
Mitochondrial myopathies: Clinical and biochemical features of 30 patients with major deletions of...
10
ORIGINAL ARTICLES Biochemical Mitochondrial Myopathles: Clinical and with Major Deletions of Muscle Mitochondrial DNA Features of 30 Patients 1. J. Hok BSc," A. E. Harding, MD,*J. M. Cooper, PhD,*t A. H. V. Schapira, MD,* A. Toscano, MD,*S J. B. Clark, DSc,% and J. A. Morgan-Hughes, MD" Analysis of mitochondrial DNA (mtDNA) in muscle and blood from 72 patients with mitochondrial myopathy showed that 30 had major deletions of a variable proportion of muscle mtDNA. All of these 30 patients presented with progressive external ophthalmoplegia and limb weakness, and 8 had the additional features of the Kearns-Sayre syndrome. Of the 42 patients without detectable muscle mtDNA deletions, 10 had progressive external ophthalmo- plegia and limb weakness, 2 had the Kearns-Sayre syndrome, 11 had limb weakness without extraocular involvement,. and 19 had multisystem disorders predominantly affecting the central nervous system. Only 2 patients with mtDNA deletions had clinically affected relatives, compared with 10 of those without deletions. In the 4 patients with polar- ographic defects exclusively involving complex I (NADH coenzyme Q reductase), the deleted protein-coding genes were confined to those for complex I subunits. Thirteen other patients with apparently identical deletions had variable clinical and biochemical features. Immunoblots of complex I polypeptides from patients with deletions were either indistinguishable from controls or showed only a mild generalized decrease in all identifiable subunits. Holt IJ, Harding AE, Cooper JM, Schapira AHV, Toscano A, Clark JB, Morgan-Hughes JA. Mitochondrial myopathies: clinical and biochemical features of 30 patients with major deletions of muscle mitochondrial DNA. Ann Neurol 1989;26:699-708 The mitochondrial myopathies and encephalomyopa- thies constitute an increasingly well-recognized group of neurological diseases characterized histologically by the presence of ragged red fibers in biopsied muscle stained according to the modified Gomori trichrome method. Within this group of disorders there is strik- ing clinical and biochemical heterogeneity. Clinical presentations include infantile lactic acidosis with fail- ure to thrive, progressive external ophthalmoplegia (PEO), proximal myopathy with weakness enhanced by exercise, and multisystem neurological syndromes mainly or exclusively affecting the central nervous sys- tem (CNS) causing seizures, ataxia, dementia, move- ment disorders, and strokelike episodes. Short stature, retinopathy, deafness, peripheral neuropathy, cardiac conduction defects, and renal and endocrine dysfunc- tion also occur [l, 27. In the majority of patients, in vitro studies of mitochondrial metabolism using polarography or enzymatic assays have shown defects of the respiratory chain, usually involving complex I or complex 111 in adults and complex IV in children El, 27. Eighteen percent of patients with morphologically defined mitochondrial myopathy have clinically af- fected relatives [3]. As maternal transmission to off- spring is eight times more frequent than paternal trans- mission [37, and occasionally large pedigrees exhibiting maternal inheritance have been observed [41, it has been suggested that these disorders may be caused by mutations of mitochondrial DNA (mtDNA) C57. Hu- man mtDNA is a circular double-stranded molecule that is 16.5 kilobases (kb) in length [GI. It is exclusively maternally transmitted 177 and codes for 13 of the 67 or so subunits of the mitochondrial respiratory chain and oxidative phosphorylation system: seven subunits of complex I; cytochrome b (complex 111); subunits I, 11, and 111 of cytochrome oxidase (complex IV); and two subunits of H+-adenosine triphosphate synthase (ATPase), subunit 6 and a subunit similar to subunit 8 of yeast ATPase [8-101. From the *University Department of Clinical Neurology, Institute of Neurology, Queen Square, and the ?Department of Biochemis- try, St. Bartholomew's Hospital Medical College, London, UK. Received Mar 30, 1989. Accepted for publication May 1, 1989. Address correspondence to Dr Harding, University Department of Clinical Neurology, Institute of Neurology, Queen Square, London WClN 3BG, UK. %Present address: Clinica Neurologica, University of Messina, Sicily, Italy. Copyright 0 1989 by the American Neurological Association 699
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Mitochondrial myopathies: Clinical and biochemical features of 30 patients with major deletions of...

ORIGINAL ARTICLES
Biochemical Mitochondrial Myopathles: Clinical and
with Major Deletions of Muscle Mitochondrial DNA
Features of 30 Patients
1. J. Hok BSc," A. E. Harding, MD,*J. M. Cooper, PhD,*t A. H. V. Schapira, MD,* A. Toscano, MD,*S J. B. Clark, DSc,% and J. A. Morgan-Hughes, MD"
Analysis of mitochondrial DNA (mtDNA) in muscle and blood from 72 patients with mitochondrial myopathy showed that 30 had major deletions of a variable proportion of muscle mtDNA. All of these 30 patients presented with progressive external ophthalmoplegia and limb weakness, and 8 had the additional features of the Kearns-Sayre syndrome. Of the 42 patients without detectable muscle mtDNA deletions, 10 had progressive external ophthalmo- plegia and limb weakness, 2 had the Kearns-Sayre syndrome, 11 had limb weakness without extraocular involvement,. and 19 had multisystem disorders predominantly affecting the central nervous system. Only 2 patients with mtDNA deletions had clinically affected relatives, compared with 10 of those without deletions. In the 4 patients with polar- ographic defects exclusively involving complex I (NADH coenzyme Q reductase), the deleted protein-coding genes were confined to those for complex I subunits. Thirteen other patients with apparently identical deletions had variable clinical and biochemical features. Immunoblots of complex I polypeptides from patients with deletions were either indistinguishable from controls or showed only a mild generalized decrease in all identifiable subunits.
Holt IJ, Harding AE, Cooper JM, Schapira AHV, Toscano A, Clark JB, Morgan-Hughes JA. Mitochondrial myopathies: clinical and biochemical features of 30 patients with major
deletions of muscle mitochondrial DNA. Ann Neurol 1989;26:699-708
The mitochondrial myopathies and encephalomyopa- thies constitute an increasingly well-recognized group of neurological diseases characterized histologically by the presence of ragged red fibers in biopsied muscle stained according to the modified Gomori trichrome method. Within this group of disorders there is strik- ing clinical and biochemical heterogeneity. Clinical presentations include infantile lactic acidosis with fail- ure to thrive, progressive external ophthalmoplegia (PEO), proximal myopathy with weakness enhanced by exercise, and multisystem neurological syndromes mainly or exclusively affecting the central nervous sys- tem (CNS) causing seizures, ataxia, dementia, move- ment disorders, and strokelike episodes. Short stature, retinopathy, deafness, peripheral neuropathy, cardiac conduction defects, and renal and endocrine dysfunc- tion also occur [l, 27. In the majority of patients, in vitro studies of mitochondrial metabolism using polarography or enzymatic assays have shown defects of the respiratory chain, usually involving complex I
or complex 111 in adults and complex IV in children El, 27.
Eighteen percent of patients with morphologically defined mitochondrial myopathy have clinically af- fected relatives [3]. As maternal transmission to off- spring is eight times more frequent than paternal trans- mission [37, and occasionally large pedigrees exhibiting maternal inheritance have been observed [41, it has been suggested that these disorders may be caused by mutations of mitochondrial DNA (mtDNA) C57. Hu- man mtDNA is a circular double-stranded molecule that is 16.5 kilobases (kb) in length [GI. It is exclusively maternally transmitted 177 and codes for 13 of the 67 or so subunits of the mitochondrial respiratory chain and oxidative phosphorylation system: seven subunits of complex I; cytochrome b (complex 111); subunits I, 11, and 111 of cytochrome oxidase (complex IV); and two subunits of H+-adenosine triphosphate synthase (ATPase), subunit 6 and a subunit similar to subunit 8 of yeast ATPase [8-101.
From the *University Department of Clinical Neurology, Institute of Neurology, Queen Square, and the ?Department of Biochemis- try, St. Bartholomew's Hospital Medical College, London, UK. Received Mar 30, 1989. Accepted for publication May 1, 1989.
Address correspondence to Dr Harding, University Department of Clinical Neurology, Institute of Neurology, Queen Square, London WClN 3BG, UK. %Present address: Clinica Neurologica, University of Messina, Sicily, Italy.
Copyright 0 1989 by the American Neurological Association 699

Early studies of m t D N A extracted from blood did not demonstrate different restriction fragment patterns between patients with mitochondrial myopathies and their unaffected relatives in the same maternal line [11, 12). Subsequently, 9 of 25 patients were found to have two populations of mtDNA in muscle, but not blood, one of which was deleted by up to 7 kb 113, 141. These observations, which demonstrated that de- fects of the mitochondrial genome may be associated with human disease, have since been confirmed 15, 163. More recently, tandem duplications of one popu- lation of mtDNAs in leukocytes from 2 patients have been reported 1171.
In this paper w e present the clinical and histochem- ical features of 30 patients with major muscle mtDNA deletions, 18 of whom were examined biochemically. The results are discussed in relation to findings in 42 patients without identifiable deletions.
Patients and Methods Patients Muscle mtDNA was analyzed from 72 patients with mito- chondrial myopathy as defined previously [2). In vitro stud- ies of mitochondrial metabolism were performed in 49 of these patients. The clinical features of many of the patients have been described in detail previously [2,18-22}. Patients numbered 1 to 43 are the same as those described by Petty and colleagues {2). Mitochondria1 DNA from 18 control subjects who were either normal or had well-defined neuro- muscular diseases unrelated to defects of mitochondrial me- tabolism was also studied. In vitro studies of mitochondrial metabolism were performed in 5 control subjects aged 25 to 60 years. Three controls had symptoms of weakness induced by exercise but were clinically normal and had no evidence of metabolic muscle disease on investigation (no lactic acidemia after exercise, and normal muscle histological and electron microscopic findings), and 2 had multisystem CNS disease later shown to have causes other than disorders of mitochondrial metabolism (intracranial calcification second- ary to small vessel disease and dominantly inherited olivo- pontocerebellar atrophy). All investigative procedures were performed with informed consent of the subjects and prior approval of the local ethical committee.
Mitochondria/ DNA Analysis DNA was isolated from blood using standard techniques 1231. For muscle from 47 patients and 6 control subjects, after intact mitochondria had been isolated from approxi- mately 5 gm of muscle by differential centrifugation for biochemical studies, the remaining pellet (containing the ma- jority of mitochondria in the sample) was incubated with 4 mg proteinase K and 1% sodium dodecyl sulphate (SDS); frozen muscle from 25 patients and 12 controls was crushed with a pestle prior to the latter step. DNA was purified by phenol and chloroform extraction. Approximately 5 p,g of DNA from blood and 1 p,g from muscle were digested with a battery of restriction endonucleases under conditions rec- ommended by the manufacturers (Northumbria Biologicals Ltd, Cramlington, UK, or Bethesda Research Laboratories,
Paisley, UK), with the addition of spermidine and bovine serum albumin. The resulting fragments were separated by agarose (0.8-1.8%) gel electrophoresis and transferred to nylon filters (Hybond-N, Amersham, Buckinghamshire, UK). HeLa cell mtDNA was nick translated 1241 or oligo- labeled 1251 with 32P-labeled deoxycytidine 5'-triphosphate and hybridized to the filters under conditions recommended by the manufacturers prior to autoradiography at - 70°C for 24 to 48 hours.
Deleted regions of muscle mtDNA from some patients were mapped by hybridizing 14 small fragments of 32P- oligolabeled HeLa cell mtDNA to DNA on filters prepared as just described { 131. In most patients the upper and lower limits of the deletions were defined by analyzing restriction site losses with reference to the published sequence of hu- man mtDNA [6]. The relative proportions of normal and abnormal muscle mtDNAs were estimated using an LKB Ultroscan XL densitometer (Pharmacia, Milton Keynes, UK).
Biochemicd Stizdies of Intact Mitochondria Approximately 5 gm of muscle was removed from the quad- riceps femoris of 49 patients (18 with deletions and 3 1 with- out) and 5 control subjects under light general anesthesia. Mitochondria were isolated within 90 minutes of biopsy us- ing a modification of the method described by Davies and colleagues [26}. The muscle was washed in ice-cold buffer (210 mM mannitol, 70 mM sucrose, 10 mM ethylenedia- minetetraacetic acid (EDTA), 50 mM Tris pH 7.4), chopped, and incubated at 4°C with trypsin (Boehringer, kwes, VK; 0.5 mg/gm muscle). After 30 minutes a fivefold excess of trypsin inhibitor type 1s (Sigma, Poole, UK) was added. The muscle was homogenized briefly (two >-second bursts) and diluted 15-fold with buffer prior to centrifugation at 2,000 g for 5 minutes. The supernatant was recentrifuged for 10 minutes at 8,000 g, and the resulting mitochondrial pellet was resuspended in 225 mM mannitol, 75 mM sucrose, 100 p,M EDTA, 10 mM Tris pH 7.2. The two centrifugation steps were repeated, and the final mitochondrial pellet was resuspended to a protein concentration of 10 to 15 mgiml. Polarographic measurements were determined at 25°C using a Clark-type oxygen electrode, and cytochrome spectra were determined at liquid nitrogen temperatures as described by Morgan-Hughes and colleagues [ 193. The isolated mitochon- dria were usually well coupled, with respiratory control ratios between 1.3 and 7.1. The latency of citrate synthase activity, a measure of the integrity of the mitochondrial membranes, was always greater than 8.
Analysis of Respiratory Complex Polypeptides Samples of muscle mitochondrial protein obtained as just described were stored in liquid nitrogen after the addition of 5 mM aminobenzamidine. Complex I polypeptides were an- alyzed as in the study by Schapira and colleagues [27]. Com- plex 111 and IV polypeptides were obtained from samples dissolved in 5% SDS, 2% mercaptoethanol, 8 M urea, and 0.25 M Tris-HC1 p H 6.8 without boiling. Proteins were sep- arated on 12 to 16% (complex I, 111) or 12 to 19% (complex IV) linear gradient SDS polyacrylamide gels as described previously 127, 281, prior to transfer to nitrocellulose by
700 Annals of Neurology Vol 26 No 6 December 1989

Case 38 I - - - ---I
Case 5 8 I- - I 1-4
I - - ) ! ---I Case 2 4
I I --- Case 3 1 I - - - - ,
I---, ----- case 83
Cases 16, 25 G 85 I--- ---I
Case 2 8 I --I
Case 7 0 1-1 1 - 4
I . ,---I c a s e 43
4 - 1 Case 2 I-t Cases 1,17,18, i4,26.27,30.44.45,48,67,68 & 69
3 1 2 4 5 6 7 8 9 I I d % I--
10 t-4 * 12 13
14 c-----(
I I I I I I I I I 0 2 4 6 8 10 12 14 16
elecuoblotting in Tris-glycine-methanol 129) for 3 hours at 70 V. Filters were quenched overnight at room temperature in a 1% mixture of milk powder in phosphate-buffered saline pH 7.4.
Immunoblotting was performed with antibodies raised in rabbits to highly purified beef heart complex I, 111, and IV holoentymes. In some cases subunit-specific antisera (to the 75,51,49, 30,24, and 13 kDa iron sulphur (FeS) proteins of complex I, the Rieske FeS protein of complex 111, and sub- units I1 and V of complex IV) were also used. Bound anti- bodies were detected using the biotin streptavidin horse- radish peroxidase system (Amersham), as described 12 7).
Histochemical Studies Quantitative histochemical studies were performed in 5 1 pa- tients. The proportion of muscle fibers showing peripheral mitochondrial accumulations (ragged red fibers) was esti- mated using the succinic dehydrogenase stain. The method of Seligman and colleagues [30} was used to demonstrate cytochrome oxidase activity. In both cases three areas were chosen at random, and 400 to 600 fibers scored, prior to calculation of the percentage of raged red and cytochrome oxidase-negative fibers.
Statistical Method Student’s t test was used to compare means of normally distributed data; all other analyses employed chi square or Fisher’s exact tests.
Results Mitochondrial DNA Analysis Deletions of mtDNA extracted from muscle were found in 30 of the 72 patients, but none of the 18 control subjects. MtDNA from blood was analyzed in 30 patients, including 14 with muscle mtDNA dele- tions, and showed a single normal-length population of
Fig 1. Linearized map of the mitochondrial genome showing (from top to bottom) extent of deleted regions in 25 patients with mitochondrial myopathy, location of coding regions (NO = NADH coenzyme Q reductme subunits, A = adenosine triphos- phate synthase, CO = cytochrome oxihse , IZS , 16s = riboso- mal R N h , 1 = transfer W A S , OH, OL = origins of heavy and light strand replication, NIC = noncoding regions, cytb = cytochrome b), and location of mitochondrial DNA probes used to map deletions in some cases; distances on bottom line are in kilo- bases. The interrupted lines at either end of the deletions repre- sent their apper and tower limits, defined by the presence or ab- sence of restriction sites.
rntDNA in each case. The muscle mtDNA deletions varied from 2.2 to 7.0 kb in length, and the proportion of deleted to normal mtDNAs ranged from 20 to 90%. The proportions of abnormal and normal mtDNAs were similar after extraction from either nu- clear pellet or isolated mitochondria in 3 patients. The possibility that some patients with less than 50% de- leted mtDNAs had tandem duplications of mtDNA El71 was excluded by showing that uncut deleted mtDNAs migrated further on electrophoresis than the normal population did.
The deletions were mapped in detail in 25 patients; all but 1 were in the 7,000 to 15,000 base pair (bp) region (Fig 1). The deleted regions included genes en- coding subunits of NADH coenzyme Q teductase, cytochrome oxidase, and adenosine triphosphate syn- thase in the majority of patients; in 1 the deletion in- volved the ribosomal RNA genes, and in another, part or all of the cytochrome b reading frame. With the possible exception of Patients 16, 25, and 85 the mus- cle mtDNA deletions always included a variable num- ber (2-8) of transfer RNA (tRNA) genes. None of the deletions definitely involved the origins of tran-
Holt et al: Mitochondrial Myopathies 701
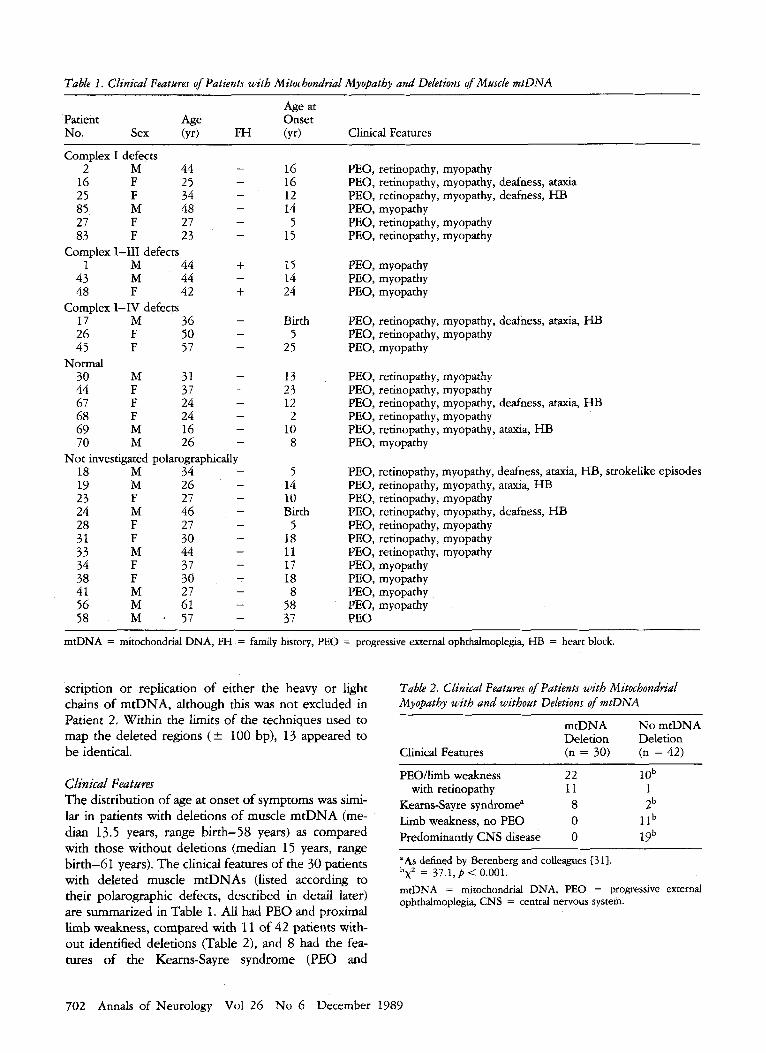
Table 1. Clinical Featuns of Patients with Mitochondrial Myopatby and Deletions of Muscle mtDNA
Age at Patient Age Onset No. Sex (yr) FH (yr) Clinical Features
Complex I defects - 2 M 44
16 F 25 25 F 34 85 M 48 27 F 27 83 F 23
- - - - -
Complex 1-111 defects 1 M 44 +
43 M 44 48 F 42 + 17 M 36 26 F 50 45 F 57
30 M 31 44 F 37 67 F 24 68 F 24
M 16 69 70 M 26
18 M 34 M 26
27 46
l9 F 24
27 23 M 28 F 31 F 30
M 44 33 F 37 34
38 F 30 41 M 27
M 61 56 58 M 1 57
-
Complex I-IV defects - - -
Normal - - - -
- -
Not investigated polarographically - - - - - - - - - - - -
16 16 12 14
5 15
15 14 24
Birth 5
25
13 23 12 2
10 8
5 14 10 Birth
5 18 I1 17 18 8
58 37
PEO, retinopathy, myopathy PEO, retinopathy, myopathy, deafness, ataxia PEO, retinopathy, myopathy, deafness, HB PEO, myopathy PEO, retinopathy, myopathy PEO, retinopathy, myopathy
PEO, myopathy PEO, myopathy PEO, myopathy
PEO, retinopathy, myopathy, deafness, ataxia, I-IB PEO, retinopathy, myopathy PEO, myopathy
PEO, retinopathy, myopathy PEO, retinopathy, myopathy PEO, retinopathy, myopathy, deafness, ataxia, HB PEO, retinopathy, myopathy PEO, retinopathy, myopathy, ataxia, HB PEO, myopathy
PEO, retinopathy, myopathy, deafness, ataxia, HB, strokelike episodes PEO, retinopathy, myopathy, ataxia, H B PEO, retinopathy, myopathy PEO, retinopathy, myopathy, deafness, HB PEO, retinopathy, myopathy PEO, retinopathy, myopathy PEO, retinopathy, myopathy PEO, myopathy PEO, myopathy PEO, myopathy PEO, myopathy PEO
mtDNA = mitochondrial DNA, FH = family history, PEO = progressive external ophthalmoplegia, HB = heart block.
scription or replication of either the heavy or light chains of mtDNA, although this was not excluded in Patient 2. Within the limits of the techniques used to map the deleted regions (k 100 bp), 13 appeared to be identical.
Clinical Feataurar The distribution of age at onset of symptoms was simi- lar in patients with deletions of muscle mtDNA (me- dian 13.5 years, range birth-58 years) as compared with those without deletions (median 15 years, range birth-61 years). The clinical features of the 30 patients with deleted muscle mtDNAs (listed according to their polarographic defects, described in detail later) are summarized in Table 1. All had PEO and proximal limb weakness, compared with 11 of 42 patients with- out identified deletions (Table 2), and 8 had the fea- tures of the Kearns-Sayre syndrome (PEO and
Table 2. Clinical Features of Patients with Mitocbondrial Myopathy with and without Deletions of mtDNA
~ ~ ~
mtDNA No mtDNA Deletion Deletion
Clinical Features (n = 30) (n = 42)
PEO/limb weakness 22 1 O b with retinopathy 11 1
Kearns-Sayre syndrome" 8 Zb Lmb weakness, no PEO 0 1 lb Predominantly CNS disease 0 19b
"As defined by Berenberg and colleagues 131).
mrDNA = mirochondrial DNA, PEO = progressive external ophthalmoplegia, CNS = central nervous system.
b 2 - x - 37.1,p < 0.001.
702 Annals of Neurology Vol 26 No 6 December 1989

Table 3 . Respiratory Activities and Cytocbrome Contents of Isolated Skeletal Muscle Mitochondria in 18 Patients with Deletions of Muscle mtDNA
State 3 Oxygen Uptake Rates (nmol/O/min/mg mt protein)
Pyruvate Succinate Ascorbate +
Cytochrome Concentrations (nmol/mg mt protein)
Patient No. (RCR) (RCR) TMPD b 61 C m3
Complex I defects 2 61" (2.6)
16 24" (2.2) 25 64" (2.8) 85 73" (3.6) 27 1 la (2.5) 83 30" (4.4)
Complex 1-111 defects 1 49" (2.2)
43 16" (2.7) 48 l l"(1.3)
Complex I-IV defects 17 12d(2.0) 26 24" (2.0) 45 46" (5.3)
NormaI 30 114 (5.0) 44 78 (6.7) 67 123 (5.4) 68 89 (5.8) 69 149 (6.8) 70 159 (7.1)
Control Range 77-129
107 (2.1) 97 (4.5)
168 (1.9) 158 (3.1) 59" (1.6)
106 (3.4)
13 a (ND) 28" (1.3)
6" (ND)
43" (1.8) 31" (2.4) 51" (3.7)
175 (1.6) 118 (1.8) 157 (5.3) 137 (2.8) 175 (2.5) 216 (2.9)
97-230
196 191 193 207 168 137"
218 260 499
100" 75" 84"
636 137" 251 278 268 418
167-485
0.34 0.35 0.43 0.43 0.42 0.34
0.67 0.53 0.40
0.45 0.20" 0.48
0.52 0.37 0.43 0.43 0.56 0.60
0.29 - 0.51
0.40" 0.63 0.41" 0.5 5" 0.40" 0.87 ND 0.79 0.49 1.00 0.49 0.65
0.48 0.70 0.41" 0.50" 0.54 0.69
0.17" 0.37" 0.23" 0.37" 1.10 1.20
0.51 0.81 0.49 0.83
0.75 0.75
0.52 0.61 0.50 0.78
0.48 - 0.60- 0.62 0.83
0.36 0.17" 0.37 0.42 0.15" 0.10"
0.57 0.34" 0.33"
0.29" 0. 14a 0.99
0.29" 0.30" 0.28" 0.28" 0.43 0.48
0.35 - 0.50
"Values below control range; see text for definition of defects. mt = mitochondrial, ND = not done, RCR = respiratory control ratios (state 3 + ADPistate 4 - ADP respiration), TMPD = tetramethyl- p henylenediamine.
retinopathy developing before the age of 20 years, as- sociated with ataxia, increased cerebrospinal fluid pro- tein, or cardiac conduction defects), as defined by Berenberg and colleagues [31}. Only 1 of the 12 pa- tients with PEO and retinopathy did not have a muscle mtDNA deletion. Deletions of muscle mtDNA were not detected in 2 patients with the Kearns-Sayre syn- drome, 11 presenting with limb weakness alone, or 19 with predominant CNS disease of whom 9 also had PEO. The differences in clinical presentation between the two groups were highly significant (x2 = 37.1, p < 0.001). The degree of disability, as classified by Petty and colleagues 123, was not significantly different be- tween the two groups of patients overall, although se- vere disability in patients with deletions was confined to those with Kearns-Sayre syndrome.
Two patients with deletions had clinically (and simi- larly) affected relatives: the daughter of the sister of Patient 1 and the daughter of Patient 48 (see Table 1). Ten of the patients without deletions (from 8 families) had clinically affected relatives, and maternal transmis- sion to offspring had occurred in 5 of these lundreds.
There was no difference in mean maternal age at the time of birth (where this was known) of patients with- out an affected relative between 16 with muscle mtDNA deletions (28.2 5 5.1 years) and 22 without deletions (26.9 f 4.9, t = 0.79, p > 0.5).
Biochemical Stadies of Intact Mitochondria The results of biochemical studies of intact mitochon- dria are shown in Table 3. A patient was defined as having a polarographic defect exclusively involving complex I if state 3 oxygen uptake rates were below the control range utilizing the NAD-linked substrates pyruvate and glutamate but were normal with succi- nate and with ascorbate plus tetramethyl-phenylene diamine (TMPD). In 2 patients state 3 oxygen uptake rates were severely reduced with NAD-linked sub- strates, but were below the control range with succi- nate (Patient 27) or ascorbate plus TMPD (Patient 83). Those with defects involving complexes I to I11 (see Table 3) had comparable reductions of state 3 oxygen uptake rates utilizing pyruvate, glutamate, and succi- nate, but ascorbate plus TMPD rates were normal. In
Holt et al: Mitochondrial Myopathies 703

Table 4. Cowehtion Between Biochemical Defects, Minimum Deleted Regions of mtDNA, and Histochemical Features of 18 Patients with Mitochondria1 Myopathy
Deleted Reading Frames (Complex) Percent Fibers Percent
Abnormal No. Deleted Patient No. mtDNA I (ND) I11 (cyt b) IV (CO) V (A) tRNAs RR cox-
Complex I defect 5" 11 1 1
6 0 18 13
7 7 7 I11 6 8 5 32 47 I11 6 3 2 13 23
1 40 3,4L,4,5 - 11,111 6 3 5 9 20
48 35 3,4L,4,5 - I11 6 8 5 4 11
111 628 5 25 39 17 55 3,4L,4,5 - 26 50 3,4L,4,5 - I11 633 5 17 26 45 50 3,4L,4,5 - I11 6 8 5 13 33
- - - - - - -
2 40 192 16 80 5,6 25 65 5,6 85 50 5 $5 27 70 3,4L,4,5 83 55 3,4L,4 -
- - - - - - - -
Complex 1-111 defects
43 40 3,4L,4,5 - 11,111 6 8 8 8 24
Complex I-IV defects
Normal 30 60 3,4L,4,5 - I11 6 8 5 12 16 44 50 3,4L,4,5 - 111 698 5 14 16 67 45 3,4L,4,5 - 111 6 8 5 5 6 68 65 3,4L,4,5 - I11 6 8 5 14 33 69 55 3,4L,4,5 - I11 6 8 5 4 5 70 20 3,4L,4,5,6 - 11,111 698 6 5 12
"Ribosomal RNA genes also deleted. mtDNA = mitochondrial DNA, tRNA = transfer RNA, RR = w e d red, Cox- = cytochrome oxidase negative, N D = NADH coenzyme Q reductase subunits, cyt b = cytochrome b, CO = cytochrome oxidase, A = adenosine triphosphate synthase.
patients with I to IV defects state 3 oxygen uptake rates were reduced with all substrates tested. In cate- gories I to 111 and I to IV the function of complexes I, 11, and 111 were not assessed individually. Polaro- graphic studies were defined as normal in 6 patients; in 1 of these (Patient 44) the respiratory rate with ascor- bate plus TMPD fell below the control range.
The results of cytochrome spectral analyses are shown in Table 3. One of the 18 patients with dele- tions studied had low cytochrome b concentrations, compared with 11 of 31 without (latter data not shown; p < 0.05, Fisher's exact test). Low concenua- tions of cytochrome aa3 were frequent, occurring in 11 of the 18 patients, including 4 of the 6 patients with normal respiratory chain activity.
As has been observed before { 2 } (see Table l), there was no clear correlation between the site of the respiratory chain defect and the clinical features. The proportion of deleted mtDNAs and the range of de- leted reading frames in patients with various biochern- ical defects is shown in Table 4, and the clinical and biochemical features of patients with apparently identi- cal deletions are summarized in Table 5.
Table 5. Clinical and Biochemical Features of 13 Patients with Apparently Identical Deletions of Muscle mtDNA (Minimum Region 8,571-13,367 bpi
Clinical Syndrome Patient No. Biochemical Defect
PEO, limb weak- ness
PEO, limb weak- ness, retinop- athy
PEO, limb weak- ness, retinop- athy, ataxia, heart block
PEO, limb weak- ness, retinop- athy, ataxia, heart block, deafness
1 45 48 26 27 30 44 68 19 69
17 18 67
Complexes 1-111 Complexes I-IV Complexes 1-111 Complexes I-IV Predominantly complex I Normal Normal Normal Not studied Normal
Complexes I-IV Not studied Normal
mtDNA = mitochondrial DNA, bp = base pains, PEO = progres- sive external ophthalmoplegia
704 Annals of Neurology Vol 26 No 6 December 1989

Table 6. Polypeptide Projles of 10 Patients with Muscle mtDNA Deletions
Patient No. Complex I Complex I11 Complex IV
Complex I defects 2 Normal ND ND
16 Generalized reduction Subunit VI deficiency Normal 25 Normal ND ND 27 Generalized reduction Normal ND
30 Normal ND ND 44 Generalized reduction ND ND 67 Normal Normal Normal 68 Normal Normal Normal 69 Normal Normal Normal 70 Normal Normal Normal
Normal polarography
mtDNA = mitochondrial DNA, ND = not done.
Anulysis of Respiratory Conzpiex Polypeptides Respiratory chain polypeptides were analyzed by SDS polyacrylamide gel electrophoresis and immunoblot- ting in 10 of the 30 patients with deletions (Table 6). Two patients (Patients 16 and 27) with complex I de- fects had mild generalized reductions of complex I polypeptides, as did a further patient with normal biochemistry. Patient 16 also appeared to have a defi- ciency of subunit VI of complex 111, although subunit- specific antiserum was not available to confirm this result. Immunoblots of complex IV subunits in 5 patients, including 4 with deleted cytochrome oxidase genes, were normal. Seven of the 21 patients without mtDNA deletions had specific subunit deficiencies 127, 321.
Histochemical Studia The proportions of ragged red and cytochrome oxi- dase-negative fibers in transverse sections of muscle in the two groups of patients are dustrated in Figure 2. There appeared to be fewer patients with deletions of muscle mtDNA with a high proportion (> 25%) of ragged red fibers, but this was not quite significant at the 5% level (Fisher’s exact test). Conversely, more patients with deletions had a b h proportion (> 5%) of cytochrome oxidase-negative fibers, and this was significant ( p < 0.05). There was only 1 patient (Pa- tient 16) with a deletion and no cytochrome-negative fibers, compared with 8 without deletions. The dele- tion in this case did not include cytochrome oxidase genes, although this was also true of 3 others, including 2 with an identical deletion, shown to have cyto- chrome oxidase-negative fibers.
Discussion This study indicates that about 40% of unselected adult patients with mitochondrial myopathy have dele- tions of muscle mtDNA. The most striking difference
between patients with deletions and those without is in their clinical features. All the patients with deletions had PEO, compared with 24% of those without dele- tions. Deletions were detected in all but 1 of the 12 patients with PEO, retinopathy, and limb weakness, and 8 of 10 with the Kearns-Sayre syndrome. In con- trast, none of the patients with limb weakness alone or adult-onset multisystem CNS disease, including those with the syndromes of myoclonic epilepsy with ragged red fibers or mitochondrial encephalopathy, lactic acidosis, and suokelike episodes, had detectable mus- cle mtDNA deletions. Similar findings were reported by Zeviani and colleagues 1163, but our results do not support their suggestion that the finding of muscle mtDNA deletions in patients with Kearns-Sayre syn- drome reinforces the concept of this syndrome as a specific disorder. The majority of our patients with deletions did not have Kearns-Sayre syndrome, and 2 patients with the syndrome did not have detectable deletions.
There appears to be some correlation between the deleted regions of mtDNA and the biochemical find- ings in these patients (see Table 4). In the 4 patients with pure complex I defects, the deletion either exclu- sively involved the ND5 and 6 genes (complex I; Pa- tients 16, 25, and 85), or the ND1 and 2 reading frames together with ribosomal RNA and tRNA genes (Patient 2). This was in contrast to the 14 pa- tients with more extensive loss of respiratory chain activity or normal polarography, in whom the deletions included reading frames for three or more subunits of complex I, one or two subunits of complex IV, and two subunits of complex V, as well as several W A S . It is interesting that the 10 patients with apparently identical deletions that were investigated biochemically had different biochemical and clinical features (see Ta- ble 5) . Five of the 6 patients with normal muscle bio- chemistry were in this group.
Holt et al: Mitochondrial Myopathies 705

14
12
10
8
6
&
2
0
14 - I 1 0 No M t DNA deletion
No o f cases
Deleted population of MtDNA
10
0 10 20 30 40 50 0 10 20 30 40 50 60
%RRF %Cox-ve
Fig 2. Histograms illnstrating percentage of ragged red Jbws (RRF) and cytochrome oxiabse-negative (Cox - ve) fibers in sec- tions of muscle from patients with mitochondria1 myopathy with deletions of muscle mitochondrial DNA (hatched columns) and those without such deletions (open columns).
There was no obvious correlation between the pro- portion of abnormal mtDNA in the muscle sampled and either clinical or biochemical severity. All but three deletions (Patients 16,25, and 85) observed here definitely involved a number of tRNAs. These dele- tions would be expected to have a detrimental effect on translation of all mitochondrially encoded subunits of the respiratory chain, unless the deleted and normal mtDNAs are physically close enough to share tRNAs within mitochondria. The functional effects of deleted mtDNAs must depend on their distribution within mitochondria, muscle fibers, and different tissues. It is also possible that the total population of mtDNAs is increased in patients with deletions; the relative pro- portion of deleted mtDNAs would have less func- tional significance in these circumstances. The differ- ent polarographic findings in patients with identical deletions may therefore reflect the absolute amount of normal, as opposed to deleted, mtDNA within indi- vidual mitochondria or its distribution in the mito- chondrial population as a whole. The variable clinical features of these patients were presumably related to the amount of normal and deleted mtDNA in muscle, brain, retinal pigment epithelium, and other tissues.
It might be argued that deletions of mtDNA repre- sent an effect-perhaps the result of a metabolically abnormal intracellular environment-rather than a cause of the disease. Points against this include the finding that the deletions do not appear to vary within individual patients and also that the proportion of ab- normal mtDNAs is not correlated with age or either clinical or biochemical severity. The functional effects
of a deleted population of mtDNAs can only be re- solved in detail by studying them in vitro, perhaps using the mitochondrial transfection techniques of Iclng and Attardi C337.
Reduced concentrations of cytochrome b, a mito- chondrial product, were seen in 11 patients without deletions and only one in the deleted group. The dele- tion did not involve the cytochrome b gene in the latter case. Cytochrome aa3 concentrations were reduced in a high proportion of patients both with and without deletions. Cytochrome a a 3 is physically associated with subunits I1 and I11 of cytochrome oxidase, both mito- chondrial products, so in the patients with deletions this could be related to impaired assembly. There was an increased incidence of cytochrome oxidase-nega- tive muscle fibers in the patients with deletions, being present in all but 1 (Patient 16); this patient’s deletion did not include cytochrome oxidase subunits but she did have reduced cytochrome au3 concentrations. Cytochrome oxidase-negative fibers were seen in the 3 other patients with deletions not involving cyto- chrome oxidase genes.
Only 1 patient with a deletion of muscle mtDNA (Patient 16) appeared to have a specific polypeptide deficiency. This involved subunit VI of complex 111, a nuclear product, in association with a polarographic defect at complex I, but subunit-specific antiserum was not available to confirm this. Mitochondrially encoded subunit I1 of cytochrome oxidase (complex IV) ap- peared to be present in normal amounts, without change in molecular mass, in the 4 patients with dele- tions studied, although the deletion did not definitely involve this reading frame in 3 of these. These results contrast with those from 7 patients without deletions that had specific deficiencies of nuclearly encoded sub- units in the functionally abnormal complex [27, 327.
The survival of deleted mtDNA molecules in mus-
706 Annals of Neurology Vol 26 No 6 December 1989

cle, but not blood, in the patients reported here is compatible with the observation that the number of muscle fibers does not increase significantly after early fetal life [34). The same probably applies to the brain in patients with CNS involvement. On the other hand, frequent cell division in leukocyte precursors should select against cells containing functionally defective mitochondria. Persistence of the deleted mtDNAs in muscle and the CNS could be enhanced by a rep- licative advantage conferred by their reduction in length. It should be noted that none of the deletions in this series definitely included the origins of replication of either the light or heavy strands of mtDNA.
The patients with mitochondrial myopathy and dele- tions of muscle mtDNA had fewer clinically affected relatives than those without deletions. The rapid cell division that occurs during early oogenesis should se- lect against functionally defective mitochondria, mak- ing transmission of deleted mtDNAs from affected mother to child relatively unlikely. It seems probable that the deletions in most patients arose as mutations during oogenesis in their mothers. We found no evi- dence that this was related to maternal age. Presuma- bly, random partitioning of the two populations of mtDNA occurs during fetal development and partly accounts for tissue specificity.
The explanation for the association between PEO and muscle mtDNA deletions is unclear. Small mus- cles, such as the external ocular muscles, are derived from only a few embryonic cells; if one of these con- tained defective mtDNA, the eventual proportion of deleted molecules would be high compared with the same situation in a large limb muscle. The functional effects of deleted mtDNAs in the external ocular mus- cles might also be expected to be considerable, as their metabolic demands must be relatively high. Never- theless, there is no reason to suspect that these muscles are more susceptible to the effects of a deletion of mtDNA than a nuclear defect of mitochondrial pro- tein synthesis or a more subtle defect of the mitochon- drial genome.
The absence of detectable muscle mtDNA deletions in about 60% of patients with mitochondrial myopathy (including maternally transmitted cases) does not ex- clude the possibility of mtDNA mutations as the basis for their disease. The limitations of restriction endo- nuclease analysis mean that deletions of less than ap- proximately 100 bp, or point mutations, have not been excluded. Less drastic rearrangements of the mito- chondrial genome, particularly point mutations as re- cently identified in Leber’s optic atrophy 1351, are probably more likely to be transmitted.
The finding of specific deficiencies of nuclear- encoded subunits in some patients with mitochondrial myopathy suggests that mutations of nuclear genes are involved in some cases 127, 32). This would be ex-
pected in view of the occasional paternal transmission of these disorders and the numerical excess of nuclear, as opposed to mitochondrial, encoded subunits in the respiratory chain. It is theoretically possible that rntDNA deletions are secondary to a nuclear defect. A mother and daughter with mitochondrial myopathy and different muscle mtDNA deletions have recently been described {36}. This observation is difficult to explain but could result from a defect of mtDNA rep- lication, a process controlled by nuclear products. The most likely molecular mechanism for mtDNA dele- tion is one of intragenomic recombination, probably occurring during replication. The recent observation of direct repeats flanking the deleted region in patients with the most common type of deletion, that seen in 13 patients reported here, suggests that this is the case C37, 381.
This work was supported by the Brain Research Trust, the Muscular Dystrophy Group of Great Britain and Northern Ireland, the Re- search Trust for Metabolic Diseases of Childhood, and the Well- come Trust.
We wish to thank Drs. G. Attardi and M. King for supplying the mtDNA probes, Drs C. I. Ragan and V. Darley-Usmar for the respiratory chain antibodies, Marjorie Ellison for technical assis- tance, and the physicians at the National Hospitals for Nervous Diseases and elsewhere for referring patients.
References 1. DiMauro S, Bonilla E, Zeviani M, ct al. Mitochondrial my-
opathies. Ann Neurol 1985;17:521-538 2. Petty RKH, Harding AE, Morgan-Hughes JA. The clinical fea-
tures of mitochondrial myopathy. Brain 1984;109:915-938 3. Harding AE, Petty RKH, Morgan-Hughes JA. Mitochondrial
myopathy: a genetic study of 71 cases. J Med Genet 1988;
4. Rosing HS, Hopkins LC, Wallace DC, et al. Maternally inher- ited mitochondrial myopathy and myoclonic epilepsy. Ann Neurol 1985;17:228-237
5. Egger J, Wilson J. Mitochondrial inheritance in a mitochon- drially mediated disease. N Engl J Med 1983;309:142-145
6. Anderson S, Bankier AT, Barrell BG, et al. Sequence and or- ganisation of the human mitochondrial genome. Nature 1981;
7. Giles RE, Blanc H, Cann HM, Wallace DC. Maternal inher- itance of human mitochondrial DNA. Proc Natl Acad Sci USA 1980;77:6715-67 19
8. Chomyn A, Mariottini P, Cleeter MWJ, et al. Six unidentified reading frames of human mitochondrial DNA encode compo- nents of the respiratory chain NADH dehydrogenase. Nature
9. Chomyn A, Mariottini P, Cleeter MWJ, ec al. Functional assign- ment of the unidentified reading frames of human mitochon- drial DNA. In: Quagliariello E et al, eds. Achievements and perspectives of mitochondrial research, vol 2: Biogenesis. Am- sterdam: Elsevier, 1985:259-2 75
10. Yatscoff RW, Goldstein S, Freedman KB. Conservation of genes coding for proteins synthesised by human mitochondria. Somatic Cell Genet 1978;4:633-645
25:528-535
290:45 7-465
1985;314:592-597
Holt et al: Mitochondrial Myopathies 707

1 1. Holt IJ, Harding AE, Morgan-Hughes JA. Mitochondrial DNA polymorphsm in mitochondrial myopathy. Hum Genet 1988; 79:53-57
12. Poulton J, Turnbull DM, Mehta AB, et al. Restriction enzyme analysis of the mitochondrial genome in mitochondrial my- opathy. J Med Genet 1988;25:600-605
13. Holt IJ, Harding AE, Morgan-Hughes JA. Deletions of mito- chondrial DNA in patients with mitochondrial myopahes. Na- ture 1988;331:717-719
14. Holt IJ, Cooper JM, Morgan-Hughes JA, Harding AE. Dele- tions of muscle mitochondrial DNA. Lancet 1988;1:1462
15. Lestienne P, Ponsot G. Kearns-Sayre syndrome with muscle mitochondrial DNA deletion. Lancet 1988;1:885
16. Zeviani M, Moraes CT, DiMauro S, et al. Deletions of mito- chondrial DNA in Kearns-Sayre syndrome. Neurology 1988;
17. Poulton J, Deadman ME, Gardiner RM. Duplications of mito- chondrial DNA in mitochondrial myopathy. Lancet 1989;l:
18. Morgan-Hughes JA, Mair WGP. Atypical muscle mitochondria in oculoskeletal myopathy. Brain 1973;96:215-224
19. Morgan-Hughes JA, Darveniza P, Kahn SN, et al. A mitochon- drial myopathy characterised by a deficiency in reducible cyto- chrome b. Brain 1977;100:617-640
20. Morgan-Hughes JA, Darveniza P, Landon DN, et al. A mito- chondrial myopathy with a deficiency of respiratory chain NADH-Co-Q reductase activity. J Neurol Sci 1979;43:27-46
21. Morgan-Hughes JA, Hayes DJ, Clark JB, et al. Mitochondrial encephalomyopathies: biochemical studies in two cases reveal- ing defects in the respiratory chain. Brain 1982;105:553-582
22. Hayes DJ, Lecky BRF, Landon DN, et al. A new mitochondrial myopathy: biochemical studies revealing a deficiency in the cytochrome b-c, complex (complex 111) of the respiratory chain. Brain 1984;107:1165-1177
23. Old JM. Fetal DNA analysis. In: Davies KE, ed. Human genetic hseases: a practical approach. Oxford: W. Press, 1986:l-17
24. kgby PWJ, Diekmann M, Rhodes C, Berg P. Labelling DNA to hgh specific activity in vitro by nick translation with DNA polymerase I. J Mol Biol 1977;113:237-351
25. Feinberg AP, Vogelstein B. A technique for radiolabelling DNA restriction endonuclease fragments to high specific activ- ity. Anal Biochem 1983;132:6-13
38:1339-1346
236-240
26. Davies KLJ, Packer L, Brooks GA. Biochemical adaptation of mitochondria, muscle and whole animal respiration to endur- ance training. Arch Biochem Biophys 1981;209:539-543
27. Schapira AHV, Cooper JM, Morgan-Hughes JA, et al. Molecu- lar basis of mitochondrial myopathies: polypeptide analysis in complex I deficiency. Lancet 1988;1:500-503
28. Laemmli UK. Cleavage of structural proteins during the assem- bly of the head of bacteriophage T4. Nature 1970;227:680-685
29. Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets. Pro- cedures and some applications. Proc Natl Acad Sci USA 1979; 76:4350-4354
30. SeLgman AM, Karavowsky MJ, Wasserkrug HL, Hawker JS. Non droplet ultrastructural demonstration of cytochrome oxi- dase activity with a polymerizing osmophilic reagent, diamino- benzidine. J Cell Biol 1968;38:1-14
31. Berenberg RA, Pellock JM, DiMauro S, et al. Lumping or split- ting? “Ophthalmoplegia-plus” or Kearns-Sayre syndrome? Ann Neurol 1977;1:37-54.
32. Morgan-Hughes JA; Schapira AHV, Cooper JM, Clark JB. Molecular defects of NADH-ubiquinone oxidoreductase (com- plex I) in mitochondrial diseases. J Bioenerg Biomembr 1988; 20:365-382
33. King MP, Attardi G. Injection of mitochondria into human cells leads to a rapid replacement of the endogenous mitochondrial DNA. Cell 1988;52:811-819
34. Stickland NC. Muscle development in the human fetus as exemplhed by m. sartorius: a quantitative study. J Anat 1981; 132~557-579
35. Wallace DC, Singh G, Lott MT, et al. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science 1988;242:1427-1430
36. Ozawa T, Yoneda M, Tan& M, et al. Maternal inheritance of deleted mitochondrial DNA in a family with mitochondrial my- opathy. Biochem Biophys Res Commun 1988;154:1240-1247
37. Schon EA, Rizzuto R, Moraes CT, et al. A direct repeat is a hotspot for large-scale deletion of human mitochondrial DNA. Science 1989;244:346-349
38. Holt IJ, Harding AE, Morgan-Hughes JA. Deletions of muscle mitochondrial DNA in mitochondrial myopathies: sequence analysis and possible mechanisms. Nucleic Acids Res 1989;17: 4465-4469
708 Annals of Neurology Vol 26 No 6 December 1989




















