
Epileptic Seizure Classification with Symmetric and Hybrid ...
Upload
independentCategory
view
0download
0
B R A I N R E S E A R C H 1 4 2 6 ( 2 0 1 1 ) 8 6 – 9 5
Ava i l ab l e on l i ne a t www.sc i enced i r ec t . com
www.e l sev i e r . com/ loca te /b ra i n res
Research Report
Metabolic gene expression changes in the hippocampus ofobese epileptic male rats in the pilocarpine model of temporallobe epilepsy
Nicole Ruiza, Luis F. Pachecoa, Bianca Farrella, Cody B. Coxb, Boris S. Ermolinskya,Emilio R. Garrido-Sanabriaa, Saraswathy Naira,⁎aDepartment of Biomedical Sciences, University of Texas at Brownsville, Brownsville, TX, USAbDepartment of Behavioral Sciences, University of Texas at Brownsville, Brownsville, TX, USA
A R T I C L E I N F O
⁎ Corresponding author at: Biomedical ScienceFax: +1 956 882 5043.
E-mail address: [email protected]
0006-8993/$ – see front matter © 2011 Elseviedoi:10.1016/j.brainres.2011.10.006
A B S T R A C T
Article history:Accepted 2 October 2011Available online 8 October 2011
Chronically epileptic male adult rats in the pilocarpine model of temporal lobe epilepsy (TLE),exhibited gross expansion of abdominal fat mass and significant weight gain several monthsafter induction of status epilepticus (SE) when compared to control rats. We hypothesized thatepileptogenesis can induce molecular changes in the hippocampus that may be associatedwith metabolism. We determined the expression levels of genes Hsd11b1, Nr3c1, Abcc8,Kcnj11, Mc4r, Npy, Lepr, Bdnf, and Drd2 that are involved in regulation of energy metabolism,in the hippocampus of age-matched control and chronic epileptic animals. Taqman-basedquantitative real time polymerase chain reaction (qPCR) and the delta–delta cycle threshold(CT) methods were used for the gene expression assays. Gene expression of Hsd11b1 (cortisolgenerating enzyme) was significantly higher in epileptic versus control rats at 24 h and2months, after induction of SE. Nr3c1 (glucocorticoid receptor) mRNA levels on the other handwere down-regulated at 24 h, 10 days and 2months, post SE. Abcc8 (Sur1; subunit of ATP-sensitive potassium (KATP) channel) was significantly down-regulated at 10 days post SE.Kcnj11 (Kir6.2; subunit of ATP-sensitive potassium (KATP) channel) was significantly up-regulated at 24 h, 1 month and 2months post SE. Thus, we demonstrated development of obesi-ty and changes in the expression ofmetabolic genes in the hippocampus during epileptogenesisin male rats in the pilocarpine model of TLE.
© 2011 Elsevier B.V. All rights reserved.
Keywords:Neuroendocrine regulationEpilepsyObesityHippocampusGlucocorticoid metabolismPilocarpine
1. Introduction
A compelling number of studies suggest that obesity may beassociated in part with a dysfunction of neural pathways. In-deed, some authors have suggested that obesity is a neurolog-ical disease more than a metabolic one (Jordan et al., 2010;O'Rahilly and Farooqi, 2006). Furthermore, obesity is a
s, University of Texas at B
(S. Nair).
r B.V. All rights reserved
common comorbidity in neurological disorders (Trevathanand Dietz, 2009). Although the comorbidity of obesity andepilep-sy has been recently demonstrated (Daniels et al., 2009; Gilliamet al., 2005) only few studies have investigated the underlyingmechanisms on the association between weight gain and ep-ilepsy (St-Pierre et al., 2009). For instance, children with newlydiagnosed, untreated epilepsy have a higher body mass index
rownsville, 80 Fort Brown, LHSB 1.824, Brownsville, TX 78520, USA.
.

87B R A I N R E S E A R C H 1 4 2 6 ( 2 0 1 1 ) 8 6 – 9 5
(BMI) than healthy children (Daniels et al., 2009). Antiepilepticdrugs (i.e. valproic acid) have also been associated with weightgain in epilepsy (Rauchenzauner et al., 2008). A recent studyreported that overweight epileptic patients treated with val-proic acid exhibit a higher risk of developing metabolic disor-ders (Verrotti et al., 2010). Scharfman et al. (2008) observed asignificant weight gain in female rats in the pilocarpinemodel of epilepsy. St-Pierre et al. observed that female Wistaralbino rats gained weight after inducing status epilepticus (SE)by administration of lithium and pilocarpine and treatingthe seizures with acepromazine (St-Pierre et al., 2009; St-Pierreand Persinger, 2005).
Several genes have been identified that play a role in neu-roendocrine regulation of obesity and associated metabolicdysfunction (Boguszewski et al., 2010). Among these, 11β-hydroxysteroid dehydrogenase Type 1 (Hsd11b1) has emergedrecently as a key player in regulation of intracellular glucocor-ticoid metabolism in the brain and peripheral tissues.Hsd11b1 encodes an enzyme responsible for converting corti-sone (11-dehydrocorticosterone in rats) into the stress hor-mone cortisol (corticosterone in rats) intracellularly in brainas well as peripheral tissues (Yau and Seckl, 2001). Up-regulation of Hsd11b1 in brain and adipose tissue is linked toobesity, metabolic dysfunction and neuronal degeneration inrodents and humans (Buren et al., 2007; Engeli et al., 2004;Loizzo et al., 2010; Nair et al., 2004). Cortisol binding to gluco-corticoid receptors (Nr3c1) in various areas of the brain (in-cluding the hippocampus, hypothalamus and pituitary) aswell as peripheral tissues, plays an important role in negativefeedback regulation of the hypothalamus–pituitary–adrenal(HPA) axis, tissue specific glucocorticoid regulation, neuronalplasticity, and electrical activity, adipogenesis, metabolicfunction and many other physiological processes (Buren etal., 2007; Dickmeis and Foulkes, 2011; Kersten, 2010). The evi-dence for the roles played by stress, glucocorticoids as wellas metabolic hormones in the function of the hippocampusincluding learning, memory, regulation of HPA axis and pe-ripheral physiology such as glucose metabolism and insulinsecretion has been comprehensively documented (Lathe,2001; Snyder et al., 2011).
ATP-binding cassette, subfamily c (CFTR/MRP), member8 (Abcc8) and potassium inwardly rectifying channel, subfam-ily j, member 11 (Kcnj11) are subunits of the ATP-sensitive po-tassium (KATP) channel originally identified in the pancreaticbeta-cells, critically involved in glucose homeostasis in diabe-tes (Bennett et al., 2010). This ATP-sensitive potassium chan-nel links glucose metabolism to electrical excitability andinsulin secretion (Bennett et al., 2010; Flanagan et al., 2009).Down-regulation of Kir6.2 (Kcnj11) in the hypothalamus hasbeen shown to correlate with neuropeptide expression in theorexigenic (hunger inducing) Npy and Agrp neurons (Gyte etal., 2007). The role of this KATP channel in neuroprotectionand improved cognitive function in the brain has recentlybeen reported (Clark et al., 2010; Judge and Smith, 2009;Slingerland et al., 2008). Mice lacking an ATP-sensitive potas-sium channel have seizures when subjected to brief periodsof hypoxia, compared to wild type mice, which revived with-out any incident (Yamada et al., 2001). Mutations in KCNJ11in humans have been linked to DEND (developmental delay,epilepsy and neonatal diabetes) syndrome, which is likely
caused by overactivity of this channel in the brain, skeletalmuscle and nerve tissue (Koster et al., 2008).
Melanocortin 4 receptor (Mc4r), (present in the brain andhypothalamus) is known to be involved in regulating food in-take (Wikberg et al., 2000). Neuropeptide Y (Npy), present inorexigenic neurons in the hypothalamus, is an orexigenic hor-mone that triggers food intake and reduces energy expendi-ture (Schwartz, 2006). It is up-regulated in the hippocampusin epilepsy because of its ability to act as a neuroprotectiveagent (Vezzani and Sperk, 2004). Dopamine receptor D2(Drd2) is known to be involved in the feeding reward systemof the brain (Davidson et al., 2007; Johnson and Kenny, 2010).Johnson and Kenny (2010) showed that rats who consume ahigh fat diet exclusively have higher reward thresholds com-pared to rats that only eat standard chow, and that high fatfed rats have lower expression of Drd2 in the striatum thancontrol rats. Brain-derived neurotrophic factor (Bdnf) is in-volved in neurogenesis. It is expressed in different parts ofthe brain including the hippocampus. Studies have shownthat Bdnf can have both pro-epileptic and antiepileptic effectsin the brain (Simonato et al., 2006). In addition, decreasedlevels of Bdnf are observed in the hippocampus, in high fatdiets and diet induced obesity models, in rodents (Yu et al.,2009). Leptin, another major neuroendocrine regulator thatnegatively regulates feeding intake, is involved in increasingBdnf levels in the hippocampus (Komori et al., 2006).
Using a pilocarpine model to study epilepsy in rats, we ob-served that the majority of the epileptic rats gain greaterweight at a faster rate compared to non-epileptic rats eventhough both groups appear to be consuming the same amountof food and fluids. We hypothesized that status epilepticus(SE) in addition to hypothalamic changes that might induceweight gain, causes molecular changes in the hippocampus,a hitherto uninvestigated region of the brain in the regulationof metabolism, energy expenditure and weight gain (Mattssonet al., 2003). The goal of this project was to determine if thegenes associated with metabolism or neuroendocrine controlof obesity (Hsd11b1, Nr3c1, Abcc8, Kcnj11, Mc4r, Npy, Lepr,Drd2 and Bdnf) are up- or down-regulated in the hippocampusof epileptic rats compared to the non-epileptic rats.
2. Results
2.1. Body weight, food and fluid intake comparisons
Differences in bodyweight gainweremeasured between controland epileptic animals for 14 weeks (Fig. 1A). The result of the re-peated measures ANOVA revealed a significant main effect ofbody weight between the two groups (F (2.97, 234.97)=18.46,p<0.001, η2=0.19) as well as a significant interaction betweenbody weight and weeks, (F (2.97, 234.97)=264.18, p<0.001,η2=0.77, indicating that the rate of weight increase differed be-tween the groups (Fig. 1B). Once it was determined that therewas a significant difference in body weight, independentsamples t-tests were used to determine at which week thetwo groups first start to differ significantly. To control forthe number of t-tests, we incorporated Bonferroni's adjust-ment and only viewed p-values less that 0.003 as significant.

250
300A
100
150
200
Control0
50Ave
rage
Foo
d In
take
(g)
Epileptic
Weeks Post Pilocarpine Treatment
350B
1 2 5 6 7 8 9 10 113 4
88 B R A I N R E S E A R C H 1 4 2 6 ( 2 0 1 1 ) 8 6 – 9 5
This revealed that the weights of the control and epilepticrats become significantly different starting at week 3 (t (98)=−3.59, p=0.001). The average weight for the control groupat week one was 219.93 g (SD=43.66) and 209.71 g (SD=44.75)for the epileptic group. At week 14, the control group had anaverage weight of 369.86 g (SD=95.69) and the epilepticgroup had an average weight of 473.42 g (SD=108.92). The ep-ileptic rats gain weight at an increased rate compared to thecontrol rats (Fig. 1B). We also observed massive expansionof abdominal adipose tissue in the epileptic animals (datanot shown). We did not observe any significant differencesin the food intake (F (2.87, 14.33)=0.451, p<0.713, η2=0.083)or in the fluid intake (F (2.39, 11.94)=0.574, p<0.607,η2=0.103) between the two groups (Figs. 2A and B).
500
600A
300
400
Control
*
0
100
200
Mea
n B
ody
Wei
ght
(g)
Epileptic
1200
1400B
Weeks Post Pilocarpine Treatment
600
800
1000
0
200
400
600
% W
eigh
t In
crea
se
Control Epileptic
1 2 3 4 5 6 7 8 9 10 11 12 13 14
1 2 3 4 5 6 7 8 9 10 11 12 13 14
Weeks Post Pilocarpine Treatment
Fig. 1 – Bodyweight comparison and percent increase ofweight in control and epileptic rats. Comparison of bodyweight between control (n=7) and epileptic (n=13) rats (Fig.1A). Repeated measures ANOVAwas used to determine ifbodyweight significantly differed between the twogroups andwhether there was an interaction between body weight andweeks. Independent samples t testswere used to determine atwhich week the groups first differ significantly. Repeatedmeasures ANOVA indicated a significant difference in bodyweight as well as a significant interaction between bodyweight and weeks. Beginning at week 3, the differencebetween the epileptic and control rats was found to bestatistically significant. *P<0.001. Fig. 1B shows the percentincrease in body weight for the control and epileptic animals.The absence of error bars for the control rats (weeks 8–14) isbecause they do not deviate from the normal adult rat weightat each of these ages.
300
150
200
250
Control Epileptic0
50
100
1 2 3 4 5 6 7 8 9 10 11A
vera
ge W
ater
Int
ake
(ml)
Weeks Post Pilocarpine Treatment
Fig. 2 – Feeding and fluid intake comparison over a 2.5 monthperiod. Comparison of food (Fig. 2A) and water intake (Fig.2B) of control rats (n=2) compared to epileptic rats (n=5 to 6).Repeated measures ANOVA was used to compare food andwater intake of control and epileptic rats over a period of2.5 months (P<0.05). There was no significant differencebetween control and epileptic rats.
2.2. Hippocampal mRNA gene expression
Statistically significant differences in gene expression weredetected between epileptic (n=20; all time points post SEgrouped together) and control (n=5) rats for Abcc8 (F(4, 20)=3.523, P=0.025) (Fig. 3A), Kcnj11 (F(4, 20)=19.889, P<0.001)(Fig. 3B), Hsd11b1 (F(4, 20)=8.528, P<0.001) (Fig. 4A) and Nr3c1(F(4, 20)=4.801, P=0.007) (Fig. 4B). Abcc8 (Fig. 3A) mRNA levelsappeared down-regulated in the post SE animals at all timepoints compared to the controls although statistically signifi-cant (P=0.012) down-regulation in expression was observedonly between the control and 10 days post SE animals.Kcnj11 (Fig. 3B) showed an initial decline in mRNA levels at24 h (P=0.016), but then increased significantly by 1 month(P=0.002) as well as 2 months (P=0.049) post SE. This up-regulation in Kcnj11 gene expression was also significant be-tween 24 h and 1 month (P<0.001) or 2 months (P<0.001),and 10 days and 1 month (P<0.001) or 2 months (P=0.010)post SE. Hsd11b1 (Fig. 4A) gene expression was up-regulatedmost significantly in the post SE animals at 24 h, when com-pared to the controls (P=0.001), 10 days (P=0.003) or 1 month(P=0.005) post SE. Hsd11b1 expression declined to almost con-trol levels by 10 days and 1 month, and started increasing by2 months (P=0.030), post SE. On the other hand, for Nr3c1(Fig. 4B), significant up-regulation was only observed at
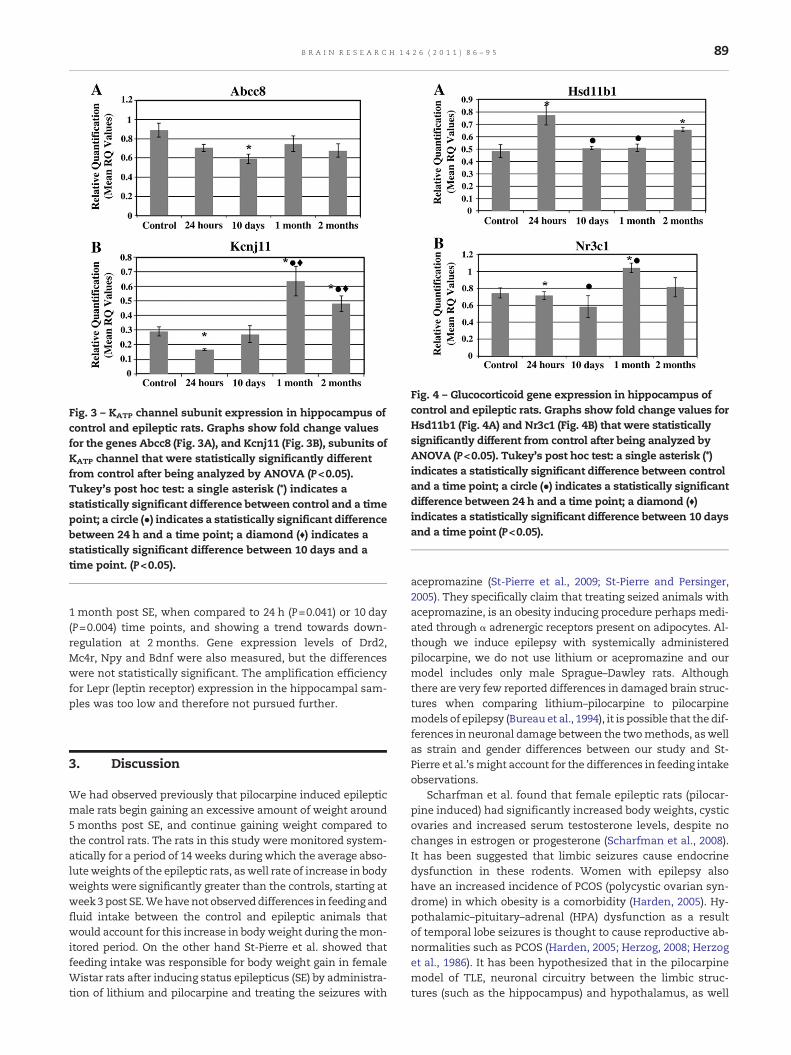
Fig. 3 – KATP channel subunit expression in hippocampus ofcontrol and epileptic rats. Graphs show fold change valuesfor the genes Abcc8 (Fig. 3A), and Kcnj11 (Fig. 3B), subunits ofKATP channel that were statistically significantly differentfrom control after being analyzed by ANOVA (P<0.05).Tukey's post hoc test: a single asterisk (*) indicates astatistically significant difference between control and a timepoint; a circle (●) indicates a statistically significant differencebetween 24 h and a time point; a diamond (♦) indicates astatistically significant difference between 10 days and atime point. (P<0.05).
Fig. 4 – Glucocorticoid gene expression in hippocampus ofcontrol and epileptic rats. Graphs show fold change values forHsd11b1 (Fig. 4A) and Nr3c1 (Fig. 4B) that were statisticallysignificantly different from control after being analyzed byANOVA (P<0.05). Tukey's post hoc test: a single asterisk (*)indicates a statistically significant difference between controland a time point; a circle (●) indicates a statistically significantdifference between 24 h and a time point; a diamond (♦)indicates a statistically significant difference between 10 daysand a time point (P<0.05).
89B R A I N R E S E A R C H 1 4 2 6 ( 2 0 1 1 ) 8 6 – 9 5
1 month post SE, when compared to 24 h (P=0.041) or 10 day(P=0.004) time points, and showing a trend towards down-regulation at 2 months. Gene expression levels of Drd2,Mc4r, Npy and Bdnf were also measured, but the differenceswere not statistically significant. The amplification efficiencyfor Lepr (leptin receptor) expression in the hippocampal sam-ples was too low and therefore not pursued further.
3. Discussion
We had observed previously that pilocarpine induced epilepticmale rats begin gaining an excessive amount of weight around5months post SE, and continue gaining weight compared tothe control rats. The rats in this study were monitored system-atically for a period of 14 weeks during which the average abso-luteweights of the epileptic rats, aswell rate of increase in bodyweights were significantly greater than the controls, starting atweek3 post SE.Wehavenot observeddifferences in feeding andfluid intake between the control and epileptic animals thatwould account for this increase in bodyweight during themon-itored period. On the other hand St-Pierre et al. showed thatfeeding intake was responsible for body weight gain in femaleWistar rats after inducing status epilepticus (SE) by administra-tion of lithium and pilocarpine and treating the seizures with
acepromazine (St-Pierre et al., 2009; St-Pierre and Persinger,2005). They specifically claim that treating seized animals withacepromazine, is an obesity inducing procedure perhaps medi-ated through α adrenergic receptors present on adipocytes. Al-though we induce epilepsy with systemically administeredpilocarpine, we do not use lithium or acepromazine and ourmodel includes only male Sprague–Dawley rats. Althoughthere are very few reported differences in damaged brain struc-tures when comparing lithium–pilocarpine to pilocarpinemodels of epilepsy (Bureau et al., 1994), it is possible that the dif-ferences in neuronal damage between the twomethods, aswellas strain and gender differences between our study and St-Pierre et al.'smight account for the differences in feeding intakeobservations.
Scharfman et al. found that female epileptic rats (pilocar-pine induced) had significantly increased body weights, cysticovaries and increased serum testosterone levels, despite nochanges in estrogen or progesterone (Scharfman et al., 2008).It has been suggested that limbic seizures cause endocrinedysfunction in these rodents. Women with epilepsy alsohave an increased incidence of PCOS (polycystic ovarian syn-drome) in which obesity is a comorbidity (Harden, 2005). Hy-pothalamic–pituitary–adrenal (HPA) dysfunction as a resultof temporal lobe seizures is thought to cause reproductive ab-normalities such as PCOS (Harden, 2005; Herzog, 2008; Herzoget al., 1986). It has been hypothesized that in the pilocarpinemodel of TLE, neuronal circuitry between the limbic struc-tures (such as the hippocampus) and hypothalamus, as well

90 B R A I N R E S E A R C H 1 4 2 6 ( 2 0 1 1 ) 8 6 – 9 5
as hormonal feedback loops that are important for HPA func-tion may be damaged (Amado and Cavalheiro, 1998).
Neuroendocrine regulation of energy metabolism in thehypothalamus is being investigated actively by many groups(Boguszewski et al., 2010; Gibson et al., 2010; McMinn et al.,2000; Neary et al., 2004; Schwartz, 2006). However not muchis known about the role of hippocampus in energy metabo-lism regulation. Recently, it has been demonstrated that asmall subset of neurons within the dentate gyrus are impor-tant for hippocampal inhibition of the HPA axis and feedbackcontrol that affect endocrine and feeding behavioral re-sponses (Snyder et al., 2011). The role of hippocampal learningand memory mechanisms in the higher order regulation offood intake has been reported and reviewed (Benoit et al.,2010; Davidson et al., 2007; Kanoski and Davidson, 2011;Squire, 2004). In the pilocarpine model of epilepsy, hippocam-pal slices from epileptic rats show lesions (Curia et al., 2008;Klitgaard et al., 2002). In this preliminary study, we focusedonly on the hippocampus region of the brain, because in tem-poral lobe epilepsy (TLE), the seizure arises in this area of thebrain (Dichter, 2009) and we were investigating the potentiallink between TLE, hippocampus and obesity. Moreover, wehad banked hippocampus cDNA samples available from con-trol and epileptic animals aged up to 2 months post SE. Pilo-carpine induces lesions in other regions of the brain (Curiaet al., 2008; Dube et al., 2000), including the hypothalamus,which have not been investigated in this preliminary study.It has been shown that the CA1 hippocampal brain region andvarious nuclei of the hypothalamus, including those areas in-volved in food intake regulation, are linked (Cenquizca andSwanson, 2006). Future studies are warranted to investigateneuroendocrine molecular changes in several areas of thebrain including the hypothalamus in TLE.
The hippocampus expresses many of the genes that are in-volved in neuroendocrine regulation of metabolism such asthe insulin receptor, leptin receptor, mineralocorticoid andglucocorticoid receptors, ATP sensitive potassium channel,NPY, BDNF etc. (Davidson et al., 2007; Herman et al., 1993;Joels and DeKloet, 1994; Kanoski et al., 2007; Karschin et al.,1997; Komori et al., 2006; Lathe, 2001; Shanley et al., 2002;Simonato et al., 2006; Vezzani et al., 1999; Zawar et al., 1999;Zhao et al., 2004). Several of these are expressed differentiallyin the hippocampus during epileptogenesis (Becker et al.,2003; Elliott et al., 2003; Gorter et al., 2006; Vezzani andSperk, 2004; Winden et al., 2011; Zagulska-Szymczak et al.,2001). Hippocampal neurogenesis has been shown to playa major role in buffering stress response which causesglucocorticoid hypersecretion and behavior that impacts en-ergy metabolism such as feeding (Snyder et al., 2011).Hippocampus-dependent learning and memory may regulateneuroendocrine mechanisms regulating appetitive and sati-ety behaviors (Davidson et al., 2007). Diet induced elevationsin peripheral cholesterol and triglyceride levels can lead tohippocampal alterations of lipid metabolism and oxidativestress (Stranahan et al., 2011) and defects in hippocampallearning (Kanoski et al., 2007; Molteni et al., 2002). In summa-ry, we hypothesize that abnormalities in the expression ofdifferent molecules that are involved in stress, metabolism,etc. in seizure-damaged hippocampal networks may eithertrigger a yet to be determined behavioral response that
affects the way animals feed or regulate energy metabolism,and contribute to metabolic dysregulation by affectinghypothalamic functions such as the HPA axis or other neuro-endocrine responses.
We demonstrate statistically significant differences in theexpression of Hsd11b1 and Nr3c1 genes in the hippocampus,between control and epileptic rats. Hsd11 expression is signif-icantly up-regulated in epileptic animals at 24 h post SE, andalthough the levels decline at 10 days and 1 month, they aresignificantly higher than control animals by 2 months postSE. Nr3c1 on the other hand is down-regulated reciprocally,as Hsd11b1 levels increase (presumably through negativefeedback regulation by increased levels of intracellular corti-sone generated by Hsd11b1) (Tomlinson et al., 2004). There isevidence that intracellular Hsd11b1 mRNA levels parallel in-tracellular cortisol/cortisone production (Buren et al., 2007).These differences in glucocorticoid metabolism gene expres-sion, suggest that at least in the hippocampus, glucocorticoidmetabolismmay be altered in epileptic animals during epilep-togenesis. It has been shown that glucocorticoid metabolismin key areas of the brain including the hippocampus, play arole in obesity, metabolic and cognitive dysfunction, aging,depression and stress response (Buren et al., 2007; Sandeepet al., 2004; Seckl and Walker, 2004; Snyder et al., 2011). Block-ing of glucocorticoid receptors during kainic acid induced SEhas been shown to reduce both structural damage to thebrain and cell death (Kaufer et al., 2004). The detrimental ef-fects of glucocorticoids were also connected with a down reg-ulation of Bdnf. The potentially elevated glucocorticoid(cortisone) levels in the hippocampus (due to up-regulationof Hsd11b1) in the epileptic animals may result in alteringneurogenesis, compared to the non-epileptic animals (Snyderet al., 2011). Neuronal plasticity and the young age (1 monthold rat) at which pilocarpine is administered may also play arole in feeding behavior (which we have not yet been able toobserve) and the development of obesity in this model of epi-lepsy as suggested previously (Davidson et al., 2009).
Kcnj11/Abcc8 complex is expressed in the hippocampus(Zawar et al., 1999). AlthoughAbcc8mRNA levelswere in generaldown-regulated, Kcnj11 levels tended to increase by 2months,post SE. At present we do not understand the significance ofthe observed differences in expression between the two sub-units of ATP sensitive K+ channel, although this protein is asso-ciated with obesity as well as cognitive function (Clark et al.,2010; Judge and Smith, 2009; Slingerland et al., 2008). Recently,upregulation of Kcnj13, an inwardly rectifying potassium chan-nel important in synaptic transmission has been demonstratedin the intrahippocampal kainate acidmodel of temporal lobe ep-ilepsy (Winden et al., 2011). The density of ATP sensitive potassi-um channels (composed of Abcc8 and Kcnj11 subunits) in theplasmamembrane connects cell membrane potential to glucosemetabolism in many cell types including hippocampal neurons(Chen andWu, 2011;Mankouri et al., 2006). Abnormal ATP sensi-tive K+ channel activity in glucose sensing POMC neurons wasshown to play a role in whole body control of glucose (Parton etal., 2007). Therefore we suggest that the differential expressionof these subunits in the hippocampus of epileptic rats may re-flect altered neuronal glucosemetabolism aswell as excitability,potentially contributing to dysfunctional energy metabolismregulation in this model.

91B R A I N R E S E A R C H 1 4 2 6 ( 2 0 1 1 ) 8 6 – 9 5
Wedid not observe differences inmRNA levels of Bdnf, Npy,Drd2 or Mc4r expression in the hippocampus of the control orepileptic animals.Wemaynot havebeen able tomeasure subtledifferences in expression, since these genes were detectable atlow levels in the hippocampus of rats, in our study. LeprmRNA levels in the hippocampus were too low to performmeaningful analyses of fold change in this study. Expressionof these genes that regulate energy regulation and body weightmay be different between humans and rodents. For example,leptin expression although easily detectable in human brain isnot detectable in mouse brain (Scott et al., 2009).
A limitation of this study is that pilocarpine causes neuronalchanges or damage in various regions of the brain including thehypothalamus, a region of the brain which is well-establishedin regulating energy metabolism during epileptogenesis (Bureauet al., 1994; Druga et al., 2003; Dube et al., 2000). Therefore it is dif-ficult to establish unequivocally the potential role of the hippo-campus in regulating energy metabolism in this complexmodel. Another limitation of this study is that seizures may in-duce acute gene expression changes that are unrelated to energymetabolism regulation. We were consistent in isolating hippo-campi in epileptic animals that had not experienced seizures inthe 2 hprior to sacrifice.Howeverwe cannot rule out thepossibil-ity of the acute effects of earlier unmonitored seizures (regardlessof the fact that onset of spontaneous seizures averaged 3–5 perweek, after the silent phase of epileptogenesis), since we didnot video monitor the animals continuously. We believe thatthe gene expression changes noted in the epileptic animals arerepresentative of chronic molecular changes in the hippocam-pus, sincewehad five animals per group for all points during epi-leptogenesis. However future studies incorporating videomonitoring of animals is warranted to differentiate acute versuschronic effects of seizures on gene expression. Moreover, geneexpression changes that are observed during epileptogenesis inthe pilocarpinemodel of TLE could be due to changes in neuronalcell population that need to bemeasured using in situ hybridiza-tion or single cell PCR in the future, but is beyond the scope of thecurrent study.
In summary, pilocarpine inducedmale epileptic rats becomeseverely obese and show significant differences in the hippo-campal expression levels of genes involved in glucocorticoidregulation and energy metabolism. We hypothesize that thesemolecular changes in the hippocampus due to epileptogenesismay reflect dysregulation of neuroendocrine mechanisms andfeedback loops regulating energy metabolism in hippocampusadditionally to the hypothalamus.
4. Experimental procedures
4.1. Induction of status epilepticus by pilocarpine
All experiments were performed in accordance with theNational Institutes of Health Guidelines for the Care and Useof Laboratory Animals and with the approval of The Universi-ty of Texas at Brownsville Institutional Animal Care andUse Committee. Sprague–Dawley rats were kept in a accli-matized temperature-controlled vivarium with water andstandard lab chow ad libitum. All efforts were made to
minimize the number of animals in the study. Chronicallyepileptic rats were obtained by the pilocarpine model oftemporal lobe epilepsy following described procedures,which is characterized by bilateral hippocampal pathology(Cavalheiro, 1995; Mello et al., 1993). At the time of perform-ing the pilocarpine induction of epilepsy, animals were ap-proximately 30–35 days (125–150 g) old. A total of forty-fiveanimals were used in our experiments. Pilocarpine was ad-ministered between 9 AM and 12 PM in an acclimatizedroom set at 20 °C. All animals received the same dose of1% methyl-scopolamine nitrate (0.1 mg/kg in saline, s.c.)(Sigma-Aldrich, St. Louis, MO) 30 min before pilocarpine ad-ministration to minimize the peripheral effects of choliner-gic stimulation (Turski et al., 1984). Animals were theninjected with 4% pilocarpine hydrochloride (Sigma-Aldrich)(350 mg/kg in saline, i.p.). After pilocarpine administration,seizures were induced by continuous clicking or clappingand rats were kept in a seizure state for 3 h. Controls werealso injected with methyl-scopolamine but received a sa-line injection instead of pilocarpine. The rats that seizedfor at least 3 h were marked as epileptic. Epileptic ratswere given an injection of diazepam to stop the seizurestate at 3 h after SE onset. During sedation, 10 ml of physi-ological saline was administered subcutaneously to rehy-drate the animals. Food was enriched with DietGel®
Recovery food (ClearH2O, Portland, ME) for at least 1 weekpost pilocarpine treatment to aid in their recovery. SE suf-fering rats were monitored for at least 4–5 h per week by atrained technician who is required to log in all seizuresthat occur in their presence and the severity of the seizure.The seizures averaged about 3–5 per week and the severityof the seizures averaged between 3 and 5 in the Racine'sscale (Racine, 1972).To investigate the time-course of geneexpression changes, age-matched control (n=5) and epilep-tic rats (n=20) were sacrificed at four different time pointsfollowing SE: 24 h (n=5), 10 days (n=5), 1 month (n=5) andmore than 2 months (n=5). Prior to sacrifice, all animalswere brought into the laboratory at least 2 h prior to tissueextraction. In that time period, we did not observe any sei-zures in any of the animals used in the study.
4.2. Monitoring weight gain
Epileptic (n=13) and control (n=7) rats were monitored dailyfor weight gain for 14 weeks. Body weights were compared inaged matched rats starting at 8 days post SE and the compar-ison continued for 14 weeks. Body weight was measured byplacing rats in a plastic bowl and weighing each rat individu-ally on a scale. Weight was recorded to the nearest gram.
4.3. Monitoring feeding and fluid intake
Ratswere allowed access to standard lab chow (Harlan 18% pro-tein rodent diet) (Harlan, Houston, TX) and water ad libitum. Atthe beginning of the week (Monday), each rat was given600 g of chow and 600 ml of tap water in two 300 ml waterbottles. Epileptic (n=5–6) and control (n=2) rats were moni-tored daily for feeding and fluid intake. Food intake was cal-culated by subtracting the leftover chow from what hadinitially been provided. Weight of the chow was recorded to

92 B R A I N R E S E A R C H 1 4 2 6 ( 2 0 1 1 ) 8 6 – 9 5
the nearest gram. Water bottles were marked with a scalemeasurement on the bottle. Fluid intake was measured andrecorded to the nearest milliliter.
4.4. RNA extraction
Thehippocampus of each ratwas excised, homogenized and theRNAwas extracted using the RNeasy Mini Kit (Qiagen, Valencia,CA). The concentration of the RNA was measured using a spec-trophotometer to check the A260/A280 ratio and the quality ofthe RNA was checked by gel electrophoresis to see if there wasany DNA contamination.
4.5. cDNA synthesis
Reverse transcription was performed using the Advantage RT-for-PCR Kit (Clontech, Mountain View, CA) following manu-facturer's instructions. 1 μg of RNA from each sample wasused to make cDNA. 1 μl of Oligo (dT)18 primer (20 μM) wasadded to each RNA sample. Briefly, RNA samples were dena-tured at 70 °C for 2 min, and immediately chilled at 4 °C anddNTP mixture (10 mM each), 5× reaction buffer (250 mM Tris–HCl, pH 8.3, 375 mM KCl, 15 mM MgCl2), recombinant RNaseinhibitor (40 U/μl), and MMLV reverse transcriptase (200 U/μl)were added. The samples were then incubated for 1 h at42 °C and for 5 min at 94 °C. Samples were diluted withDEPC-treated water and stored at −80 °C.
4.6. qPCR
Gene expression was measured by quantitative PCR (qPCR)using TaqMan gene expression assays (Applied Biosystems,Carlsbad, CA). The gene expression assays used were Hsd11b1(hydroxysteroid (11-beta) dehydrogenase 1) [Rn00690787_m1,amplicon length=94 bp, RefSeq: NM_017080.2], Nr3c1 (glucocor-ticoid receptor) [Rn01405584_m1, amplicon length=75 bp,RefSeq: NM_012576.2], Kcnj11 (potassium inwardly-rectifyingchannel, subfamily J, member 11) [Rn01764077_s1, ampliconlength=65 bp, RefSeq: NM_031358.3], Abcc8 (ATP-binding cas-sette, subfamily C [CFTR/MRP], member 8) [Rn00564778_m1,amplicon length=70 bp, RefSeq: NM_013039.2], Mc4r (melano-cortin 4 receptor) [Rn01491866_s1, amplicon length=156 bp,RefSeq: NM_013099.2], Drd2 (dopamine receptor D2)[Rn01418275_m1, amplicon length=85 bp, RefSeq: NM_012547.1], Lepr (leptin receptor) [Rn01433205, amplicon length=94 bp,RefSeq:NM_012596.1], Npy (neuropeptide Y) [Rn01410146_m1,amplicon length=130 bp, RefSeq: NM_012614.1] and Bdnf(brain derived neurotrophic factor) [Rn02531967_s1, ampliconlength=142 bp, RefSeq: NM_012513.3]. The endogenous controlwas Gapdh (glyceraldehyde 3 phosphate dehydrogenase)[Rn99999916_s1, amplicon length=87 bp, RefSeq: NM_017008.3]. We have shown in a previous study that Gapdh expressionis stable across samples from control and epileptic rats sacri-ficed at different time points (Ermolinsky et al., 2008). Amplifi-cation efficiency curves for all gene expression assays wererun prior to performing the actual experiments. The curvesallowed quantitation of cDNA such that amplification efficien-cies were similar between the target gene and Gapdh. Amplifi-cation efficiencies of all gene expression assays were alsochecked prior to experimentation, with the sample designated
as the calibrator, using seven point dilution series. The StepOne Plus Real-Time PCR System (Applied Biosystems, Carlsbad,CA) was used to run the amplification efficiency curves and theexperimental plates. Each cDNA sample was run in triplicateand no template control wells were included on each plate tocheck for contamination. In a few cases, outlier samples froma triplicate series were excluded from final analysis. The qPCRreaction mix consisted of sterile water, Universal PCR MasterMix (20 μmol/l) (2× concentration, contains AmpliTaq GoldDNA Polymerase, AmpErase UNG, dNTPs with dUTP, ROX pas-sive reference and optimized buffer components) (Applied Bio-systems, Carlsbad, CA) and TaqMan gene expression assay(20× mix). The Real-time PCR program included a holdingstage at 50 °C for 2 min and 95 °C for 10 min, and cycling 40times at 95 °C for 15 s, and 60 °C for 1 min. The Delta–DeltaComparative Threshold method was used to quantify the foldchange between the samples.
4.7. Statistical analysis
SPSS Statistics 17.0 software was used for all analyses. Repeat-ed measures analysis of variance (ANOVA) was used to ana-lyze the body weight, food intake and fluid intake data todetermine if there was a statistically significant difference be-tween the two groups. The Greenhouse–Geisser calculationwas used to correct the degrees of freedom as Mauchly's testindicated that the assumption of sphericity had been violated(p>.05). When conducting t-tests subsequent to the repeatedmeasures ANOVA, the Bonferroni correction was also usedto control for type I error (p values less than 0.003 were consid-ered significant). Histograms, Q–Q plots, and the Shapiro–Wilktest were used to determine if the data for each gene was nor-mally distributed. The data that were not normally distributedwere transformed using the Box–Cox transformation method.One way ANOVA was used to determine if the changes in themRNA levels were significant in between control and epilepticanimals (all time points post SE grouped together). Tukey'spost hoc test was used to compare gene expression betweencontrol and each of the different time points post SE, as wellas between the post SE time points. In these cases, p<0.05was considered significant.
Disclosure of conflicts of interest
The authors have no conflicts of interest to disclose.
Author contributions
All authors have approved the final article.Nicole Ruiz performed body weight, feeding and fluid in-
takemeasurements, RNA isolations from tissues, qPCR, statis-tical analyses, intellectual input on the project, and writingthe manuscript and preparation of figures.
Luis F. Pacheco performed pilocarpine induction of epilep-sy, sacrifice of animals, dissection, and review of manuscript.
Bianca Farrell performed pilocarpine procedure, weighing,monitoring and maintenance of rats, statistical analysis ofbody weight, and review of manuscript.

93B R A I N R E S E A R C H 1 4 2 6 ( 2 0 1 1 ) 8 6 – 9 5
Cody B. Cox provided statistical expertise and input in allthe statistical analyses using in this study.
Boris S. Ermolinsky provided intellectual input and techni-cal assistance on qPCR assay and analyses, and review ofmanuscript.
Emilio R Garrido-Sanabria contributed intellectual input onthe project, and provided hippocampal cDNA samples, statis-tical analysis, and reviewed and edited the manuscript.
Saraswathy Nair was responsible for initiating the projectand providing intellectual input and funds for the designand execution of this work. She also helped in data interpreta-tion, writing and editing of the manuscript.
Acknowledgments
This work was supported by intramural funds obtained by Dr.Nair at the University of Texas at Brownsville.
R E F E R E N C E S
Amado, D., Cavalheiro, E.A., 1998. Hormonal and gestationalparameters in female rats submitted to the pilocarpine modelof epilepsy. Epilepsy Res. 32, 266–274.
Becker, A.J., Chen, J., Zien, A., Sochivko, D., Normann, S.,Schramm, J., Elger, C.E., Wiestler, O.D., Blumcke, I., 2003.Correlated stage- and subfield-associated hippocampal geneexpression patterns in experimental and human temporallobe epilepsy. Eur. J. Neurosci. 18, 2792–2802.
Bennett, K., James, C., Hussain, K., 2010. Pancreatic beta-cellK-ATP channels: hypoglycaemia and hyperglycaemia. Rev.Endocr. Metab. Disord. 11, 157–163.
Benoit, S.C., Davis, J.F., Davidson, T.L., 2010. Learned and cognitivecontrols of food intake. Brain Res. 1350, 71–76.
Boguszewski, C.L., Paz, G., Velloso, L.A., 2010. Neuroendocrinebody weight regulation: integration between fat tissue,gastrointestinal tract, and the brain. Endokrynol. Pol. 61,194–206.
Bureau, Y.R.J., Peredery, O., Persinger, M.A., 1994. Concordance ofquantitative damage within the diencephalon andtelencephalon following systemic pilocarpine (380 mg/kg) orlithium (3 mEq/kg)/pilocarpine (30 mg/kg) induced seizures.Brain Res. 648, 265–269.
Buren, J., Bergstrom, S.A., Loh, E., Soderstrom, I., Olsson, T., Mattsson,C., 2007. Hippocampal 11beta-hydroxysteroid dehydrogenase type1 messenger ribonucleic acid expression has a diurnal variabilitythat is lost in the obese Zucker rat. Endocrinology 148, 2716–2722.
Cavalheiro, E.A., 1995. The pilocarpine model of epilepsy. Ital. J.Neurol. Sci. 16, 33–37.
Cenquizca, L.A., Swanson, L.W., 2006. Analysis of directhippocampal cortical field CA1 axonal projections todiencephalon in the rat. J. Comp. Neurol. 497, 101–114.
Chen, B.S., Wu, S.N., 2011. Functional role of the activity ofATP-sensitive potassium channels in electrical behavior ofhippocampal neurons: experimental and theoretical studies. J.Theor. Biol. 272, 16–25.
Clark, R.H., McTaggart, J.S., Webster, R., Mannikko, R., Iberl, M.,Sim, X.L., Rorsman, P., Glitsch, M., Beeson, D., Ashcroft, F.M.,2010. Muscle dysfunction caused by a K-ATP channel mutationin neonatal diabetes is neuronal in origin. Science 329, 458–461.
Curia, G., Longo, D., Biagini, G., Jones, R.S.G., Avoli, M., 2008. Thepilocarpine model of temporal lobe epilepsy. J. Neurosci.Methods 172, 143–157.
Daniels, Z.S., Nick, T.G., Liu, C., Cassedy, A., Glauser, T.A., 2009.Obesity is a common comorbidity for pediatric patients withuntreated, newly diagnosed epilepsy. Neurology 73, 658–664.
Davidson, T.L., Chan, K., Jarrard, L.E., Kanoski, S.E., Clegg, D.J.,Benoit, S.C., 2009. Contributions of the hippocampus andmedial prefrontal cortex to energy and body weight regulation.Hippocampus 19, 235–252.
Davidson, T.L., Kanoski, S.E., Schier, L.A., Clegg, D.J., Benoit, S.C.,2007. A potential role for the hippocampus in energy intakeand body weight regulation. Curr. Opin. Pharmacol. 7, 613–616.
Dichter, M.A., 2009. Emerging concepts in the pathogenesis ofepilepsy and epileptogenesis. Arch. Neurol. 66, 443–447.
Dickmeis, T., Foulkes, N.S., 2011. Glucocorticoids and circadianclock control of cell proliferation: at the interface betweenthree dynamic systems. Mol. Cell. Endocrinol. 331, 11–22.
Druga, R., Kubova, H., Suchomelova, L., Haugvicova, R., 2003.Lithium/pilocarpine status epilepticus-induced neuropathologyof piriform cortex and adjoining structures in rats isage-dependent. Physiol. Res. 52, 251–264.
Dube, C., Boyet, S., Marescaux, C., Nehlig, A., 2000. Progressivemetabolic changes underlying the chronic reorganization ofbrain circuits during the silent phase of the lithium–pilocarpinemodel of epilepsy in the immature and adult rat. Exp. Neurol.162, 146–157.
Elliott, R.C., Miles, M.F., Lowenstein, D.H., 2003. Overlappingmicroarray profiles of dentate gyrus gene expression duringdevelopment- and epilepsy-associated neurogenesis and axonoutgrowth. J. Neurosci. 23, 2218–2227.
Engeli, S., Bohnke, J., Feldpausch, M., Gorzelniak, K., Heintze, U.,Janke, J., Luft, F.C., Sharma, A.M., 2004. Regulation of 11 beta-HSDgenes in human adipose tissue: influence of central obesity andweight loss. Obes. Res. 12, 9–17.
Ermolinsky, B., Otalora, L.F.P., Arshadmansab, M.F., Zarei, M.M.,Garrido-Sanabria, E.R., 2008. Differential changes inmGlu2 andmGlu3 gene expression following pilocarpine-induced statusepilepticus: a comparative real-time PCR analysis. Brain Res.1226, 173–180.
Flanagan, S.E., Clauin, S., Bellanne-Chantelot, C., de Lonlay, P.,Harries, L.W., Gloyn, A.L., Ellard, S., 2009. Update of mutationsin the genes encoding the pancreatic beta-cell K-ATP channelsubunits Kir6.2 (KCNJ11) and sulfonylurea receptor 1 (ABCC8)in diabetes mellitus and hyperinsulinism. Hum. Mutat. 30,170–180.
Gibson, C.D., Carnell, S., Ochner, C.N., Geliebter, A., 2010.Neuroimaging, gut peptides and obesity: novel studies of theneurobiology of appetite. J. Neuroendocrinol. 22, 833–845.
Gilliam, F.G., Mendiratta, A., Pack, A.M., Bazil, C.W., 2005. Epilepsyand common comorbidities: improving the outpatient epilepsyencounter. Epileptic Disord. 7, S27–S33.
Gorter, J.A., van Vliet, E.A., Aronica, E., Breit, T., Rauwerda, H., daSilva, F.H.L., Wadman, W.J., 2006. Potential newantiepileptogenic targets indicated by microarray analysis in arat model for temporal lobe epilepsy. J. Neurosci. 26,11083–11110.
Gyte, A., Pritchard, L.E., Jones, H.B., Brennand, J.C., White, A., 2007.Reduced expression of the K-ATP channel subunit, kir6.2, isassociated with decreased expression of neuropeptide Y andagouti-related protein in the hypothalami of Zucker diabeticfatty rats. J. Neuroendocrinol. 19, 941–951.
Harden, C.L., 2005. Polycystic ovaries and polycystic ovarysyndrome in epilepsy: evidence for neurogonadal disease.Epilepsy Curr 5, 142–146.
Herman, J.P., Watson, S.J., Chao, H.M., Coirini, H., Mcewen, B.S., 1993.Diurnal regulation of glucocorticoid receptor andmineralocorticoid receptormessenger-RNAs in rat hippocampus.Molecular and Cellular Neuroscience 4, 181–190.
Herzog, A.G., 2008. Disorders of reproduction in patients withepilepsy: primary neurological mechanisms. Seizure-EuropeanJournal of Epilepsy 17, 101–110.

94 B R A I N R E S E A R C H 1 4 2 6 ( 2 0 1 1 ) 8 6 – 9 5
Herzog, A.G., Seibel, M.M., Schomer, D.L., Vaitukaitis, J.L.,Geschwind, N., 1986. Reproductive endocrine disorders in menwith partial seizures of temporal-lobe origin. Arch. Neurol. 43,347–350.
Joels, M., DeKloet, E.R., 1994. Mineralocorticoid and glucocorticoidreceptors in the brain — implications for ion permeability andtransmitter systems. Prog. Neurobiol. 43, 1–36.
Johnson, P.M., Kenny, P.J., 2010. Dopamine D2 receptors inaddiction-like reward dysfunction and compulsive eating inobese rats. Nat. Neurosci. 13, 635–641.
Jordan, S.D., Konner, A.C., Bruning, J.C., 2010. Sensing the fuels:glucose and lipid signaling in the CNS controlling energyhomeostasis. Cell. Mol. Life Sci. 67, 3255–3273.
Judge, S.I., Smith, P.J., 2009. Patents related to therapeuticactivation of K(ATP) and K(2P) potassium channels forneuroprotection: ischemic/hypoxic/anoxic injury and generalanesthetics. Expert Opin. Ther. Pat. 19, 433–460.
Kanoski, S.E., Davidson, T.L., 2011. Western diet consumption andcognitive impairment: links to hippocampal dysfunction andobesity. Physiol. Behav. 103, 59–68.
Kanoski, S.E., Meisel, R.L., Mullins, A.J., Davidson, T.L., 2007. Theeffects of energy-rich diets on discrimination reversal learningand on BDNF in the hippocampus and prefrontal cortex of therat. Behav. Brain Res. 182, 57–66.
Karschin, C., Ecke, C., Ashcroft, F.M., Karschin, A., 1997.Overlapping distribution of K-ATP channel-forming Kir6.2subunit and the sulfonylurea receptor SUR1 in rodent brain.FEBS Lett. 401, 59–64.
Kaufer, D., Ogle, W.O., Pincus, Z.S., Clark, K.L., Nicholas, A.C.,Dinkel, K.M., Dumas, T.C., Ferguson, D., Lee, A.L., Winters,M.A., Sapolsky, R.M., 2004. Restructuring the neuronal stressresponse with anti-glucocorticoid gene delivery. Nat. Neurosci.7, 947–953.
Kersten, S., 2010. Regulation of nutrient metabolism andinflammation. Results Probl. Cell Differ. 52, 13–25.
Klitgaard, H., Matagne, A., Vanneste-Goemaere, J., Margineanu,D.G., 2002. Pilocarpine-induced epileptogenesis in the rat:impact of initial duration of status epilepticus onelectrophysiological and neuropathological alterations.Epilepsy Res. 51, 93–107.
Komori, T., Morikawa, Y., Nanjo, K., Senba, E., 2006. Induction ofbrain-derived neurotrophic factor by leptin in the ventromedialhypothalamus. Neuroscience 139, 1107–1115.
Koster, J.C., Cadario, F., Peruzzi, C., Colombo, C., Nichols, C.G.,Barbetti, F., 2008. The G53D mutation in Kir6.2 (KCNJ11) isassociated with neonatal diabetes and motor dysfunction inadulthood that is improved with sulfonylurea therapy. J. Clin.Endocrinol. Metab. 93, 1054–1061.
Lathe, R., 2001. Hormones and the hippocampus. J. Endocrinol.169, 205–231.
Loizzo, S., Campana, G., Vella, S., Fortuna, A., Galietta, G.,Guarino, I., Costa, L., Capasso, A., Renzi, P., Frajese, G.V.,Franconi, F., Loizzo, A., Spampinato, S., 2010. Post-natalstress-induced endocrine and metabolic alterations in miceat adulthood involve different pro-opiomelanocortin-derivedpeptides. Peptides 31, 2123–2129.
Mankouri, J., Taneja, T.K., Smith, A.J., Ponnambalam, S.,Sivaprasadarao, A., 2006. Kir6.2 mutations causing neonataldiabetes prevent endocytosis of ATP-sensitive potassiumchannels. EMBO J. 25, 4142–4151.
Mattsson, C., Lai, M., Noble, J., McKinney, E., Yau, J.L., Seckl, J.R.,Walker, B.R., 2003. Obese Zucker rats have reducedmineralocorticoid receptor and 11 beta-hydroxysteroiddehydrogenase type 1 expression in hippocampus— implicationsfor dysregulation of the hypothalamic–pituitary–adrenal axis inobesity. Endocrinology 144, 2997–3003.
McMinn, J.E., Baskin, D.G., Schwartz, M.W., 2000. Neuroendocrinemechanisms regulating food intake and body weight. Obes.Rev. 1, 37–46.
Mello, L.E., Cavalheiro, E.A., Tan, A.M., Kupfer, W.R., Pretorius, J.K.,Babb, T.L., Finch, D.M., 1993. Circuit mechanisms of seizures inthe pilocarpine model of chronic epilepsy: cell loss and mossyfiber sprouting. Epilepsia 34, 985–995.
Molteni, R., Barnard, R.J., Ying, Z., Roberts, C.K., Gomez-Pinilla, F.,2002. A high-fat, refined sugar diet reduces hippocampalbrain-derived neurotrophic factor, neuronal plasticity, andlearning. Neuroscience 112, 803–814.
Nair, S., Lee, Y.H., Lindsay, R.S., Walker, B.R., Tataranni, P.A.,Bogardus, C., Baier, L.J., Permana, P.A., 2004. 11beta-Hydroxysteroid dehydrogenase Type 1: geneticpolymorphisms are associated with Type 2 diabetes in PimaIndians independently of obesity and expression in adipocyteand muscle. Diabetologia 47, 1088–1095.
Neary, N.M., Goldstone, A.P., Bloom, S.R., 2004. Appetiteregulation: from the gut to the hypothalamus. Clin. Endocrinol.60, 153–160.
O'Rahilly, S., Farooqi, I.S., 2006. Genetics of obesity. PhilosophicalTransactions of the Royal Society B-Biological Sciences 361,1095–1105.
Parton, L.E., Ye, C.P., Coppari, R., Enriori, P.J., Choi, B., Zhang, C.Y.,Xu, C., Vianna, C.R., Balthasar, N., Lee, C.E., Elmquist, J.K.,Cowley, M.A., Lowell, B.B., 2007. Glucose sensing by POMCneurons regulates glucose homeostasis and is impaired inobesity. Nature 449, 228–232.
Racine, R.J., 1972. Modification of seizure activity by electricalstimulation. 2. Motor seizure. Electroencephalogr. Clin.Neurophysiol. 32, 281–294.
Rauchenzauner, M., Habertandt, E., Scholl-Burgi, S., Karall, D.,Schoenherr, E., Tatarczyk, T., Engl, J., Laimer, M., Luef, G.,Ebenbichler, C.F., 2008. Effect of valproic acid treatment onbody composition, leptin and the soluble leptin receptor inepileptic children. Epilepsy Res. 80, 142–149.
Sandeep, T.C., Yau, J.L.W., MacLullich, A.M.J., Noble, J., Deary, I.J.,Walker, B.R., Seckl, J.R., 2004. 11 beta-Hydroxysteroiddehydrogenase inhibition improves cognitive function inhealthy elderly men and type 2 diabetics. Proc. Natl. Acad. Sci.U. S. A. 101, 6734–6739.
Scharfman, H.E., Kim, M., Hintz, T.M., MacLusky, N.J., 2008.Seizures and reproductive function: insights from female ratswith epilepsy. Ann. Neurol. 64, 687–697.
Schwartz, M.W., 2006. Central nervous system regulation of foodintake. Obesity 14, 1S–8S.
Scott, M.M., Lachey, J.L., Sternson, S.M., Lee, C.E., Elias, C.F.,Friedman, J.M., Elmquist, J.K., 2009. Leptin targets in the mousebrain. J. Comp. Neurol. 514, 518–532.
Seckl, J.R., Walker, B.R., 2004. 11 beta-Hydroxysteroid dehydrogenasetype 1 as a modulator of glucocorticoid action: from metabolismto memory. Trends Endocrinol. Metab. 15, 418–424.
Shanley, L.J., O'Malley, D., Irving, A.J., Ashford, M.L., Harvey, J.,2002. Leptin inhibits epileptiform-like activity in rathippocampal neurones via PI 3-kinase-driven activation of BKchannels. Journal of Physiology-London 545, 933–944.
Simonato, M., Tongiorgi, E., Kokaia, M., 2006. Angels and demons:neurotrophic factors and epilepsy. Trends Pharmacol. Sci. 27,631–638.
Slingerland, A.S., Hurkx,W., Noordam, K., Flanagan, S.E., Jukema,J.W., Meiners, L.C., Bruining, G.J., Hattersley, A.T.,Hadders-Algra, M., 2008. Sulphonylurea therapy improvescognition in a patient with the V59M KCNJ11 mutation.Diabet. Med. 25, 277–281.
Snyder, J.S., Soumier, A., Brewer, M., Pickel, J., Cameron, H.A., 2011.Adult hippocampal neurogenesis buffers stress responses anddepressive behaviour. Nature 476, 458–461.
Squire, L.R., 2004. Memory systems of the brain: a brief history andcurrent perspective. Neurobiol. Learn. Mem. 82, 171–177.
St-Pierre, L.S., Bubenik, G.A., Parker, G.H., Persinger, M.A., 2009.Insidious weight gain in prepubertal seized rats treated withan atypical neuroleptic: the role of food consumption, fluid

95B R A I N R E S E A R C H 1 4 2 6 ( 2 0 1 1 ) 8 6 – 9 5
consumption, and spontaneous ambulatory activity. EpilepsyBehav. 14, 288–292.
St-Pierre, L.S., Persinger, M.A., 2005. Extreme obesity in female ratsfollowing prepuberal induction of lithium–pilocarpine seizuresanda single injectionof acepromazine. EpilepsyBehav. 7, 411–418.
Stranahan, A.M., Cutler, R.G., Button, C., Telljohann, R., Mattson,M.P., 2011. Diet-induced elevations in serum cholesterol areassociated with alterations in hippocampal lipid metabolismand increased oxidative stress. J. Neurochem. 118, 611–615.
Tomlinson, J.W., Walker, E.A., Bujalska, I.J., Draper, N., Lavery,G.G., Cooper, M.S., Hewison, M., Stewart, P.M., 2004. 11beta-Hydroxysteroid dehydrogenase type 1: a tissue-specificregulator of glucocorticoid response. Endocr. Rev. 25, 831–866.
Trevathan, E., Dietz, W.H., 2009. Obesity in neurology practice: acall to action. Neurology 73, 654–655.
Turski, W.A., Cavalheiro, E.A., Bortolotto, Z.A., Mello, L.M.,Schwarz, M., Turski, L., 1984. Seizures produced by pilocarpinein mice: a behavioral, electroencephalographic andmorphological analysis. Brain Res. 321, 237–253.
Verrotti, A., Manco, R., Agostinelli, S., Coppola, G., Chiarelli, F.,2010. The metabolic syndrome in overweight epileptic patientstreated with valproic acid. Epilepsia 51, 268–273.
Vezzani, A., Sperk, G., 2004. Overexpression of NPY and Y2receptors in epileptic brain tissue: an endogenousneuroprotective mechanism in temporal lobe epilepsy?Neuropeptides 38, 245–252.
Vezzani, A., Sperk, G., Colmers, W.F., 1999. Neuropeptide Y:emerging evidence for a functional role in seizure modulation.Trends Neurosci. 22, 25–30.
Wikberg, J.E.S., Muceniece, R., Mandrika, I., Prusis, P., Lindblom, J.,Post, C., Skottner, A., 2000. New aspects on the melanocortinsand their receptors. Pharmacol. Res. 42, 393–420.
Winden, K.D., Karsten, S.L., Bragin, A., Kudo, L.C., Gehman, L.,Ruidera, J., Geschwind, D.H., Engel Jr., J., 2011. A systems level,functional genomics analysis of chronic epilepsy. PLoS One 6,e20763.
Yamada, K., Ji, J.J., Yuan, H.J., Miki, T., Sato, S., Horimoto, N.,Shimizu, T., Seino, S., Inagaki, N., 2001. Protective role ofATP-sensitive potassium channels in hypoxia-inducedgeneralized seizure. Science 292, 1543–1546.
Yau, J.L.W., Seckl, J.R., 2001. 11 beta-Hydroxysteroid dehydrogenasetype I in the brain; thickening the glucocorticoid soup. Mol.Psychiatry 6, 611–614.
Yu, Y., Wang, Q., Huang, X.F., 2009. Energy-restricted pair-feedingnormalizes low levels of brain-derived neurotrophicfactor/tyrosine kinase B mRNA expression in the hippocampus,but not ventromedial hypothalamic nucleus, in diet-inducedobese mice. Neuroscience 160, 295–306.
Zagulska-Szymczak, S., Filipkowski, R.K., Kaczmarek, L., 2001.Kainate-induced genes in the hippocampus: lessons fromexpression patterns. Neurochem. Int. 38, 485–501.
Zawar, C., Plant, T.D., Schirra, C., Konnerth, A., Neumcke, B., 1999.Cell-type specific expression of ATP-sensitive potassiumchannels in the rat hippocampus. Journal of Physiology-London514, 327–341.
Zhao, W.Q., Chen, H., Quon, M.J., Alkon, D.L., 2004. Insulin and theinsulin receptor in experimental models of learning andmemory. Eur. J. Pharmacol. 490, 71–81.
Copyright © 2022 FDOKUMEN




















