
eso de um adulto é uma variável aleatória com distribuição nor
Upload
independentCategory
view
0download
0
Journal of PathologyJ Pathol 2014; 233: 238–246Published online 21 May 2014 in Wiley Online Library(wileyonlinelibrary.com) DOI: 10.1002/path.4356
ORIGINAL PAPER
Mutant p53 accumulation in human breast cancer is not anintrinsic property or dependent on structural or functionaldisruption but is regulated by exogenous stress and receptor statusPavla Bouchalova,1 Rudolf Nenutil,1 Petr Muller,1 Roman Hrstka,1 M Virginia Appleyard,2 Karen Murray,2Lee B Jordan,3 Colin A Purdie,3 Philip Quinlan,2 Alastair M Thompson,2,4 Borivoj Vojtesek1 and Philip J Coates5*
1 Regional Centre for Applied Molecular Oncology, Masaryk Memorial Cancer Institute, Brno, Czech Republic2 Clinical Research Centre, Dundee Cancer Centre, University of Dundee, Ninewells Hospital and Medical School, Dundee, UK3 Department of Pathology, Ninewells Hospital and Medical School, Dundee, UK4 Department of Surgical Oncology, MD Anderson Cancer Center, Houston, TX, USA5 Tayside Tissue Bank, Jacqui Wood Cancer Centre, University of Dundee, Ninewells Hospital and Medical School, Dundee, UK
*Correspondence to: PJ Coates, Tayside Tissue Bank, Jacqui Wood Cancer Centre, University of Dundee, Ninewells Hospital and Medical School,Dundee DD1 9SY, UK. E-mail: [email protected]
AbstractMany human cancers contain missense TP53 mutations that result in p53 protein accumulation. Although generallyconsidered as a single class of mutations that abrogate wild-type function, individual TP53 mutations mayhave specific properties and prognostic effects. Tumours that contain missense TP53 mutations show variablep53 stabilization patterns, which may reflect the specific mutation and/or aspects of tumour biology. We usedimmunohistochemistry on cell lines and human breast cancers with known TP53 missense mutations and assessedthe effects of each mutation with four structure–function prediction methods. Cell lines with missense TP53mutations show variable percentages of cells with p53 stabilization under normal growth conditions, rangingfrom approximately 50% to almost 100%. Stabilization is not related to structural or functional disruption, butagents that stabilize wild-type p53 increase the percentages of cells showing missense mutant p53 accumulationin cell lines with heterogeneous stabilization. The same heterogeneity of p53 stabilization occurs in primary breastcancers, independent of the effect of the mutation on structural properties or functional disruption. Heterogeneousaccumulation is more common in steroid receptor-positive or HER2-positive breast cancers and cell lines than intriple-negative samples. Immunohistochemcal staining patterns associate with Mdm2 levels, proliferation, gradeand overall survival, whilst the type of mutation reflects downstream target activity. Inhibiting Mdm2 activityincreases the extent of p53 stabilization in some, but not all, breast cancer cell lines. The data indicate thatmissense mutant p53 stabilization is a complex and variable process in human breast cancers that associates withdisease characteristics but is unrelated to structural or functional properties. That agents which stabilize wild-typep53 also stabilize mutant p53 has implications for patients with heterogeneous mutant p53 accumulation, wheretherapy may activate mutant p53 oncogenic function.Copyright © 2014 Pathological Society of Great Britain and Ireland. Published by John Wiley & Sons, Ltd.
Keywords: p53; mutation; breast cancer; immunohistochemistry; therapy
Received 3 January 2014; Revised 13 March 2014; Accepted 21 March 2014
No conflicts of interest were declared.
Introduction
TP53 gene mutations are common in human tumoursand many studies have attempted to correlate p53 muta-tion status and prognosis, with disappointing results[1–4]. Tumours containing mutant p53 are usually con-sidered as a single class and are compared to tumoursthat retainwild-type (wt) p53. However, individual TP53mutations differentially influence the overall functionalproperties of the mutant protein and certain missensep53 mutant proteins have oncogenic functions, in con-trast to the tumour-suppressor function of wtp53 or theloss of function caused by nonsense mutations or TP53
loss [3,4]. Clinically, discriminating between specificTP53 mutations or mutation types provides prognosticinformation in certain human malignancies [5–10].One consequence of a missense mutation in TP53 is
accumulation of the mutant protein, allowing mutationdetection by immunohistochemical staining; tumoursshowing a high percentage of p53-positive cells are clas-sified as containing mutant p53. This simple approachis inaccurate because of the highly variable numbers ofcells that stain for p53 in tumours with wtp53 or mutantp53, confounding the results of many studies [1,11–14].On the other hand, immunohistochemical studies sug-gest that missense mutant p53 is not always inherently
Copyright © 2014 Pathological Society of Great Britain and Ireland. J Pathol 2014; 233: 238–246Published by John Wiley & Sons, Ltd. www.pathsoc.org.uk www.thejournalofpathology.com

Mutant p53 stabilization in breast cancer 239
stabilized in tumour cells, and the extent of protein accu-mulation may therefore reflect the specific type of muta-tion or some other aspect of the tumour’s pathobiology.Simple explanations for these observations would bethat the immunohistochemical data are due to artifactsof fixation and processing, loss of the mutant allele fromsome tumour cells, or gain of mutation as a later eventduring clonal evolution. These explanations are unlikely,because heterogeneity of staining is not localized to spe-cific areas of the tumour.More plausible explanations are that staining patterns
reflect the intrinsic nature of the mutation, where somemutant proteins are inherently more stable than others,or where some mutations are particularly disruptive tofunction and the mutant proteins are consequently lessable to induce their own destruction thorough transcrip-tional regulation of Mdm2 [15]. Indeed, the site andnature of the mutation in p53 results in proteins withnative (folded) or denatured (unfolded) structures thathave stabilization and degradation pathways differentfrom those of wtp53 [16–18]. TP53 mutations havealso been classified as disruptive or non-disruptive tofunction, depending on mutation site and type [8], andcan be given a severity score from structural/functionalcalculations [19]. Therefore, variations in mutant p53levels may relate to the inherent stability/activity ofthe individual mutant, which would allow informationregarding the class of mutation by immunohistochem-ical staining. A more recent hypothesis derived fromexperimental studies of transgenic mice and zebrafish isthat tumour-specific factors are responsible for mutantp53 stabilization [20–24]. In mice containing only amutant Tp53 gene, p53 accumulation is not seen innon-tumour cells under normal conditions, but occursin spontaneously developing tumours [20]. Factors thatstabilize wtp53 (DNA damage, lack of Mdm2 and onco-genic stress) also stabilize mutant p53 in non-tumourcells. Thus, levels of mutant p53 in human tumours maybe symptomatic of specific aspects of tumour pathobi-ology, such as genetic instability or the precise onco-genic events that have occurred. These hypotheses arenot mutually exclusive and the extent of mutant p53 sta-bilization may represent a combination of intrinsic prop-erties together with endogenous or exogenous stresses.To clarify the reasons for variability of mutant p53 stabi-lization, we investigated the relationships between spe-cific mutations, cellular stress and p53 levels in a panelof cell lines and primary human breast cancers contain-ing different TP53 missense mutations.
Materials and methods
Tissue cultureHuman cell lines with known p53 mutational statuswere obtained from ATCC (Table 1) and cul-tured in the recommended medium containing 10%fetal calf serum (FCS), 0.3 mg/ml L-glutamineand 1% penicillin–streptomycin (Invitrogen, Life
Table 1. Immunohistochemical and structural analysis of 17human cell lines for TP53 mutation classification
Cell line MutationStaining(%)a X-rayb NMRb Disruptivec Severityd
HOS R156P 99.3 U (1.84) U (1.64) No 71.75BT549 R249S 98.3 ? (0.67) U (1.50) Yes 95.70SKUT-1 R175H 97.0 n.a. U (0.97) No 92.55A431 R273H 96.8 F (0.00) F (−0.04) No 87.26HT-29 R273H 93.2 F (0.00) F (−0.04) No 87.26DLD-1 S241F 92.8 F (0.06) F (0.52) Yes 82.44MB231 R280K 92.4 F (0.34) n.a. No 71.45SW837 R248W 91.6 F (0.23) ? (0.64) Yes 83.31BT20 K132Q 87.5 U (1.82) U (1.29) No 75.40MB468 R273H 86.5 F (0.00) F (−0.04) No 87.26CAMA-1 R282T 86.3 ? (0.61) U (0.30) No 94.99RD R282W 86.0 F (−1.11) F (−0.72) No 92.37HT3 G245V 86.2 ? (0.51) F (1.45) Yes 111.3SKBR3 R175H 80.5 n.a. U (0.97) No 92.55MOLT4 R248Q 62.4 F (0.21) F (0.02) Yes 62.49BT474 E285K 62.0 U (1.21) U (1.16) No 74.72T47D L194F 53.4 F (−1.15) ? (0.56) No 66.05
aThe mean percentage of p53-positive cells.bThe folding scores of mutant p53 predicted from the X-ray crystallography orNMR structures. A higher score indicates a more unfolded structure. U, predictedto be a largely unfolded protein (score > 0.7); F, predicted to adopt a foldedstructure (score < 0.5); n.a., not available from the structure; ?, no predictionmade (score 0.5–0.7).cClassification according to whether or not the mutation disrupts function.dMutation severity score. A cut-off value of 60.18 discriminates severe fromnon-severe mutations; all mutations are classed as severe.
Technologies, Paisley, UK) at 37∘C and 10% CO2. Cellswere exposed to UV light (40 J/m2 using a Stratalinker1800) or Roscovitine (10 μM; Sigma-Aldrich, DorsetUK) and collected 16 h later [25]. T47D, BT474 andSKBR3 cells were also treated with the Mdm2 inhibitorNutlin-3 (10 μM; Sigma-Aldrich) for 24 h. To producecell pellets, cells were fixed in 4% neutral bufferedformalin, resuspended in 1.5% molten agarose andprocessed into paraffin wax using standard histologicalprocedures. Alternatively, cells grown directly ontoglass slides were fixed in methanol:acetone (50:50) at−20∘C for 10 min, air-dried overnight and stored at−80∘C. For colony formation, cells were dissociatedinto single cells through 21g needles, checked for theabsence of doublets by microscopy before plating atcloning density on glass slides, and fixed after coloniesof > 20 cells had formed (7–10 days).
Human tumour samplesA series of 240 untreated primary breast cancers takenat a resectional surgery in Dundee were collected. Por-tions of tissue were used for diagnostic histopathologyand separate tumour pieces were dissected by a spe-cialist breast pathologist and stored frozen for analysisof TP53 DNA sequence, using the Roche Amplichip(Roche Diagnostics). Patient details have been reportedpreviously [26]. A second cohort of 55 triple-negativebreast cancers was examined by direct Sanger sequenc-ing and p53 immunohistochemistry. Microarrays wereprepared in the Tayside Tissue Bank from diagnostichistopathology blocks, using six 0.6 mmm cores/cancer.The use of the tissues was approved by local ethical
Copyright © 2014 Pathological Society of Great Britain and Ireland. J Pathol 2014; 233: 238–246Published by John Wiley & Sons, Ltd. www.pathsoc.org.uk www.thejournalofpathology.com

240 P Bouchalova et al
review and all patients provided informed consent for thecollection and use of their tumour material for research.For this study, only those tumours identified as contain-ing mutant p53 were analysed.
Immunohistochemistry and analysis of stainingpatternsSections and cells were stained for p53 using rabbitpolyclonal CM1 antiserum and the mouse monoclonalantibody DO-1. Immunoreactivity was detected byavidin–biotin complex peroxidase staining (Elite ABCKit, Vector Laboratories, Peterborough, UK) or byEnVision+ System–HRP (Dako Denmark A/S) withdiaminobenzidine as chromogen, as described [11,27].For cell lines, the percentage of cells showing p53immunostaining was calculated by examination ofat least 10 independent microscope fields. p53 stain-ing patterns in tumours were described as showing‘heterogeneous’ or ‘homogeneous’ staining by visualexamination of the images by two investigators withexperience of p53 immunohistochemistry (RN andPJC). Homogeneous staining was defined as > 90% oftumour cells positive for nuclear p53 staining, whereasheterogeneous staining was defined by > 10% of nucleishowing an absence of p53 staining. Variable intensityof staining in individual cells is not taken into accountwhen considering heterogeneity – a simple positiveand negative staining for each tumour cell was used. Inpractice, tumours either show a very small number ofnegative cells (<5%) or > 20% negative cells, so thatthe discrimination of homogeneity and heterogeneityis easily achieved. Breast cancer microarrays werealso stained for Ki67 (Dako), Cdkn1a/p21 and Mdm2[11,27]. The latter two were scored by the QuickScoremethod, where intensity of staining is recorded on ascale of 0–3 and percentages of positive cells are scoredon a scale of 0–6, and the two values multiplied togive scores in the range 0–18 [28]. Only the percentagescale is recorded for Ki67. Oestrogen receptor (ER),progesterone receptor (PR) and the HER2 status ofbreast cancers were assessed by immunohistochemistryand in situ hybridization, as previously reported [29,30].
Subclassification of p53 mutation typesby sequence dataUsing the nucleotide sequence data from each cell lineand tumour, we classified TP53 mutations using fourdifferent approaches. (a) We predicted the effect ofthe mutation on the relative ability of the protein tofold into a wild-type conformation using the wtp53structures derived from X-ray crystallography and (b)those derived from NMR data, calculating the structuraleffects of individual mutations with PoPMuSic [31].This algorithm produces a score for folding and allowsclassification as either unfolded or folded. The data areapplicable only to the core regions of p53 and predic-tions are lacking for some individual amino acid residuesin either the NMR-derived structure or the X-ray
structure. Because the NMR and X-ray data sometimesprovide different folding predictions, we analysed eachmethod separately. Non-sense mutations (stop codonor frame-shift) that lead to an absence of protein arenot classified as folded or unfolded. (c) Mutations wereclassified as disruptive or non-disruptive, based on thedegree of disturbance to the structure of p53 in complexwith DNA [8]. In brief, disruptive mutations produce anon-similar amino acid within the L2 and L3 loops ofp53 or introduce a stop codon into the coding sequence.This scheme classifies some TP53 hotspot mutations asnon-disruptive, due to similarity between wild-type andmutant amino acids, eg the common R175H and R273Hmutations [8]. (d) The fourth system uses 12 weightedparameters related to structure, stability and activity toassess the effect of the mutation through the PREDMUTalgorithm [19] at: http://www.ifm.liu.se/bioinfo. Thecombined effects are reported as a severity score andclassified as severe if the score is > 60.18 [19].
StatisticsFor cell line data, Pearson correlation coefficients wereused for comparing staining patterns with PoPMu-SiC or PREDMUT scores and non-paired two-wayt-tests, assuming unequal variance for disruptive versusnon-disruptive mutants. Statistical significance of asso-ciations of clinical data with staining patterns, foldedstate, disruptive nature or severity was assessed usingtwo-tailed p values (Fisher’s exact test), using the Inspireprogramme [32], or Kaplan–Meier for survival. Pearsoncoefficients or ANOVA were used for continuous vari-ables (folding and severity scores).
Results
p53 staining patterns in human cancer cell linesImmunostaining for p53 was optimized to detect a lowlevel of p53 in normal tissues and cell lines with wtp53[11,27], such that an absence of staining indicatesnon-sense or frame-shift TP53 mutation or loss of bothalleles. In 17 human cell lines that contain p53 missensepoint mutations, the percentage of p53-positive cellsvaried between 99.3% and 53.4% (Figure 1, Table 1).Increasing the antibody concentration two-fold did notdecrease the proportion of negatively-stained cells.Scoring p53 in multiple different colonies of T47D,SKBR3 and BT474 cells derived from low-densityplating showed that each colony contained a mixtureof positively stained and unstained cells in the sameproportions as in bulk cultures.Cell lines were classified as expressing p53 mutants
that adopt folded or unfolded conformations, on thebasis of X-ray or NMR-derived structures of the p53core domain. The percentages of p53-positive cellsdid not correlate with either the folding state or thefolding scores of the mutations derived from theNMR- (r2 = 0.082) or X-ray (r2 = 0.016) -derived
Copyright © 2014 Pathological Society of Great Britain and Ireland. J Pathol 2014; 233: 238–246Published by John Wiley & Sons, Ltd. www.pathsoc.org.uk www.thejournalofpathology.com
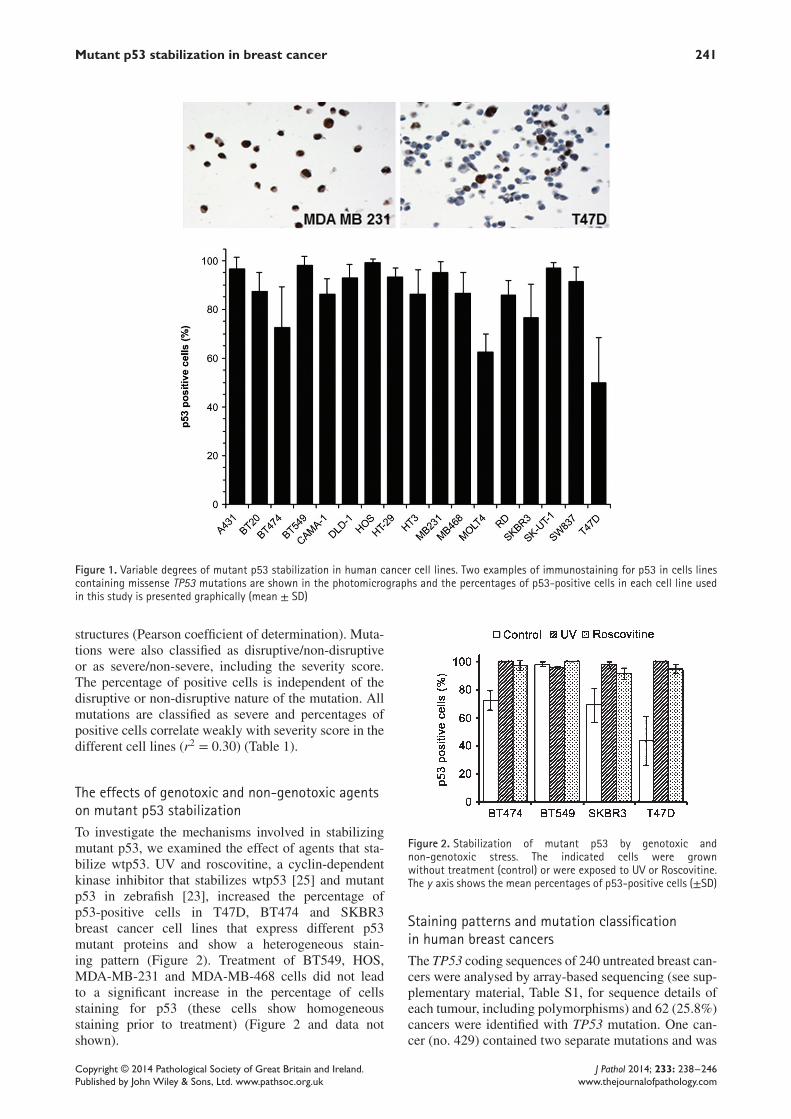
Mutant p53 stabilization in breast cancer 241
Figure 1. Variable degrees of mutant p53 stabilization in human cancer cell lines. Two examples of immunostaining for p53 in cells linescontaining missense TP53 mutations are shown in the photomicrographs and the percentages of p53-positive cells in each cell line usedin this study is presented graphically (mean ± SD)
structures (Pearson coefficient of determination). Muta-tions were also classified as disruptive/non-disruptiveor as severe/non-severe, including the severity score.The percentage of positive cells is independent of thedisruptive or non-disruptive nature of the mutation. Allmutations are classified as severe and percentages ofpositive cells correlate weakly with severity score in thedifferent cell lines (r2 = 0.30) (Table 1).
The effects of genotoxic and non-genotoxic agentson mutant p53 stabilizationTo investigate the mechanisms involved in stabilizingmutant p53, we examined the effect of agents that sta-bilize wtp53. UV and roscovitine, a cyclin-dependentkinase inhibitor that stabilizes wtp53 [25] and mutantp53 in zebrafish [23], increased the percentage ofp53-positive cells in T47D, BT474 and SKBR3breast cancer cell lines that express different p53mutant proteins and show a heterogeneous stain-ing pattern (Figure 2). Treatment of BT549, HOS,MDA-MB-231 and MDA-MB-468 cells did not leadto a significant increase in the percentage of cellsstaining for p53 (these cells show homogeneousstaining prior to treatment) (Figure 2 and data notshown).
Figure 2. Stabilization of mutant p53 by genotoxic andnon-genotoxic stress. The indicated cells were grownwithout treatment (control) or were exposed to UV or Roscovitine.The y axis shows the mean percentages of p53-positive cells (±SD)
Staining patterns and mutation classificationin human breast cancersThe TP53 coding sequences of 240 untreated breast can-cers were analysed by array-based sequencing (see sup-plementary material, Table S1, for sequence details ofeach tumour, including polymorphisms) and 62 (25.8%)cancers were identified with TP53 mutation. One can-cer (no. 429) contained two separate mutations and was
Copyright © 2014 Pathological Society of Great Britain and Ireland. J Pathol 2014; 233: 238–246Published by John Wiley & Sons, Ltd. www.pathsoc.org.uk www.thejournalofpathology.com

242 P Bouchalova et al
not included in subsequent analyses. In the remain-ing 61 patients, mutations were classified as produc-ing folded or unfolded proteins, based on either X-ray-or NMR-derived structures; as causing a disruptive ornon-disruptive mutation, and their severity scores werecalculated. Since all mutations are classified as severe,cancers were divided into groups with severity scoresabove the median value (81.63) or ≤ this value.There were 12 frame-shift or nonsense mutations
that could not be classified as folded or unfolded muta-tions or given a severity score but were all classed asdisruptive mutations. One mutation in exon 10 couldnot be given a severity score and is not included in thestructural data, and individual mutations do not havedata for their structural effect using X-ray crystal orNMR data. Immunohistochemical staining patterns forp53 could not be classified in three cancers, due to lackof sufficient tumour tissue in the microarray, and the12 nonsense/frame-shift mutations were all unstainedfor p53, as expected, leaving a group of 46 cancers thatcontain missense TP53 mutations for classification ofstaining pattern (31 with heterogeneous p53 stainingand 15 with homogeneous staining; see Figure 3 forexamples of staining patterns). In those cancers withheterogeneous p53 staining, all cores showed a similarpattern, where positively stained tumour cells weremixed with negatively stained tumour cells in eachtissue core (six cores/cancer were used, taken fromdifferent areas of the tumour).
Association of mutation typeswith clinicopathological dataClinicopathological data include HER2, ER, PR, num-ber of nodes containing tumour, invasive grade, tumoursize, disease-free survival and immunohistochemcalscores for Ki67, Mdm2 and p21. The associations ofp53 mutation classifications are summarized in Table 2(see supplementary material, Table S2, for full detailsof each cancer). There were no associations betweenthe predicted classes of missense mutations in terms ofprotein folding, functional disruption or severity scorewithin this tumour set, and p53 immunohistochemicalstaining patterns were independent of the mutationtype, based on these sequence-based classifications(Table 2). Mutations predicted to adopt a folded struc-ture associated with higher expression of p21 but notwith any other clinicopathological variable. Using theseverity score as a continuous variable, or after clas-sification into two groups based on the median score,revealed no associations with clinicopathological fac-tors. The homogeneous staining pattern associated withhigher Ki67 score (p = 0.0079), lower Mdm2 levels,PR-negative cancers (p = 0.0003) and ER-negativecancers (p = 0.03). None of eight grade 2 tumoursshowed a homogeneous staining pattern compared to15 of 37 grade 3 tumours (p = 0.038), and tumourswith heterogeneous staining showed improved sur-vival by Kaplan–Meier analysis (p = 0.036; log rank;Mantel–Cox).
The association of mutant p53 staining pattern withER/PR status was confirmed in a separate group oftriple-negative breast cancers. Of 27 patients identifiedwith TP53 mutations (details given in Table S3, seesupplementary material) from 55 tumours sequenced,four contained non-sense or frame-shift mutation and21of the 23 tumours with missense mutation showedhomogeneous staining (91%). Thus, from the combinedanalysis of all patients included in this study, 35 of48 PR− cancers with missense TP53 mutations showedhomogeneous staining, whereas 19 of 20 PR+ tumourswith missense TP53 mutations showed heterogeneousstaining (p = 2.44 × 10−7). We also noticed that threeof four triple-negative cell lines showed homogeneousmutant p53 accumulation, whilst all four breast cancercell lines that expressed ER, PR or amplified HER2showed heterogeneous p53 staining (see supplementarymaterial, Table S4).
Mdm2 influences mutant p53 stabilization in somebut not all breast cancer cell linesIn view of the correlation between high levels of Mdm2and the heterogeneity of mutant p53 accumulation seenin human breast cancer samples, we treated three breastcancer cell lines that showed heterogeneous p53 stainingwith Nutlin, an inhibitor ofMdm2 [33]. Nutlin treatmentincreased the percentage of cells containing stabilizedmutant p53 in the two steroid receptor-positive cell lines,T47D and BT474 (p < 0.01 for each), but not in SKBR3HER2-amplified cells (Figure 4).
Discussion
Numerous studies have attempted to employ TP53mutation as a predictive or prognostic biomarker inhuman cancer, with generally disappointing results.However, subclassification of TP53 mutations mayprovide more accurate prediction than a simple mutantversus non-mutant classification [5–8]. In breast can-cer, a large phase 3 trial showed that TP53 status wasprognostic for overall survival but did not predict taxanesensitivity [34], which may relate to different prognosticand predictive impacts in luminal and triple-negativecancers [35,36]. Thus, the emerging picture is of vari-able effects of TP53 status, depending on mutation typeand tumour type.In immunohistochemical studies, mutant p53 sta-
bilization is often heterogeneous within an individualtumour, for unknown reasons [11–14,37]. We demon-strated a similarly variable accumulation of mutantp53 in cell lines that contained different TP53 mis-sense mutations. The finding of stable staining patternsin cell lines indicates that different mutant p53 sta-bilization patterns in primary tumours are not animmunohistochemical artifact of fixation or pro-cessing. We also showed that individual colonies ofheterogeneously stained cell lines retained the same
Copyright © 2014 Pathological Society of Great Britain and Ireland. J Pathol 2014; 233: 238–246Published by John Wiley & Sons, Ltd. www.pathsoc.org.uk www.thejournalofpathology.com
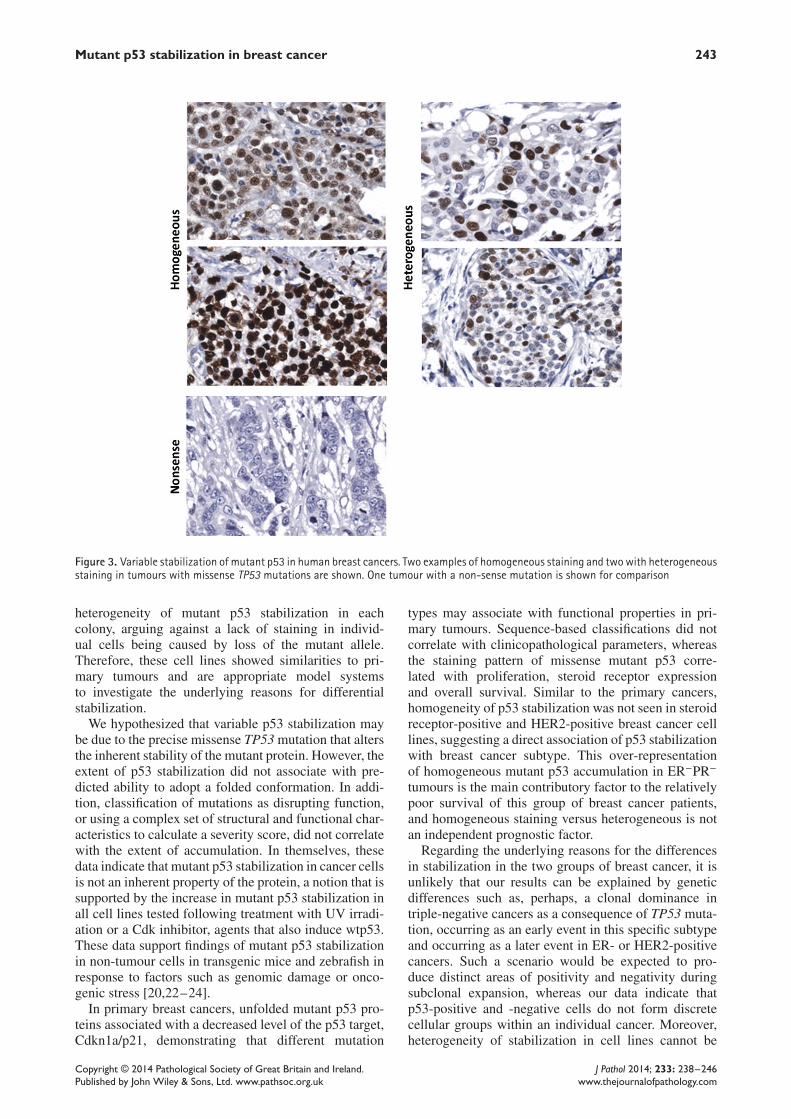
Mutant p53 stabilization in breast cancer 243
Figure 3. Variable stabilization of mutant p53 in human breast cancers. Two examples of homogeneous staining and twowith heterogeneousstaining in tumours with missense TP53 mutations are shown. One tumour with a non-sense mutation is shown for comparison
heterogeneity of mutant p53 stabilization in eachcolony, arguing against a lack of staining in individ-ual cells being caused by loss of the mutant allele.Therefore, these cell lines showed similarities to pri-mary tumours and are appropriate model systemsto investigate the underlying reasons for differentialstabilization.We hypothesized that variable p53 stabilization may
be due to the precise missense TP53mutation that altersthe inherent stability of the mutant protein. However, theextent of p53 stabilization did not associate with pre-dicted ability to adopt a folded conformation. In addi-tion, classification of mutations as disrupting function,or using a complex set of structural and functional char-acteristics to calculate a severity score, did not correlatewith the extent of accumulation. In themselves, thesedata indicate that mutant p53 stabilization in cancer cellsis not an inherent property of the protein, a notion that issupported by the increase in mutant p53 stabilization inall cell lines tested following treatment with UV irradi-ation or a Cdk inhibitor, agents that also induce wtp53.These data support findings of mutant p53 stabilizationin non-tumour cells in transgenic mice and zebrafish inresponse to factors such as genomic damage or onco-genic stress [20,22–24].In primary breast cancers, unfolded mutant p53 pro-
teins associated with a decreased level of the p53 target,Cdkn1a/p21, demonstrating that different mutation
types may associate with functional properties in pri-mary tumours. Sequence-based classifications did notcorrelate with clinicopathological parameters, whereasthe staining pattern of missense mutant p53 corre-lated with proliferation, steroid receptor expressionand overall survival. Similar to the primary cancers,homogeneity of p53 stabilization was not seen in steroidreceptor-positive and HER2-positive breast cancer celllines, suggesting a direct association of p53 stabilizationwith breast cancer subtype. This over-representationof homogeneous mutant p53 accumulation in ER−PR−
tumours is the main contributory factor to the relativelypoor survival of this group of breast cancer patients,and homogeneous staining versus heterogeneous is notan independent prognostic factor.Regarding the underlying reasons for the differences
in stabilization in the two groups of breast cancer, it isunlikely that our results can be explained by geneticdifferences such as, perhaps, a clonal dominance intriple-negative cancers as a consequence of TP53 muta-tion, occurring as an early event in this specific subtypeand occurring as a later event in ER- or HER2-positivecancers. Such a scenario would be expected to pro-duce distinct areas of positivity and negativity duringsubclonal expansion, whereas our data indicate thatp53-positive and -negative cells do not form discretecellular groups within an individual cancer. Moreover,heterogeneity of stabilization in cell lines cannot be
Copyright © 2014 Pathological Society of Great Britain and Ireland. J Pathol 2014; 233: 238–246Published by John Wiley & Sons, Ltd. www.pathsoc.org.uk www.thejournalofpathology.com
244 P Bouchalova et al
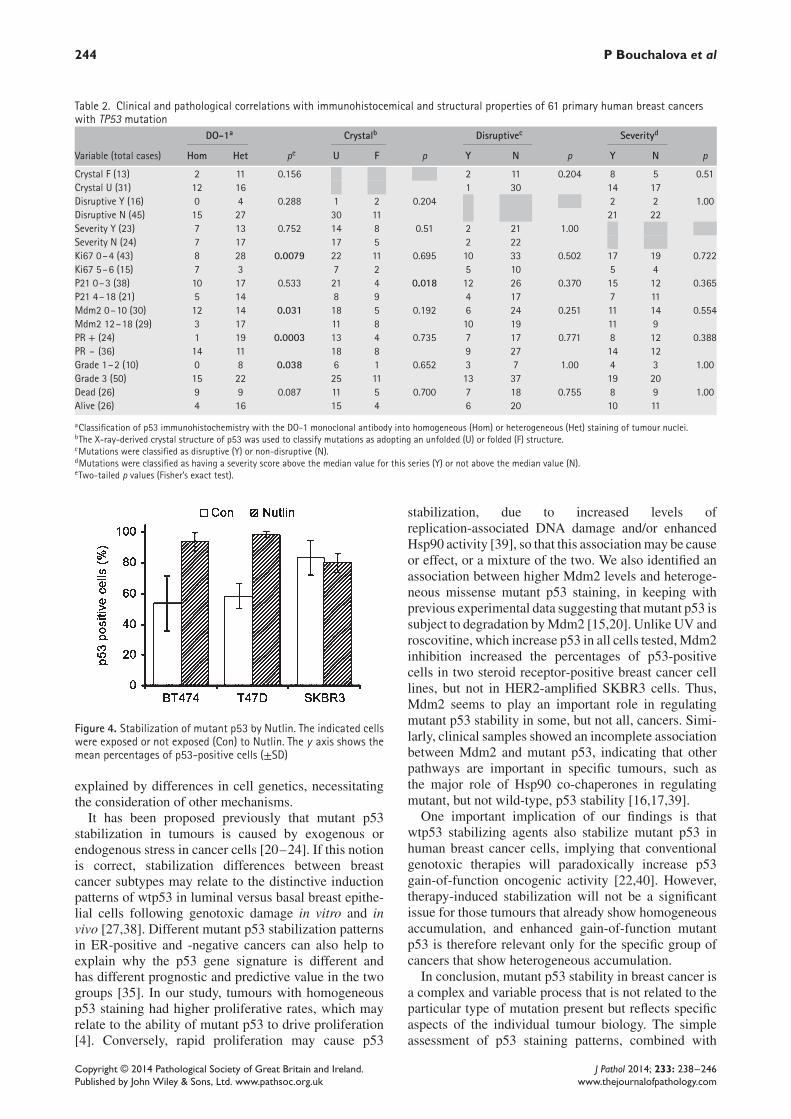
Table 2. Clinical and pathological correlations with immunohistocemical and structural properties of 61 primary human breast cancerswith TP53 mutation
DO-1a Crystalb Disruptivec Severityd
Variable (total cases) Hom Het pe U F p Y N p Y N p
Crystal F (13) 2 11 0.156 2 11 0.204 8 5 0.51Crystal U (31) 12 16 1 30 14 17Disruptive Y (16) 0 4 0.288 1 2 0.204 2 2 1.00Disruptive N (45) 15 27 30 11 21 22Severity Y (23) 7 13 0.752 14 8 0.51 2 21 1.00Severity N (24) 7 17 17 5 2 22Ki67 0–4 (43) 8 28 0.0079 22 11 0.695 10 33 0.502 17 19 0.722Ki67 5–6 (15) 7 3 7 2 5 10 5 4P21 0–3 (38) 10 17 0.533 21 4 0.018 12 26 0.370 15 12 0.365P21 4–18 (21) 5 14 8 9 4 17 7 11Mdm2 0–10 (30) 12 14 0.031 18 5 0.192 6 24 0.251 11 14 0.554Mdm2 12–18 (29) 3 17 11 8 10 19 11 9PR + (24) 1 19 0.0003 13 4 0.735 7 17 0.771 8 12 0.388PR – (36) 14 11 18 8 9 27 14 12Grade 1–2 (10) 0 8 0.038 6 1 0.652 3 7 1.00 4 3 1.00Grade 3 (50) 15 22 25 11 13 37 19 20Dead (26) 9 9 0.087 11 5 0.700 7 18 0.755 8 9 1.00Alive (26) 4 16 15 4 6 20 10 11
aClassification of p53 immunohistochemistry with the DO-1 monoclonal antibody into homogeneous (Hom) or heterogeneous (Het) staining of tumour nuclei.bThe X-ray-derived crystal structure of p53 was used to classify mutations as adopting an unfolded (U) or folded (F) structure.cMutations were classified as disruptive (Y) or non-disruptive (N).dMutations were classified as having a severity score above the median value for this series (Y) or not above the median value (N).eTwo-tailed p values (Fisher’s exact test).
Figure 4. Stabilization of mutant p53 by Nutlin. The indicated cellswere exposed or not exposed (Con) to Nutlin. The y axis shows themean percentages of p53-positive cells (±SD)
explained by differences in cell genetics, necessitatingthe consideration of other mechanisms.It has been proposed previously that mutant p53
stabilization in tumours is caused by exogenous orendogenous stress in cancer cells [20–24]. If this notionis correct, stabilization differences between breastcancer subtypes may relate to the distinctive inductionpatterns of wtp53 in luminal versus basal breast epithe-lial cells following genotoxic damage in vitro and invivo [27,38]. Different mutant p53 stabilization patternsin ER-positive and -negative cancers can also help toexplain why the p53 gene signature is different andhas different prognostic and predictive value in the twogroups [35]. In our study, tumours with homogeneousp53 staining had higher proliferative rates, which mayrelate to the ability of mutant p53 to drive proliferation[4]. Conversely, rapid proliferation may cause p53
stabilization, due to increased levels ofreplication-associated DNA damage and/or enhancedHsp90 activity [39], so that this associationmay be causeor effect, or a mixture of the two. We also identified anassociation between higher Mdm2 levels and heteroge-neous missense mutant p53 staining, in keeping withprevious experimental data suggesting that mutant p53 issubject to degradation byMdm2 [15,20]. Unlike UV androscovitine, which increase p53 in all cells tested,Mdm2inhibition increased the percentages of p53-positivecells in two steroid receptor-positive breast cancer celllines, but not in HER2-amplified SKBR3 cells. Thus,Mdm2 seems to play an important role in regulatingmutant p53 stability in some, but not all, cancers. Simi-larly, clinical samples showed an incomplete associationbetween Mdm2 and mutant p53, indicating that otherpathways are important in specific tumours, such asthe major role of Hsp90 co-chaperones in regulatingmutant, but not wild-type, p53 stability [16,17,39].One important implication of our findings is that
wtp53 stabilizing agents also stabilize mutant p53 inhuman breast cancer cells, implying that conventionalgenotoxic therapies will paradoxically increase p53gain-of-function oncogenic activity [22,40]. However,therapy-induced stabilization will not be a significantissue for those tumours that already show homogeneousaccumulation, and enhanced gain-of-function mutantp53 is therefore relevant only for the specific group ofcancers that show heterogeneous accumulation.In conclusion, mutant p53 stability in breast cancer is
a complex and variable process that is not related to theparticular type of mutation present but reflects specificaspects of the individual tumour biology. The simpleassessment of p53 staining patterns, combined with
Copyright © 2014 Pathological Society of Great Britain and Ireland. J Pathol 2014; 233: 238–246Published by John Wiley & Sons, Ltd. www.pathsoc.org.uk www.thejournalofpathology.com

Mutant p53 stabilization in breast cancer 245
p53 pathway analysis for mutant and wtp53 tumours[11,37,41], may have benefit in identifying p53 func-tionality in human cancer and for identifying patientsin whom therapy may inadvertently cause activation ofmutant p53 oncogenic function.
Acknowledgements
We thank all of the women who kindly donated tis-sue to the Tayside Tissue Bank for research purposes,and the nurses, clinicians and other staff involved. Weare grateful to the Tayside Tissue Bank for preparingTMAs and for immunostaining. This study was sup-ported by the Czech Republic (Grant No. IGA MZCR NT/13794-4/2012), the European Regional Devel-opment Fund and the State Budget of the Czech Repub-lic (RECAMO; Grant Nos CZ.1.05/2.1.00/03.0101 andMH CZ-DRO MMCI 00209805) and Breast CancerResearch (Scotland).
Author contributions
PB carried out cell culture and immunohistochemicalanalysis of cell lines; RN analysed p53 staining in pri-mary breast cancer samples, performed statistical analy-sis in cell lines and primary material and was involved inthe study concept and design; PM performed structuralpredictions of mutant p53 folding and was involved inthe design of the study; RH was involved in interpreta-tion of cell line data and participated in study design;MVA and KM performed cell culture experiments andanalysis of p53 stabilization; LBJ and CAP selected tis-sues for TMA construction and analysis of TP53 muta-tion and performed immunohistochemical analysis onbreast cancer material; PQ collated clinicopathologicaland immunostaining data and performed statistical anal-yses; AMT was involved in project design and coor-dination and writing the manuscript; BV was involvedin the concept of the study, study design and coordina-tion, and writing the manuscript; and PJC was involvedin study concept, design and coordination, analysis ofp53 immunostaining, categorization of mutant types andwriting the manuscript. All authors read and approvedthe final manuscript.
Abbreviations
ER, oestrogen receptor; HER2, human epidermalgrowth factor receptor-2; NMR, nuclear magnetic reso-nance; PR, progesterone receptor; wtp53, wild-type p53
References1. Munro AJ, Lain S, Lane DP. p53 abnormalities and outcomes in col-
orectal cancer: a systematic review. Br J Cancer 2005; 92: 434–444.2. Soussi T, Wiman KG. Shaping genetic alterations in human cancer:
the p53 mutation paradigm. Cancer Cell 2007; 12: 303–312.3. Zilfou JT, Lowe SW. Tumor suppressive functions of p53. Cold
Spring Harb Perspect Biol 2009; 1: a001883.
4. Goh AM, Coffill CR, Lane DP. The role of mutant p53 in humancancer. J Pathol 2011; 223: 116–126.
5. Hashimoto T, Tokuchi Y, Hayashi M, et al. p53-null mutationsundetected by immunohistochemical staining predict a poor outcomewith early-stage non-small cell lung carcinomas. Cancer Res 1999;59: 5572–5577.
6. OlivierM, Langerod A, Carrieri P, et al. The clinical value of somaticTP53 genemutations in 1794 patients with breast cancer.Clin CancerRes 2006; 12: 1157–1167.
7. Ozcelik H, Pinnaduwage D, Bull SB, et al. Type of TP53 mutationand ERBB2 amplification affects survival in node-negative breastcancer. Breast Cancer Res Treat 2007; 105: 255–265.
8. Poeta ML, Manola J, Goldwasser MA, et al. TP53 mutations andsurvival in squamous-cell carcinoma of the head and neck. N Engl
J Med 2007; 357: 2552–2561.9. Young KH, Leroy K, Moller MB, et al. Structural profiles of TP53
gene mutations predict clinical outcome in diffuse large B-celllymphoma: an international collaborative study. Blood 2008; 112:3088–3098.
10. Robles AI, Harris CC. Clinical outcomes and correlates of TP53mutations and cancer. Cold Spring Harb Perspect Biol 2010; 2:a001016.
11. Nenutil R, Smardova J, Pavlova S, et al. Discriminating functionaland non-functional p53 in human tumours by p53 and MDM2immunohistochemistry. J Pathol 2005; 207: 251–259.
12. Hall PA, Lane DP. p53 in tumour pathology: can we trust immuno-histochemistry? – Revisited! J Pathol 1994; 172: 1–4.
13. Wynford-Thomas D. p53 in tumour pathology: can we trust immuno-cytochemistry? J Pathol 1992; 166: 329–330.
14. Thompson AM, Anderson TJ, Condie A, et al. p53 allele losses,mutations and expression in breast cancer and their relationship toclinico-pathological parameters. Int J Cancer 1992; 50: 528–532.
15. Midgley CA, Lane DP. p53 protein stability in tumour cells isnot determined by mutation but is dependent on Mdm2 binding.Oncogene 1997; 15: 1179–1189.
16. Muller P, Ceskova P, Vojtesek B. Hsp90 is essential for restoringcellular functions of temperature-sensitive p53 mutant protein butnot for stabilization and activation of wild-type p53: implications forcancer therapy. J Biol Chem 2005; 280: 6682–6691.
17. Lukashchuk N, Vousden KH. Ubiquitination and degradation ofmutant p53.Mol Cell Biol 2007; 27: 8284–8295.
18. Muller P, Hrstka R, Coomber D, et al. Chaperone-dependent sta-bilization and degradation of p53 mutants. Oncogene 2008; 27:3371–3383.
19. Carlsson J, Soussi T, Persson B. Investigation and prediction of theseverity of p53mutants using parameters from structural calculations.FEBS J 2009; 276: 4142–4155.
20. Terzian T, Suh YA, Iwakuma T, et al. The inherent instability ofmutant p53 is alleviated by Mdm2 or p16INK4a loss. Genes Dev2008; 22: 1337–1344.
21. Jackson JG, Post SM, Lozano G. Regulation of tissue- andstimulus-specific cell fate decisions by p53 in vivo. J Pathol 2011;223: 127–136.
22. Suh YA, Post SM, Elizondo-Fraire AC, et al.Multiple stress signalsactivate mutant p53 in vivo. Cancer Res 2011; 71: 7168–7175.
23. Lee KC, Goh WL, Xu M, et al. Detection of the p53 responsein zebrafish embryos using new monoclonal antibodies. Oncogene2008; 27: 629–640.
24. Guo L, Liew HP, Camus S, et al. Ionizing radiation induces adramatic persistence of p53 protein accumulation and DNA damagesignaling in mutant p53 zebrafish. Oncogene 2013; 32: 4009–4016.
25. Kotala V, Uldrijan S, Horky M, et al. Potent induction of wild-typep53-dependent transcription in tumour cells by a syntheticinhibitor of cyclin-dependent kinases. Cell Mol Life Sci 2001; 58:1333–1339.
Copyright © 2014 Pathological Society of Great Britain and Ireland. J Pathol 2014; 233: 238–246Published by John Wiley & Sons, Ltd. www.pathsoc.org.uk www.thejournalofpathology.com

246 P Bouchalova et al
26. Baker L, Quinlan PR, Patten N, et al. p53 mutation, deprivation andpoor prognosis in primary breast cancer. Br J Cancer 2010; 102:719–726.
27. Coates PJ, Appleyard MV, Murray K, et al. Differential contextualresponses of normal human breast epithelium to ionizing radiation ina mouse xenograft model. Cancer Res 2010; 70: 9808–9815.
28. Detre S, Saclani Jotti G, Dowsett M. A ‘quickscore’ method forimmunohistochemical semiquantitation: validation for oestrogenreceptor in breast carcinomas. J Clin Pathol 1995; 48: 876–878.
29. Purdie CA, Baker L, Ashfield A, et al. Increased mortality inHER2-positive, oestrogen receptor positive invasive breast cancer: apopulation-based study. Br J Cancer 2010; 103: 475–481.
30. Purdie CA, Jordan LB, McCullough JB, et al. HER2 assessment oncore biopsy specimens using monoclonal antibody CB11 accuratelydetermines HER2 status in breast carcinoma. Histopathology 2010;56: 702–707.
31. Kwasigroch JM, Gilis D, Dehouck Y, et al. PoPMuSiC, rationallydesigning point mutations in protein structures. Bioinformatics 2002;18: 1701–1702.
32. Quinlan PR, Thompson A, Reed C. Inspire: an integrated agentbased system for hypothesis generation within cancer datasets. InIEEE/WIC/ACM International Conference on Web Intelligence andIntelligent Agent Technology, 2008; 587–590.
33. Klein C, Vassilev LT. Targeting the p53–MDM2 interaction to treatcancer. Br J Cancer 2004; 91: 1415–1419.
34. Bonnefoi H, Piccart M, Bogaerts J, et al. TP53 status for predictionof sensitivity to taxane versus non-taxane neoadjuvant chemotherapyin breast cancer (EORTC 10994/BIG 1–00): a randomised phase 3trial. Lancet Oncol 2011; 12: 527–539.
35. Coutant C, Rouzier R, Qi Y, et al. Distinct p53 gene signaturesare needed to predict prognosis and response to chemotherapy inER-positive and ER-negative breast cancers. Clin Cancer Res 2011;17: 2591–2601.
36. De Laurentiis M. Optimum chemotherapy regimen for early breastcancer. Lancet Oncol 2011; 12: 514–515.
37. Abdel-Fatah TM, Powe DG, Agboola J, et al. The biological, clinicaland prognostic implications of p53 transcriptional pathways in breastcancers. J Pathol 2010; 220: 419–434.
38. Huper G, Marks JR. Isogenic normal basal and luminal mammaryepithelial isolated by a novel method show a differential response toionizing radiation. Cancer Res 2007; 67: 2990–3001.
39. Muller P, Ruckova E, Halada P, et al. C-terminal phosphorylation ofHsp70 and Hsp90 regulates alternate binding to co-chaperones CHIPand HOP to determine cellular protein folding/degradation balances.Oncogene 2013; 32: 3101–3110.
40. Prives C, White E. Does control of mutant p53 by Mdm2 complicatecancer therapy? Genes Dev 2008; 22: 1259–1264.
41. Thompson AM, Lane DP. p53 transcriptional pathways in breastcancer: the good, the bad and the complex. J Pathol 2010; 220:401–403.
SUPPLEMENTARY MATERIAL ON THE INTERNETThe following supplementary material may be found in the online version of this article:
Table S1. TP53 sequence data of 240 breast cancers
Table S2. Detailed clinicopathological information for 61 breast cancers with TP53 mutations
Table S3. Immunohistocemical classification of 27 triple-negative breast cancers with TP53 mutations
Table S4. Receptor status and p53 immunohistochemical stabilization patterns of eight breast cancer cell lines
Copyright © 2014 Pathological Society of Great Britain and Ireland. J Pathol 2014; 233: 238–246Published by John Wiley & Sons, Ltd. www.pathsoc.org.uk www.thejournalofpathology.com
Copyright © 2022 FDOKUMEN




















