laser scanning confocalmicroscope with 488- and 561-nm excitation lines (LeicaMicrosystems, Wetzlar, Germany). The images were acquiredusing sequential scanning to avoid channel cross talk. Thedouble stainings for Syb (with 3-amino-9-ethylcarbazoleperoxidase substrate) and CD68 (with Alexa Fluor 488conjugates) were visualized using an Olympus AX70 micro-scope (Olympus Optical, Co., Hamburg, Germany).
Volume EstimationsCavalieri estimates of the volumes of selected brain
regions were made in Nissl-stained sections using Stereo-Investigator software (MicroBrightField, Inc., Williston, VT).Briefly, an appropriately spaced sampling grid (150 Km forcortex, thalamus, corpus callosum, cerebellar gray matter, andwhite matter; 100 Km for hippocampus and striatum; 50 Kmfor the deep cerebellar nuclei), was randomly superimposedover sections, and the number of points covering each centralnervous system (CNS) region was counted from 1 hemi-sphere, and the corresponding regional volume was calculatedas described previously (12). All cerebellar regions weremeasured from 12 consecutive sections, thus, not representingthe total volume of these regions, but giving a good estimateof differences between CTSDj/j and control mice.
Thickness MeasurementsCortical thickness measurements were made in Nissl-
stained sections through different areas of cerebral cortex,including prefrontal cortex, primary motor cortex (M1), barrel-field area of the primary somatosensory cortex (S1BF), andprimary visual cortex, as defined by Paxinos and Franklin (13).This analysis was performed by drawing 10 lines through thecortex to 3 adjacent sections in which the examined corticalarea was present, with the length of each perpendicular lineextending from the white matter to the pial surface measuredusing ImageJ software. Similarly, the thicknesses of individ-ual cortical laminae were measured in M1 and S1BF from 3adjacent Nissl-stained sections, with individual measuresobtained for laminae I, IV (S1BF only), V and VI, whereasa combined measure was taken for laminae II and III.
Threshold Analysis of Syb, Syp, SNAP25, andSyx Thalamus
The optical density of Syb, Syp, SNAP25, and Syximmunoreactivity was assessed using a semiautomatedthresholding image analysis system (Image Pro Plus; MediaCybernetics, Silver Springs, MD). Analysis was performed
blind to genotype, as previously described (12). Forty non-overlapping images were captured through the thalamus on 3consecutive sections via a live video camera (JVC, 3CCD,KY-F55B), mounted onto a Zeiss Axioplan microscope usinga �40 objective, and saved as JPEGs. All parameters,including lamp intensity, video camera setup, and calibration,were maintained constantly throughout image capturing.
Images were subsequently analyzed using Image ProPlus software and an appropriate threshold that selected theforeground immunoreactivity above background. Thisthreshold was then applied as a constant to all subsequentimages analyzed per batch of animals and reagent used todetermine the specific area of immunoreactivity for eachantigen in thalamus. Each field measured 120 Km wide, witha height of 90 Km. Therefore, the total area compiled from40 fields in thalamus corresponded to 432,000 Km2. Macroswere recorded to transfer the data to an Microsoft Excelspreadsheet for statistical analysis. Data were plotted graphi-cally as the mean percentage area of immunoreactivity perfield T SEM.
Line Profile Analysis of ImmunohistochemicalStainings
The distribution of immunoreactivity for GFAP, F4/80, and SYP was analyzed in S1BF using a line profilemethod. Images were captured using a DAGE-MTI CCD-100 camera (DAGE-MTI Inc., Michigan City, IN) from 3adjacent sections of S1BF. Each image was 1,300 � 1,030pixels at a resolution of 150 dots per inch. These imageswere filtered in ImageJ software (Wayne Rasband, NationalInstitutes of Health) using Gaussian blur filter with radiusof 8 pixels. Ten lines were drawn from lamina I to laminaVI upon every image, and the relative pixel brightnesswas measured along the length of each line using a scale of1 to 255, where white = 1 and black = 255. Data fromthese line profiles from individual animals were exportedto an Excel spreadsheet for subsequent analysis. The lengthof each individual line was normalized to correct forcortical atrophy. Finally, the brightness value in each pixelwas inverted to calculate the intensity of the staining be-cause the staining intensity (SI) is inversely correlated tothe brightness of the image. A histogram composed of100 bins was then calculated, with each bin representing1% of the length of the original line, and the SI in eachbin representing the corresponding mean SI value. The re-sults are expressed as percentage difference between theCTSDj/j and control mice as follows: 100% � (SICTSDj/j j
SICTSD+/+) / SICTSD+/+.
Measurement of Total Neuronal NumberStereoInvestigator software was used to obtain optical
fractionator estimates of the number of Nissl-stainedsomatosensory relay neurons in the ventral posterior thala-mic nucleus (VPM/VPL), PV-positive inhibitory neurons inthe reticular thalamic (Rt) nucleus, and Nissl-stained corticalneurons in laminae IV and VI of S1BF. These opticalfractionator estimates were obtained as previously described(14), with a random starting section chosen, followed byevery sixth Nissl-stained or PV-stained section thereafter.
Partanen et al J Neuropathol Exp Neurol � Volume 67, Number 1, January 2008
� 2007 American Association of Neuropathologists, Inc.18
Copyright © 2007 by the American Association of Neuropathologists, Inc. Unauthorized reproduction of this article is prohibited.
Dow
nloaded from https://academ
ic.oup.com/jnen/article/67/1/16/2916840 by guest on 27 July 2022

Only neurons with a clearly identifiable nucleus were sampled,and all counts were performed using a �100 oil objective (NA1.4). The following sampling scheme was applied to eachregion of interest: VPM/VPL dissector frame, 74 � 45 Km;grid area, 175 � 175 Km; Rt dissector frame, 74 � 24 Km;grid area, 100 � 100 Km; S1BF lamina IV and lamina VIdissector frame, 41 � 26 Km; grid area, 175 � 175 Km.
Data AnalysisStatistical significance (p) of the altered mean values
of the structural, threshold, line profile, and neuron numberdata was analyzed with SPSS v12.0 (SPSS, Inc., Chicago,IL) and Microsoft Excel programs using Student t-test. Thevolume data were expressed in cubic millimeters andthickness values in micrometers. The SIs of the line profileanalysis were expressed in arbitrary SI units (see above).Values of all quantitative results are represented as mean TSEM. The mean coefficient of error for all individual opticalfractionator and Cavalieri estimates was calculated accord-ing to the method of Gundersen and Jensen (15) and was lessthan 0.08 in all these analyses.
Electron MicroscopyMice were anesthetized using subcutaneous injections
of ketamine (Orion Pharma, Espoo, Finland; 75Y100 mg/kg)and killed by perfusion fixation with 0.1 mol/L phosphatebuffer containing 4% paraformaldehyde and 2.5% glutaralde-hyde. Brains were removed and immersed in fresh fixative for24 hours, washed, and stored in 0.1 mol/L phosphate bufferbefore cutting free-floating 70-Km coronal sections. Sectionscontaining regions of interest (VPM/VPL and S1BF) werepostfixed in 1% osmium tetroxide in 0.1 mol/L phosphatebuffer for 45 minutes. After dehydration through an ascendingseries of ethanol and propylene oxide, all sections wereembedded on glass slides in Durcupan resin. Regions ofinterest (~1 � 1 mm) were then cut from a randomly selectedsection using a scalpel and glued onto a resin block. Ultrathinsections (~60 nm) were cut and collected on formvar-coatedgrids (Agar Scientific, Stansted, UK), stained with uranylacetate and lead citrate in an LKB BUltrostainer,[ and thenquantitatively assessed in a Philips CM12 transmissionelectron microscope. A timed-counting protocol was used toestimate synaptic density in regions of interest, as describedpreviously (16). Briefly, ultrathin sections containing theregion of interest were placed on coded grids, and the numberof synaptic profiles observed during a 15-minute analysisperiod were recorded for 2 separate counting sessions pergrid. The total distance traveled over each section during eachcounting period was measured to ensure that a similar totalarea had been included in each analysis. Analysis wasperformed blind to genotype. Raw data were collated usingExcel spreadsheets, and statistical tests were performed usingGraphpad Prism software.
Western BlottingSynaptic protein complexes were analyzed from the
cerebral cortices and thalami of control and CTSDj/j mice byhomogenizing the tissue in 40 mmol/L Tris-HCl, pH 8.8,containing protease inhibitors. Homogenates were then
centrifuged at 1,700 � g for 10 minutes at 4-C, and theprotein concentration was determined using a bicinchoninicacid protein kit (Interchim, Montlu0on, France). Undernonreducing conditions, 20 Kg from each homogenate wassubjected to 12% sodium dodecyl sulfateYpolyacrylamide gelelectrophoresis gel. The gels were blotted onto PVDFmembranes, blocked with 4% (wt/vol) bovine serum albuminin TBS containing 0.1% Tween 20 and incubated with theprimary antibodies overnight at 4-C. Immunoreactive bandswere visualized by enhanced chemiluminescence afterincubation with goat anti-mouse immunoglobulin G coupledto horseradish peroxidase (Bio-Rad, Hercules, CA; 1:4000).
RESULTS
CTSDj/j Brains Show Widespread AtrophyMacroscopically, the brains of CTSDj/j mice seemed
relatively normal but proportionately smaller than those ofcontrols at the age of 24 days. Consistent with thisobservation, Nissl-stained sections revealed widespreadatrophy of the CNS in the affected mice (Fig. 1A). Cavalieriestimates of regional volume showed significant effects uponboth white and gray matter of CTSDj/j mice: Of allstructures, the corpus callosum was the most significantlyaffected in mutant mice, being reduced by 32% comparedwith controls (Fig. 1B). Each cerebral region examined,including the striatum (j22%), thalamus (j22%), hippo-campus (j19%), and cortex (j16%), displayed a markedlydecreased volume in CTSDj/j mice, as did the deepcerebellar nuclei (j24%) and cerebellar white matter(j22%) (Fig. 1B). Of all structures in mutant mice, thecerebellar gray matter was least affected (j12%) (Fig. 1B).
Cortical Thinning in CTSDj/j Mice is the Resultof Lamina-Specific Changes
To determine whether CTSD deficiency affected corticalsubfields that serve different functions to a similar extent, wesurveyed the thickness of prefrontal cortex, M1, S1BF, andvisual cortex, and also measured the individual laminarthicknesses within these regions (Fig. 2A). Statisticallysignificant thinning was evident in most cortical subfieldsbut occurred to different extents in S1BF (j12%), visualcortex (j11%), M1 (j6%), and PF (not significant) (Fig. 2B).
These changes in overall cortical thickness wereaccompanied by distinct effects on the thickness of individ-ual laminae, which occurred to different extents in eachcortical subfield. In M1 of CTSDj/j mice, laminae I, V, andVI were of normal thickness, whereas the combined thick-ness of laminae II and III was significantly reduced (by 15%)compared with controls (Fig. 2B). In S1BF, the thickness ofsuperficial laminae I to III and lamina V were indistinguish-able between genotypes. In contrast, laminae IV (j20%)and VI (j23%) were markedly and significantly thinner inCTSDj/j mice compared with controls (Fig. 2B).
Localized Microglial Activation is Accompaniedby Widespread Astrocytosis in CTSDj/j Mice
Staining for F4/80 revealed that microglial activationshowed extreme regional accentuations in the brain of
J Neuropathol Exp Neurol � Volume 67, Number 1, January 2008 Thalamus and Synapses in CTSD Deficiency
� 2007 American Association of Neuropathologists, Inc. 19
Copyright © 2007 by the American Association of Neuropathologists, Inc. Unauthorized reproduction of this article is prohibited.
Dow
nloaded from https://academ
ic.oup.com/jnen/article/67/1/16/2916840 by guest on 27 July 2022

CTSDj/j mice, being most pronounced within the thalamusand olivary nuclei. F4/80-immunoreactive microglia werepresent in the ventrolateral part of the anteroventral thalamicnucleus, and in the VPM/VPL and posterior thalamic nuclei
and the lateral superior olive, rostral, and dorsal periolivaryregion, but were virtually absent from adjacent nuclei(Fig. 3A). In addition, F4/80-positive microglia were presentin the substantia nigra and also formed 2 densely staining
FIGURE 2. Cortical lamina-specific thinning in cathepsin D (CTSD)j/j mice. (A) Nissl-stained sections from 2 representativecortical regions, primary barrel field of primary somatosensory cortex (S1BF), and primary motor cortex (M1) reveal thesignificant atrophy of these regions in CTSDj/j compared with controls+/+. (B, upper panel) Measurements of total corticalthickness show significant atrophy of S1BF, primary motor cortex (M1), and primary visual cortex (V1), but not prefrontal cortexin CTSDj/j mice. (B, lower panels) Measurements of individual laminar thickness, using the boundaries depicted in (A), indicatethe different lamina-specific effects in S1BF and M1. Thickness data are expressed as mean cortical thickness (in micrometers TSEM). *, p e 0.05; †, p e 0.000. Scale bar = (A) 500 Km.
FIGURE 1. Regional atrophy of the cathepsin D (CTSD)j/j CNS. (A) Low-power micrographs of Nissl-stained parasagittalsections reveal the overall atrophy of the CNS in CTSDj/j mice compared with controls+/+. (B) Cavalieri estimates of regionalvolume indicate the extent of atrophy in grey and white matter structures of CTSDj/j mice compared with controls. Data areexpressed as mean regional volume in Km3 T SEM. *, p e 0.05; †, p e 0.001; ‡, p e 0.000. Scale bar = (A) 2,000 Km.
Partanen et al J Neuropathol Exp Neurol � Volume 67, Number 1, January 2008
� 2007 American Association of Neuropathologists, Inc.20
Copyright © 2007 by the American Association of Neuropathologists, Inc. Unauthorized reproduction of this article is prohibited.
Dow
nloaded from https://academ
ic.oup.com/jnen/article/67/1/16/2916840 by guest on 27 July 2022
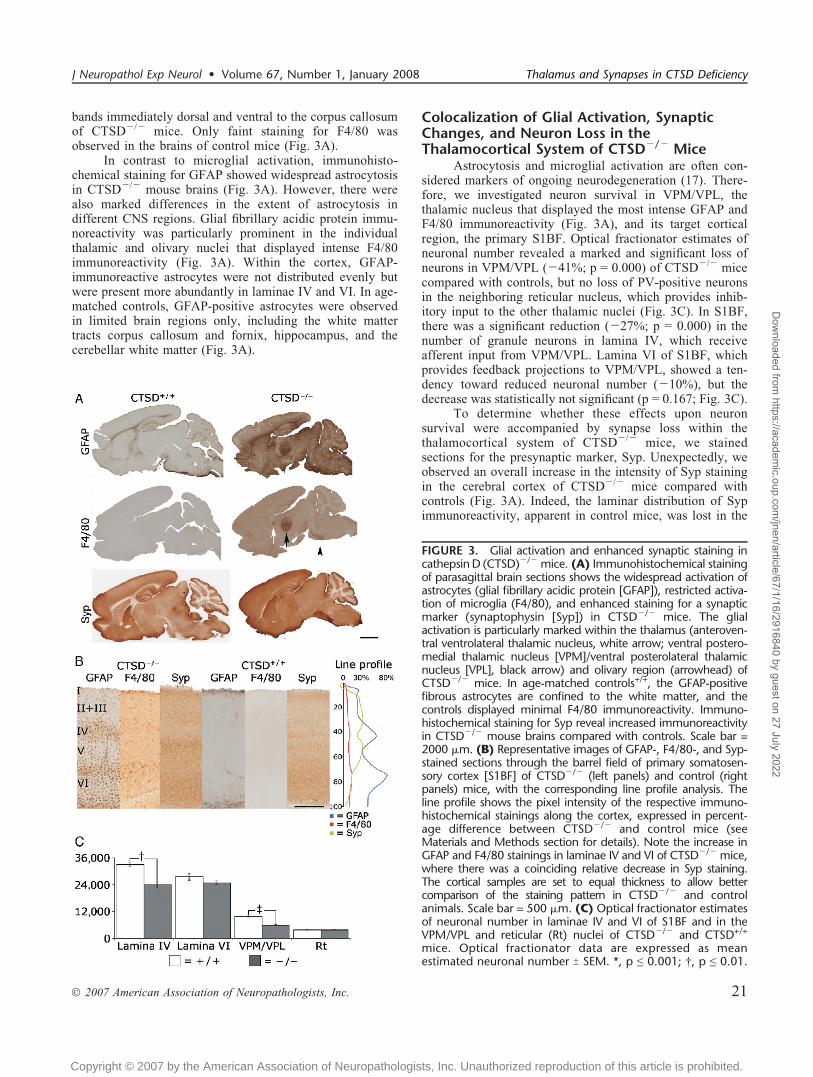
bands immediately dorsal and ventral to the corpus callosumof CTSDj/j mice. Only faint staining for F4/80 wasobserved in the brains of control mice (Fig. 3A).
In contrast to microglial activation, immunohisto-chemical staining for GFAP showed widespread astrocytosisin CTSDj/j mouse brains (Fig. 3A). However, there werealso marked differences in the extent of astrocytosis indifferent CNS regions. Glial fibrillary acidic protein immu-noreactivity was particularly prominent in the individualthalamic and olivary nuclei that displayed intense F4/80immunoreactivity (Fig. 3A). Within the cortex, GFAP-immunoreactive astrocytes were not distributed evenly butwere present more abundantly in laminae IV and VI. In age-matched controls, GFAP-positive astrocytes were observedin limited brain regions only, including the white mattertracts corpus callosum and fornix, hippocampus, and thecerebellar white matter (Fig. 3A).
Colocalization of Glial Activation, SynapticChanges, and Neuron Loss in theThalamocortical System of CTSDj/j Mice
Astrocytosis and microglial activation are often con-sidered markers of ongoing neurodegeneration (17). There-fore, we investigated neuron survival in VPM/VPL, thethalamic nucleus that displayed the most intense GFAP andF4/80 immunoreactivity (Fig. 3A), and its target corticalregion, the primary S1BF. Optical fractionator estimates ofneuronal number revealed a marked and significant loss ofneurons in VPM/VPL (j41%; p = 0.000) of CTSDj/j micecompared with controls, but no loss of PV-positive neuronsin the neighboring reticular nucleus, which provides inhib-itory input to the other thalamic nuclei (Fig. 3C). In S1BF,there was a significant reduction (j27%; p = 0.000) in thenumber of granule neurons in lamina IV, which receiveafferent input from VPM/VPL. Lamina VI of S1BF, whichprovides feedback projections to VPM/VPL, showed a ten-dency toward reduced neuronal number (j10%), but thedecrease was statistically not significant (p = 0.167; Fig. 3C).
To determine whether these effects upon neuronsurvival were accompanied by synapse loss within thethalamocortical system of CTSDj/j mice, we stainedsections for the presynaptic marker, Syp. Unexpectedly, weobserved an overall increase in the intensity of Syp stainingin the cerebral cortex of CTSDj/j mice compared withcontrols (Fig. 3A). Indeed, the laminar distribution of Sypimmunoreactivity, apparent in control mice, was lost in the
FIGURE 3. Glial activation and enhanced synaptic staining incathepsinD (CTSD)j/j mice. (A) Immunohistochemical stainingof parasagittal brain sections shows the widespread activation ofastrocytes (glial fibrillary acidic protein [GFAP]), restricted activa-tion of microglia (F4/80), and enhanced staining for a synapticmarker (synaptophysin [Syp]) in CTSDj/j mice. The glialactivation is particularly marked within the thalamus (anteroven-tral ventrolateral thalamic nucleus, white arrow; ventral postero-medial thalamic nucleus [VPM]/ventral posterolateral thalamicnucleus [VPL], black arrow) and olivary region (arrowhead) ofCTSDj/j mice. In age-matched controls+/+, the GFAP-positivefibrous astrocytes are confined to the white matter, and thecontrols displayed minimal F4/80 immunoreactivity. Immuno-histochemical staining for Syp reveal increased immunoreactivityin CTSDj/j mouse brains compared with controls. Scale bar =2000 Km. (B) Representative images of GFAP-, F4/80-, and Syp-stained sections through the barrel field of primary somatosen-sory cortex [S1BF] of CTSDj/j (left panels) and control (rightpanels) mice, with the corresponding line profile analysis. Theline profile shows the pixel intensity of the respective immuno-histochemical stainings along the cortex, expressed in percent-age difference between CTSDj/j and control mice (seeMaterials and Methods section for details). Note the increase inGFAP and F4/80 stainings in laminae IV and VI of CTSDj/j mice,where there was a coinciding relative decrease in Syp staining.The cortical samples are set to equal thickness to allow bettercomparison of the staining pattern in CTSDj/j and controlanimals. Scale bar = 500 Km. (C) Optical fractionator estimatesof neuronal number in laminae IV and VI of S1BF and in theVPM/VPL and reticular (Rt) nuclei of CTSDj/j and CTSD+/+
mice. Optical fractionator data are expressed as meanestimated neuronal number T SEM. *, p e 0.001; †, p e 0.01.
J Neuropathol Exp Neurol � Volume 67, Number 1, January 2008 Thalamus and Synapses in CTSD Deficiency
� 2007 American Association of Neuropathologists, Inc. 21
Copyright © 2007 by the American Association of Neuropathologists, Inc. Unauthorized reproduction of this article is prohibited.
Dow
nloaded from https://academ
ic.oup.com/jnen/article/67/1/16/2916840 by guest on 27 July 2022
mutant mice (Fig. 3A). To further investigate this redistrib-ution of Syp expression and its potential relationship to thelamina-specific activation of glial cells, we used line profileanalysis to compare the intensity and distribution of Syp,GFAP, and F4/80 immunoreactivity in S1BF, which receivesafferent input from VPM/VPL. In S1BF of control mice,GFAP and F4/80 staining were virtually absent, and Sypstaining increased gradually toward the deeper parts of thecortex (Fig. 3B). In contrast, within the S1BF of CTSDj/j
mice, GFAP staining increased in a dorsoventral manner andexhibited 2 distinct intensity peaks, similar to F4/80 staining(Fig. 3B). Synaptophysin staining, although generally ele-vated in CTSDj/j mice, also showed 2 bands of relativelyreduced SI, which coincided perfectly with the bands ofincreased GFAP SI in laminae IV and VI (Fig. 3B). Thesedata indicate that in the S1BF of CTSDj/j mice, glial
activation is accompanied by redistribution of the synapticmarker, Syp, in a lamina-specific manner.
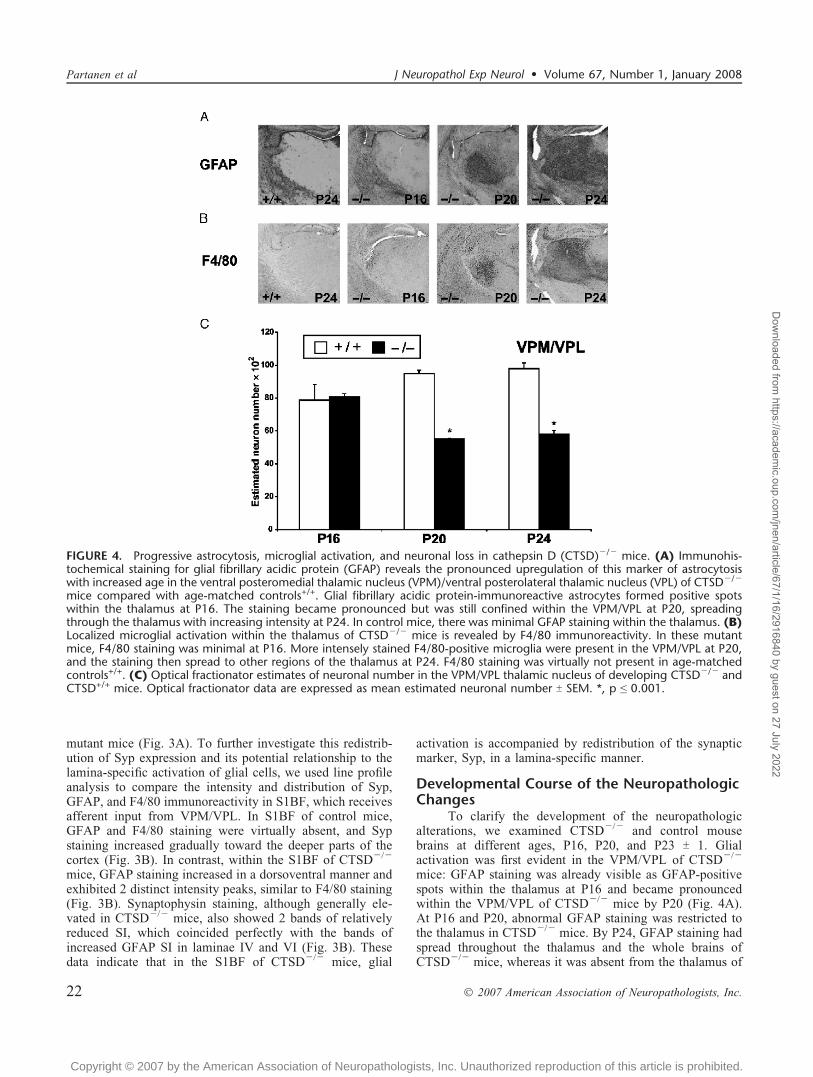
Developmental Course of the NeuropathologicChanges
To clarify the development of the neuropathologicalterations, we examined CTSDj/j and control mousebrains at different ages, P16, P20, and P23 T 1. Glialactivation was first evident in the VPM/VPL of CTSDj/j
mice: GFAP staining was already visible as GFAP-positivespots within the thalamus at P16 and became pronouncedwithin the VPM/VPL of CTSDj/j mice by P20 (Fig. 4A).At P16 and P20, abnormal GFAP staining was restricted tothe thalamus in CTSDj/j mice. By P24, GFAP staining hadspread throughout the thalamus and the whole brains ofCTSDj/j mice, whereas it was absent from the thalamus of
FIGURE 4. Progressive astrocytosis, microglial activation, and neuronal loss in cathepsin D (CTSD)j/j mice. (A) Immunohis-tochemical staining for glial fibrillary acidic protein (GFAP) reveals the pronounced upregulation of this marker of astrocytosiswith increased age in the ventral posteromedial thalamic nucleus (VPM)/ventral posterolateral thalamic nucleus (VPL) of CTSDj/j
mice compared with age-matched controls+/+. Glial fibrillary acidic protein-immunoreactive astrocytes formed positive spotswithin the thalamus at P16. The staining became pronounced but was still confined within the VPM/VPL at P20, spreadingthrough the thalamus with increasing intensity at P24. In control mice, there was minimal GFAP staining within the thalamus. (B)Localized microglial activation within the thalamus of CTSDj/j mice is revealed by F4/80 immunoreactivity. In these mutantmice, F4/80 staining was minimal at P16. More intensely stained F4/80-positive microglia were present in the VPM/VPL at P20,and the staining then spread to other regions of the thalamus at P24. F4/80 staining was virtually not present in age-matchedcontrols+/+. (C) Optical fractionator estimates of neuronal number in the VPM/VPL thalamic nucleus of developing CTSDj/j andCTSD+/+ mice. Optical fractionator data are expressed as mean estimated neuronal number T SEM. *, p e 0.001.
Partanen et al J Neuropathol Exp Neurol � Volume 67, Number 1, January 2008
� 2007 American Association of Neuropathologists, Inc.22
Copyright © 2007 by the American Association of Neuropathologists, Inc. Unauthorized reproduction of this article is prohibited.
Dow
nloaded from https://academ
ic.oup.com/jnen/article/67/1/16/2916840 by guest on 27 July 2022
control mice (Fig. 4A). Microglial activation, as indicated byF4/80 staining, was minimal in CTSDj/j mice at P16 butbecame more evident in VPM/VPL at P20, and by P24, ithad spread also to other thalamic nuclei (Fig. 4B). In controlmice, F4/80 staining was practically absent in all of thesetime points (Fig. 4B). Based on these data, it seems thatactivation of astrocytes slightly precedes that of microglia inthe thalamus of CTSDj/j mice. The number of neuronswithin the VPM/VPL was significantly reduced in CTSDj/j
mice at P20 and P24 but not at P16 (Fig. 4C), indicating thatloss of neurons coincided with microglial activation in thisthalamic nucleus.
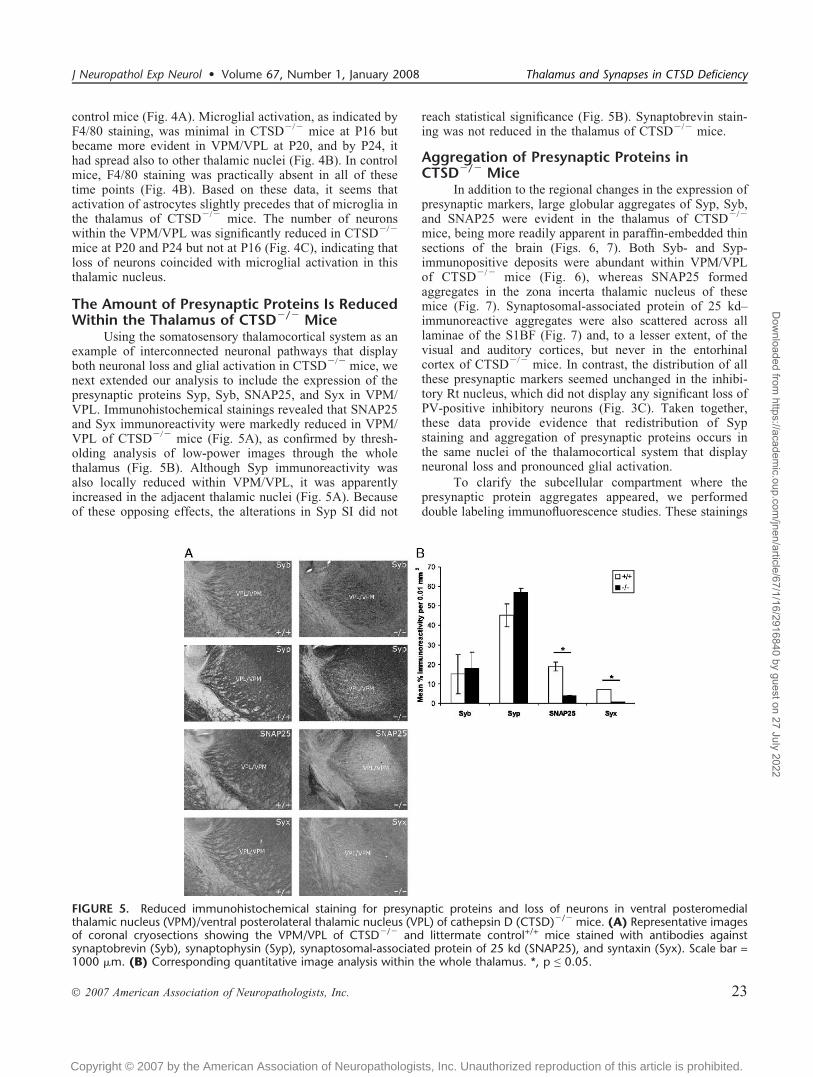
The Amount of Presynaptic Proteins Is ReducedWithin the Thalamus of CTSDj/j Mice
Using the somatosensory thalamocortical system as anexample of interconnected neuronal pathways that displayboth neuronal loss and glial activation in CTSDj/j mice, wenext extended our analysis to include the expression of thepresynaptic proteins Syp, Syb, SNAP25, and Syx in VPM/VPL. Immunohistochemical stainings revealed that SNAP25and Syx immunoreactivity were markedly reduced in VPM/VPL of CTSDj/j mice (Fig. 5A), as confirmed by thresh-olding analysis of low-power images through the wholethalamus (Fig. 5B). Although Syp immunoreactivity wasalso locally reduced within VPM/VPL, it was apparentlyincreased in the adjacent thalamic nuclei (Fig. 5A). Becauseof these opposing effects, the alterations in Syp SI did not
reach statistical significance (Fig. 5B). Synaptobrevin stain-ing was not reduced in the thalamus of CTSDj/j mice.
Aggregation of Presynaptic Proteins inCTSDj/j Mice
In addition to the regional changes in the expression ofpresynaptic markers, large globular aggregates of Syp, Syb,and SNAP25 were evident in the thalamus of CTSDj/j
mice, being more readily apparent in paraffin-embedded thinsections of the brain (Figs. 6, 7). Both Syb- and Syp-immunopositive deposits were abundant within VPM/VPLof CTSDj/j mice (Fig. 6), whereas SNAP25 formedaggregates in the zona incerta thalamic nucleus of thesemice (Fig. 7). Synaptosomal-associated protein of 25 kdYimmunoreactive aggregates were also scattered across alllaminae of the S1BF (Fig. 7) and, to a lesser extent, of thevisual and auditory cortices, but never in the entorhinalcortex of CTSDj/j mice. In contrast, the distribution of allthese presynaptic markers seemed unchanged in the inhibi-tory Rt nucleus, which did not display any significant loss ofPV-positive inhibitory neurons (Fig. 3C). Taken together,these data provide evidence that redistribution of Sypstaining and aggregation of presynaptic proteins occurs inthe same nuclei of the thalamocortical system that displayneuronal loss and pronounced glial activation.
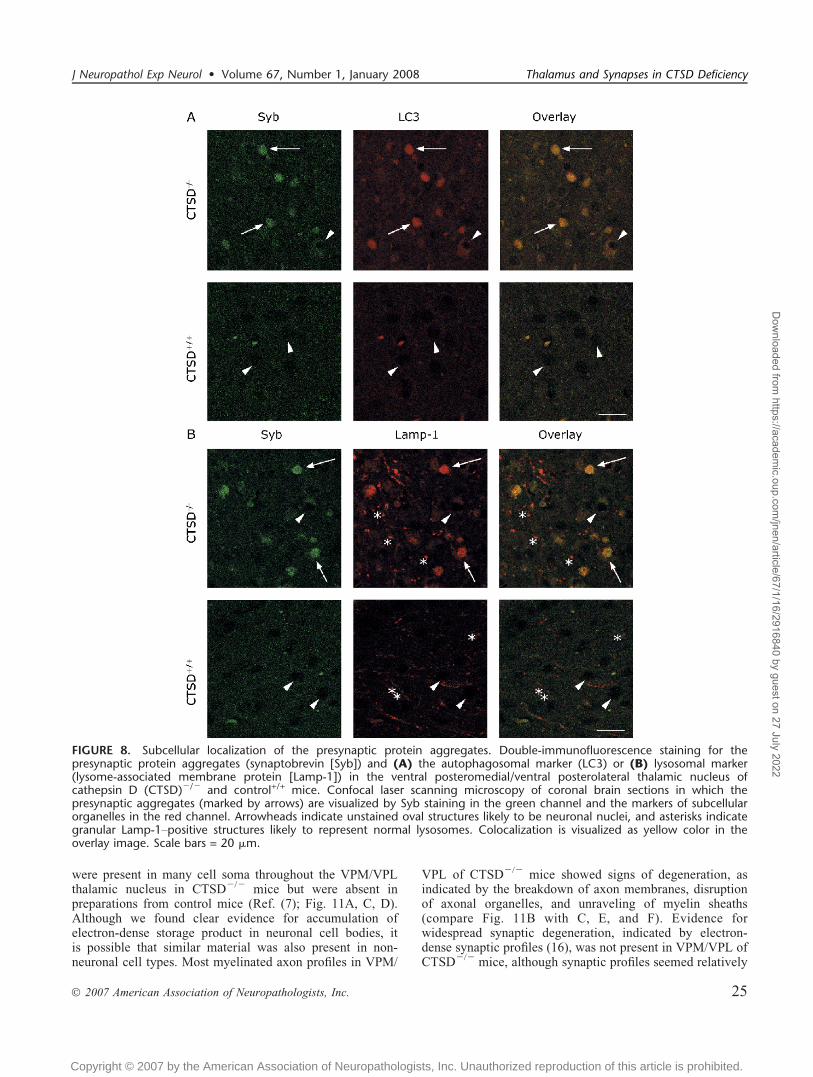
To clarify the subcellular compartment where thepresynaptic protein aggregates appeared, we performeddouble labeling immunofluorescence studies. These stainings
FIGURE 5. Reduced immunohistochemical staining for presynaptic proteins and loss of neurons in ventral posteromedialthalamic nucleus (VPM)/ventral posterolateral thalamic nucleus (VPL) of cathepsin D (CTSD)j/j mice. (A) Representative imagesof coronal cryosections showing the VPM/VPL of CTSDj/j and littermate control+/+ mice stained with antibodies againstsynaptobrevin (Syb), synaptophysin (Syp), synaptosomal-associated protein of 25 kd (SNAP25), and syntaxin (Syx). Scale bar =1000 Km. (B) Corresponding quantitative image analysis within the whole thalamus. *, p e 0.05.
J Neuropathol Exp Neurol � Volume 67, Number 1, January 2008 Thalamus and Synapses in CTSD Deficiency
� 2007 American Association of Neuropathologists, Inc. 23
Copyright © 2007 by the American Association of Neuropathologists, Inc. Unauthorized reproduction of this article is prohibited.
Dow
nloaded from https://academ
ic.oup.com/jnen/article/67/1/16/2916840 by guest on 27 July 2022

revealed good colocalization of the Syb-positive aggregateswith the autophagosomal marker LC3 (Fig. 8A) and thelysosomal marker Lamp-1 (Fig. 8B) in the VPM/VPL ofCTSDj/j mice, suggesting that the presynaptic proteinaggregates reside in autophagosomes/autophagolysosomesin CTSDj/j mice. Moreover, there was also a pool of smallvesicular Lamp-1Ypositive structures devoid of Syb staining,indicating that the normal lysosomes did not containpresynaptic protein aggregates in CTSDj/j mice (Fig. 8B).
As an attempt to establish whether the presynapticprotein aggregates resided in microglial cells, we double-stained paraffin sections of the CTSDj/j mouse brains forSyb and the microglia-specific marker CD68 (Fig. 9A, B).There was no overlap between these stainings, indicatingthat Syb-positive protein aggregates did not reside in theactivated microglial cells (Fig. 9C).
CTSD Deficiency Leads to Changes in theAmount of Syp/Syp Complex
On the basis of immunohistochemical evidence forabnormal aggregates of presynaptic proteins in CTSDj/j
mice, we next studied the presynaptic protein complexes byimmunoblotting. Of the examined proteins, Syb, Syx, andSNAP25 together form the SNARE complex needed fordocking and release of synaptic vesicles from the presynap-tic terminals, and Syp is likely to have a regulatory role inthis process via formation of a Syp/Syb-heterodimer (18).The amount of the SNARE complex (73 kd) seemed
unchanged in the cortex and thalamus of CTSDj/j mice(Fig. 10). However, Syp/Syb-complex (56 kd), whichregulates the availability of Syb for formation of SNAREcomplexes, was far more abundant in the brains of CTSDj/j
mice than in control mice (Fig. 10). Accordingly, the amountof the Syp-homodimer (76 kd) was reduced in the brains ofthe affected mice compared with controls, particularly in thethalamus (Fig. 10). No changes in the abundance of themonomeric forms of Syb, Syp, SNAP25, or Syx wereobserved between the affected and control animals (Fig. 10and data not shown). Taken together, these data show, at themolecular level, that the presynaptic protein machineryassociated with synaptic transmission is affected inCTSDj/j mice and emphasize the importance of presynap-tic events in the pathogenesis of CTSD deficiency.
Ultrastructural Evidence for AxonalDegeneration and Synaptic Loss in VPM/VPL,but not S1BF, of CTSDj/j Mice
Finally, we examined synaptic and axonal morphologyin the thalamocortical system at an ultrastructural level. Asexpected, accumulations of electron-dense storage product
FIGURE 6. Aggregation of immunoreactive clusters of syn-aptobrevin (Syb) and synaptophysin (Syp) in the thalamus ofcathepsin D (CTSD)j/j mouse. Representative coronal paraf-fin sections of the thalamus of CTSDj/j and littermatecontrol+/+ mice immunohistochemically stained for (A, C, E)Syb and (B, D) Syp. Immunohistochemical staining revealsthe accumulation of immunoreactive clusters of Syp and Sybin ventral posterolateral thalamic nucleus (VPL) and in ventralposteromedial thalamic nucleus (VPM). (E) In the control+/+
mice, such aggregates were not detected. Scale bars = (A, B)50 and (C–E) 10 Km.
FIGURE 7. Synaptosomal-associated protein of 25 kd(SNAP25) staining is clustered in the somatosensory cortexand thalamus of cathepsin D (CTSD)j/j mice. Representativecoronal paraffin sections of the barrel field of primarysomatosensory cortex (S1BF) and thalamus (Thal) of CTSDj/j
mice and littermate controls+/+, immunohistochemicallystained for SNAP25 reveals accumulation of SNAP25-positiveaggregates in S1BF and dorsal sector of zona incerta (ZID)/ventral sector of zona incerta (ZIV) of Thal in CTSD-deficientmice (CYF) but not in controls (A, B). Medial leminiscus (mL)between ZID/ZIV and ventral posterolateral thalamic nucleus(VPL)/ventral posteromedial thalamic nucleus (VPM) areindicated in pictures (B, D). Note the absence of SNAP25-immunoreactive aggregates in VPM/VPL. Scale bars = (A–D)50 and (E, F) 10 Km.
Partanen et al J Neuropathol Exp Neurol � Volume 67, Number 1, January 2008
� 2007 American Association of Neuropathologists, Inc.24
Copyright © 2007 by the American Association of Neuropathologists, Inc. Unauthorized reproduction of this article is prohibited.
Dow
nloaded from https://academ
ic.oup.com/jnen/article/67/1/16/2916840 by guest on 27 July 2022
were present in many cell soma throughout the VPM/VPLthalamic nucleus in CTSDj/j mice but were absent inpreparations from control mice (Ref. (7); Fig. 11A, C, D).Although we found clear evidence for accumulation ofelectron-dense storage product in neuronal cell bodies, itis possible that similar material was also present in non-neuronal cell types. Most myelinated axon profiles in VPM/
VPL of CTSDj/j mice showed signs of degeneration, asindicated by the breakdown of axon membranes, disruptionof axonal organelles, and unraveling of myelin sheaths(compare Fig. 11B with C, E, and F). Evidence forwidespread synaptic degeneration, indicated by electron-dense synaptic profiles (16), was not present in VPM/VPL ofCTSDj/j mice, although synaptic profiles seemed relatively
FIGURE 8. Subcellular localization of the presynaptic protein aggregates. Double-immunofluorescence staining for thepresynaptic protein aggregates (synaptobrevin [Syb]) and (A) the autophagosomal marker (LC3) or (B) lysosomal marker(lysome-associated membrane protein [Lamp-1]) in the ventral posteromedial/ventral posterolateral thalamic nucleus ofcathepsin D (CTSD)j/j and control+/+ mice. Confocal laser scanning microscopy of coronal brain sections in which thepresynaptic aggregates (marked by arrows) are visualized by Syb staining in the green channel and the markers of subcellularorganelles in the red channel. Arrowheads indicate unstained oval structures likely to be neuronal nuclei, and asterisks indicategranular Lamp-1Ypositive structures likely to represent normal lysosomes. Colocalization is visualized as yellow color in theoverlay image. Scale bars = 20 Km.
J Neuropathol Exp Neurol � Volume 67, Number 1, January 2008 Thalamus and Synapses in CTSD Deficiency
� 2007 American Association of Neuropathologists, Inc. 25
Copyright © 2007 by the American Association of Neuropathologists, Inc. Unauthorized reproduction of this article is prohibited.
Dow
nloaded from https://academ
ic.oup.com/jnen/article/67/1/16/2916840 by guest on 27 July 2022

scarce in these mice. Nevertheless, there was evidence ofBpinching[ of presynaptic nerve terminals from postsynapticspines by nonneuronal cell types (possibly an astrocyte;
Fig. 11G). Qualitative analyses suggesting a paucity ofsynapses in VPM/VPL were supported by quantitativeassessment of synapse number, showing a markedly reducednumber of synapses in the affected animals compared withcontrols (p G 0.005; 2-tailed t-test; Fig. 11H). Thus, the lackof degenerating synaptic profiles in VPM/VPL was mostlikely because synaptic degeneration and subsequentremoval of all synaptic debris in this nucleus was essentiallycomplete at the time point examined.
In contrast to VPM/VPL, ultrastructural examinationof S1BF from the same CTSDj/j mice showed very fewchanges compared with control preparations. Accumulationsof electron-dense storage product in cell soma were rare andmuch less prominent than in VPM/VPL. There was noevidence of axonal degeneration, and most synapses seemednormal (Fig. 11B). Quantitative assessment of synapsenumber in lamina IV of S1BF revealed no differencebetween CTSDj/j and control mice (p 9 0.05; 2-tailedt-test). However, a couple of examples of degeneratingsynaptic profiles were identified (data not shown), suggest-ing that pathologic changes at the level of axons andsynapses were only beginning to occur in S1BF during theperiod when axonal degeneration and synapse loss wereprevalent in the VPM/VPL nucleus of the thalamus.
FIGURE 9. Immunochemical staining of the presynapticprotein aggregates and activated microglia in the thalamus ofcathepsin D (CTSD)j/j mouse. Coronal paraffin sections of thethalamus of CTSDj/j mouse were first stained for synapto-brevin (Syb), which was visualized as red color using theperoxidase substrate 3-amino-9-ethylcarbazole (A, arrows).After this, the sections were stained for the microglial markerCD68 using fluorescent Alexa Fluor 488 antibody conjugates(B, arrowheads). An overlay of the images with pseudocolors(C) shows no overlap of Syb-positive protein aggregates(arrows) and activated microglial cells (arrowheads). Scalebar = 20 Km.
FIGURE 10. Western blot analysis of presynaptic proteins incathepsin D (CTSD)j/j and control mouse brains. Represen-tative results of immunoblotting analyses indicated no appa-rent changes in the steady-state levels of the SNARE complex(73 kd) in the cortex and Thal of CTSDj/j mice (n = 4)compared with controls+/+, probed here with antibody againstsynaptobrevin (Syb) (upper panel). Immunoblotting with Sybantibody revealed an abnormally high amount of synapto-physin (Syp)/Syb complex (56 kd) in the cortex and thalamus(Thal) of mutant mice (middle panel). Immunoblotting withantibody against Syp confirmed the finding and also indicateda markedly decreased level of Syp homodimer (76 kd) inCTSDj/j mice compared with controls (lower panel).
Partanen et al J Neuropathol Exp Neurol � Volume 67, Number 1, January 2008
� 2007 American Association of Neuropathologists, Inc.26
Copyright © 2007 by the American Association of Neuropathologists, Inc. Unauthorized reproduction of this article is prohibited.
Dow
nloaded from https://academ
ic.oup.com/jnen/article/67/1/16/2916840 by guest on 27 July 2022
DISCUSSION
Neuropathologic Findings in CTSDj/j MiceResemble Those in Human CongenitalNCL Patients
In the present study, we have used CTSDj/j mice toreveal novel molecular and cellular aspects underlying thepathogenesis of CTSD deficiencies. We observed structuralalterations and atrophy of various brain regions, includingboth white and gray matter in CTSDj/j mouse brains. Thesefindings were in good agreement with the extreme andgeneralized brain atrophy previously observed in human andovine congenital NCL caused by CTSD deficiency (5, 6, 19).In accordance with the remarkable loss of myelin and axonsin the subcortical white matter of patients with congenitalNCL (19), the corpus callosum showed the highest degree ofatrophy in CTSDj/j mice. Astrocytosis in brains of the
affected mice was severe and generalized, similar to that inCTSDj/j human patients and lambs (5, 6, 19). Despite thesesimilarities, the degree of brain atrophy and neuronal loss ismarkedly milder in CTSDj/j mice than in CTSDj/j humansor sheep, possibly reflecting functional and structural differ-ences in the brain between these species. In comparison withmouse models of other NCL types, however, this mousemodel of CTSD deficiency shows the most severe phenotypeand brain atrophy, thus being parallel to the relativephenotypes of the corresponding human NCL diseases.
The Somatosensory Thalamocortical SystemShows Severe Degenerative Changes inCTSDj/j Mice
A striking feature of CTSDj/j mice was the remark-ably pronounced microglial activation within VPM/VPLnucleus of the thalamus. In further analyses, this thalamicnucleus was revealed as a particular focus of degenerativechanges, exhibiting not only intense glial activation but alsoneuronal loss, redistribution of presynaptic proteins, axonaldegeneration, and loss of synapses. The regional specificityof these changes was evident because they did not occur inthe neighboring thalamic nuclei. Although thalamic patholo-gy in CTSDj/j mice has already been reported (11), thesevere involvement of discrete nuclei within this region wasunexpected because the cerebral cortex has been reported tobe the most severely affected brain region in both human
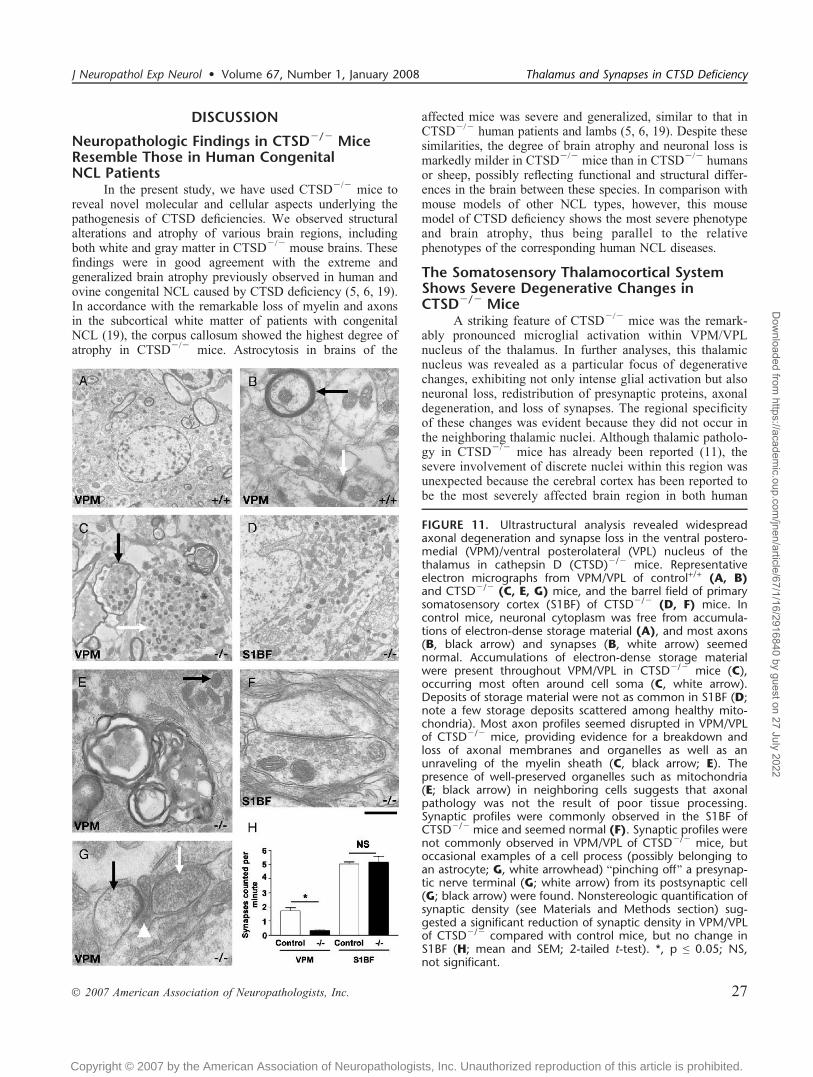
FIGURE 11. Ultrastructural analysis revealed widespreadaxonal degeneration and synapse loss in the ventral postero-medial (VPM)/ventral posterolateral (VPL) nucleus of thethalamus in cathepsin D (CTSD)j/j mice. Representativeelectron micrographs from VPM/VPL of control+/+ (A, B)and CTSDj/j (C, E, G) mice, and the barrel field of primarysomatosensory cortex (S1BF) of CTSDj/j (D, F) mice. Incontrol mice, neuronal cytoplasm was free from accumula-tions of electron-dense storage material (A), and most axons(B, black arrow) and synapses (B, white arrow) seemednormal. Accumulations of electron-dense storage materialwere present throughout VPM/VPL in CTSDj/j mice (C),occurring most often around cell soma (C, white arrow).Deposits of storage material were not as common in S1BF (D;note a few storage deposits scattered among healthy mito-chondria). Most axon profiles seemed disrupted in VPM/VPLof CTSDj/j mice, providing evidence for a breakdown andloss of axonal membranes and organelles as well as anunraveling of the myelin sheath (C, black arrow; E). Thepresence of well-preserved organelles such as mitochondria(E; black arrow) in neighboring cells suggests that axonalpathology was not the result of poor tissue processing.Synaptic profiles were commonly observed in the S1BF ofCTSDj/j mice and seemed normal (F). Synaptic profiles werenot commonly observed in VPM/VPL of CTSDj/j mice, butoccasional examples of a cell process (possibly belonging toan astrocyte; G, white arrowhead) Bpinching off[ a presynap-tic nerve terminal (G; white arrow) from its postsynaptic cell(G; black arrow) were found. Nonstereologic quantification ofsynaptic density (see Materials and Methods section) sug-gested a significant reduction of synaptic density in VPM/VPLof CTSDj/j compared with control mice, but no change inS1BF (H; mean and SEM; 2-tailed t-test). *, p e 0.05; NS,not significant.
J Neuropathol Exp Neurol � Volume 67, Number 1, January 2008 Thalamus and Synapses in CTSD Deficiency
� 2007 American Association of Neuropathologists, Inc. 27
Copyright © 2007 by the American Association of Neuropathologists, Inc. Unauthorized reproduction of this article is prohibited.
Dow
nloaded from https://academ
ic.oup.com/jnen/article/67/1/16/2916840 by guest on 27 July 2022

and ovine CTSD deficiency (5, 6, 19). Experiments duringthe course of the disease indicated that, indeed, it was thethalamic VPM/VPL in which the glial activation was firstseen in CTSDj/j mice, with the earliest signs of astroglialactivation being visible at P16, followed by marked acti-vation of both astrocytes and microglia, as well as signif-icantly reduced neuronal number at P20. Previously, thevariable involvement of individual thalamic nuclei has beennoted at autopsy of infantile NCL patients (20), magneticresonance images of patients with different forms of NCL(21), and in mouse models of infantile and juvenile NCL(22, 23), suggesting that the thalamus is an important centerin which the pathologic processes may originate in multipleforms of NCL.
Pathologic alterations in VPM/VPL of CTSDj/j micewere accompanied by degenerative changes in the function-ally associated laminae IV and VI of S1BF cortex. Lineprofile analysis revealed novel evidence for lamina-specificglial activation in CTSDj/j mice. The most intense glialactivation occurred in laminae IV and VI of S1BF cortex,the same laminae showing alterations in Syp expression.Lamina IV of S1BF also showed significant neuronal lossand thinning in CTSDj/j mice, whereas lamina VI wassignificantly reduced in thickness but showed only a mildtendency toward reduced neuronal number, suggesting thatthe packaging of neurons may be abnormal in lamina VI.Although this spatial correlation between the glial activationand neuronal loss may simply reflect activation of glial cellsduring neurodegeneration, it may alternatively reflect theinfluence of astrocytes upon synaptic efficacy (24, 25, 26).Taken together, these data indicate that the somatosensorythalamocortical system is a particular focus of neurodege-nerative events in CTSDj/j mice.
The thalamus forms a gateway from the peripheralsensory organs to the cortex. Somatosensory information isnormally relayed from VPM/VPL to lamina IV neurons inS1BF. This input is regulated by cortical neurons in laminaVI, which project back to the thalamus. The presence ofneurodegenerative changes in these interconnected pathwaysraises the possibility of a dysfunction within the thalamo-cortical circuitry of CTSDj/j mice. Despite no loss ofinhibitory neurons in the Rt nucleus of affected mice, theloss of feedback from neurons in lamina VI may decreasethe modulatory influence of S1BF upon VPM/VPL. Thismay result in a greater excitatory input from VPM/VPL toits target neurons in the cortex and cause excitotoxic effectsin lamina IV. Although the axonal pathology observed inVPM/VPL is consistent with this suggestion, the functionalconsequences of these alterations remain unclear. As such, itwill be important to test somatosensory thalamocorticalcircuitry functions in CTSDj/j mice. Interestingly, elevatedsomatosensory evoked potentials have been reported inpatients with the Finnish variant of late-infantile NCL(CLN5) (27), reflecting ongoing dysfunction within thesomatosensory thalamocortical system.
Synaptic Alterations in CTSDj/j MiceUnexpectedly, immunoreactivity for Syp, a synaptic
vesicle membrane protein, was increased in the cortical and
subcortical brain regions of CTSDj/j mice as comparedwith controls. However, this did not apply to VPM/VPL,where the staining for Syp and 2 other synaptic markers,SNAP25 and Syx, was markedly decreased, as might beexpected with the observed loss of neurons and synapses inthis brain region. Interestingly, synaptic loss in CTSDj/j
mice was evident only in VPM/VPL, but not in S1BF,suggesting that the degenerative changes in the thalamusprecede those in the cortex.
Another distinctive feature of CTSDj/j mice was theabnormal accumulation of presynaptic proteins: Syp- andSyb-immunoreactive aggregates were confined to VPM/VPL, whereas SNAP25-positive aggregates were morewidely distributed in the affected mice. As shown by doublelabeling experiments, the Syb-positive protein aggregateslocalized within the autophagosomal/autophagolysosomalcompartment. It is unclear whether these aggregates mayhave a pathogenic role in CTSD deficiency, but vesicularstalling and generation of cytosolic or intravesicular aggre-gates in axons may be associated with blocked axonaltransport in a variety of neurodegenerative conditions (28).Indeed, our ultrastructural evidence of gross axonal pathol-ogy with disrupted organelles and unraveled myelin sheathswithin VPM/VPL of CTSDj/j mice would be compatiblewith the suggestion of axonal transport deficits. Previously,Koike et al (29) reported axonal pathology and accumulationof autophagosomes in cortical neurons as well as corpuscallosum of CTSDj/j mice and suggested that axonaltransport may be impeded in these mice. Although we wereunable to confirm the type of cells in which the aggregatesresided, the distribution of these aggregates was clearlydistinct from that of microglial cells, thus indirectlyimplying that the presynaptic protein aggregates may residewithin neurons. Thus, the involvement of presynaptic proteinaccumulation within autophagosomes and its potential con-sequences to axonal transport and signaling in CTSDj/j
mice warrants further examination.Synaptic transmission requires the release of neuro-
transmitters from synaptic vesicles by regulated exocytosis.This is facilitated by formation of the SNARE complexcomposed of Syb, SNAP25, and Syx (18). Synaptophysinparticipates the regulation of this process by forming a Syp/Syb-heterodimer, which then controls the ability of Syb toenter the SNARE complex (18, 30, 31). Thus, the increasedamount of Syb/Syp-heterodimer in the cortex and thalamusof CTSDj/j mice may be associated with excessive neuro-nal activity, which could occur during seizures in these mice(7). Indeed, elevated level of Syp/Syb-complex has beenreported in the rat kindling model of epilepsy (32).Alternatively, the abnormally high levels of the Syp/Syb-heterodimer can result from a failure of this complex todissociate normally. Dissociation of the heterodimer pre-cedes the formation of the SNARE complex and iscontrolled by neuronal activity via a poorly understoodcalcium-dependent mechanism (18, 33). Although the Syp/Syb-dimer can be experimentally dissociated by proteolyticcleavage using tetanus toxin (18), the proteases responsiblefor the dissociation under physiologic conditions areunknown, and it remains to be seen whether CTSD or other
Partanen et al J Neuropathol Exp Neurol � Volume 67, Number 1, January 2008
� 2007 American Association of Neuropathologists, Inc.28
Copyright © 2007 by the American Association of Neuropathologists, Inc. Unauthorized reproduction of this article is prohibited.
Dow
nloaded from https://academ
ic.oup.com/jnen/article/67/1/16/2916840 by guest on 27 July 2022

endosomal/lysosomal proteases may have a role in thisprocess. The presynaptic protein complexes may also betargeted to the autophagosomal compartment for degrada-tion, and the Syb-containing aggregates arise from a failureof this autophagic process in CTSDj/j mice. Together,these data provide the first direct in vivo evidence forsynaptic abnormalities in CSTD deficiency at the tissue,molecular, and ultrastructural levels. Our findings are also inline with previous in vitro studies, suggesting that synapticmalfunction in infantile NCL is reflected by a reducedsynaptic vesicle pool size in primary neuronal cultures of theCLN1 knockout mouse (34).
In conclusion, we report a series of localized patho-logic changes in brains of CTSDj/j mice, particularlypronounced within the thalamocortical pathways betweenVPM/VPL and laminae IV and VI of the S1BF. Thesechanges included not only loss of neurons and synapses butalso axonal degeneration, redistribution of presynapticmarkers, and abnormal accumulation of the synaptic Syp/Syb complex. The present data suggest that presynapticmodulation may be disrupted in CTSDj/j mice, leading toaggregation of presynaptic proteins within autophagosomalcompartment and, further, to axonal and neuronal degenera-tion in CTSD deficiencies. These data also emphasize therole of CTSD, a lysosomal protease, in influencing thepresynaptic organization.
ACKNOWLEDGMENTSThe authors thank Sirkka Kanerva and Steve Mitchell
for expert assistance with immunoblots and electron micro-scopy, as well as Drs. Mikko Liljestrom and Mika Hukkanenat the Institute of Biomedicine/Cell Imaging Unit, Universityof Helsinki, for assistance with fluorescent microscopy. Prof.Takashi Ueno is gratefully acknowledged for the LC3antiserum and Dr. Ulla Lahtinen for critical reading of thearticle.
REFERENCES1. Press EM, Porter RR, Cebra J. The isolation and properties of a
proteolytic enzyme, Cathepsin D, from bovine spleen. Biochem J 1960;74:501Y51
2. Faust PL, Kornfield S, Cirgwin JM. Cloning and sequence analysis ofcDNA for human cathepsin D. Proc Natl Acad Sci U S A 1985;82:4910Y4
3. Glondu M, Coopman P, Laurent-Matha V, Garcia M, Rochefort H,Liaudet-Coopman E. A mutated cathepsin-D devoid of its catalyticactivity stimulates the growth of cancer cells. Oncogene 2001;20:6920Y9
4. Jaattela M, Cande C, Kroemer G. Lysosomes and mitochondria in thecommitment to apoptosis: A potential role for cathepsin D and AIF.Cell Death Differ 2004;11:135Y6
5. Tyynela J, Sohar I, Sleat DE, et al. A mutation in the ovine cathepsin Dgene causes a congenital lysosomal storage disease with profoundneurodegeneration. EMBO J 2000;19:2786Y92
6. Siintola E, Partanen S, Stromme P, et al. Cathepsin D deficiencyunderlies congenital human neuronal ceroid-lipofuscinosis. Brain 2006;129:1438Y45
7. Koike M, Nakanishi H, Saftig P, et al. Cathepsin D deficiency induceslysosomal storage with ceroid lipofuscin in mouse CNS neurons. JNeurosci 2000;20:6898Y906
8. Saftig P, Hetman M, Schmahl W, et al. Mice deficient for the lysosomalproteinase cathepsin D exhibit progressive atrophy of the intestinalmucosa and profound destruction of lymphoid cells. EMBO J 1995;14:3599Y608
9. Haltia M. The neuronal ceroid-lipofuscinoses. J Neuropathol ExpNeurol 2003;62:1Y13
10. Tyynela J. Neuronal ceroid-lipofuscinoses. In: Saftig P. ed. Lysosomes.Georgetown, TX: Landes Bioscience/Eurekah.com, 2005:82Y99
11. Nakanishi H, Zhang J, Koike M, et al. Involvement of nitric oxidereleased from microglia-macrophages in pathological changes ofcathepsin DYdeficient mice. J Neurosci 2001;21:7526Y33
12. Bible E, Gupta P, Hofmann SL, Cooper JD. Regional and cellularneuropathology in the palmitoyl protein thioesteraseY1 null mutantmouse model of infantile neuronal ceroid lipofuscinosis. Neurobiol Dis2004;16:346Y59
13. Paxinos G, Franklin KBJ. The Mouse Brain in Stereotaxic Coordinates,2nd ed. San Diego, CA: Academic Press, 2001
14. Pontikis CC, Cotman SL, Macdonald ME, Cooper JD. Thalamocorticalneuron loss and localized astrocytosis in the Cln3(Deltaex7/8) knock-inmouse model of Batten disease. Neurobiol Dis 2005;20:823Y36
15. Gundersen HJ, Jensen EB. The efficiency of systematic sampling instereology and its prediction. J Microsc 1987;147:229Y63
16. Gillingwater TH, Ingham CA, Parry KE, et al. Delayed synapticdegeneration in the CNS of Wlds mice after cortical lesion. Brain 2006;129:1546Y56
17. Raivich G, Bohatschek M, Kloss CU, Werner A, Jones LL, KreutzbergGW. Neuroglial activation repertoire in the injured brain: Gradedresponse, molecular mechanisms and cues to physiological function.Brain Res Brain Res Rev 1999;30:77Y105
18. Reisinger C, Yelamanchili SV, Hinz B, et al. The synaptopysin/synap-tobrevin complex dissociates independently of neuroexocytosis. JNeurochem 2004;90:1Y8
19. Garborg I, Torvik A, Hals J, Tangsrud SE, Lindemann R. Congenitalneuronal ceroid lipofuscinosis. A case report. Acta Pathol MicrobiolImmunol Scand [A] 1987;95:119Y25
20. Haltia M, Rapola J, Santavuori P. Infantile type of so-called neuronalceroid-lipofuscinosis. Acta Neuropathol (Berl) 1973;26:157Y70
21. Autti T, Raininko R, Vanhanen SL, Santavuori P. Magnetic resonancetechniques in neuronal ceroid lipofuscinoses and some other lysosomaldiseases affecting the brain. Curr Opin Neurol 1997;10:519Y24
22. Kielar C, Maddox L, Bible E, et al. Successive neuron loss in thethalamus and cortex in a mouse model of infantile neuronal ceroidlipofuscinosis. Neurobiol Dis 2007;25:150Y62
23. Weimer JM, Custer AW, Benedict JW, et al. Visual deficits in a mousemodel of Batten disease are the result of optic nerve degeneration andloss of dorsal lateral geniculate thalamic neurons. Neurobiol Dis 2006;22:284Y93
24. Oliet SH, Piet R, Poulain DA. Control of glutamate clearance andsynaptic efficacy by glial coverage of neurons. Science 2001;292:923Y6
25. Piet R, Poulain DA, Oliet SH. Modulation of synaptic transmissionby astrocytes in the rat supraoptic nucleus. J Physiol Paris 2002;96:231Y6
26. Volterra A, Meldolesi J. Astrocytes, from brain glue to communicationelements: The revolution continues. Nat Rev Neurosci 2005;6:626Y40
27. Lauronen L, Huttunen J, Kirveskari E, et al. Enlarged SI and SIIsomatosensory evoked responses in the CLN5 form of neuronal ceroidlipofuscinosis. Clin Neuropysiol 2002;113:1491Y500
28. Goldstein LS. Do disorders of movement cause movement disordersand dementia? Neuron 2003;40:415Y25
29. Koike M, Shibata M, Waguri S, et al. Participation of autophagy instorage of lysosomes in neurons from mouse models of neuronalceroid-lipofuscinoses (Batten disease). Am J Pathol 2005;167:1713Y28
30. Edelmann L, Hanson PI, Chapman ER, Jahn R. Synaptobrevin bindingto synaptophysin: A potential mechanism for controlling the exocytoticfusion machine. EMBO J 1995;14:224Y31
31. Tarsa L, Goda Y. Synaptophysin regulates activity-dependent synapseformation in cultured hippocampal neurons. Proc Natl Acad Sci U S A2002;22:1012Y6
32. Hinz B, Becher A, Mitter D, et al. Activity-dependent changes of thepresynaptic synaptophysin-synaptobrevin complex in adult rat brain.Eur J Cell Biol 2001;80:615Y9
33. Daly C, Ziff EB. Ca2+-dependent formation of a dynamin-synaptophysincomplex. J Biol Chem 2002;277:9010Y5
34. Virmani T, Gupta P, Liu X, Kavalali ET, Hofmann SL. Progressivelyreduced synaptic vesicle pool size in cultured neurons derived fromneuronal ceroid lipofuscinosisY1 knockout mice. Neurobiol Dis 2005;20:314Y23
J Neuropathol Exp Neurol � Volume 67, Number 1, January 2008 Thalamus and Synapses in CTSD Deficiency
� 2007 American Association of Neuropathologists, Inc. 29
Copyright © 2007 by the American Association of Neuropathologists, Inc. Unauthorized reproduction of this article is prohibited.
Dow
nloaded from https://academ
ic.oup.com/jnen/article/67/1/16/2916840 by guest on 27 July 2022
Copyright © 2022 FDOKUMEN




















