
HIV-1 Diversity in the Envelope Glycoproteins: Implications for Viral Entry Inhibition
Upload
independentCategory
view
0download
0
Pergamon 0163-7258(94)00074-3
Pharmac. Ther. Vol. 65, pp. 41W2, 1995 Copyright ‘a 1995 Elsevier Science Ltd
Printed in Great Britain. All rights reserved 0163-7258195 $29.00
Associate Editor: S. PESTKA
MECHANISMS OF VIRAL INHIBITION BY INTERFERONS
SANTO LANDOLFO,* GIORGIO GRIBAUDO, ALESSANDRA ANGERETTI and MARISA GARIGLIO Institute of Microbiology, Medical School of Torino, Unicersity of Torino,
and Immunogenetics and Experimental Oncology Center, C.N.R., Torino, Italy
Abstract-Interferons (IFNs) are a family of related proteins grouped in four species (r,fi, ;I and w) according to their cellular origin, inducing agents and antigenic and functional properties. Their binding to specific receptors leads to the activation of signal transduction pathways that stimulate a defined set of genes, whose products are eventually responsible for the IFN antiviral effects. Their action against viruses is a complex phenomenon. It has been reported that IFNs restrict virus growth at the levels ofpenetration, uncoating, synthesis of mRNA, protein synthesis and assembly. This review will attempt to evaluate evidence of the involvement of the IFN-inducible proteins in the expression of the antiviral state against RNA or DNA viruses.
Keywords-Interferons, viruses, transcriptional activation, antiviral state, virus escape
1. 2. 3. 4. 5.
6.
7.
CONTENTS
Introduction The Interferon Receptors Signal Transduction Transcriptional Activation Proteins Involved in the Molecular Mechanisms of the Antiviral State Induced by Interferons 5.1. 2’,5’-Oligoadenylate synthetases 5.2. Double-stranded RNA-dependent protein kinase 5.3. Mx proteins Induction of the Antiviral State and its Regulation 6.1. RNA viruses
6.1.1. Picornaviruses 6.1.2. Vesicular Stomatitis virus 6.1.3. Influenza virus 6.1.4. Retroviruses 6.1.5. Reoviruses
6.2. DNA viruses 6.2.1. Vaccinia virus 6.2.2. Herpesviruses 6.2.3. Hepatitis B virus 6.2.4. Simian virus 40
Defenses of Viruses Against Interferons Acknowledgements References
416 416 417 418
421 421 421 422 423 423 424 424 425 426 427 428 428 429 430 431 432 434 434
*Corresponding author. Abbreviations-CAT, chloramphenicol acetyl transferase; CE. chicken embryo cells; CMV, cytomegalo-
virus; DG. diacylglycerol; dsRNA, double-stranded RNA; EBV, Epstein-Barr virus; eIF-2, eukaryotic peptide chain initiation factor 2; EMC, encephalomyocarditis virus; HBV, hepatitis B virus; HIV, human immunodeficiency virus; HSV, herpes simplex virus; Hu, human; IE, immediately-early; IFN, interferon; ISGF, interferon-stimulated gene factor; ISRE, interferon-stimulated response element; MCMV. murine cytomegalovirus; MSV, murine sarcome virus; Mu, murine; MuLV, murine leukemia virus; PKR, double-stranded RNA-dependent protein kinase; RSV, rous sarcoma virus; STATS, signal transducers and activators of transcription; SV40. simian virus 40; VA, virus-associated RNA; VSV, vesicular stomatitis virus.

416 S. Landolfo t’t nl
I. INTRODUCTION
Interferons (IFNs) constitute a heterogeneous family of polypeptides, initially discovered by virtue of their ability to interfere with viral replication and now known to modulate a variety of physiological responses (Isaacs and Lindenmann, 1957; Stewart, 1979; Pestka rt cd., 1987; Pfeffer. 1987; De Maeyer and De Maeyer-Guignard, 1988). Their genomic organization and protein structure, type of inducing agents and producer cells serve to distinguish four varieties: IFN-a, IFN-w, IFN-1 and IFN-7. The human (Hu)-IFN-x family consists of at least 18 genes, 6 of which are pseudogenes. There are 6 Hu-IFN-to genes, 5 of which are pseudogenes. Only a single gene, encoding what was previously called “fibroblast I FN”. has been characterized for I FN-fi. These three subtypes are placed in the Type I IFN superfamily because their genes are all clustered in the short arm of chromosome 9 and do not contain intervening sequences. In contrast, Hu-IFN-y is encoded by a single copy gene. containing three introns, and located on chromosome 12. IFN-;, is designated as Type 11 IFN. Hu-IFN-p genes encode proteins of 165 or 166 amino acids, whereas IFN-(10 genes code for proteins of 172 amino acids. The degree of sequence similarity among the members of the Hu-IFN-2 subfamily varies by an average of 8%. whereas the divergence between the IFN-x and IFN-(1, subfamilies is of the order of 30%. Hu-IFN-P consists of a single polypeptide of 166 amino acids that displays a level of sequence similarity of 30 o/o with IFN-X. In contrast to IFN-x and IFN-(r,, IFN-/l is N-glycosylated at position 180. which accounts for some functional differences observed between IFN-fl and IFN-X. Hu-IFN-7 differs significantly from Type I IFNs and they share no obvious sequence similarity. It consists of a protein of 166 amino acids. with two potential N-glycosylation sites at position 28 and 100.
At least 12 nonallelic IFN-‘2 genes have been identified in the mouse (Mu) (De Maeyer and De Maeyer-Guignard. 1988). They encode polypeptides of 166 or 167 amino acids and their general structure is comparable to that of Hu-IFN. The single copy Mu-IFN-/3 gene encodes a mature protein of 16 1 amino acids with three potential N-glycosylation sites at position 29, 69 and 76. and displays a degree of sequence similarity of 63% at the level of coding sequence with the Hu-IFN-P. Mu-IFN-a and Mu-IFN-/3 genes are also clustered (mouse chromosome 4). The Mu-IFN-y gene is organized in four exons and three introns like its human counterpart. Mature Mu-IFN-7 has 136 amino acids with two potential N-glycosylation sites.
Another protein, the ovine trophoblast protein 1 (the major secreted protein of the preimplantation conceptus in sheep) recently has been included in the IFN system under the term IFN-T (Cross and Roberts, 1991). It induces the antiviral state, but its production does not seem to require the stimulus of viral infection. Its major role appears to reside in the control of maternal recognition during pregnancy and in the preparation of the endometrium for implantation.
2. THE INTERFERON RECEPTORS
The biological actions of IFNs are initiated by interactions of their molecules with specific high-affinity receptors on the plasma membrane (Pestka rt crl., 1987; De Maeyer and De Maeyer- Guignard, 1988). The binding of IFNs to cells has been studied in many tissue culture cells from both humans and mice. Both IFN-r and IFN-p bind to the same high-affinity species-specific receptors, designated Type I receptors. Type I receptors are relatively few (2.5 x 10’ binding sites!‘cell) and bind the ligand with high affinity (&lO~” M). The Hu-IFN-xl’/j receptor is encoded by a gene on chromosome 21. In cross-linking experiments. the Hu-IFN-Type I receptor appears to consist of a glycoprotein with an apparent molecular weight of 110 kDa. UzC et nl. (1990) used a gene transfer approach to clone a cDNA that appears to code for at least one component of the Hu-IFN-r receptor. This cDNA encodes a glycoprotein of 557 amino acids with an N-terminal hydrophobic region and a single transmembrane-spanning domain. When mouse cells express Hu-IFN-a receptor cDNA, they become slightly sensitive to the antiviral activity of certain Hu-IFN-s(-B2 (or &x8), but not to others, suggesting either that the receptors for IFN-a/fi are members of a heterogeneous family or that additional species-specific components are needed for the signal transduction. Subsequent work showed that an IFN-c~ binding moiety was a component for all the IFN-x//I species tested, but that additional components were necessary for a completely functional receptor (Uze et (II., 1992). The

Mechanism of viral inhibition by interferons 417
heterogeneous structure of IFN-Type I receptors is also indicated by the finding that Type 2 complement receptors, present on B lymphoma cells, bind to IFN-c( (Delcayre et al., 1991).
IFN-y acts through the Type II IFN receptor, which is encoded by a gene on chromosome 6 (q16-q22) in the human and by chromosome 10 in the murine system (Rashidbaigi rt ul., 1986; Mariano et al.. 1987; Jung et al., 1987; Aguet et al., 1988; Kumar et al., 1989; Cofano ct al.. 1990; Farrar and Schreiber, 1993). Genes for both human and murine receptor molecules have been cloned and appear to code for a glycoprotein with a molecular weight of 95 and 80 kDa, respectively. When transfected in cells of heterologous species, the IFN-Type II receptor cDNA does not transduce the activation signal to the nucleus, suggesting that other components are required to activate cellular second messengers (Jung et al., 1987, 1988, 1990). These additional components appear to be encoded by a gene on human chromosome 21 (Jung et al., 1987, 1988) or on chromosome 16 in the mouse (Hibino et NI.. 1991). Expression of human-mouse chimeric receptors, produced by interchanging the extracellular. transmembrane or intracellular domains, revealed that the site of the species-specific interactions between the receptor and the human ancessory protein is in the extracellular domain (Hibino ct al.. 1992). The cloning of the gene and cDNA of their ancessory factor (AF-1) has been described (Soh et rd.. 1993, 1994). Results suggest that the ancessory factors comprise a family of such factors needed for different activities (Soh et al., 1993; Cook et al., 1994). The nature of this transduction signal is unknown. However, evidence suggests that IFN-y-induced receptor dimerization may be of physiologic significance (Farrar and Schreiber, 1993; Cook et c/I.*).
3. SIGNAL TRANSDUCTION
The signal transduction pathways activated by the IFN-receptor interaction that are responsible for gene expression and protein synthesis are still controversial. Two apparently contrasting models have been proposed. The first proposed that IFN signaling is mediated by activation of second messengers that stimulate the transcriptional activation of IFN-inducible genes (Pfeffer and Tan, 1991). The second model suggests a direct activation of a transcription complex in the cytoplasm, with no requirement for second messengers (Levy and Darnell, 1990; Kerr and Stark, 199 1; Williams, 1991; Pellegrini and Schindler, 1993). These models, however, are not mutually exclusive, and a review of the literature suggests that they can be reconciled.
Early reports demonstrated that IFN-z or IFN-j? induces a rapid and transient increase of diacylglycerol (DG) and/or phosphatidic acid in different human cell lines (Yap et rrl., 1986a.b; Pfeffer ct N/.. 1990). The magnitude of these increases correlated with two biological activities. namely cell growth arrest and induction of the antiviral state. It has not been shown that IFN-r:fl induce inositolphospholipid turnover in Daudi or HeLa cells. In the latter, however, IFN-sr rapidly activates phospholipase A, which in turn accumulates DG (Pfeffer et cd., 1990; Hannigan and Williams, 199 I ). In some cell lines, therefore, IFN-(/Ps appear to induce early changes in lipid metabolism, and exploit DG as a second messenger.
A number of reports suggest that specific protein kinases are involved in the IFN signaling process (Pfeffer and Tan, 1991). Pretreatment of cells with selective inhibitors of protein kinase C. such as staurosporine or H7. or with 2-aminopurine, an inhibitor of H, RNA-dependent protein kinase. inhibited gene expression by IFN-z (Faltynek et al., 1989). IFN-CYS selectively and transiently activate the protein kinase C c isoform in sensitive, but not in resistant. Daudi cells. This finding is very important in the light of previous results showing that IFN-‘2 does not raise intracellular Caz+. The c isoform does not require Ca’* for its activity. In mouse 3T3 cells. a transient activation of phospholipase AZ giving rise to rapid formation of arachidonic acid has been observed following IFN-r treatment. Inhibiting phospholipase A1 activity with p-bromophenacyl bromide specifcally blocked the binding of nuclear factors to the IFN-stimulated regulatory element (ISRE) of the 2’.5’-oligoadenylate synthetase (Hannigan and Williams, 1991). A scheme in which both selective PKC < isoform and phospholipase A’ are necessary for expression of IFN-r activities can thus be envisaged.
*Cook. J. R.. Wang. P., Schwartz, B., Mirochnitchenko, 0.. Garotta, G. and Pestka. S. Construction of a modified human IFN-7 receptor with enhanced biological function. Manuscript in preparation.

418 S. Landolfo et ul
2&j=~G vyk2. JAKl. JAKZ,...)
Fig. 1. Activation of signal transducers and transcription factors regulating the expression of IFN-inducible genes.
In contrast to the need for global changes in second messenger concentrations, Levy and Darnell (1990) have proposed a model of IFN-dependent transcriptional activation in which signal transduction to the nucleus does not involve second messengers. The signal generated by interaction of IFN-c( or IFN-11 with their receptors reaches a defined set of genes in the nucleus only through a series of specific protein-protein interactions that generate a transcriptionally active complex. This complex, called IFN-stimulated gene factor 3 (ISGF-3) is present in the cytoplasm in a latent form, becomes activated within a few minutes after IFN binding to cell surface receptors and migrates to the nucleus, where it binds to the ISRE of inducible genes (Levy et al., 1988, 1989; Fu et al., 1992) (Fig. 1). However, it is clear from the results of Cook et al. (1994) that activation of the transcription factor GAF (~91) by Hu-IFN-y is not sufficient for generation of antiviral activity.
4. TRANSCRIPTIONAL ACTIVATION
Depending on the type of cell, a few hours lie between initial interaction of IFNs with their receptors and the appearance of cellular proteins regulating their biological action. This ‘lag period’ is the black box to which investigators have addressed their efforts in the last 20 years. It has been found that the IFN response has two phases (Fig. 2). The first, including the lag period, takes place a few minutes after IFN treatment, is mediated by regulatory genes, is cycloheximide resistant and involves the modification of pre-existing cellular factors. In the second, transcription of structural genes increases gradually a few hours after treatment, requires active protein synthesis and terminates with the appearance of cellular proteins that are very heterogeneous functionally and structurally.
Studies with cDNA and genomic clones coding for IFN-inducible proteins revealed that gene expression is mainly regulated at the transcriptional level. Some investigators (Larner et al., 1984; Friedman et ul., 1984) have used nuclear transcription assays to show that in isolated nuclei, RNA synthesis of some IFN-regulated genes (e.g. pIF-I, pfF-2, 1-8) doubles in 5-10 min after exposure to IFN-(r/p, reaches the maximum (lo- to 50-fold) between 30 and 180 min, and decreases after 446 hr to reach basal levels at 12-24 hr. However. if IFN is removed after 8 hr. the transcription rate falls rapidly below the basal levels. Transcription of Class I major histocompatibility antigens. fl-microglobulin and metallothionen Zfcl genes following IFN-r/P treatment is independent ofprotein synthesis, since it takes place even in the presence of cycloheximide and is blocked by #xx-amanitin, indicating that this transcription requires RNA polymerase II (Friedman and Stark, 1985).
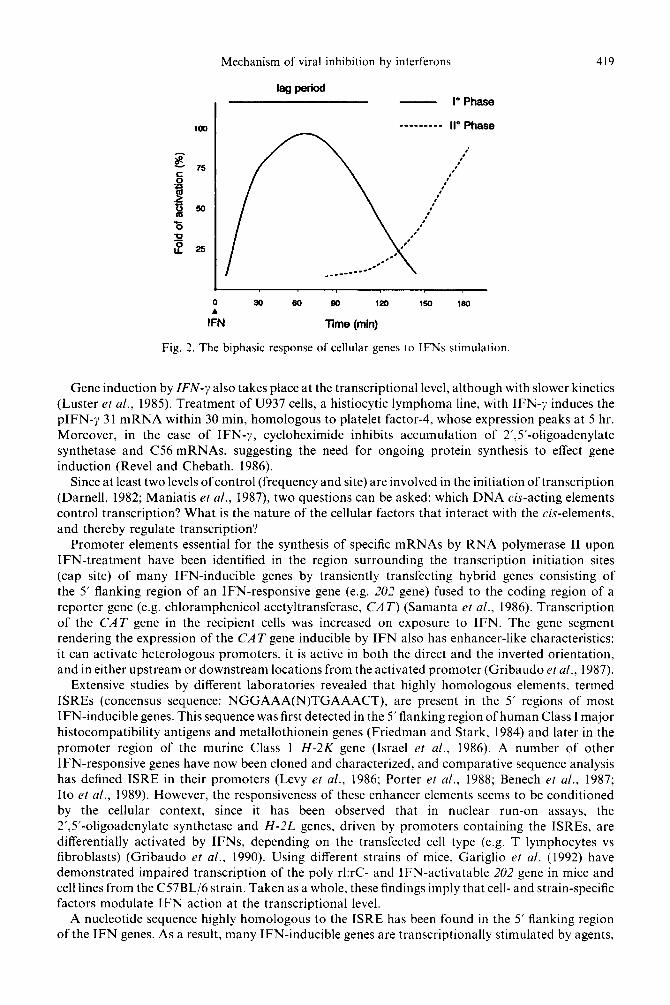
Mechanism of viral inhibition by interferons 419
lag period - I” Phase
_________ 11” Phase
0 30 60 90 120 150 180
A Time (min)
Fig. 2. The biphasic response of cellular genes to IFNs stimulation
Gene induction by IFN-y also takes place at the transcriptional level, although with slower kinetics (Luster et al., 1985). Treatment of U937 cells, a histiocytic lymphoma line, with IFN-7 induces the pIFN-y 3 1 mRNA within 30 min, homologous to platelet factor-4, whose expression peaks at 5 hr. Moreover, in the case of IFN-y, cycloheximide inhibits accumulation of 2’,5’-oligoadenylate synthetase and C56 mRNAs, suggesting the need for ongoing protein synthesis to effect gene induction (Revel and Chebath, 1986).
Since at least two levels ofcontrol (frequency and site) are involved in the initiation of transcription (Darnell, 1982; Maniatis et al., 1987) two questions can be asked: which DNA c&acting elements control transcription? What is the nature of the cellular factors that interact with the &-elements, and thereby regulate transcription?
Promoter elements essential for the synthesis of specific mRNAs by RNA polymerase II upon IFN-treatment have been identified in the region surrounding the transcription initiation sites (cap site) of many IFN-inducible genes by transiently transfecting hybrid genes consisting of the 5’ flanking region of an IFN-responsive gene (e.g. 202 gene) fused to the coding region of a reporter gene (e.g. chloramphenicol acetyltransferase, CAT) (Samanta et al., 1986). Transcription of the CAT gene in the recipient cells was increased on exposure to IFN. The gene segment rendering the expression of the CAT gene inducible by IFN also has enhancer-like characteristics: it can activate heterologous promoters, it is active in both the direct and the inverted orientation, and in either upstream or downstream locations from the activated promoter (Gribaudo et al., 1987).
Extensive studies by different laboratories revealed that highly homologous elements, termed ISREs (concensus sequence: NGGAAA(N)TGAAACT), are present in the 5’ regions of most IFN-inducible genes. This sequence was first detected in the 5’ flanking region of human Class I major histocompatibility antigens and metallothionein genes (Friedman and Stark, 1984) and later in the promoter region of the murine Class 1 H-2K gene (Israel et al., 1986). A number of other IFN-responsive genes have now been cloned and characterized, and comparative sequence analysis has defined ISRE in their promoters (Levy et al., 1986; Porter ef al., 1988; Benech et al., 1987;
Ito et al., 1989). However, the responsiveness of these enhancer elements seems to be conditioned by the cellular context, since it has been observed that in nuclear run-on assays, the 2’,5’-oligoadenylate synthetase and H-2L genes, driven by promoters containing the ISREs, are differentially activated by IFNs, depending on the transfected cell type (e.g. T lymphocytes vs fibroblasts) (Gribaudo et a/., 1990). Using different strains of mice, Gariglio et a/. (1992) have demonstrated impaired transcription of the poly rl:rC- and IFN-activatable 202 gene in mice and cell lines from the C57BL/6 strain. Taken as a whole, these findings imply that cell- and strain-specific factors modulate IFN action at the transcriptional level.
A nucleotide sequence highly homologous to the ISRE has been found in the 5’ flanking region of the IFN genes. As a result, many IFN-inducible genes are transcriptionally stimulated by agents,

420 S. Landolfo et al
such as viruses and double-stranded RNA (dsRNA). Differences between the molecular mechanisms exploited by viruses that induce IFN-r/P and dsRNA account for the finding that the sets of genes induced by these agents do not completely overlap (Landolfo et d., 1993).
IFN-inducible genes are divided into three major groups according to the location of the ISRE in their promoters. In the first, a TATAA element is located at -30 nt from the transcription start site, and the ISRE is further upstream from this element. At least three types of DNA- protein complexes (termed ISGFs) have been identified following IFN-x treatment: (I) those activated within a few minutes without requiring protein synthesis (ISGF3 or E factor. depending on the terminology used); (2) those activated in a few hours and requiring protein synthesis (ISGF? or M factor. and the G factor inducible by IFN-y); (3) constitutive factors (ISGFI). also known as C. present in both IFN-treated and untreated cells (Levy and Darnell. 1990: Kerr and Stark. 199 1).
The second group includes the TATAA-lacking genes. i.e. the human and murine 2’..5’A synthetase gene. in which the ISRE is located where the TATAA element is located in the TATAA-containing genes. At least six binding activities (termed Cl-C6) have been identified so far. and are thought to be directly involved in the formation of the basic transcription initiation machinery (Yan and Tamm, 1990, 1991).
Finally, in a third type of gene. such as the 200 cluster family, the ISRE (designated GA box), similar but not identical to the ISREs found in other stimulatable genes, is located in the tirst exon at approximately t-40 nt downstream from the first of about 14 transcription starting sites (Samanta ct (I/.. 1986; Gribaudo ct al., 1987). In addition to a few constitutive DNA-protein complexes. an inducible retarded complex, designated GAbfl, has been identified upon IFN-x treatment (Gariglio ct rd.. 1994).
Some insights into the signal transduction pathways exploited by IFNs to activate inducible genes have been provided by the use of naturally or deliberately established mutant cell lines partially responsive to IFNs (Pellegrini et al., 1989; McKendry ef NI., 1991; Miiller L’I ul.. 1993). One such recessive mutant line (designated as 11.1) is resistant to IFN-c(. but partially responsive to IFN-/j and fully sensitive to IFN-7. Using transfection with genomic DNA in conjunction with a powerful back-selection. a cosmid capable of complementing the mutant I I,1 was found. It contains a whole message encoding a nonreceptor protein tyrosine kinase designated Tyk2. Three other mutants have now been complemented with a family of protein tyrosine kinases, designated Jakl and Jak2 (tyrosine-kinase of the Janus family). The picture that is now emerging from these mutant lines is the following: Tyk2 is uniquely involved in the IFN-r.l/j pathway and Jak2 in the IFN-;$ pathway, whereas Jak I is essential for both pathways. Activation of Jak results in phosphorylation of latent cytoplasmic proteins termed STATS (signal transducers and activators of transcription) (Ihle ct d., 1994).
The primary positive regulator of IFN-r activity. ISGF3, consists of a 9 11’84 kDa protein and a 113 kDa protein. termed p9 1 or Stat 1 r, ~84 or Stat l/1 and ~113 or Stat2, respectively. The sequences of the 91 and 84 kDa proteins are identical, except that the 38 COOH-terminal amino acids of the 91 kDa protein are absent in the 84 kDa protein (Levy rt al., 1989; Schindler ct ~1.. 1992a.b).
The DNA binding component of the ISGF3 complex is a 49 kDa protein, ~48. Sequence analysis of this protein showed it to be a member of a family of DNA binding proteins that includes IRFI. IRF2 and ICSBP (Veals et ul., 1992). Upon activation, the ISGF3 complex promptly moves into the nucleus where it binds to the ISREs and switches on transcription of inducible genes (Levy and Darnell. 1990). Although the use of IFN-resistant mutant cell lines. along with the complementation of their defects, has been shown to be a powerful tool in clarifying the signal transduction cascade proceeding from Ii&and binding to the ISGF3 interaction with the ISRE, the subsequent events that take place after ISRE-ISGF3 interaction and which lead to the activation of the basal transcription machinery still remain obscure.
The transacting factors so far described appear to be specifically activatable by IFN-2. An IFN-;* activatable factor has been shown to bind the IFN-;I-activation site of the human gene encoding a 67 kDa cytoplasmic guanylate-binding protein (Decker rt (I/.. 1991). Inactive Stat91 in the cytoplasms of untreated cells is a monomer and upon IFN-;--induced phosphorylation. it forms a stable IFN-;, activatable factor homodimer, capable of binding to a specific IFN-;,-activation site element. Dimerization of Stat91 is mediated through SH-2 phosphoryl peptide interactions (Shuai et cd.. 1994).

Mechanism of viral inhibition by interferons 421
5. PROTEINS INVOLVED IN THE MOLECULAR MECHANISMS OF THE ANTIVIRAL STATE INDUCED BY INTERFERONS
The characteristics and mechanisms of action of the IFN-induced proteins with documented roles in establishment of the antiviral state have been extensively reviewed by Staeheli (1990) Samuel (199 1) and Sen and Ransohoff (1993). In the following sections, we will briefly review their principal features.
5.1. 2’,5’-Oligoadenylate Synthetases
The 2’,5’-oligoadenylate system (2,5A) is composed of at least three enzymatic activities that synthesize and degrade 2,5A oligomers: 2’,5’-oligoadenylate synthetases, 2,5A-dependent RNAase and 2’,5’-phosphodiesterase. The 2’,5’-oligoadenylate synthetase are IFN-induced enzymes, which, if activated by the cofactor, dsRNA, polymerize ATP into 2’,5’-linked oligomers of adenosine with general formula 2’,5’ pppA(pA), , where n is between 2 and 15. The only known function of these 2’,5’-oligoadenylates (2,5A) is to activate a latent ribonuclease responsible for degradation of viral and cellular single-stranded RNA. In most cells, 2,5A are degraded to AMP and ATP by 2’,5’-phosphodiesterase (Lengyel, 1982; Hovanessian, 1991; Sen and Lengyel, 1992).
Human and mouse 2’,5’-oligoadenylate synthetase are a group of isoenzymes with different intracellular locations and requirements for dsRNA activation. They are coded by different genes or produced by differential splicing. In human cells, four immunologically related polypeptides (40, 46, 69 and 100 kDa), displaying 2’,5’-A synthetase activity, have been identified (Chebath et al., 1987a; Hovanessian et al., 1987; Marie and Hovanessian, 1992).
The second key enzyme of this pathway is a latent IFN-inducible endoribonuclease known as 2,5A-dependent RNAase, formerly designated RNAse L or F. The binding of 2,5A activates this enzyme to cleave single-stranded RNA primarily after UA, UC, UU and UG residues, producing 3’-phosphate-terminated products (Lengyel, 1987). Human and murine cDNAs for the 2,5A-dependent RNAase have been cloned recently and the recombinant proteins produced. The nucleotide and predicted amino acid sequences of both enzymes were determined, resulting in open reading frames encoding proteins of about 80 kDa. Alignment of the murine and human forms of 2,5A-dependent RNAase indicates 65% sequence similarity between overlapping regions. A limited sequence similarity with RNAase E, an endoribonuclease from Escherichia coli implicated in the control of mRNA stability and rRNA processing, was also found (Zhou et al., 1993).
Although a wider significance of the 2,5A pathway has been proposed in the light of evidence of its involvement in cellular pre-mRNA splicing, and in the IFN-dependent control ofcell proliferation and differentiation in hematopoietic cells, its antiviral activities are the best documented. Data indicate that it inhibits picornavirus replication: in studies in which constitutive expression of a transfected 2’,5’-oligoadenylate synthetasecDNA was obtained, replication ofencephalomyocarditis (EMC) and mengo viruses was impaired, whereas that of vesicular stomatitis virus (VSV) and Herpes simplex virus (HSV)-2 was not affected (Chebath et al., 1987b; Rysiecki et al., 1989). In IFN-treated, EMC virus (EMCV) infected cells. 2’,5’-oligoadenylate synthetase is activated by binding of partially dsRNA intermediate produced in the course of the normal picornavirus replication cycle (Gribaudo ct N/.. 1991). Strong inhibition of human immunodeficiency virus (HIV)-1 replication has also been observed in cells stably transfected with a 2’,5’-oligoadenylate synthetase expression vector driven by the HIV-l long terminal repeats (Schroder et al., 1990).
5.2. Double-stranded RNA-dependent Protein Kinase
IFN treatment induces the synthesis of a 68 kDa polypeptide that displays a protein kinase activity when activated by binding to dsRNA. single-stranded RNA with double-stranded regions. or some polyanions (Hovanessian. 1989; Katze, 1992). This kinase is now called dsRNA-dependent protein kinase (PKR). The activation step is characterized by an autophosphorylation event of several serine and threonine residues of the kinase protein, which requires interaction of two molecules of the enzyme with a single dsRNA molecule (Mathews and Shenk, 1991). The phosphorylated PKR can phosphorylate other proteins. but not other molecules of latent PKR. Thus, autocatalytic activation

422 S. Landolfo et al.
is avoided. The principal substrate of activated PKR is serine 51 of the cc-subunit of eukaryotic peptide chain initiation factor 2 (eIF-2) a complex protein consisting of three subunits (a, p and y) and responsible for attachment of the initiator tRNA to the ribosome. To perform this step, eIF-2 binds GTP and interacts with the methionyl-tRNA to form a ternary complex, which then combines with the ribosome. Upon the release of the initiator tRNA, eIF-2 complexed with GDP, instead of GTP, is released from the ribosome. To begin a new initiation cycle, GDP should be replaced by GTP by the guanine nucleotide exchange factor (eIF-2B). If the N-subunit of eIF-2 is phosphorylated, the efficiency of the exchange reaction is reduced, since a tight complex is formed with guanine nucleotide exchange factor. This becomes sequestered and unable to support further peptide chain initiation, so that initiation of protein synthesis is inhibited (Mathews and Shenk, 1991).
Human PKR has been cloned and shown to contain all the conserved domains of serine/ threonine protein kinases. The only identified dsRNA-binding domain is located in the amino-terminal region (Meurs et al., 1990; Feng et al., 1992). Several roles have been suggested for PKR. On its transfection into murine cells, the constitutive expression of wild-type human PKR cDNA confers a partial resistance to infection with EMCV. The replicating viral RNA is the most probable activating agent. The phenotype of the transfected cells also revealed a decreased rate of cellular protein synthesis (Chong et al., 1992; Meurs et al., 1992).
A transformed phenotype has been obtained recently by transfecting a mutant, but not the wild-type, human PKR cDNA encoding a polypeptide devoid of eIF-2 kinase activity. In addition, injection of these transformed cells in nude mice produced large tumors (Koromilas et al., 1992; Meurs et al., 1993). Taken as a whole, these findings indicate that: (1) mutations in the PKR may convert the wild-type molecule into a tumorigenic protein that is catalytically inactive, but can inhibit the activity of the endogenous protein and (2) while the mechanism of the antitumor activities of wild-type PKR remains to be established, this IFN-inducible enzyme may behave as a tumor-suppressor gene (Lengyel, 1993).
5.3. Mx proteins
Type I IFN treatment of vertebrate cells can induce the synthesis of a family of related proteins called Mx proteins (Pavlovic and Staeheli, 1991; Staeheli et al., 1993). Molecular cloning of their cDNA permitted their constitutive expression and showed that each is sufficient to confer resistance to some RNA viruses in the absence of IFN (Staeheli et al., 1986). The Mx system was originally characterized by the finding that the resistance of A2G mice to influenza virus is genetically determined: in the cells of this strain, Type I IFN induces the synthesis of a nuclear protein, Mx- 1, which inhibits virus replication (Arnheiter et al., 1990; Kolb et al., 1992; Zurcher et al., 1992). The sensitivity of most inbred strains of mice to influenza virus was subsequently demonstrated to be a consequence of the presence in their genome of defective M-u-1 genes incapable of coding for the functional protein. Mx-1 acts by impairing the primary transcription of a viral genome (Krug et al., 1985). Its target seems to be the PB2 subunit of the influenza virus polymerase complex: it has been suggested that Mx-I either forms a complex with PB2 or competes with PB2 for its normal site in the active polymerase complex (Pavlovic et al., 1992). A second Mx gene, M.u-2, has been identified in the mouse genome. Its function remains unclear.
In rat cells, three Mx proteins (Mx-1, Mx-2 and Mx-3) induced by Type I IFN have been identified. Nuclear Mx-I inhibits replication of the influenza virus, whereas cytoplasmic Mx-2 only confers resistance to VSV. Mx-3 seems devoid of antiviral activity (Pavlovic and Staeheli, 1991).
In human cells, IFNs induce the synthesis of two Mx-related proteins, Mx-A and Mx-B. Mx-A is a cytoplasmic protein, and its expression results in inhibition of the replication of both influenza virus and VSV (Pavlovic et al., 1990). This anti-VSV activity is abolished by a single amino acid substitution near the C-terminus of the protein. The mutated protein still confers resistance to the influenza virus. However, when Mx-A is localized to the nucleus by fusion with a heterologous nuclear translocation signal, it acts like murine Mx-1 by impairing primary transcription of the influenza virus (Pavlovic et al., 1992).
All the Mx proteins contain a GTP-binding domain. Purified recombinant Mx-1 and Mx-A proteins display a GTPase activity that requires a high concentration of substrate for the maximal rate of hydrolysis. GTP binding and hydrolyzing activity are thought to be required for the

Mechanism of viral inhibition by interferons 423
interaction of Mx proteins with components of the viral polymerase complex (Melen et uf., 1992;
Staeheli et al., 1993). The mechanism(s) by which the Mx proteins mediate resistance to VSV and the influenza virus
in the cytoplasm are still a matter of debate. Inhibition of some late steps in the viral cycle has been proposed following the discovery of Mx-homologous proteins, namely the yeast Vps, responsible for vacuolar protein sorting, and the rat dynamin DlOO, a microtubule-associated ATPase (Rothman et al., 1990). The antiviral activities of Mx proteins thus may be exerted by altering protein trafficking, and so interfering with normal protein transport through the Golgi network to the membrane.
Despite these constant homologies, however, some Mx proteins localize within the nucleus, others in the cytoplasm. Furthermore, mouse Mx-1 is effective only against influenza, rat Mx-2 only against VSV, human Mx-A against both. Rat Mx-3 seems devoid of activity. Further work is required to explain these differences and create a clear picture of the antiviral activity of Mx proteins.
6. INDUCTION OF THE ANTIVIRAL STATE AND ITS REGULATION
The restriction of virus growth by IFNs is associated with a variety of physiological changes, some of which could be assigned to specific IFN-induced proteins. Therefore, it has become axiomatic that the antiviral state should be regarded as the result of activities mediated by the several IFN-induced proteins (Fig. 3). In the following sections, we will review some of the molecular mechanisms exploited by IFNs to inhibit the replication of RNA, as well as DNA viruses.
6.1. RNA Viruses
RNA viruses, such as picorna- or togaviruses, contain a positive or plus (+) strand. After uptake by the host cell, they bind to ribosomes and are translated into a polyprotein, giving rise to the different viral proteins. Of these, the viral polymerase transcribes (-) strands from the (+) strands; these are used for further synthesis of (+) strands. Other RNA viruses, such as myxoviruses, rhabdoviruses and arenaviruses, contain (-) strands that must first be transcribed into (+) strands to serve as mRNAs. Negative RNA viruses contain their own polymerase and begin their replication cycle by transcribing the viral genome into monocistronic mRNAs. Retroviruses contain the enzyme reverse transcriptase in the virion and begin their infectious cycle by transcribing the viral genome into DNA, which integrates as provirus in the cellular DNA. Lastly, reoviruses have a double-layered capsid, which contains 10 or 11 segments of dsRNA. They start their replication cycle by removal of the virion outer shell, thus activating the RNA-dependent RNA polymerase. RNA positive-sense
IFlU-? R
Fig. 3. An overview of the antiviral state induced by IFNs and its related mechanisms.

424 S. Landolfo et nl
transcripts induce the production of proteins and are also a template for the production of antisense strands, which remain associated with the positive-sense strand (Roizman, 1990).
We will now discuss the earlier and recent results concerning the mechanisms exploited by IFNs to inhibit the replication of some RNA viruses in infected cells.
6.1.1. Picorrzmirusrs
During infection with picornaviruses, the host cell protein synthesis is shut down transiently. as with mengo virus, or totally abolished with polioviruses. and with high M.O.I. of EMCV (Falcoff and Sanceau, 1979; Munoz and Carrasco, 1981, 1983). A great body of evidence indicates that the main stage ofpicornavirus multiplication affected by IFNs is viral protein synthesis, and is a result of greatly increased degradation of viral and cellular RNAs. The principal enzymatic system involved in this degradation process is the 2’.5’-oligoadenylate synthetase/RNase L system. although in some cases. the PKR system also seems to play a role. The first evidence of 2’,5’-oligoadenylate synthetase activity in rim was provided by Williams PI al. (1979) who demonstrated 2.5A products in IFN-treated EMCV-infected L cells. Other studies, using IFN-treated HeLa cells, indicated that inhibition of EMCV RNA accumulation parallels induction of 2’.5’-oligoadenylate synthetase before the onset of IFN-induced PKR activity (Whitaker-Dowling and Youngner, 1987).
A chemically synthesized analog of natural 2’,5’A-oligonucleotides inhibits activation of the 2’.5’A-dependent RNase L in IFN-treated mouse L cells by EMCV, rRNA cleavage. and partially restores EMCV RNA synthesis and virus yields (Watling et al.. 1985). The most compelling evidence that this system plays a key role in picornavirus resistance comes from cell lines permanently transfected with cDNA encoding these enzymes (Chebath et 01.. 1987b). Constitutivc expression of a cDNA encoding the human 40 kDa form of 2.5A synthetase in transfected Chinese hamster ovary cells inhibited mengo virus, but not VSV or HSV-2 multiplication, and also impaired EMCV replication in other human cell lines (Rysiecki et (I/., 1989). Moreover. in HeLa cells infected with EMCV. the 40 kDa form of 2.5A synthetase is activated by binding of dsRNA of viral origin (Gribaudo ct trl., 1991). When a dominant negative mutant of 2’,5’A_dependent RNase L, which retains 2’,5’A binding capacity, but lacks RNase activity, was expressed at high levels in murine cells, it prevented specific rRNA cleavage in response to 2,5A transfection and rendered cells unresponsive to the antiviral activity of IFN-x;‘B for EMCV (Hassel et al., 1993).
PKR. the other dsRNA-dependent enzyme, also plays a role in the IFN-mediated resistance against picornaviruses. Coinfection of IFN-treated mouse L cells with vaccinia and EMC viruses caused an up to IOOO-fold increase of EMCV compared with cells infected with EMCV alone (Whitaker-Dowling and Youngner, 1983. 1984). Since PKR activity is inhibited by a vaccinia virus-encoded factor (Akkaraju et (11.. 1989). growth of EMCV was permitted, despite activation of the 2’,5’-oligoadenylate synthetase/RNase L pathway. Further evidence of the involvement of both IFN-induced dsRNA-dependent enzymes in picornavirus inhibition comes from the use of mutant cell lines bearing altered 2’.5’-oligoadenylate synthetase expression patterns (Kumar it trl., 1987. 198Xb; Lewis, 1988). In some of these mutants. IFN-induced EMCV protection was observed in the absence of a functional 2’.5’-oligoadenylate synthetase,lRNase L pathway, suggesting involvement of the PKR system. In contrast, in a few clones the IFN-induced antiviral state against EMCV was observed in the absence of both activities. Thus, it is amply clear that the antiviral action of IFN against picornavirus is mainly carried out by the 2’.5’-oligoadenylate synthetase;RNase L system and partly by the PKR pathway. However, additional. still unidentified IFN-induced proteins may make a further contribution to picornavirus inhibition.
Its dramatic sensitivity to IFNs has made VSV the prime choice for assaying antiviral activity. Definition of the biochemical pathways responsible for VSV inhibition in IFN-treated cells. however, still remains obscure. There is compelling evidence that the host cell and the IFN type influence the stage of the VSV multiplication cycle in which inhibition occurs. Inhibition of viral uptake due to reduced rates of pinocytosis has been observed in human, mouse and chick cells treated with homologous IFN-x or IFN-/I (Wilcox et rd., 1983; Whitaker-Dowling and Youngner. 1986). These

Mechanism of viral inhibition by interferons 425
results are in line with the finding that IFNs modulate the fluidity of the cell surface and, therefore, inhibit the generation of pinocytic vesicles (Pfeffer et al., 1980). Efficient VSV adsorption and penetration, in fact, has been noted in cell lines insensitive to IFN (Belkowski and Sen, 1987).
Early studies by Marcus and colleagues (197 1) demonstrated that pretreatment of primary CE cells with IFN-c(/fl reduces primary transcription of VSV by 60&70%. Manders et al. (1972) then found a reduction to about 20% in IFN-n-treated human fibroblasts. Reduction to about one-quarter of primary viral RNA synthesis was found in human amnion U cells treated with IFN-7 (Ulker and Samuel, 1985). More recently Belkowski and Sen (1987) have demonstrated a reduction to about 90% of primary VSV transcripts in human and mouse infected cells pretreated with IFN-c(. The exact mechanisms responsible for this inhibition are still undefined.
It has been observed that VSV nucleocapsids purified from IFN-treated cells, but not from untreated cells, are associated with RNase activity (Kumar et al., 1988a). Specific VSV degradation, therefore, decreases the amount of VSV RNA templates available for primary transcription. An additional mechanism, operative at an early VSV multiplication step, may stem from the Mx-A protein, since it reduced more than 50-fold the pools of VSV proteins and RNAs when expressed constitutively in Swiss 3T3 mouse cells in the absence of IFN (Pavlovic et al., 1990). RNA reduction was also observed in cells infected in the presence of cycloheximide, suggesting that Mx-A interferes with normal VSV mRNA synthesis either by inhibiting the activity of the viral polymerase complex or by reducing the stability of the VSV mRNAs.
A drastic inhibition of VSV mRNA translation may also account for the impaired VSV replication in IFN-treated cells. This conclusion can be drawn from the findings of Masters and Samuel (1983) who compared viral RNA and protein pools in untreated and IFN-cc-treated amnion U cells. These authors estimated a 90% average reduction of translation of VSV mRNAs. The mechanisms activated by IFNs to impair VSV mRNA translation appear to operate at different steps. Coinfection of mouse L cells with VSV and vaccinia viruses abrogated IFN-cl/p-induced resistance to VSV (Paez and Esteban, 1984). Since vaccinia virus encodes a protein, SKIF, which inhibits activation of the PKR (Akkaraju et al., 1989) this enzyme seems to be specifically involved in the blockade of VSV mRNA translation.
However. other mechanisms can be envisaged. One possibility is that IFN reduces the methylation of VSV mRNA. Up to 60% of VSV mRNAs with unmethylated cap structures were translated inefficiently itz vitro (De Ferra and Baglioni. 1981, 1983). However, this reduction is not enough to account for the drastic inhibition of VSV protein synthesis observed in parallel. In addition, blockade of VSV mRNA methylation has not been observed in other cell lines, such as mouse L cells (Whitaker-Dowling and Youngner, 1983) human amnion U cells (Ball and White, 1978) and primary CE cells (Masters and Samuel, 1983).
Another explanation of impaired VSV mRNA translation has been postulated by Sahni and Samuel (1986). On transfecting COS cells with a VSV cDNA encoding the VSV G protein, they found that, although IFN did not change the concentration of VSV mRNAs, the amount of VSV G protein was reduced to one-fifth, implying that VSV mRNAs contain secondary structures causing their specific degradation by an IFN-induced cellular factor. However, since it subsequently has been demonstrated that transient transfection of COS cells with unrelated plasmids activates PKR, these findings perhaps can be attributed to nonspecific activation of PKR leading to VSV mRNAs’ degradation.
IFN has also been reported to operate at a later step in the VSV growth cycle, namely the virion assembly process. due to specific inhibition of the incorporation of G and M proteins (Maheshwari ct al.. 1980; Jay et al., 1983). Further work with other cell lines and IFN concentrations has failed to confirm these results (Olden et al., 1982; Faltynek and Baglioni, 1983). Thus, the ability of IFNs to operate at this late stage of VSV replication is uncertain.
6.1.3. It~fluenzrr Virus
The influenza virus was the first virus employed to detect the antiviral activity of IFNs (Stewart, 1979). However, the mechanisms induced by IFNs to block influenza virus replication have proved hard to define and contradictory explanations have been offered. Operation in an early transcription step has been proposed (Bean and Simpson. 1973). as well as during an intermediate

426 S. Landolfo et al.
step between primary and secondary transcriptions, such as viral protein synthesis (Repik et al., 1974).
The way in which an antiviral state is induced has been rendered clearer by the discovery of the mouse M.u-I gene, which regulates expression of the genetically determined, IFN-mediated influenza virus resistance phenotype (Haller, 1981). Work on cells derived from mice congenic for the Mx system has demonstrated that IFN-induced proteins other than Mx protein do not play any role in the IFN-mediated resistance against influenza virus (Staeheli, 1990). Early studies by Horisberger et al. (1980) showed that in Mx’ cells, IFN acts at a stage between virion uncoating and the beginning of protein synthesis. Later, Meyer and Horisberger (1984) concluded that the action of IFN mediated by Mx protein occurred mainly after the synthesis of primary transcripts, the probable target being mRNA translation, since comparable amounts of primary influenza virus transcripts were found in IFN-treated Mx’ or Mx- cells. In contrast, Krug et al. (1985) having failed to detect significant levels of influenza virus-specific transcripts in IFN-treated Mx+ cells, proposed that Mx protein presumably blocks the onset of influenza virus transcription. Similar conclusions were reached by Ransohoff et al. (1985) who found that in cycloheximide-treated bovine cells, IFN-inhibited primary transcripts of polyadenylated influenza viral RNAs present 7 hr after infection.
Action of Mx protein against influenza virus has been elucidated partly by constitutively expressing Mx-A or murine Mx-1 in mouse Swiss 3T3 cells, followed by infection with influenza A virus (Pavlovic et al., 1990). Indirect immunofluorescence showed that Mx-1 has a nuclear localization, whereas Mx-A and Mx-B are located in the cytoplasm. In addition, Mx-A conferred resistance to influenza, as well as to VSV. Pavlovic et al. (1992) later demonstrated that in infected cells expressing Mx-A protein, all viral mRNAs were normally synthesized, accumulated in the nucleus and efficiently transported into the cytoplasm. However, viral protein synthesis and genome amplification were strongly inhibited, whereas the concentrations of the primary transcripts encoding the three influenza virus polymerase proteins, PBl, PB2 and PA, were strongly reduced. Altogether these results demonstrate that the mouse Mx-1 protein inhibits the primary transcription of influenza virus, whereas Mx-A interferes with a subsequent step.
6.1.4. Retroairuses
IFNs inhibit the production of retroviruses by both acutely and chronically infected cells (Stewart, 1979; De Maeyer and De Maeyer-Guignard, 1988). It is abundantly clear that they operate at more than one level, depending on the retrovirus and the cell line involved.
Salzberg et al. (1983) measured the amount of cytoplasmic viral RNA 45 min after infection and found a marked reduction in the penetration of Moloney murine leukemia virus (MuLV) in host cells treated with IFN-a/p. Nevertheless, viral adsorption was comparable between untreated and IFN-treated cells. Since IFNs may modulate the structure and function of the cell membrane, their antiviral effect could be due to changes in the properties of subcellular structures involved in the pinocytosis process, as observed for VSV.
Cogent evidence has also been provided that IFNs may modify late stages of the Moloney MuLV cycle growth, such as virus maturation. and lead to the production of noninfectious C-type particles (Pitha et al., 1979, 1980). Although the morphology and 70 S RNA content do not differ in the presence or absence of IFN treatment, the released noninfectious particles contain env, gag and other proteins, the most prominent being an 85 kDa glycoprotein different from gp71. The presence of this protein appears to correlate with loss of infectivity by the MuLV virions.
Aboud and Hassan (1983) found an approximately 95% inhibition of Moloney MuLV release after IFN treatment of chronically infected mouse 3T3 cells. Accumulation of intracellular viral structures lacking RNA genome suggested that IFN resulted in defective virus assembly and hence, that Moloney MLV maturation is its main target of action. However, still some disagreement exists as to whether intracellular synthesis and processing of viral proteins are affected by IFN.
A late step in virus cycle growth also appears to be affected by IFN in the case of Friend MuLV. Riggin and Pitha (1982) found that the synthesis of proviral DNA and its subsequent integration into the cell genome were not altered by IFNs. Reduced virus production presumably was due to a defect of a later step such as virion maturation.

Mechanism of viral inhibition by interferons 427
In contrast to the above retroviruses, murine sarcoma virus (MSV) appears to be affected during early steps. Pretreatment of mouse 3T3 cells with IFN inhibited the transforming capability of Kirsten MSV (Avery et al., 1980). Since this occurred only a few hours after infection and no proviral DNA was detectable, it was concluded that synthesis or integration of proviral DNA was blocked by IFN. Inhibition of MSV integration in the host cell DNA after IFN treatment has been observed by Huleihel and Aboud (1983). Translocation of viral DNA into the nucleus was delayed in IFN-treated rat kidney cells. Moreover, this DNA failed to integrate in the host genome.
A late step in the life-cycle of other retroviruses appears to be affected by IFNs. In the case of mouse mammary tumor virus, the rates of viral DNA and protein synthesis were not inhibited by IFN (Sen and Sarkar, 1980). However, IFN-treated cells displayed an increased amount of virus particles trapped at the cell surface, indicating a block in the last step of virus morphogenesis. Similar results have also been reported by Chatterjee and Hunter (1987) who found that IFN treatment of human or monkey cells chronically infected with Mason-Pfizer monkey virus decreased the production of infectious particles by more than 95%, whereas they were normal in the cytoplasm compartment. At least two steps of the Rous sarcoma virus (RSV) growth cycle are affected by IFNs. Pretreatment of chick embryo cells with IFN inhibited virus multiplication. RSV-specific proteins, RNAs as well as viral DNA, were reduced significantly a few days after infection, suggesting that IFN blocks a step between penetration and onset of viral DNA synthesis (Strube et al., 1982; Zens et al., 1989). In chronically RSV-infected chick embryo cells, IFN did not reduce the viral RNA and protein synthesis, but strongly diminished the production of infectious particles, suggesting that a late step was affected (Zens et al., 1989).
Induction of the antiviral state by IFNs towards HIV, because of its complexity and the extensive literature deserves separate treatment. The main findings on this subject are summarized in the next section. Several mechanisms have been postulated over the last 5 years. Kornbluth et al. (1989) and Meylan et al. (1993) examined the ability of IFN-a, -/I or -y to inhibit the multiplication of HIV in cultured human macrophages. These cells were fully protected from the cytopathic effect, no viral RNA or DNA was detectable in their lysates and no infectious virions or p24 core antigen were released into the supernatants. Since IFN treatment was highly effective even a few days after HIV infection, the authors concluded that an intermediate step, such as proviral DNA synthesis and integration, or a later step in virion maturation is the major target of IFN action. Using the polymerase chain reaction and a set of primers detecting complete proviral DNA, Shirdzi and Pitha (1993) demonstrated that in CEM-174 cells or peripheral blood lymphocytes, IFN-c( interferes with the initiation of HIV reverse transcription and inhibits provirus formation.
In contrast to these observations, Hansen et uf. (1992) found that the levels of HIV DNA, RNA, or p24 antigen and reverse transcriptase activity in T-cell cultures treated with IFN-c( were comparable to those in control cultures. Quantitation of gpl20 by immunogold particle analysis revealed a marked depletion of envelope glycoprotein in virions released from IFN-treated cells, causing a loss of infectivity. These authors suggested that IFN’s target in HIV maturation is the assembly of gp120 onto mature viral particles.
Of the cellular proteins induced de nova by IFN-a or IFN-y, RBP9-27 was shown to bind RNA in vitro and inhibit HIV expression after transfection into human cells (Constantoulakis et al., 1993). It primarily inhibited Rev-dependent post-transcriptional steps of viral gene expression, suggesting that it blocks HIV replication by antagonizing Rev function. Emilie et al. (1992) demonstrated that IFN-7 inhibits the transactivation of HIV long terminal repeats during viral infection, and it antagonizes that effect in HT4LacZ-1 cells by interacting with the activity of cellular factors.
6.1.5. Reoviruses
Early studies of the effect of IFN on reoviruses revealed that they reduce both virus mRNA and dsRNA in mouse L cells (Gauntt, 1972; Vassef et al., 1973; Gupta et a’.. 1974). Investigation of the entire reovirus growth cycle showed that adsorption, penetration and conversion to subviral particles were resistant to IFN. Accumulation of viral mRNA was only slightly reduced, whereas the synthesis of early reovirus proteins was inhibited by up to 75%. Late mRNA, dsRNA accumulation and progeny formation were inhibited up to 95% (Wiebe and Joklik, 1975; Gupta et al., 1982). It was

428 S. Landolfo 6’1 ul
concluded that transcription by the virion-associated polymerase was not impaired significantly and that translation of the reovirus primary transcripts was the main target of IFN.
The exact mechanisms through which IFN inhibits early reovirus mRNA translation are still uncertain, despite several recent studies. The methylation pattern of reovirus mRNA cap structures was reduced after IFN treatment, and extracts from IFN-treated cells were less efficient methylators of reovirus mRNAs lacking methylated cap structures (Desrosiers and Lengyel. 1979). However, in mouse L cells, cap methylation was only slightly reduced by IFN treatment, yet the reovirus yield was reduced by more than 98% (Desrosiers and Lengyel, 1979). Therefore, the extent to which alteration by IFN of the mRNA methylation pattern restricts reovirus growth remains to be established. An alternative mechanism based on PKR activation has been proposed by Gupta ct cd. (1982). IFN treatment of mouse L cells, followed by reovirus infection, activates PKR in t~iw by phosphorylating the eIF-2. NIH 3T3 cells lacking the (2’,5’A),,-dependent ribonuclease system were still sensitive to IFN action towards reovirus infection. suggesting a prominent role of PKR in the production of an antiviral state. Samuel and Kunston (1982) found that induction and decay of PKR kinase activation correlated with the onset of the antiviral state. Moreover, IFN-treated cells infected with reovirus serotype 2 contained an increased amount of phosphorylated eIF-2. Further support for the role of the PKR comes from the findings of Imami and Jacobs (1988), that the a3 protein of reovirus serotype 1 inhibits the activation of PKR by dsRNA, and that this could be overcome by an excess of dsRNA. Since serotype I contains more PKR inhibitory activity than serotypc 3, and is more resistant to IFN, the authors concluded that PKR is the key enzyme in IFN-mediated resistance against reoviruses.
In human cells, the situation is quite different. Nilsen et 01. (1982) demonstrated that in HeLa cells, inhibition of reovirus mRNA accumulation correlated with the activation of the 2’.5’-oligoadenylate synthetase/RNase L system, as evaluated by the increased levels of intracellular 2,5A products, as well as the increased degradation of host mRNA and rRNA. Moreover, in human amnion U cells. activation of PKR by IFN is accompanied by a strong antiviral state against VSV, but a poor antiviral response to reovirus. Altogether, these results suggest that IFN exploits different mechanisms against reovirus, depending on the type of cell and the species.
6.2. DNA Viruses
Most DNA viruses (except poxviruses) replicate in the nucleus of infected cells, where they utilize the transcriptional machinery to produce their mRNAs, and in some cases, use cellular enzymes to duplicate their genomes.
Although several in Go studies and clinical trials have demonstrated that IFNs limit severity of viral infections, it is generally thought that many DNA viruses are relatively resistant to their actions. This resistance, at least as far as IFN-mediated inhibition of viral protein translation is concerned. could be related, with the exception of poxviruses. to the negligible amount of dsRNA synthesized during infection. In addition, several lines of research suggest that most DNA viruses have evolved molecular mechanisms to counteract the antiviral state induced by IFNs. Results illustrating the mechanisms of the IFN-mediated antiviral activities towards DNA viruses will be discussed in the following sections.
Vaccinia virus, the laboratory prototype of poxviruses. is a large virus that replicates in the cytoplasm of susceptible host cells, and has received the most attention in investigations of the clrects of IFNs on the DNA viruses. Joklik and Merigan ( 1966) and Metz and Esteban (1972) showed that vaccinia virus replication is extremely sensitive to IFN in some L-cell strains: pretreatment with Type 1 IFN resulted in severe inhibition of both viral and cellular protein synthesis. Similar restrictions were observed in IFN-treated primary chicken embryo (CE) cells (Jungwirth et 01.. 1972; Esteban and Metz. 1973). Curiously, despite the fact that viral protein synthesis has been indicated as the principal stage of vaccinia replication inhibited by IFN. early viral mRNA transcription was enhanced in IFN-treated cells. Blockade of vaccinia virus protein synthesis was correlated with an increase of 2.5A levels and a rapid and extensive degradation of ribosomal RNA in IFN-treated.

Mechanism of viral inhibition by interferons 429
vaccinia-infected L-cells (Esteban et al., 1984; Goswami and Sharma, 1984). These findings, along with the observation of viral-specific dsRNA production in vaccinia-infected cells (Colby and Duesberg, 1969) suggested that the 2,5A pathway, activated in response to IFN, is involved in inhibition of vaccinia replication. As in mammalian host systems, Grim et uf. (1987) found in IFN-treated vaccinia-infected CE cells that early virus mRNAs, though synthesized at the same rates as control infected cells, failed to accumulate to normal levels. Moreover. after extraction from cells, viral mRNAs showed extensive degradation as the result of 2,5A-dependent RNase activation. Interestingly, the hallmarks of the phenotype observed when vaccinia virus infection is inhibited by IFN (high concentrations of 2,5A, breakdown of viral and ribosomal RNA, cessation of viral and host protein synthesis) recently have been shown to be virtually identical to those observed during infection by vaccinia virus temperature-sensitive mutants bearing an abortive late phenotype (Cohrs et al.. 1989; Pacha et a/., 1990). These data suggest that viral gene products may interact with the component of the host 2,5A pathway during the normal virus life-cycle.
In contrast with what is observed in L- and CE-cells, replication of vaccinia virus is relatively resistant to IFN in certain cell lines: IFN-treatment of human (HeLa, MRCS, Hep-2) as well as monkey (CVI) or mouse (L929) lines has little or no effect on viral multiplication (Rice and Kerr, 1984). In these lines, high levels of 2,5A have been reported to accumulate, even when virus replication is not inhibited, and. besides authentic 2,5A oligomers, nonphosphorylated cores and additional unidentified compounds have been isolated (Rice et al.. 1984). The presence among the latter products of an inhibitor(s) of the 2,5A-dependent RNase activation have been suggested to explain the unresponsiveness of these lines (Rice et al., 1985).
To conclude, inhibition of vaccinia virus replication by IFN is thought to be due mainly to a block in viral protein synthesis. There is circumstantial evidence for a crucial role of the 2,5A pathway in the establishment of this antiviral state. However, for other poxviruses. namely the ectromelia virus, recently, Karupiah et al. (1993) reported that the IFN-y-induced inhibition of its replication is mediated by the induction of nitric oxide synthase, as demonstrated by the constitutive expression of nitric oxide synthase cDNA in human cells.
6.2.2. Herpewiruses
Herpesviruses cause serious infections, both acute and latent, in immunocompetent and immunocompromised hosts. The protection offered by IFN depends on the type of virus and the cell line employed.
IFN-induced inhibition of the growth of HSV- 1 occurs in different cell lines and is characterized by a strong reduction of immediate-early (IE) viral-specific mRNAs expression. The IFN action, at this level. takes place between the uptake of viral DNA in the nucleus of infected cells and the onset of IE gene transcription (Mittnacht er al., 1988; Oberman and Panet. 1988; Klotzbucher et cd.. 1990). LaMarco and McKnight (1989) and De Stasio and Taylor (1990) using an IE promoter-driven reporter gene, found strong transactivation of the marker gene in cells transfected with such a construct and subsequently infected with HSV- 1, though this was abolished when the transfected cells were treated with IFNs before viral infection. This phenomenon has been correlated, by replacing the HSV- 1 infection with a construct constitutively expressing VP 16. with attentuation of the activity of this protein in cells treated with IFN. VP16, the major virion-transactivating protein encoded by the HSV-1 genome, is synthesized as a late polypeptide and assembled into the virion particle. Its activity is needed for the activation of IE gene transcription, and the transactivation event occurs via the formation of a protein complex containing VP16 and cellular transcription factors. Inhibition of this complex may be carried out by an as yet unknown IFN-induced protein that directly affects its formation or modifies its correct assembly.
IFN also acts in late stages of the HSV-I cycle: a block of viral morphogenesis was observed by Chatterjee et al. (1985). who later reported a strong reduction of the synthesis of two glycoproteins (gB and gD) in human as well as monkey cells treated with Type I IFN (Chatterjee and Whitley, 1989). These data, therefore, suggest that synthesis of anomalous viral particles and reduction of virion release act synergistically with the inhibition and IE gene transcription to render cells less permissive to HSV-1 growth.

430 S. Landolfo et al.
Murine cytomegalovirus (MCMV) has been widely studied as a model for human CMV infections, since it resembles the human virus in many aspects of its biology, replication and pathogenesis. We recently undertook an investigation of the mode of IFN action against MCMV. Martinotti et al. (1993) reported that Type I IFN treatment of murine embryo fibroblasts derived from genetically resistant, but not from susceptible, mouse strains reduced the rate of MCMV replication. Further examination of the three classes of viral mRNAs, which are expressed in a specific, time-restricted manner, revealed that the IE transcripts failed to accumulate to normal levels in IFN-treated cells. The IE proteins in MCMV correspond to nonstructural viral gene products synthesized very early in the virus cycle and directly involved in the regulation of viral gene expression throughout infection. Identification of IE gene transcription as the principal step in IFN action against MCMV has now been confirmed by the results of Gribaudo et al. (1993) showing that the uptake and transport of viral particles in IFN-a-treated and MCMV-infected NIH 3T3 cells were apparently unaffected, whereas the transcriptional activity of the IE region was greatly reduced. Since transcription of this viral segment is regulated by a strong enhancer (Dorsch-Hasler et al., 1985) when the activity of a marker gene driven by these enhancer sequences was examined in transfected fibroblasts, a significant reduction was observed upon IFN treatment in either the absence or the presence of virus infection. These data indicate that IFN inhibits both the basal and the transactivated activity of IE enhancer. Moreover, the use of deletion mutants revealed that putative IFN-c( responsive elements mediating the negative response are scattered through the enhancer sequences. Since such negative regulation occurs in the absence of viral infection, in contrast to what is observed in the HSV- 1 system, IFN-a-treatment may result in a decrease in the activity of cellular transcription factors, such as NF-KB, whose cognate binding sites occur several times within the IE enhancer.
Although there are similiarities between murine and human CMV, different effects of IFN-n were observed on human CMV gene expression. Stinski et al. (1982) reported inhibition of IE protein synthesis as the major effect of IFN treatment, since the levels of the corresponding transcripts in control and treated cells were similar. Therefore, they proposed that the decrease in the amount of early virus RNAs was a consequence of the reduced availability of IE proteins needed for regulation of early transcription. This discrepancy could be due to differences in the genomic organization of the IE enhancer region of murine and human CMV: in the latter, putative IFN-r responsive elements may be missing.
62.3. Hepatitis B Virus
Several studies have shown that IFN-CY improves the course and natural history of chronic hepatitis B, and it has been approved and licensed for this purpose in most countries (Braken et al., 1992; Martin and Friedman, 1992). Its efficacy in acute viral hepatitis B, however, is not clear. Moreover, IFN-y does not induce sustained inhibition of hepatitis B virus (HBV) replication. The molecular mechanisms exploited by IFN in restricting HBV growth are still poorly characterized, and their establishment h vitro has proved difficult, since culture systems for such virus replication are not available. Therefore, several recent studies have been performed in human hepatoma cells stably or transiently transfected with HBV DNA. These cell lines produce replicative intermediates and mature viruses and are a useful tool for the investigation of the effect of IFNs on HBV replication. Hayashi and Koike (1989) used this system to compare the activities of IFN-c(, p and 1’ on HBV replication in a stably HepG2-transfected cell line. They found that all IFNs inhibited HBV replication as shown by the amount of HBV DNAs from intracellular core particles. Despite their different mechanisms in eliciting cellular responses, the inhibitory effects of Type I and II IFNs were similar with regard to their marked reduction in the levels of both the minus and the plus strand of HBV DNA. Unexpectedly, IFNs had no effects on the level of HBV DNA in virus particles from the culture medium, nor on the amount of the two major HBV transcripts of 3.6 and 2.2 kb or on HBsAg and HBcAg/HBeAg levels. Therefore, it has been proposed that IFNs inhibit some step in the pregenome RNA-primed assembly. A different picture can be drawn from the study by Tur-Kaspa e/ (11. (1990) of the effects of IFN on expression of a marker gene under the control of HBV regulatory sequences and by determining the steady-state levels of viral mRNAs in HBV DNA-transfected HepG2 cells. IFN-c(, but not IFN-;), inhibited the marker gene expression in cells transfected with constructs containing the HBV enhancer linked to either HBV or Simian virus 40 (SV40) promoters. Moreover,

Mechanism of viral inhibition by interferons 431
IFN-a resulted in a decrease of total HBV mRNA in HBV-transfected HepG2 cells actively producing viruses. In this case, the effect of IFN-a is mainly thought to take place at the level of HBV transcription, probably by IFN-induced or modified cellular proteins that bind to the HBV enhancer and partially inhibit its activity (Tur-Kaspa et al., 1990). In line with these findings, Caselmann et al. (1992) reported that IFN-ct2b or -/? transiently inhibit replication of HBV, together with a decrease in the amount of HBV mRNA and surface and early antigen intracellular levels. Gilles e/ al. (1992) also reported that in transgenic mice carrying the integrated HBV genome, a single intraperitoneal injection of IFN-a, as well as TNF-c(, negatively regulates hepatic 2.1 kb steady-state HBV mRNA, showing that IFN-c( may affect HBV gene expression in uivo (Kakumu ef al., 1992).
To conclude, since the primary effect of IFN on HBV replication apparently is dependent on the assay system used, further experiments are necessary to establish the molecular and pathophysiological significance of the negative regulation of HBV gene expression by IFN-r.
62.4. Simian Virus 40
SV40 is a papovavirus that multiplies in and lyses monkey cells, whereas it cannot replicate in human and murine cells. However, SV40 can transform these nonpermissive cell types. Viral genomes in the transformed cells are integrated in the host chromosomal DNA and express viral-specific early mRNAs and proteins. Several studies have revealed appreciable protection against the cytopathic effects of SV40 in monkey cells exposed to IFN before infection. Inhibition of viral replication is characterized by strong reduction of both early and late SV40 mRNA expression, as well as viral-specific protein synthesis (Oxman and Levin, 1971; Mozes and Defendi, 1978: Brennan and Stark, 1983). Similar inhibition of early mRNAs and protein accumulation has been observed in mouse and human cells treated with IFN before infection with SV40 (Mozes and Defendi, 1979; Yamamoto et ul., 1975). However, IFN treatment of murine or human cells transformed by SV40 does not decrease the viral-specific macromolecular synthesis, nor suppress the neoplastic phenotype. This unresponsiveness is specific with regard to SV40 gene expression, since SV40-transformed cells develop a normal antiviral state against VSV following exposure to IFN. The presence of constitutively expressed T antigen, as in SV40-transformed COS cells, has been correlated with the lack of IFN-induced inhibition of SV40 replication observed in these cells (Brennan and Stark, 1983). Therefore, a critical role of T antigen in counteracting IFN action has been suggested to explain the different effects of IFN on SV40-infected and on SV40-transformed nonpermissive cells. To further investigate the phenomenon, Garcia-Blanc0 et al. (1985) infected SV40-transformed human and mouse cell lines with a viable SV40 deletion mutant (d11263), which codes for a truncated form of T antigen. The results obtained by first treating these cells lines with human or mouse Type I IFN, and then infecting with the SV40 deletion mutant, showed that accumulation of the truncated T antigen (coded by the infecting virus) was inhibited by IFN, whereas expression of the endogenous T antigen (specified by the integrated SV40 DNA) was not. Apparently, the molecular mechanisms induced by IFN and responsible for inhibition of viral gene expression may discriminate. within the same cell, between expression of a gene in the viral genome and that of one integrated into a host chromosome.
As to the antiviral mechanisms in permissive cells, Yamamoto et al. (1975) reported a decrease of viral inhibition with multiplicity of infection. In addition, when infections were carried out with naked SV40 DNA, instead of with virions, a reduced IFN effectiveness was reported. These authors, therefore, suggested that uncoating of the viral DNA was blocked in IFN-treated cells. In line with this hypothesis. Brennan and Stark (1983), using a T antigen temperature-sensitive mutant, which cannot replicate SV40 DNA or negatively regulate early SV40 mRNA synthesis at the nonpermissive temperature, demonstrated that an antiviral state against SV40 cannot be maintained in the presence of T antigen, and that the activity of IFN against this mutant does not decrease with increasing multiplicity of infection at the nonpermissive temperature, whereas infection with naked mutant DNA is still insensitive to the IFN-mediated blockade of early rival RNA synthesis. They suggested that early viral transcription blockade may be a consequence of defective uncoating of the infecting particles in the IFN-treated cells. Transcription of infecting DNA not completely free ofcoat proteins cannot proceed as in the control cells.
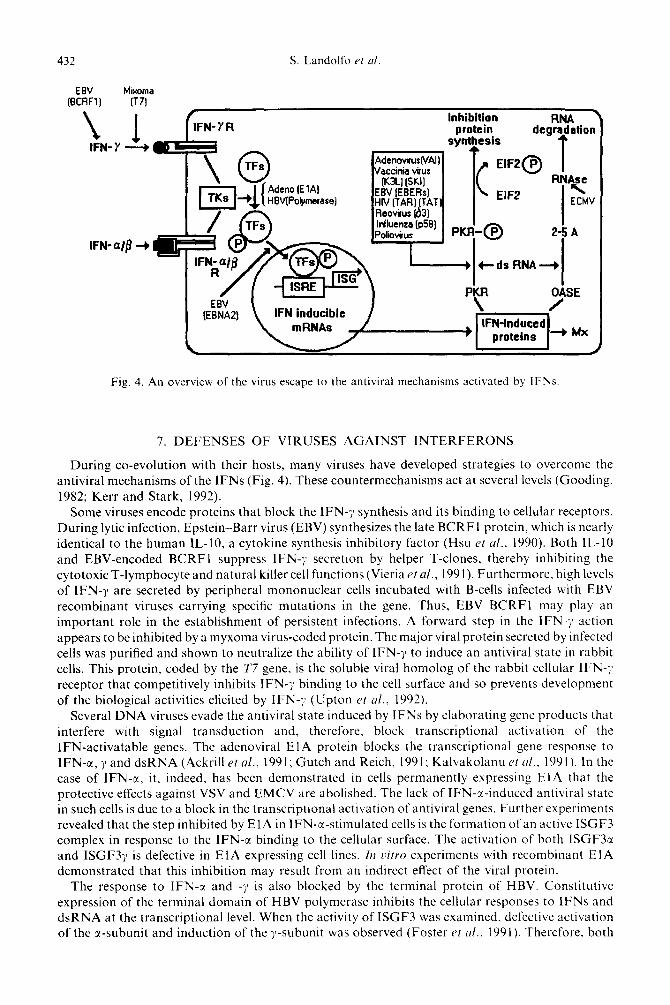
432 S. Landolfo et al
EBV [BCRFl)
Mixoma IT71
Inhibition protein
synthesis
RNA T degradation
‘(TFs)\
Fig. 4. An overview of the virus escape to the antiviral mechanisms activated by IFNs.
7. DEFENSES OF VIRUSES AGAINST INTERFERONS
During co-evolution with their hosts, many viruses have developed strategies to overcome the antiviral mechanisms of the IFNs (Fig. 4). These countermechanisms act at several levels (Gooding. 1982; Kerr and Stark, 1992).
Some viruses encode proteins that block the IFN-9 synthesis and its binding to cellular receptors. During lytic infection, Epstein-Barr virus (EBV) synthesizes the late BCRF I protein. which is nearly identical to the human IL-lo, a cytokine synthesis inhibitory factor (Hsu et ul., 1990). Both IL-10 and EBV-encoded BCRFl suppress IFN-y secretion by helper T-clones, thereby inhibiting the cytotoxic T-lymphocyte and natural killer cell functions (Vieria rt al., 1991). Furthermore, high levels of IFN-11 are secreted by peripheral mononuclear cells incubated with B-cells infected with EBV recombinant viruses carrying specific mutations in the gene. Thus, EBV BCRFl may play an important role in the establishment of persistent infections. A forward step in the IFN-;I action appears to be inhibited by a myxoma virus-coded protein. The major viral protein secreted by infected cells was purified and shown to neutralize the ability of IFN-~1 to induce an antiviral state in rabbit cells. This protein, coded by the T7 gene. is the soluble viral homolog of the rabbit cellular IFN-1 receptor that competitively inhibits IFN-y binding to the cell surface and so prevents development of the biological activities elicited by IFN-;I (Upton et al., 1992).
Several DNA viruses evade the antiviral state induced by IFNs by elaborating gene products that interfere with signal transduction and, therefore, block transcriptional activation of the IFN-activatable genes. The adenoviral ElA protein blocks the transcriptional gene response to IFN-cr, y and dsRNA (Ackrill PI al., 1991; Gutch and Reich, 1991; Kalvakolanu et cl/., 1991). In the case of IFN-c(. it, indeed, has been demonstrated in cells permanently expressing ElA that the protective effects against VSV and EMCV are abolished. The lack of IFN-r-induced antiviral state in such cells is due to a block in the transcriptional activation of antiviral genes. Further experiments revealed that the step inhibited by ElA in IFN-a-stimulated cells is the formation of an active ISGF3 complex in response to the IFN-cx binding to the cellular surface. The activation of both ISGF3a and ISGF3y is defective in ElA expressing cell lines. In z>itro experiments with recombinant ElA demonstrated that this inhibition may result from an indirect effect of the viral protein.
The response to IFN-c( and -7 is also blocked by the terminal protein of HBV. Constitutive expression of the terminal domain of HBV polymerase inhibits the cellular responses to IFNs and dsRNA at the transcriptional level. When the activity of ISGF3 was examined, defective activation of the u-subunit and induction of the y-subunit was observed (Foster et ul.. 1991). Therefore, both

Mechanism of viral inhibition by interferons 433
adeno and hepatitis B viruses impair assembly of the active ISGF3 complex, which mediates the
IFN-induced transcription of several cellular genes. A step downstream from ISGF3 formation seems to be inhibited in EBV-immortalized
lymphoblastoid cell lines resistant to the antiproliferative effects of IFN-c(. The presence of EBNA2, a viral-encoded transcription factor influencing several viral and host genes, is responsible for the lack of IFN-sr responsiveness, since cells immortalized by a virus deletion mutant devoid of this gene are sensitive to growth inhibition by IFN-u (Aman and Von Gabain, 1990). In cells expressing the EBNA2 protein, activation of IFN-responsive promoters is impaired in spite of the correct formation of the ISGF3 complex, therefore demonstrating that further steps in the IFN-mediated transcriptional response are affected by the viral protein (Kanda et al., 1992).
As an example of the different strategies adopted to resist the action of IFN, some viruses have evolved molecular mechanisms impairing the activity of the dsRNA-dependent IFN-inducible enzymes. The best characterized of these counteracting pathways are those inhibiting PKR activity, which is blocked in cells infected with a variety of viruses. Several viral products, including nucleic acids and proteins, interfere with PKR activation by dsRNA. The earliest identified mechanism was the adenovirus-coded virus-associated (VA) RNAs: viral mutants unable to produce VA RNAs are sensitive to the antiviral activities of IFNs, compared with the wild-type virus (Kitajewski et (11.. 1986). Viral resistance is related to such short (160 nt) RNAs produced late in infection that bind PKR, since they possess a double-stranded hairpin structure. This binding prevents activation of PKR by dsRNA, thus overcoming the inhibition of protein synthesis initiation that occurs in cells infected with adenoviruses lacking VA genes. It requires the apical stem of the molecule, which contains a base-paired stem, while the region involved in the inhibitory function is the complex central domain characterized by a stem-loop structure (Mathews and Shenk, 1991).
PKR can also bind the transacting responsive region RNA, a viral genome segment with a stem-loop structure produced by HIV-l. The final effect is still controversial, since both activation and inhibition of PKR have been reported (Gunnery et al., 1990). Moreover, HIV TAT protein binds to PKR and inhibits its activation, thus contributing to the inhibition of the enzyme that occurs during HIV-l infection (Roy et al., 1989). A further example of a viral transcript that counteracts PKR activity comes from the finding that EpsteinBarr virus small RNAs devoid of apparent effects on latent or lytic infection, bind PKR because of their similarity to adeno VA1 RNAs and block its activation (Clarke et al., 1990, 1991).
The resistance of vaccinia virus to IFN is related to the production of proteins with activities against PKR. The specific kinase inhibitory factor inhibits PKR activation by sequestering dsRNA (Whitaker-Dowling and Younger, 1983, 1984, 1986). More recently, a dsRNA-binding protein, encoded by the E3L gene, was demonstrated to inhibit the PKR activity by competing for binding to dsRNA (Watson et al., 1991; Chang et al., 1992; Davies et a/., 1993). Another vaccinia virus protein, K3L, an 8%amino acid polypeptide with a 28% sequence similarity to the N-terminus of eIF2-r (the main substrate of PKR), decoys the kinase activity (Beattie et a/., 1991; Davies ef N/., 1991).
In cells infected with reo, influenza and polio virus, the activity of the PKR pathway may be inhibited through different mechanisms. The reo a3 protein, a multifunctional zinc metallo-polypeptide of the outer viron capsid, has dsRNA-binding activity, and thus, can compete with PKR (Imani and Jacobs, 1988). In the case of cells infected with influenza virus, the activity of PKR is down-regulated by a cellular PKR inhibitor (Katze et al., 1986, 1988; Lee et d., 1990). This inhibitor. referred to as ~58 based on its molecular weight of 58,000, is released from a specific inhibitor upon virus infection and inhibits PKR autophosphorylation, as well as eIF2-2 phosphorylation (Katze, 1992; Lee et al., 1992). However, the mechanism of this inhibition remains to be elucidated. A different mechanism seems to be employed by the polio virus to inhibit PKR activity: this IFN-inducible enzyme is proteolyzed in polio-infected cells by a cellular protease, since the polio-encoded protease activities are not involved (Black et a/., 1989). This protease requires viral-specific dsRNA. Its target PKR domain is localized in the N-terminus and overlaps the dsRNA-binding domain (Black et al., 1993).
Turning lastly to the 2’,5’-oligoadenylate synthetase/RNAse L pathway, it has been observed that RNase L is inactivated in EMCV-infected cells, but such inactivation can be reversed by prior IFN treatment. The mechanisms of this effect. however, should be determined. Furthermore, Cayley et <I/.

434 S. Landolfo et al.
(1984) observed in cells infected with HSV the production of 2,5A analogs that can impair the activation of RNase L by authentic 2.5A.
A~knolcled~~nzentsThe authors’ research is supported by grants from the Italian National Research Council (C.N.R., P.F. ‘FAT.MA‘ and ‘A.C.R.O.‘) and A.I.R.C. We are grateful to M. Raviola for excellent secretarial support.
REFERENCES
Aboud, M. and Hassan, Y. (1983). Accumulation and breakdown of RNA-deficient intracellular virus particles in interferon-treated NIH 3T3 cells chronically producing moloney murine leukemia virus. J. Virol. 45: 489-495.
Ackrill, A. M.. Foster. F. R., Laxton, C. D.. Flavell, D. M., Stark, G. R. and Kerr. I. M. (1991) Inhibition of the cellular response to inteferons by products of the adenovirus type 5 E/A oncogene. Nucl. ,4cids Res. 19: 4387~-4393.
Aguet, M.. Dembic. Z. and Merlin. G. (1988) Molecular cloning and expression of the human interferon-; receptor. Cell 55: 273-280.
Akkaraju. G., Whitaker-Dowling, P., Youngner. J. S. and Jagus, R. (1989) Vaccinia specific kinase inhibitory factor prevents translational inhibition by double-stranded RNA in rabbit reticulocyte lysate. J. hiol. Chem. 264: 10321~10325.
Aman, P. and Von Gabain. A. (1990) An Epstein-Barr virus immortalization associated gene segment interferes specifically with the IFN-induced antiproliferative response in human B-lymphoid cell lines. EMBO J. 9: 147-152.
Arnheiter. H., Skuntz, S.. Noteborn. M.. Chang. S. and Meier. E. (1990) Transgenic mice with intracellular immunity lo influenza virus. Cell 62: 5 l-61.
Avery, R. J.. Norton, J. D.. Jones, J. S., Burke, D. C. and Morris, A. G. (1980) Interferon inhibits transformation by murine sarcoma viruses before integration of provirus. Nature 288: 93-95.
Ball. L. A. and White, C. N. (1978) Effect of interferon pretreatment on coupled transcription and translation in cell-free extracts of primary chick embryo cells. Virology 84: 496-508.
Bean, W. J., Jr and Simpson, R. W. (1973) Primary transcription of the influenza virus genome in permissive cells. Virology, 56: 646-65 1.
Beattie, E., Battaglia, J. and Paoletti, E. (1991) Vaccinia virus-encoded eIF-2x homologue abrogates the antiviral effect of interferon. Viro/ogj. 183: 419-422.
Belkowski. L. S. and Sen. G. C. (1987) Inhibition of vesicular stomatitis viral mRNA synthesis by interferons. J. Viral. 61: 653-660.
Benech, P., Vigneron, M.. Peretz. D., Revel. M. and Chebath, J. (1987) Interferon-responsive regulatory elements in the promoter of the human 2’.5’-oligo(A)synthetase gene. Molec Cell. Biol. 7: 4498-4504.
Black, T. L., Safer, B.. Hovanessian. A. and Katze, M. G. (1989) The cellular 68,000 M, protein kinase is highly autophosphorylated and activated yet significantly degraded during poliovirus infection: implications for translational regulation. J. Vim/. 63: 2244-2251.
Black. T. L.. Barber. G. N. and Katze. M. G. (1993) Degradation of the interferon-induced 68.000-M, protein kinase by poliovirus requires RNA. J. Viral. 67: 791&800.
Braken, J. B., Koopmans, P. P.. Van Munster. I. P. and Gribnau, F. W. (1992) Current status of interferon alpha in the treatment of chronic hepatitis B. Phrrm. Week/d. Sci. 44: 167-173.
Brennan. M. B. and Stark. G. R. (1983) Interferon pretreatment inhibits simian virus 40 infections by blocking the onset of early transcription. Cell 44: 8 1 I- 8 16.
Caselmann, W. H., Meyer, M.. Scholz, S., Hofschneider. P. H. and Koshy, R. (1992) Type I interferons inhibit hepatitis B virus replication and induce hepatocellular gene expression in cultured liver cells. J. Ir~/clc,t. &s. 166: 966 -971.
Cayley. P.. Davies. J.. McCullagh. K. and Kerry, I. (1984) Activation of the ppp(A2’p)nA system in interferon-treated. herpes simplex virus infected cells and evidence for novel inhibitors of the ppp(A2’p)nA-dependent RNase. Eur. J. Biochem. 143: 165--l 74.
Chang. H. W., Watson. J. C. and Jacobs, B. L. (1992) The E3L gene of vaccinia virus encodes an inhibitory of the interferon-induced, double-stranded RNA-dependent protein kinase. Proc. mm. Aud. Sci. U.S.A 89: 4825-4829.
Chatterhee, S. and Hunter. E. (1987) Recombinant human interferons inhibit replication of mason-Pfizer monkey virus in primate cells. Virology 157: 548-551.
Chatterjee. S. and Whitley, R. J. (1989) Effect of recombinant hybrid human interferon on replication and morphogenesis of HSV- I in monkey cells. C’irus Res. 12: 33-42.
Chatterjee, S., Hunter. E. and Whitley, R. (1985) Effect of cloned human interferons on protein synthesis and morphogenesis of herpes simplex virus. J. Viral. 56: 419-425.
Chebath, J., Benech. P., Hovanessian. A. G., Galabru. J. and Revel. M. (1987a) Four different forms of IFN-induced 2’-5’ oligoadenylate synthetase identified by immunoblotting in human cells. J. hi&. C/KW. 262: 3852 3857.

Mechanism of viral inhibition by interferons 435
Chebath, J., Benech, P., Revel, M. and Vigneron, M. (1987b) Constitutive expression of (2’-5’)oligo A synthetase confers resistance to picornavirus infection. Nature 330: 587-588.
Chong, K. L.. Feng, L., Schappert, K., Meurs, E., Donahue, T. F., Friesen, J. D., Hovanessian, A. G. and Williams, B. R. G. (1992) Human ~68 kinase exhibits growth suppression in yeast and homology to the translational receptor GCNZ. EMBO J. 11: 1553-1562.
Clarke, P. A., Sharp, N. A. and Clemens, M. J. (1990) Translational control by the Epstein-Barr virus small RNA EBER-1. Reversal of the double-stranded RNA-induced inhibition of protein synthesis in reticulocyte lysates. Eur. .I Biochem. 193: 635-641.
Clarke, P. A., Schwemmle, M., Schickinger, J., Hilse, K. and Clemens, M. J. (1991) Binding of Epstein-Barr virus small RNA EBER-1 to the double-stranded RNA-activated protein kinase DAI. Nucl. Acids Res. 19: 243-248.
Cofano, F., Moore, S. K., Tanaka, S., Yuhki, N., Landolfo, S. and Appella, E. (1990) Affinity purification, peptide analysis and cDNA sequence of the mouse interferon-y receptor. J. biol. Chem. 265: 4064-4071.
Cohrs, R. J., Condit, R. C., Pacha, R. F., Thompson, C. L. and Sharma, 0. K. (1989) Modulation of ppp(A2’p),A_dependent RNase by a temperature-sensitive mutant of vaccinia virus. J. Viral. 63: 948-95 1.
Colby, C. and Duesberg, P. H. (1969) Double-stranded RNA in vaccinia virus infected cells. Nature 222: 940-942.
Constantoulakis, P., Campbell, M., Felber, B. K., Nasioulas, G., Afonina, E. and Pavlakis, G. N. (1993) Inhibition of Rev-mediated HIV-l expression by an RNA binding protein encoded by the interferon-inducible 9-27gene. Science 259: 1314-1318.
Cook, J. R., Emanuel, S. L., Donnelly, R. J., Soh, J., Mariano, T. M., Schwartz, B., Rhee, S. and Pestka, S. (1994) Sublocalization of the human interferon-gamma receptor accessory factor gene and characterization of accessory factor activity by yeast artificial chromosomal fragmentation. J. hiol. Chem. 269, in press.
Cross, J. C. and Roberts, R. M. (1991) Constitutive and trophoblast-specific expression of a class of bovine interferon genes. Proc. natn. Acad. Sci. U.S.A. 88: 3817-3821.
Darnell, J. E., Jr (1982) Variety in the level of gene control in eukaryotic cells. Nature 297: 365-371. Davies, M. V., Elroy-Stein, O., Jagus, R., Moss, B. and Kaufmann, R. J. (1991) The vaccinia virus K3L
gene product potentiates translation by inhibiting double-stranded RNA-activated protein kinase and phosphorylation of the alpha subunit of eukaryotic initiation factor 2. J. Viro/. 66: 1943-1950.
Davies, M. V., Chang, H.-W., Jacobs, B. L. and Kaufman, R. J. (1993) The E3L and K3L vaccinia virus gene products stimulate translation through inhibition of the double-stranded RNA-dependent protein kinase by different mechanisms. J. Virol. 67: 1688-1692.
Decker, T., Lew, J. D., Mirkovitch, J. and Darnell, J. E., Jr (1991) Cytoplasmic activation of GAF, and IFN-y-regulated DNA binding factor. EMBO .I. 10: 927-932.
De Ferra, F. and Baglioni, C. (1981) Viral messenger RNA unmethylated in the 5’-terminal guanosine in interferon-treated HeLa cells infected with vesicular stomatitis virus. Viriologj, 112: 426-435.
De Ferra, F. and Baglioni, C. (1983) Increase in adenosylhomocysteine concentration in interferon-treated HeLa cells and inhibition of methylation of vesicular stomatitis mRNA. J. hiol. Chem. 258: 2118-2121.
Delcayre, A. X., Salas, F., Mathur, S., Kovats, K., Lotz, M. and Lernhardt, W. (1991) Epstein-Barr virus/complement C3d receptor is an interferon t( receptor. EMBO J. 10: 919-926.
De Maeyer. E. and De Maeyer-Guignard, J. (1988) Inteyferons and Other Regulatory C_vtokines. John Wiley and Sons. New York.
Desrosiers. R. C. and Lengyel, P. (1979) Impairment of reovirus mRNA (CAP), methylation in interferon-treated mouse L929 cells. Biochim. biophys. Acta 562: 471-480.
De Stasio, P. R. and Taylor, M. W. (1990) Specific effect of interferon on the herpes simplex virus type 1 transactivation event. J. Viral. 64: 2588-2593.
Dorsch-Hasler, K., Keil, G. M., Weber, F., Jasin, M., Schaffner, W. and Koszinowski, U. H. (1985) A long and complex enhancer activates transcription of the gene coding for the highly abundant immediate early mRNA in murine cytomegalovirus. Proc. nom. Acad. Sci. U.S.A. 82: 8325-8329.
Emilie, D., Maillot. M. C., Nicolas, J. F., Fior, R. and Galanaud, P. (1992) Antagonistic effect of interferon-gamma on tat-induced transactivation of HIV long terminal repeat. J. biol. Chem. 267: 20565-20570.
Esteban, M. and Metz, D. H. (1973) Inhibition of early vaccinia virus protein synthesis in interferon-treated chicken embryo fibroblasts. J. gen. Viral. 20: 11 l-l 15.
Esteban, M.. Benavente, J. and Paez, E. (1984) Effect of interferon on integrity of vaccinia virus and robosomal RNA in infected ceils. Virology 134: 40-5 1.
Falcoff, R. and Sanceau, J. (1979) Mengovirus-induced inhibition of cell protein synthesis and subsequent recovery of cellular activity in interferon-pretreated cells. Virolog?, 98: 433-438.
Faltynek, C. R. and Baglioni, C. (1983) Treatment of human cells with interferon has no effect on the glycosylation of viral and cellular proteins. Virologjs 127: 225-229.
Faltynek, C. R., Princler. G. L., Gusella, G. L., Varesio, L. and Radzioch. D. (1989) A functional protein kinase C is required for induction of 2-5A synthetase by recombinant interferon-rA in Daudi cells. J. hiol. Chem. 264: 14305-14311.
Farrar, M. A. and Schreiber. R. D. (1993) The molecular cell biology of interferon-y and its receptor. Annu. Rec. Immunol. 11: 571-611.

436 S. Landolfo et al
Feng, G-S., Chong. K.. Kumar, A. and Williams, B. R. G. (1992) Identification of the dsRNA binding domains in the interferon-induced ~68 kinase. Proc. natn. Acad. Sci. U.S.A. 89: 544775451.
Foster, G. R.. Ackrill. A. M., Goldin, R. D., Kerr, I. M.. Thomas, H. C. and Stark, G. R. ( IY9 1) Expression of the terminal protein region of hepatitis B virus inhibits cellular responses to interferons r and 7 and double-stranded RNA. Proc. tzattz. Acad. Sci. U.S.A. 88: 2888-2892.
Friedman. R. L. and Stark. G. R. (1985) Interferon induced transcription of HLA and metallothionein genes containing homologous upstream sequences. Nature 314: 6377639.
Friedman. R. L.. Manly. S. P., McMahon, M.. Kerr. I. and Stark, G. R. (1984) Transcriptional and post-transcriptional regulation of interferon induced gene expression in human cells, c‘ei/ 38: 7455755.
Fu, X-Y., Schindler, C., Improta. T., Aebersold, R. and Darnell. J. E., Jr (1992) The proteins of ISGF-3, the interferon a-induced transcriptional activator, define a gene family involved in signal transduction. Proc. ttattt. ilcad. Sci. U.S.A. 89: 7840-7843.
Garcia-Bianco, M. A.. Ghosh, P. K., Jayaram, B. M.. Ivory, S., Lebowitz, P. and Lengyel, P. (1985) Selectivity of interfero:l action in simian virus 40-transformed cells superinfected with simian virus 40. J. Viral. 53: 8933898.
Gariglio. M ., Panico, S., Cavallo. G.. Divaker, C., Lengyel, P. and Landolfo, S. (I 992) Impaired transcription of the poly rl:rC- and interferon-activatable 202 gene in mice and cell lines from the C57BLi6 strain. Virolog), 187: 115~_123.
Gariglio, M.. Gaboli. M., Mana, C.. Ying, G.-G., Gribaudo, G.. Cavallo, R. and Landolfo, S. (1994) Characterization of nuclear factors involved in 202 gene induction by interferon-a in murine leukemia cells. ELW. J. Bioclicni 221: 731-739.
Gamut. C. J. (I 972) Effect of interferon on synthesis of ssRNA in reovirus type 3-infected L cell cultures. Bioch~w~. hioph!~s. I&s. Conttnun. 47: 122881236.
Gilles. P. N., Fey. G. and Chisari, F. V. (1992) Tumor necrosis factor alpha negatively regulates hepatitis B virus gene expression in transgenic mice. J. Vim/. 66: 3955-3960.
Gooding. L. R. (1992) Virus proteins that counteract host immune defenses. C’eN 71: 5-7. Goswami. B. and Sharma, 0. K. (1984) Degradation of rRNA in interferon-treated vaccinia virus infected cells.
J. hiol. Chcttt. 259: 1371ll374. Gribaudo. G., Toniato, E., Engel, D. A. and Lengyel, P. (1987) Interferons as gene activators. Characteristics
of an interferon-activatable enhancer. J. Biol. Chetn. 262: 11878811883. Gribaudo. G., Gariglio M., Cavallo. G. and Landolfo, S. (1990) Cell and type specificity of interferon action.
Unusual characteristics of the transcriptional control of gene expression by interferon-gamma in T cells. Eur. J. h7r~7uriol. 20: 12433 1249.
Gribaudo. G., Lembo. D.. Cavallo. G.. Landolfo. S. and Lengyel, P. (1991) Interferon action: binding of viral RNA to the 40-kilodalton 2’-5’ oligoadenylate synthetase in interferon-treated HeLa cells infected with encephalomyocarditis virus. J. Viral. 65: 174881757.
Gribaudo, G.. Ravaglia. S.. Caliendo. A.. Cavallo, R., Gariglio. M., Martinotti, M. G. and Landolfo, S. (1993) Interferons inhibit onset of murine cytomegalovirus immediate-early gene transcription. l’irologv 197: 303 311.
Griin. J.. Kroon, E.. Zoller. B., Krempien, U. and Jungwirth. C. (1987) Reduced steady-state levels of vaccinia virus-specific early mRNAs in interferon-treated chick embryo fibroblasts. Virology 158: 28-33.
Gunnery. S., Rice, A. P., Robertson, H. D. and Mathews, M. B. (1990) Tat-responsive region RNA of human immunodeficiency virus 1 can prevent activation of the double-stranded RNA-activated protein kinase. Proc,. ttctftt. Acrrd. Sci. U.S.A. 87: 8687.-8691.
Gupta. S. L.. Graziadei, W. D.. Weideli. H., Sopori, M. L. and Lengyel, P. (1974) Selective inhibition of viral protein accumulation in interferon-treated cells: nondiscriminate inhibition of the translation of added viral and cellular messenger RNAs in their extracts. Virology. 57: 49-63.
Gupta. S. L.. Holmes, S. L. and Mehra, L. L. (1982) Interferon action against reovirus: activation of interferon-induced protein kinase in mouse L929 cells upon reovirus infection. Virolog~~ 120: 4955499.
Gutch. M. J. and Reich, N. C. (1991) Repression ofthe interferon signal transduction pathway by the adenovirus ElA oncogene Proc,. tzrtttt. Ar,ad. Sci. U.S.A. 88: 791337917.
Hailer. 0. (I 98 I ) Natural resistance to tumors and viruses. Curr. 7-0~. Microhiol. Ittmuttol. 92: 25 -52. Hannigan. G. E. and Williams, B. R. G. (1991) Signal transduction by interferon-alpha through arachidonic
acid metabolism. Scicncc~ 251: 2044207. Hansen, B. D.. Nara. P. L.. Maheshwari. R. K.. Sidhu. G. S.. Bernbaum. J. G., Hoekzema. D.. Meltzen, M. S.
and Gendelman. H. E. (1992) Loss of infectivity by progeny virus from alpha interferon-treated human immunodefciency virus type l-infected T cells is associated with defective assembly of envelope gpl20. J. Vir-ol. 66: 7543-7548.
Hassel. B. A.. Zhou. A.. Sotomayor. C., Maran. A. and Silverman. R. H. (1993) A dominant negative mutant of 2-5A dependent RNase suppresses antiproliferative and antiviral effects of interferon. EMBO J. 8: 329773304.
Hayashi. Y. and Koike, K. (1989) Interferon inhibits hepatitis B virus replication in a stable expression system of transfected viral DNA. J. Vim/. 63: 2936-2940.
Hibino. Y., Mariano. T. M., Kumar, C. S.. Kozak, C. A. and Pestka, S. (1991) Expression and reconstitution of a biologically active mouse interferon gamma receptor in hamster cells. J. IGo/. Chrm. 266: 694886951.
Hibino. Y.. Kumar. C. S.. Mariano. T. M.. Lai, D. and Pestka. S. (1992) Chimeric interferon gamma receptors

Mechanism of viral inhibition by interferons 437
demonstrate that an accessory factor required for activity interacts with the extracellular domain. J. biol. Clrem. 267: 3741-3749.
Horisberger, M. A., Haller, 0. and Arnheiter, H. (1980) Interferon-dependent genetic resistance to influenza virus in mice: virus replication in macrophages is inhibited at an early step. J. gen. Viral. 50: 205210.
Hovanessian, A. G. (1989) The double-stranded RNA-activated protein kinase induced by interferons: dsRNA-PK. J. Interjeron Res. 9: 641-647.
Hovanessian, A. G. (199 1) Interferon-induced and double-stranded RNA-activated enzymes; a specific protein kinase and 2’-5’-oligoadenylate synthetases. J. Interferon Res. 11: 199-205.
Hovanessian, A. G., Laurent, A. G., Chebath, J., Galabru, J., Robert, N. and Svab, J. (1987) Identification of 69-kd and IOO-kd forms of 2-5A synthetases in interferon-treated human cells by specific monoclonal antibodies. EMBO J. 6: 1273-1280.
Hsu, D-H., de Waal Malefyt, R., Fiorentino, D. F., Dang, M-N., Vieira, P., de Vries, J., Spits, H., Mosmann, R. R. and Moore, K. W. (1990) Expression of interleukin-10 activity by Epstein-Barr virus protein BCRFI. Science 250: 830-832.
Huleihel, M. and Aboud, M. (1983) Inhibition of retrovirus DNA supercoiling in interferon-treated cells. J. Viral 48: 120-126.
Ihle, J. N., Witthuln, B. A., Quelle, F. W., Yamamoto, K., Thierfelder, W. E., Kreider, B. and Silvennoinen, 0. (1994) Signaling by the cytokine receptor superfamily: JAKs and STATS. Z’IBS 19: 222-227.
Imani, F. and Jacobs, B. L. (1988) Inhibitory activity for the interferon-induced protein kinase is associated with the reovirus serotype 1 sigma 3 protein. Proc. natn. Acad. Sci. U.S.A. 85: 7887-7891.
Isaacs, A. and Lindenmann, J. (1957) Virus interference 1. The interferon. Proc. R. Sot. Ser. B 147: 2588267.
Israel, A., Kimura, A., Fournier, A., Fellous, M. and Kourilsky, P. (1986) Interferon response sequence potentiates activity of an enhancer in the promoter region of a mouse H-2 gene. Nature 322: 743-746.
Ito, N., Gribaudo, G., Toniato, E., Thakur, A., Yagi, Y., Barbosa, J., Kamarck, M., Ruddle, F. H. and Lengyel, P. (1989) Interferons as gene activators: identification of an interferon responsive “GA box” in a murine gene. In: UCLA S_vmposium on Growth Inhibitory and Cytotoxic Polypeptides, pp. 1699178, Sporn, M. (ed.) A. R. Liss, New York.
Jay, F. T.. Dawood, M. R. and Friedman, R. M. (1983) Interferon induces the production of membrane protein-deficient and infectivity-defective vesicular stomatitis virions through interference in the virion assembly process. J. gen. Virol64: 707-712.
Joklik, W. K. and Merigan, T. (1966) Concerning the mechanism of action of interferon. Proc. natn. Acad. Sci. U.S.A. 56: 558-565.
Jung, V., Rashidbaigi, A., Jones, C., Tischfield, J. A., Shows, T. B. and Pestka, S. (1987) Human chromosomes 6 and 21 are required for sensitivity to human interferon gamma. Proc. natn. Acad. Sci. U.S.A. 84: 415 l-41 55.
Jung, V., Jones, C., Rashidbaigi, A., Geyer, D. D., Morse, H. G., Wright, R. B. and Pestka. S. (1988) Chromosome mapping of biological pathways by fluorescence-activated cell sorting and cell fusion: the human interferon gamma receptor as a model system. Som. Ccl1 Mol. Gen. 14: 583-592.
Jung, V., Jones, C., Kumar, C. S., Stefanos, S., O’Connell, S. and Pestka, S. (1990) Expression and reconstitution of a biologically active human interferon gamma receptor in hamster cells. J. biol. Chem. 265: 1827-1830.
Jungwirth, C., Horak, I., Bodo, J., Lindner, J. and Schultze, B. (1972) The synthesis of pox-virus-specific RNA in interferon-treated cells. Virology 48: 59-70.
Kakumu. S., Ito, Y., Wakita, T., Yoshioka, K., Ishikawa, T., Takayanagi, M., Higashi, Y. and Yang, Z. Q, (1992) Effects of transforming growth factor-beta 1 against the inhibitory action of interferon on DNA synthesis and viral replication in hepatitis B virus DNA-transfected cell. J. Med. Virol. 38: 62-66.
Kalvakolanu, D. V. R., Bandyopadhyay, S. K., Harter, M. L. and Sen, G. C. (1991) Inhibition of interferon- inducible gene expression by adenovirus ElA proteins: block in transcriptional complex formation. Proc. natn. Acad. Sci. U.S.A. 88: 7459-7463.
Kanda, K., Decker, T., Aman, P., Wahlstrom, M., von Gabain, A. and Kallin, B. (1992) The EBNA2-related resistance towards alpha interferon (IFN-(r) in Burkitt’s lymphoma cells effects induction of IFN-induced genes but not the activation of transcription factor ISGF-3. Molec. Cell. Biol. 12: 4930-4936.
Karupiah. G., Xie, Q-W., Buller, R. M. L.. Nathan, C., Duarte, C. and MacMicking, J. D. (1993) Inhibition of viral replication by interferon-y-induced nitric oxide synthase. Science 261: 1445-1448.
Katze, M. G. (1992) The war against the interferon-induced dsRNA-activated protein kinase: can viruses win? J. Interferon Res. 12: 241-248.
Katze, M. G., Detjen, B. M., Safer, B. and Krug, R. M. (1986) Translational control by influenza virus: suppression of the kinase that phosphorylates the alpha subunit of initiation factor eIF-2 and selective translation of influenza viral mRNAs. Molec. CeN. Biol. 6: 1741-1750.
Katze. M. G., Tomita, J., Black, T., Krug, R. M., Safer, B. and Hovanessian, A. G. (1988) Influenza virus regulates protein synthesis during infection by repressing the autophosphorylation and activity of the cellular 68,000 M protein kinase. J. Virol. 62: 3710-3717.
Kerr, I. M. and Stark, G. R. (1991) The control of interferon-inducible gene expression. FEBS Let?. 285: 194-198.
Kerr, I. M. and Stark, G. R. (1992) The antiviral effects of the interferons and their inhibition. J. Interjeron Res. 12: 237-240.
Kitajewski, J., Schneider, R. J., Safer, B., Munemitsu, S. M., Samuel, C. E., Thimmappay, B. and Shenk, T.

438 S. Landolfo et al
(1986) Adenovirus VA1 RNA antagonizes the antiviral action of interferon by preventing activation of the interferon-induced eIF-2r kinase. Cell 45: 195-200.
Klotzbucher. A., Mittnacht, S., Kirchner, H. and Jacobsen, H. (1990) Different effects of IFN-;I and IFN-c(/fi on immediate early gene expression of HSV-1. Virolog_v 179: 4877491.
Kolb, E.. Laine, E., Strehler, D. and Staeheli, P. (1992) Resistance to influenza virus infection of Mx transgenic mice expression Mx protein under the control of two constitutive promoters. J. Virol. 66: 1709-1716.
Kornbluth, R. S., Oh, P. S., Munis, J. R., Cleveland, P. H. and Richman, D. D. (1989) Interferon and bacterial lipopolysaccharide protect macrophages from productive infection by human immunodeficiency virus in vitro. J. <~-up. Med. 169: 1137-1151.
Koromilas, A. E., Roy, S., Barber, G. N., Katze, M. G. and Sonenberg, N. (1992) Malignant transformation by a mutant of the IFN-inducible dsRNA-dependent protein kinase. Science 257: 168551689.
Krug, R. M., Shaw, M., Broni, B., Shapiro, G. and Haller, 0. (1985) Inhibition of influenza viral messenger RNA synthesis in cells expressing the interferon-induced Mx gene product. J. Viral. 56: 201-206.
Kumar, R., Tiwari, R. K., Kusari, J. and Sen, G. C. (1987) Clonal derivatives of the RD-114 cell line differ in their antiviral and gene-inducing responses to interferons. J. Virol. 61: 2727-2732.
Kumar, R., Chattopadhyay, D., Banerjee, A. K. and Sen. G. C. (1988a) Ribonuclease activity is associated with subviral particles isolated from interferon-treated vesicular stomatitis virus-infected cells, J. Viral. 62: 641-643.
Kumar, R., Choubey, D.. Lengyel, P. and Sen, G. C. (1988b) Studies on the role of the 2’-5’-oligoadenylate synthetase-RNase L pathway in beta interferon-mediated inhibition of encephalomyocarditis virus replication. J. Viral. 62: 3175-3181.
Kumar, C. S., Mulhukumaran, G., Frost, L. J., Noe, M., Ahn, Y. H., Mariano, T. M. and Pestka, S. (1989) Molecular characterization of the murine interferon-7 cDNA. J. hiol. Chem. 264: 17939-17946.
LaMarco, K. L. and McKnight, S. L. (1989) Purification of a set of cellular polypeptides that bind to the purine-rich cis-regulatory element of herpes simplex virus immediate early genes. Genes Der. 3: 1372-1383.
Landolfo. S., Gariglio, M., Gribaudo, G. and Garotta, G. (1994) Double-stranded RNAs as gene activators. In: Progress it1 Molecular and Subcellular Biolog>,. Vol. 14. pp. 15-27, Muller, W. H. G. and Schroder, H. C. (eds) Springer Verlag, Heidelberg.
Larner. A. C., Jonak, G., Cheng, Y-S. E., Korant. B., Knight E and Darnell, J. E.. Jr (1984) Transcriptional induction of two genes in human cells by p-interferon. Proc. natn. Acud. Sci. U.S.A. 81: 673336737.
Lee, T. G., Tomita. J.. Hovanessian. A. G. and Katze. M. G. (1990) Purification and partial characterization of a cellular inhibitor of the interferon-induced protein kinase of 68,000 M, from influenza virus-infected cells. Proc. natn. Awd. Sci. U.S.A 87: 620886212.
Lee, T. G.. Tomita, J., Hovanessian, A. G. and Katze, M. G. (1992) Characterization and regulation of the 58,000 dalton cellular inhibitor of the interferon-induced, dsRNA activated protein kinase. J. hiol. C’hem. 267: 14238814243.
Lengyel. P. (1982) Biochemistry of interferons and their actions. Annu. Rev. Biochem 51: 251-282. Lengyel. P. (1987) Double-stranded RNA and interferon action. 1. hzter&ron Res. 7: 51 I-519. Lengyel, P. (1993)Tumorsuppressor genes: news about the interferon connection. Proc. nutn. Acad. Sci. U.S.A.
90: 5893-5895. Levy. D. and Darnell, J. E. (1990) Interferon-dependent transcriptional activation: signal transduction without
second messengers involvement‘? Nerv Biologist 2: 923-928. Levy, D. E.. Larner. A., Chaudhuri, A., Babiss, L. E. and Darnell, J. E. (1986) Interferon-stimulated
transcription: isolation of an inducible gene and identification of its regulatory region. Proc. tzutn. Acad. Sci. U.S.A. 83: 8929-8933.
Levy, D. E.. Kessler, D. S., Pine, R. Reich, N. and Darneil, J. E. Jr (1988) Interferon-induced nuclear factors that bind a shared promoter element correlate with positive and negative transcriptional control. Gene Der~. t: 383 393.
Levy. D. E., Kessler. D. S., Pine, R. and Darnell, J. E.. Jr (1989) Cytoplasmic activation of ISGF3. the positive regulator of interferon-a-stimulated transcription, reconstituted in t,itro. Gene Det>. 3: 136221371.
Lewis, J. A. (1988) Induction of an antiviral state by interferon in the absence of elevated levels of 2,5-oligo(A) synthetase and eIF-2 kinase. Virology, 162: 118127.
Loh, J. E., Schindler, C., Ziemiecki, A., Harpur. A. G.. Wilks. A. F. and Flavell, R. A. (1994) Mutant cell lines unresponsive to alpha/beta and gamma interferon are defective in tyrosine phosphorylation of ISGF-3~ components. Molec. Cell. Biol. 14: 2170-2179.
Luster, A. D.. Unkeless. J. C. and Ravetch, J. V. (1985) Gamma-interferon transcriptionally regulates an early-response gene containing homology to platelet proteins. Nature 315: 672-676.
Maheshwari, R. K., Banerjee, D. K., Waechter, C. J.. Olden, K. and Friedman. R. M. (1980) Interferon treatment inhibits glycosylation of a viral protein. Nature 287: 454-456.
Manders, E. K., Tilles, J. G. and Huang, A. S. (1972) Interferon-mediated inhibition of virion-detected transcription. Virology 49: 573-58 1.
Maniatis. T., Goodbourn. S. and Fischer, J. A. (1987) Regulation of inducible and tissue-specific gene expression. Science 236: 1237-1245.
Marcus, P. I., Engelhardt, D. L.. Hunt. J. M. and Sekellick, M. (1971) Interferon action: inhibition of vesicular stomatitis virus RNA synthesis induced by virion-bound polymerase. Scienc,e 174: 5933598.

Mechanism of viral inhibition by interferons 439
Mariano, T. M., Kozak, C. A., Langer, J. A. and Pestka, S. (1987) The mouse immune interferon receptor gene is located on chromosome 10. J. biol. Chem. 262: 5812-5814.
Marie, 1. and Hovanessian, A. G. (1992) The 69-kDa 2-5A synthetase is composed of two homologous and adjacent functional domains. J. biol. Chem. 267: 9933-9939.
Martin, P. and Friedman, L. S. (1992) Therapies for hepatitis B virus: current status and future possibilities. Adu. e.up. Med. Biol. 312: 11 l-120.
Martinotti, M. G., Gribaudo, G., Ganglio, M., Caliendo, A., Lembo, D., Angeretti, A., Cavallo, R. and Landolfo, S. (1993) Effect of interferon-a on immediate early gene expression of murine cytomegalovirus. J. Interjkron Res. 13: 105p109.
Masters, P. S. and Samuel, C. E. (1983) Mechanism of interferon action: inhibition of vesicular stomatitis virus replication in human amnion U cells by cloned human leukocyte interferon II. Effect on viral macromolecular synthesis. J. biol. Chem. 258: 12026-12033.
Mathews, M. B. and Shenk, T. (1991) Adenovirus virus-associated RNA and translation control. J. Viral. 65: 5657-5662.
McKendry, R., John, J., Flavell, D., Miiller, M., Kerr, I. M. and Stark, G. R. (1991) High-frequency mutagenesis of human cells and characterization of a mutant unresponsive to both a and y interferons. Proc. natn. Acad. Sci. U.S.A. 88: 11455-l 1459.
Melen, K., Ronni, T., Broni, B., Krug, R. M., von Bonsdorff, C. H. and Julkunen, I. (1992) Interferon-induced Mx proteins form oligomers and contain a putative leucine zipper. J. biol. Chem 267: 25898-25907.
Metz, D. M. and Esteban, M. (1972) Interferon inhibits viral protein synthesis in L cells infected with vaccinia virus. Nature 238: 385-388.
Meurs, E., Chong, K., Galabru, J., Thomas, N. S. B., Kerr, I., Williams, B. R. G. and Hovanessian, A. G. (1990) Molecular cloning and characterization of the human double-stranded RNA-activated protein kinase induced by interferon. Cell 62: 379-390.
Meurs, E. F., Watanabe, Y., Kadereit, S., Barber, G. N. Katze, M. G., Chong, K., Williams, B. R. G. and Hovanessian, A. G. (1992) Constitutive expression of human double-stranded RNA-activated ~68 kinase in murine cells mediates phosphorylation of eukaryotic initiation factor 2 and partial resistance to encephalomyocarditis virus growth. J. Viral. 66: 580445814.
Meurs, E. F., Galabru, J., Barber, G. N., Katze, M. G. and Hovanessian, A. G. (1993) Tumor suppressor function of the interferon-induced double-stranded RNA-activated protein kinase. Proc. natn. Acad. Sci. U.S.A. 90: 232-236.
Meyer, T. and Horisberger, M. A. (1984) Combined action of mouse alpha and beta interferon in influenza-infected macrophage carrying the resistance gene Mx. J. Viral. 49: 709-716.
Meylan, P. R., Guatelli, J. C., Munis, J. R., Richman, D. D. and Kornbluth, R. S. (1993) Mechanisms for the inhibition of HIV replication by interferons-alpha-beta, and -gamma in primary human macrophages. Virology 193: 1388148.
Mittnacht, S., Straub, P., Kirchner, H. and Jacobsen, H. (1988) Interferon treatment inhibits onset of herpes simplex virus immediate-early transcription. Virology 164: 201-210.
Mozes, L. W. and Defendi, V. (1979) The differential effect of interferon on T antigen-production in simian virus 40-infected or transformed cells. Virology 93: 558-568.
Miiller. M., Laxton, C., Briscoe. J., Schindler, C., Improta, T., Darnell, J. E., Jr, Stark, G. R. and Kerr. I. M. (1993) Complementation of a mutant cell line: central role of the 91 kDa polypeptide of ISGF3 in the interferon-a and -y signal transduction pathways. EMBO J. 12: 4221-4228.
Munoz, A. and Carrasco, L. (1981) Protein synthesis and membrane integrity in interferon-treated HeLa cells infected with encephalomyocarditis virus. J. gen. Virol. 56: 153-162.
Munoz, A. and Carrasco, L. (1983) Effect of interferon treatment on blockade of protein synthesis induced by poliovirus infection. Eur. J. Biochem. 137: 623-629.
Nilsen, T. W., Maroney, P. A. and Baglioni, C. (1982) Synthesis of (2’-5’) oligoadenylate and activation of an endoribonuclease in interferon-treated HeLa cells infected with reovirus. J. Viral. 42: 103991045.
Oberman, F. and Panet, A. (1988) Inhibition of transcription of herpes simplex virus immediate-early genes in interferon-treated human cells. J. gen. Virol. 69: 116771177.
Olden, K., Bernard, B. A., Turner, W. and White, S. L. (1982) Effect of interferon on protein glycosylation and comparison with tunicamycin. Nature 300: 290-292.
Oxman, M. N. and Levin, M. J. (1971) Interferon and transcription of early virus-specific RNA in cells infected with simian virus 40. Proc. natn. Acad. Sci. U.S.A. 68: 299-302.
Pacha, R. F., Meis, R. J. and Condit, R. C. (1990) Structure and expression of the vaccinia virus gene which prevents virus-induced breakdown of RNA. J. Virol. 64: 3853-3863.
Paez, E. and Esteban, M. (1984) Resistance of vaccinia virus to interferon is related to an interference phenomenon between the virus and the interferon system. Virology 134: 12-28.
Pavlovic, J. and Staeheli, P. (1991) The antiviral potentials of Mx proteins. J. Interjkron Res. 11: 215-219. Pavlovic, J., Zurcher, T., Haller, 0. and Staeheh, P. (1990) Resistance to influenza virus and vesicular stomatitis
virus conferred by expression of human MxA protein. J. Viral. 64: 3370-3375. Pavlovic, J., Haller, 0. and Staeheh, P. (1992) Human and mouse Mx proteins inhibit different steps of the
influenza virus multiplication cycle. J. Viral. 66: 256442569. Pellegrini, S. and Schindler, C. (1993) Early events in signalling by interferons. TIBS 18: 338-342.

440 S. Landolfo et al.
Pellegrini, S., John, J., Shearer, M.. Kerr, I. M. and Stark, G. R. (1989) Use of a selectable marker regulated by alpha interferon to obtain mutations in the signaling pathway. Molec. Cell. Biol. 9: 4605-4612.
Pestka, S., Langer, J. A., Zoon. K. C. and Samuel, C. E. (1987) Interferons and their actions. Annu. Rec. Biochem. 56: 727~771.
Pfeffer, L. M. (1987) Mechanisms of Intet$wm Actions. CRC Press, Boca Raton. Pfeffer. L. M. and Tan. Y H. (199 1) Do second messengers play a role in interferon signal transduction? TIBS
16: 321-323. Pfeffer, L. M.. Wang, E. and Tamm, I. (1980) Interferon effects on microfilament organization, cellular
fibronectin distribution, and cell mobility in human fibroblasts. J. Cell Biol. 85: 9915. Pfeffer, L. M., Strulovici, B. and Saltiel, A. R. (1990) Interferon-a selectively activates the /I isoform of protein
kinase C through phosphatidylcholine hydrolysis. Proc. natn. Acad. Sci. U.S.A. 87: 6537-6541. Pitha. P. M., Wivel. N. A., Fernie, B. F. and Harper, H. P. (1979) Effect of interferon on MuLV infection. IV.
Formation of noninfections virus in chronically infected cells. J. gen. Virol. 42: 467-480. Pitha. P. M., Fernie. B.. Maldarelli, F., Hattman, T. and Wivel, N. A. (1980) Effect of interferon on mouse
leukemia virus (MuLV). V. Abnormal proteins on virious of Rauscher MuLV produced in the presence of interferon. J. gen. Virnl. 46: 977110.
Porter, A. C. G.. Chernajovsky, Y.. Dale, T. C., Gilbert, C. S., Stark, G. R. and Kerr, 1. M. (1988) Interferon response element of the human gene 6-16. EMBO J. 7: 85-92.
Ransohoff. R. M., Maroney, P. A.. Nayak, D. P., Chambers, T. M. and Nilsen, T. W. (1985) Effect of human alpha A interferon on influenza virus replication in MDBK cells. J. Virol. 56: 1049.-1052.
Rashidbaigi. A.. Langer, J. A., Jung, V., Jones. C., Morse, H. G., Tischfield, J., Trill, J. J.. Jung, H.-F. and Pestka, S. (1986) The gene for the human immune interferon receptor is located on chromosome 6. Proc. m/17. Acud. Sci. U..S.A 83: 384-388.
Repik. P.. Flamand, A. and Bishop, D. H. L. (1974) Effect of interferon upon the primary and secondary transcription of vesicular stomatitis and influenza viruses. J. Virol. 14: 1169-I 178.
Revel. M. and Chebath, J. (1986) Interferon-activated genes. TIBS 11: 66-70. Rice, A. P. and Kerr. I. M. (1984) Interferon-mediated, double-stranded RNA-dependent protein kinase is
inhibited in extracts from vaccinia virus-infected cells. J. Virol. 50: 229-236. Rice. A. P.. Roberts. W. K. and Kerr, I. M. (1984) 2-5A accumulates to high levels in interferon-treated, vaccinia
virus-infected cells in the absence of any inhibition of virus replication. J. Viral. 50: 220-228. Rice. A. P., Kerr, S. M.. Roberts, W. K.. Brown, R. E. and Kerr, 1. M. (1985) Novel 2’-5’-oligoadenylates
synthesized in interferon-treated, vaccinia virus-infected cells. J. Virot’. 56: 1041~-1044. Riggin. C. H. and Pitha. P. M. (1982) Effect of interferon on the exogenous friend murine leukemia virus
infection. b’irolog), 118: 202-213. Roizman. B. (1990) Multiplication of viruses. An overview. In: Fields Virology, 2nd edn, pp. 87-94. Fields. B.
N. and Knipe, D. M. (eds) Raven Press Ltd, New York. Rothman, J. H., Raymond. C. K. Gilbert, T.. O’Hara. P. J. and Stevens, T. H. (1990) A putative GTP binding
protein homologous to vertebrate IFN-inducible Mx proteins performs an essential function in yeast vacuolar protein sorting. Cc// 61: 106331074.
Roy. S., Katze. M. G., Parkin, N. T.. Edery, I., Hovanessian, A. G. and Sonenberg, N. (1989) Control oftheinterferoninduced68kilodalton protein kinase by theHIV-1 tatgeneproduct. Science247: 1216-1219.
Rysiecki. G.. Gewert, D. R. and Williams, B. R. G. (1989) Constitutive expression of a 2’.5’-oligoadenylate synthetase cDNA results in increased antiviral activity and growth suppression. J. Intetfkron Res. 9: 6499657.
Sahni, G. and Samuel. C. E. (1986) Mechanism of interferon action. Expression of vesicular stomatitis virus G gene in transfected cos cells is inhibited by interferon at the level of protein synthesis. J. hiol. Chenz. 261: 16764416768.
Salzberg. S., Shurtz, R.. Feder. J. and Aboud, M. (1983) Effect of interferon on the penetration of murine leukemia virus and binding of its genome RNA to polybosomes at the early stage of infection. Arc/u Viral. 78: 267-278.
Samanta. H.. Engel. D. A.. Chao, H. M., Thakur, A.. Garcia-Bianco, M. A. and Lengyel, P. (lY86) lnterferons as gene activators. J. hiol. Chem. 261: 11849-l 1858.
Samuel. C. E. (199 1) Antiviral actions of interferon: interferon-regulated cellular proteins and their surprisingly selective antiviral activities. Virolog)) 183: l-l 1.
Samuel, C. E. and Knutson, G. S. (1982) Mechanism of interferon action. Kinetics of decay of the antiviral state and protein phosphorylation in mouse fibroblasts treated with natural and cloned interferons. J. hiol. Chrm. 257: 1 1796-I 180 I.
Schindler. C., Fu. X-Y . . Improta. T., Aebersold, R. and Darnell, J. E. (1992a) Proteins of transcription factor ISGF-3: one gene encodes the 91 and 84 kDA ISGF-3 proteins that are activated by interferon-r. Proc. natn. Awd. Sci. U.S.A. 89: 783667839.
Schindler. C., Shuai. K., Prezioso. V. R. and Darnell, J. E. (1992b) Interferon-dependent tyrosine phosphorylation of a latent cytoplasmic transcription factor. Science 257: 809-8 15.
Schroder, H. C., Ugarkovic, D.. Merz, H., Kuchino, Y., Okamoto. T. and Muller. W. E. (1990) Protection of HeLa-T4 cells against human immunodeficiency virus (HIV) infection after stable transfection with HIV LTR 2’-5’-oligoadenylate svnthetase hybrid gene. FASEB J. 4: 3 124-2 130.
Sen. G. C. and Lengyel,-P. (1992) The interferons system. A bird’s eye view of its biochemistry. J. bio/. Chem. 267: 50 17-5020.

Mechanism of viral inhibition by interferons 441
Sen, G. C. and Ransohoff, R. M. (1993) Interferon-induced antiviral actions and their regulation. Adr. Virus Res. 42: 57-102.
Sen, G. C. and Sarkar. N. H. (1980) Effects of interferon on the production of murine mammary tumor virus by mammary tumor cells in culture. Virology 102: 431-443.
Shirazi, Y. and Pitha, P. M. (1993) Interferon alpha-mediated inhibition of human immundeficiency virus type 1 provirus synthesis in T-cells. Virology 193: 303-312.
Shuai, K., Horvath, C. M., Tsai Huang, L. H., Quereshi, S. A., Cowburn, D. and Darnell, J. E. (1994) Interferon activation of the transcription factor Stat91 involves dimerization through Sh2-phosphotyrosyl peptide interactions. C’eN 76: 821-828.
Soh, J., Donnelly, R. J., Mariano, T. M., Cook, J. R., Schwartz, B. and Pestka, S. (1993) Identification of a yeast artificial chromosome clone encoding an accessory factor for the human interferon y receptor: evidence for multiple accessory factors. Proc. natn. Acad. Sci. U.S.A. 90: 8737-8741.
Soh, J., Donnelly, R. J., Kotenko, S., Mariano, T. M., Cook, J. R., Wang, N., Emanuel, S. L., Schwartz, B., Miki, T. and Pestka, S. (1994) Identification and sequence of an accessory factor required for activation of the human interferon gamma receptor. Cell 76: in press.
Staeheli, P. (1990) Interferon-induced proteins and the antiviral state. Adc. Virus Res. 38: 147p200. Staeheli, P., Hailer, O., Boll, W., Lindenmann, J. and Weissmann, C. (1986) Mx protein: constitutive expression
in 3T3 cells transformed with cloned Mx cDNA confers selective resistance to influenza virus. Cell44: 1477158. Staeheli, P., Pitossi. F. and Pavlovic. J. (1993) Mx proteins: GTPases with antiviral activity. Trends Cell Biol.
3: 268-272. Stewart, W. E., II (1979) The Intwferon System Springer-Veriag, New York. Stinski, M. F., Thonsen, D. R. and Rodriguez, J. E. (1982) Synthesis of human cytomegalovirus-specified RNA
and proteins in interferon-treated cells at early times after infection. J. gen. Virol. 60: 261-270. Strube. W., Strube, M., Kroath, H., Jungwirth, C., Bodo, G. and Graft, T. (1982) Interferon inhibits
establishment of fibroblast infection with avian retroviruses. J. Inter/&on Res. 2: 37-49. Tur-Kaspa. R.. Teicher, L.. Laub, O., Itin, A., Dagan, D.. Bloom, B. R. and Shafritz, D. A. (1990) Alpha
interferon suppresses hepatitis B virus enhancer activity and reduces viral gene transcription. J. Viral. 64: 1821-1824.
Ulker, N. and Samuel, C. E. (1985) Mechanism of interferon action: inhibition of vesicular stomatitis virus replication in human amnion U cells by cloned human q-interferon. II. Effect of viral macromolecular synthesis. J. hiol. Chem. 260: 4324-4330.
Upton, C., Mossman, K. and McFadden, G. (1992) Encoding of a homolog of the IFN-7 receptor by myxoma virus. Science 258: 136991372.
Uze, G., Lutfalla, G. and Gresser, I. (1990) Genetic transfer of a functional human interferon alpha receptor into mouse cells: cloning and expression of its cDNA. Cell 60: 225-234.
Uze, G., Lutfalla. G., Bandu. M. T., Proudhon, D. and Mogensen, K. E. (1992) Behaviour of a cloned murine alpha beta receptor expressed in homospecific or heterospecific background. Proc. natn. Acud. Sci. b’.S.il. 89: 4774-4778.
Vassef, A.. Beaud, G.. Paucker, K. and Lengyel, P. (1973) Interferon assay based on the inhibition of double-stranded reovirus RNA accumulation in mouse L cells. J. gen. Viral. 19: 81-87.
Veals, S. A., Schindler, C., Leonard, D.. Fu, X-Y., Aebersold, R. and Darnell. J. E. (1992) Subunit of an alpha-interferon-responsive transcription factor is related to interferon regulatory factor and Myb families of DNA-binding proteins. Molec. Cell. Biol. 12: 3315-3324.
Vieira, P., de Waal-Malefyt, R., Dang, M. N.. Johnson, K. E., Kasteiein, R., Fiorentino. D. F.. deVries, J. E.. Roncarolo, M. G.. Mosmann. T. R. and Moore, W. (1991) Isolation and expression of human cytokine synthesis inhibitory factor cDNA clones: homology to Epstein-Barr virus open reading frame BCRFI. Proc,. nutn. Aud. Sci. U.S.A. 88: 117221176.
Watling, D., Serafinowska, H. T., Reese, C. B. and Kerr, I. M. (1985) Analogue inhibitor of 2-5A action: effect on the interferon-mediated inhibition of encephalomyocarditis virus replication. EMBO J. 4: 431-436.
Watson, J. C.. Chang, H. W. and Jacobs. B. L. (1991) Characterization of a vaccinia virus encoded double-stranded RNA binding protein that may be involved in the inhibition of the double stranded RNA dependent protein kinase. Virology 185: 2066216.
Whitaker-Dowling, P. and Youngner, J. S. (1983) Vaccinia rescue of VSV from interferon-induced resistance: reversal of translation block and inhibition of protein kinase activity. Virolog), 131: 128-136.
Whitaker-Dowling, P. and Youngner, J. S. (1984) Characterization of a specific kinase inhibitory factor produced by vaccinia virus which inhibits the interferon-induced protein kinase. Virolog!, 137: 171-181.
Whitaker-Dowling, P. and Youngner. J. S. (1986) Vaccinia-mediated rescue ofencephalomyocarditis virus from the inhibitory effects of interferon. Virology, 152: 50-57.
Whitaker-Dowling, P. and Younger, J. S. (1987) Antiviral effects ofinterferon in different virus-host cell systems. In: Mechunisms of’htet@on Action. 1st edn, pp. 83-98, Pfeffer, L. M. (ed.) CRC Press, Inc., Boca Raton.
Wiebe, M. E. and Joklik, W. K. (1975) The mechanism of inhibition of reovirus replication by interferon, Virology, 66: 229-240.
Wilcox, D. K., Whitaker-Dowling. P. A.. Youngner. J. S. and Widnell, C. C. (1983) Interferon treatment inhibits pinocytosis. Molec. Cell. Biol. 3: 1533-1536.
Williams, B. R. G. (1991) Signal transduction and transcriptional regulation of interferon-r-stimulated genes. J. Interferon Res. 11: 2077213.

442 S. Landolfo et al
Williams, B. R. G., Golgher, R. R., Brown, R. E., Gilbert, C. S. and Kerr, 1. M. (1979) Natural occurrence of 2-5A in interferon-treated EMC virus-infected L cells. Nature 282: 5822586.
Yamamoto, K., Yamaguchi, N. and Oda, K. (1975) Mechanism of interferon-induced inhibition of early simian virus 40 (SV40) functions. Virology 68: 58-70.
Yan, C. and Tamm, I. (1990) Protein factors that bind to the murine 2’.5’-oligoadenylate synthetase ME-12 gene 5’ upstream regulatory region. J. biol. Gem. 265: 20188-20194.
Yan, C. and Tamm, I. (1991) Molecular cloning and characterization of interferon cc/p response element binding factorsofthemurine(2’-5’)oligoadenylatesynthetase ME-12gene. Proc. mtn. Acad. Sci. U.S.A. 88: 144-148.
Yap, W. H., Teo, T. S., McCoy, E. and Tan, Y. H. (1986a) Rapid and transient rise in diacylglycerol concentration in Daudi cells exposed to interferon. Proc. natn. Acad. Sci. U.S.A. 83: 7765-7769.
Yap, W. H., Teo, T. S. and Tam, Y. H. (1986b) An early event in the interferon-induced transmembrane signalhng process. Science 234: 3555357.
Zens, W., Degen, H. J., Bernekow, A., Gelderblom, H. and Jungwirth, C. (1989) Two interferon sensitive steps in the replication cycle of rous sarcoma virus. Virology 171: 535-542.
Zhou, A., Hassel, B. A. and Silverman, R. H. (1993) Expression cloning of 2-SA-dependent RNAase: a uniquely regulated mediator of interferon action. Cell 72: 7533765.
Zurcher, T., Pavlovic, J. and Staeheli, P. (1992) Nuclear localization of mouse Mxl protein is necessary for inhibition of influenza virus. J. Virol. 66: 5059-5066.
Copyright © 2022 FDOKUMEN




















