
Spatial effects in real networks: Measures, null models, and applications

Cell, Vol. 75, 59-72, October 8, 1993, Copyright © 1993 by Cell Press
Mice Carrying Null Mutations of the Genes Encoding Insulin-like Growth Factor I (Igf.1) and Type 1 IGF Receptor (Igflr) Jeh-Ping Liu,* Julie Baker,* Archibald S. Perkins,l" Elizabeth J. Roberteon,*$ and Arglrle Efetratiadie* *Department of Genetics and Development Columbia University New York, New York 10032 tDepartment of Pathology Yale University School of Medicine New Haven, Connecticut 06510
Summary
Newborn mice homozygous for a targeted disruption of insulin-like growth factor gene 1 (Igf.1) exhibit a growth deficiency similar in severity to that previously observed in viable Igf.2 null mutants (60% of normal blrthweight). Depending on genetic background, some of the Ig f . l ( - / - ) dwarfs die shortly after birth, while others survive and reach adulthood. In contrast, null mutants for the Igf l r gene die invariably at birth of respiratory failure and exhibit a more severe growth deficiency (45% normal size). In addition to general- ized organ hypoplasla in Ig f l r ( - / - ) embryos, including the muscles, and developmental delays In ossification, deviations from normalcy were observed in the central nervous system and epidermis. Ig f . l ( - / - ) / Ig f l r ( - / - ) double mutants did not differ in phenotype from Ig f l r ( - / - ) single mutants, while in Igf .2(-) / Igf l r(- / -) and Igf . l ( - / - ) / Igf .2(-) double mutants, which are phe- notypioally identical, the dwarfism was further exacer- bated (30% normal size). The roles of the IGFs in mouse embryonic development, as revealed from the phenotyplc differences between these mutants, are discussed.
Introduction
The family of insulin-like growth factors (IGFs) and their cognate receptors and binding proteins (IGFBPs) consists of two ligands (IGF-I and IGF-II) that are structu rally homol- ogous to proinsulin, two receptors (types 1 and 2; IGF1R and IGF2R), and at least six IGFBPs (for reviews, see LeRoith, 1991; Schofield, 1992). Because the IGFs are produced by many tissues, it is thought that they function in an autocrine/paracrine fashion, although they may also act as classical hormones, since they circulate in the plasma in association with IGFBPs.
IGF-I mediates many of the effects of growth hormone postnatally (see Daughaday, 1989; Isaksson et al., 1991; Lowe, 1991; Han and Hill, 1992) and is thought to have a dual function, acting as both a mitogen and a differentia- tion factor, according to the results of in vitro studies using
:[:Present address: Biological Laboratories, Harvard University, Cam- bridge, Massachusetts 02138.
a variety of cell lines, primary cultures, and tissue explants (reviewed by Sara and Hall, 1990; Lowe, 1991; Hart and Hill, 1992). The essential growth-promoting function of IGF-II, which is restricted to the period of embryogenesis in the mouse, was revealed from our results of targeted mutagenesis (DeChiara et al., 1990, 1991). These results also showed that the Igf-2 gene is subject to parental im- printing; the paternal Igf-2 allele is expressed, while the maternal allele is silent in most tissues. Thus, the heterozy- gous progeny carrying a paternally derived mutated Igf-2 gene (Igf-2(p-) mutants) and the homozygous Igf-2(-/-) mutants are phenotypically indistinguishable: they are via- ble and fertile proportionate dwarfs, with a body weight 60% that of wild-type littermates. In contrast, when the disrupted Igf-2 allele is transmitted maternally, the off- spring are phenotypically normal.
In vitro studies have demonstrated that the two specific cell surface receptors with which the IGFs interact are structurally unrelated (reviewed by Roth, 1988; Czech, 1989; Neely et al., 1991; Nissley et al., 1991; Moxham and Jacobs, 1992; Kornfeld, 1992). One of these receptors, IGF1 R, which resembles the insulin receptor, is a disulfide- linked heterotetrameric (a21~2) transmembrane glycopro- tein, with extracellular ligand-binding and intracellular ty- rosine kinase domains (UIIrich et al., 1986). Construction and expression of chimeric Igflr cDNAs defined a cyste- ine-rich segment of the extracellular a subunit as the bind- ing region for both the IGF-I and IGF-II ligands (Gustafson and Rutter, 1990; Zhang and Roth, 1991). Recent binding studies using NIH 3T3 fibroblasts transfected with a hu- man Igflr cDNA showed that IGFIR binds IGF-I with 15- to 20-fold higher affinity than IGF-II (Germain-Lee et al., 1992).
The second IGF receptor, IGF2R, is a single-chain poly- peptide devoid of tyrosine kinase activity. It binds IGF-II avidly but recognizes IGF-I barely, if at all (see Nissley et al., 1991; Kornfeld, 1992). In mammals, IGF2R also serves as the cation-independent mannose 6-phosphate receptor involved in lysosomal enzyme targeting (Morgan et al., 1987). The bifunctional mammalian IGF2R/cation-inde- pendent mannose 6-phosphate receptor is encoded by a gene (Igf2r) that is imprinted but in a reciprocal fashion to Igf-2 (the expressed allele is maternal; Barlow et al., 1991). IGF2R has an apparent role in IGF-II turnover (Oka et al., 1985; Kiess et al., 1987; Nolan et al., 1990), but despite some suggestive evidence (see Nishimoto et al., 1991) it remains unclear whether it participates in a signal- ing pathway. Blocking of IGF2R by antibodies in various cultured cells did not inhibit the mitogenic effect of IGF-II (Mottola and Czech, 1984; Kiess et al., 1987), while analo- gous blocking of IGF1R did impair IGF-II function (Furla- netto et al., 1987; Conover et al., 1986). Thus, it is thought that the biological effects of both IGF-I and IGF-II are medi- ated via interactions with IGFIR.
The widespread pattern of Igflr gene expression during rodent embryogenesis (Bondy et al., 1990, 1992), in con- junction with the results of RNAase protection assays

Cell 60
A endogenous Igf-1 I -
t B E B H
I I f I
t a rge t ing vec tor . . . . tk
targeted Igf-1
6.2 kb
Bs p B F
I l l I ' S ~ ' E 4
X i ~ i t t
, 4 I I noo
. Es
i i, F neo L
7.3 k.b I I
probe
B 31
1 I H E I I ,
X
C E
K E E E
I I 1 kb
TI T=T~ E M m
]
Figure 1. Targeting of the Mouse Igf-1 Locus (A) Restriction map of the wild-type/gf-1 gene (top) in the region containing exon 4 (E4) that was mutagenized by targeting (bottom) using the indicated replacement vector (middle). The restriction sites are: BamHI (B), EcoRI (E), Hindllt(H), Spel (S), BspEI (Bs), Pvul (P), and Kpnl (K). (13) Amino acid sequence of mouse IGF-I, showing the portion of the polypeptide (ellipse) encoded by the segment of E4 that was eliminated from the mutated gene. Arrows indicate tyrosine residues important for binding to IGF1R (see text). An arrowhead denotes the corresponding position of an intron between exons 3 and 4 of Igf-l. (C) Southern analysis of Hindlll-digasted genomic DNA from control ES cells (E) and three different targeted clones (1"1 -T3). The sizes of fragments (in kb) hybridiz- ing to the probe shown in the bottom of (A) are indicated. The size markers (lane M) are Hindlll fragments of phage ). DNA.
showing that the level of steady-state/gflr mRNA detected in various rat embryonic tissues declines significantly post- natally (Werner et al., 1989), im plied that IG F1 R is involved in prenatal development. An embryonic function of IGF-I was also suggested by studies on Igf-1 gene expression during rodent embryogenesis (Bondy et al., 1990, 1992; Ayer-LeLievre et al., 1991; Streck and Pintar, 1992; Streck et al., 1992).
To expand our previous study on IGF-II and determine the roles of IGF-I and IGF1R in vivo, we disrupted the cognate genes by targeted mutagenesis, examined the mutant phenotypes, and then ascertained genetically whether the IGF-I and IGF-II signal transduction occurs via IGF1R in the mouse embryo.
Results
Mutagenesis of the Igf.1 and Igflr Loci Mature IGF-I is a 70 amino acid, single-chain polypeptide, consisting of domains S (amino acids 1-29), C (amino acids 30--41), A (amino acids 42-62), and D (amino acids 63-70), whose folding is stabilized by three intrachain di- sulfide bridges (see Figure 1B). Experiments analyzing the binding affinities of synthetic IGF-I analogs to IGF1R have identified tyrosines 24, 31, and 60 as critical residues for the ligand-receptor interaction (Bayne et al., 1990). The last four amino acids of domain B, and all the re- maining domains, are encoded by a single exon (exon 4; Hall et al., 1992). To generate a null mutation, we designed a targeting vector that would delete a portion of exon 4 (Figure 1A). Successful targeting would abolish the func-
tion of IGF-I, because of the elimination of residues 51- 70, including the important Tyr-60, and removal of two of the disulfide bonds (Figure 1B).
To generate a null mutation of the Ig f l r gene, we de- signed a targeting vector that would delete part of exon 3 (Abbott et al., 1992), which encodes the major portion of the cysteine-rich ligand-binding domain (Figure 2A).
To construct replacement vectors for mutagenesis and identification of targeted embryonic stem (ES) cells by ap- plication of a positive-negative selection protocol (Man- sour et al., 1988), we used fragments of genomic clones and transcriptionally competent cassettes of the bacterial neomycin-resistance gene (neo) and the herpes simplex virus thymidine kinase gene (tk). The final constructs (de- scribed in detail in Experimental Procedures) are shown in Figures 1A and 2C. tn each case, linearized replacement vector DNA was introduced into recipient ES ceils by elec- troporation, and the cells were seeded and selected with drugs on feeder layers of STO fibroblasts. Successful tar- geting events were determined by polymerase chain reac- tion (PCR) assays and verified by Southern analyses (Fig- ures 1C and 2D). Targeted clones with a euploid karyotype were injected into host blastocysts from the MF1 and C57BL/6J mouse strains to generate germline chimeras.
After transmission of the mutations, intercrosses be- tween heterozygous progeny, either Ig f - l (+ / - ) or Ig f l r (+/-), yielded corresponding homozygous mutants. The same heterozygotes were also used in a breeding program that included/gf-2(p-) m utants (DeChiara et al., 1990), to obtain all possible combinations of double mutants. The phenotypic manifestations of the mutations are described

Igf-1 and Igf lr Null Mutations 61
A I-- a-subunit _-II-ii - ~-subuni= --I
E E E E E EI~ E2 I ~ E ~ E6 ~E7~ E8 ~Eg ~Et 0~El1~ EI2~E131 E1$~1 E1 1~2 E21
A ¢ CA A signal cysteina-dch prot~lyUc tran=membrane Wrosln e klnue paptlde domain processing domain domain
IRe (RKRR)
I I
B g a a t t e a c a g a c g g t a a g a t c t a c g c a g a t t t c a g a g g a g g t ~ t g e t t t t c t a g c c a g t c t c c g g t a c g c c a c g c t g ECORZ (M)
gtgg tggcagcgga tgacccaggggaggc tg tg tagccca tca tcc t taccaccc tc tc tccc tc tg tcc tcacagTG C P S V C G K R A C T E N N E C C H P E C L G S C H
TGCCC~GTGTGTGCGGG~GCGAGCCTGCACCGAG~C~CGAGTGC~CCACCCGGAGTGCCTGGGCA~TGCCAC P~II
T P D D N T T C V A C R H Y Y Y K G V C V P A C P P ACACCGGACGAC~CACAACCTGCGTGGCCTGCAGACACTACTACTACAAAGGCGTGTGTGTGCCTGCCTGCCCGCCT G T Y R F E G W R C V D R D F C A N I P N A E S S D
G,3CACCTACAGGTTCGAGGGC~GCGCTGTGTGGATCGCGATT~TGCGCC~CATCCCCAACGCTGAGAGCAGTGAC S D G F V I H D D E C M Q E C P S G F I R N S T Q ( S )
~GGATGGCTTCGTTA~CACGACGATGAGTGCATGCAGGAGTG~CCTCAGGCTTCATCCGCAACAGCACCCAGAG
g t c a g c g g c t c t t g t c c c t a g g c c c a g g a g g g t t c t c t g c t g c a t a g c c c c t a a g t g t a g g a c c t c c c a g g g g t t c a c AvrZI
ccagatg~gtctgtcagagggtgatggctgagcttgttttaatgc~aatcttggagtggttgggtctgtttcagcttg
gcttttt~atttctaaactttgagggtgccttgctagc
D C
P---A -4."b. .... i Y Y f I o P,. Io
E3
X ~',.,X I 1 ~ 1 | tk --=,,-~.__.~
E3 pGEM3Z
EE 1 B/ H I'~¢ B T , ,
E3 E4 I" . . . . . . . . . . . . . . . . "1
3.2kb I l k b I m Probe
Figure 2. Targeting of the Mouse Igflr Locus (A) Diagram of the structure of the IGF1R pre- cursor (adapted from UIIrich et al., 1986). The segments encoded by exons 1-21 (El-E21), as assigned for the human Igflr gene (Abbott et al., 1992), are indicated. (B) Nucleotide se- quence of the targeted mouse Igflr exon 3, encoding the major portion of the cysteine-rich domain shown in (/%). (C) Restriction map of the wild-type Igflr locus (top) in the region of exons 3 and 4 (E3 and E4) that was targeted (bottom) using a replacement vector (middle). The re- striction sites are: Hindlll (H), EcoRI (E), Hincll (Hc), Pvull (P), Avrll (A), BamHI (B), Sail (S), and Xhol (X). BIA is a BamHI/Avrll hybrid site. (D) Southern analysis of Hincll-digested geno- mic DNA from a nontargeted clone (lane N) and seven different targeted ES cell lines (all other lanes). The sizes of fragments (in kb) hybridiz- ing to the probe shown in the bottom of (C) are indicated. The size markers (lane M) are Hindlll fragments of phage ~. DNA.
below, beginning with the most pleiotropic/gflr mutation, while a detailed developmental analysis of the impact of the mutations on growth kinetics is presented in the ac- companying paper (Baker et al., 1993 [this issue of Ce//]).
Ig f l r ( - I - ) Mutants: Dwarfism and Neonatal Lethality The heterozygous Igf l~+/-) progeny derived from male germline chimeras did not exhibit any discernible pheno- type, while in several litters of offspring derived from inter- crosses between animals heterozygous for the mutation, the severely growth-deficient neonates that we recovered were always inviable. The birthweight of these pups was approximately 45% of that of their wild-type littermates (Figure 3C). Subsequent genotyping showed that these inviable dwarfs were homozygous Igfl"~-I-) mutants.
In contrast with the invariable phenotypo of Ig f l~- I - ) mutants, which was manifested in both inbred and outbred genetic backgrounds and in animals derived from 2 inde- pendently targeted clones, the F2 heterozygotes were phenotypically normal (Figure 3A), regardless of the pater- nal or maternal transmission of the mutated allele. Surpris- ingly, quantitative Northern analysis of poiy(A) ÷ RNA from E16.5 and E18.5 whole embryos, using an Igflr exon 3 probe, has indicated reproducibly that there is no differ- ence in the level of steady-state Igflr mRNA between her-
erozygous mutants and wild-type littermates (data not shown). The mechanism compensating for the reduction of the gene dosage by half is unknown. Parallel analysis of RNA from Igf l r (- I -) mutant embryos was negative for hybridization, as expected.
To ascertain whether the targeted Igflr gene repre- sented a null allele, we cultured primary embryonic fibro- blasts from E14.5 wild-type, Igflr(+l-), and Igf l r (- I -) lit- termates and assayed for the presence of functional IGF1R in a membrane protein fraction that was purified by binding to wheat germ agglutinin (see Oemar et al., 1991). Neither cross-linking of biotinylated IGF-I to the a subunit of IGFIR nor ligand-dependent autophosphoryla- tion of the IGF1R I~ subunit was observed with the wheat germ agglutinin-bound fraction from the Igf l r (- I -) mu- tants, while positive results were obtained with the two control extracts (data not shown).
Igf l r ( - / - ) Mutants: Anatomical and Histological Analyses and Comparison with Igf.2(p-) Mutants Respiratory Failure at Birth Close monitoring at birth indicated that the Igf l r (- I -) dwarfs were born alive but, despite visible efforts to breathe, became cyanotic and died within minutes. Lung tissue dissected from several mutant neonates obtained from independent litters failed to float on water, indicating

Cell 62
C
I/
R
Figure 3. Mutant Mice (A) Genotyped E17.5 embryos of the same litter: wild-type (w), heterozygous (h), and homozygous (r) for the Igflr gene mutation. The phenotypically indistinguishable wild-type and heterozygous embryos differ from the homozygous mutants not only in size, but also in the appearance of the skin, which is significantly more opaque. (B and C) Litters of genotyped E16.5 embryos and newborn mice, respectively, including four phenotypes: wild-type (W) animals, Igf-2(p-) mutants (11), Igflr(-/-) mutants (R), and Igf-2(p-)/Igflr(-/-) double mutants (II/R). ((3) A litter of E17.5 embryos: wild-type ON), Igf-l(-/-) mutant (I), Igfo20o-) mutant (11), and Igf.l(-I-)llgf-2(p-) double mutant (1111). The translucent skin of the double mutant is notable. The photographic enlargements of the panels are not proportional to one another.
that air never reached the alveoli. The primary cause of this respiratory failure is unknown at present. Histopatho- logical examination revealed no particular lung abnormali- ties that could explain the observed atelectasis that leads to asphyxia. Thus, no structural block was evident in the bronchi, bronchioles, or upper respiratory tract, while the alveolar epithelium in neonates and E16.5-E18.5 homozy- gous mutant embryos was indistinguishable from that of normal littermates (Figures 4A and 4B). Moreover, immu- nostaining for surfactant apoprotein in sections of mutant and wild-type E18.5 embryos and parallel quantitation by Western analysis (using an antibody provided by G. Singh) yielded indistinguishable results (data not shown). This suggests that the inability to breathe cannot be attributed to a primary inability of the lungs to expand. Muscle Hypoplasla The reduced body weight of these mutant mice seems to be a consequence of a decrease in tissue cell number (hypoplasia) and not in cell size, since comparisons of cell dimensions in muscle, liver, and lung between E17.5 wild-type and I g f l r ( - I - ) embryos revealed no statistically significant differences (data not shown). It is possible that
the failure of the mutants to breathe is due to muscle hypo- plasia. Examination of a number of muscle groups in E16.5-E18.5 wild-type and mutant littermates showed that the mutation had affected the number of myocytes. For example, each of the three layers of the anterolateral ab- dominal muscle was 4 to 8 cells thick in wild-type embryos, while the number of cells did not exceed 1 or 2 in mutant littermates (Figure 4C). A similar reduction in the number of myocytes in mutant animals was also observed in mus- cles of the neck and limbs and in respiratory muscles (inter- costal muscles and diaphragm; Figures 4D and 4E). How- ever, we do not yet have an accurate assessment of whether the hypoplasia of muscles is comparable to the generalized hypoplasia observed in all organs of these growth-deficient embryos or if it is disproportionately se- vere. We noted that the muscle cells of the mutants appeared to be normal, according to limited qualitative assays for the expression of muscle-specific markers (Figure 4). Impact of the Mutation on the Nervous System Examination of the central nervous system in I g f l r ( - I - ) mutants revealed a morphological deviation from wild type
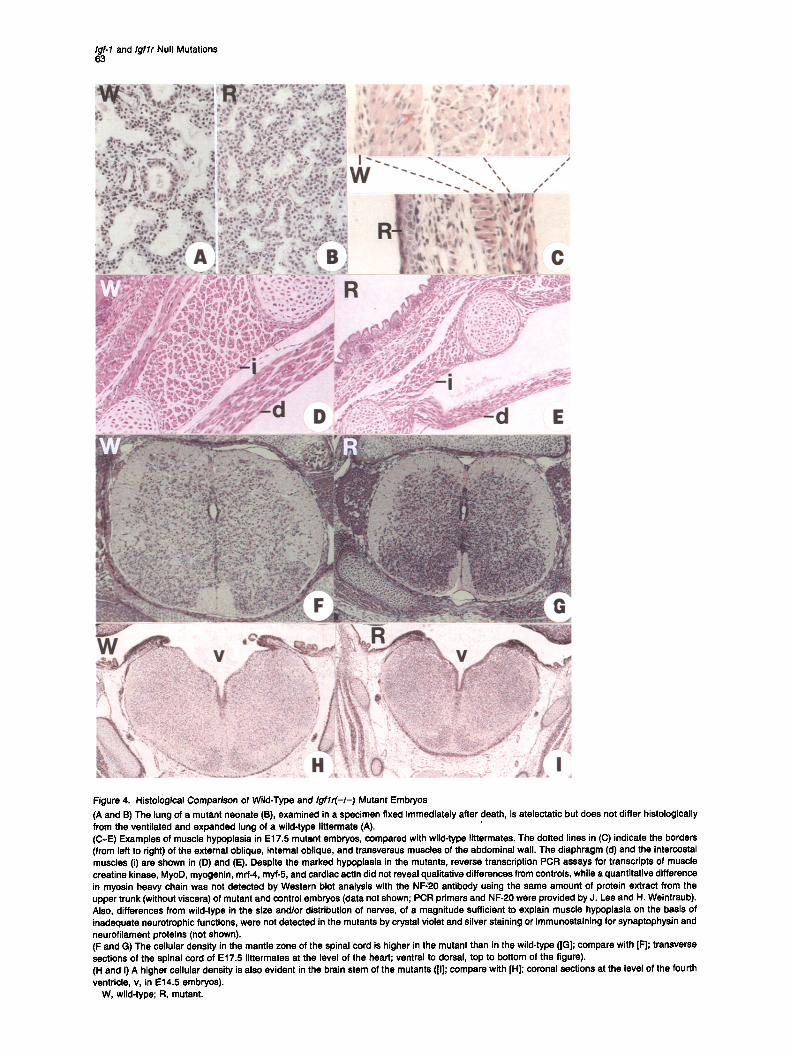
Igf-1 and Igflr Null Mutations 63
Figure 4. Histological Comparison of Wild-Type and Igf lr(-I-) Mutant Embryos (A and 13) The lung of a mutant neonate (B), examined in a specimen fixed immediately after death, is atelectatic but does not differ histologically from the ventilated and expanded lung of a wild-type littermate (A). (C-E) Examples of muscle hypoplasia in E17.5 mutant embryos, compared with wild-type littermates. The dotted lines in (C) indicate the borders (from left to right) of the external oblique, internal oblique, and transversus muscles of the abdominal wall. The diaphragm (d) and the intercostal muscles (i) are shown in (D) and (E). Despite the marked hypeplasia in the mutants, reverse transcription PCR assays for transcripts of muscle creatine kinase, MyoD, myogenin, mrf-4, myf-5, and cardiac actin did not reveal qualitative differences from controls, while a quantitative difference in myosin heavy chain was not detected by Western blot analysis with the NF;20 antibody using the same amount of protein extract from the upper trunk (without viscera) of mutant and control embryos (data not shown; PCR primers and NF-20 were provided by J. Lee and H. Weintraub). Also, differences from wild-type in the size and/or distribution of nerves, of a magnitude sufficient to explain muscle hypoplasia on the basis of inadequate neurotrophic functions, were not detected in the mutants by crystal violet and silver staining or immunostaining for synaptophysin and neurofilament proteins (not shown). (F and G) The cellular density in the mantle zone of the spinal cord is higher in the mutant than in the wild-type (IG]; compare with IF]; transverse sections of the spinal cord of E17.5 littermates at the level of the heart; ventral to dorsal, top to bottom of the figure). (H and I) A higher cellular density is also evident in the brain stem of the mutants ([I]; compare with [H]; coronal sections at the level of the fourth ventricle, v, in E14.5 embryos).
W, wild-type; R, mutant.

Cell 64
Figure 5. Skin Histology (A-D) Histological comparisons of the epidermis in E16.5 littermates (sections at the level of the kidney, dorsal to the spinal cord); wild-type embryo ON), Igflr(-/-) mutant (R), Igf-2(p-.-) mutant (It), and Igf.2(p-)/Igflr(-/-) double mutant (ll/R). (E and F) Wild-type and Igf lr(-I-) mutant littermates are also compared at E18.5 (sections at the level of the liver, dorsal to the spinal cord). Of the strata of the epidermis (b, basale; s, spinosum; g, granulosum; and c, corneum), the spinous layer is particularly thin in the mutant. ((3 and H) These panels are analogous to sections (E) and (F), respectively, but at a lower magnification, to show the difference in hair follicle (h) number and spacing between wild-type and Igf lr(-I-) littermates (sections of the abdominal wall at the level of the intestine). (J-M) Comparison of the epidermis in E18.5 littermates: wild-type embryo (W), Igf-1 (-I-) mutant (I), Igf-2(p-) mutant (11), and Igf-l(-/-)/Igf-2(p-) double mutants (I/ll) (level of sections as in [A-DI). The strata of the epidermis do not differ between the wild-type and the two single mutants, but in the double mutant the epidermis is underdeveloped and is charectedzed by a very thin spinous layer.
that was not detected in Igf-2(p-) mutants. In transverse sections of the spinal cord of E14.5-E18.5 I g f l r ( - / - ) em- bryos at various levels, we observed that the cellular den- sity in the mantle zone was significantly higher in mutant than in wild-type littermates (Figures 4F and 4G). Prelimi- nary counting of cells has suggested that the ratio of mu- tant to wild-type cell densities is progressively reduced with developmental age, being 1.7, 1.5, and 1.2, at E16.5, E17.5, and E18.5, respectively. At the current level of anal- ysis, we think that the increase in cell density corresponds not to an absolute increase in cell number, but rather to crowding of neural cells in the mutants, resulting from a reduction in the amount of the surrounding neuropil (i.e., neuronal fibers and cytoplasm of neuroglial cells). More limited data suggest that the increase in cell density also occurs in the brainstem of the mutant mice (Figures 4H and 41).
While a detailed histopathological analysis of the ner- vous system of I g f l r ( - I - ) mutants is pending, the following preliminary observation is notable (S. Newman and J. Goldman, personal communication). Culturing of E18.5 forebrain cells from I g f l r ( - / - ) embryos and wild-type con- trois has indicated that significantly fewer oligodendrocyte progenitors of the mutants develop to a stage that can be recognized by the specific 01 marker (Sommer and Schachner, 1981). Impact of the Mutation on Skin Development The I g f l r ( - I - ) mutant embryos (see Figures 3A and 3B) and neonates (see Figure 3C) differed in appearance from their wild-type littermates because their skin, instead of being opaque, was significantly more translucent, Histo- logical examination revealed that of the strata of the epi- dermis (basale, spinosum, granulosum, and corneum), the stratum spinosum was extremely thin in the mutants, con-

Igf-1 and Igflr Null Mutations 65
i
II-- i i ! a,
!
el;. , . , ,
Pl
e14.5 e15.5 ~.= :.tBil~e16.5 e17i5 J_,____ I N
~ . t "~:" "
i
N N
Figure 6. Bone Development In the first three rows, the first appearance of ossification centers is compared between wild-type embryos (W), Igf-2(p-) mutants (11), and Igflr (- /-) mutants (R). Examples of developmental delays are indicated (also see text). Each of the developmental ages at which littermates were examined is shown at the bottom of the third row (N denotes neonates). The skeletons of Igf-2(p-)/Igflr(-I-) double mutant (II/R) littarmates (E18.5 embryo and neonate) are shown in the fourth row (left), for comparison, The oss!fication centers of the frontal (f) and parietal (p) bones, and also of the nasal (n) bone, are present in the wild-type E14.5 and E16.5 specimens, respectively, although they are not evident in this reproduction (arrowheads without lettering). The ossification centers of the interparietal (i) and supraoccipital (s) bones are as indicated. In Igf lr(-I-) mutants, a 1 day delay in ossification was observed in the bones of the trunk and of the extremities, except for the following bones: clavicle, ribs, femur, hyoid bone, body of the cervical and lumbar vertebrae, diaphyses of the radius and the ulna, metacarpals, and. metatarsals (2 day delay). In forelimbs and hindlimbs, ossification of the digits, which in wild-type embryos appears at E 17.5, was not observed in Igf lr(-I-) neonates. In Igf-2(p-) mutants, a one-day developmental delay was noted in hyoid, cervical and lumbar vertebrae, radius, ulna, femur, digits, nasal bone, and all cranial bones, apart from the frontal and temporal bones. In the examined specimes of double mutants (IIIR), ossification of the supraoccipital bone was detected only at birth, while in wild-type controls and in single Igf-2(p-) and Igf lr(-I-) mutants, ossification occurred at E16.5, E17.5, and E18.5, respectively. The newborn double mutant did not exhibit commencement of ossification in the interparietal bone, the body of the cervical vertebrae, the metacarpals, metatarsals, and digits. The skeletons of 3 newborn littermates, Igf-1(-/-) mutant (I), Igf-2(p-) mutant (11), and Igf-l(-I-)llgf-2(p-) double mutant (1111), are shown in the fourth row (right).

Cell 66
Table 1. Survival Frequency of Igf-l(-I-) Mutants
Homozygous Survivors (% Mice Mutants Neonatal of homozygous
Cross Litters Born (% of total) Deaths mutants)
1291Sv x 1291Sv 19 102 20 (20%) 18 2 (10%) C57BL/6J x 129/Sv 20 148 32 (22%) 27 5 (16%) MF1 x 1291Sv 9 87 22 (25%) 7 15 (68%)
The expected frequency of 25% homozygous mutants was also observed in embryos. Among 138 genotyped embryos from 22 litters, 33 (24%) were wild type, 71 (51%) were heterozygotes, and 34 (25%) were homozygotes.
sisting of fewer cells than in normal animals (compare Figures 5A and 5B and also Figures 5E and 5F). Moreover, in the skin of mutant embryos, there was a marked de- crease in the absolute number of hair follicles, which were smaller and more widely spaced than in wild-type controls (Figures 5G and 6H). These differences from wild-type were not detected in Igf-2(p-) mutants (Figure 5C; com- pare with Figure 5A). Delayed Bone Development We performed a detailed developmental analysis of the appearance of ossification centers in E14.5-E18.5 em- bryos and neonates by comparing the skeletons of Igflr ( - / - ) mutants, Igf-2(p-) mutants, and wild-type littermates after staining with alcian blue and alizarin red (Figure 6; McLeod, 1980). To simplify the description of this analysis, we did not take into account the degree to which ossifica- tion had progressed, but rather used a qualitative criterion, scoring for the first appearance of particular ossification centers.
We observed that the ossification centers of cranial and facial bones appeared later in Igf lr(-I-) embryos than in wild-type controls after a lag of about 2 embryonic days, with the exception of the interparietal bone, which exhib- ited an even longer delay ( - 4 days; Figure 6). The devel- opmental delay in the bones of the trunk and extremities was 1-2 days (Figure 6).
In the same comparison performed between Igf-2(p-) mutants and wild-type controls, either the timing in the appearance of ossification centers in the mutants was nor- mal or the delay in ossification did not exceed I embryonic day (see Figures 6).
Igf. l(- I-) Mutants: Dwarfism and Variable Survival The heterozygous Igf-1(+/-) progeny of male germline chi- meras did not display any obvious phenotypic difference from wild-type littermates, while in the litters obtained from intercrossas of these heterozygotes, about 25% of the recovered neonates exhibited a birthweight approximately 60% of norma~. Subsequent genotyping showed that With- out exception these dwarf mice were Igf-l(-I-) mutants, while the heterozygous F2 progeny were phenotypically normal, regardless of the paternal or maternal transmis- sion of the mutated allele.
Some of the newborn Igf-l(-I-) mutants were found dead. However, close monitoring of births revealed that these animals were born alive and that most of them were able to breathe. Observations of several litters indicated that neonatal death usually occurred between 15 rain and
6 hr after birth. The cause of this neonatal lethality is un- known.
Interestingly, some of the Igf-l(-I-) mutants survived to adulthood, at a variable frequency that depended on genetic background (Table 1). Thus, when inbred 1291Sv Igf-l(+l-) mice or (C57BL/6J x 129/Sv) F1 hybrids were intercrossed, only 10% and 16% of the homozygous mu- tant progeny survived, respectively. In contrast, 68% of the Igf-l(-I-) mutant offspring obtained from crosses be- tween (MF1 x 129/Sv) F1 hybrids reached adulthood. Pooled sera from adult Igf- l(-I-) mutants and control sera from wild-type mice were examined for the presence of immunoreactive IGF-I (serum is the richest source for this polypeptide; see Daughaday and Rotwein, 1989). Mea- surements by radioimmunoassay using a polyclonal anti- body against human IGF-I indicated that the serum level of IGF-I was below detection limits in the mutants (data not shown). The postnatal growth of surviving Igf- l(-I-) mutants is described in the accompanying paper (Baker et al., 1993).
Because of the delays in ossification and the underde- velopment of the epidermis observed in Igf lr(-I-) mu- tants, the same tissues were examined in Igf-l(-/-) mu- tants for comparison. Their epidermis, like that of the Igf-2(p-) mutants, appeared normal (see Figures 5K and 5L), while ossification was only slightly delayed and did not differ from that of Igf-2(p-) mutants (Figure 6, bottom).
Igflr(-I-) Mutants and Igf. l(- /-) l lgflr(- /-) Double Mutants Are Phenotypically Indistinguishable We reasoned that if IGF-I interacts exclusively with IGF1 R, the phenotype of Igf- l(-I-) l lgf lr ( - / - ) double mutants should be indistinguishable from that of the Igflr ( - I - ) single mutation. Our prediction about this genetic cross proved true. From appropriate crosses, we obtained litters of offspring that included such single and double mutant siblings, all of which died immediately after birth of respira- tory failure and were phenotypically indistinguishable in terms of birthweight (45% of normal) and bone develop- ment (data not shown). This is strong genetic evidence that the IGF.I ligand does not utilize any receptor other than IGF1R.
Phenotype of Igf.2(p-)llgflr(-I-) Double Mutants Using the same rationale described above for IGF-I, we next examined genetically the potential IGF-II-IGFIR in- teraction in vivo. For this purpose, we intercrossed mice carrying the Igflr and Igf-2 mutations and relied on meiotic

Igf-1 and Igflr Null Mutations 67
recombination events to generate Igf-2(p-)l lgflr(-I-) dou- ble mutants, since both the Igflr and Igf-2 genes, although not closely linked, reside on mouse chromosome 7 (in the center and distal regions of the chromosome, respectively, at a genetic distance of approximately 41 cM; see Zemel et al., 1992; Hillyard et al., 1993).
The newborn mice obtained from these matings (de- scribed in detail in Experimental Procedures) fell into four distinct phenotypic categories (see Figure 3C): wild-type animals, viable dwarfs with body weight 60°/0 of normal (Igf-2(p-) mutants), inviable dwarfs with 450/0 normal weight (Igr lr(- I-) mutants), and dwarfs with 30% normal weight, which also died immediately after birth of respiratory fail- ure. Genotyping showed that the animals in the last cate- gory were Igf-2(p-)l lgflr(-/-) double mutants.
The significantly more pronounced dwarfism observed in double mutants, in comparison with Igf l r (- I -) mutants, did not fulfill the prediction of the genetic cross, but rather provided evidence that an unknown receptor(s), which we call XR, is involved in IGF-II growth-promoting signal trans- duction. Thus, we could not distinguish from these results alone whether IGF-II interacts in vivo exclusively with XR or binds to both XR and IGF1R.
When Igf-2(p-)l lgflr(-I-) double mutant embryos (see Figure 3B) were examined histologically, the results did not reveal any underdevelopment of the epidermis beyond that observed in Igf l r (- I -) single mutants (see Figure 5D; compare with Figure 5B). Moreover, the central nervous system of the double mutants exhibited the same high cellular density observed in Igf l r (- I -) mutants (data not shown). However, the delays in bone development were somewhat more pronounced in two specimens of Igf- 2(p-) l lgf lr(-I-) double mutants (see Figure 6, bottom) than in Igf l r (- I -) mutants. This difference can be attrib- uted to developmental variability (see below).
Phenotype of Igf. l(- I -) l lgf.2(p-) Double Mutants To investigate further the developmental roles of IGF-I and IGF-II, we generated mice null for both Igf-1 and Igf-2 (see Experimental Procedures). F2 progeny were obtained from matings between males heterozygous for both the Igf-1 and Igf-2 mutations and females heterozygous for the Igf-I mutation. Among these newborn mice, in addition to wild-type animals and the expected Igf-2(p-) and Igf- 1(-/-) mutants (both about 60% of normal weight), we obtained severely growth-deficient progeny with 30% nor- mal body weight that died shortly after birth of respiratory failure. According to genotyping, these inviable 30°/0 nor- mal offspring were Igf-l(-/-)/Igf-2(p-) double mutants. These results (see also Discussion) indicate that absence of both IGF-I and IGF-II results in a compound phenotype of dwarfism and neonatal lethality that is indistinguishable from the phenotype of Igf.2(p-)l lgflr(-/-) double mutants.
When E17.5 and E18.5 Igf-l(-/-)/Igf-2(p-) double mutant embryos (see Figure 3D) were examined histologically, analysis of the epidermis (see Figure 5M) showed that it exhibits the same underdevelopment in the stratum spino- sum as that observed in Igf l r (- / -) mutants and Igf-2(p-)/ Igf l r (- I -) double mutants. Moreover, the skeletons of Igf- l(-I-) l lgf-2(p-) double mutant neonates (see Figure 6,
bottom) exhibited the same developmental delays in the ossification of particular bones as the Igf l r (- / -) mutants. A comparison between Igf-l(-I-)/Igf-2(p-) and Igf-2(p-)l Igf l r (- / -) double mutant littermates did not reveal differ- ences in ossification delays in some litters, while in other litters, we observed further retardation in the latter mutants that can apparently be ascribed to developmental vari- ability.
Discussion
A major conclusion from our genetic data is that IGF-I, in addition to its previously known postnatal role, is also involved in embryonic processes of growth and, poten- tially, differentiation. This is direct evidence demonstrating that IGF-I has a continuous function throughout develop- ment and is not merely a factor that assumes postnatally the growth promoting role played by IGF-II during rodent embryogenesis. An additional important conclusion of this genetic study is that Igflr is an essential gene, since we demonstrated that, at least in mice, the presence of func- tional IGF1R is indispensable for normal embryonic devel- opment and survival after birth. Notably, the neonatal le- thality observed in Igf l r (- I -) mutant mice is invariable and independent of the genetic background of the tested strains. Finally, on the basis of direct and indirect evidence (see below), we conclude that IGF1R mediates the in vivo signaling of both IGF ligands, while IGF-II utilizes an addi- tional receptor.
In Vivo Signaling of the IGFs via IGFIR Our comparisons between the phenotypes of the single and double mutants studied (summarized in Table 2) show the Igf-l(-/-)/Igf-2(p-) and Igf-2(p-)/Igflr(-/-) double mu- tants to be identical. Thus, within the framework of the examined mutational effects, all ligand-receptor interac- tions have been accounted for. Moreover, the data allow the interpretation that both the IGF-I and IGF-II ligands interact with IGFIR in embryos, as they do in cell culture (see Introduction). In the case of IGF-I, the evidence is direct and unequivocal because of the phenotypic identity between Igf l r (- / -) single mutants and Igf- l (- /-) / Igf l r ( - / - ) double mutants, demonstrating that IGF-I interacts exclusively with IGF1R. However, the phenotype of Igf-1 ( - / - ) mutants is less severe than that of Igf l r (- / -) mutants and Igf-1(-I-)/Igflr(-I-) double mutants. From this com- parison, one can deduce that IGF1R should also interact with IGF-II. Direct evidence that this is indeed the case was derived from the study of developmental growth kinet- ics (see Baker et al., 1993). Finally, from the comparison showing the dwarfism of Igf-2(p-)/Igflr(-/-) double mu- tants to be significantly more pronounced than that of ei- ther single mutation, it was inferred that IGF-II also inter- acts with an unknown receptor (XR; discussed further by Baker et al., 1993).
Because of the phenotypic disparity between the invari- able neonatal lethality of the Igf l r (- I -) mutants and the viability of the Igf-2(p-) mutants, the indispensable func- tions impaired by the receptor mutation must be mostly attributable to a disruption of the IGF-I-IGF1R interaction.

Cell 68
Table 2. Summary of Mutant Phenotypes
Birthweight Neonatal Delayed Ossification Underdeveloped Genotype (% of normal) Lethality (embryos or neonates) Epidermis (embryos)
Igf-l(-I-) 60 ± ± - /gf-2¢o-) 60 - ± - Igflr(-/-) 45 + + + Igf-l(-I-)llgf-2(p-) 30 + + + Igf-l(-I-)llgflr(-t-) 45 + + ND Igf-2(p-)llgflr(-/-) 30 + + +
ND, not determined.
However, the concomitant absence of an IGF-II-IGF1R interaction may be aggravating the I g f l r ( - I - ) phenotype, since some Ig f - l ( - I - ) mutants survive to adulthood, while the death of the remaining neonates is not due to respira- tory failure in the majority of cases.
Variations in expressivity, manifested as variability ei- ther in phenotypic effects or in survival, have also been observed in other mouse mutations, for example, in null mutations of the genes Wnt-1 (Thomas et al., 1991), Hox- 3.1 (Le Mouellic et al., 1992), and c-los (Johnson et al., 1992). This phenomenon is often attributed to differences in genetic background and random assortment of alleles of modifying genes affecting a phenotype (see Thomas et al., 1991). The fact that the variability in neonatal lethality observed in Igf-1(-I-) mutants is mouse strain-dependent is consistent with this view. Interestingly, an analogous dependence of neonatal lethality on genetic background was previously noted in mice carrying the diminutive (dm) dwarfing mutation (Stevens and Mackensen, 1958). Thus, to account for the apparent discrepancy between the I g f l r ( - / - ) and I g f - l ( - / - ) phenotypes, we propose that as long as IGF1R is present, IGF-II is sometimes able to com- pensate for unknown but essential functions of the absent IGF-I that are variably affected by other factors (modifiers). On the other hand, the absence of both IGF-I and IGF-II or the ablation of IGF1R results in perinatal lethality.
Is IGF-I Involved in Cell Differentiation? As the cause of the neonatal lethality of I g f l r ( - I - ) mutants remains obscure, it is not possible at present to assess whether the yet unidentified indispensable functions that IGF-I presumably cannot fulfill in the absence of IGF1R are distinct from its mitogenic activity and are related to embryonic cell differentiation. Preliminary data that we cite (see Results) are consistent with the possibility that IGF-I acts as a survival factor for oligodendrocyte precursors in embryos, as it does in culture (Barres et, al., 1992). Never- theless, to the degree that the mutations have been ana- lyzed, the plausible candidacy of IGF-I as an embryonic differentiation factor is only a useful working hypothesis, because we did not detect any phenotypic abnormalities that could be considered differentiation defects in a strict sense. Thus, we did not observe recognizable pathological changes in any tissue, such as disorganization, while the major apparent differences from wild-type that we de- tected can largely be interpreted as due to developmental delays.
In regard to bone development, only a slight retardation
in the degree of ossification was noted in I g f - l ( - I - ) neo- nates, indistinguishable from that observed in Igf-2 null mutants, while neither of these mutations alone had any convincingly recognizable effect on the development of the epidermis. The previously mentioned hypothesis that, in the presence of intact IGF1 R, each ligand can compen- sate to a significant degree for the absence of the other can also explain the visible differences between the Igf-1 and Igf-2 null mutants and the more severe phenotypes manifested in I g f l r ( - I - ) and all double mutants. In the latter cases, our observations indicate that the bone differ- entiation program is not perturbed per se, but rather pro- ceeds at a slower pace. This type of retardation might not be exactly analogous to the difference from wild-type observed in the developing skin of these mutants. Overall, the/gf l r ( - I - ) and the double mutant embryos resemble younger wild-type specimens in the timing of appearance of ossification centers, while their epidermis never ac- quires wild-type morphology in terms of numbers of spi- nous cell layers and hair follicles.
Skin development illustrates an apparent correlation be- tween the pattern of Ig f l r gene expression and the pheno- typic effects of the corresponding mutation. IGF1R local- ized exclusively in the basal layer of human skin has been detected by immunostaining (Krane et al., 1991), and Northern analysis has demonstrated the presence of Ig f l r mRNA in cultured human keratinocytes (Tavakkol et al., 1992). Interestingly, keratinocytes could grow in a defined medium in the presence of a combination of epidermal growth factor and IGF-I, while neither growth factor alone was effective (Krane et al., 1991). In rat embryos, in situ hybridization analyses have shown that both Igf-1 and Igf-2 transcripts are present in the dermis but not the epidermis (Bondy et al., 1990; Ayer-LeLievre et al., 1991). From these observations, in conjunction with our in vivo data, we spec- ulate that during skin development, IGF ligands produced in the dermis might participate in epidermal cell prolifera- tion in a paracrine fashion by binding to IGFIR present on the surface of cells in the basal layer.
The Igf.1 and Igflr Genes Are Not Imprinted In contrast with null mutants, progeny heterozygous for the Igf-1 and Ig f l r mutations are phenotypically normal, regardless of the parental legacy of their mutated allele. Thus, unlike the Igf.2 and Igf2r loci, which are subject to reciprocal parental imprinting (DeChiara et al., 1991; Barlow et al., 1991), the/gf-1 and Ig f l r genes are not im- printed. In a previous study using reverse transcription

Igf-1 and Igflr Null Mutations 69
P C R assays , / g f l r t ransc r ip ts w e r e d e t e c t e d in normal ,
bu t not p a r t h e n o g e n e t i c , m o u s e e m b r y o s f rom the 8-cel l
s t age o n w a r d s , l ead ing to the s u g g e s t i o n tha t the I g f l r g e n e is impr in ted ( R a p p o l e e et aL, 1992). In v iew o f ou r
resul ts , th is in te rp re ta t ion is not tenab le .
Experimental Procedures
Construction of Replacement Veotora To construct a vector for targeting of the Igf-1 gene, we first screened a mouse genomic library with a rat IGF-I cDNA (J. Welsh and A. E., unpublished data) and isolated a phage ~. clone containing a 14.4 kb DNA fragment that included exon 4 of the Igf- 1 gsne, as demonstrated by restriction mapping and partial sequencing data that agreed with previous partial characterizations of this genomic region (Mathews et al,, 1986; Tollefsan et al., 1989). A replacement vector was then constructed from subcloned genomic fragments in several steps. The final product (cloned into pBluascript SK+; Stratagene) consists of a 1.85 kb XhoI-Hindlll fragment from plasmid pMCltk (tk cassette; provided by K. Thomas and M. Capecchi), a 1.0 kb SpeI-BspEI frag- ment of Igfol genomic sequence (ending after the first 74 bp of exon 4), a neo cassette (a 1.1 kb XhoI-BamHI fragment from pMClpola; Stratagsne), and a 5.7 kb PvuI-Kpnl/of- / fragment (beginning 7 bp after the 5' splice site of the intron downstream of exon 4). Thus, a BspEI-Pvul Igf- 1 fragment (119 bp) was replaced with the neo cassette, which was positioned in the targeting vector in the same transcriptional orientation as the tk cassette and the remnant of exon 4.
To construct a vector for Igflr gene targeting, a mouse genomic library was screened with a 380 bp probe isolated from the rat rig F1R-4 cDNA clone 0Nerner et al., 1989) that included the last 240 bp of exon 3 and the first 140 bp of exon 4 (from a Pvull site to the end of the available sequence). We isolated s phage ;L clone containg a 17 kb DNA fragment that included exon 3 (313 bp) of t he /g f l r gene, as shown by restriction mapping and partial DNA sequencing (see Figure 2B). From subcloned gsnomic fragments, a replacement vector was constructed (into pGEM3Z; Promega Corporation), consisting of a 5' genomic fragment (4.5 kb, from a Sail site to a Pvull site, converted to a Xhol site by attachment of a linker) that retained the first 73 bp of exon 3; the 1.1 kb XhoI-BamHf neo cassette; a downstream Igflr genomic fragment (AvrlI-BamHI; -0 .75 kb); and the 1.85 kb Xhol- Hindlll tk cassette. Thus, a PvulI-Avrit Igflr fragment containing the 3' terminal portion of exon 3 (240 bp) and 17 bp of the downstream intron sequence were replaced with the neo cassette positioned in the same transcriptional orientation as the tk cassette and the endogenous /gfl r gsne.
Gene Targeting in ES Cells CCE ES cells (passages 11 and 13 for Igf-1 and Igflrtargeting, respec- tively) were grown to 80%-90% confluence on mitomycin C-treated G418-resistant STO cell feeder layers (Robertson, 1987) in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum, 10% newborn calf serum, and 0.1 mM 13-mercaptoethanol at 37°C, 5% CO2. A total of 8 (Igf- 1) and 25 {Igflr) electroporations were performed, using per experiment 2 x 107 to 2.5 x 107 cells in 0.5 ml of phosphate- buffered saline (Specialty Media) and 20 p.g of Sacll4inearized Igf-1 replacement vector DNA or 16 p.g of Sall-lineerized Igflr replacement vector DNA in a 0.4 cm cuvette of a Bio-Rad gene pulser set at 220 V, 960 p.F. Cells from each cuvette were then equally'distributed on three 10 cm feeder plates. G418 (200 p.g/ml effective concentration; Sigma) was added 48 hr after plating. For double selection, gancyclovir (2 pM) was added to some of the plates 72 hr after the addition of G418. Resistant ES clones were picked into 24-microwell plates coated with feeder STO fibroblasts. After expansion, each clone was split into 2 wells. One of these samples was frozen, while the other was used for DNA analysis. On average, in the Igf.1 experiment, 33% of the G418-resistant colonies were also resistant to gancyclovir, and 1.2% of these were homologous recombinants (3 clones, all possessing a normal complement of chromosomes). In the Igflr experiment, resis- tance to both drugs was observed in 18% of the G418-resistant colo- nies, and 1.7% of these were homologous recombinants (9 clones). Six of the targeted clones had a euploid karyotype.
ES Celt DNA Analysis After aspiration of the medium and washing with phosphete-bufferad saline, 500 p.J of lysis buffer (50 mM Tds--HCI [pH 7.5], 100 mM NaCI, 50 mM EDTA, 0.5 mM spermidine, 5 mM dithiothraitol, 1% SDS, and 400 pg/ml proteinase K) was added to each well of the plates containing confluent ES cells. The lysate was transferred to a 1.5 ml Eppendorf tube and incubated at 55°C overnight. Cell lysatss from 10 clones (40 pl each) were pooled, extracted with phenol and chloroform, and the DNA was precipitated after addition of 100 ~l of 10 M ammonium acetate and 400 p.I of isopropanol. The pellet was resuspended in 400 id of Tris-EDTA, and 1 p.I of this solution was used for PCR analysis. Each PCR reaction contained 50 pmol of each primer, 200 pM dNTPs, 10 mM Tris-HCI (pH 8.3), 50 mM KCI, 1.5 mM MgCI~, 0,1 mg/ml gelatin, and 1.25 U of Taq polymerase (Boehringer Mannheim) in a final volume of 50 Id.
The assays for Igf.1 targeting were performed with the primers 5'-TGCGCTGACAGCCGGAACAC-3' (neo; located at a distance of 450 bp from the beginning of the cassette) and 5'-CTGCCACT- TAGCTCTCTGCC-3' (Igt-1 locus sequence; lying on genomic DNA, upstream of the 5' Spel site present on the replacement vector). PCR cycling was for 1 rain at 94°C, 1 rain at 60°C, and 1 rain at 72°C for 25 cycles. A PCR product of correct size for the distance between the primers (1.9 kb), visualized after Southern blotting, was detected in 3 of 29 analyzed groups of clones.
The assays for Igflr targeting were performed with the primers 5'-CAGGACATAGCGTTGGCTACCC-3' (neo) and 5'-GGACCTTCTA- CAAGGTGGGGAC-3' (Igflr locus sequence; lying on genomic DNA, downstream of the 3' end of the sequence present in the replacement vector). PCR cycling was for 1 rain at 94°C, 1 rain at 70°C, and 1.5 rain at 74°C for 30 cycles, After the addition to each tube of 60 pmol of each primer, 12 nmol of each of the dNTPs, and 1.5 U of Taq polymerase, the amplification was continued for 30 more cycles. A 30 pJ aliquot of each PCR reaction was analyzed by electrophoresis on a 1% agarose gel. A PCR product of correct size (1.0 kb), visualized by ethidium bromide staining, was detected in 9 of 53 analyzed groups of clones.
Following identification of individual positive clones by PCR, South- ern analysis was used to verify their authenticity.
B lu tocye t Injections Blastocysts were flushed at 3.5 days postcoitum from the uterine horns of naturally mated C57BI.J6J and MF1 females in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum. Approximately 15 ES cells of targeted clones possessing a normal karyotype were injected into each blastocoel, and groups of 6-12 blastocysts were transferred into pseudopregnant females, as described (Bradley, 1987). Seven germline chimeras of both host strains transmitting the mutated Igf-1 gene were obtained from one of the injected clones, while in the Igflr case, two of the injected clones yielded eight germline transmitters.
Genotyplng For genotyping of animals by Southern analysis, DNA was prepared as described (Hogan et al., 1986) from the yolk sac of E9.5--E16.5 embryos, the tails of E17.5 and E18.5 embryos and neonates, and the tail tip of 2- to 4-week-old mice. For probes we used a 440 bp EcoRI-Spel fragment located on genomic DNA upstream of the 5'/gf-1 fragment present in the corresponding targeting vector (see Figure 1A) and a 460 bp BamHI-HinclJ fragment located on genomic DNA downstream of the 3' end of the Igflr sequence represented in the corresponding vector (see Figure 1C).
Histological and Anatomical Analyses For histopethology, E16.5-E18.5 embryos were dissected from anes- thetized females and perfused individually through the left ventricle of the heart with 4% paraformaldehyde in 0.12 M phosphate buffer. After perfusion, the embryos were fixed in 4% paraformaldehyde for 2 hr and then transferred to phosphate-buffered saline and processed for paraffin sectioning. Younger embryos (E12.5--E15.5) were dis- sected and fixed in 4% paraformaldehyde at 4°C overnight. Sections (6-8 mm) were stained with hematoxylin and eosin.
For skeletal analysis, embryos were eviscerated, fixed in ethanol at room temperature for 4 days, transferred to acetone for 4 days, and

Cell 70
Table 3. Results of RII/rll (Female) x Riilrll (Male) Crosses
A. Expected Frequencies of Gametes and Offspring"
Maternal Gametes
Paternal Gametes RII (0,5) rll (0.5)
Nonrecombinant Rii (0.3) Rll/Rii (0.15) rlllRii (0.15) rll (0,3) RIIIrll (0.15) rll/rll (0.15)
Recombinant RII (0.2) RIIIRII (0.10) rlllRII (0.10) rii (0.2) RII/rii (0.10) rll/rii (0.10)
B. Data b
Genotype Number of Embryos Observed Frequency Expected Frequency Phenotype of Newborn Mice
RIIIRII 53 t 159 0.11 I 0.32 0.10
RII/rll ~ 106 0.21 0.25 J rlllRII
RIIIRii 71 ) 195 0.14 ) 0.39 0.15 RII/rii ~. 124 0.25 0.25
J rll/Rii
rlllrll 86 0.17 0.15
rlllrii 60 0.12 0.10
t 0.35
t 0.40
Normal
Dwarfism (60% of normal size)
Dwarfism (45% of normal size) Neonatal lethality
Dwarfism (30% of normal size) Neonatal lethality
• The expected frequency of recombinant paternal gametes is 40% (the genetic distance between Igflr and Igf-2 on chromosome 7 is 41 cM). b The data (500 genotyped embryos) are from 62 litters derived from imbred and outbred crosses.
then stained with alizarin red S and alcian blue 8GS at 37°C for 3-5 days. The tissues were cleared with 1% KOH, and the skeletons were stored in glycerol, as described (McLeod, 1980).
Genetic Crosses To generate Igf-2(p-)/Igflr(-/-) double mutant mice, we had to rely on meiotic recombination events, since these genes both reside on chromosome 7, at a distance of 41 cM apart (see Hillyard et al., 1993). To simplify the description of matings initiated for this purpose, and of the genotypes of offspring, we will denote the wildotype and mutated versions of the Igf lr gene as R and r (for receptor), respectively, while II and ii will be the corresponding symbols for the wild-type and mutated Igf-2 gene.
First, we crossed females homozygous for the Igf-2 mutated gene (Rii/Rii) with males heterozygous for the Igf lr mutated gene (RII/rll), to obtain among the F1 progeny Riilrll animals. In all subsequent crosses, RIIIrll females were mated with Riilrll males so that all of the F2 progeny receiving the paternal ii gene would be Igf-2(p-) mutants, owing.to parental imprinting. At the same time, we expected that 20% of the paternal gametes would be rii recombinants (see Table 3), which, in combination with rll maternal gametes, would yield the desired dou- ble mutants (rlllrii). Table 3 shows that the possible combinations of paternal and maternal (nonrecombinantand recombinant) gametes can yield six genotypes, which, as it turned out, corresponded to four (instead of three) phenotypes. Genotyping of 500 embryos from 62 litters of RIIIrll x Rii/rll matings indicated that the frequency distribu- tion of genotypes was in excellent agreement with the expectation (Table 3).
To generate Igf- l(-I-) l lgf-2(p-) double mutants, we first crossed females homozygous for the/gf-2 mutated gene with males heterozy- gous for the Igf-1 mutated gene, to obtain, among the F1 progeny, males heterozygous for both mutated loci. Such males were then mated to heterozygous Igf- l(+l-) females, so that all of the F2 progeny receiving the mutated Igf-2 gene would be Igf-2(p-) mutants, owing to parental imprinting. The fraction of the latter progeny that were homozygous for the Igf-1 mutation corresponded to the desired double mutants.
To generate offspring with combinations of mutations present in the same litters, we first crossed females heterozygous for the Igf l r mutation with males heterozygous for both the Igf-I and/gf-2 muta- tions. Among the progeny, we kept and then crossed males heterozy- gous for all three mutations and females heterozygous for both Igf-1 and Igfl r.
Acknowledgments
We thank S. Newman and J. Goldman for communicating unpublished data; J. Krueger for help in interpreting skin histology; C. T. Roberts and D. LeRoith for providing an Igf l rcDNA probe; J. Lee and H. Wein- traub for providing PCR primers and antibodies for assays of muscle gene expression; G. Singh for providing a surfactant apoprotsin anti- body; N. Jenkins for use of facilities; and R. Axel, C. Bondy, T. OeChi- ara, E. Fuchs, T. Jassal, V. E. Pspaioannou, J. Pintar, and S. Zeitlin for discussions and advice. This work was supported by grants to A. E. and E. J. R. from the National Institutes of Health. J.-Po L. was supported by an award from the Markey Charitable Trust to the Colum- bia University Center for Molecular Toxicology and Nutrition. J. B. was supported by a training grant on hormone research from the National Institute of Diabetes and Digestive and Kidney Diseases. E. J. R. was supported by the Raymond and Beverly Sackler Foundation.
Received April 28, 1993; revised July 20, 1993.
References
Abbott, A. M., Bueno, R., Pedrini, M. T., Murray, J. M., and Smith, R. J. (1992). Insulin-like growth factor I receptor gene structure. J. Biol. Chem. 267, 10759-10763. Ayer-LeLievre, C. St~hlbom, P,-A., and Sara, V. R. (1991). Expression of IGFol and -II mRNA in the brain and craniofacial region of the rat fetus. Development 111, 105-115. Baker, J., Liu, J.-P., Robertson, E. J., and Efstratiadis, A. (1993). Role of insulin-like growth factors in embryonic and postnatal growth. Cell 75 (this issue).

Igf-1 and Igflr Null Mutations 71
Barlow, D. P., StOger, R., Hermann, B. G., Saito, K., and Schweifer, N. (1991). The mouse insulin-like growth factor type-2 receptor is im- printed and closely linked to the Tree locus. Nature 349, 84-87. Barres, B. A., Hart, I. K., Colas, H. S. R., Burne, J. F., Voyvodic, J. T., Richardson, W. D., and Raft, M. C. (1992). Cell death and control of cell survival in the oligodendrocyte lineage. Cell 70, 31-46. Bayne, M. L., Applebaum, J., Chicchi, G. G., Miller, R. E., and Cascieri, M. A. (1990). The roles of tyrosines 24, 31, and 60 in the high affinity binding of insulin-like growth factor-I to the type 1 insulin-like growth factor receptor. J. Biol. Chem. 265, 15648-15652. Bondy, C. A., Warner, H., Roberts, C. T., and LeRoith, D. (1990). Cellular pattern of insulin-like growth factor-I (IGF-I) and type I IGF receptor gene expression in early organogenesis: comparison with IGF-II gene expression. Mol. Endocrinol. 4, 1386-1398. Bondy, C., Warner, H., Roberts, C. T., and LeRoith, D. (1992). Cellular pattern of type-I insulin-like growth factor receptor gene expression during maturation of the rat brain: comparison with insulin-like growth factors I and I1. Neuroscience 46, 909-923. Bradley, A. (1987). Production and analysis of chimeric mice. In Terato- carcinomas and Embryonic Stem Cells, E. J. Robertson, ed. (Oxford, England: IRL Press), pp. 131-151. Conover, C. A., Misra, P., Hintz, R. L., and Rosenfeld, R. G. (1986). Effect of an anti-insulin-like growth factor I receptor antibody on insulin- like growth factor II stimulation of DNA synthesis in human fibroblasts. Biochem. Biophys. Res. Commun. 139, 501-508. Czech, M. P. (1989). Signal transmission by the insulin-like growth factors. Cell 59, 235-238. Daughaday, W. H. (1989). A personal history of the origin of the soma- tomedin hypothesis and recent challenges to its validity. Perspect. Biol. Med. 32, 194-211. Daughaday, W. H., and Rotwein, P. (1989). Insulin-like growth factors I and I1: peptide, messenger ribonucleic acid and gene structures, serum, and tissue concentrations. Endocr. Rev. 10, 66-91. DeChiara, T. M., Efstratiadis, A., and Robertson, E. J. (1990). A growth- deficiency phenotype in heterozygous mice carrying an insulin-like growth factor II gene disrupted by targeting. Nature 345, 76-80. DeChiara, T. M., Robertson, E. J., and Efstratiadis, A. (1991). Parental imprinting of the mouse insulin-like growth factor II gene. Cell 64, 849- 859. Furlanetto, R. W., DiCarlo, J. N., and Wisehart, C. (1987). The type II insulin-like growth factor receptor does not mediate deoxyribonucleic acid synthesis in human fibroblasts. J. Clin. EndocrinoL Metab. 64, 1142-1149. Germain-Lee, E. L., Janicot, M., Lammers, R., UIIrich, A., and Casella, S. J. (1992). Expression of the type I insulin-like growth factor receptor with low affinity for insulin-like growth factor I1. Biochem. J. 281,413-- 417. Gustafson, T. A., and Rutter, W. J. (1990). The cysteine-rich domains of the insulin and insulin-like growth factor I receptors are primary determinants of hormone binding specificity. J. Biol. Chem. 266, 18663-18667. Hall, L. J., Kajimoto, Y., Bichell, D., Kim, S.-W., James, P. L., Counts, D., Nixon, L. J., Tobin, G., and Rotwein, P. (1992). Functional analysis of the rat insulin-like growth factor I gene and identification of an IGF-I gene promoter. DNA Cell Biol. 11,301-313. Han, V. K. M., and Hill, D. J. (1992). The involvement of insulin-like growth factors in embryonic and fetal development. In The Insulin-like Growth Factors: Structure and Biological Functions, P. N. Schofield, ed. (Oxford, England: Oxford University Press), pp. 178-219.
Hillyard, A. L., Doolittle, D. P., Davisson, M. T., and Roderick, T. H. (1993). Locus map of the mouse with comparative map points of human on mouse. Mouse Genome 91, 25. Hogan, B., Costantini, F., and Lacy, E. (1986). Manipulating the Mouse Embryo: A Laboratory Manual (Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press).
Isaksson, O. G. P., Ohlsson, C., Nilsson, A., Isgaard, J., and Lindahl, A. (1991). Regulation of cartilage growth by growth hormone and insu- lin-like growth factor I. Pediatr. Nephrol. 5, 451-453.
Johnson, R. S., Spiegelman, B. M., and Papaioannou, V. (1992). Pleio-
tropic effects of a null mutation in the c-fos proto-oncogene. Cell 71, 577-586. Kiess, W., Haskell, J. F., Lee, L., Greenstein, L. A., Miller, B. E., Aar- ons, A. L., Rechler, M. M., and Nissley, S. P. (1987). An antibody that blocks insulin-like growth factor (IGF) binding to the type II IGF receptor is neither an agonist nor an inhibitor of IGF-stimulated biological re- sponsss in L6 myoblasts. J. Biol. Chem. 262, 12745-12751. Kornfeld, S. (1992). Structure and function of the mannose 6-phos- phatelinsulinlike growth factor II receptors. Annu. Rev. Biochem. 61, 307-330. Krane, J. E., Murphy, D. P., Carter, D. M., and Krueger, J. G. (1991). Synergistic effects of epidermal growth factor (EGF) and insulin-like growth factor Ilsomatomedin C (IGF-I) on keratinocyte proliferation may be mediated by IGF-I transmodulation of the EGF receptor. J. Invest. Dermatol. 96, 419-424. Le Mouellic, H., Lallemand, Y., and BrQlet, P. (1992). Homeosis in the mouse induced by a null mutation in the Hox-3.1 gene. Cell 69, 251- 264. LeRoith, D., ed. (1991). Insulin-like Growth Factors: Molecular and Cellular Aspects (Boca Raton, FL: CRC Press). Lows, W. L. (1991 ). Biological actions o1 the insulin-like growth factors. In Insulin-like Growth Factors: Molecular and Cellular Aspects, D. Le- Roith, ed. (Boca Raton, FL: CRC Press), pp. 49-85. Mansour, S. L., Thomas, K. R., and Capecchi, M. R. (1988). Disruption of the proto-oncogene int-2 in mouse embryo-derived stem cells: a general strategy for targeting mutations to non-selectable genes. Na- ture 336, 348-352. Mathews, L. S., Norstedt, G., and Palmiter, R. D. (1986). Regulation of insulin-like growth factor I gene expression by growth hormone. Proc. Natl. Acad. Sci. USA 83, 9343-9347. McLeod, M. J. (1980). Differential staining of cartilage and bone in whole tissue fetuses by alcian blue and alizarin red S. Teratology 22, 299-301. Morgan, D. O., Edman, J. C., Standring, D. R., Fried, V. A., Smith, M. C., Roth, R. A., and Rutter, W. J. (1987). Insulin-like growth factor II receptor as a multifunctional binding protein. Nature 329, 301-307. Mottola, C., and Czech, M. P. (1984). The type II insulin-like growth factor receptor does not mediate increased DNA synthesis in H-35 hepatoma cells. J. Biol. Chem. 259, 12705-12713. Moxham, C., and Jacobs, S. (1992). Insulin-like growth factor recep- tors. In The Insulin-like Growth Factors: Stucture and Biological Func- tions, P. N. Schofield, ed. (Oxford, England: Oxford University Press), pp. 80-110. Neely, E. K., Beukers, M. W., Oh, Y., Cohen, P., and Rosenfeld, R. G. (1991). Insulin-like growth factor receptors. Acta Paediatr. Scand. (Suppl.) 372, 116-123. Nishimoto, I., Murayama, Y., and Okamoto, T. (1991). Signal transduc- tion mechanism of IGF-II/man-6-P receptor. In Modern Concepts of Insulin-like Growth Factors, E. M. Spencer, ed. (New York: Elsevier), pp. 517-522. Nissley, P., Kiess, W., and Skier, M. M. (1991). The insulin-like growth factor II/mannose 6-phosphate receptor. In Insulin-like Growth Fac- tors: Molecular and Cellular Aspects, D. LeRoith, ed. (Boca Raton, FL: CRC Press), pp. 111-150.
Nolan, C. M., Kyls, J. W., Watanabe, H., and Sly, W. S. (1990). Binding of insulin-like growth factor II (IGF-II) by human cation-independent mannose 6-phosphate receptorllGF-II receptor expressed in receptor- deficient mouse L cells. Cell Regul. 1,197-213.
Oemar, B. S., Foellmer, H. G., Hodgdon-Anandant, L., and Rosenz- weig, S. A. (1991). Regulation of insulin-like growth factor I receptors in diabetic masangial cells. J. Biol. Chem. 266, 2369-2373.
Oka, Y., Rozek, L. M., and Czech, M. P. (1985). Direct demonstration of rapid insulin-like growth factor II receptor internalization and recy- cling in rat adipocytas: insulin stimulates 1251 insulin-like growth factor II degradation by modulating the IGF-II receptor recycling process. J. Biol. Chem. 260, 9435-9442.
Rappolee, D. A., Sturm, K. S., Behrendtsen, O., Schultz, G. A., Ped- ersen, R. A., end Werb, Z. (1992). Insulin-like growth factor II acts through an endogenous growth pathway regulated by imprinting in

Cell 72
early embryos. Genes Dev. 6, 939-952. Robertson, E. J. (1987). Embryo-derived stem cell lines. In Teratocarci- nomas and Embryonic Stem Cells: A Practical Approach, E. J. Robert- son, ed. (Oxford, England: IRL Press), pp. 71-112. Roth, R. A. (1988). Structure of the receptor for insulin-like growth factor I1: the puzzle amplified. Science 239, 1269-1271. Sara, V. R., and Hall, K. (1990). Insulin-like growth factors and their binding proteins. Physiol. Rev. 70, 591-614. Schofield, P. N., ed. (1992). The Insulin-like Growth Factors: Structure and Biological Functions (Oxford, England: Oxford University Press). Sommer, I., and Schachner, M (1981). Monoclonal antibodies (01 to 04) to oligodendrocyte cell surfaces: an immunocytological study in the central nervous system. Dev. Biol. 83, 311-327. Stevens, L. C., and Mackensen, J. A. (1958). The inheritance and expression of a mutation in the mouse affecting blood formation, the axial skeleton, and body size. J. Hered. 49, 153-160. Streck, R. D., and Pintar, J. E. (1992). The embryonic pattern of rat insulin-like growth factor-I gene expression suggests a role in induction and early growth of the liver. Endocrinology 131, 2030-2032. Streck, R. D., Wood, T. L., Hsu, M.-S., and Pintar, J. E. (1992). Insulin- like growth factor I and I I and insulin-like growth factor binding protein-2 RNAs are expressed in adjacent tissues within rat embryonic and fetal limbs. Dev. Biol. 151,586-596. Tavakkol, A., Elder, J. T., Griffiths, C. E. M., Cooper, K. D., Talwar, H., Fisher, G. J., Keane, K. M., Foltin, S. K., and Voorhees, J. J. (1992). Expression of growth hormone receptor, insulin-like growth factor 1 (IGF-1) and IGF-1 receptor mRNA and proteins in human skin. J. In- vest. Dermatol. 99, 343-349. Thomas, K. R., Musci, T. S., Neumann, P. E., and Capecchi, M. R. (1991). Swaying is a mutant allele of the proto-oncogene Wnt-l. Cell 67, 969-976. Tollefsen, S. E., Lajara, R., McCusker, R. H., Clemmons, D. R., and Rotwein, P. (1989). Insulin-like growth factors (IGF) in muscle develop- ment. J. Biol. Chem. 264, 13810-13817. UIIrich, A., Gray, A., Tam, A. W., Yang-Feng, T., Tsubokawa, M., Col- lins, C., Henzel, W., Le Bon, T., Kathuria, S., Chen, E., Jacobs, S., Francke, U., Ramachandran, J., and Fujita-Yamaguchi, Y. (1986). In- sulin-like growth factor I receptor primary structure: comparison with insulin receptor suggests structural determinants that define functional specificity. EMBO J. 6, 2503-2512. Warner, H., Woloschak, M., Adamo, M., Shen-Orr, Z., Roberts, C. T., and LeRoith, D. (1989). Developmental regulation of the rat insulin-like growth factor I receptor gene. Proc. Natl. Acad. Sci. USA 86, 7451- 7455. Zemel, S., Bartolomei, M. S., and Tilghman, S. M. (1992). Physical linkage of two mammalian imprinted genes, H19 and insulin-like growth factor 2. Nature Genet. 2, 61-65. Zhang, B., and Roth, R. A. (1991). Binding properties of chimeric insu- lin receptors containing the cysteine-rich domain of either the insulin- like growth factor I receptor or the insulin receptor related receptor. Biochemistry 30, 5113-5117.
Copyright © 2022 FDOKUMEN




















