
Peroxynitrite-induced p38 MAPK pro-apoptotic signaling in enterocytes

www.elsevier.com/locate/jhep
Journal of Hepatology 47 (2007) 325–337
Modulation of MAPK pathways and cell cycle by replicating hepatitisB virus: Factors contributing to hepatocarcinogenesisq
Ruth Chin1, Linda Earnest-Silveira1, Bernd Koeberlein2, Susanne Franz3,Hanswalter Zentgraf3, Xuebin Dong4,5, Eric Gowans4,5,
C.-Thomas Bock2, Joseph Torresi1,*
1Department of Medicine, CCREID, Royal Melbourne Hospital, University of Melbourne, Post Office, Parkville, Vic. 3050, Australia2Department of Molecular Pathology, University Hospital of Tuebingen, Germany
3Applied Tumor Virology, German Cancer Research Center (DKFZ), Heidelberg, Germany4Burnet Institute, AMREP, Prahran, Vic., Australia
5Department of Microbiology, Monash University, Vic., Australia
Background/Aims: Chronic infection with the hepatitis B virus (HBV) is strongly associated with the development of
hepatocellular carcinoma but the mechanism by which this occurs is unknown. Numerous studies have focused on the
HBV X protein showing that it activates signal transduction pathways while few have investigated these changes in
HBV-replicating hepatocytes.
Methods: We utilized the recombinant adenovirus system to deliver a replication competent HBV genome into Huh7 and
primary marmoset hepatocytes (PMH) to examine the effects of active viral replication on the regulation of Ras-ERK sig-
nal transduction and related pathways.Results: Huh7 cells and PMHs replicating HBV demonstrated significant upregulation in phosphorylated ERK, Akt, c-
myc together with increased p53, cyclin B1 and p21cip1 expression and cell cycle progression to G2 phase in the absence of
increased cell proliferation. Phosphorylation of the key cell survival kinase, Akt, was significantly increased, resulting in
increased serine phosphorylation of the downstream target, GSK3-b.
Conclusions: These results demonstrated simultaneous activation of the MAP Kinase and Akt pathways in HBV-repli-
cating hepatocytes that resulted in dysregulation in the control of cell cycle progression and which help explain the early
pathogenic mechanisms that underlie malignant transformation associated with chronic hepatitis B infection.
� 2007 European Association for the Study of the Liver. Published by Elsevier B.V. All rights reserved.
Keywords: Hepatitis; Carcinoma; Cell signaling
0168-8278/$32.00 � 2007 European Association for the Study of the Liver. Published by Elsevier B.V. All rights reserved.
doi:10.1016/j.jhep.2007.03.025
Received 3 December 2006; received in revised form 5 March 2007; accepted 6 March 2007; available online 18 April 2007
Associate Editor: F. Zoulimq The authors who have taken part in this study declared that they have no relationship with the manufacturers of the product involved either in the
past or present and did not receive funding from the manufacturers to carry out their research.* Corresponding author. Tel.: +61 3 8344 3262; fax: +61 3 93471863.
E-mail addresses: [email protected] (R. Chin), [email protected] (L. Earnest-Silveira), [email protected] (B.Koeberlein), [email protected] (S. Franz), [email protected] (H. Zentgraf), [email protected] (X. Dong), [email protected] (E. Gowans), [email protected] (C.-Thomas Bock), [email protected] (J. Torresi).
Abbreviations: HBV, hepatitis B virus; MAPK, mitogen activated protein kinases; ERK, extracellular signal-regulated kinases; SAPK/JNK,stress-activated protein kinases/NH2-terminal-Jun kinases.

326 R. Chin et al. / Journal of Hepatology 47 (2007) 325–337
1. Introduction
The mechanism by which HBV results in HCC devel-opment is not well understood and the risk of develop-ing HCC remains significantly elevated despitesuppression of viral replication by antiviral therapyand after clearance of serum HBeAg and HBV viremia[1–3].
The HBx protein of HBV has been proposed as animportant oncogenic stimulus for the development ofHCC in chronically infected individuals [4–7]. Howeverseveral studies have reported divergent effects of HBx onp53-, Fas- and TNF-a-[8–10], c-myc-, and Ras-mediatedapoptosis [11–13] and transactivation of IL-8 [14–16].Our understanding of how HBV causes HCC remainsa challenge and the frequent discrepancies in experimen-tal findings may reflect limitations in duplicating cellularchanges that occur in natural infection. The HBV large(LHB) and middle (MHB) envelope proteins have alsobeen shown to contribute to hepatocarcinogenesis ofHBV [17,18], further emphasizing the role of viral fac-tors, other than HBx, and active HBV replication asmediators of HCC development.
To address the role of active HBV replication onhepatocarcinogenesis we used a recombinant adenovirussystem to deliver a replication competent HBV genomerather than single viral genes into primary marmosethepatocytes, and contrasted the changes to cell signalingpathways induced by HBV infection with those seen inthe human hepatoma cell line, Huh7.
2. Materials and methods
2.1. Plasmids
A 1.5· full length replication competent HBV (genotype A, subtypeadw2) [17] was subcloned into pAdTrack (provided by B Vogelstein,Howard Hughes Medical Centre, Baltimore) which was pre-digestedwith HindIII and EcoRV (Promega). The plasmid pAdTrack-HBVwtwas digested with PmeI and transformed into AdEasier-1 cells by elec-troporation (Bio-Rad Gene Pulser). Clones (AdEasy-HBV) were trans-formed into Top 10F 0 cells (Invitrogen) and confirmed by DNAsequencing. An AdEasy-GFP control expressing green fluorescent pro-tein (GFP) only was also produced.
2.2. Antibodies
Primary antibodies used were total p44/p42 MAP Kinase, phos-pho-p44/p42 MAP Kinase (Thr202/Tyr204), total Akt, phospho-Akt(Thr308), total c-myc, phospho-c-myc (Thr58/Ser62), phospho-GSK3-ß (Ser9), cyclin D1, cyclin A, cyclin B1, p21-Waf1/Cip1, cdc2,pan-actin (all by Cell Signaling Technology) and p53 antibody(DKZF, Heidelberg, Germany).
2.3. Cell lines
Huh7 and 293T cells were grown in Dulbecco’s modified Eagle’smedium (DMEM) (Invitrogen) supplemented with 10% fetal calfserum (FCS) and streptomycin 50 lg/ml at 37 �C in 5% CO2.
Primary marmoset hepatocyte (PMH) cultures were prepared fromthe common marmoset, Callithrix jacchus, according to a previouslydescribed method [19].
2.4. Production of recombinant adenoviruses
The rAdHBV virions were produced by transfection of pAdEasy-HBVwt into semi-confluent 293T cells and amplified by serial pas-saging in culture. For the initial production of the first passage ofrAdHBV virions, 293T cells were transfected with PacI-linearizedpAdEasy-HBVwt DNA using Fugene 6 Transfection Reagent(Roche) according to manufacturer’s instruction and transfectionsmonitored by GFP expression. Cells were collected at day 5 post-transfection, centrifuged at 1500g for 5 min, resuspended in 2 mlof fresh media and lysed by freeze-thawing. Lysates were centrifugedat 3000g for 15 min and supernatants containing rAdHBV virionswere filtered with a 0.22 lm filter (Millipore) and stored in 500 llaliquots at �70 �C. High titre virus stocks were prepared by serialpassaging in 293T cells. The titre of the final virus stock was deter-mined by infecting Huh7 cells and analyzing for GFP expression byflow cytometry 48 h post infection (PI) (see below). Titres wereexpressed as GFP expressing units (GEU) and an MOI of 1 wasdefined as the viral dilution that resulted in >99% GEUs. A recom-binant adenovirus expressing GFP alone, rAdGFP, was produced inthe same manner to serve as a control virus.
For infections with rAdHBV or rAdGFP virions, Huh7 cells grownin 6-well tissue culture plates (Nunc) and at 70% confluency wereinfected with rAdHBV or rAdGFP at a multiplicity of infection(MOI) of 1.0. For PMH cells, infections were performed with rAdHBVor rAdGFP at an MOI of 1.0 on confluent cells (day 3 post-plating).Mock-infected controls were treated with PBS alone.
2.5. Detection of HBV DNA
Cells and culture media supernatants were collected 24 or 72 h PIfor PMH and Huh7 cells, respectively, for analyses of HBV DNA rep-licative intermediates as described previously [20]. Total cell lysateswere collected 24 or 96 h PI for PMH and Huh7 cells, respectively,for analyses of HBV cccDNA using real time PCR as described byBowden et al. [21]. The ß-Globin gene was used as an internal controlstandard and levels of HBV cccDNA were expressed as copies ofcccDNA/genome equivalent (cp/GEq).
2.6. Analysis by Western immunoblotting
Huh7 cells were harvested 72 h PI while PMHs were harvested 24 hPI using 200 ll of ice-cold protein cell lysis buffer (20 mM Tris–HCl(pH 7.5), 150 mM sodium chloride, 1 mM sodium EDTA, 1 mMEGTA, 1% Triton 2.5 mM sodium pyrophosphate, 1 mM b-glycero-phosphate, 1 mM sodium vanadate, 1 mM sodium molybdate, 5 mMsodium fluoride) supplemented with complete protease inhibitor cock-tail (Roche). To assess the effect of PDGF stimulation, cell monolayerwas incubated with 10 ng/ml of PDGF for 30 min prior to harvest.Lysates were clarified by centrifugation and the supernatants, repre-senting cytoplasmic and membrane fractions, were collected. The pel-let, representing the nuclear fraction, was redissolved in protein lysisbuffer containing 0.5% SDS. Total cellular fractions were preparedby adding SDS (final concentration 0.5%) to 100 ll of non-clarified celllysates. Thirty micrograms of protein from the total cell and cytoplas-mic fractions and total amount of the nuclear fractions were dissolvedin Laemmli buffer (50 mM Tris (pH 6.8), 2% SDS, 5% 2-mercap-toethanol, 8% glycerol, 0.01% bromophenol blue), resolved by a dena-turing 12% SDS–PAGE and transferred to Hybond-C Extramembrane (Amersham BioSciences). Membranes were blocked with5% skim milk powder in TBST (20 mM Tris, pH 7.4, 50 mM sodiumchloride, 0.5% Tween 20) at 4 �C overnight, followed by incubationwith primary antibody in TBST-5% skim milk powder at 4 �C over-night. Membranes were washed with TBST followed by incubationwith secondary antibody (1:1000 dilution of anti-mouse or anti-rabbit)(DAKO Cytomation) in TBST-5% skim milk powder at room temper-ature for 1 h, washed, followed by ECL (Amerham BioSciences).

Fig. 1. Infection of Huh7 and PMH cells with rAdHBV virions results in active viral replication. Huh7 cells and PMH were infected with rAdHBV
virions. GFP expression was determined by fluorescence microscopy (a and e) and flow cytometry (b and f). Autoradiographs of intracellular and secreted
HBV replicative DNA intermediates, relaxed circular (RC), double stranded linear (L) and single stranded (SS) at day 3 PI for Huh7 cells and day 1 PI
for PMH, respectively (c and g). Serum starvation of rAdHBV-infected cells resulted in inhibition of HBV replication (d).
R. Chin et al. / Journal of Hepatology 47 (2007) 325–337 327
Experiments were performed in triplicate, unless otherwise stated.Immunoblots were analyzed with a Bio-Rad GS710 Scanner withQUANTITY ONE 4.1.0 software package (Bio-Rad), and Prism 4.0(GraphPad Software).
2.7. Flow cytometry for GFP expression, cell cycle
analysis
Triplicate sets of PMH and Huh7 cells were infected with serialdilutions of rAdHBV or rAdGFP, collected at 24 and 48 h, respec-tively, washed twice with PBS and fixed in 1.0 ml of BD Cytofix
Buffer (BD Pharmingen). Expression of green fluorescence wasmeasured by flow cytometry using a FACS Calibur (BectonDickinson).
For cell cycle analysis, triplicate sets of Huh7 cells were infectedwith rAdHBV or rAdGFP, collected at 24, 36 and 72 h PI, washed twicewith PBS and fixed with 70% ethanol (in PBS) at �20 �C overnight.Cells were pelleted by centrifugation at 1500g, resuspended in 0.1%BSA in PBS, incubated with 20 lg/ml RNAseA (Roche), at 37 �C for30 min, then stained with 5 lg/ml propidium iodide (Sigma–Aldrich)for 30 min in the dark. Flow cytometry was performed with the FACSCalibur (Becton Dickinson), and cell cycle phases analyzed with Flow-Jo Version 4.6.2. Apoptosis was determined by scoring Sub-G1 cellsdisplaying pyknotic nuclei using flow cytometry.
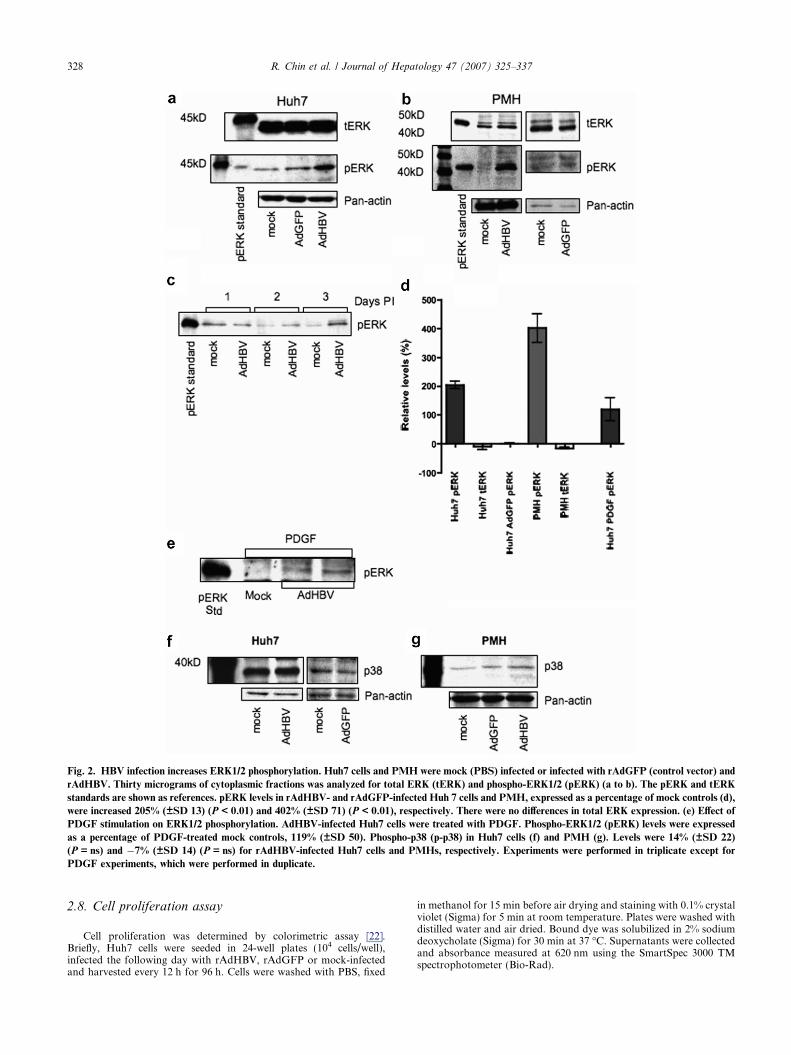
Fig. 2. HBV infection increases ERK1/2 phosphorylation. Huh7 cells and PMH were mock (PBS) infected or infected with rAdGFP (control vector) and
rAdHBV. Thirty micrograms of cytoplasmic fractions was analyzed for total ERK (tERK) and phospho-ERK1/2 (pERK) (a to b). The pERK and tERK
standards are shown as references. pERK levels in rAdHBV- and rAdGFP-infected Huh 7 cells and PMH, expressed as a percentage of mock controls (d),
were increased 205% (±SD 13) (P < 0.01) and 402% (±SD 71) (P < 0.01), respectively. There were no differences in total ERK expression. (e) Effect of
PDGF stimulation on ERK1/2 phosphorylation. AdHBV-infected Huh7 cells were treated with PDGF. Phospho-ERK1/2 (pERK) levels were expressed
as a percentage of PDGF-treated mock controls, 119% (±SD 50). Phospho-p38 (p-p38) in Huh7 cells (f) and PMH (g). Levels were 14% (±SD 22)
(P = ns) and �7% (±SD 14) (P = ns) for rAdHBV-infected Huh7 cells and PMHs, respectively. Experiments were performed in triplicate except for
PDGF experiments, which were performed in duplicate.
328 R. Chin et al. / Journal of Hepatology 47 (2007) 325–337
2.8. Cell proliferation assay
Cell proliferation was determined by colorimetric assay [22].Briefly, Huh7 cells were seeded in 24-well plates (104 cells/well),infected the following day with rAdHBV, rAdGFP or mock-infectedand harvested every 12 h for 96 h. Cells were washed with PBS, fixed
in methanol for 15 min before air drying and staining with 0.1% crystalviolet (Sigma) for 5 min at room temperature. Plates were washed withdistilled water and air dried. Bound dye was solubilized in 2% sodiumdeoxycholate (Sigma) for 30 min at 37 �C. Supernatants were collectedand absorbance measured at 620 nm using the SmartSpec 3000 TMspectrophotometer (Bio-Rad).

Fig. 3. HBV infection increases phosphorylated Akt. Huh7 cells and
PMH were mock (PBS) infected or infected with rAdGFP and rAdHBV.
Thirty micrograms of total cellular fractions was analyzed for phospho-
Akt (pAkt). (a) pAkt levels at days 1 and 3 PI in rAdHBV-infected
Huh7. (b) pAkt levels at day 1 PI in rAdGFP- and rAdHBV-infected
PMH. (c) pAkt levels were increased in rAdHBV-infected Huh7 cells,
206% (±SD 22) (P < 0.05) and PMH, 260% (±SD 120) (P < 0.01),
respectively. Total Akt levels remained unchanged. Levels were
expressed as % relative to mock controls. Experiments were performed
in triplicate.
Fig. 4. HBV infection increases inactive GSK3-ß. Huh7 cells and PMH
were mock (PBS) infected or infected with rAdGFP and rAdHBV.
Thirty micrograms of total cellular fractions was analyzed for phospho-
Ser-9- GSK3-ß (a and b) and levels expressed as a percentage relative to
mock controls (c). Levels were increased by 67% (±SD 21) (P < 0.05)
and 255% (±SD 63) P < 0.05, for rAdHBV-infected Huh7 cells and
PMH, respectively. (d) PDGF stimulation of rAdHBV-infected Huh7
cells indicating decreased Ser-9-GSK3-ß phosphorylation compared to
PDGF-stimulated mock controls. Experiments were performed in
triplicate except for PDGF experiments, which were performed in
duplicate.
R. Chin et al. / Journal of Hepatology 47 (2007) 325–337 329
3. Results
3.1. HBV replication in Huh7 cells and PMH
Although Huh7 cells were known to be permissive forHBV replication this was not known for PMH. Infec-tion of both Huh7 cells and PMH with rAdHBVresulted in expression of GFP in >95% of cells(Fig. 1a, b, e and f). Intra- and extra-cellular HBVDNA replication intermediates (relaxed circular, doublestranded linear and single stranded) were readily detect-able 72 h PI for Huh7, increasing thereafter until day 6(Fig. 1c), and 24 h PI for PMH cells (Fig. 1g). HBVcccDNA was detected by quantitative real time PCR
in both Huh7 cells and PMH. The median (±SD) levelof HBV cccDNA was higher in PMH (2459 ± 549 cp/GEq) compared to Huh7 cells (1962 ± 748 cp/GEq).Because FCS is a growth factor and may stimulate

Fig. 5. HBV infection does not affect p53 while increases p21cip1. Huh7 and PMH were mock (PBS) infected or infected with rAdGFP (control vector)
and rAdHBV. Thirty micrograms of total cellular fractions was analyzed for p53 (a and b) and p21cip1 (c–e). Total p21cip1 levels in rAdHBV-infected cells
were expressed as a percentage of mock controls (e). Levels of p53 were increased 12% (±SD 24; P = ns) in rAdHBV infected Huh7 cells and 63% (±SD
7; P = 0.0038) relative to mock controls. For Huh7 cells p21cip1 levels were increased by 42% (±SD 20) (P = 0.02) and in infected PMH by 83% (±SD 30)
(P = 0.014). Experiments were performed in triplicate.
330 R. Chin et al. / Journal of Hepatology 47 (2007) 325–337
signaling pathways, at 48 h PI, rAdHBV infected Huh7cells were deprived of serum to assess the effect of serumstarvation on viral replication. Deprivation of FCS for24–48 h was associated with a significant decline inHBV DNA (Fig. 1d) and therefore all subsequent exper-iments were performed in the presence of 10% FCS andcomparisons made between rAdHBV-infected cells andrAdGFP and mock-infected cells.
3.2. HBV stimulates the MAPK signaling pathway
In order to examine the effects of HBV on the Ras-ERK pathway cell lysates of mock (PBS), rAdGFPand rAdHBV-infected Huh7s were examined for expres-sion of total and phospho-ERK1/2. Infection of Huh7cells with rAdHBV resulted in a 205% (±SD 13) upreg-ulation of ERK1/2 phosphorylation (pERK) compared
to mock controls (P < 0.01) while the levels of totalERK remained unchanged (Fig. 2a and d). The upregu-lation of pERK was sustained over a 3-day period(154% ± SD 46; P = 0.0023) (Fig. 2c). To determinethat the observed effects were not simply a consequenceof HBV replication in malignantly transformed Huh7cells we repeated the experiments in PMH. Infection ofPMH with rAdHBV resulted in a 402% (±SD 71)increase in the level of pERK compared to mock con-trols (P < 0.01) (Fig. 2b and d). Infection with rAdGFPdid not result in increased expression of total or pERK.
rAdHBV-infected Huh7 cells were also more sensitiveto PDGF stimulation which resulted in a 119% (±SD57) increase in pERK compared to mock controls(Fig. 2e). Phosphorylated p38 MAPK was not signifi-cantly altered in rAdHBV-infected Huh7 (14% ±SD22) and PMHs (�7% ±SD 14) (P = ns) (Fig. 2f and g).

Fig. 6. HBV infection increases phospho-c-myc. Huh7 cells and PMH
were mock (PBS) infected or infected with rAdGFP (control vector) and
rAdHBV. Thirty micrograms of total cellular fractions was analyzed for
total and phospho-c-myc (p-c-myc) (a and b). p-c-myc in rAdHBV-
infected Huh7 cells and PMH were expressed as a percentage relative to
mock controls (c), 63% (±SD 4) (P = 0.05) and 125% (±SD 35)
(P = 0.004), respectively. There were no differences in total c-myc
expression. Experiments were performed in triplicate.
R. Chin et al. / Journal of Hepatology 47 (2007) 325–337 331
3.3. HBV stimulates the Akt pathway
Upregulation of Ras-ERK also feeds into the PI3Keffector pathway. We therefore investigated the effectof HBV replication on PI3K-Akt signaling. Infectionof Huh7 cells with rAdHBV resulted in a 206% (±SD
22) increase in the level of phospho-Akt (pAkt) com-pared to mock controls (P < 0.05) (Fig. 3a and c). Therewas no difference in pAkt expression in rAdGFP-infected cells compared to mock controls (results notshown). Similarly, infection of PMH with rAdHBVresulted in significant increase in pAkt levels of 260%(±SD 120) (P < 0.01) (Fig. 3b and c). In order to deter-mine that the upregulation of pAkt had a biologicallyfunctional role, we investigated the effect of pAkt onthe phosphorylation of GSK3-b, a downstream sub-strate of Akt. In both Huh7 cells and PMH replicatingHBV the phospho-Ser9-GSK3-b increased by 67%(±SD 21) (P < 0.05) (Fig. 4a and c) and 255% (±SD63) (P < 0.05) (Fig. 4b and c), respectively, comparedto mock controls. Furthermore, stimulation with thegrowth factor, PDGF, a known activator of GSK3-ß,resulted in an increase in pGSK3-ß levels in mock-controls but not in rAdHBV-infected Huh7 cells(Fig. 4d).
We also investigated the effect of HBV inducedupregulation of Akt on p53 and found that p53 levelswere increased in rAdHBV-infected Huh7 cells (43%±SD 21) and PMH (63% ±SD 7) (P = 0.0038) com-pared to mock controls (Fig. 5a and b). Total intracellu-lar levels of p21 were also increased both in rAdHBV-infected Huh7 cells, 42% (±SD 20) (P = 0.02) andPMHs, 83% (±SD 30) (P = 0.014) (Fig. 5c–e).
3.4. HBV modulates c-myc
Induction of c-myc protein phosphorylation is Ras-dependent [23], and can be rapidly induced in responseto mitogenic stimulation [24]. In rAdHBV-infectedHuh7 cells and PMH, we found increased expressionof phospho-c-myc (p-c-myc), 63% (±SD 4.0)(P = 0.05) and 125% (±SD 35) (P = 0.004), respectively,compared to mock controls, while total c-myc levelswere unchanged (Fig. 6).
3.5. HBV stimulates cyclins A and B
In order to investigate the effects of HBV replicationon the cell cycle, intracellular levels of cyclins D1, A andB1 were determined in rAdHBV-infected Huh7 cells andPMH compared to those of rAdGFP-infected and mockcontrols. In the total cell lysates of rAdHBV-infectedHuh7 cells, cyclin D1, A and B1 levels were increased,28% (±SD 18) (P = 0.05), 53% (±SD 30) (P = 0.05),and 110% (±SD 26) (P = 0.013), respectively (Fig. 7a–c). In rAdHBV-infected PMHs, neither cyclin D1 norcyclin A was significantly increased compared to mockcontrols (Fig. 7d). However, intranuclear cyclin B1was strongly upregulated in both rAdHBV-infectedHuh7 as well as PMH, 105% (±SD 25) (P = 0.05) and419% (±SD 138) (P = 0.0015), respectively (Fig. 7b, c,e and f), while cytoplasmic cyclin B1 levels were reduced

Fig. 7. HBV infection increases cyclin D1, A and B1 proteins. Huh7 cells and PMH were mock (PBS) infected or infected with rAdGFP or rAdHBV. For
Huh7 cells: (a) cytoplasmic and total cellular fractions (30 lg) were analyzed for total cyclin D1; (b), total cellular fractions (30 lg) were analyzed for
total cyclin A and total and nuclear fractions for cyclin B1; (c), total cyclin D1, cyclin A and cyclin B1 levels from total cellular fractions and nuclear
cyclin B1 of rAdHBV-infected cells were expressed relative to mock controls: cyclin D1, 28% (±SD 18) (P = 0.05); cyclin A, 53% (±SD 30) (P = 0.05)
and cyclin B1, 110% (±SD 26) (P = 0.013). For PMH: (d) total cellular fractions (30 lg) were analyzed for total cyclin D1 and cyclin A. (e) Nuclear and
cytoplasmic fractions (30 lg) were analyzed for Cyclin B1; (f), total cyclin B1 levels from rAdHBV-infected PMH were expressed relative to mock
controls: cytoplasmic fraction, �29% (±SD 6) (P = 0.0015) and nuclear fraction, 419% (±SD 138) (P = 0.0015). Experiments were performed in
triplicate.
332 R. Chin et al. / Journal of Hepatology 47 (2007) 325–337
by 29% (±SD 6) (P = 0.0015) in PMH (Fig. 7e) andHuh7 cells (data not shown) compared to mock controls(Fig. 7e and f). In addition to the increase in cyclin B1
we also observed an increase in cdc2 levels in rAdHBV-infected (69% ±SD 5) compared to mock-infectedcells (P < 0.0001) (Fig. 7b). There were no significant

Fig. 8. HBV infected Huh7 cells have increased progression to G2 phase. Flow cytometry histograms of DNA content of mock (PBS)-, rAdGFP- and
rAdHBV-infected Huh7 cells. Total DNA content from triplicate experiments was determined by propidium iodide staining at days 1 (a), 2 (b) and 3 (c)
PI, and the % of cells in G1, S and G2 phases of the cell cycle was determined using the FlowJo software program. Experiments were performed in
triplicate and data presented include means and SD.
Fig. 9. Cell proliferation is not increased in rAdHBV-infected cells. Huh
7 cells were infected with either rAdGFP or rAdHBV and cellular
proliferation determined over a 90 h time course by colorimetric assay.
Proliferation of AdHBV-infected cells was not significantly different to
rAdGFP or mock-infected cells.
R. Chin et al. / Journal of Hepatology 47 (2007) 325–337 333
differences in cyclin D1, A or B1 observed in rAdGFP-infected cells compared to mock controls.
3.6. HBV stimulates cell cycle progression to G2 phase
The effect of active viral replication on total cellularDNA synthesis in rAdHBV-, rAdGFP- and PBS(mock)-infected Huh7 cells was determined at days 1,2 and 3 PI (Fig. 8). The proportion of rAdHBV-infectedcells in G1 and S phases over days 1–3 was not signifi-cantly different from mock or rAdGFP-infected con-trols. However, the proportion of cells in G2 phase atday 3 was significantly increased in rAdHBV-infectedHuh7 cells, 32% (±SD 1.0) compared to mock controls,(8% ±SD 5.0, P < 0.03) and rAdGFP infected cells,(15% ±SD 1.0) (Fig. 8c), indicating that HBV replica-tion stimulated initiation of cell DNA synthesis. How-ever, despite this cellular proliferation was notsignificantly increased (Fig. 9) and the sub-G1 cell pop-ulation indicative of pyknotic nuclei characteristic of

Fig. 10. HBV infected cells have reduced apoptosis. Huh 7 cells were
infected with either rAdGFP or rAdHBV and the proportion of apoptotic
cells determined by propidium iodide staining at days 1 and 3 PI and
analyzing sub-G1 cells by flow cytometry. Experiments were performed
in duplicate and results are expressed as means and SD of the fold
reduction in apoptosis relative to mock (uninfected cells).
334 R. Chin et al. / Journal of Hepatology 47 (2007) 325–337
apoptosis was reduced (Fig. 10) in HBV infected cellscompared to rAdGFP and mock infected cells. Togetherwith the increase in the levels of p21cip1 (Fig. 5b), cdc2(Fig. 7b) and intranuclear cyclin B1 (Fig. 7b and e) thesefindings are compatible with G2 arrestin rAdHBV-infected cells.
4. Discussion
Dysregulation of the signal transduction pathwayssuch as Ras-MAPK signaling, IRS1/IGF pathways,NFkB, cell cycle, Wnt/b-catenin and apoptotic path-ways have all been implicated in the development ofHBV-associated carcinogenesis [7,25–30]. However, itis difficult to derive a unifying theme to explain the pre-cise mechanism(s) linking the initiating oncogenic stim-ulus with subsequent changes in signal transduction.We sought to examine how HBV infection, rather thanthe expression of a single viral protein, affected the cellcycle and also influenced related pathways in an inte-grated manner to trigger a succession of cellular eventsthat might promote oncogenesis. To achieve this we uti-lized the recombinant adenovirus system to deliver areplication competent HBV genome into primary mar-moset hepatocytes to approximate intracellular eventsthat follow natural infection and contrasted these find-ings with that of the human hepatoma cell line, Huh7.We demonstrated that PMH supported HBV replicationin vitro, including the production of HBV cccDNA, andthat HBV-replicating Huh7 cells and PMH expressedhigher levels of pERK, pAkt, p-c-myc, p21cip1, p53, cyc-lins D1, A and B1 (Fig. 11). Furthermore, in HBV-rep-licating Huh7 cells were arrested in G2 phase (Fig. 8).These effects were not mediated by the adenoviral
expression system used to deliver the HBV genome asinfection with rAdenoGFP did not significantly alterany of the above cellular parameters, consistent withthe findings of others who have utilized recombinantadenoviral delivery systems to study the effects of viralproteins on cell signaling events [22,31,32] and cell cycleregulatory proteins including p21cip1, p27kip1, cyclins orthe activity of cyclin dependent kinases [33]. AlthoughHuh7 cells are a transformed cell line the pattern ofchanges in MAPK and Akt signaling and cell cycle reg-ulatory proteins was remarkably similar to that weobserved in HBV infected PMH, suggesting that Huh7cells are a suitable cell line for further studies of themechanisms of HBV associated hepatocarcinogenesisand apoptosis.
Previous studies, mainly utilizing the isolated expres-sion of HBx protein in cell culture systems and the HBxgene in transgenic mice, have been invaluable in deter-mining which signaling pathways may be involved inthe multi-step carcinogenic process. However, numerousdiscrepancies have been reported in these studies and theuse of neoplastic cell lines to study oncogenic mecha-nisms also seems counter-intuitive. We were able toovercome this by using primary primate-derivedhepatocytes.
Akt is a serine/threonine kinase whose principal func-tion is to facilitate growth-mediated signal transductionpathways involved in cell cycle and is involved in exten-sive cross-talk between several signaling pathways [34](Fig. 11). The PI(3)K/Akt signaling pathway may in facthave a central role in driving hepatocyte transformation[35]. We found, in both Huh7 cells and PMH infectedwith HBV, that phosphorylated Akt levels were signifi-cantly increased. Also, of the numerous targets thatAkt acts upon, we examined the effects on GSK3-ßphosphorylation and p21cip1. Consistent with the upreg-ulation of pAkt and pERK, HBV-infected cells demon-strated a significant increase in p-Ser9-GSK3-ß (theinactive form of GSK3-ß).
GSK3-ß is constitutively active in resting cells and isa key enzyme responsible for the regulation and turn-over of cyclin D1 [24,36]. In HBV-infected cells, there-fore, the Akt induced inhibition of GSK3-ß would beexpected to increase cyclin D1 levels. Furthermore, thepresence of increased levels of inactive GSK3-ß (i.e., p-Ser9-GSK3-ß) would also be expected to prevent thedegradation of c-myc [23,24]. The increased p-c-myclevels in HBV-replicating cells may reflect not only Ras-ERK-mediated stimulation but also decreased degrada-tion resulting from the inactivation of GSK3-ß (Fig. 11).
In both HBV infected Huh7 cells and PMH we foundelevations in both p53 and its downstream substratep21cip1 levels. p21cip1 has an important role in mediatingcell cycle arrest at the G1/S and G2/M boundaries ofcell cycle by both p53 dependent [37,38] and indepen-dent [33,39] mechanisms. It is absent in cells during S

Fig. 11. Simplified schematic representation of the effect of HBV replication on the Ras and PI3K/Akt pathways. The diagram shows how integration of
multiple cellular networks might contribute to cell growth and survival. Arrowheads in black (fi) indicate either pathway activation or increase/decreased
protein expression. The symbol (a) indicates pathway inhibition. [This figure appears in colour on the web.]
R. Chin et al. / Journal of Hepatology 47 (2007) 325–337 335
phase [38] but in G2 phase the intranuclear accumula-tion of cyclins A and B1 is accompanied by the de-novosynthesis and accumulation of p21cip1[39]. The accumu-lation of p21cip1 in G2 is ERK dependent and serves todelay cell cycle progression to mitosis in order to protectcells against DNA damaging agents and apoptosis[33,39,40]. The increased p53, p21cip1, cdc2 levels andintranuclear accumulation of cyclin B1 observed inHuh7 cells are all consistent with G2 arrest. This wasfurther supported by the cell cycle analysis of infectedHuh7 cells which showed that cells infected with HBVdid not progress beyond G2 phase (Fig. 9). In contrastto Huh7 cells PMH have a limited capacity to divide.Consequently, comparative cell cycle analyses withHBV infected PMH as G2 arrest in these primary hepa-tocytes may have represented little more than the inabil-ity of these cells to readily enter mitosis rather than aneffect of HBV infection. Nevertheless, like HBV infectedHuh7 cells infected PMH revealed changes consistent
with G2 arrest, namely increased p53, p21cip1, cdc2 lev-els and intranuclear cyclin B1. The ability of HBV toproduce G2 arrest may provide the virus with a survivaladvantage since viral replication is increased in both G1and G2 phases of cell cycle and reduced during S phase[41]. Interestingly, HBx protein has also been shown toincrease the levels of p21cip1 in hepatocytes and therebyreduce cell cycle progression [42]. However, the role ofHBx on p21cip1 expression in the context of HBV repli-cation is not clear. We have therefore generated arecombinant adenovirus encompassing an infectiousclone of HBV that harbors an X-gene deletion (datanot shown) in order to clarify the role of HBx onERK and Akt signaling pathways and cell cycleprogression.
Although dysregulated cell signaling events are a fea-ture of HBV-infected cells and provide a catalyst formalignant transformation, the precise mechanisms arelikely to be far more complex than our findings alone

336 R. Chin et al. / Journal of Hepatology 47 (2007) 325–337
would suggest. Defining the role of HBV replication inhepatocarcinogenesis will be essential for the develop-ment of effective therapeutic strategies.
Acknowledgements
The AdEasy system was generously provided by BVogelstein (Howard Hughes Medical Institute, Balti-more). We thank Dr Scott Bowden and Ms KathyJackson, Victorian Infectious Diseases Reference Labo-ratory, North Melbourne, Victoria, Australia, for kindlyperforming quantitative real time PCR analyses forHBV cccDNA. This work was supported by grants ofthe Deutsche Krebshilfe, ‘‘Dr. Mildred-Scheel-Stiftungfuer Krebsforschung’’, Grant No. 10-2142-Bo1.
References
[1] Yuen MF, Yuan HJ, Wong DK, Yuen JC, Wong WM, Chan AO,et al. Prognostic determinants for chronic hepatitis B in Asians:therapeutic implications. Gut 2005;54:1610–1614.
[2] Lampertico P, Vigano M, Manenti E, Iavarone M, Lunghi G,Colombo M. Five years of sequential LAM to LAM+ADVtherapy suppresses HBV replication in most HBEAG-negativecirrhotics, preventing decompensation but not hepatocellularcarcinoma. Hepatology 2005;42:983A.
[3] Chen C-J, Yang H-I, Jun S, Jen C-L, You S-L, Lu S-N, et al.Risk of hepatocellular carcinoma across a biological gradient ofserum hepatitis B virus DNA level. JAMA 2006;295:65–73.
[4] Pahl HL. Activators and target genes of Rel/NF-kappaBtranscription factors. Oncogene 1999;18:6853–6866.
[5] Feitelson MA. Parallel epigenetic and genetic changes in thepathogenesis of hepatitis virus-associated hepatocellular carci-noma. Cancer Lett 2005.
[6] Bouchard MJ, Wang L, Schneider RJ. Activation of focaladhesion kinase by hepatitis B virus HBx protein: multiplefunctions in viral replication. J Virol 2006;80:4406–4414.
[7] Branda M, Wands J. Signal transduction cascades and hepatitis Band C related hepatocellular carcinoma. Hepatology2006;43:891–902.
[8] Elmore LW, Hancock AR, Chang SF, Wang XW, Chang S,Callahan CP, et al. Hepatitis B virus X protein and p53 tumorsuppressor interactions in the modulation of apoptosis. Proc NatlAcad Sci USA 1997;94:14707–14712.
[9] Park US, Park SK, Lee YI, Park JG. Hepatitis B virus-X proteinupregulates the expression of p21waf1/cip1 and prolongs G1 fi Stransition via a p53-independent pathway in human hepatomacells. Oncogene 2000;19:3384–3394.
[10] Pan J, Duan LX, Sun BS, Feitelson MA. Hepatitis B virus Xprotein protects against anti-Fas-mediated apoptosis in humanliver cells by inducing NF-kappa B. J Gen Virol 2001;82:171–182.
[11] Su F, Schneider RJ. Hepatitis B virus HBx protein sensitizes cellsto apoptotic killing by tumor necrosis factor alpha. Proc NatlAcad Sci USA 1997;94:8744–8749.
[12] Terradillos O, Billet O, Renard CA, Levy R, Molina T, Briand P,et al. The hepatitis B virus X gene potentiates c-myc-induced liveroncogenesis in transgenic mice. Oncogene 1997;14:395–404.
[13] Kim YC, Song KS, Yoon G, Nam MJ, Ryu WS. Activated rasoncogene collaborates with HBx gene of hepatitis B virus totransform cells by suppressing HBx-mediated apoptosis. Onco-gene 2001;20:16–23.
[14] Mahe Y, Mukaida N, Kuno K, Akiyama M, Ikeda N, Matsu-shima K, et al. Hepatitis B virus X protein transactivates human
interleukin-8 gene through acting on nuclear factor kB andCCAAT/enhancer-binding protein-like cis-elements. J Biol Chem1991;266:13759–13763.
[15] Su F, Theodosis CN, Schneider RJ. Role of NF-kappaB and mycproteins in apoptosis induced by hepatitis B virus HBx protein. JVirol 2001;75:215–225.
[16] Park SG, Lee T, Kang HY, Park K, Cho KH, Jung G. Theinfluence of the signal dynamics of activated form of IKK on NF-kappaB and anti-apoptotic gene expressions: a systems biologyapproach. FEBS Lett 2006;580:822–830.
[17] Bock CT, Tillmann HL, Maschek HJ, Manns MP. A PreSmutation isolated from a patient with chronic hepatitis B infectionleads to virus retention and misassembly. Gastroenterology1997;113:1976–1982.
[18] Hsieh YH, Su IJ, Wang HC, Chang WW, Lei HY, Lai MD, et al.Pre-S mutant surface antigens in chronic hepatitis B virusinfection induce oxidative stress and DNA damage. Carcinogen-esis 2004;25:2023–2032.
[19] Premkumar A, Dong X, Haqshenas G, Gage PW, Gowans EJ.Amantadine inhibits the function of an ion channel encoded byGB virus-B but fails to inhibit virus replication. Antivir Ther2006;11:289–295.
[20] Chin R, Shaw T, Torresi J, Sozzi V, Trautwein C, Bock T, et al.In vitro susceptibilities of wild-type or drug-resistant hepatitis Bvirus to (�)-beta-D-2,6-diaminopurine dioxolane and 2 0-fluoro-5-methyl-beta-L-arabinofuranosyluracil. Antivir Chem Chemother2001;45:2495–2501.
[21] Bowden S, Jackson K, Littlejohn M, Locarnini S. Quantificationof HBV covalently closed circular DNA from liver tissue by real-time PCR. Methods Mol Med 2004;95:41–50.
[22] Siavoshian S, Abraham JD, Kieny MP, Schuster C. HCV core,NS3, NS5A and NS5B proteins modulate cell proliferationindependently from p53 expression in hepatocarcinoma cell lines.Arch Virol 2004;149:323–336.
[23] Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR.Multiple Ras-dependent phosphorylation pathways regulate mycprotein stability. Genes Dev 2000;14:2501–2514.
[24] Massague J. G1 cell-cycle control and cancer. Nature2004;432:298–306.
[25] Benn J, Schneider RJ. Hepatitis B virus HBx protein deregulatescell cycle checkpoint controls. Proc Natl Acad Sci USA1995;92:11215–11219.
[26] Benn J, Su F, Doria M, Schneider RJ. Hepatitis B virus HBxprotein induces transcription factor AP-1 by activation ofextracellular signal-regulated and c-Jun N-terminal mitogen-activated protein kinases. J Virol 1996;70:4978–4985.
[27] Tarn C, Lee S, Hu Y, Ashendel C, Andrisani OM.Hepatitis B virus X protein differentially activates RAS-RAF-MAPK and JNK pathways in X-transforming versusnon-transforming AML12 hepatocytes. J Biol Chem2001;276:34671–34680.
[28] Han HJ, Jung EY, Lee WJ, Jang KL. Cooperative repression ofcyclin-dependent kinase inhibitor p21 gene expression by hepatitisB virus X protein and hepatitis C virus core protein. FEBS Lett2002;518:169–172.
[29] Cha MY, Kim CM, Park YM, Ryu WS. Hepatitis B virus Xprotein is essential for the activation of Wnt/beta-catenin signal-ing in hepatoma cells. Hepatology 2004;39:1683–1693.
[30] Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKbetacouples hepatocyte death to cytokine-driven compensatory pro-liferation that promotes chemical hepatocarcinogenesis. Cell2005;121:977–990.
[31] Qadri I, Iwahashi M, Capasso JM, Hopken MW, Flores S,Schaack J, et al. Induced oxidative stress and activated expressionof manganese superoxide dismutase during hepatitis C virusreplication: role of JNK, p38 MAPK and AP-1. Biochem J2004;378:919–928.

R. Chin et al. / Journal of Hepatology 47 (2007) 325–337 337
[32] Bataller R, Paik YH, Lindquist JN, Lemasters JJ, Brenner DA.Hepatitis C virus core and nonstructural proteins induce fibrogeniceffects in hepatic stellate cells. Gastroenterology 2004;126:529–540.
[33] Niculescu AB, Chen X, Smeets M, Hengst L, Prives C, Reed SI.Effects of p21Cip1/Waf1 at both the G1/S and the G2/M cell cycletransitions: pRb is a critical determinant in blocking DNAreplication and in preventing endoreduplication. Mol Cell Biol1998;18:629–643.
[34] Scheid MP, Woodgett JR. PKB/AKT: functional insights fromgenetic models. Nat Rev Mol Cell Biol 2001;2:760–768.
[35] Coutant A, Rescan C, Gilot D, Loyer P, Guguen-Guillouzo C,Baffet G. PI3K-FRAP/mTOR pathway is critical for hepatocyteproliferation whereas MEK/ERK supports both proliferation andsurvival. Hepatology 2002;36:1079–1088.
[36] Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthasekinase-3beta regulates cyclin D1 proteolysis and subcellularlocalization. Genes Dev 1998;12:3499–3511.
[37] Bates S, Ryan KM, Phillips AC, Vousden KH. Cell cycle arrestand DNA endoreduplication following p21Waf1/Cip1 expression.Oncogene 1998;17:1691–1703.
[38] Dulic V, Stein GH, Farahi Far D, Reed SI. Nuclear accumulationof p21Cip1 at the onset of a role at the G2/M-phase transition.Mol Cell Biol 1998;18:546–557.
[39] Dangi S, Chen FM, Shapiro P. Activation of extracellular signal-regulated kinase (ERK) in G2 phase delays mitotic entry throughp21cip1. Cell Prolif 2006;39:261–279.
[40] Deacon K, Mistry P, Chernoff J, Blank JL, Patel R. p38mitogen-activated protein kinase mediates cell death and p21-activated kinase mediates cell survival during chemotherapeu-tic drug-induced mitotic arrest. Mol Biol Cell2003;14:2071–2087.
[41] Ozer A, Khaoustov VI, Mearns M, Lewis DE, Genta RM,Darlington GJ, et al. Effect of hepatocyte proliferation andcellular DNA synthesis on hepatitis B virus replication. Gastro-enterology 1996;110:1519–1528.
[42] Qiao L, Leach K, McKinstry R, Gilfor D, Yacoub A, Park JS,et al. Hepatitis B virus X protein increases expression of p21(Cip-1/WAF1/MDA6) and p27(Kip-1) in primary mouse hepatocytes,leading to reduced cell cycle progression. Hepatology2001;34:906–917.
Copyright © 2022 FDOKUMEN




















