
AZFc deletions and spermatogenic failure: a population-based survey of 20,000 Y chromosomes
Upload
khangminh22Category
view
2download
0
Genotype-phenotype correlation of contiguous gene deletions of SLC6A8, BCAP31 and ABCD1
2.2
van de Kamp JM, Errami A, Howidi M, Anselm I, Winter S, Phalin-Roque J, Osaka H, van Dooren SJM,
Mancini GM, Steinberg SJ, Salomons GS
Submitted

50
Chapter 2 Male patients
ABSTRACT
The BCAP31 gene is located between SLC6A8, associated with X-linked creatine
transporter deficiency, and ABCD1, associated with X-linked adrenoleukodystrophy.
Deletions involving both ABCD1 and BCAP31 cause neonatal lethal “contiguous ABCD1
DXS1357E deletion syndrome” (CADDS). Deletions of SLC6A8 are associated with an
atypical severe phenotype which might be explained by involvement of BCAP31. However,
the deletion size in most reported patients is unknown. Recently, isolated defects of
BCAP31 were found to cause severe developmental delay with deafness and dystonia.
We characterized the breakpoints in eight patients with deletions of SLC6A8, BCAP31,
and/or ABCD1 and studied the genotype-phenotype correlations in these patients and
two previously reported patients with known deletion size. Contiguous gene deletions
involving BCAP31 caused profound developmental delay, failure to thrive, hearing loss, and
childhood death, probably mainly due to loss of BCAP31. Only deletions involving both
BCAP31 and ABCD1 were associated with hepatic cholestasis and death before one year
which might be explained by synergistic effects of loss of ABCD1 and BCAP31. Remarkably,
a patient with isolated deletion of the 3’end of SLC6A8 had a similar phenotype as seen in
SLC6A8-BCAP31 deletions but without deafness. This might be caused by disturbance of a
regulatory element between SLC6A8 and BCAP31.

Contiguous gene deletions of SLC6A8, BCAP31 and ABCD1
51
2.2
INTRODUCTION
Two disease genes, SLC6A8 and ABCD1, lie close together on chromosome Xq28.
Loss-of-function mutations in SLC6A8 cause X-linked creatine transporter deficiency
(CRTR-D), which is characterized by severely reduced creatine on 1H-magnetic resonance
spectroscopy (1H-MRS) of the brain. Male patients present with intellectual disability with
severe speech delay, behavioral problems, and seizures. The diagnosis is further based on
increased creatine/creatinine ratio in urine and/or deficient creatine uptake in cultured
fibroblasts.1 Loss-of-function mutations in ABCD1 cause X-linked adrenoleukodystrophy
(X-ALD), which is characterized by reduced β-oxidation of very long chain fatty acid
(VLCFA), demyelination of white matter, and adrenal cortex atrophy with a wide range
of phenotypic expression. The most severe, childhood cerebral, form has an onset
after three years of age and is characterized by neurological deterioration starting with
behavioral problems and learning deficits and progressing to total disability and death. The
characteristic elevations of VLCFA in plasma are already present in the first weeks of life.2
BCAP31 is located, in reverse orientation, between SLC6A8 and ABCD1. In 2002 three
patients with deletions of ABCD1 extending into the neighboring BCAP31 (DXS1357E)
were described.2 They had increased VLCFA as in X-ALD but otherwise a severe neonatal
presentation with profound hypotonia and development delay, hepatic cholestasis, and
death in the first year of live. The syndrome was named “contiguous ABCD1 DXS1357E
deletion syndrome” (CADDS). The exact extent of the deletions was not determined.
In 2012 a fourth CADDS patient with a similar phenotype was described with a large
deletion containing (part of) BCAP31, ABCD1, PLXNB3, SRPK3, IDH3G, SSR4, and PDZD4.3
Centromeric to BCAP31, large deletions involving SLC6A8 were reported in three
patients with a more severe presentation than in classic CRTR-D with pronounced
hypotonia and development delay, severe failure to thrive, and dystonia or choreathethoid
movements.4, 5 In one of these patients the deletion extended into BCAP31.5 However, the
exact deletion size was unknown in the other two.4
A role was suggested for BCAP31 in the atypical and severe presentation of deletions
of ABCD1 or SLC6A8 extending into BCAP31. Just recently, loss-of-function mutations
in BCAP31 were found to cause severe motor and intellectual disability, dystonia,
sensorineural deafness, failure to thrive, and early death.6
Conclusions regarding the contribution of the separate genes in contiguous gene
deletions involving SLC6A8, BCAP31, and/or ABCD1 were hampered by the fact that
the deletion size was unclarified in five2, 4 of the seven reported deletion patients. We

52
Chapter 2 Male patients
characterized the breakpoints in these five patients and provide an update of the patients
who were alive at the time of the previous report.4, 5 In addition, we describe two new
patients with a BCAP31- ABCD1 deletion (CADDS) and one patient with an isolated partial
SLC6A8 deletion. We discuss the genotype-phenotype correlations in these eight patients
and the two patients of whom the deletion size was already well characterized.3, 5
MATERIALS AND METHODS
Materials and Patients
DNA was isolated from blood or cultured fibroblasts of eight patients with suspected
deletions of SLC6A8 and/or ABCD1. Three patients were suspected of SLC6A8 deletions
and five patients of ABCD1 deletions based on clinical and biochemical features and the
absence of polymerase chain reaction (PCR) products of the involved gene. Case reports
of two of the patients with SLC6A8 deletions were previously reported.4 Three of the
patients with ABCD1 deletions were previously reported and involvement of BCAP31 was
already shown by PCR analysis of a marker in this gene.2
Breakpoint analysis
Multiplex ligation-dependent probe amplification (MLPA) using the P049 kit with probes
for several exons of SLC6A8, BCAP31, ABCD1, and neighboring genes, was performed to
confirm the deletions and to estimate their size. To narrow down the regions of the
breakpoint, PCRs of about 200 base pairs in intervals of approximately 5-10 kilobase were
designed flanking the deleted MLPA probes. Finally, long-range PCR over the breakpoint
was performed followed by DNA sequencing to reveal the exact breakpoints. All primers
were designed with a high specificity for the X chromosome, as a paralogous gene region
occurs on chromosome 16.7
RNA analysis of BCAP31
RNA was isolated from fibroblasts of patient 2-6 and the patient previously reported
by Osaka et al.5 Subsequently cDNA was synthesized using oligodT. To amplify specific
regions of the transcripts (i.e. exons 1-8, exons 1-4, and exons 5-8), primers were
designed in exons 1, 4, 5 and 3’UTR of the BCAP31 gene.

Contiguous gene deletions of SLC6A8, BCAP31 and ABCD1
53
2.2
RESULTS
Breakpoint analysis
The breakpoints were sequenced by long-range PCR in seven of the eight patients
(Supplementary file). Long-range PCR was unsuccessful in patient 6. However, the region
of the breakpoint was narrowed down by MLPA analysis showing the presence of exon
8 but deletion of exon 7 of ABCD1 and exon 5 of BCAP31. A specific PCR in exon 8 of
BCAP31 indicated its presence and thus the breakpoint was located between exons 7
and 8 of ABCD1 and between exons 5 and 8 of BCAP31. Two patients had isolated partial
SLC6A8 deletions and eight a contiguous gene deletion involving BCAP31 and SLC6A8 and/
or ABCD1 (Figure 1, Table 1).
Figure 1. Location and size of the deletions on Xq28. Deletions are depicted with black bars. The exact breakpoint in patient 6 is unknown and the uncertainty regarding the involvement of exon 6 and 7 of BCAP31 in the deletion is depicted by a gray bar.
RNA analysis of BCAP31
RT-PCR detected BCAP31 transcripts of exons 1-8 in RNA isolated from fibroblasts of
patient 2. In RNA from fibroblasts of patient 3 and the patient reported by Osaka et al.5
only a transcript of exons 1-4 was detected. In patients 4-6 no BCAP31 transcripts were
detected (Supplementary file).
Genotype-phenotype correlation
The clinical features of the patients are summarized in Table 1. The patients with contiguous
gene deletions involving BCAP31 shared many features: profound developmental delay,
severe failure to thrive, sensorineural hearing loss, and childhood death. Seizures occurred

54
Chapter 2 Male patients
Table 1. Clinical features.
Patient 1 2 Osaka et al. 2012
3 4 5 6 7 8 Iwasa et al. 2012
CRTR-D BCAP31 XL-ALD
Deletion size 2.1 kb 4.9 kb 19 kb 40 kb 110 kb 64 kb 34-42 kb 50 kb 31 kb 90 kb mutation mutation mutation
Involved genes SLC6A8 SLC6A8 SLC6A8, BCAP31
PNCK, SLC6A8, BCAP31
PNCK, SLC6A8, BCAP31, ABCD1, PLXNB3, SRPK3
SLC6A8, BCAP31, ABCD1
BCAP31, ABCD1 BCAP31, ABCD1 BCAP31, ABCD1 BCAP31, ABCD1, PLXNB3, SRPK3, IDH3G, SSR4, PDZD4
SLC6A8 BCAP31 ABCD1
Age 40 years Died 8 years septic shock
9 years Died 8 years unknown cause
Died < 5 months
Died 4 months LF, RF
Died 8 months Died 11 months LF, GI bleeding
Died 4 months RF, GI bleeding
Died 8 months pneumonia, sepsis
Normal live expectancy
Death 7 months-24 yearsa
Average death at 9.4 years
Development Walking at 2 years, speaks single words
Smiles, eye contact, no milestones attained
Profound delay, no head control
Some eye contact, no milestones attained
? Profound delay Delayed, smiles, alert and active at 4 months, sedated at 7 months.
Profound delay Profound delay No milestones attained
Mild-severe delay, walking at mean age of 2 years
No milestones attained or only head controla
Early development normal, onset neurological deterioration > 3 years
Motor symptoms
- Profound axial hypotonia, hypertonic limbs, quadriplegia
Hypertonic Profound neonatal hypotonia
Hypertonic Profound neonatal hypotonia
Hypotonia Profound neonatal hypotonia
Profound neonatal hypotonia
Hypotonia Mild hypotonia Pyramidal signs, quadriplegia
Neurological deterioration > 3 years
Extrapyramidal - Dystonia from 3 months; severe choreoathetosis from 4-5 years
Severe dystonia and athetosis from 4 months
Severe choreoathetosis from 3 years
? - - Frequent episodes of opisthotonus, bruxism
- - Some patients, mild
Severe dystonia -
Seizures (onset) 2 years - 4 years status 4 years ? 2 months - - 2 months - +/- +/- -
FTT - ++b ++b ++b ? ++(with IUGR)
++b ++ ++ ++(with IUGR)c
+/- ++ -
Microcephaly - -d ? -d ? ? -d ? ? +++e - +f -
Hepatic - - Mildly elevated liver transaminases
Transient elevated liver transaminases
Cholestasis Cholestasis Cholestasis Cholestasis Cholestasis Cholestasis - Transient elevated liver transaminases
-
Congenital SNHL
- g - + + + ? ? + + + - + -
Opthalmological - Strabismus Does not persuit objects
Pigmentary retinopathy at 3 years
? ? - Cataract ? Does not see Strabismus Strabismus, optic atrophy
-
Dysmorphic Mild - - +h ? - Mildi - - +j Mild atypical +/- -
Brain MRI nd T2 hyperintensities periatrial WM, thin CC
T2 hyperintensities GP, decreased volume WM, myelination delay, very thin CC; cerebellar vermian atrophy
T2 hyperintensities in BG, myelination delay, thin CC
Mild focal dilatation left Sylvian fissure, immature myelination
Myelination delay
No abnormalities
nd Diffuse increased T1 hyperintensities WM
Enlarged ventricles, thin CC, thin WM, myelination delayk
Mild myelination delay, T2 hyperintensities, thin CC, enlarged ventricles, cerebral/ cerebellar atrophy
Periventricular hypomyelination, cerebral/cerebellar atrophy
Predominantly parieto-occipital WM abnormalities
Adrenal nd nd nd nd ? Small adrenal glands
nd nd nd Adrenal hypoplasy
nd nd Addison’s disease

Contiguous gene deletions of SLC6A8, BCAP31 and ABCD1
55
2.2
Table 1. Clinical features.
Patient 1 2 Osaka et al. 2012
3 4 5 6 7 8 Iwasa et al. 2012
CRTR-D BCAP31 XL-ALD
Deletion size 2.1 kb 4.9 kb 19 kb 40 kb 110 kb 64 kb 34-42 kb 50 kb 31 kb 90 kb mutation mutation mutation
Involved genes SLC6A8 SLC6A8 SLC6A8, BCAP31
PNCK, SLC6A8, BCAP31
PNCK, SLC6A8, BCAP31, ABCD1, PLXNB3, SRPK3
SLC6A8, BCAP31, ABCD1
BCAP31, ABCD1 BCAP31, ABCD1 BCAP31, ABCD1 BCAP31, ABCD1, PLXNB3, SRPK3, IDH3G, SSR4, PDZD4
SLC6A8 BCAP31 ABCD1
Age 40 years Died 8 years septic shock
9 years Died 8 years unknown cause
Died < 5 months
Died 4 months LF, RF
Died 8 months Died 11 months LF, GI bleeding
Died 4 months RF, GI bleeding
Died 8 months pneumonia, sepsis
Normal live expectancy
Death 7 months-24 yearsa
Average death at 9.4 years
Development Walking at 2 years, speaks single words
Smiles, eye contact, no milestones attained
Profound delay, no head control
Some eye contact, no milestones attained
? Profound delay Delayed, smiles, alert and active at 4 months, sedated at 7 months.
Profound delay Profound delay No milestones attained
Mild-severe delay, walking at mean age of 2 years
No milestones attained or only head controla
Early development normal, onset neurological deterioration > 3 years
Motor symptoms
- Profound axial hypotonia, hypertonic limbs, quadriplegia
Hypertonic Profound neonatal hypotonia
Hypertonic Profound neonatal hypotonia
Hypotonia Profound neonatal hypotonia
Profound neonatal hypotonia
Hypotonia Mild hypotonia Pyramidal signs, quadriplegia
Neurological deterioration > 3 years
Extrapyramidal - Dystonia from 3 months; severe choreoathetosis from 4-5 years
Severe dystonia and athetosis from 4 months
Severe choreoathetosis from 3 years
? - - Frequent episodes of opisthotonus, bruxism
- - Some patients, mild
Severe dystonia -
Seizures (onset) 2 years - 4 years status 4 years ? 2 months - - 2 months - +/- +/- -
FTT - ++b ++b ++b ? ++(with IUGR)
++b ++ ++ ++(with IUGR)c
+/- ++ -
Microcephaly - -d ? -d ? ? -d ? ? +++e - +f -
Hepatic - - Mildly elevated liver transaminases
Transient elevated liver transaminases
Cholestasis Cholestasis Cholestasis Cholestasis Cholestasis Cholestasis - Transient elevated liver transaminases
-
Congenital SNHL
- g - + + + ? ? + + + - + -
Opthalmological - Strabismus Does not persuit objects
Pigmentary retinopathy at 3 years
? ? - Cataract ? Does not see Strabismus Strabismus, optic atrophy
-
Dysmorphic Mild - - +h ? - Mildi - - +j Mild atypical +/- -
Brain MRI nd T2 hyperintensities periatrial WM, thin CC
T2 hyperintensities GP, decreased volume WM, myelination delay, very thin CC; cerebellar vermian atrophy
T2 hyperintensities in BG, myelination delay, thin CC
Mild focal dilatation left Sylvian fissure, immature myelination
Myelination delay
No abnormalities
nd Diffuse increased T1 hyperintensities WM
Enlarged ventricles, thin CC, thin WM, myelination delayk
Mild myelination delay, T2 hyperintensities, thin CC, enlarged ventricles, cerebral/ cerebellar atrophy
Periventricular hypomyelination, cerebral/cerebellar atrophy
Predominantly parieto-occipital WM abnormalities
Adrenal nd nd nd nd ? Small adrenal glands
nd nd nd Adrenal hypoplasy
nd nd Addison’s disease
56
Chapter 2 Male patients
Patient 1 2 Osaka et al. 2012
3 4 5 6 7 8 Iwasa et al. 2012
CRTR-D BCAP31 XL-ALD
Other symptoms
Episodes of high fever, hydronephrosis
Hydronephrosis Thymus hypoplasia
Unexplained episodic fever
Urinary cr/rn 1.84 3.8 6.24 1.7 nd nd nd nd nd nd 1.5-6.3 (<10 yrs); 0.74-4.8 (>10yrs)
nd nd
VLCFA C26:0 (ug/ml) C26:1 (ug/ml) C26/22 C24/22
nd Normal Normal Normal Iincreased: 4.082 2.583 0.193 1.798
Increased: 2.98 2.34 0.18 1.85
Increased (fibroblasts)
Increased: 2.09 2.02 0.16 1.56
Increased: 2.48 1.86 0.21 1.65
Increased: 0.496 3.29
nd nd Increased: 1.18+/-0.53 0.19+/-0.05 0.07+/-0.04 1.49+/-0.45
Other biochemical abnormalities
Possible mitochondrial dysfunctionl, elevated NH3 during febrile episodes
Possible mitochondrial dysfunctionm, elevated NH3 during febrile episodes
Mildly elevated NH3, normal lactate and organic acids
Possible mitochondrial dysfunction (rare)n
Reference 1 1, 4 (pat 1), 22 5 4 (pat 2) Unpublished 2 (pat 3) Unpublished 2 (pat 1) 2 (pat 2) 3 1 6 2
a one exceptional patient is alive at 13 yrs and attained sitting, autonomous wheelchair and simple sign language; b height and weight about -3 to -4 SD; c height and weight -6 SD; d head circumference about -1.5 to -2.5 SD; e head circumference -10 SD; f head circumference between -2 and -5 SD; g normal audiogram at 4 yrs, mild low frequency hearing loss at 36 years; h thin cupped ears, deep set eyes, prominent nasal bridge, high-arched palate with submucous cleft, 5th finger clinodactyly; i frontal bossing, midface hyoplasia, pointed chin; j flat orbital edge, hypoplastic nose, micrognathia, light brown hair, a salmon patch on forehead, inguinal hernia, micropenis, cryptorchidism, arachnodactyly, club feet, hirsute back; k brain autopsy showed hypomyelination, secondary hypoxic-ischemic changes, heterotopia and dysplasia of inferior olivary nuclei; l transient elevated lactate, increased excretion tricarboxylic acid (TCA) intermediates, ethyl-malonic acid and 3-methylglutaconic acid but normal respiratory chain complex activities and elctron microscopy of mitochondria in muscle biopsy; m transient elevated plasma and CSF lactate, increased excretion TCA intermediates, decreased activity respiratory chain complex I, III and IV in muscle biopsy; n reported in one patient with SLC6A8 mutation.Abbreviations: BG = basal ganglia, CC = corpus callosum, Cr/Crn = creatine to creatinine ratio, FFT = failure to thrive, GI= gastro intestinal, GP = globus pallidus, LF= liver failure, MRI = magnetic resonance spectroscopy, nd = not detected, RF=respiratory failure, SNHL= sensorineural hearing loss, VLCFA = very long chain fatty acids, WM = white matter, ? = unknown, - = absent, + = present
Table 1. Continued
in some. The patients with deletions involving both BCAP31 and ABCD1 (n=6) all developed
cholestatic liver disease and died in the first year of life. It is not documented whether
the cause of death was related to liver failure in all cases. In contrast, the patients with
deletions of SLC6A8 and BCAP31 but not ABCD1 (n=2) did not develop cholestatic liver
disease, survived until at least 6 years and developed severe dystonia and choreoathethosis
after 3 years. The patient with an isolated deletion of exons 8-13 of SLC6A8 had the

Contiguous gene deletions of SLC6A8, BCAP31 and ABCD1
57
2.2
Patient 1 2 Osaka et al. 2012
3 4 5 6 7 8 Iwasa et al. 2012
CRTR-D BCAP31 XL-ALD
Other symptoms
Episodes of high fever, hydronephrosis
Hydronephrosis Thymus hypoplasia
Unexplained episodic fever
Urinary cr/rn 1.84 3.8 6.24 1.7 nd nd nd nd nd nd 1.5-6.3 (<10 yrs); 0.74-4.8 (>10yrs)
nd nd
VLCFA C26:0 (ug/ml) C26:1 (ug/ml) C26/22 C24/22
nd Normal Normal Normal Iincreased: 4.082 2.583 0.193 1.798
Increased: 2.98 2.34 0.18 1.85
Increased (fibroblasts)
Increased: 2.09 2.02 0.16 1.56
Increased: 2.48 1.86 0.21 1.65
Increased: 0.496 3.29
nd nd Increased: 1.18+/-0.53 0.19+/-0.05 0.07+/-0.04 1.49+/-0.45
Other biochemical abnormalities
Possible mitochondrial dysfunctionl, elevated NH3 during febrile episodes
Possible mitochondrial dysfunctionm, elevated NH3 during febrile episodes
Mildly elevated NH3, normal lactate and organic acids
Possible mitochondrial dysfunction (rare)n
Reference 1 1, 4 (pat 1), 22 5 4 (pat 2) Unpublished 2 (pat 3) Unpublished 2 (pat 1) 2 (pat 2) 3 1 6 2
a one exceptional patient is alive at 13 yrs and attained sitting, autonomous wheelchair and simple sign language; b height and weight about -3 to -4 SD; c height and weight -6 SD; d head circumference about -1.5 to -2.5 SD; e head circumference -10 SD; f head circumference between -2 and -5 SD; g normal audiogram at 4 yrs, mild low frequency hearing loss at 36 years; h thin cupped ears, deep set eyes, prominent nasal bridge, high-arched palate with submucous cleft, 5th finger clinodactyly; i frontal bossing, midface hyoplasia, pointed chin; j flat orbital edge, hypoplastic nose, micrognathia, light brown hair, a salmon patch on forehead, inguinal hernia, micropenis, cryptorchidism, arachnodactyly, club feet, hirsute back; k brain autopsy showed hypomyelination, secondary hypoxic-ischemic changes, heterotopia and dysplasia of inferior olivary nuclei; l transient elevated lactate, increased excretion tricarboxylic acid (TCA) intermediates, ethyl-malonic acid and 3-methylglutaconic acid but normal respiratory chain complex activities and elctron microscopy of mitochondria in muscle biopsy; m transient elevated plasma and CSF lactate, increased excretion TCA intermediates, decreased activity respiratory chain complex I, III and IV in muscle biopsy; n reported in one patient with SLC6A8 mutation.Abbreviations: BG = basal ganglia, CC = corpus callosum, Cr/Crn = creatine to creatinine ratio, FFT = failure to thrive, GI= gastro intestinal, GP = globus pallidus, LF= liver failure, MRI = magnetic resonance spectroscopy, nd = not detected, RF=respiratory failure, SNHL= sensorineural hearing loss, VLCFA = very long chain fatty acids, WM = white matter, ? = unknown, - = absent, + = present
same severe presentation with death at 8 years as the two patients with SLC6A8-BCAP31
deletions but without sensorineural hearing loss. In contrast, the patient with an isolated
deletion of exons 5-12 of SLC6A8 had a phenotype consistent with classic CRTR-D.
DISCUSSION
The phenotype of patients with contiguous gene deletions involving BCAP31 differed
substantially from the phenotype of respectively isolated loss of SLC6A8 (causing CRTR-D)
and ABCD1 (causing XL-ALD), suggesting an important role for BCAP31.
BCAP31 encodes B-cell-receptor associated protein 31 (BAP31), a 28-kDa (246 amino
acid) integral membrane protein that is localized in the endoplasmatic reticulum (ER)
membrane.8, 9 It is a protein-sorting factor that associates with newly synthesized integral
membrane proteins and controls their very different fates (egress, retention, survival,
and degradation).10 However, BAP31 is also involved in apoptosis, participating in ER-
mitochondrial apoptosis signaling. The mitochondrial fission protein Fission 1 (Fis1) interacts
with BAP31 at the ER, forming a platform for recruitment and activation of procaspase-8
during Fas-mediated apoptosis.11, 12 BCAP31 is cleaved by caspase-8, generating a p20
BAP31 that remains integrated in the membrane.9, 13 p20 induces apoptosis9 by causing
a rapid transmission of ER calcium into the mitochondria leading to mitochondrial

58
Chapter 2 Male patients
recruitment of dynamin-like protein 1 (Dlp1), which results in mitochondrial fission.13
In contrast, full-length BAP31 inhibits Fas-mediated apoptosis.14, 15 BAP31 also associates
with components of the cytoskeleton actomyosin complex, suggesting that BAP31 may
play a role in the structural organization of the cytoplasm.16
Recently, loss-of-function mutations in BCAP31 were found in seven individuals from
three families presenting with severe motor and intellectual disability, early dystonia,
sensorineural deafness, hypomyelination, failure to thrive, and early death. Fibroblasts
of the affected individuals showed altered ER morphology and disorganized Golgi but
contrary to expectation no massive accumulation of unfolded proteins or exacerbated cell
death.6 The profound developmental delay, sensorineural hearing loss, failure to thrive,
and childhood death in the patients with contiguous gene deletions involving BCAP31 are
very similar to the consequences of isolated BCAP31 defects and confirm the association
of loss between BCAP31 and this phenotype.
Neonatal hepatic cholestasis leading to liver failure and death in the first year was
restricted to the patients with deletions involving both ABCD1 and BCAP31 and is not
described in isolated ABCD1 defects2 nor in isolated BCAP31 defects.6 Only transient
increases of liver transaminases, mainly during febrile episodes, were found in patients
with deletions of BCAP31-SLC6A8 and were also described in patients with isolated BCAP31
defects.6 Therefore the hepatic cholestasis appears to result from the combined loss of
BCAP31 and ABCD1. Loss of BCAP31 might aggravate the peroxisomal disturbance caused
by loss of ABCD1. BAP31 interacts with Fis111 which is, together with dynamin-like protein
1 (Dlp1), implicated in both mitochondrial and peroxisomal fission.17 A heterozygous
dominant negative mutation in Dlp1 caused a defect of fission of both mitochondria and
peroxisomes manifesting with abnormally formed mitochondria and peroxisomes in a
neonate who died at one month of age with abnormal brain development, optic atrophy,
lactic academia and mildly elevated plasma VLCFA.17 However, peroxisomes of normal
size and number were reported in patients with BCAP31-ABCD1 deletions.2, 11 Otherwise,
VLCFAs are (potentially) toxic in CNS and adrenal glands18 and BAP31 deficient cells
might be more sensitive to these effects due to deregulated apoptosis.
Severe dystonia and choreoathethosis only occurred in patients with SLC6A8-BCAP31
deletions. However, dystonia is probably related to loss of BCAP31 because it was also
described in isolated BCAP31 defects and the patients with BCAP31-ABCD1 deletions died
before these symptoms usually appear.

Contiguous gene deletions of SLC6A8, BCAP31 and ABCD1
59
2.2
Remarkably, the patient with an isolated deletion of SLC6A8 exons 8-13 had the same
severe phenotype as patients with deletions or mutations of BCAP31 but without the
hearing loss or increases of liver transaminases. We detected BCAP31 mRNA in fibroblasts
of this patient, which argues against an effect on BCAP31 transcription. Alternatively, the
severe phenotype might be due to the complete loss of SLC6A8. However, deletion of
exons 5-12 of SLC6A8 was not associated with this severe presentation. Therefore, only
deletions extending beyond the 3’end of SLC6A8 seem associated with a severe phenotype
which suggests a disturbance of regulatory elements in the non-coding region between
SLC6A8 and BCAP31 leading to a comparable phenotype as in BCAP31 defects but without
hearing loss. Interestingly, one of the three described isolated defects of BCAP31 in fact
concerned a deletion of exon 8 including BCAP31 and part of the 3’UTR of SLC6A8.
Normal cerebral creatine levels were found in this patient suggesting that SLC6A8 was not
involved in the phenotype although SLC6A8 mRNA in patient fibroblasts was reduced.6
In three patients additional genes were involved in the deletion and might have
contributed to their phenotype. The patient reported by Iwasa,3 with the largest deletion,
had very profound microcephaly which was not found in any of the other patients and
might be due to genes deleted only in this patient and possibly also in patient 4 of whom
limited clinical information is known. PLXNB3 might contribute to the cerebral features
as it is most abundantly expressed in the brain and is a potent stimulator of neurite
outgrowth.19 In support of this, a patient with a deletion of exon 3-10 of ABCD1 and
exons 1-2 of PLXNB3 presented with convulsions from 2 years, predominantly occipital
white mater abnormalities at 3 years, and possible developmental delay.20 PNCK which was
involved in patient 3 and 4 is also mainly expressed in the brain.21 No isolated defects of
this gene have been reported so far.
In conclusion we confirm that BCAP31 deficiency is associated with profound
developmental delay, sensorineural hearing loss, failure to thrive, severe dystonia,
increases of liver transaminases, and childhood death. However, only deletions involving
both BCAP31 and ABCD1 are associated with hepatic cholestasis and death in the first
year, probably due to synergic effects of loss of these two genes. Isolated deletions of
SLC6A8 extending beyond the 3’end are associated with a severe phenotype comparable
to the phenotype in loss of BCAP31 but without the hearing loss. This might be caused by
disturbance of a regulatory element between SLC6A8 and BCAP31.

60
Chapter 2 Male patients
ACKNOWLEDGMENTS
The authors are grateful to Vincent Lauffer, Warsha Kanhai, Lorna Pope, Ben Nota and
Joe Ndika for excellent laboratory assistance.

Contiguous gene deletions of SLC6A8, BCAP31 and ABCD1
61
2.2
SUPPLEMENTARY FILE
Direct sequencing of the breakpoints
The deletion notation is according to GRCh37 Build (hg19).
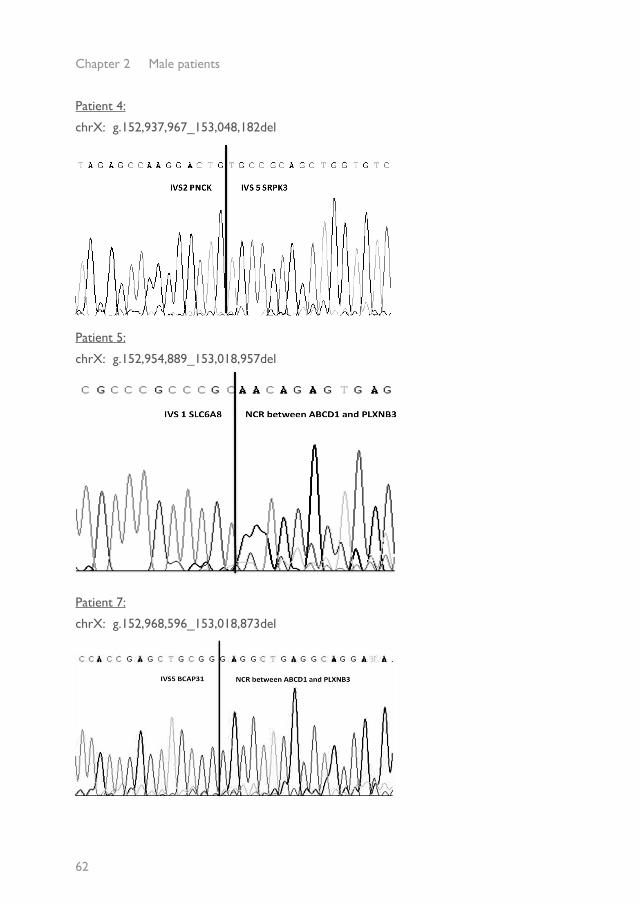
Patient 1:
chrX: g.152,958,196_152,960,341 del
Patient 2:
chrX: g.152,959,229_152,964,178 del
Patient 3:
chrX: g.152,932,965_152,972,594 del
62
Chapter 2 Male patients
Patient 4:
chrX: g.152,937,967_153,048,182del
Patient 5:
chrX: g.152,954,889_153,018,957del
Patient 7:
chrX: g.152,968,596_153,018,873del

Contiguous gene deletions of SLC6A8, BCAP31 and ABCD1
63
2.2
Patient 8:
chrX: g.152,973,240_153,004,372del
RT-PCR of BCAP31
RT-PCR of transcripts of BCAP31 of exon 1-4 and exon 1-8 in RNA isolated from
fibroblasts of patient 2-6 (2-6), patient reported by Osaka et al 2012 (O), normal control
(C) and normal control without reverse transcriptase (-RT), showing the presence of
exon 1-4 in patient 2 and 3 and the patient reported by Osaka et al 2012 and exon 1-8
in patient 2.

64
Chapter 2 Male patients
REFERENCES1. van de Kamp JM, Betsalel OT, Mercimek-Mahmutoglu S et al. Phenotype and genotype in 101 males
with X-linked creatine transporter deficiency. J Med Genet 2013 Jul;50(7):463-72.
2. Corzo D, Gibson W, Johnson K et al. Contiguous deletion of the X-linked adrenoleukodystrophy gene (ABCD1) and DXS1357E: a novel neonatal phenotype similar to peroxisomal biogenesis disorders. Am J Hum Genet 2002 Jun;70(6):1520-31.
3. Iwasa M, Yamagata T, Mizuguchi M et al. Contiguous ABCD1 DXS1357E deletion syndrome: Report of an autopsy case. Neuropathology 2013 Jun;33(3):292-8.
4. Anselm IA, Alkuraya FS, Salomons GS et al. X-linked creatine transporter defect: a report on two unrelated boys with a severe clinical phenotype. J Inherit Metab Dis 2006 Feb;29(1):214-9.
5. Osaka H, Takagi A, Tsuyusaki Y et al. Contiguous deletion of SLC6A8 and BAP31 in a patient with severe dystonia and sensorineural deafness. Mol Genet Metab 2012 May;106(1):43-7.
6. Cacciagli P, Sutera-Sardo J, Borges-Correia A et al. Mutations in BCAP31 Cause a Severe X-Linked Phenotype with Deafness, Dystonia, and Central Hypomyelination and Disorganize the Golgi Apparatus. Am J Hum Genet 2013 Sep 5;93(3):579-86.
7. Eichler EE, Lu F, Shen Y et al. Duplication of a gene-rich cluster between 16p11.1 and Xq28: a novel pericentromeric-directed mechanism for paralogous genome evolution. Hum Mol Genet 1996 Jul;5(7):899-912.
8. Mosser J, Sarde CO, Vicaire S, Yates JR, Mandel JL. A new human gene (DXS1357E) with ubiquitous expression, located in Xq28 adjacent to the adrenoleukodystrophy gene. Genomics 1994 Jul 15;22(2):469-71.
9. Ng FW, Nguyen M, Kwan T et al. p28 Bap31, a Bcl-2/Bcl-XL- and procaspase-8-associated protein in the endoplasmic reticulum. J Cell Biol 1997 Oct 20;139(2):327-38.
10. Wang B, Heath-Engel H, Zhang D et al. BAP31 interacts with Sec61 translocons and promotes retrotranslocation of CFTRDeltaF508 via the derlin-1 complex. Cell 2008 Jun 13;133(6):1080-92.
11. Iwasawa R, Mahul-Mellier AL, Datler C, Pazarentzos E, Grimm S. Fis1 and Bap31 bridge the mitochondria-ER interface to establish a platform for apoptosis induction. EMBO J 2011 Feb 2;30(3):556-68.
12. Wang B, Nguyen M, Chang NC, Shore GC. Fis1, Bap31 and the kiss of death between mitochondria and endoplasmic reticulum. EMBO J 2011 Feb 2;30(3):451-2.
13. Breckenridge DG, Stojanovic M, Marcellus RC, Shore GC. Caspase cleavage product of BAP31 induces mitochondrial fission through endoplasmic reticulum calcium signals, enhancing cytochrome c release to the cytosol. J Cell Biol 2003 Mar 31;160(7):1115-27.
14. Nguyen M, Breckenridge DG, Ducret A, Shore GC. Caspase-resistant BAP31 inhibits fas-mediated apoptotic membrane fragmentation and release of cytochrome c from mitochondria. Mol Cell Biol 2000 Sep;20(18):6731-40.
15. Wang B, Nguyen M, Breckenridge DG et al. Uncleaved BAP31 in association with A4 protein at the endoplasmic reticulum is an inhibitor of Fas-initiated release of cytochrome c from mitochondria. J Biol Chem 2003 Apr 18;278(16):14461-8.
16. Ducret A, Nguyen M, Breckenridge DG, Shore GC. The resident endoplasmic reticulum protein, BAP31, associates with gamma-actin and myosin B heavy chain. Eur J Biochem 2003 Jan;270(2):342-9.

Contiguous gene deletions of SLC6A8, BCAP31 and ABCD1
65
2.2
17. Waterham HR, Koster J, van Roermund CWT, Mooyer PAW, Wanders RJA, Leonard JV. A lethal defect of mitochondrial and peroxisomal fission. N Engl J Med 2007 Apr 26;356(17):1736-41.
18. Galea E, Launay N, Portero-Otin M et al. Oxidative stress underlying axonal degeneration in adrenoleukodystrophy: a paradigm for multifactorial neurodegenerative diseases? Biochim Biophys Acta 2012 Sep;1822(9):1475-88.
19. Hartwig C, Veske A, Krejcova S, Rosenberger G, Finckh U. Plexin B3 promotes neurite outgrowth, interacts homophilically, and interacts with Rin. BMC Neurosci 2005;6:53.
20. Matsumoto T, Miyake N, Watanabe Y et al. X-linked adrenoleukodystrophy with partial deletion of ALD due to fusion with the neighbor gene, PLXNB3. Am J Med Genet A 2005 Oct 15;138A(3):300-2.
21. Gardner HP, Rajan JV, Ha SI et al. Cloning, characterization, and chromosomal localization of Pnck, a Ca(2+)/calmodulin-dependent protein kinase. Genomics 2000 Jan 15;63(2):279-88.
22. Howidi M, Parsons H. A case of creatine transporter deficiency in a young child with choreoathetosis. Curr Pediatr Res 2010;14(2):102-105.

Copyright © 2022 FDOKUMEN




















