
Eroid-lipofuscinosis (Batten disease) - Massey Research Online
Upload
independentCategory
view
1download
0
1 3
Acta NeuropatholDOI 10.1007/s00401-014-1262-6
OrIgINAl PAPer
Common pathobiochemical hallmarks of progranulin‑associated frontotemporal lobar degeneration and neuronal ceroid lipofuscinosis
Julia K. Götzl · Kohji Mori · Markus Damme · Katrin Fellerer · Sabina Tahirovic · Gernot Kleinberger · Jonathan Janssens · Julie van der Zee · Christina M. Lang · Elisabeth Kremmer · Jean‑Jacques Martin · Sebastiaan Engelborghs · Hans A. Kretzschmar · Thomas Arzberger · Christine Van Broeckhoven · Christian Haass · Anja Capell
received: 15 January 2014 / revised: 13 February 2014 / Accepted: 13 February 2014 © Springer-Verlag Berlin Heidelberg 2014
grN-associated FTlD-TDP (FTlD-TDP/grN), but also those which are characteristic for NCl and lysosomal impairment. In Grn(−/−) mice the lysosomal proteins cathepsin D (CTSD), lAMP (lysosomal-associated mem-brane protein) 1 and the NCl storage components saposin D and subunit c of mitochondrial ATP synthase (SCMAS) were all found to be elevated. Moreover, these mice display increased levels of transmembrane protein (TMeM) 106B, a lysosomal protein known as a risk factor for FTlD-TDP pathology. In line with a potential pathological overlap of FTlD and NCl, Ctsd(−/−) mice, a model for NCl, show elevated levels of the FTlD-associated proteins grN and TMeM106B. In addition, pathologically phosphorylated TDP-43 occurs in Ctsd(−/−) mice to a similar extent as in
Abstract Heterozygous loss-of-function mutations in the progranulin (GRN) gene and the resulting reduction of grN levels is a common genetic cause for frontotem-poral lobar degeneration (FTlD) with accumulation of TAr DNA-binding protein (TDP)-43. recently, it has been shown that a complete grN deficiency due to a homozy-gous GRN loss-of-function mutation causes neuronal ceroid lipofuscinosis (NCl), a lysosomal storage disorder. These findings suggest that lysosomal dysfunction may also contribute to some extent to FTlD. Indeed, Grn(−/−) mice recapitulate not only pathobiochemical features of
Electronic supplementary material The online version of this article (doi:10.1007/s00401-014-1262-6) contains supplementary material, which is available to authorized users.
J. K. götzl · K. Mori · K. Fellerer · g. Kleinberger · C. M. lang · C. Haass (*) · A. Capell (*) Adolf-Butenandt Institute, Biochemistry, ludwig-Maximilians-University Munich, Schillerstrasse 44, 80336 Munich, germanye-mail: [email protected]
A. Capell e-mail: [email protected]
J. K. götzl Institute of Neuroscience, Technical University Munich, 80802 Munich, germany
M. Damme Department of Biochemistry, Christian-Albrechts-University Kiel, 20498 Kiel, germany
S. Tahirovic · e. Kremmer · T. Arzberger · C. Haass german Center for Neurodegenerative Diseases (DZNe) Munich, 80336 Munich, germany
g. Kleinberger · J. Janssens · J. van der Zee · C. Van Broeckhoven Department of Molecular genetics, Neurodegenerative Brain Disease group, VIB, 2610 Antwerp, Belgium
g. Kleinberger · J. Janssens · J. van der Zee · J.-J. Martin · S. engelborghs · C. Van Broeckhoven Institute Born-Bunge, University of Antwerp, 2610 Antwerp, Belgium
g. Kleinberger · C. Haass Munich Cluster for Systems Neurology (SyNergy), 80336 Munich, germany
e. Kremmer Institute of Molecular Immunology, Helmholtz Center Munich, german research Center for environmental Health (gmbH), 81377 Munich, germany
H. A. Kretzschmar · T. Arzberger Center for Neuropathology and Prion research, ludwig-Maximilians-University Munich, 81377 Munich, germany
T. Arzberger Department of Psychiatry and Psychotherapy, ludwig-Maximilians-University Munich, 80336 Munich, germany

Acta Neuropathol
1 3
Grn(−/−) mice. Consistent with these findings, some NCl patients accumulate pathologically phosphorylated TDP-43 within their brains. Based on these observations, we searched for pathological marker proteins, which are char-acteristic for NCl or lysosomal impairment in brains of FTlD-TDP/grN patients. Strikingly, saposin D, SCMAS as well as the lysosomal proteins CTSD and lAMP1/2 are all elevated in patients with FTlD-TDP/grN. Thus, our findings suggest that lysosomal storage disorders and grN-associated FTlD may share common features.
Keywords Frontotemporal lobar degeneration (FTlD) · Progranulin (grN) · TDP-43 · Neuronal ceroid lipofuscinosis (NCl) · Cathepsin D · lysosome · Neurodegeneration
Introduction
Frontotemporal lobar degeneration (FTlD) is the second most frequent form of dementia in people under the age of 65 years with 30–50 % of the patients having a positive family history [28, 62, 64]. The pathological hallmarks of the major variant of FTlD are intracellular ubiquitin and TAr DNA-binding protein (TDP)-43 positive inclusions [3, 49]. This subtype of FTlD is therefore designated FTlD-TDP [42] distinguishing it from other types of FTlD such as FTlD-tau, FTlD-FUS (fused in sarcoma) [43] or FTlD-DPr [45]. In FTlD-TDP patients, TDP-43, a DNA- and rNA-binding protein involved in transcription and splicing, is hyperphosphorylated, proteolytically processed and frequently mislocalized to the cytoplasm [3, 31, 36, 49]. The majority of FTlD-TDP causing mutations were identified in the GRN gene [4, 16], which account for up to 20 % of familial FTlD-TDP cases [24, 27]. Of the 69 path-ogenic mutations reported to date (http://www.molgen.vib-ua.be/FTDMutations/) [17], most are loss-of-function mutations leading to GRN haploinsufficiency [27], which result in a severe reduction of grN levels in tissues and biological fluids of patients [20, 25, 67]. Additionally, mis-sense mutations also lead to reduced functional grN by impairing secretion or misfolding [46, 63, 77, 80].
While lysosomal dysfunction and impaired autophagy have been discussed to contribute to neurodegenerative diseases such as Alzheimer’s disease and Parkinson’s dis-ease (reviewed in [51, 58]), less evidence exists for lysoso-mal malfunction in FTlD-TDP. However, recently it was reported that a complete loss of grN surprisingly results in adult-onset neuronal ceroid lipofuscinosis (NCl), a neu-ronal lysosomal storage disease (lSD) [68]. In that study, two siblings were shown to carry a homozygous dele-tion of four base pairs in the GRN gene (c.813_816del), which leads to a frameshift and a premature termination
of translation. The families of the parents were distantly related and in both families some cases with dementia without clear diagnosis were reported [68]. The identi-cal heterozygous mutation was previously shown to cause FTlD-TDP [5, 41, 87]. NCl, a fatal disorder with pro-gressive neuronal loss, is the most common cause of neu-rodegeneration in children and young adults. NCl is a clinically and genetically heterogeneous disorder, with autosomal recessive inheritance, in most NCl variants (reviewed in [35]). lipofuscin, an autofluorescent lipopig-ment, is commonly found in all forms of NCl. Depending on the NCl variant, the major protein components of lipo-fuscin are either the sphingolipid activator proteins (sapo-sin) A and D or subunit c of mitochondrial ATP synthase (SCMAS) [19, 73]. Interestingly, lipofuscin accumulation [1, 54, 83] as well as accumulation of ubiquitin [1, 26, 54, 83, 85, 86] and p62 [83] positive protein aggregates have been detected in Grn knockout mice. These data provided some evidence that reduced grN might be associated with a dysfunction of the autophagosomal–lysosomal sys-tem, even before the linkage of grN to NCl was known. Besides the fact that a total loss of grN results in NCl, rare FTlD-causing mutations in the charged multivesicu-lar body protein 2B (CHMP2B) gene [66] or in the valosin-containing protein (VCP) gene [81, 82] may further suggest a lysosomal involvement in FTlD [72, 74]. In addition, TMeM106B, a recently identified risk factor for grN-associated FTlD-TDP (FTlD-TDP/grN), is localized in late endosomes and lysosomes [7, 13, 40]. It was suggested that TMEM106B variants confer a risk for FTlD-TDP by increased expression levels of TMeM106B [76]. elevated TMeM106B mrNA and protein levels were confirmed in GRN mutation carriers [13, 21]. Furthermore, studies in cellular model systems have provided evidence that over-expression of TMeM106B leads to lysosomal impairment and might influence grN expression [7, 13]. In addition, grN levels are influenced by sortilin, a member of the vac-uolar protein sorting (Vps) 10 receptor family. Most likely sortilin is responsible for uptake and clearance of extracel-lular grN, by facilitating its transport to lysosomes [33].
In this study, we aimed to investigate potential similari-ties between NCl and FTlD-TDP. Our findings suggest that lysosomal storage disorders and FTlD may share common pathological mechanisms and could belong to two extreme ends of one disease spectrum.
Materials and methods
Human brain tissue
Frontal cortical brain tissue from six FTlD-TDP patients of a Belgian GRN-mutation founder family (grN

Acta Neuropathol
1 3
IVS1+5g>C) [8, 16], six control cases without neurologic pathology and one case with familial adult NCl [44, 50] were provided from the Antwerp Brain Bank (Institute Born-Bunge at the University of Antwerp, Antwerp, Bel-gium). The london Neurodegenerative Disease Brain Bank and Brains for Dementia research provided frontal corti-cal brain tissue from two juvenile NCl patients, one NCl type 2 (ClN2) patient caused by deficiency of the lysoso-mal enzyme tripeptidyl peptidase 1 (TPP-1), and four nor-mal control cases. In addition, paraffin-embedded frontal cortical sections from three NCl cases and one control were provided. Frontal cortical brain tissue of three ClN2 cases and one normal control cases without neurologic pathology were obtained from the Human Brain and Spi-nal Fluid resource Center, los Angeles, and brain tissue of four ClN2 cases were distributed by the Batten Disease registry, genetic Services and Specialty Clinical labora-tories, NYS Institute for Basic research in Developmental Disabilities. Detailed information on pathology and clinical representation are summarized in Supplementary Tables 1 and 2.
Mouse tissue
Mice were killed by CO2 inhalation according to animal handling laws. Brain tissues were dissected from the Grn knockout mouse strain with deletion of Grn exons 2–13 [37] and the Ctsd knockout mouse strain [60].
Antibodies
The following antibodies were used for immunoblotting: mouse monoclonal anti-β-actin antibody (Sigma-Aldrich; 1:10,000–20,000), rabbit polyclonal anti-calnexin antibody (Stressgen; 1:2,000), mouse monoclonal anti-α-tubulin antibody (Sigma-Aldrich; 1:5,000–1:40,000), rabbit poly-clonal anti-TDP43 antibody (ProteinTech group; 1:1,000), rat monoclonal anti-phospho TDP-43 (Ser409/Ser410) antibody clone 1D3 (1:10) [48], rat monoclonal anti-C-ter-minal TDP-43 antibody clone 2H4 (1:50) generated against amino acid residues 404–413 of human TDP-43, rat mono-clonal anti-TMeM106B antibody clone 6F2 (1:50) [40], goat polyclonal anti-cathepsin D antibody (Santa Cruz Biotechnology; 1:500), rat monoclonal anti-grN anti-body clone 8H10 (1:50) raised against the C-terminus of mouse grN (amino acid residues 580–597), mouse mon-oclonal anti-hlAMP1 antibody clone H4A3 (Santa Cruz Biotechnology; 1:500), mouse monoclonal anti-hlAMP2 antibody clone H4B4 (Santa Cruz Biotechnology; 1:500), goat polyclonal anti-saposin D antibody (1:1,000) [38], rabbit anti-gFAP (glial fibrillary acidic protein) antibody (Dako; 1:5,000) and mouse monoclonal anti-gAPDH anti-body (Invitrogen life Technologies; 1:10,000). Secondary
antibodies were horseradish peroxidase-conjugated donkey anti-goat Igg (Santa Cruz Biotechnology; 1:5,000), anti-mouse Igg (Promega; 1:10,000), anti-rabbit Igg (Promega; 1:10,000), goat anti-rat Igg + IgM (l+M) (Dianova; 1:5,000), and generated mouse anti-rat Igg2c (1:1,000). For immunohistochemistry the following antibodies were used: rat monoclonal anti-mlAMP1 antibody clone 1D4B (developed by J. Thomas August, distributed by Develop-mental Studies Hybridoma Bank, NICHD, maintained by the University of Iowa, Department of Biology; 1:200), mouse anti-hlAMP1 clone H4A3 (developed by J. Thomas August and James e.K. Hildreth, distributed by Develop-mental Studies Hybridoma Bank; 1:2,000), rabbit poly-clonal anti-IBA1 antibody (Wako Chemicals; 1:200), rab-bit anti-SCMAS antibody (1:5,000) [59], goat polyclonal anti-saposin D antibody (1:200,000) [38], goat polyclonal anti-cathepsin D antibody (Santa Cruz Biotechnology; 1:200,000), rabbit polyclonal anti-TMeM106B antibody clone N2077 (1:5,000) [13], rat monoclonal anti-phospho TDP-43 (Ser409/Ser410) antibody clone 1D3 (1:50) [48], anti-HlA-DP/DQ/Dr (Cr3/43) antibody (Dako; 1:100) and mouse anti-p62 antibody (BD Biosciences; 1:1,000). Secondary antibodies directed to rat or rabbit Igg were conjugated to Alexa Fluor 488 or Alexa Fluor 555 (Invit-rogen life Technologies; 1:200). The DCS Super Vision 2 HrP-Polymer-Kit for mouse/rabbit antibodies (DCS Innovative Diagnostik-Systeme) and rabbit anti-goat anti-body (Dako; 1:1,000) as a bridging antibody were used for human sections.
Biochemical analysis and immunoblotting
Aliquots of crunched frozen brain tissue were used for sequential biochemical protein extraction. All buffers were freshly supplemented with protease inhibitor cocktail (Sigma-Aldrich) and phosphatase inhibitor (roche Applied Science). First, the brain material was homogenized in high salt (HS) buffer (0.5 M NaCl, 10 mM Tris–HCl pH 7.5, 5 mM eDTA, 1 mM DTT, 10 % sucrose) to extract solu-ble, non-transmembrane proteins, such as grN, CTSD, gFAP, prosaposin and soluble TDP-43. HS-soluble pro-teins were separated by centrifugation at 15,000×g, 4 °C for 30 min. To extract transmembrane proteins (lamp1/2, TMeM106B, calnexin) and less soluble non-transmem-brane proteins (prosaposin, saposin D, TDP-43, pTDP-43), the HS-insoluble protein pellet was further extracted with rIPA buffer (150 mM NaCl, 20 mM Tris–HCl pH 7.4, 1 % NP40, 0.05 % Triton X-100, 0.5 % sodium-desoxycholate, 2.5 mM eTDA), followed by centrifugation at 150,000×g, 4 °C for 45 min. Finally, insoluble proteins (TDP-43, pTDP-43) were further extracted with urea buffer (30 mM Tris–HCl pH 8.5, 7 M urea, 2 M thiourea, 4 % CHAPS) and centrifuged at 150,000×g, 4 °C for 45 min. The protein

Acta Neuropathol
1 3
concentrations of HS and rIPA fractions were determined by BCA protein assay (Pierce, Thermo Scientific). The urea fractions were adjusted according to protein concentra-tion of the corresponding rIPA fractions. equal amounts of protein were separated by SDS-PAge and transferred onto polyvinylidene difluoride membranes (Immobilon-P, Merck Millipore) if not stated otherwise. For TMeM106B detection, sample preparation and separation by urea SDS-PAge was performed as previously described [40]. For detection of saposin D, separated protein samples were transferred onto nitrocellulose blotting membrane (Protran BA85, ge Healthcare lifesciences) followed by boiling the membrane in phosphate buffered saline (PBS). For pro-tein detection, membranes were probed with the indicated primary antibodies followed by horseradish peroxidase-conjugated secondary antibody. Bound antibodies were detected with eCl (Amersham Western Blotting Detection reagent, ge Healthcare lifesciences) or eCl Plus (Pierce eCl Plus Western Blotting Substrate, Thermo Scientific). Images were acquired with the lAS-4,000 image reader (Fujifilm life Science) and quantitatively analyzed using the Multi-gauge V3.0 software (Fujifilm life Science).
Immunohistochemistry
Brains of the Grn mice were fixed with 4 % paraformalde-hyde (PFA) in PBS for 48 h. After fixation, the tissue was equilibrated in 30 % sucrose in PBS at 4 °C, embedded in Shandon M-1 embedding Matrix (Thermo Scientific), frozen on dry ice and stored at −80 °C. For immunohisto-chemistry, 25 μm frozen tissue sections were cut, treated with 10 mM sodium citrate, pH 6 at 95 °C for 20 min, washed with 0.5 % Triton X-100 in PBS, blocked with 5 % goat serum (Invitrogen life Technologies) and 0.5 % Triton X-100 in PBS for 1 h and subsequently incubated overnight with primary antibodies diluted in blocking solution. The sections were incubated with appropriate secondary anti-bodies. To block the lipofuscin autofluorescence, sections were incubated with 0.1 % Sudan black B (Sigma-Aldrich) in 70 % ethanol for 20 min. Confocal images were acquired with an lSM 710 confocal microscope (Carl Zeiss Micro-Imaging) in sequential scanning mode using a Plan-Apochromat 40×/1.4 oil objective and the ZeN 2011 soft-ware package (black edition, Carl Zeiss MicroImaging). Paraffin-embedded human sections were deparaffinized with xylene and ethanol. Antigen retrieval was performed by microwaving in citrate buffer pH 6 for 18 min. endog-enous peroxidase was blocked with 5 % H2O2 in methanol for 15 min. Then sections were settled in PBS with 0.02 % Brij35 (Sigma-Aldrich). After blocking with 2 % FBS in PBS for 5 min, the respective primary antibody was applied to the sections overnight at 4 °C. When the first antibody was derived from goat, rabbit anti-goat antibody (Dako)
was used as bridging antibody for 1 h. After brief rinsing with 0.02 % Brij35 in PBS, antibody binding was detected and visualized with DCS Super Vision 2 HrP-Polymer-Kit for mouse/rabbit antibodies (DCS Innovative Diagnostik-Systeme) using DAB as chromogen. For phosphorylated TDP-43 (clone 1D3) and HlA-DP/DQ/Dr (clone Cr3/43) staining, automated staining with the Benchmark system (roche Applied Science) was performed according to man-ufacturer’s protocol. Counterstaining for cellular structures was performed with haemalum. Microscopic images were obtained using a BX41 microscope with a 40×/0.65 objec-tive (Olympus) and cellSens Ver 1.6 software (Olympus).
Statistical analysis
For statistical analysis the unpaired, two-tailed student t test or the Mann–Whitney test was performed. All statis-tical analyses were performed using the graphPad Prism 5.04 program (graphPad Software) and statistical signifi-cance was set at *P < 0.05, **P < 0.01, and ***P < 0.001.
Results
grN deficiency in mice recapitulates pathobiochemical features of FTlD-TDP/grN and NCl
We studied expression levels of FTlD- and NCl-associ-ated proteins in Grn(−/−) mice. Grn(−/−) mice geneti-cally recapitulate the complete absence of grN as it was found in two NCl siblings with a homozygous GRN loss-of-function mutation [68]. We therefore expected that these mice could serve as a bonafide model for NCl with grN deficiency. Indeed similar to NCl patients, Grn(−/−) mice show accumulation of lipofuscin in brain [1, 54, 83]. Since lipofuscin accumulation was seen earliest in the 7- to 8-month-old Grn(−/−) mice and is increasing with age [1, 54], we decided to analyze grn(−/−) mice from the age of 3–4 months up to 20–24 months. One of the two pro-teins known to accumulate to high levels in a subset of NCl patients, namely in CTSD (ClN10) and palmitoyl protein thioesterase (PPT) 1 (ClN1) associated NCl, is saposin D [35]. We therefore analyzed saposin D levels in Grn(−/−), Grn(+/−) and Grn(+/+) mouse brains dur-ing ageing. Significant saposin D elevation occurred at 12 months of age and lead to a 4.5-fold increase in 20- to 24-month-old Grn(−/−) mice compared to Grn(+/+) and Grn(+/−) mice (Fig. 1a; Supplementary Fig. 1). Moreover, cathepsin D (CTSD), a lysosomal aspartic protease of the pepsin superfamily, which is elevated in lSD variants [14, 79], was already significantly elevated in the 3- to 4-month-old Grn(−/−) mice (Supplementary Fig. 2a). During age-ing both, the proenzyme and the mature form of CTSD

Acta Neuropathol
1 3
Fig. 1 Grn(−/−) mice recapitulate pathobiochemical features of NCl and FTlD-TDP. Grn(+/+), Grn(+/−) and Grn(−/−) mouse brains at the age of 20–24 months were sequentially extracted with HS and rIPA buffer. The rIPA fractions were probed for prosapo-sin and saposin D (a) and TMeM106B (e). The HS fractions were probed for the lysosomal proteinase CTSD, with an antibody that recognizes the proenzyme (CTSDpro) and mature form (CTSDmat) of CTSD, circle indicates a non-specific band (b). Actin (HS) and calnexin (rIPA) were used as loading controls. The graphs (a, b, e) represent the quantification of protein levels by measuring the chemi-luminescence signal on the immunoblots. Data were normalized to the corresponding mean value of Grn(+/+) mice and are shown
as mean ± SD. For statistical analysis the unpaired t test was used to compare Grn(−/−) to Grn(+/+) mice (*P < 0.05; **P < 0.01; ***P < 0.001). c Brain sections of 20- to 24-month-old Grn(+/+), Grn(+/−) and Grn(−/−) mice were stained for lAMP1, IBA1 and DAPI. The scale bar represents 20 μm. d Grn(+/+), grn(+/−) and Grn(−/−) mouse brains at the age of 16–18 months were sequen-tially extracted with HS, rIPA and urea buffer. Urea fractions were probed with the antibody directed to the carboxyl-terminus of TDP-43, recognizing full-length TDP-43 as well as phosphorylated TDP-43 (pTDP-43) (d, upper panel), and with the phospho-specific anti-body for TDP-43 (d, lower panel). Actin was used to verify equal loading

Acta Neuropathol
1 3
increased up to 10-fold in 20- to 24-month-old Grn(−/−) mice in comparison to Grn(+/+) and Grn(+/−) mice (Fig. 1b). Increased expression of CTSD was at least in part caused by enhanced transcription, since we observed a 3-fold increase of Ctsd mrNA at 20–24 months (Supple-mentary Fig. 2b). Finally, Grn(−/−) mice also showed ele-vated levels of the lysosomal protein lAMP1 primarily in microglia as shown by co-localization with IBA1 (Fig. 1c). Thus, the Grn(−/−) mice recapitulate several key features typical for NCl. However, these mice not only show NCl-like pathobiochemistry, but also some features known to be characteristic for FTlD-TDP. Consistent with previous finding [83, 85, 86], insoluble and pathologically phospho-rylated TDP-43 accumulates in the brains of these mice at age of 16–18 months (Fig. 1d). Interestingly, TMeM106B a risk factor for FTlD-TDP/grN and shown to be elevated in FTlD-TDP/grN patients [9, 13], accumulates already in young Grn(−/−) mice at age of 3–4 months and was further elevated up to 3-fold at 20–24 months (Fig. 1e; Sup-plementary Fig. 3a). elevation of TMeM106B protein is not due to increased transcription of Tmem106b (Supple-mentary Fig. 3b). Similar results as described above were obtained for a second independent Grn knockout mouse model (Supplementary Fig. 4) [83]. Immunohistochemis-try confirmed an increase of CTSD and saposin D in cor-tex, hippocampus and thalamus of aged Grn(−/−) mice (16 months) (Supplementary Fig. 5). In addition, subunit c of mitochondrial ATP synthase (SCMAS), the other major storage protein in most NCl variants is also elevated in all analyzed brain regions of Grn(−/−) mice (Supplementary Fig. 5).
A mouse model for NCl recapitulates pathobiochemical features of FTlD-TDP/grN
Based on our findings in Grn(−/−) mice, we speculated that NCl mouse models might present with pathobiochem-ical features of FTlD-TDP. Ctsd(−/−) mice [60] provide an excellent tool for studying NCl [39] caused by CTSD loss-of-function mutations [22, 65] since they develop severe neurological symptoms with accumulation of stor-age material, dysmyelination, neurodegeneration and pre-mature death [39, 53]. CTSD deficiency leads to the most severe variant of NCl and infants die within the first days after birth [22, 66]. Although the course of degeneration is milder in mice, Ctsd(−/−) mice die prematurely at the age of 26 (+/−1) days [39, 60], while Ctsd(+/−) mice have a normal life span and no obvious phenotype [39].
By analyzing brain lysates of Ctsd(+/+), (+/−), and (−/−) mice at the age of 21–24 days, we confirmed the accumulation of prosaposin in Ctsd(−/−) mice [34] (Fig. 2a). In addition, saposin D, the major storage com-ponent of CTSD-associated NCl patients, strongly
accumulated in Ctsd(−/−) mice (Fig. 2a). Immunohis-tochemistry confirmed increased expression of saposin D in all analyzed brain regions (Supplementary Fig. 6). Strikingly, Ctsd(−/−) mice also showed a robust increase of grN and TMeM106B levels (Fig. 2b, c). Close to the terminal stage Ctsd(−/−) mice have elevated numbers of activated microglial cells in various brain regions [47, 53]. In line with previous findings [55, 69], microglial cells are a source of increased grN expression (Supplemen-tary Fig. 6). Interestingly, the increase of grN protein is partially mediated by elevated Grn transcript levels in Ctsd(−/−) mice (Supplementary Fig. 7a), whereas the increased levels of TMeM106B are caused by posttran-scriptional mechanisms (Supplementary Fig. 7b).
Next, we investigated whether insoluble, phosphoryl-ated TDP-43, as detected in Grn(−/−) mice (Fig. 1d) and found as a biochemical marker in human FTlD-TDP brain [31, 49] (see also Fig. 4), accumulates in Ctsd(−/−) mice. Indeed, we found that pathologically phosphorylated TDP-43 selectively accumulated in brains of Ctsd(−/−) mice (Fig. 2d). Thus these findings further confirm an overlap of pathobiochemical features of FTlD and NCl in the stud-ied mouse model.
Phosphorylated TDP-43 in brains of NCl patients
To search for FTlD-associated TDP-43 pathology in humans, we analyzed brains of NCl patients diagnosed with ClN2, juvenile NCl and familial adult NCl (Sup-plementary Table 2). Immunohistochemically, these cases should accumulate the most common NCl storage protein SCMAS [35] which could indeed be confirmed for the ana-lyzed cases (#21, 22, 23) (Fig. 3a). Furthermore, biochemi-cal analyses showed that saposin D, CTSD and lAMP2 were all significantly elevated in NCl brains (Fig. 3b). Consistent with a potential pathological overlap of FTlD-TDP/grN and NCl, phosphorylated TDP-43 was detected in 9 out of 11 patients, although it remained largely soluble (Fig. 3b). Interestingly, case #20 and particularly case #26, which showed the highest levels of soluble phosphoryl-ated TDP-43 showed a significant accumulation of insolu-ble phosphorylated TDP-43. In addition, similar to the depletion of soluble TDP-43 in FTlD-TDP/grN patients [3, 49], NCl patients had significantly reduced levels of the soluble TDP-43 holoprotein (Fig. 3b). In case #26 the strongest reduction of soluble TDP-43 was observed, which is in line with the striking increase of insoluble phosphoryl-ated TDP-43 in the brain of this patient (Fig. 3b). Finally, in the nine cases with accumulation of phosphorylated TDP-43 the strongest astrogliosis was observed (Fig. 3b). Thus, human patients with lysosomal dysfunction show at least to some extent FTlD-related TDP-43 pathology, although deposition of TDP-43 could not be detected (data

Acta Neuropathol
1 3
not shown). The latter is likely due to the short life span of the NCl patients. In contrast to the mouse model for NCl, no increase of TMeM106B was observed (Fig. 3b), which may be related to the massive neuronal cell death, which could lead to a selective reduction of the neuronally expressed TMeM106B.
Increase of NCl-associated lysosomal proteins in brains of FTlD-TDP/grN patients
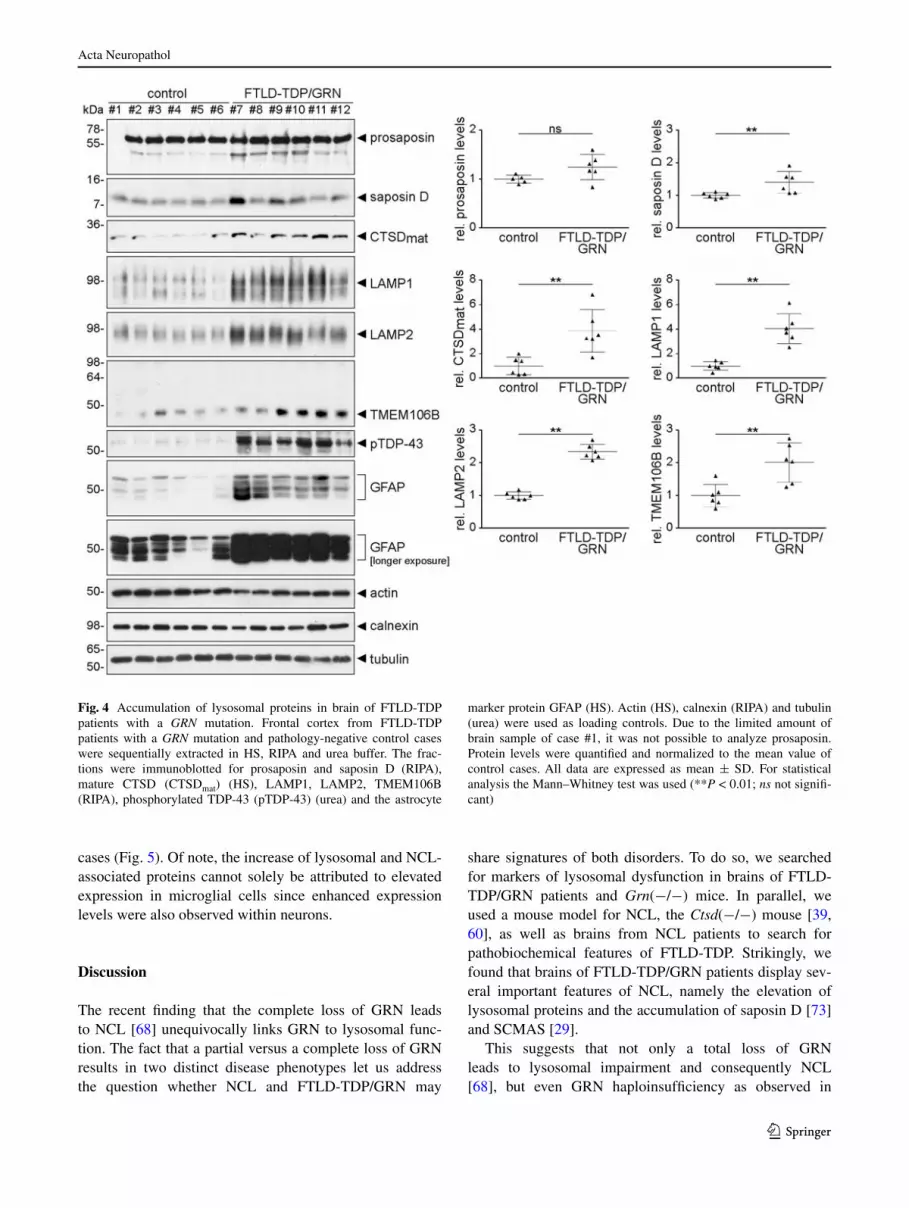
If lysosomal dysfunction contributes to the pathogenesis of FTlD-TDP/grN, one would expect proteins that are typi-cally elevated in brains of NCl patients to be increased in FTlD-TDP/grN cases. We therefore investigated brains of FTlD-TDP/grN patients and healthy controls for levels of saposin D, a protein known to accumulate in two NCl vari-ants and in Grn (−/−) mice as shown above. Indeed, sapo-sin D was also significantly elevated in brains of FTlD-TDP/grN patients (Fig. 4). Moreover, the lysosomal
hydrolase CTSD and the lysosomal membrane proteins lAMP1 and lAMP2 were also significantly elevated (~4-fold, ~4-fold, and ~2.5-fold, respectively) in brains of GRN mutation carriers compared to control brains (Fig. 4). TMeM106B and pathologically phosphorylated TDP-43 were also elevated as expected for FTlD-TDP/grN cases (Fig. 4). In addition, all FTlD-TDP/grN patients showed an increase of gFAP expression due to astrogliosis (Fig. 4). Thus, FTlD-TDP/grN patients apparently share known pathobiochemical features with NCl patients (see Fig. 3).
To further confirm these findings, we performed immu-nohistochemistry on sections of FTlD-TDP/grN brains. elevated levels of saposin D could be detected in granular structures within the cytoplasm of neurons and glial cells (Fig. 5). Since saposin D as a major storage component has only been described for a subset of NCl patients, we additionally stained brains of FTlD-TDP/grN patients for the other more common NCl storage component SCMAS. Interestingly, staining intensities for SCMAS were higher
Fig. 2 Ctsd(−/−) mice, an NCl model, recapitulate pathobiochemical features for FTlD-TDP. Brains of 21- to 24-day-old Ctsd(+/+), Ctsd(+/−) and Ctsd(−/−) mice were sequentially extracted with HS, rIPA and urea buffer. HS extracts were immunoblotted for the proenzyme (CTSDpro) and the mature form (CTSDmat) of CTSD (a) and grN (b). Note, that the two youngest Ctsd(−/−) mice (21 days of age) loaded in the last two lanes show the lowest levels of grN expression. Prosaposin, saposin D (a), and TMeM106B (c) were detected in rIPA extracts. Actin (HS) and calnexin (rIPA) confirmed equal loading. Pro-tein levels were quantified and normalized to the mean value of Ctsd(+/+) and Ctsd(+/−) mice. The data are shown as mean ± SD. For statistical analysis the unpaired t test was used to compare Ctsd(−/−) to control mice (**P < 0.01; ***P < 0.001). d Urea fractions were probed for insoluble full-length TDP-43 and phospho-rylated TDP-43 (pTDP-43) with antibodies directed to the carboxyl-terminus and the phos-phorylation site Ser409/Ser410 as marker of FTlD-TDP pathology. Tubulin was used as loading control

Acta Neuropathol
1 3
in neurons of FTlD-TDP/grN patients when compared to neurons of control cases (Fig. 5). Thus, both stor-age components of NCl variants were elevated in brains of FTlD-TDP/grN patients. In line with the biochemi-cal data, staining intensities for neuronal and glial CTSD, lAMP1 and, as shown recently [9, 13], for intraneuronal
TMeM106B were elevated in FTlD-TDP/grN patients. Intracellular inclusions showed FTlD-TDP typical p62 and phosphorylated TDP-43 immunoreactivity, reflect-ing the enhanced biochemically detected phosphorylated TDP-43 levels. Furthermore, stainings for HlA revealed numerous activated microglial cells in FTlD-TDP/grN
Fig. 3 Phosphorylated TDP-43 in NCl patients. a Immunohisto-chemical detection of SCMAS in frontal cortex of NCl patients (case #20, 21, 22) and control case #14. Scale bar represents 50 μm. b Frontal cortical tissue from NCl patients diagnosed with ClN2 (#18–21 and #25–28), juvenile NCl (#22, 23), adult NCl (#24), and control cases (#13–17) was sequentially extracted in HS, rIPA and urea buffer. The fractions were immunoblotted for saposin D (rIPA), mature CTSD (CTSDmat) (HS), lAMP2, TMeM106B (rIPA), phos-
phorylated TDP-43 at Ser409/Ser410 (pTDP-43) (HS, urea), and TDP-43 (HS). gAPDH (HS), actin (rIPA, urea) were used as loading controls. Protein levels were quantified and normalized to the mean value of control cases in gel 1. All data are expressed as mean ± SD. For statistical analysis the Mann–Whitney test was used (*P < 0.05; **P < 0.01; ns not significant). Note that pTDP-43 and gFAP could not be quantified due to absence of signals in controls
Acta Neuropathol
1 3
cases (Fig. 5). Of note, the increase of lysosomal and NCl-associated proteins cannot solely be attributed to elevated expression in microglial cells since enhanced expression levels were also observed within neurons.
Discussion
The recent finding that the complete loss of grN leads to NCl [68] unequivocally links grN to lysosomal func-tion. The fact that a partial versus a complete loss of grN results in two distinct disease phenotypes let us address the question whether NCl and FTlD-TDP/grN may
share signatures of both disorders. To do so, we searched for markers of lysosomal dysfunction in brains of FTlD-TDP/grN patients and Grn(−/−) mice. In parallel, we used a mouse model for NCl, the Ctsd(−/−) mouse [39, 60], as well as brains from NCl patients to search for pathobiochemical features of FTlD-TDP. Strikingly, we found that brains of FTlD-TDP/grN patients display sev-eral important features of NCl, namely the elevation of lysosomal proteins and the accumulation of saposin D [73] and SCMAS [29].
This suggests that not only a total loss of grN leads to lysosomal impairment and consequently NCl [68], but even grN haploinsufficiency as observed in
Fig. 4 Accumulation of lysosomal proteins in brain of FTlD-TDP patients with a GRN mutation. Frontal cortex from FTlD-TDP patients with a GRN mutation and pathology-negative control cases were sequentially extracted in HS, rIPA and urea buffer. The frac-tions were immunoblotted for prosaposin and saposin D (rIPA), mature CTSD (CTSDmat) (HS), lAMP1, lAMP2, TMeM106B (rIPA), phosphorylated TDP-43 (pTDP-43) (urea) and the astrocyte
marker protein gFAP (HS). Actin (HS), calnexin (rIPA) and tubulin (urea) were used as loading controls. Due to the limited amount of brain sample of case #1, it was not possible to analyze prosaposin. Protein levels were quantified and normalized to the mean value of control cases. All data are expressed as mean ± SD. For statistical analysis the Mann–Whitney test was used (**P < 0.01; ns not signifi-cant)

Acta Neuropathol
1 3
Fig. 5 Increase of the major NCl storage components and of lysosomal proteins in brains of FTlD-TDP/grN patients. Immunohistochemical detec-tion of saposin D, SCMAS, CTSD, lAMP1, TMeM106B, phosphorylated TDP-43 (pTDP-43), p62 and HlA-DP/DQ/Dr (HlA) in frontal cortex of FTlD-TDP/grN patients (case #10, 11) and control case #6. The NCl storage proteins sapo-sin D and SCMAS accumulate in FTlD-TDP/grN patients. While saposin D is elevated in neurons and glial cells, SCMAS is mainly elevated in neurons. In FTlD-TDP/grN cases expression levels of lysosomal proteins lAMP1 and CTSD are elevated in neurons, but also in glial cells when compared to the control case. Staining intensities for TMeM106B are enhanced in neurons of FTlD-TDP/grN cases. Furthermore, FTlD-TDP/grN cases show characteristic pTDP-43- and p62-positive cytoplasmic inclusions. Neurons indicated by arrowheads are shown at a higher magnification in the pictures inserted. Scale bars represent 50 and 10 μm in the inset

Acta Neuropathol
1 3
FTlD-TDP/grN may confer a risk for lysosomal dysfunc-tion. The significant accumulation of lysosomal proteins and saposin D observed in brains of FTlD-TDP/grN patients can be recapitulated in Grn(−/−) mice, but not in Grn(+/−) mice. The reason why grN haploinsufficiency in mice is insufficient to cause features of NCl or FTlD-TDP is unknown (current study, [1, 37, 54]), but one may speculate that the relative short life span of mice in com-parison to humans prevents age-dependent neuropathol-ogy. In addition, the variability in age at onset in patients with grN haploinsufficiency suggests that additional genetic or environmental factors play a role for developing FTlD-TDP.
Our findings raise the question whether the increase of lysosomal proteins and the accumulation of saposin D and SCMAS, which resemble certain lysosomal diseases, are the cause or the consequence of neuronal degeneration and death in FTlD-TDP. At least for Alzheimer’s disease, an early increase of CTSD has been reported [12] and lysoso-mal dysfunction is discussed as a primary and early cause for neurodegeneration [52].
In line with these findings, Grn(−/−) mice showed sig-nificantly increased lysosomal proteins long before patho-logical signatures of FTlD-TDP could be detected. CTSD and the lysosomal membrane protein TMeM106B were already elevated at 3–4 months (Supplementary Figs. 2a, 3a), an age where no astrogliosis and microgliosis [1, 26, 54, 83, 86] as well as no pathological TDP-43 phos-phorylation was observed [83, 85, 86] (data not shown). evidence for lysosomal impairment in FTlD-TDP/grN patients is provided by the elevated levels of CTSD and of lysosomal membrane proteins, including lAMP1, lAMP2, and TMeM106B. That the increase of lysoso-mal proteins is simply achieved by an increased number of activated microglial cells is unlikely, since we found increased lysosomal proteins not only in microglial cells, but also in neurons (Fig. 5). Particularly TMeM106B is widely expressed in neurons [9, 13] and therefore probably increased independent of microgliosis (Fig. 5). Specifically, the increase of the latter protein is supportive for a lyso-somal component in FTlD-TDP/grN, as TMeM106B, recently identified as lysosomal membrane protein [7, 13, 40], has been associated with a significant increase for the risk to develop FTlD [15, 21, 76, 78]. Whether these pro-teins are increased as a response to lysosomal stress is cur-rently unclear. However, we find it more likely that these membrane proteins are simply increased due to the enlarge-ment of lysosomes unable to cope with the accumulation of proteins destined for lysosomal break down. This is in line with the robust increase of TMeM106B in Ctsd(−/−) mice, a model of NCl with dramatic enlargement of lys-osomes [39]. On the other hand, the equal increase of both, proenzyme and mature CTSD in Grn(−/−) mice, indicates
fairly normal maturation of CTSD and thus suggests nor-mal lysosomal acidification. In Grn(−/−) mice CTSD is elevated on mrNA [1, 70, 83] and on protein level. ele-vated Ctsd mrNA might indicate an active up-regulation of CTSD to compensate increased lysosomal cargo accu-mulation. This is consistent with the recent finding of increased mrNA levels of other lysosomal enzymes [70] in Grn(−/−) mice. Here, the authors suggested that mTOrC1 (mammalian target of rapamycin complex 1) is less active in Grn(−/−) mice, which as a consequence leads to trans-location of TFeB (transcription factor eB) to the nucleus and therefore to increased expression of lysosomal genes regulated via a CleAr (coordinated lysosomal expression and regulation) element [70]. Whether CTSD elevation is beneficial or harmful remains to be further addressed. Both scenarios have been described. There is growing evidence that CTSD can promote cell survival in neurodegenera-tive disorders [10, 56, 84], but it has also been shown that increased expression of lysosomal hydrolases could result in lysosomal destabilization and leakage of lysosomal content into the cytosol followed by increased apoptosis (reviewed in [6, 57]) and neuronal vulnerability [2].
In addition, we also searched for pathobiochemical fea-tures of FTlD-TDP in Ctsd(−/−) mice, a model for NCl. These mice have a severe CNS phenotype and a shortened life span ultimately leading to death after 3–4 weeks [60]. They accumulate SCMAS [39] and prosaposin [34]. Sapo-sin D, a cleavage product of prosaposin and the major stor-age component in patients with a loss-of-function mutation in CTSD [65], has so far not been detected in Ctsd(−/−) mice [34]. Therefore, it has been speculated that CTSD might be required for saposin D generation in mice [34]. Although CTSD is involved in processing of prosapo-sin, it is not sufficient to liberate mature saposin D [32]. In line with that, we show that Ctsd(−/−) mice accumu-late both prosaposin and saposin D. Strikingly, we found phosphorylated TDP-43 and elevated levels of TMeM106B in Ctsd(−/−) mice and at least some brains of patients with NCl show increased levels of soluble phosphoryl-ated TDP-43. In addition, grN protein and mrNA levels are significantly elevated in brain extracts of Ctsd(−/−) mice. However, we cannot exclude that the high grN expression is triggered by inflammation, since grN levels increase due to inflammatory conditions in activated micro-glial cells [69] and microgliosis is present throughout the brain at terminal stage in Ctsd(−/−) mice [47, 53]. In con-trast, we previously observed a robust increase of grN by impaired lysosomal acidification due to inhibition of vacu-olar ATPase [11] and grN transcription is supposed to be regulated by TFeB [70], the master regulator of expression of lysosomal proteins [61]. Therefore, the increase of grN in Ctsd(−/−) mice might also represent a rescue effort in response to lysosomal dysfunction. This is also consistent

Acta Neuropathol
1 3
with the finding that the complete loss of grN-function in humans causes NCl [68]. However, the changes of phos-phorylated TDP-43, grN and TMeM106B are not entirely identical in the analyzed NCl patients and Ctsd(−/−) mice. Whereas phosphorylated TDP is mostly insoluble in Ctsd(−/−) mice, it remains soluble in NCl patients (with the interesting exception of case #20 and #26). It is tempt-ing to speculate that TMeM106B, the genetic risk factor for FTlD-TDP/grN and as recently shown for C9orf72 expansion associated FTlD [23, 75], affect TDP-43 pathol-ogy in NCl patients, as it has been shown for C9orf72 cases [75]. Therefore, it might be interesting to determine the risk alleles of TMeM106B in NCl cases in future stud-ies and address the questions whether the genetic modifier of FTlD also influence the disease progression of NCl. In comparison to Ctsd(−/−) mice, the lack of an increase of neuronally expressed TMeM106B in NCl brain may be caused by the massive neuronal loss seen in NCl patients (reviewed in [35]). One should also keep in mind that the NCl brains investigated harbored no loss-of-function mutation in the CTSD gene (ClN10). Therefore, we can-not exclude that the robust increase of TMeM106B in Ctsd(−/−) mice is a direct result of the CTSD loss, and not of the lysosomal dysfunction.
Together, our findings suggest that lysosomal impair-ment in NCl and lSD might be accompanied by patho-logical alterations typically observed in FTlD-TDP patients and vice versa. This is supported by the finding that CHMP2B [74] and VCP [72] mutations apparently also affect lysosomal function. Moreover, accumulating evi-dence in Alzheimer’s and Parkinson’s disease suggest that dysfunction in the endosomal-autophagic-lysosomal sys-tem contributes to disease progression (reviewed in [18, 30, 52, 71]). Thus, lysosomal dysfunction might be a general feature of neurodegeneration.
Acknowledgments We thank Paul Saftig (Christian-Albrechts Uni-versity of Kiel) for providing the Ctsd knockout mouse strain, Masugi Nishihara (University of Tokyo) for the grn knockout mouse strain, Konrad Sandhoff (University of Bonn, Kekulé-Institut für Organis-che Chemie und Biochemie) for the saposin D antibody, elizabeth Neufeld (University of California los Angeles) for the SCMAS anti-body and Virginia lee and Alice Chen-Plotkin (University of Penn-sylvania) for the TMeM106B antibody clone N2077. We are grate-ful to Andrea Wenninger-Weinzierl and Brigitte Kraft for excellent technical assistance and to Dorothee Dormann for critical reading of the manuscript and valuable comments. This work was supported by the european research Council under the european Union’s Seventh Framework Programme (FP7/2007–2013)/erC grant Agreement No. 321366-Amyloid (advanced grant to C.H.) and the Competence Net-work for Neurodegenerative Diseases (KNDD) of the Bundesministe-rium für Bildung und Forschung (BMBF to C.H.). J.K.g. is supported by the rTg1373 (DFg) and C.M.l. by the Hans & Ilse Breuer foun-dation. The Antwerp site was in part funded by the Metlife Founda-tion Award (to C.V.B.), the Belgian Science Policy Office Interuni-versity Attraction Poles Program, the Medical Foundation Queen elisabeth, the Foundation for Alzheimer research (SAO/FrA),
Flemish government initiated Methusalem excellence program, the Agency for Innovation by Science and Technology (IWT) Flanders, the research Foundation Flanders (FWO) and the University of Ant-werp research Fund. We acknowledge the contribution of genetic data by I. gijselinck and M. Cruts, clinical and pathological data by the neurologists T. Van langenhove, P. Cras, P.P. De Deyn and immu-nohistochemical data, autopsy brain samples and sections by the neu-ropathologist by A. Sieben. We are also grateful for the support by the research nurses at VIB, the personnel at the neurological depart-ments at the Antwerp University Hospital and ZNA Middelheim, the Antwerp Biobank at the Institute Born-Bunge and the genetic Ser-vice Facility at VIB. J.J. receives a Ph.D. fellowship of the IWT. We acknowledge the london Neurodegenerative Disease Brain Bank and Brains for Dementia research, the Human Brain and Spinal Fluid resource Center, VA West los Angeles Healthcare Center, los Angeles, CA 90073, which is sponsored by NINDS/NIMH, National Multiple Sclerosis Society and the Veterans Affairs West los Ange-les Healthcare Center for providing brain tissue. We thank the Batten Disease registry, genetic Services and Specialty Clinical laborato-ries, NYS Institute for Basic research in Developmental Disabilities.
References
1. Ahmed Z, Sheng H, Xu YF, lin Wl, Innes Ae, gass J, Yu X, Wuertzer CA, Hou H, Chiba S, Yamanouchi K, leissring M, Petrucelli l, Nishihara M, Hutton Ml, Mcgowan e, Dickson DW, lewis J (2010) Accelerated lipofuscinosis and ubiquitina-tion in granulin knockout mice suggest a role for progranulin in successful aging. Am J Pathol 177(1):311–324. doi:10.2353/ajpath.2010.090915
2. Amritraj A, Wang Y, revett TJ, Vergote D, Westaway D, Kar S (2013) role of cathepsin D in U18666A-induced neuronal cell death: potential implication in Niemann–Pick type C disease pathogenesis. J Biol Chem 288(5):3136–3152. doi:10.1074/jbc.M112.412460
3. Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, Oda T (2006) TDP-43 is a component of ubiquitin-positive tau-negative inclu-sions in frontotemporal lobar degeneration and amyotrophic lat-eral sclerosis. Biochem Biophys res Commun 351(3):602–611. doi:10.1016/j.bbrc.2006.10.093
4. Baker M, Mackenzie Ir, Pickering-Brown SM, gass J, rade-makers r, lindholm C, Snowden J, Adamson J, Sadovnick AD, rollinson S, Cannon A, Dwosh e, Neary D, Melquist S, rich-ardson A, Dickson D, Berger Z, eriksen J, robinson T, Zehr C, Dickey CA, Crook r, Mcgowan e, Mann D, Boeve B, Feldman H, Hutton M (2006) Mutations in progranulin cause tau-nega-tive frontotemporal dementia linked to chromosome 17. Nature 442(7105):916–919. doi:10.1038/nature05016
5. Benussi l, Binetti g, Sina e, gigola l, Bettecken T, Meitinger T, ghidoni r (2008) A novel deletion in progranulin gene is associ-ated with FTDP-17 and CBS. Neurobiol Aging 29(3):427–435. doi:10.1016/j.neurobiolaging.2006.10.028
6. Boya P, Kroemer g (2008) lysosomal membrane permeabiliza-tion in cell death. Oncogene 27(50):6434–6451. doi:10.1038/onc.2008.310
7. Brady OA, Zheng Y, Murphy K, Huang M, Hu F (2013) The fron-totemporal lobar degeneration risk factor, TMeM106B, regulates lysosomal morphology and function. Hum Mol genet 22(4):685–695. doi:10.1093/hmg/dds475
8. Brouwers N, Nuytemans K, van der Zee J, gijselinck I, engel-borghs S, Theuns J, Kumar-Singh S, Pickut BA, Pals P, Der-maut B, Bogaerts V, De Pooter T, Serneels S, Van den Broeck M, Cuijt I, Mattheijssens M, Peeters K, Sciot r, Martin JJ, Cras

Acta Neuropathol
1 3
P, Santens P, Vandenberghe r, De Deyn PP, Cruts M, Van Broe-ckhoven C, Sleegers K (2007) Alzheimer and Parkinson diagno-ses in progranulin null mutation carriers in an extended founder family. Arch Neurol 64(10):1436–1446. doi:10.1001/archneur.64.10.1436
9. Busch JI, Martinez-lage M, Ashbridge e, grossman M, Van Deerlin VM, Hu F, lee VM, Trojanowski JQ, Chen-Plotkin AS (2013) expression of TMeM106B, the frontotemporal lobar degeneration-associated protein, in normal and diseased human brain. Acta Neuropathol Commun 1(1):36
10. Butler D, Hwang J, estick C, Nishiyama A, Kumar SS, Baveghems C, Young-Oxendine HB, Wisniewski Ml, Charalam-bides A, Bahr BA (2011) Protective effects of positive lysosomal modulation in Alzheimer’s disease transgenic mouse models. PloS One 6(6):e20501. doi:10.1371/journal.pone.0020501
11. Capell A, liebscher S, Fellerer K, Brouwers N, Willem M, lam-mich S, gijselinck I, Bittner T, Carlson AM, Sasse F, Kunze B, Steinmetz H, Jansen r, Dormann D, Sleegers K, Cruts M, Herms J, Van Broeckhoven C, Haass C (2011) rescue of progranulin deficiency associated with frontotemporal lobar degeneration by alkalizing reagents and inhibition of vacuolar ATPase. J Neurosci 31(5):1885–1894. doi:10.1523/JNeUrOSCI.5757-10.2011
12. Cataldo AM, Barnett Jl, Berman SA, li J, Quarless S, Bursz-tajn S, lippa C, Nixon rA (1995) gene expression and cellular content of cathepsin D in Alzheimer’s disease brain: evidence for early up-regulation of the endosomal-lysosomal system. Neuron 14(3):671–680
13. Chen-Plotkin AS, Unger Tl, gallagher MD, Bill e, Kwong lK, Volpicelli-Daley l, Busch JI, Akle S, grossman M, Van Deerlin V, Trojanowski JQ, lee VM (2012) TMeM106B, the risk gene for frontotemporal dementia, is regulated by the microrNA-132/212 cluster and affects progranulin path-ways. J Neurosci 32(33):11213–11227. doi:10.1523/JNeUrOSCI.0521-12.2012
14. Cluzeau CV, Watkins-Chow De, Fu r, Borate B, Yanjanin N, Dail MK, Davidson CD, Walkley SU, Ory DS, Wassif CA, Pavan WJ, Porter FD (2012) Microarray expression analysis and identi-fication of serum biomarkers for Niemann-Pick disease, type C1. Hum Mol genet 21(16):3632–3646. doi:10.1093/hmg/dds193
15. Cruchaga C, graff C, Chiang HH, Wang J, Hinrichs Al, Spiegel N, Bertelsen S, Mayo K, Norton JB, Morris JC, goate A (2011) Association of TMeM106B gene polymorphism with age at onset in granulin mutation carriers and plasma granulin protein levels. Arch Neurol 68(5):581–586. doi:10.1001/archneurol.2010.350
16. Cruts M, gijselinck I, van der Zee J, engelborghs S, Wils H, Pirici D, rademakers r, Vandenberghe r, Dermaut B, Martin JJ, van Duijn C, Peeters K, Sciot r, Santens P, De Pooter T, Mat-theijssens M, Van den Broeck M, Cuijt I, Vennekens K, De Deyn PP, Kumar-Singh S, Van Broeckhoven C (2006) Null mutations in progranulin cause ubiquitin-positive frontotemporal demen-tia linked to chromosome 17q21. Nature 442(7105):920–924. doi:10.1038/nature05017
17. Cruts M, Theuns J, Van Broeckhoven C (2012) locus-specific mutation databases for neurodegenerative brain diseases. Hum Mutat 33(9):1340–1344. doi:10.1002/humu.22117
18. ebrahimi-Fakhari D, Wahlster l, Mclean PJ (2012) Protein deg-radation pathways in Parkinson’s disease: curse or blessing. Acta Neuropathol 124(2):153–172. doi:10.1007/s00401-012-1004-6
19. elleder M, Sokolova J, Hrebicek M (1997) Follow-up study of subunit c of mitochondrial ATP synthase (SCMAS) in Batten disease and in unrelated lysosomal disorders. Acta Neuropathol 93(4):379–390
20. Finch N, Baker M, Crook r, Swanson K, Kuntz K, Surtees r, Bisceglio g, rovelet-lecrux A, Boeve B, Petersen rC, Dick-son DW, Younkin Sg, Deramecourt V, Crook J, graff-radford Nr, rademakers r (2009) Plasma progranulin levels predict
progranulin mutation status in frontotemporal dementia patients and asymptomatic family members. Brain 132(Pt 3):583–591. doi:10.1093/brain/awn352
21. Finch N, Carrasquillo MM, Baker M, rutherford NJ, Coppola g, Dejesus-Hernandez M, Crook r, Hunter T, ghidoni r, Benussi l, Crook J, Finger e, Hantanpaa KJ, Karydas AM, Sengdy P, gonzalez J, Seeley WW, Johnson N, Beach Tg, Mesulam M, Forloni g, Kertesz A, Knopman DS, Uitti r, White Cl 3rd, Caselli r, lippa C, Bigio eH, Wszolek ZK, Binetti g, Macken-zie Ir, Miller Bl, Boeve BF, Younkin Sg, Dickson DW, Petersen rC, graff-radford Nr, geschwind DH, rademakers r (2011) TMeM106B regulates progranulin levels and the penetrance of FTlD in grN mutation carriers. Neurology 76(5):467–474. doi:10.1212/WNl.0b013e31820a0e3b
22. Fritchie K, Siintola e, Armao D, lehesjoki Ae, Marino T, Pow-ell C, Tennison M, Booker JM, Koch S, Partanen S, Suzuki K, Tyynela J, Thorne lB (2009) Novel mutation and the first pre-natal screening of cathepsin D deficiency (ClN10). Acta Neuro-pathol 117(2):201–208. doi:10.1007/s00401-008-0426-7
23. gallagher MD, Suh e, grossman M, elman l, McCluskey l, Van Swieten JC, Al-Sarraj S, Neumann M, gelpi e, ghetti B, rohrer JD, Halliday g, Van Broeckhoven C, Seilhean D, Shaw PJ, Frosch MP, Alafuzoff I, Antonell A, Bogdanovic N, Brooks W, Cairns NJ, Cooper-Knock J, Cotman C, Cras P, Cruts M, De Deyn PP, Decarli C, Dobson-Stone C, engelborghs S, Fox N, galasko D, gearing M, gijselinck I, grafman J, Hartikainen P, Hatanpaa KJ, Highley Jr, Hodges J, Hulette C, Ince Pg, Jin lW, Kirby J, Kofler J, Kril J, Kwok JB, levey A, lieberman A, llado A, Martin JJ, Masliah e, McDermott CJ, McKee A, Mclean C, Mead S, Miller CA, Miller J, Munoz Dg, Murrell J, Paulson H, Piguet O, rossor M, Sanchez-Valle r, Sano M, Schneider J, Silbert lC, Spina S, van der Zee J, Van langenhove T, War-ren J, Wharton SB, White Iii Cl, Woltjer rl, Trojanowski JQ, lee VM, Van Deerlin V, Chen-Plotkin AS (2014) TMeM106B is a genetic modifier of frontotemporal lobar degeneration with C9orf72 hexanucleotide repeat expansions. Acta Neuropathol 127(3):407–418. doi:10.1007/s00401-013-1239-x
24. gass J, Cannon A, Mackenzie Ir, Boeve B, Baker M, Adamson J, Crook r, Melquist S, Kuntz K, Petersen r, Josephs K, Pickering-Brown SM, graff-radford N, Uitti r, Dickson D, Wszolek Z, gonzalez J, Beach Tg, Bigio e, Johnson N, Weintraub S, Mesu-lam M, White Cl 3rd, Woodruff B, Caselli r, Hsiung gY, Feld-man H, Knopman D, Hutton M, rademakers r (2006) Mutations in progranulin are a major cause of ubiquitin-positive frontotem-poral lobar degeneration. Hum Mol genet 15(20):2988–3001. doi:10.1093/hmg/ddl241
25. ghidoni r, Benussi l, glionna M, Franzoni M, Binetti g (2008) low plasma progranulin levels predict progranulin mutations in frontotemporal lobar degeneration. Neurology 71(16):1235–1239. doi:10.1212/01.wnl.0000325058.10218.fc
26. ghoshal N, Dearborn JT, Wozniak DF, Cairns NJ (2012) Core features of frontotemporal dementia recapitulated in progranulin knockout mice. Neurobiol Dis 45(1):395–408. doi:10.1016/j.nbd.2011.08.029
27. gijselinck I, Van Broeckhoven C, Cruts M (2008) granulin muta-tions associated with frontotemporal lobar degeneration and related disorders: an update. Hum Mutat 29(12):1373–1386. doi:10.1002/humu.20785
28. graff-radford Nr, Woodruff BK (2007) Frontotemporal demen-tia. Semin Neurol 27(1):48–57. doi:10.1055/s-2006-956755
29. Hall NA, lake BD, Dewji NN, Patrick AD (1991) lysosomal storage of subunit c of mitochondrial ATP synthase in Batten’s disease (ceroid-lipofuscinosis). Biochem J 275(Pt 1):269–272
30. Harris H, rubinsztein DC (2012) Control of autophagy as a ther-apy for neurodegenerative disease. Nat rev Neurol 8(2):108–117. doi:10.1038/nrneurol.2011.200

Acta Neuropathol
1 3
31. Hasegawa M, Arai T, Nonaka T, Kametani F, Yoshida M, Hashi-zume Y, Beach Tg, Buratti e, Baralle F, Morita M, Nakano I, Oda T, Tsuchiya K, Akiyama H (2008) Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann Neurol 64(1):60–70. doi:10.1002/ana.21425
32. Hiraiwa M, Martin BM, Kishimoto Y, Conner ge, Tsuji S, O’Brien JS (1997) lysosomal proteolysis of prosaposin, the pre-cursor of saposins (sphingolipid activator proteins): its mecha-nism and inhibition by ganglioside. Arch Biochem Biophys 341(1):17–24. doi:10.1006/abbi.1997.9958
33. Hu F, Padukkavidana T, Vaegter CB, Brady OA, Zheng Y, Mac-kenzie Ir, Feldman HH, Nykjaer A, Strittmatter SM (2010) Sortilin-mediated endocytosis determines levels of the fronto-temporal dementia protein, progranulin. Neuron 68(4):654–667. doi:10.1016/j.neuron.2010.09.034
34. Jabs S, Quitsch A, Kakela r, Koch B, Tyynela J, Brade H, glat-zel M, Walkley S, Saftig P, Vanier MT, Braulke T (2008) Accu-mulation of bis(monoacylglycero)phosphate and gangliosides in mouse models of neuronal ceroid lipofuscinosis. J Neurochem 106(3):1415–1425. doi:10.1111/j.1471-4159.2008.05497.x
35. Jalanko A, Braulke T (2009) Neuronal ceroid lipo-fuscinoses. Biochim Biophys Acta 1793(4):697–709. doi:10.1016/j.bbamcr.2008.11.004
36. Janssens J, Van Broeckhoven C (2013) Pathological mechanisms underlying TDP-43 driven neurodegeneration in FTlD-AlS spectrum disorders. Hum Mol genet. doi:10.1093/hmg/ddt349
37. Kayasuga Y, Chiba S, Suzuki M, Kikusui T, Matsuwaki T, Yamanouchi K, Kotaki H, Horai r, Iwakura Y, Nishihara M (2007) Alteration of behavioural phenotype in mice by targeted disruption of the progranulin gene. Behav Brain res 185(2):110–118. doi:10.1016/j.bbr.2007.07.020
38. Klein A, Henseler M, Klein C, Suzuki K, Harzer K, Sandhoff K (1994) Sphingolipid activator protein D (sap-D) stimulates the lysosomal degradation of ceramide in vivo. Biochem Biophys res Commun 200(3):1440–1448. doi:10.1006/bbrc.1994.1612
39. Koike M, Nakanishi H, Saftig P, ezaki J, Isahara K, Ohsawa Y, Schulz-Schaeffer W, Watanabe T, Waguri S, Kametaka S, Shibata M, Yamamoto K, Kominami e, Peters C, von Figura K, Uchiy-ama Y (2000) Cathepsin D deficiency induces lysosomal stor-age with ceroid lipofuscin in mouse CNS neurons. J Neurosci 20(18):6898–6906
40. lang CM, Fellerer K, Schwenk BM, Kuhn PH, Kremmer e, edbauer D, Capell A, Haass C (2012) Membrane orienta-tion and subcellular localization of transmembrane protein 106B (TMeM106B), a major risk factor for frontotempo-ral lobar degeneration. J Biol Chem 287(23):19355–19365. doi:10.1074/jbc.M112.365098
41. le Ber I, Camuzat A, Hannequin D, Pasquier F, guedj e, rov-elet-lecrux A, Hahn-Barma V, van der Zee J, Clot F, Bakchine S, Puel M, ghanim M, lacomblez l, Mikol J, Deramecourt V, lejeune P, de la Sayette V, Belliard S, Vercelletto M, Meyrig-nac C, Van Broeckhoven C, lambert JC, Verpillat P, Campion D, Habert MO, Dubois B, Brice A, French research network on FF-M (2008) Phenotype variability in progranulin mutation car-riers: a clinical, neuropsychological, imaging and genetic study. Brain 131(Pt 3):732–746. doi:10.1093/brain/awn012
42. Mackenzie Ir, Neumann M, Bigio eH, Cairns NJ, Alafuzoff I, Kril J, Kovacs gg, ghetti B, Halliday g, Holm Ie, Ince Pg, Kamphorst W, revesz T, rozemuller AJ, Kumar-Singh S, Akiy-ama H, Baborie A, Spina S, Dickson DW, Trojanowski JQ, Mann DM (2009) Nomenclature for neuropathologic subtypes of fron-totemporal lobar degeneration: consensus recommendations. Acta Neuropathol 117(1):15–18. doi:10.1007/s00401-008-0460-5
43. Mackenzie Ir, Neumann M, Bigio eH, Cairns NJ, Alafu-zoff I, Kril J, Kovacs gg, ghetti B, Halliday g, Holm Ie, Ince Pg, Kamphorst W, revesz T, rozemuller AJ, Kumar-Singh S,
Akiyama H, Baborie A, Spina S, Dickson DW, Trojanowski JQ, Mann DM (2010) Nomenclature and nosology for neuropatho-logic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol 119(1):1–4. doi:10.1007/s00401-009-0612-2
44. Martin JJ, Ceuterick C (1997) Adult neuronal ceroid-lipofuscino-sis—personal observations. Acta Neurol Belg 97(2):85–92
45. Mori K, Weng SM, Arzberger T, May S, rentzsch K, Kremmer e, Schmid B, Kretzschmar HA, Cruts M, Van Broeckhoven C, Haass C, edbauer D (2013) The C9orf72 ggggCC repeat is translated into aggregating dipeptide-repeat proteins in FTlD/AlS. Science 339(6125):1335–1338. doi:10.1126/science.1232927
46. Mukherjee O, Wang J, gitcho M, Chakraverty S, Taylor-rein-wald l, Shears S, Kauwe JS, Norton J, levitch D, Bigio eH, Hatanpaa KJ, White Cl, Morris JC, Cairns NJ, goate A (2008) Molecular characterization of novel progranulin (grN) muta-tions in frontotemporal dementia. Hum Mutat 29(4):512–521. doi:10.1002/humu.20681
47. Nakanishi H, Zhang J, Koike M, Nishioku T, Okamoto Y, Komi-nami e, von Figura K, Peters C, Yamamoto K, Saftig P, Uchiyama Y (2001) Involvement of nitric oxide released from microglia-macrophages in pathological changes of cathepsin D-deficient mice. J Neurosci 21(19):7526–7533
48. Neumann M, Kwong lK, lee eB, Kremmer e, Flatley A, Xu Y, Forman MS, Troost D, Kretzschmar HA, Trojanowski JQ, lee VM (2009) Phosphorylation of S409/410 of TDP-43 is a consistent feature in all sporadic and familial forms of TDP-43 proteinopathies. Acta Neuropathol 117(2):137–149. doi:10.1007/s00401-008-0477-9
49. Neumann M, Sampathu DM, Kwong lK, Truax AC, Micse-nyi MC, Chou TT, Bruce J, Schuck T, grossman M, Clark CM, McCluskey lF, Miller Bl, Masliah e, Mackenzie Ir, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, lee VM (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314(5796):130–133. doi:10.1126/science.1134108
50. Nijssen PC, Ceuterick C, van Diggelen OP, elleder M, Martin JJ, Teepen Jl, Tyynela J, roos rA (2003) Autosomal dominant adult neuronal ceroid lipofuscinosis: a novel form of NCl with granular osmiophilic deposits without palmitoyl protein thioester-ase 1 deficiency. Brain Pathol 13(4):574–581
51. Nixon rA (2013) The role of autophagy in neurodegenerative disease. Nat Med 19(8):983–997. doi:10.1038/nm.3232
52. Nixon rA, Yang DS (2011) Autophagy failure in Alzheimer’s disease-locating the primary defect. Neurobiol Dis 43(1):38–45. doi:10.1016/j.nbd.2011.01.021
53. Partanen S, Haapanen A, Kielar C, Pontikis C, Alexander N, Inki-nen T, Saftig P, gillingwater TH, Cooper JD, Tyynela J (2008) Synaptic changes in the thalamocortical system of cathepsin D-deficient mice: a model of human congenital neuronal ceroid-lipofuscinosis. J Neuropathol exp Neurol 67(1):16–29. doi:10.1097/nen.0b013e31815f3899
54. Petkau Tl, Neal SJ, Milnerwood A, Mew A, Hill AM, Orban P, gregg J, lu g, Feldman HH, Mackenzie Ir, raymond lA, leav-itt Br (2012) Synaptic dysfunction in progranulin-deficient mice. Neurobiol Dis 45(2):711–722. doi:10.1016/j.nbd.2011.10.016
55. Petkau Tl, Neal SJ, Orban PC, MacDonald Jl, Hill AM, lu g, Feldman HH, Mackenzie Ir, leavitt Br (2010) Progranulin expression in the developing and adult murine brain. J Comp Neurol 518(19):3931–3947. doi:10.1002/cne.22430
56. Qiao l, Hamamichi S, Caldwell KA, Caldwell gA, Yacoubian TA, Wilson S, Xie Zl, Speake lD, Parks r, Crabtree D, liang Q, Crimmins S, Schneider l, Uchiyama Y, Iwatsubo T, Zhou Y, Peng l, lu Y, Standaert Dg, Walls KC, Shacka JJ, roth KA, Zhang J (2008) lysosomal enzyme cathepsin D protects against alpha-synuclein aggregation and toxicity. Mol Brain 1:17. doi:10.1186/1756-6606-1-17

Acta Neuropathol
1 3
57. repnik U, Stoka V, Turk V, Turk B (2012) lysosomes and lysoso-mal cathepsins in cell death. Biochim Biophys Acta 1824(1):22–33. doi:10.1016/j.bbapap.2011.08.016
58. rubinsztein DC (2006) The roles of intracellular protein-degra-dation pathways in neurodegeneration. Nature 443(7113):780–786. doi:10.1038/nature05291
59. ryazantsev S, Yu WH, Zhao HZ, Neufeld eF, Ohmi K (2007) lysosomal accumulation of SCMAS (subunit c of mitochon-drial ATP synthase) in neurons of the mouse model of muco-polysaccharidosis III B. Mol genet Metab 90(4):393–401. doi:10.1016/j.ymgme.2006.11.006
60. Saftig P, Hetman M, Schmahl W, Weber K, Heine l, Mossmann H, Koster A, Hess B, evers M, von Figura K et al (1995) Mice deficient for the lysosomal proteinase cathepsin D exhibit pro-gressive atrophy of the intestinal mucosa and profound destruc-tion of lymphoid cells. eMBO J 14(15):3599–3608
61. Sardiello M, Palmieri M, di ronza A, Medina Dl, Valenza M, gennarino VA, Di Malta C, Donaudy F, embrione V, Polishchuk rS, Banfi S, Parenti g, Cattaneo e, Ballabio A (2009) A gene network regulating lysosomal biogenesis and function. Science 325(5939):473–477. doi:10.1126/science.1174447
62. Seelaar H, rohrer JD, Pijnenburg YA, Fox NC, van Swieten JC (2011) Clinical, genetic and pathological heterogeneity of fron-totemporal dementia: a review. J Neurol Neurosurg Psychiatry 82(5):476–486. doi:10.1136/jnnp.2010.212225
63. Shankaran SS, Capell A, Hruscha AT, Fellerer K, Neumann M, Schmid B, Haass C (2008) Missense mutations in the progranu-lin gene linked to frontotemporal lobar degeneration with ubiq-uitin-immunoreactive inclusions reduce progranulin production and secretion. J Biol Chem 283(3):1744–1753. doi:10.1074/jbc.M705115200
64. Sieben A, Van langenhove T, engelborghs S, Martin JJ, Boon P, Cras P, De Deyn PP, Santens P, Van Broeckhoven C, Cruts M (2012) The genetics and neuropathology of frontotemporal lobar degeneration. Acta Neuropathol 124(3):353–372. doi:10.1007/s00401-012-1029-x
65. Siintola e, Partanen S, Stromme P, Haapanen A, Haltia M, Mae-hlen J, lehesjoki Ae, Tyynela J (2006) Cathepsin D deficiency underlies congenital human neuronal ceroid-lipofuscinosis. Brain 129(Pt 6):1438–1445. doi:10.1093/brain/awl107
66. Skibinski g, Parkinson NJ, Brown JM, Chakrabarti l, lloyd Sl, Hummerich H, Nielsen Je, Hodges Jr, Spillantini Mg, Thus-gaard T, Brandner S, Brun A, rossor MN, gade A, Johannsen P, Sorensen SA, gydesen S, Fisher eM, Collinge J (2005) Muta-tions in the endosomal eSCrTIII-complex subunit CHMP2B in frontotemporal dementia. Nat genet 37(8):806–808. doi:10.1038/ng1609
67. Sleegers K, Brouwers N, Van Damme P, engelborghs S, gijselinck I, van der Zee J, Peeters K, Mattheijssens M, Cruts M, Vandenberghe r, De Deyn PP, robberecht W, Van Broeck-hoven C (2009) Serum biomarker for progranulin-associated frontotemporal lobar degeneration. Ann Neurol 65(5):603–609. doi:10.1002/ana.21621
68. Smith Kr, Damiano J, Franceschetti S, Carpenter S, Canafoglia l, Morbin M, rossi g, Pareyson D, Mole Se, Staropoli JF, Sims KB, lewis J, lin Wl, Dickson DW, Dahl HH, Bahlo M, Berko-vic SF (2012) Strikingly different clinicopathological phenotypes determined by progranulin-mutation dosage. Am J Hum genet 90(6):1102–1107. doi:10.1016/j.ajhg.2012.04.021
69. Tanaka Y, Matsuwaki T, Yamanouchi K, Nishihara M (2013) exacerbated inflammatory responses related to acti-vated microglia after traumatic brain injury in progranu-lin-deficient mice. Neuroscience 231:49–60. doi:10.1016/j.neuroscience.2012.11.032
70. Tanaka Y, Matsuwaki T, Yamanouchi K, Nishihara M (2013) Increased lysosomal biogenesis in activated microglia and
exacerbated neuronal damage after traumatic brain injury in pro-granulin-deficient mice. Neuroscience 250C:8–19. doi:10.1016/j.neuroscience.2013.06.049
71. Tofaris gK (2012) lysosome-dependent pathways as a unifying theme in Parkinson’s disease. Mov Disord 27(11):1364–1369. doi:10.1002/mds.25136
72. Tresse e, Salomons FA, Vesa J, Bott lC, Kimonis V, Yao TP, Dantuma NP, Taylor JP (2010) VCP/p97 is essential for matu-ration of ubiquitin-containing autophagosomes and this func-tion is impaired by mutations that cause IBMPFD. Autophagy 6(2):217–227
73. Tyynela J, Palmer DN, Baumann M, Haltia M (1993) Storage of saposins A and D in infantile neuronal ceroid-lipofuscinosis. FeBS lett 330(1):8–12
74. Urwin H, Authier A, Nielsen Je, Metcalf D, Powell C, Froud K, Malcolm DS, Holm I, Johannsen P, Brown J, Fisher eM, van der Zee J, Bruyland M, Consortium Fr, Van Broeckhoven C, Collinge J, Brandner S, Futter C, Isaacs AM (2010) Disruption of endocytic trafficking in frontotemporal dementia with CHMP2B mutations. Hum Mol genet 19(11):2228–2238. doi:10.1093/hmg/ddq100
75. van Blitterswijk M, Mullen B, Nicholson AM, Bieniek KF, Heck-man Mg, Baker MC, Dejesus-Hernandez M, Finch NA, Brown PH, Murray Me, Hsiung gY, Stewart H, Karydas AM, Finger e, Kertesz A, Bigio eH, Weintraub S, Mesulam M, Hatanpaa KJ, White Iii Cl, Strong MJ, Beach Tg, Wszolek ZK, lippa C, Caselli r, Petrucelli l, Josephs KA, Parisi Je, Knopman DS, Petersen rC, Mackenzie Ir, Seeley WW, grinberg lT, Miller Bl, Boylan KB, graff-radford Nr, Boeve BF, Dickson DW, rademakers r (2014) TMeM106B protects C9OrF72 expan-sion carriers against frontotemporal dementia. Acta Neuropathol 127(3):397–406. doi:10.1007/s00401-013-1240-4
76. Van Deerlin VM, Sleiman PM, Martinez-lage M, Chen-Plotkin A, Wang lS, graff-radford Nr, Dickson DW, rademakers r, Boeve BF, grossman M, Arnold Se, Mann DM, Pickering-Brown SM, Seelaar H, Heutink P, van Swieten JC, Murrell Jr, ghetti B, Spina S, grafman J, Hodges J, Spillantini Mg, gilman S, lieber-man AP, Kaye JA, Woltjer rl, Bigio eH, Mesulam M, Al-Sarraj S, Troakes C, rosenberg rN, White Cl 3rd, Ferrer I, llado A, Neumann M, Kretzschmar HA, Hulette CM, Welsh-Bohmer KA, Miller Bl, Alzualde A, lopez de Munain A, McKee AC, gear-ing M, levey AI, lah JJ, Hardy J, rohrer JD, lashley T, Mac-kenzie Ir, Feldman HH, Hamilton rl, Dekosky ST, van der Zee J, Kumar-Singh S, Van Broeckhoven C, Mayeux r, Vonsattel JP, Troncoso JC, Kril JJ, Kwok JB, Halliday gM, Bird TD, Ince Pg, Shaw PJ, Cairns NJ, Morris JC, Mclean CA, DeCarli C, ellis Wg, Freeman SH, Frosch MP, growdon JH, Perl DP, Sano M, Bennett DA, Schneider JA, Beach Tg, reiman eM, Woodruff BK, Cummings J, Vinters HV, Miller CA, Chui HC, Alafuzoff I, Hartikainen P, Seilhean D, galasko D, Masliah e, Cotman CW, Tunon MT, Martinez MC, Munoz Dg, Carroll Sl, Marson D, riederer PF, Bogdanovic N, Schellenberg gD, Hakonarson H, Trojanowski JQ, lee VM (2010) Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat genet 42(3):234–239. doi:10.1038/ng.536
77. van der Zee J, le Ber I, Maurer-Stroh S, engelborghs S, gijselinck I, Camuzat A, Brouwers N, Vandenberghe r, Sleegers K, Hannequin D, Dermaut B, Schymkowitz J, Campion D, San-tens P, Martin JJ, lacomblez l, De Pooter T, Peeters K, Matthei-jssens M, Vercelletto M, Van den Broeck M, Cruts M, De Deyn PP, rousseau F, Brice A, Van Broeckhoven C (2007) Mutations other than null mutations producing a pathogenic loss of pro-granulin in frontotemporal dementia. Hum Mutat 28(4):416. doi:10.1002/humu.9484
78. van der Zee J, Van langenhove T, Kleinberger g, Sleegers K, engelborghs S, Vandenberghe r, Santens P, Van den Broeck M,

Acta Neuropathol
1 3
Joris g, Brys J, Mattheijssens M, Peeters K, Cras P, De Deyn PP, Cruts M, Van Broeckhoven C (2011) TMeM106B is associ-ated with frontotemporal lobar degeneration in a clinically diag-nosed patient cohort. Brain 134(Pt 3):808–815. doi:10.1093/brain/awr007
79. Vitner eB, Dekel H, Zigdon H, Shachar T, Farfel-Becker T, eilam r, Karlsson S, Futerman AH (2010) Altered expression and distribution of cathepsins in neuronopathic forms of gau-cher disease and in other sphingolipidoses. Hum Mol genet 19(18):3583–3590. doi:10.1093/hmg/ddq273
80. Wang J, Van Damme P, Cruchaga C, gitcho MA, Vidal JM, Seijo-Martinez M, Wang l, Wu JY, robberecht W, goate A (2010) Pathogenic cysteine mutations affect progranulin function and production of mature granulins. J Neurochem 112(5):1305–1315. doi:10.1111/j.1471-4159.2009.06546.x
81. Watts gD, Thomasova D, ramdeen SK, Fulchiero eC, Mehta Sg, Drachman DA, Weihl CC, Jamrozik Z, Kwiecinski H, Kaminska A, Kimonis Ve (2007) Novel VCP mutations in inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia. Clin genet 72(5):420–426. doi:10.1111/j.1399-0004.2007.00887.x
82. Watts gD, Wymer J, Kovach MJ, Mehta Sg, Mumm S, Darvish D, Pestronk A, Whyte MP, Kimonis Ve (2004) Inclusion body myopathy associated with Paget disease of bone and frontotem-poral dementia is caused by mutant valosin-containing protein. Nat genet 36(4):377–381. doi:10.1038/ng1332
83. Wils H, Kleinberger g, Pereson S, Janssens J, Capell A, Van Dam D, Cuijt I, Joris g, De Deyn PP, Haass C, Van Broeckhoven C,
Kumar-Singh S (2012) Cellular ageing, increased mortality and FTlD-TDP-associated neuropathology in progranulin knockout mice. J Pathol 228(1):67–76. doi:10.1002/path.4043
84. Yang DS, Stavrides P, Mohan PS, Kaushik S, Kumar A, Ohno M, Schmidt SD, Wesson D, Bandyopadhyay U, Jiang Y, Pawlik M, Peterhoff CM, Yang AJ, Wilson DA, St george-Hyslop P, Westa-way D, Mathews PM, levy e, Cuervo AM, Nixon rA (2011) reversal of autophagy dysfunction in the TgCrND8 mouse model of Alzheimer’s disease ameliorates amyloid pathologies and mem-ory deficits. Brain 134(Pt 1):258–277. doi:10.1093/brain/awq341
85. Yin F, Banerjee r, Thomas B, Zhou P, Qian l, Jia T, Ma X, Ma Y, Iadecola C, Beal MF, Nathan C, Ding A (2010) exaggerated inflammation, impaired host defense, and neuropathology in pro-granulin-deficient mice. J exp Med 207(1):117–128. doi:10.1084/jem.20091568
86. Yin F, Dumont M, Banerjee r, Ma Y, li H, lin MT, Beal MF, Nathan C, Thomas B, Ding A (2010) Behavioral deficits and pro-gressive neuropathology in progranulin-deficient mice: a mouse model of frontotemporal dementia. FASeB J 24(12):4639–4647. doi:10.1096/fj.10-161471
87. Yu Ce, Bird TD, Bekris lM, Montine TJ, leverenz JB, Steinbart e, galloway NM, Feldman H, Woltjer r, Miller CA, Wood eM, grossman M, McCluskey l, Clark CM, Neumann M, Danek A, galasko Dr, Arnold Se, Chen-Plotkin A, Karydas A, Miller Bl, Trojanowski JQ, lee VM, Schellenberg gD, Van Deerlin VM (2010) The spectrum of mutations in progranulin: a collabora-tive study screening 545 cases of neurodegeneration. Arch Neurol 67(2):161–170. doi:10.1001/archneurol.2009.328
Copyright © 2022 FDOKUMEN














