
C-TAK1 Regulates Ras Signaling by Phosphorylating the MAPK Scaffold, KSR1
11
Molecular Cell, Vol. 8, 983–993, November, 2001, Copyright 2001 by Cell Press C-TAK1 Regulates Ras Signaling by Phosphorylating the MAPK Scaffold, KSR1 half of KSR1 contains a putative kinase-like domain. Based on this finding together with genetic epistasis experiments, it was initially predicted that KSR1 might be a protein kinase that acts upstream of, or in parallel to, Raf-1 (Therrien et al., 1995). Although some reports Ju ¨ rgen Mu ¨ ller, 1 Ste ´ phane Ory, 1 Terry Copeland, 1 Helen Piwnica-Worms, 2 and Deborah K. Morrison 1,3 1 Regulation of Cell Growth Laboratory Center for Cancer Research NCI-Frederick Frederick, Maryland 21702 have suggested that mammalian KSR1 can phosphory- 2 Howard Hughes Medical Institute and late Raf-1 (Zhang et al., 1997b; Xing and Kolesnick, Department of Cell Biology and Physiology 2001), several observations are inconsistent with the Washington University School of Medicine idea that KSR1 has intrinsic kinase activity. First, all St. Louis, Missouri 63110 mammalian KSR1 proteins lack a critical lysine residue in the catalytic domain that is normally required for the phosphotransfer reaction (Therrien et al., 1995; Mu ¨ ller et al., 2000). Second, mutagenesis of residues predicted Summary to be important for kinase activity does not impair the function of C. elegans KSR (Stewart et al., 1999). Third, Kinase suppressor of Ras (KSR) is a conserved com- expression of the isolated kinase-like domain of KSR1 ponent of the Ras pathway that interacts directly with results in dominant inhibition of Ras signaling rather than MEK and MAPK. Here we show that KSR1 translocates constitutive activation of the pathway, as was initially from the cytoplasm to the cell surface in response predicted based on the model that KSR1 is a kinase to growth factor treatment and that this process is required for Ras signaling (Therrien et al., 1996; Yu et regulated by Cdc25C-associated kinase 1 (C-TAK1). al., 1997; Joneson et al., 1998). Significantly, KSR1 has C-TAK1 constitutively associates with mammalian been found to interact with proteins that do possess KSR1 and phosphorylates serine 392 to confer 14-3-3 kinase activity, including Raf-1, MEK, and MAPK (Ther- binding and cytoplasmic sequestration of KSR1 in un- stimulated cells. In response to signal activation, the rien et al., 1996; Denouel-Galy et al., 1997; Xing et al., phosphorylation state of S392 is reduced, allowing the 1997; Yu et al., 1997). Both MEK and MAPK associate KSR1 complex to colocalize with activated Ras and directly with KSR1, while the interaction with Raf-1 ap- Raf-1 at the plasma membrane, thereby facilitating the pears to be indirect. MEK binding is mediated by the phosphorylation reactions required for the activation kinase-like domain and is required for both the positive of MEK and MAPK. function of full-length KSR1 and the dominant inhibitory activity of the isolated kinase-like domain (Stewart et al., Introduction 1999; Mu ¨ ller et al., 2000). Taken together, these findings suggest that KSR1 itself lacks enzymatic activity and instead serves as a docking platform for the authentic The Ras GTPase is a critical regulator of cellular prolifer- kinase components of the Ras/MAPK cascade. ation and differentiation in multicellular organisms. A To further elucidate the putative MAPK scaffolding major route by which Ras transmits many cellular signals is through the sequential activation of the cytoplasmic function of KSR1, we sought to identify proteins that serine/threonine kinases Raf-1, MEK, and MAPK (Mar- are associated with KSR1 and might therefore regulate shall, 1996; Wittinghofer, 1998; Shields et al., 2000). De- the biological activity of KSR1 or its associated MAPK spite the fact that the primary components of this path- components. Here, we report that Cdc25C-associated way, their functional role, and their epistatic relationship kinase 1 (C-TAK1) is a new member of the KSR1 com- to one another have been characterized in considerable plex. C-TAK1 constitutively associates with the N termi- detail, there are still several aspects of Ras signal trans- nus of KSR1 and is responsible for the phosphorylation mission that are not fully understood. For example, it of KSR1 at serine 392 (S392), a site that mediates binding has been well established that localization of Ras and to 14-3-3 proteins. We show that KSR1 is a cytoplasmic Raf-1 to the plasma membrane is essential for pathway protein that rapidly translocates to the plasma mem- activation under normal conditions (Leevers et al., 1994; brane in response to growth factor treatment, thereby Magee and Marshall, 1999; Bar-Sagi, 2001), but it is relocalizing associated MEK proteins to the membrane unclear whether MEK and MAPK are also recruited to and promoting MEK activation by Raf-1. Strikingly, mu- the membrane and if so, what regulatory mechanisms tation of S392 and disruption of 14-3-3 binding to this might control these events. In this respect, several ge- site results in the constitutive localization of KSR1 at netically identified components of the Ras pathway, in- the plasma membrane and enhanced Ras/MAPK pathway cluding kinase suppressor of Ras (KSR), might partici- activity, suggesting that phosphorylation of S392 by pate in such events, although their exact roles in Ras C-TAK1 functions to retain KSR1 in the cytosol of quies- signaling have not yet been fully elucidated. cent cells. These findings identify C-TAK1 as a regulator Determining the function of KSR1 has been particularly of the MAPK scaffolding function of KSR1 and under- problematic, due largely to the fact that the C-terminal score the emerging significance of regulated intracellu- lar scaffolds that promote the assembly of multiprotein signaling complexes. 3 Correspondence: [email protected]
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of C-TAK1 Regulates Ras Signaling by Phosphorylating the MAPK Scaffold, KSR1

Molecular Cell, Vol. 8, 983–993, November, 2001, Copyright 2001 by Cell Press
C-TAK1 Regulates Ras Signalingby Phosphorylatingthe MAPK Scaffold, KSR1
half of KSR1 contains a putative kinase-like domain.Based on this finding together with genetic epistasisexperiments, it was initially predicted that KSR1 mightbe a protein kinase that acts upstream of, or in parallelto, Raf-1 (Therrien et al., 1995). Although some reports
Jurgen Muller,1 Stephane Ory,1 Terry Copeland,1
Helen Piwnica-Worms,2 and Deborah K. Morrison1,3
1 Regulation of Cell Growth LaboratoryCenter for Cancer ResearchNCI-FrederickFrederick, Maryland 21702 have suggested that mammalian KSR1 can phosphory-2 Howard Hughes Medical Institute and late Raf-1 (Zhang et al., 1997b; Xing and Kolesnick,Department of Cell Biology and Physiology 2001), several observations are inconsistent with theWashington University School of Medicine idea that KSR1 has intrinsic kinase activity. First, allSt. Louis, Missouri 63110 mammalian KSR1 proteins lack a critical lysine residue
in the catalytic domain that is normally required for thephosphotransfer reaction (Therrien et al., 1995; Mulleret al., 2000). Second, mutagenesis of residues predictedSummaryto be important for kinase activity does not impair thefunction of C. elegans KSR (Stewart et al., 1999). Third,Kinase suppressor of Ras (KSR) is a conserved com-expression of the isolated kinase-like domain of KSR1ponent of the Ras pathway that interacts directly withresults in dominant inhibition of Ras signaling rather thanMEK and MAPK. Here we show that KSR1 translocatesconstitutive activation of the pathway, as was initiallyfrom the cytoplasm to the cell surface in responsepredicted based on the model that KSR1 is a kinaseto growth factor treatment and that this process isrequired for Ras signaling (Therrien et al., 1996; Yu etregulated by Cdc25C-associated kinase 1 (C-TAK1).al., 1997; Joneson et al., 1998). Significantly, KSR1 hasC-TAK1 constitutively associates with mammalianbeen found to interact with proteins that do possessKSR1 and phosphorylates serine 392 to confer 14-3-3kinase activity, including Raf-1, MEK, and MAPK (Ther-binding and cytoplasmic sequestration of KSR1 in un-
stimulated cells. In response to signal activation, the rien et al., 1996; Denouel-Galy et al., 1997; Xing et al.,phosphorylation state of S392 is reduced, allowing the 1997; Yu et al., 1997). Both MEK and MAPK associateKSR1 complex to colocalize with activated Ras and directly with KSR1, while the interaction with Raf-1 ap-Raf-1 at the plasma membrane, thereby facilitating the pears to be indirect. MEK binding is mediated by thephosphorylation reactions required for the activation kinase-like domain and is required for both the positiveof MEK and MAPK. function of full-length KSR1 and the dominant inhibitory
activity of the isolated kinase-like domain (Stewart et al.,Introduction 1999; Muller et al., 2000). Taken together, these findings
suggest that KSR1 itself lacks enzymatic activity andinstead serves as a docking platform for the authenticThe Ras GTPase is a critical regulator of cellular prolifer-kinase components of the Ras/MAPK cascade.ation and differentiation in multicellular organisms. A
To further elucidate the putative MAPK scaffoldingmajor route by which Ras transmits many cellular signalsis through the sequential activation of the cytoplasmic function of KSR1, we sought to identify proteins thatserine/threonine kinases Raf-1, MEK, and MAPK (Mar- are associated with KSR1 and might therefore regulateshall, 1996; Wittinghofer, 1998; Shields et al., 2000). De- the biological activity of KSR1 or its associated MAPKspite the fact that the primary components of this path- components. Here, we report that Cdc25C-associatedway, their functional role, and their epistatic relationship kinase 1 (C-TAK1) is a new member of the KSR1 com-to one another have been characterized in considerable plex. C-TAK1 constitutively associates with the N termi-detail, there are still several aspects of Ras signal trans- nus of KSR1 and is responsible for the phosphorylationmission that are not fully understood. For example, it of KSR1 at serine 392 (S392), a site that mediates bindinghas been well established that localization of Ras and to 14-3-3 proteins. We show that KSR1 is a cytoplasmicRaf-1 to the plasma membrane is essential for pathway protein that rapidly translocates to the plasma mem-activation under normal conditions (Leevers et al., 1994; brane in response to growth factor treatment, therebyMagee and Marshall, 1999; Bar-Sagi, 2001), but it is relocalizing associated MEK proteins to the membraneunclear whether MEK and MAPK are also recruited to and promoting MEK activation by Raf-1. Strikingly, mu-the membrane and if so, what regulatory mechanisms tation of S392 and disruption of 14-3-3 binding to thismight control these events. In this respect, several ge- site results in the constitutive localization of KSR1 atnetically identified components of the Ras pathway, in- the plasma membrane and enhanced Ras/MAPK pathwaycluding kinase suppressor of Ras (KSR), might partici- activity, suggesting that phosphorylation of S392 bypate in such events, although their exact roles in Ras C-TAK1 functions to retain KSR1 in the cytosol of quies-signaling have not yet been fully elucidated. cent cells. These findings identify C-TAK1 as a regulator
Determining the function of KSR1 has been particularly of the MAPK scaffolding function of KSR1 and under-problematic, due largely to the fact that the C-terminal score the emerging significance of regulated intracellu-
lar scaffolds that promote the assembly of multiproteinsignaling complexes.3 Correspondence: [email protected]

Molecular Cell984
Figure 1. Detection of KSR1-Associated Kinases
(A) Cos cells expressing either WT KSR1 or the various KSR1 deletion mutants were left untreated (�) or treated for 5 min with EGF (�) priorto lysis. The KSR1 proteins were immunoprecipitated from the lysates using Pyo antibody, and immune complex kinase assays were performed.Labeled proteins were separated by SDS-PAGE, transferred to nitrocellulose, and visualized by autoradiography (left panel). The membranewas then probed with Pyo antibody to verify the expression of the transfected proteins (right panel) and reprobed with antibodies againstMAPK and MEK (lower left panels). WT KSR1 and the deletion mutants are schematically depicted.(B) KSR1 immunoprecipitates were prepared as in (A), and immune complex kinase assays were performed with the addition of purified Raf-1.The membrane was probed with a Raf-1 antibody to verify that equal amounts of Raf-1 were present in the reactions and reprobed with anantibody recognizing phosphorylated and activated MEK (P-MEK).
Results consistent with other studies that have failed to demon-strate any catalytic activity for this protein (Muller et al.,2000).Detection of a Protein Kinase that Phosphorylates
the N-Terminal Domain of KSR1 Next, to determine if the phosphorylation of the KSR1proteins could be attributed to MEK or MAPK, we re-To investigate whether other protein kinases in addition
to MEK and MAPK directly associate with KSR1, we probed the KSR1 immunoprecipitates for the presenceof these known KSR1-interacting kinases (Figure 1A). Inperformed immune complex kinase assays using full-
length wild-type (WT) KSR1 and various KSR1 deletion agreement with previous studies (Cacace et al., 1999;Muller et al., 2000), MEK constitutively associated withmutants. KSR1 proteins were immunoprecipitated from
untreated or EGF-treated Cos cells that had been lysed WT KSR1 and the C-terminal C�542 protein, while MAPKinteracted with WT KSR1 and the N-terminal N�539 pro-in a buffer containing 1% NP-40. The KSR1 immune
complexes were then washed extensively with NP-40 tein in an EGF-inducible manner. Neither MEK norMAPK, however, was detected in the immunoprecipi-lysis buffer that contained 1 M NaCl, and incubated with
[�-32P]ATP to detect any associated kinases that might tates of the N�424 protein, implying that this mutantis phosphorylated by a previously unidentified KSR1-phosphorylate proteins present in the KSR1 complex.
As shown in Figure 1A, the proteins detectably phos- associated kinase.phorylated in these assays were WT KSR1 and the trun-cated N-terminal proteins, N�539 and N�424. Since the Raf-1 Phosphorylates and Activates
KSR1-Associated MEKN-terminal mutants lack the putative kinase domain, thephosphorylation of these proteins is presumably due to A recent study has reported that KSR1 isolated under
the conditions described above is capable of phosphor-the presence of associated protein kinases. Further-more, the phosphorylation of N�424 was not increased ylating Raf-1 on threonine 269 (Xing and Kolesnick,
2001). Previously, however, we were unable to detectby EGF treatment, in contrast to WT KSR1 and N�539,suggesting that KSR1 is a substrate for both inducible KSR1-dependent phosphorylation of Raf-1 when KSR1
was isolated under more stringent conditions using lysisand constitutive kinase activities. Interestingly, no phos-phorylated proteins were observed in the samples con- buffers that contain SDS (Michaud et al., 1997; Muller
et al., 2000). Furthermore, the results presented in Figuretaining the isolated KSR1 kinase-like domain (C�542),

Phosphorylation and Regulation of KSR1 by C-TAK1985
treated cells and contained one major phosphopeptideeluting in fraction 23 and one minor phosphopeptideeluting in fraction 7. These phosphopeptides were alsopresent in the full-length WT protein; however, WT KSR1contained additional phosphopeptides eluting in frac-tions 41 and 53 that were not observed in the N�424profiles. The phosphopeptides unique to WT KSR1 wereonly detected in EGF-treated cells and appear to repre-sent sites phosphorylated by kinases whose activity orKSR1 association is induced by EGF, such as that ob-served for MAPK.
To precisely map the residues of the N�424 proteinthat are phosphorylated, peptides isolated in fractions23 and 7 were subjected to phosphoamino acid analysisand N-terminal sequencing by Edman degradation (Fig-ure 2). From this analysis, we were able to determinethat the peptide contained in fraction 23 was phosphory-lated on serine 392 (S392), while the peptide found infraction 7 was phosphorylated on serine 297 (S297). Toverify the identification of these sites, N�424 proteinscontaining serine to alanine mutations at positions 297and 392 were phosphorylated in immune complex ki-nase assays and analyzed as described above. The re-sulting HPLC profiles revealed that the N�424 proteinFigure 2. The N�424-Associated Kinase Phosphorylates KSR1 atmutated at S392 lacked the major peptide eluting inS392fraction 23, while the protein mutated at S297 lackedKSR1 proteins labeled in immune complex kinase assays were di-the minor peak eluting in fraction 7 (Figure 2). Thus,gested with trypsin, and the tryptic phosphopeptides were sepa-
rated by HPLC. The profile of radioactivity collected in the HPLC S392 and, to a much lesser extent, S297 are the KSR1column fractions is shown for WT KSR1, N�424, and N�424 proteins residues phosphorylated by the N�424-associatedcontaining serine to alanine mutations at amino acid residues 297 kinase.(S297A) and 392 (S392A). The number under the Edman degredationcolumn indicates the cycle in which 32P counts were released. The
The C-TAK1 Protein Kinase Constitutively Associatesasterisk indicates the residue phosphorylated.
with the N Terminus of KSR1S392 and S297 have previously been identified as invivo sites of KSR1 phosphorylation that mediate the
1A indicate that KSR1 isolated according to Xing and interaction between KSR1 and 14-3-3 dimers (CacaceKolesnick (2001) is still complexed with several protein et al., 1999). Known protein kinases that have been re-kinases. Therefore, to determine whether KSR1 isolated ported to phosphorylate their substrates on residuesunder these less stringent conditions or any of the KSR1- generating 14-3-3 binding sites include Akt (Zimmer-associated kinases can phosphorylate Raf-1, we added mann and Moelling, 1999), C-TAK1 (Peng et al., 1998),purified Raf-1 to the immune complex kinase assays. and the checkpoint kinases Chk1 and Chk2/Cds1 (PengAs shown in Figure 1B, no increase in Raf-1 phosphory- et al., 1997; Sanchez et al., 1997; Zeng et al., 1998). Inlation was observed in any of the KSR1-containing sam- addition, members of the AGC kinase family such asples. In contrast, we found that MEK bound to either protein kinase C (PKC), protein kinase A (PKA), andWT KSR1 or the C�542 mutant was efficiently phosphor- p90Rsk have been found to phosphorylate substratesylated on its activating serine residues (S217/S221) by at sites contained within an RxxS sequence (Pearsonpurified Raf-1. Taken together, these results do not sup- and Kemp, 1991), which is the motif surrounding bothport the model that KSR1 is a Raf-1 kinase, and instead the S392 and S297 sites. When KSR1 immunoprecipi-indicate that KSR1 facilitates signal transmission be- tates were probed for the presence of these kinases,tween Raf-1 and MEK. C-TAK1 was detected in samples containing N�424 but
not in those containing C�542. Akt, Chk1, PKC, p90Rsk,and the catalytic subunit of PKA were not observed inIdentification of the KSR1 Residues Phosphorylated
by the N�424 KSR1-Associated Kinase any of the KSR1 samples (Figure 3A and data notshown). C-TAK1 also associated with WT KSR1, but notTo provide insight as to the identity of the kinase that
phosphorylates the N�424 protein in vitro, we initiated with the N�320 or N�249 proteins, and the binding ofC-TAK1 to either the WT or N�424 proteins was notexperiments to determine the residue(s) of KSR1 phos-
phorylated by this kinase. KSR1 proteins labeled as de- dependent on or induced by EGF treatment (Figures 3Band 3C). The biological relevance of the interaction wasscribed in Figure 1A were isolated from the gel matrix
and digested with trypsin. The resulting tryptic phospho- indicated by the coimmunoprecipitation of endogenousC-TAK1 and KSR1 in mouse brain (Figure 3C), a tissuepeptides were then separated using reversed phase
high-pressure liquid chromatography (HPLC). As de- expressing high levels of KSR1 protein and in whichendogenous complex formation among KSR1, MEK,picted in Figure 2, the HPLC profiles were identical for
the N�424 protein isolated from either untreated or EGF- MAPK, and 14-3-3 has been detected (Muller et al.,
Molecular Cell986
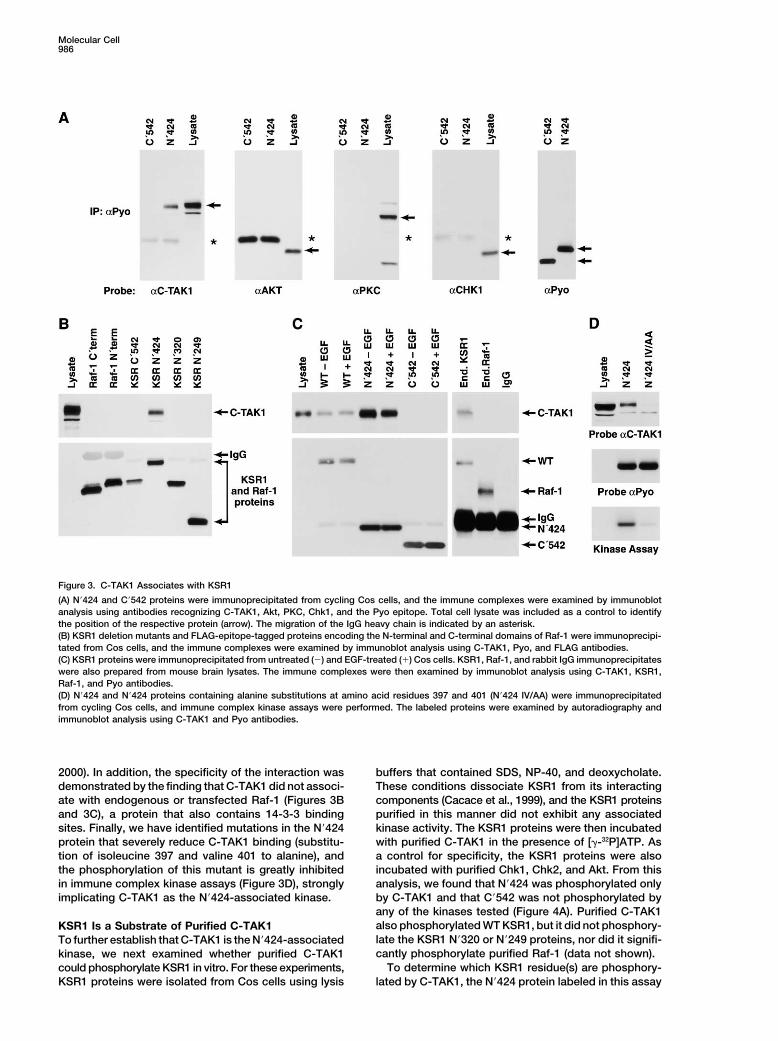
Figure 3. C-TAK1 Associates with KSR1
(A) N�424 and C�542 proteins were immunoprecipitated from cycling Cos cells, and the immune complexes were examined by immunoblotanalysis using antibodies recognizing C-TAK1, Akt, PKC, Chk1, and the Pyo epitope. Total cell lysate was included as a control to identifythe position of the respective protein (arrow). The migration of the IgG heavy chain is indicated by an asterisk.(B) KSR1 deletion mutants and FLAG-epitope-tagged proteins encoding the N-terminal and C-terminal domains of Raf-1 were immunoprecipi-tated from Cos cells, and the immune complexes were examined by immunoblot analysis using C-TAK1, Pyo, and FLAG antibodies.(C) KSR1 proteins were immunoprecipitated from untreated (�) and EGF-treated (�) Cos cells. KSR1, Raf-1, and rabbit IgG immunoprecipitateswere also prepared from mouse brain lysates. The immune complexes were then examined by immunoblot analysis using C-TAK1, KSR1,Raf-1, and Pyo antibodies.(D) N�424 and N�424 proteins containing alanine substitutions at amino acid residues 397 and 401 (N�424 IV/AA) were immunoprecipitatedfrom cycling Cos cells, and immune complex kinase assays were performed. The labeled proteins were examined by autoradiography andimmunoblot analysis using C-TAK1 and Pyo antibodies.
2000). In addition, the specificity of the interaction was buffers that contained SDS, NP-40, and deoxycholate.These conditions dissociate KSR1 from its interactingdemonstrated by the finding that C-TAK1 did not associ-
ate with endogenous or transfected Raf-1 (Figures 3B components (Cacace et al., 1999), and the KSR1 proteinspurified in this manner did not exhibit any associatedand 3C), a protein that also contains 14-3-3 binding
sites. Finally, we have identified mutations in the N�424 kinase activity. The KSR1 proteins were then incubatedwith purified C-TAK1 in the presence of [�-32P]ATP. Asprotein that severely reduce C-TAK1 binding (substitu-
tion of isoleucine 397 and valine 401 to alanine), and a control for specificity, the KSR1 proteins were alsoincubated with purified Chk1, Chk2, and Akt. From thisthe phosphorylation of this mutant is greatly inhibited
in immune complex kinase assays (Figure 3D), strongly analysis, we found that N�424 was phosphorylated onlyby C-TAK1 and that C�542 was not phosphorylated byimplicating C-TAK1 as the N�424-associated kinase.any of the kinases tested (Figure 4A). Purified C-TAK1also phosphorylated WT KSR1, but it did not phosphory-KSR1 Is a Substrate of Purified C-TAK1late the KSR1 N�320 or N�249 proteins, nor did it signifi-To further establish that C-TAK1 is the N�424-associatedcantly phosphorylate purified Raf-1 (data not shown).kinase, we next examined whether purified C-TAK1
To determine which KSR1 residue(s) are phosphory-could phosphorylate KSR1 in vitro. For these experiments,KSR1 proteins were isolated from Cos cells using lysis lated by C-TAK1, the N�424 protein labeled in this assay

Phosphorylation and Regulation of KSR1 by C-TAK1987
for N�424 phosphorylated in immune complex kinaseassays (Figure 2) and contained one major peptide elut-ing in fraction 23 and one minor peptide eluting in frac-tion 7. Sequencing of these peptides together with sub-sequent mutant analysis (data not shown) revealed thatC-TAK1 phosphorylates KSR1 primarily on S392, withweak phosphorylation being observed at S297. Basedon these findings and the results presented in Figure3, we conclude that C-TAK1 is the N�424-associatedkinase.
In Vivo Regulation of KSR1 Phosphorylation at S392To investigate whether the phosphorylation state of theKSR1 S297 or S392 sites is regulated in vivo, WT KSR1proteins were metabolically labeled with [32P]orthophos-phate, digested with trypsin, and examined by HPLCanalysis (Figure 4C). Consistent with previous reports(Cacace et al., 1999; Volle et al., 1999), we found thatS297 and S392 (eluting in fractions 7 and 23, respec-tively) were major sites of KSR1 phosphorylated in qui-escent cells. When cells were treated with EGF, thephosphorylation state of S297 remained unchanged,while the phosphorylation of S392 was significantly re-duced (Figure 4C). Although EGF treatment did inducethe phosphorylation of a peptide eluting in fraction 21,this fraction does not contain the S392 peptide, as thispeak was still observed in the HPLC profile of KSR1containing mutations at the S297 and S392 sites. Thus,these findings indicate that S392 is dephosphorylatedfollowing growth factor treatment. In the experimentsdescribed above, the in vitro kinase activity of C-TAK1and its association with KSR1 were not significantlyaffected by EGF treatment. Therefore, the dephosphory-lation of S392 in vivo does not appear to be the resultof C-TAK1 inactivation or dissociation and is likely tobe due to the activation of an unidentified phosphatasethat does not stably associate with the KSR1 complex.
Mutation of S392 Localizes the KSR1 Complexto the Plasma MembraneAs 14-3-3 binding has been shown to regulate the sub-cellular localization of the C-TAK1 substrate Cdc25C(Kumagai and Dunphy, 1999; Peng et al., 1997, 1998;Yang et al., 1999), we next examined the role of S392phosphorylation and, consequently, 14-3-3 binding with
Figure 4. In Vitro and In Vivo Phosphorylation of KSR1 on S392 respect to KSR1 subcellular localization. For these stud-(A) N�424 and C�542 KSR1 proteins were incubated with recombi- ies, full-length KSR1 proteins were generated in whichnant C-TAK1, Akt, Chk1, and Chk2 in the presence of [�-32P]ATP. S392 alone or S392 and S297 were mutated to alaninePhosphorylated KSR1 proteins were separated by SDS-PAGE,
residues (termed S392A and S297A/S392A mutants, re-transferred to nitrocellulose membranes, and quantitated using aspectively). The intracellular localization of WT KSR1phosphoimager. The activities of Chk1 and Chk2 were confirmed
using the CHKtide substrate, and the activity of Akt was demon- and the mutant KSR1 proteins was then examined instrated using the Crosstide substrate (data not shown). Cos cells by indirect immunofluorescence. As shown in(B) N�424 phosphorylated by C-TAK1 in vitro was digested with Figure 5A, WT KSR1 was localized in the cytoplasmtrypsin and examined by HPLC analysis. of quiescent cells, but rapidly translocated to the cell(C) Cos cells expressing WT or S297A/S392A KSR1 were labeled in
periphery in response to EGF treatment. Strikingly, wevivo with [32P]orthophosphate and either left untreated or werefound that significant amounts of the S392A proteinstreated with EGF prior to cell lysis. KSR1 proteins were immunopre-
cipitated, digested with trypsin, and examined by HPLC analysis. were constitutively localized to the plasma membraneThe values for the cpm incorporated into the S392 site are 12,461 even in the absence of growth factor stimulation. Thecpm from untreated cells and 6,017 cpm from EGF-treated cells. S297A/S392A mutant exhibited the same staining pat-
tern as the S392 protein, indicating that the dissociationof 14-3-3 from the S392 site had the same effect aswas subjected to trypsin digestion and HPLC analysis.
As shown in Figure 4B, the HPLC profile of N�424 phos- completely eliminating 14-3-3 binding by mutation ofboth the S297 and S392 sites. Similar results were ob-phorylated by C-TAK1 was identical to that observed

Molecular Cell988
and S392 must be disrupted to reduce 14-3-3 dimerbinding to KSR1. Equivalent levels of MEK were foundin all the KSR1 immunoprecipitates; however, increasedlevels of MAPK were observed in the S392A and S297A/S392A samples. In EGF-treated cells, the levels of MAPKassociating with the S392A and S297A/S392A mutantswere higher than those observed for WT KSR1. In addi-tion, even though MAPK was not observed in KSR1immunoprecipitates from untreated cells, low levels ofMAPK were detected in S392A and S297A/S392A sam-ples. Therefore, while the constitutive interaction be-tween KSR1 and MEK is not affected by mutation ofthe serine sites, the association of KSR1 with MAPK isenhanced, indicating that 14-3-3 binding and/or mem-brane localization may influence the accessibility of theFxFP MAPK binding site on KSR1.
KSR1 Proteins Mutated at S392 Have EnhancedBiological ActivityTo measure the biological activity of the S392A andS297A/S392A proteins, we used the meiotic maturationof Xenopus oocytes as an assay system (Therrien et al.,1996). In this system, expression of WT KSR1 cooper-ates with activated Ras to accelerate oocyte maturation,while expression of the isolated KSR1 kinase-like do-main blocks Ras signaling. When we examined the effectof the S392A or S297A/S392A proteins in this assay,we found that expression of these proteins alone was
Figure 5. Effect of S392 Mutation on KSR1 Subcellular Localization, insufficient to induce oocyte maturation; however, inComplex Formation, and Biological Activity comparison to WT KSR1, both mutant proteins demon-(A) Cos cells expressing WT, S392A, or S297A/S392A KSR1 were strated an enhanced ability to augment Ras-dependentleft untreated or were treated with EGF. The intracellular localization maturation (Figure 5C). In addition, while equivalentof the KSR1 proteins was then determined by indirect immunofluo-
amounts of the WT and mutant KSR1 proteins wererescence using Pyo antibody.expressed in the oocytes, increased levels of activated(B) KSR1 proteins were immunoprecipitated from untreated (�) orMAPK were detected in oocytes coexpressing Ras andEGF treated (�) Cos cells, and the immune complexes were exam-
ined by immunoblot analysis using Pyo, MEK, MAPK, and 14-3-3 either the S392A or S297A/S392A mutant (Figure 5C).antibodies. These findings suggest that by accelerating the activa-(C) Xenopus oocytes were injected with RNA encoding either the tion of MAPK, the KSR1 proteins defective in S392 phos-WT, S392A, or S297A/S392A KSR1 proteins and activated RasV12.
phorylation have an increased ability to augment RasGerminal vesicle breakdown (GVBD) was scored 5 hr following Rassignaling.injection. Oocyte lysates were prepared and examined by immunoblot
analysis using Pyo and phospho-MAPK antibodies (P-MAPK).
Colocalization of KSR1, MEK, and Activated MAPKThe findings presented above are consistent with C-TAK1acting as a negative regulator of the KSR1 scaffoldingfunction. To further investigate this idea, we examinedtained when the intracellular localization of the KSR1
proteins was examined in NIH/3T3 cells (data not the intracellular localization of MEK in Cos cells express-ing either WT, S392A, or S297A/S392A KSR1 proteinsshown). Thus, the dephosphorylation of the S392 site
correlates with the translocation of KSR1, and mutation (Figure 6A). Consistent with the constitutive associationbetween MEK and KSR1, MEK appeared to colocalizeof S392 constitutively localizes KSR1 to the plasma
membrane. with KSR1 in all cells examined. MEK was found in thecytoplasm of quiescent cells expressing WT KSR1, butTo further investigate the effect of S392 phosphoryla-
tion on KSR1 function, we compared the ability of the could be detected at the cell surface following EGFtreatment. Moreover, MEK was already localized to theWT, S392A, and S297A/S392A proteins to interact with
components of the KSR1 complex (Figure 5B). As ex- plasma membrane prior to growth factor addition in cellsexpressing either the S392A or S297A/S392A proteins.pected, 14-3-3 was present in the immunoprecipitates
of WT KSR1 and S392A, but was not detected in those Next, we examined the intracellular localization of acti-vated MAPK using antibodies that specifically recognizeof the S297A/S392A mutant, further confirming that
S297 and S392 mediate the binding of KSR1 to 14-3-3 the activated, phosphorylated form of MAPK (Figure 6B).Prior to EGF treatment, no specific staining could bedimers. The levels of 14-3-3 bound to S392A were not
significantly different than those bound to WT KSR1, detected in any of the KSR1-expressing cells (data notshown), indicating that although significant amounts ofand the amount of 14-3-3 bound to WT KSR1 did not
change in response to EGF treatment, consistent with the S392A and S297A/S392A proteins are localized atthe plasma membrane of resting cells, growth factorprevious findings that the interaction with both S297
Phosphorylation and Regulation of KSR1 by C-TAK1989
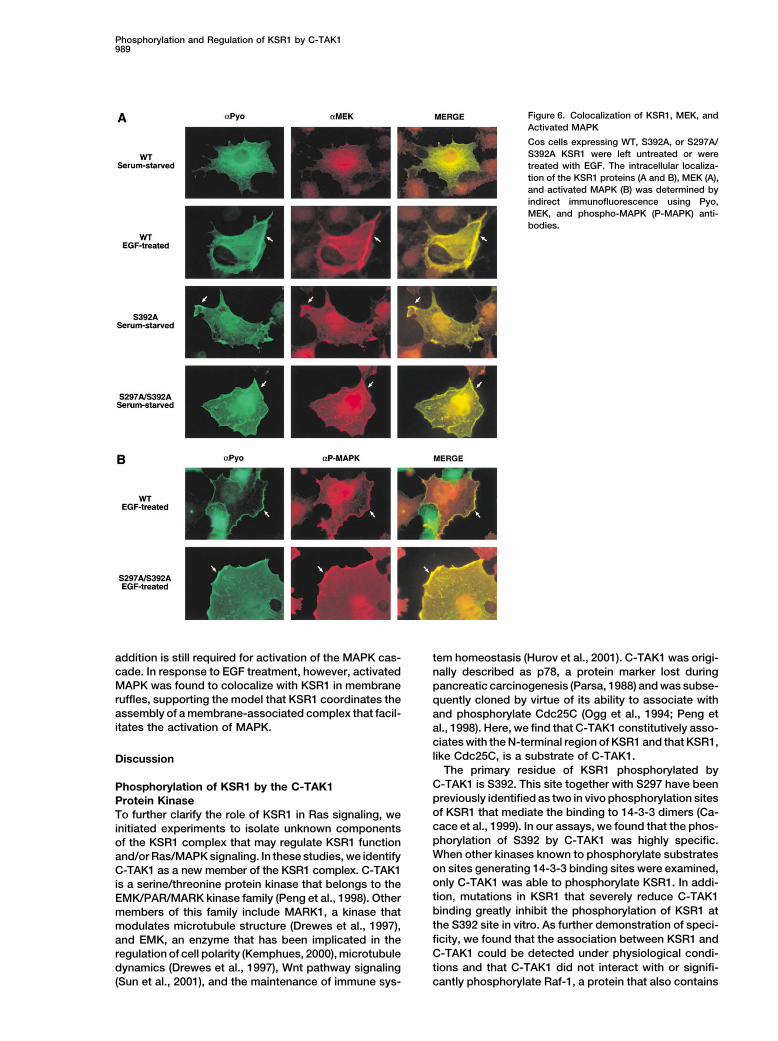
Figure 6. Colocalization of KSR1, MEK, andActivated MAPK
Cos cells expressing WT, S392A, or S297A/S392A KSR1 were left untreated or weretreated with EGF. The intracellular localiza-tion of the KSR1 proteins (A and B), MEK (A),and activated MAPK (B) was determined byindirect immunofluorescence using Pyo,MEK, and phospho-MAPK (P-MAPK) anti-bodies.
addition is still required for activation of the MAPK cas- tem homeostasis (Hurov et al., 2001). C-TAK1 was origi-cade. In response to EGF treatment, however, activated nally described as p78, a protein marker lost duringMAPK was found to colocalize with KSR1 in membrane pancreatic carcinogenesis (Parsa, 1988) and was subse-ruffles, supporting the model that KSR1 coordinates the quently cloned by virtue of its ability to associate withassembly of a membrane-associated complex that facil- and phosphorylate Cdc25C (Ogg et al., 1994; Peng etitates the activation of MAPK. al., 1998). Here, we find that C-TAK1 constitutively asso-
ciates with the N-terminal region of KSR1 and that KSR1,like Cdc25C, is a substrate of C-TAK1.Discussion
The primary residue of KSR1 phosphorylated byC-TAK1 is S392. This site together with S297 have beenPhosphorylation of KSR1 by the C-TAK1previously identified as two in vivo phosphorylation sitesProtein Kinaseof KSR1 that mediate the binding to 14-3-3 dimers (Ca-To further clarify the role of KSR1 in Ras signaling, wecace et al., 1999). In our assays, we found that the phos-initiated experiments to isolate unknown componentsphorylation of S392 by C-TAK1 was highly specific.of the KSR1 complex that may regulate KSR1 functionWhen other kinases known to phosphorylate substratesand/or Ras/MAPK signaling. In these studies, we identifyon sites generating 14-3-3 binding sites were examined,C-TAK1 as a new member of the KSR1 complex. C-TAK1only C-TAK1 was able to phosphorylate KSR1. In addi-is a serine/threonine protein kinase that belongs to thetion, mutations in KSR1 that severely reduce C-TAK1EMK/PAR/MARK kinase family (Peng et al., 1998). Otherbinding greatly inhibit the phosphorylation of KSR1 atmembers of this family include MARK1, a kinase thatthe S392 site in vitro. As further demonstration of speci-modulates microtubule structure (Drewes et al., 1997),ficity, we found that the association between KSR1 andand EMK, an enzyme that has been implicated in theC-TAK1 could be detected under physiological condi-regulation of cell polarity (Kemphues, 2000), microtubuletions and that C-TAK1 did not interact with or signifi-dynamics (Drewes et al., 1997), Wnt pathway signaling
(Sun et al., 2001), and the maintenance of immune sys- cantly phosphorylate Raf-1, a protein that also contains

Molecular Cell990
Figure 7. Model for KSR1 Function
KSR1 is localized in the cytoplasm of quies-cent cells with MEK bound to its kinase-likedomain and a dimer of 14-3-3 bound to theS297 and S392 phosphorylation sites. C-TAK1constitutively associates with the N-terminalregion of KSR1 to maintain the phosphoryla-tion of S392 and, consequently, the bindingof 14-3-3 to this site (the regulated site,“Reg”). In response to input signals such asgrowth factor treatment, the S392 site ofKSR1 becomes dephosphorylated (by an un-known mechanism), perhaps exposing theKSR1 C1 domain and the FxFP MAPK bindingsite. As a result, the KSR1 complex translo-cates to the plasma membrane, colocalizingMEK with its upstream activator Raf-1 anddownstream target MAPK.
14-3-3 binding motifs. While we did observe very weak to the cell periphery following growth factor treatment.Moreover, we found that the changes in the subcellularphosphorylation of S297 in vitro, it is not clear whetherlocalization of KSR1 correlated with changes in the inthis is an authentic target site for C-TAK1 phosphoryla-vivo phosphorylation state of the S392 site. Both S297tion. The observation that purified C-TAK1 did not phos-and S392 were highly phosphorylated in quiescent cells;phorylate the KSR1 N�320 mutant, which contains thehowever, the phosphorylation of S392 was significantlyS297 site but cannot bind C-TAK1, suggests that if C-TAK1reduced following growth factor treatment. It is impor-is bound to KSR1 it may phosphorylate S297 at lowtant to note that, although we did not detect a changelevels in vitro simply because the sequence motif sur-in the amount of 14-3-3 bound to KSR1 in response torounding S297 is similar to that of the S392 site.growth factor treatment, this observation is consistentwith previous findings that 14-3-3 binds as a dimer and
Regulation of Intracellular Protein that the interaction with both the S297 and S392 sitesLocalization by C-TAK1 must be disrupted to reduce the overall association withPrior to this study, the only known substrates of C-TAK1 14-3-3 (Cacace et al., 1999). Strikingly, we found thatwere Cdc25C and the tyrosine phosphatase PTPH1. proteins mutated at either the S392 site alone or mutatedC-TAK1 phosphorylates S216 of Cdc25C (Peng et al., at both the S297 and S392 sites were constitutively1998) and S359 of PTPH1 (Zhang et al., 1997a), sites on located at the plasma membrane. Thus, the close corre-both proteins that confer binding to 14-3-3. For Cdc25C, lation between S392 phosphorylation and the subcellu-it is known that the phosphorylation of S216 plays an lar localization of the protein strongly suggests that theimportant regulatory role (Peng et al., 1997; Graves et S392 site is the regulated 14-3-3 binding site of KSR1.al., 2001). In interphase, 14-3-3 binding to the S216 site As has been shown for the 14-3-3-mediated cyto-helps to retain Cdc25C in the cytoplasm. At the start plasmic retention of Cdc25C, we propose that 14-3-3of mitosis, S216 is dephosphorylated, resulting in the binding to the S392 site may mask an amino acid se-release of 14-3-3 and the exposure of a nuclear localiza- quence or protein domain that is involved in the intracel-tion sequence that enables Cdc25C to translocate to lular targeting of KSR1. The S297 and S392 sites thatthe nucleus where it dephosphorylates and activates mediate the binding of 14-3-3 are located on either sideCdc2. The activity of C-TAK1 has been found to be of the conserved cysteine-rich C1 domain of KSR1. Forconstant throughout the cell cycle, and it is thought that other proteins such as PKC and Raf-1, C1 domains haveC-TAK1 is the cytoplasmic kinase that keeps Cdc25C been found to interact with membrane-bound lipids/phosphorylated at S216 when the cell is not undergoing proteins and to be required for the stable membranemitosis (Peng et al., 1998). However, in response to association of these molecules (Hurley et al., 1997). Re-DNA damage, Chk1 is a nuclear kinase that becomes cently, our laboratory has solved the solution structureactivated and can phosphorylate Cdc25C on Ser216, of the KSR1 C1 domain and has found that it containsenabling Cdc25C to be exported from the nucleus (Peng a likely binding site for membrane-bound lipids/proteinset al., 1997; Sanchez et al., 1997). Regardless of the and that it is essential for the translocation of KSR1 tokinase that phosphorylates Cdc25C, the functional con- the plasma membrane (M. Zhou and D.K.M., unpublishedsequence of S216 phosphorylation and 14-3-3 binding data). Therefore, the release of 14-3-3 from the S392 siteis to keep Cdc25C sequestered in the cytoplasm. may result in the exposure of the C1 domain, which in
Based on our findings, we propose that phosphoryla- turn may mediate the membrane targeting of KSR1.tion of the KSR1 S392 site by C-TAK1 plays an analo-gous regulatory role in modulating the intracellular local- KSR1 Functions as a Scaffoldization of KSR1 (depicted in Figure 7). By indirect for the Ras/MAPK Pathwayimmunofluorescence, we found that KSR1 was localized The model that KSR1 functions as a MAPK scaffold first
emerged when KSR1 was found to interact with the kinasein the cytoplasm of resting cells but rapidly translocated

Phosphorylation and Regulation of KSR1 by C-TAK1991
components of the MAPK cascade. MEK has been shown served feature of these signaling pathways. From thisstudy, we find that C-TAK1 plays a critical role in regulat-to constitutively associate with KSR1, and, consistent
with this observation, we found that MEK colocalized ing the scaffolding function of KSR1 by phosphorylatingKSR1 at a site that confers 14-3-3 binding, thus seques-with WT KSR1 in the cytoplasm of quiescent cells and
at the plasma membrane of growth factor-treated cells. tering the KSR1 complex in the cytoplasm in the ab-sence of cell signaling. In response to growth factorSince activated Ras induces the translocation of Raf-1
to the plasma membrane, this finding suggests that a treatment, the phosphorylation state of the KSR1 S392site is reduced by an as yet unidentified mechanism,primary function of KSR1 may be to transport MEK to
the intracellular location where its upstream activator leading to the translocation of KSR1 and its associatedproteins to the plasma membrane.Raf-1 is found. Therefore, in unstimulated cells, C-TAK1-
mediated phosphorylation of the KSR1 S392 site may Regulated intracellular localization is an emergingtheme in signal transduction and cell cycle regulation,be required to keep the KSR1/MEK complex constrained
to the cytoplasm in an inactive state, a mechanism that and proteins that control this process are likely to playpivotal roles in both normal and pathological conditions.is deactivated upon signal reception. In support of this
model, MEK was constitutively localized to the plasma In quiescent cells, key molecules involved in signaltransduction and cell cycle regulation must be kept inmembrane in cells expressing the S392A and S297A/
S392A KSR1 proteins. an inactive state. Therefore, proteins such as C-TAK1and 14-3-3, which contribute to the maintenance of inac-The constitutive membrane localization of the KSR1
complex, however, appears to be insufficient to activate tive protein conformations, would be expected to beimportant cellular regulators. The regulation of Ras/the MAPK cascade and instead seems to prime the
pathway for accelerated MAPK activation once input MAPK signaling by C-TAK1 further illustrates the use ofcomplex regulatory mechanisms in fundamental cellularsignals are received. In cells expressing either the S392A
or S297A/S392A KSR1 proteins, activated MAPK was processes. Our results emphasize how phosphorylationcan influence the cell biological parameters of key pro-detected only in membrane ruffles after growth factor
treatment. In addition, the KSR1 mutants were unable teins rather than simply modulating the activity of indi-vidual components of a signaling cascade.to promote maturation of Xenopus oocytes on their own,
but in the presence of activated Ras, they exhibitedExperimental Proceduresan enhanced ability to accelerate MAPK activation and
oocyte maturation. Thus, while an important function ofAntibodies and Reagents
KSR1 may be to colocalize MEK with its activator Raf-1, The phospho-MAPK, phospho-MEK, and AKT antibodies were ob-constitutive localization of the KSR1/MEK complex at tained from Cell Signaling (Beverly, MA). The MEK antibody wasthe membrane would still require other signals to induce purchased from Transduction Laboratories (Lexington, KY), the
FLAG eptitope antibody was from Sigma (St. Louis, MO), and anti-the membrane localization and activation of Raf-1. Abodies recognizing the Pyo epitope and KSR1 have been previouslymore rapid activation of the MAPK cascade would bedescribed (Cacace et al., 1999; Therrien et al., 1996). Antibodiesexpected, however, if the KSR1/MEK complex was al-directed against MAPK, Chk1, PKC, PKA, and p90Rsk were obtained
ready preassembled at the membrane. from Santa Cruz Biotechnology (Santa Cruz, CA). The C-TAK1 pro-Significantly, we found that MEK bound to KSR1 could tein and antibody were previously described in Peng et al. (1998).
be efficiently phosphorylated and activated by Raf-1, The Chk1, Chk2, and Akt purified kinases, as well as the CHKtide andCrosstide substrates were purchased from Upstate Biotechnologyindicating that KSR1 does not interfere with the ability(Lake Placid, NY).of Raf-1 to phosphorylate MEK and may instead present
MEK in a conformation that is conducive for phosphory-Generation of DNA Constructslation by Raf-1. These findings further support the ideaConstructs encoding WT-KSR1 and the KSR1 deletion mutants have
that a primary function of KSR1 is to localize MEK with been previously described (Therrien et al., 1996). The full-lengthactivated Raf-1 at the plasma membrane and then to WT construct encodes amino acids 1–873, the C-terminal domainprovide a platform for the subsequent phosphorylation construct C�542 starts at amino acid 542, and the N-terminal domain
constructs N�539, N�424, N�320, and N�249 encode amino acidsrequired for Ras/MAPK signal transmission. This model1–539, 1–424, 1–320, and 1–249, respectively. Point mutations werefor KSR1 function would also explain why the kinase-likeintroduced into KSR1 by site-directed mutagenesis (QuickChangedomain of KSR1 acts in a dominant inhibitory mannerkit; Stratagene, La Jolla, CA). All mutations were confirmed by DNA
(Joneson et al., 1998; Therrien et al., 1996; Yu et al., sequencing.1997). Since these proteins contain the MEK bindingregion but lack sequences required for membrane local- Cell Culture, Transfection, Metabolic Labeling,
and Coimmunoprecipitation Assaysization, they would be expected to sequester MEK inCos cells were cultured in Dulbecco’s modified Eagle mediumthe cytosol and thereby short-circuit the activation of(DMEM) supplemented with 10% fetal calf serum at 37�C in 5%MAPK.CO2. Plasmid DNAs were transfected using Fugene reagent (Roche,Indianapolis, IN) according to the manufacturer’s instructions.
Conclusion Transfected cells were either incubated for 48 hr in fully supple-mented medium or serum starved for 18 hr prior to growth factorIn summary, we present evidence that KSR1 functionsaddition. Cells were then lysed in NP-40 lysis buffer (20 mM Trisas a regulated MAPK scaffold for the Ras pathway.[pH 8.0]; 137 mM NaCl; 10% glycerol; 1% NP-40; 0.15 U/ml aprotinin;Functionally equivalent scaffolding proteins have also1 mM PMSF; 20 �M leupeptin; 5 mM sodium vanadate) and KSR1been identified for the mammalian JNK pathway and forproteins were immunoprecipitated. After extensive washing in NP-
MAPK modules in yeast (Whitmarsh and Davis, 1998), 40 lysis buffer containing 1 M NaCl, the samples were examinedindicating that the coordinated assembly of MAPK pro- directly by SDS-PAGE and immunoblotting or were analyzed in im-
mune complex kinase assays. For metabolic labeling experiments,teins with their activators may be an evolutionarily con-

Molecular Cell992
transfected Cos cells were incubated for 4–6 hr at 37�C in phos- cal reading of the manuscript. We also acknowledge Neil Michaudfor valuable input in the initial phase of this project.phate-free DMEM containing 2.5% dialyzed calf serum and 1 mCi of
[32P]orthophosphate (Amersham, Piscataway, NJ) per ml of labelingmedium. EGF stimulation was performed as described above, and Received July 9, 2001; revised September 18, 2001.cells were washed twice with ice cold Tris-buffered saline (TBS)(137 mM NaCl; 20 mM Tris [pH 7.4]) prior to lysis for 20 min at 4�C
Referencesin radioimmunoprecipitation assay (RIPA) buffer (NP-40 lysis bufferthat contains 0.5% sodium deoxycholate and 0.1% sodium dodecyl
Bar-Sagi, D. (2001). A Ras by any other name. Mol. Cell. Biol. 21,sulfate [SDS]).1441–1443.
Cacace, A.M., Michaud, N.R., Therrien, M., Mathes, K., Copeland,Immune Complex Kinase AssaysT., Rubin, G.M., and Morrison, D.K. (1999). Identification of constitu-Transfected Cos cells were lysed in NP-40 lysis buffer and KSR1tive and ras-inducible phosphorylation sites of KSR1: implicationsproteins were immunoprecipitated. The immunoprecipitates werefor 14-3-3 binding, mitogen-activated protein kinase binding, andwashed five times with NP-40 lysis buffer containing 1 M NaCl. AfterKSR1 overexpression. Mol. Cell. Biol. 19, 229–240.a final wash in 30 mM Tris (pH 7.4), the immune complexes were
incubated in kinase buffer (100 mM Tris [pH 7.4]; 25 mM �-Glycerol- Denouel-Galy, A., Douville, E.M., Warne, P.H., Papin, C., Laugier,phosphate; 5 mM EGTA; 1 mM DTT; 1 mM sodium vanadate; 60 �M D., Calothy, G., Downward, J., and Eychene, A. (1997). Murine KsrATP; 10 mM MgCl2) containing 20 �Ci of [�-32P]ATP. After incubation interacts with MEK and inhibits Ras-induced transformation. Curr.for 30 min at 30�C, the assays were terminated by the addition of Biol. 8, 46–55.gel sample buffer. The samples were resolved by SDS-PAGE, the Drewes, G., Ebneth, A., Preuss, U., Mandelkow, E.M., and Mandel-phosphoproteins were visualized by autoradiography, and the kow, E. (1997). MARK, a novel family of protein kinases that phopho-amount of 32P incorporated into the proteins was quantitated using rylate mictotuble-associated proteins and trigger microtubule dis-a phosphor-imager and ImageQuant software (Molecular Dynamics, ruption. Cell 89, 297–308.Piscataway, NJ). In experiments where KSR1 was used as an in
Graves, P.R., Lovly, C.M., Uy, G.L., and Piwnica-Worms, H. (2001).vitro substrate, the KSR1 proteins were isolated from RIPA lysates,Localization of human Cdc25C is regulated both by nuclear exportwashed as described above, and then incubated in kinase bufferand 14-3-3 protein binding. Oncogene 20, 1839–1851.containing the appropriate purified protein kinases.Hurley, J.H., Newton, A.C., Parker, P.J., Blumberg, P.M., and Nishi-zuka, Y. (1997). Taxonomy and function of C1 protein kinase CPhosphorylation Site Mappinghomology domains. Protein Sci. 6, 477–480.After separation by SDS-PAGE, 32P-labeled proteins were trans-Hurov, J.B., Stappenbeck, T.S., Zmasek, C.M., Ranganath, S.H.,ferred to nitrocellulose membranes and visualized by autoradiogra-White, L.S., Russell, J.H., Chan, A.C., Murphy, K.M., and Piwnica-phy. Membrane pieces containing the phosphoproteins were ex-Worms, H. (2001). Immune cell dysfunction and autoimmune diseasecised and blocked with 1.5% PVP-40 for 1 hr at 37�C. The boundin mice lacking the EMK/Par1 protein kinase. Mol. Cell. Biol. 21,KSR1 proteins were then subjected to enzymatic digestion with3206–3219.trypsin. The resulting tryptic phosphopeptides were separated and
eluted by reversed-phase HPLC as previously described (Cacace Joneson, T., Fulton, J.A., Volle, D.J., Chaika, O.V., Bar-Sagi, D., andet al., 1999). HPLC fractions containing peaks of radioactivity were Lewis, R.E. (1998). Kinase suppressor of Ras inhibits the activationsubjected to phosphoamino acid analysis and semiautomated Ed- of extracellular ligand-regulated (ERK) mitogen-activated proteinman degradation as previously described (Morrison et al., 1993). (MAP) kinase by growth factors, activated Ras, and Ras effectors.
J. Biol. Chem. 273, 7743–7748.Immunofluorescence Leevers, S.J., Paterson, H.F., and Marshall, C.J. (1994). RequirementCos cells seeded onto 18 mm glass coverslips were transfected for Ras in Raf activation is overcome by targeting Raf to the plasmawith the appropriate KSR1 constructs. Forty-eight hours after trans- membrane. Nature 369, 411–414.fection, serum starved cells were either left untreated or were treated
Kemphues, K. (2000). PARsing embryonic polarity. Cell 101,for 7 min with EGF. The cells were then washed once with phosphate345–348.buffered saline (PBS) and fixed in freshly prepared 4% paraformal-Kumagai, A., and Dunphy, W.G. (1999). Binding of 14-3-3 proteinsdehyde/PBS for 10 min at 25�C. Following two washes with PBS,and nuclear export control the intracellular localization of the mitoticthe cells were permeabilized for 5 min with 0.1% Triton X-100 ininducer Cdc25. Genes Dev. 13, 1067–1072.PBS. The cells were washed again with PBS and blocked for 1 hr
in 3% bovine serum albumin (BSA) in PBS. Following an incubation Magee, T., and Marshall, C. (1999). New insights into the interactionfor 1 hr at 25�C in the appropriate antibody, the cells were washed of Ras with the plasma membrane. Cell 98, 9–12.four times with PBS and incubated with either anti-mouse or anti-
Marshall, C.J. (1996). Ras effectors. Curr. Opin. Cell Biol. 2, 197–204.rabbit Alexa dye secondary antibody (Molecular Probes, Eugene,
Michaud, N.R., Therrien, M., Cacace, A., Edsall, L.C., Spiegel, S.,OR) diluted 1:1000 in blocking buffer for 45 min at 25�C, protectedRubin, G.M., and Morrison, D.K. (1997). KSR stimulates Raf-1 activityfrom light. After four more washes in PBS, the coverslips werein a kinase-independent manner. Proc. Natl. Acad. Sci. USA 94,washed in distilled water and mounted in Prolong antifade medium12792–12796.(Molecular Probes).Morrison, D.K., Heidecker, G., Rapp, U.R., and Copeland, T.D.(1993). Identification of the major phosphorylation sites of the Raf-1RNA Transcription, Oocyte Injection, and Analysiskinase. J. Biol. Chem. 268, 17309–17316.Capped RNA was transcribed using the Message Machine kit (Am-
bion, Austin, TX). Buffer or RNA (30 ng) encoding the various KSR1 Muller, J., Cacace, A.M., Lyons, W.E., McGill, C.B., and Morrison,constructs was injected into stage VI oocytes as described (Therrien D.K. (2000). Identification of B-KSR1, a novel brain-specific isoformet al., 1996). After approximately 12 hr, the oocytes were injected of KSR that functions in neuronal signaling. Mol. Cell. Biol. 20, 5529–with RasV12 RNA and were subsequently scored for GVBD, as evi- 5539.denced by the appearance of a white spot at the animal pole. For
Ogg, S., Gabrielli, B., Piwnica-Worms, H., Peng, C.Y., Graves, P.R.,biochemical analysis, oocytes were lysed (10 �l of RIPA buffer perThoma, R.S., Byrnes, M.J., Wu, Z., and Stephenson, M.T. (1994).oocyte) by trituration with a pipette tip. Lysates were cleared byPurification of a serine kinase that associates with and phosphory-centrifugation at 14,000 � g for 5 min at 4�C.lates human Cdc25C on serine 216. J. Biol. Chem. 269, 30461–30469.
Parsa, I. (1988). Loss of a Mr 78,000 marker in chemically inducedAcknowledgmentstransplantable carcinomas and primary carcinoma of human pan-creas. Cancer Res. 48, 2265–2272.We thank Dan Ritt for excellent technical assistance and Monica
Murakami, Marion Lohrum, Mark Fortini, and Martina Pyrski for criti- Pearson, R., and Kemp, B. (1991). Protein kinase phosphorylation

Phosphorylation and Regulation of KSR1 by C-TAK1993
site sequences and consensus specificity motifs: tabulations. Meth-ods Enzymol. 200, 62–81.
Peng, C.-Y., Graves, P.R., Thomas, R.S., Wu, Z., Shaw, A.S., andPiwnica-Worms, H. (1997). Mitotic and G2 checkpoint control: regu-lation of 14-3-3 protein binding by phosphorylation of Cdc25c onserine 216. Science 277, 1501–1505.
Peng, C.Y., Graves, P.R., Ogg, S., Thoma, R.S., Byrnes, M.J., Wu, Z.,Stephenson, M.T., and Piwnica-Worms, H. (1998). C-TAK1 proteinkinase phosphorylates human Cdc25C on serine 216 and promotes14-3-3 protein binding. Cell Growth Differ. 9, 197–208.
Sanchez, Y., Wong, C., Thoma, R., Richman, R., Wu, Z., Piwnica-Worms, H., and Elledge, S. (1997). Conservation of the Chk1 check-point pathway in mammals: linkage of DNA damage to Cdk regula-tion through Cdc25. Science 277, 1497–1501.
Shields, J., Pruitt, K., McFall, A., Shaub, A., and Der, C. (2000).Understanding Ras: ‘it ain’t over ‘til it’s over’. Trends Cell Biol. 10,147–154.
Stewart, S., Sundaram, M., Zhang, Y., Lee, J., Han, M., and Guan,K.-L. (1999). Kinase suppressor of Ras forms a multiprotein signalingcomplex and modulates MEK localization. Mol. Cell. Biol. 19, 5523–5534.
Sun, T.Q., Lu, B., Feng, J.J., Reinhard, C., Jan, Y.N., Fantl, W.J., andWilliams, L.T. (2001). PAR-1 is a dishevelled-associated kinase anda positive regulator of Wnt signalling. Nat. Cell Biol. 3, 628–636.
Therrien, M., Chang, H.C., Solomon, N.M., Karim, F.D., Wassarman,D.A., and Rubin, G.M. (1995). KSR, a novel protein kinase requiredfor RAS signal transduction. Cell 83, 879–888.
Therrien, M., Michaud, N.R., Rubin, G.M., and Morrison, D.K. (1996).KSR modulates signal propagation within the MAPK cascade. GenesDev. 10, 2684–2695.
Volle, D.J., Fulton, J.A., Chaika, O.V., McDermott, K., Huang, H.,Steinke, L.A., and Lewis, R.E. (1999). Phosphorylation of the kinasesuppressor of ras by associated kinases. Biochemistry 38, 5130–5137.
Whitmarsh, A., and Davis, R. (1998). Structural organization of MAP-kinase signaling modules by scaffold protein in yeast and mammals.Trends Biochem. Sci. 23, 481–485.
Wittinghofer, A. (1998). Signal transduction via Ras. Biol. Chem. 379,933–937.
Xing, H., Kornfeld, K., and Muslin, A.J. (1997). The protein kinaseKSR interacts with 14-3-3 protein and Raf. Curr. Biol. 7, 294–300.
Xing, H.R., and Kolesnick, R. (2001). Kinase suppressor of Ras sig-nals through Thr269 of c-Raf-1. J. Biol. Chem. 276, 9733–9741.
Yang, J., Winkler, K., Yoshida, M., and Kornbluth, S. (1999). Mainte-nance of G2 arrest in the Xenopus oocyte: a role for 14-3-3-mediatedinhibition of Cdc25 nuclear import. EMBO J. 18, 2174–2183.
Yu, W., Fantl, W.J., Harrowe, G., and Williams, L.T. (1997). Regulationof the MAP kinase pathway by mammalian Ksr through direct inter-action with MEK and ERK. Curr. Biol. 8, 56–64.
Zeng, Y., Forbes, K.C., Wu, Z., Moreno, S., Piwnica-Worms, H., andEnoch, T. (1998). Replication checkpoint requires phosphorylationof the phosphatase Cdc25 by Cds1 or Chk1. Nature 395, 507–510.
Zhang, S.H., Kobayashi, R., Graves, P.R., Piwnica-Worms, H., andTonks, N.K. (1997a). Serine phosphorylation-dependent associationof the band 4.1-related protein-tyrosine phosphatase PTPH1 with14-3-3beta protein. J. Biol. Chem. 272, 27281–27287.
Zhang, Y., Yao, B., Delikat, S., Bayoumy, S., Lin, X.-H., Basu, S.,McGinley, M., Chan-Hui, P.-Y., Lichenstein, H., and Kolesnick, R.(1997b). Kinase suppressor of Ras is ceramide-activated proteinkinase. Cell 89, 63–72.
Zimmermann, S., and Moelling, K. (1999). Phosphorylation and regu-lation of Raf by Akt (protein kinase B). Science 286, 1741–1744.




















