
Liposomal and Viral Vectors for Gene Therapy of the Central Nervous System
13
Current Drug Targets - CNS & Neurological Disorders, 2005, 4, 453-465 453 Liposomal and Viral Vectors for Gene Therapy of the Central Nervous System Maria C. Pedroso de Lima* ,a,b M. Teresa Girão da Cruz a,b , Ana L.C. Cardoso a,b , Sérgio Simões b,c and Luís Pereira de Almeida b,c a Department of Biochemistry, Faculty of Sciences and Technology, University of Coimbra, Apartado 3126, 3001- 401 Coimbra, Portugal b Center for Neuroscience and Cell Biology, University of Coimbra, 3004-517 Coimbra, Portugal c Laboratory of Pharmaceutical Technology, Faculty of Pharmacy, University of Coimbra, 3000-295 Coimbra, Portugal Abstract: Due to the presence of the blood-brain barrier, the central nervous system (CNS) is not easily accessible to systemically delivered macromolecules with therapeutic activity such as growth factors, cytokines or enzymes. Therefore, the expression of exogenously administered genes in the brain has been proposed for a wide variety of inherited and acquired diseases of the CNS, for which classical pharmacotherapy is unavailable or not easily applicable. Gene therapy to the CNS has been the target of a great number of studies aiming at finding a viable therapeutic strategy for the treatment of neurological disorders. This approach has already been used as a promising tool for brain protection and repair from neuronal insults and degeneration in several animal models, and is currently being applied in clinical trials. The choice of an appropriate vector system for transferring the desired gene into the affected brain area is an important issue for developing a safe and efficient gene therapy approach for the CNS. In this review, we focus on the various types of vectors that have been used for gene delivery into the CNS. Particular emphasis is given to their mode of preparation, biological activity, safety and in vivo behavior. Examples illustrating the potential of both viral and non-viral vectors in therapeutic applications to brain disorders are provided. In addition, the use of lentiviral vectors for in vivo modeling of genetic disorders of the CNS is discussed. Keywords: Gene therapy, gene delivery, non-viral vectors, liposomes, viral vectors, lentivirus, CNS, brain. 1. INTRODUCTION therapeutic molecule in the brain, upon transfer of genes coding for protective molecules. Typically, the goal is to replace or increase the production of an endogenous brain protein that is dysfunctional or scarce (not only diffusible secreted molecules but also non-secreted molecules). Another strategy is to provide an entirely new property to target cells, such as the delivery of suicide genes into brain tumors or to provide therapy for dominant diseases by reducing the expression of defective genes, using antisense, RNA interference or ribozyme technologies. Gene therapy can be defined as the introduction of nucleic acids into cells to modify the course of a medical condition or disease [1]. Initially proposed for the treatment of monogenic diseases [1], gene therapy is now recognized to be a “novel form of drug delivery”, offering a multitude of different strategies for the treatment of both inherited and acquired diseases [2]. During the last years, gene transfer techniques evolved from application in molecular and cellular biology into a broad field of experiments in mammals, including non- human primates and clinical trials. The recent success of gene therapy for severe combined immunodeficiency (SCID) [3-5] suggests that its more widespread use to CNS disorders can also become a reality in the near future. Nevertheless, the brain is a complex, differentiated structure, in terms of cell and neuronal circuitry, protected by the skull, which restricts its direct access. The blood brain barrier (BBB) limits systemic access into the brain, and the post-mitotic, non-dividing nature of most neuronal cells limits the delivery of nucleic acids to the cell nucleus. Finally, the need to deliver therapeutic molecules into specific neuronal populations, within target regions, and in appropriate therapeutic doses, is a further rate-limiting issue to the clinical implementation of gene therapy for neurological disorders. In this context, the development of adequate gene delivery systems is required, this being one of the major challenges in the gene therapy research field. Delivery of therapeutic genes into the central nervous system (CNS) can potentially protect against neuronal insults and degeneration, by expression of neurotrophic factors, anti-oxidant or anti-apoptotic molecules. Therefore, gene therapy appears as a valid promising alternative to conventional therapeutic approaches, allowing the development of strategies aiming at locally producing a Currently, there are two main approaches for gene transfer into the CNS, the in vivo and ex vivo strategies. In the ex vivo approach, neuronal or non-neuronal cells, either from the host or from other sources, are genetically engineered in *Address correspondence to the author at the Department of Biochemistry, Faculty of Sciences and Technology, University of Coimbra, Apartado 3126, 3001-401 Coimbra, Portugal; Tel: + 351-239 820 190; Fax: +351-239 853 607; E-mail: [email protected] 1568-007X/05 $50.00+.00 © 2005 Bentham Science Publishers Ltd.
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Liposomal and Viral Vectors for Gene Therapy of the Central Nervous System

Current Drug Targets - CNS & Neurological Disorders, 2005, 4, 453-465 453
Liposomal and Viral Vectors for Gene Therapy of the Central NervousSystem
Maria C. Pedroso de Lima*,a,b M. Teresa Girão da Cruza,b, Ana L.C. Cardosoa,b,Sérgio Simõesb,c and Luís Pereira de Almeidab,c
aDepartment of Biochemistry, Faculty of Sciences and Technology, University of Coimbra, Apartado 3126, 3001-401 Coimbra, PortugalbCenter for Neuroscience and Cell Biology, University of Coimbra, 3004-517 Coimbra, PortugalcLaboratory of Pharmaceutical Technology, Faculty of Pharmacy, University of Coimbra, 3000-295 Coimbra,Portugal
Abstract: Due to the presence of the blood-brain barrier, the central nervous system (CNS) is not easily accessibleto systemically delivered macromolecules with therapeutic activity such as growth factors, cytokines or enzymes.Therefore, the expression of exogenously administered genes in the brain has been proposed for a wide variety ofinherited and acquired diseases of the CNS, for which classical pharmacotherapy is unavailable or not easilyapplicable. Gene therapy to the CNS has been the target of a great number of studies aiming at finding a viabletherapeutic strategy for the treatment of neurological disorders. This approach has already been used as apromising tool for brain protection and repair from neuronal insults and degeneration in several animal models,and is currently being applied in clinical trials.
The choice of an appropriate vector system for transferring the desired gene into the affected brain area is animportant issue for developing a safe and efficient gene therapy approach for the CNS. In this review, we focus onthe various types of vectors that have been used for gene delivery into the CNS. Particular emphasis is given totheir mode of preparation, biological activity, safety and in vivo behavior. Examples illustrating the potential ofboth viral and non-viral vectors in therapeutic applications to brain disorders are provided. In addition, the use oflentiviral vectors for in vivo modeling of genetic disorders of the CNS is discussed.
Keywords: Gene therapy, gene delivery, non-viral vectors, liposomes, viral vectors, lentivirus, CNS, brain.
1. INTRODUCTION therapeutic molecule in the brain, upon transfer of genescoding for protective molecules. Typically, the goal is toreplace or increase the production of an endogenous brainprotein that is dysfunctional or scarce (not only diffusiblesecreted molecules but also non-secreted molecules). Anotherstrategy is to provide an entirely new property to target cells,such as the delivery of suicide genes into brain tumors or toprovide therapy for dominant diseases by reducing theexpression of defective genes, using antisense, RNAinterference or ribozyme technologies.
Gene therapy can be defined as the introduction ofnucleic acids into cells to modify the course of a medicalcondition or disease [1]. Initially proposed for the treatmentof monogenic diseases [1], gene therapy is now recognizedto be a “novel form of drug delivery”, offering a multitudeof different strategies for the treatment of both inherited andacquired diseases [2].
During the last years, gene transfer techniques evolvedfrom application in molecular and cellular biology into abroad field of experiments in mammals, including non-human primates and clinical trials. The recent success ofgene therapy for severe combined immunodeficiency (SCID)[3-5] suggests that its more widespread use to CNSdisorders can also become a reality in the near future.
Nevertheless, the brain is a complex, differentiatedstructure, in terms of cell and neuronal circuitry, protectedby the skull, which restricts its direct access. The bloodbrain barrier (BBB) limits systemic access into the brain,and the post-mitotic, non-dividing nature of most neuronalcells limits the delivery of nucleic acids to the cell nucleus.Finally, the need to deliver therapeutic molecules intospecific neuronal populations, within target regions, and inappropriate therapeutic doses, is a further rate-limiting issueto the clinical implementation of gene therapy forneurological disorders. In this context, the development ofadequate gene delivery systems is required, this being one ofthe major challenges in the gene therapy research field.
Delivery of therapeutic genes into the central nervoussystem (CNS) can potentially protect against neuronalinsults and degeneration, by expression of neurotrophicfactors, anti-oxidant or anti-apoptotic molecules. Therefore,gene therapy appears as a valid promising alternative toconventional therapeutic approaches, allowing thedevelopment of strategies aiming at locally producing a
Currently, there are two main approaches for gene transferinto the CNS, the in vivo and ex vivo strategies. In the exvivo approach, neuronal or non-neuronal cells, either fromthe host or from other sources, are genetically engineered in
*Address correspondence to the author at the Department ofBiochemistry, Faculty of Sciences and Technology, University of Coimbra,Apartado 3126, 3001-401 Coimbra, Portugal; Tel: + 351-239 820 190; Fax:+351-239 853 607; E-mail: [email protected]
1568-007X/05 $50.00+.00 © 2005 Bentham Science Publishers Ltd.
454 Current Drug Targets - CNS & Neurological Disorders, 2005, Vol. 4, No. 4 de Lima et al.
vitro to produce a therapeutic protein, and subsequentlytransplanted into the host. Various cell types from skinfibroblasts and Schwann cells to modified cell lines havebeen used in CNS transplantation [6]. Nevertheless, celllines are prone to tumor formation, and primary cells aredifficult to produce in sufficient amounts and with adequateprotein expression levels. Stem cells, particularly embryonicstem cells, can be expanded and engineered to produce thetransgene of interest and are therefore a promising source ofcells for transplantation. A variant of ex-vivo gene therapyinvolves the use of cells encapsulated into a semi-permeablemembrane that allows diffusion not only of therapeuticmolecules and metabolites out of the membrane, but also ofnutrients and oxygen for life support of the cells within thedevice [6,7]. Therefore, even cells from non-human sourcescan be implanted into the host, because the membrane willconfer protection from the immune system. The in vivoapproach involves the direct insertion of genetic materialinto the target cells, either by systemic delivery or by in situinjection into the brain tissue.
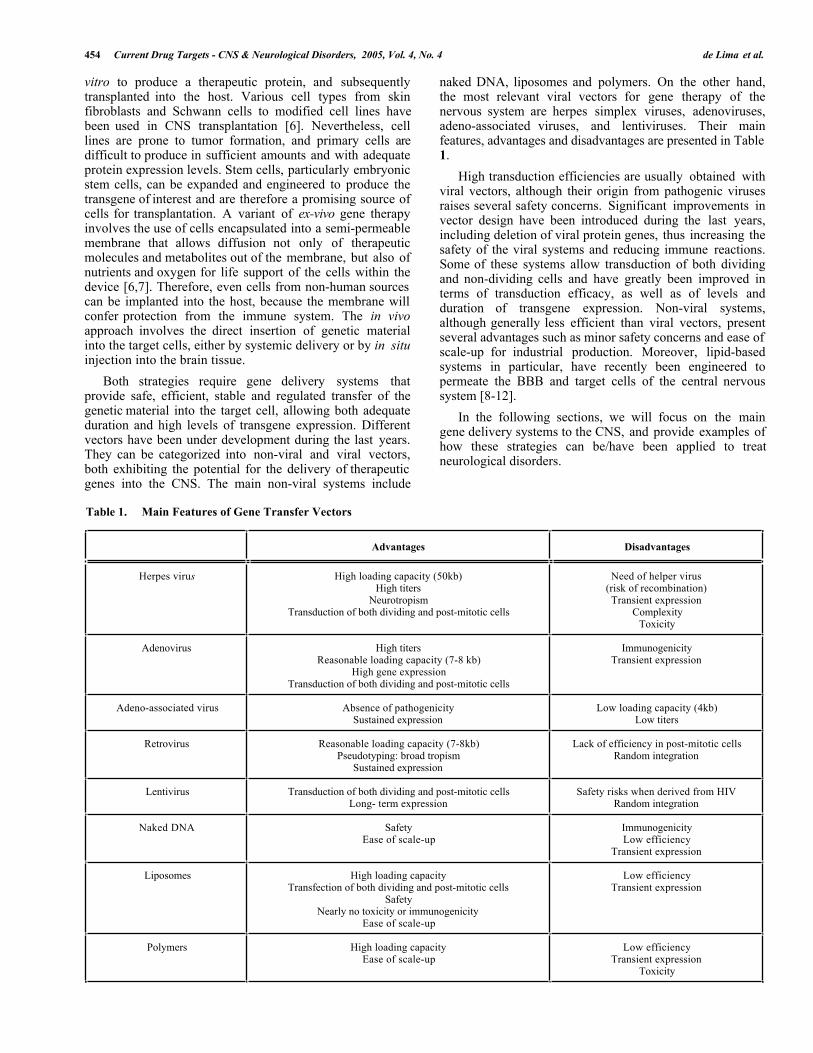
naked DNA, liposomes and polymers. On the other hand,the most relevant viral vectors for gene therapy of thenervous system are herpes simplex viruses, adenoviruses,adeno-associated viruses, and lentiviruses. Their mainfeatures, advantages and disadvantages are presented in Table1.
High transduction efficiencies are usually obtained withviral vectors, although their origin from pathogenic virusesraises several safety concerns. Significant improvements invector design have been introduced during the last years,including deletion of viral protein genes, thus increasing thesafety of the viral systems and reducing immune reactions.Some of these systems allow transduction of both dividingand non-dividing cells and have greatly been improved interms of transduction efficacy, as well as of levels andduration of transgene expression. Non-viral systems,although generally less efficient than viral vectors, presentseveral advantages such as minor safety concerns and ease ofscale-up for industrial production. Moreover, lipid-basedsystems in particular, have recently been engineered topermeate the BBB and target cells of the central nervoussystem [8-12].
Both strategies require gene delivery systems thatprovide safe, efficient, stable and regulated transfer of thegenetic material into the target cell, allowing both adequateduration and high levels of transgene expression. Differentvectors have been under development during the last years.They can be categorized into non-viral and viral vectors,both exhibiting the potential for the delivery of therapeuticgenes into the CNS. The main non-viral systems include
In the following sections, we will focus on the maingene delivery systems to the CNS, and provide examples ofhow these strategies can be/have been applied to treatneurological disorders.
Table 1. Main Features of Gene Transfer Vectors
Advantages Disadvantages
Herpes virus High loading capacity (50kb)High titers
NeurotropismTransduction of both dividing and post-mitotic cells
Need of helper virus(risk of recombination)Transient expression
ComplexityToxicity
Adenovirus High titersReasonable loading capacity (7-8 kb)
High gene expressionTransduction of both dividing and post-mitotic cells
ImmunogenicityTransient expression
Adeno-associated virus Absence of pathogenicitySustained expression
Low loading capacity (4kb)Low titers
Retrovirus Reasonable loading capacity (7-8kb)Pseudotyping: broad tropism
Sustained expression
Lack of efficiency in post-mitotic cellsRandom integration
Lentivirus Transduction of both dividing and post-mitotic cellsLong- term expression
Safety risks when derived from HIVRandom integration
Naked DNA SafetyEase of scale-up
ImmunogenicityLow efficiency
Transient expression
Liposomes High loading capacityTransfection of both dividing and post-mitotic cells
SafetyNearly no toxicity or immunogenicity
Ease of scale-up
Low efficiencyTransient expression
Polymers High loading capacityEase of scale-up
Low efficiencyTransient expression
Toxicity

Liposomal and Viral Vectors for Gene Therapy Current Drug Targets - CNS & Neurological Disorders, 2005, Vol. 4, No. 4 455
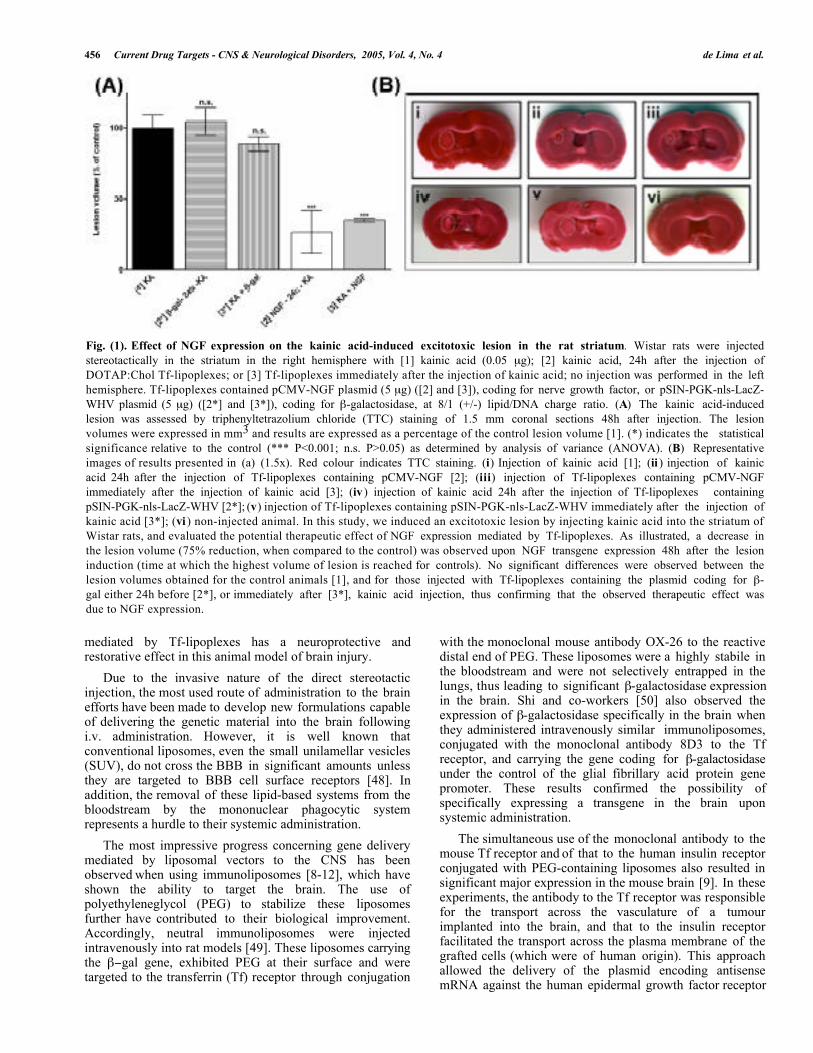
SYSTEMS FOR GENE DELIVERY TO THE CNS Among non-viral vectors, lipid-based systems, includinglipoplexes, have been the most widely used for gene therapy.Lipoplexes are formed upon electrostatic interactions ofplasmid DNA with cationic liposomes, resulting in aheterogeneous mixture of particles [27-31], which enter thecells by endocytosis [32-36]. Lipoplexes have severalattractive features as vectors for gene transfer: (1) they arenon-immunogenic and non-toxic at therapeutic doses; (2)they can transfect post-mitotic, non-dividing cells, includingneurons; (3) they can be used to deliver multiple genes ofany type of nucleic acids (linear or super-coiled); (4) they arerelatively simple to prepare and can be administered into thebody by several different routes; (5) the persistingextrachromosomal state of delivered plasmids reduces thepossibility that transfected cells would undergo neoplastictransformation. Although some lipid formulations fromcationic liposomes to non-liposomal formulations such asFugene™ were successfully employed to transfect neuronalcells in vitro [37-40], or even organotypic cultures of brainslices [41], the in vivo application of these systems has beenusually characterized by a relatively low efficiency of genetransfer to the CNS. However, several efforts have beenmade to overcome this disadvantage. Imaoka and co-workers[42] stereotactically injected lipoplexes containing thesynthetic cationic lipid dioctadecylamidoglycyl spermineand the gene coding for tyrosine hydroxylase (TH) into thestriatum of Parkinson’s disease (PD) animal models, andverified a significant behavioral recovery of the animals. Zouand co-workers [43] also showed that intraventricularinjection of DC-Chol containing lipoplexes, carrying thegene coding for nerve growth factor (NGF), resulted inreduction of the damage caused by traumatic brain injury,namely in the cholinergic neurons. More recently, Hagiharaand co-workers [44] successfully employed an anionicliposomal formulation, HVJ-AVE, whose membrane mimicsthe HIV envelope enriched with fusogenic proteins of thehemagglutinating virus of Japan. Upon stereotactic injectionof the HVJ-AVE/LacZ complexes coding for β-galactosidase,the authors observed transfection not only of cells close tothe injection site, but also in an area as far as 15 mm rostraland caudal. Hayashi and co-workers [45] also used an HJV-liposome complex to deliver a hepatocyte growth factor(HGF, a neurotrophic and anti-apoptotic factor) into thesubarachnoid space, via intrathecal injection through thecisterna magna. Positive staining was achieved in the cortex,hippocampus and choroids plexus of gerbils, andtransfection of the HGF gene was more effective than that ofrecombinant HGF in preventing neuronal cell death after anischemia-induced lesion. In our laboratory, we have exploredthe potential of transferrin-associated cationic liposome/DNAcomplexes (Tf-lipoplexes) to promote gene delivery toneuronal cells. Following demonstration of the feasibility ofTf-lipoplexes to mediate efficient transfection of the β-galactosidase reporter gene both in vitro [46] and in vivo[47], we evaluated their potential to deliver a therapeuticgene. We presented evidence of the successful delivery andexpression of the NGF encoding gene in the rodent brainupon intrastriatal injection [47]. Our results indicate that Tf-lipoplexes allow a significant enhancement of transfectionactivity as compared to plain lipoplexes, and that Tf-lipoplex-mediated NGF transgene expression attenuates themorphological damages of the kainic acid-induced lesion(Fig. 1). These findings show that NGF gene transfer
2. Non-Viral VectorsThe simplest approach of gene delivery involves the
direct injection of naked DNA into the tissue, or itssystemic administration, which leads to expression andoccasional integration into the chromosomes of thetransfected cells. However, due to the fast degradation bynucleases, the levels of expression are usually low andlimited to a restricted area [13]. Bombardment of goldparticles coated with plasmid DNA (“bio-ballistic”) has beenused to improve the efficiency of DNA delivery, and hasprovided high levels of gene expression in glial cells andneurons [14,15]. Although gene delivery is observed only insuperficial layers of the injected tissue, the high velocityparticles can damage the neuronal tissue [16]. An alternativeapproach involves electroporation through application ofhigh-voltage electrical pulses to facilitate cellpermeabilization, following naked DNA injection.Nevertheless, due to high mortality of cells following high-voltage exposure, this procedure is difficult to be applied invivo, and its use is so far restricted to neuronal cells inculture [17-19].
Another method for gene delivery that has beenextensively studied involves the use of cationic polymericsystems (polyplexes). Cationic polymers (e.g. poly-lysine,spermidine, poly-ethylenimine) efficiently bind andcondense DNA, and can be injected intracerebrally [20-22].Although they show a high ability to diffuse from theinjection site, the duration of expression is relatively short.Recently, a new generation of biocompatible controlledrelease polymers, based on polyphosphoesters, has beenintroduced for nucleic acid delivery. These polymers arebiodegradable and can be designed for long-term DNArelease, while exhibiting low cytotoxicity and improvedsoft-tissue response as compared to other commonly usedpolymer-based carriers [23]. Poly-L-lysine has also beenused in modified iron oxide nanoparticles (IONP-PLL),aiming at improving their ability to bind DNA [24]. Theseparticles succeeded in transferring DNA through the BBB toglial cells without disrupting the barrier, although themechanisms involved in this process and in the IONP-PLL-mediated gene delivery to cells are not completely clear.
Another new approach for DNA delivery to the nervoussystem consists of multifunctional proteins constructed bycombining bioactive protein domains from different origins.The sum of these independent elements offers the promise ofsupplying condensed DNA, promoting cell binding andinternalization, endosome disruption and nuclear targetingactivities to the referred vehicles, while lacking most of theinconvenience of viral material. Arís and co-workers [25]developed a non-viral vector based on an engineered β-galactosidase protein, taking advantage of its nuclearlocalization motifs, by introducing an Arg-Gly-Asp (RGD)cell internalization peptide, and a poly-lysine tail with DNAcondensing properties, which rendered this protein conjugateefficient for gene delivery to cultured cells. This vector wasalso used in vivo to mediate gene delivery by intracerebralinjection to the intact and damaged post-natal rat brain,achieving widespread gene delivery without triggering aninflammatory reaction [26].
456 Current Drug Targets - CNS & Neurological Disorders, 2005, Vol. 4, No. 4 de Lima et al.
Fig. (1). Effect of NGF expression on the kainic acid-induced excitotoxic lesion in the rat striatum. Wistar rats were injectedstereotactically in the striatum in the right hemisphere with [1] kainic acid (0.05 µg); [2] kainic acid, 24h after the injection ofDOTAP:Chol Tf-lipoplexes; or [3] Tf-lipoplexes immediately after the injection of kainic acid; no injection was performed in the lefthemisphere. Tf-lipoplexes contained pCMV-NGF plasmid (5 µg) ([2] and [3]), coding for nerve growth factor, or pSIN-PGK-nls-LacZ-WHV plasmid (5 µg) ([2*] and [3*]), coding for β-galactosidase, at 8/1 (+/-) lipid/DNA charge ratio. (A) The kainic acid-inducedlesion was assessed by triphenyltetrazolium chloride (TTC) staining of 1.5 mm coronal sections 48h after injection. The lesionvolumes were expressed in mm3 and results are expressed as a percentage of the control lesion volume [1]. (*) indicates the statisticalsignificance relative to the control (*** P<0.001; n.s. P>0.05) as determined by analysis of variance (ANOVA). (B) Representativeimages of results presented in (a) (1.5x). Red colour indicates TTC staining. (i) Injection of kainic acid [1]; (ii ) injection of kainicacid 24h after the injection of Tf-lipoplexes containing pCMV-NGF [2]; (iii) injection of Tf-lipoplexes containing pCMV-NGFimmediately after the injection of kainic acid [3]; (iv ) injection of kainic acid 24h after the injection of Tf-lipoplexes containingpSIN-PGK-nls-LacZ-WHV [2*]; (v) injection of Tf-lipoplexes containing pSIN-PGK-nls-LacZ-WHV immediately after the injection ofkainic acid [3*]; (vi ) non-injected animal. In this study, we induced an excitotoxic lesion by injecting kainic acid into the striatum ofWistar rats, and evaluated the potential therapeutic effect of NGF expression mediated by Tf-lipoplexes. As illustrated, a decrease inthe lesion volume (75% reduction, when compared to the control) was observed upon NGF transgene expression 48h after the lesioninduction (time at which the highest volume of lesion is reached for controls). No significant differences were observed between thelesion volumes obtained for the control animals [1], and for those injected with Tf-lipoplexes containing the plasmid coding for β-gal either 24h before [2*], or immediately after [3*], kainic acid injection, thus confirming that the observed therapeutic effect wasdue to NGF expression.
mediated by Tf-lipoplexes has a neuroprotective andrestorative effect in this animal model of brain injury.
with the monoclonal mouse antibody OX-26 to the reactivedistal end of PEG. These liposomes were a highly stabile inthe bloodstream and were not selectively entrapped in thelungs, thus leading to significant β-galactosidase expressionin the brain. Shi and co-workers [50] also observed theexpression of β-galactosidase specifically in the brain whenthey administered intravenously similar immunoliposomes,conjugated with the monoclonal antibody 8D3 to the Tfreceptor, and carrying the gene coding for β-galactosidaseunder the control of the glial fibrillary acid protein genepromoter. These results confirmed the possibility ofspecifically expressing a transgene in the brain uponsystemic administration.
Due to the invasive nature of the direct stereotacticinjection, the most used route of administration to the brainefforts have been made to develop new formulations capableof delivering the genetic material into the brain followingi.v. administration. However, it is well known thatconventional liposomes, even the small unilamellar vesicles(SUV), do not cross the BBB in significant amounts unlessthey are targeted to BBB cell surface receptors [48]. Inaddition, the removal of these lipid-based systems from thebloodstream by the mononuclear phagocytic systemrepresents a hurdle to their systemic administration.
The simultaneous use of the monoclonal antibody to themouse Tf receptor and of that to the human insulin receptorconjugated with PEG-containing liposomes also resulted insignificant major expression in the mouse brain [9]. In theseexperiments, the antibody to the Tf receptor was responsiblefor the transport across the vasculature of a tumourimplanted into the brain, and that to the insulin receptorfacilitated the transport across the plasma membrane of thegrafted cells (which were of human origin). This approachallowed the delivery of the plasmid encoding antisensemRNA against the human epidermal growth factor receptor
The most impressive progress concerning gene deliverymediated by liposomal vectors to the CNS has beenobserved when using immunoliposomes [8-12], which haveshown the ability to target the brain. The use ofpolyethyleneglycol (PEG) to stabilize these liposomesfurther have contributed to their biological improvement.Accordingly, neutral immunoliposomes were injectedintravenously into rat models [49]. These liposomes carryingthe β−gal gene, exhibited PEG at their surface and weretargeted to the transferrin (Tf) receptor through conjugation

Liposomal and Viral Vectors for Gene Therapy Current Drug Targets - CNS & Neurological Disorders, 2005, Vol. 4, No. 4 457
(HEGFR) gene into the brain tumor, which resulted in a100% increase of the mouse lifespan when compared tocontrol animals. Zhang and co-workers [10] also presentedevidence for widespread β-galactosidase expression in theprimate brain after i.v. injection of pegylatedimmunoliposomes coupled to the monoclonal antibody tothe human insulin receptor. More recently, the authors alsodemonstrated the ability of similar liposomes coupled to themonoclonal antibody to the Tf receptor and containing thegene coding for TH, to normalize striatal TH activity afteri.v. delivery into lesioned rats (whose TH activity was 98%depleted), and to reverse the induced motor impairment [12].Overall, these findings represent major breakthroughs in thedevelopment of non-viral systems for gene therapy to theCNS.
non-lytic, state of latency [62], which can be used forsustained expression of transgenes. A protective effect ofHSV-mediated neurotrophin gene transfer in cisplatin-induced neuropathy has recently been shown [63].3.2. Adenoviral-Derived Vectors
Adenoviruses represent a widely used group of genetherapy vectors. For gene transfer purposes, the vectors aregenerally made replication incompetent by deletion of theearly gene 1 (E1A). Further deletions increase the spaceavailable for the transgene insertion up to 7 to 8 Kb.Packaging cell lines are generally used for vector production.Very high titers, of up to 1010 cfu/ml (colony forming unitsper milliliter) can be achieved, which is one of the mainadvantages of this type of vector over other viral vectors.Besides this aspect, adenoviruses are able to transduce non-dividing cells and the transgene is expressed at high levels.Nevertheless, the genetic material remains episomal, therebybeing transiently expressed and not being duplicated duringcell replication. Moreover, the adenoviral E2 protein causesimmune and inflammatory reactions, which althoughpotentially useful in cancer therapy, represents a problem formost other therapeutic targets. Recently, “gutless” adenoviralvectors, which do not carry genes encoding viral proteins,have become available, thus decreasing the inflammatoryresponse to this class of vectors [64-68]. Nevertheless,inflammation is not suppressed because the viral particle byitself causes an inflammatory reaction [69].
3. Viral VectorsViruses are small but complex parasites that must use
host cellular machinery in order to replicate. Throughmutation and natural selection during the course ofevolution, viruses became highly adapted and efficacious todeliver their own genome to host cells. Gene therapyapproaches using viral vectors take advantage of the viralability/“know how” to deliver therapeutic genes to cells,while eliminating the genes implicated in the replication orthe pathogenicity of wild type viruses.
Viral vectors can be divided into recombinant anddefective viral vectors depending on the remaining fractionof the viral genome [51]. Initially, most vectors belonged tothe recombinant group. By homologous recombination, oneor more genes necessary for viral replication were replaced bythe transgene, preserving a substantial fraction of the viralgenome (particularly in adenoviral vectors). Nowadays, mostvectors belong to the defective group (also designated as“gutless”), and do not contain genes coding for viralproteins. The remaining viral sequences in the vectors arecis-acting elements required for viral particle packaging andassembly, integration into the genome and transcriptionaland post-transcriptional processing. Finally, an internalexpression cassette with a mammalian promoter and the geneof interest are integrated into the vector [52].
Adenoviruses were first used for CNS gene delivery in1993 [70-74]. The fact that wild-type viruses cause onlymild respiratory illness in humans, contributed to the overallidea that these were rather safe vectors. Unfortunately, therecent death of a patient, Jesse Gelsinger, during a phase 1clinical trial using an adenoviral-derived vector, clearlydemonstrated that the immune response to the vector issomehow unpredictable. The cause of death is probablyrelated to an immune reaction. Although any immunereaction in the CNS is likely to be milder as compared withperipheral administration due to the purportedimmunoprivileged condition of the CNS, this incidentstrongly reinforces the idea that before initiating a clinicaltrial, exhaustive preclinical studies must be performed toprovide a careful assessment of the safety issues.3.1. Herpes Simplex-Derived Vectors3.3. Adeno-Associated Viral Vectors (AAV Vectors) Direct in vivo gene delivery mediated by viral vectors was
performed for the first time with Herpes simplex virus(HSV)-derived vectors [53,54]. Herpes simplex are doublestranded 150 kb DNA viruses encoding 70 to 80 genes, andare able to transduce a wide range of both dividing and non-dividing cell populations. The infection is common inhumans causing primarily cold sores, and occasionallyencephalitis and other severe conditions, being either lytic orlatent and persisting for years [55]. In Herpes vectors, thetransgene remains episomal after transduction, which leadsto transient expression. The main advantage of Herpessimplex-based vectors is their high loading capacity, 30 to50 kb, the highest among viral vectors [56]. In addition,HSV infects both neurons and glial cells, although it showsgreater efficiency in transducing neurons [57-59], aninteresting property for gene therapy of the nervous system.In neurons, the virus is transported retrogradely alongneurites to the cell body [60,61], and maintains a life-long,
AAV vectors are very promising vectors since nopathology associated with the virus infection has ever beenreported [75]. AAV are very small (around 20 nm) with anicosahedral shape, surrounded by spikes. A single DNAstrand constitutes the AAV genome, the virus needing ahelper virus to complete its replication cycle. AAV infectsboth dividing and non-dividing cells, including neurons ormuscle cells and the derived AAV vectors allow integrationand sustained expression of transgenes without causingtoxicity or immune response. Nevertheless, their small sizerestricts use to the delivery of small transgenes of up to 5Kb [76]. The wild-type virus specifically integrates in aparticular site in chromosome 19, a property that is lostupon engineering of the vector.
AAV vectors were first used for CNS gene therapy in1994 [77,78] and since then, several studies demonstratedthat they are promising gene therapy vectors. In an elegant

458 Current Drug Targets - CNS & Neurological Disorders, 2005, Vol. 4, No. 4 de Lima et al.
study, AAV-mediated delivery of the genes encoding humanTH and GTP cyclohydrolase 1 (GTPCH1) into the striatumof dopamine deficient mice restored feeding behavior,partially improved locomotor activity and prevented lethality[79]. In a similar approach, by delivering the genes encodingenzymes involved in dopamine synthesis (tyrosinehydroxylase, aromatic-L-amino-acid decarboxylase, and GTPCH1), AAV vectors mediated dopamine expression in thestriatum, resulting in behavioral recovery of 6-hydroxydopamine-lesioned parkinsonian rats [80]. In analternative approach, using a rat model of PD, glial cell line-derived growth factor (GDNF) gene transfer promotedfunctional regeneration in the lesioned nigrostriatal system[81]. More recently, GDNF delivered by AAV was shown toprovide neuroanatomical and behavioral protection in the 3-nitropropionic acid rat model of Huntington’s disease [82].A Phase 1 clinical trial with AAV vectors has recently beeninitiated. In this trial, the vectors were introduced into thesubthalamic nucleus, an area that is overactive in PD and issometimes surgically removed for treatment of late stagedisease. Local expression of the transgene glutamic aciddecarboxylase is expected to increase production of γ -aminobutyric acid, locally inhibiting neuronal activity, inorder to control the hyperkinetic movement of PD.
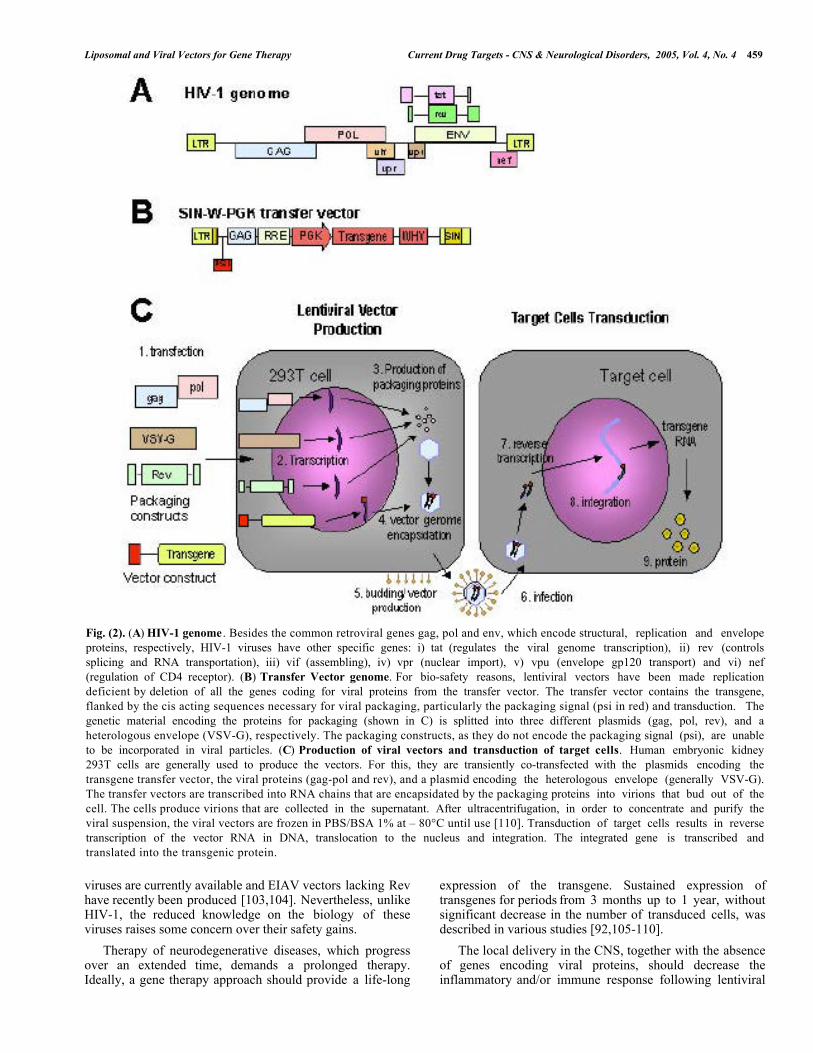
encountered by other vectors. The lentivirus pre-integrationcomplex allows docking to the nuclear membrane andtranslocation through the nuclear pore in an energydependent fashion, due to the presence of nuclear localizationsignals in several viral proteins [62,93,94]. Lentiviralvectors were shown to be particularly efficient for CNSdelivery. Integration of the genetic material allows asustained, long-term expression, without the need forrepeated injection of the vectors, which could induce ahumoral response to the viral antigens [95]. The vector has alarge cloning capacity (9 kb), which allows the transfer ofmost transgenes of interest. The particularly devastatingnature of the disease caused by HIV-1 imposes safetymeasures in vector design. Deletion of the viral genes thatare not essential for gene delivery is part of the standardprocedure for development of viral vectors (Fig. 2). The factthat the vectors do not carry any genes encoding viralproteins, which could trigger a cellular immune response,contributes, together with chromosomal integration, to thelong-term transgene expression [95].3.4.1. Design of a Biosafe Lentiviral Vector
A fundamental aspect essential for vector safety is toavoid the spread of the virus by replication. For thispurpose, genes encoding viral proteins were deleted from thetransfer vector and the genetic material was splitted intodifferent plasmids encoding the virion packaging proteins(gag, pol), a heterologous envelope (VSV-G), rev protein(controls splicing and RNA transport), and the transgeneexpression cassette, respectively (Fig. 2). The packagingplasmids contain the genes encoding all viral proteins intrans, with deletion of vpu, env, nef, vif and vpr [96], andmore importantly, the packaging signal and adjacentsequences. Absence of the cis acting sequences in thepackaging constructs blocks packaging, reverse transcriptionand integration of the transcripts derived from the packagingplasmid and ultimately blocks replication of the producedviral vectors. The wild-type HIV-1 envelope gene env hasbeen deleted and replaced by a gene coding for theheterologous envelope protein G from the vesicularstomatitis virus (VSV-G), which is expressed from aseparate plasmid. This new envelope increases the vectorphysical stability, thus facilitating the concentration stepsand ultimately, the production of high virus titers [95].Finally, the transfer vector contains the transgenedownstream of a constitutive promoter with the cis actingsequences necessary for packaging, reverse transcription andintegration [97].
AAV vectors have also been used in models ofamyotrophic lateral sclerosis (ALS). Bcl-2 encoding AAVvectors increased motoneuron survival and neuromuscularfunction in transgenic ALS mice [83]. Also in a knockoutmouse for mucopolysaccharidosis type VII, AAV genedelivery rescued the phenotype with a significant correctionof brain lesions [84]. Furthermore, AAV vectors have shownpromising effects in epilepsy models. Accordingly, AAV-mediated expression of the inhibitory neuroactive peptidegalanin, coupled to fibronectin, attenuated in vivo focalseizure sensitivity and prevented kainic acid-induced hilarcell death [85]. Moreover, anticonvulsant andantiepileptogenic effects have been described upon AAV-mediated neuropeptide Y expression in the rat hippocampus[86].
The technology of gene silencing by RNA interferencehas been explored with AAV vectors. AAV vectors encodingshort hairpin RNAs were shown to be able to silence mutantataxin-1 expression in a transgenic mouse model ofspinocerebellar ataxia type 1 (SCA1) [87]. The vectorsmediated inhibition of polyglutamine-inducedneurodegeneration suggesting that a similar strategy could beused in other polyQ (poly glutamine) disorders.
Besides their main role as therapeutic carriers, AAVvectors have been used to generate in vivo experimentalmodels of disease, particularly of Alzheimer’s,polyglutamine, Parkinson’s and epileptic diseases [88-91].
Further biosafety developments included the productionof self-inactivating viral vectors. Deletion of part of the3’LTR leads to a vector, which after reverse transcription hasan inactive 5’ LTR promoter and therefore, can no longer betranscribed into a functional vector. This avoids oncogeneactivation and vector rescue after integration into the targetgenome and simultaneously eliminates LTR interferencewith the transgene promoter, thereby increasing itsexpression [98,99]. Human embryonic kidney 293T cells aregenerally used to produce the vectors. Another line ofresearch has been focused on developing lentiviral vectorsfrom either equine (EIAV) [100,101] or feline lentivirus(FIV) [102], which are not pathogenic in the humanorganism. Minimal versions of these non-primate derived
3.4. Lentiviral VectorsThe HIV-1 virus, a member of the lentiviral group of
retroviruses, is able to infect and transduce macrophages andlymphocytes, both being terminally differentiated, non-dividing cells. This unique property has been used byBlomer and co-workers [92] to produce HIV-1 derived vectorin order to transduce non-dividing cells. They succeeded intransducing both growth-arrested cells in vitro, as well asneurons in vivo, overcoming the problems that had been
Liposomal and Viral Vectors for Gene Therapy Current Drug Targets - CNS & Neurological Disorders, 2005, Vol. 4, No. 4 459
Fig. (2). (A) HIV-1 genome . Besides the common retroviral genes gag, pol and env, which encode structural, replication and envelopeproteins, respectively, HIV-1 viruses have other specific genes: i) tat (regulates the viral genome transcription), ii) rev (controlssplicing and RNA transportation), iii) vif (assembling), iv) vpr (nuclear import), v) vpu (envelope gp120 transport) and vi) nef(regulation of CD4 receptor). (B) Transfer Vector genome. For bio-safety reasons, lentiviral vectors have been made replicationdeficient by deletion of all the genes coding for viral proteins from the transfer vector. The transfer vector contains the transgene,flanked by the cis acting sequences necessary for viral packaging, particularly the packaging signal (psi in red) and transduction. Thegenetic material encoding the proteins for packaging (shown in C) is splitted into three different plasmids (gag, pol, rev), and aheterologous envelope (VSV-G), respectively. The packaging constructs, as they do not encode the packaging signal (psi), are unableto be incorporated in viral particles. (C) Production of viral vectors and transduction of target cells. Human embryonic kidney293T cells are generally used to produce the vectors. For this, they are transiently co-transfected with the plasmids encoding thetransgene transfer vector, the viral proteins (gag-pol and rev), and a plasmid encoding the heterologous envelope (generally VSV-G).The transfer vectors are transcribed into RNA chains that are encapsidated by the packaging proteins into virions that bud out of thecell. The cells produce virions that are collected in the supernatant. After ultracentrifugation, in order to concentrate and purify theviral suspension, the viral vectors are frozen in PBS/BSA 1% at – 80°C until use [110]. Transduction of target cells results in reversetranscription of the vector RNA in DNA, translocation to the nucleus and integration. The integrated gene is transcribed andtranslated into the transgenic protein.
viruses are currently available and EIAV vectors lacking Revhave recently been produced [103,104]. Nevertheless, unlikeHIV-1, the reduced knowledge on the biology of theseviruses raises some concern over their safety gains.
expression of the transgene. Sustained expression oftransgenes for periods from 3 months up to 1 year, withoutsignificant decrease in the number of transduced cells, wasdescribed in various studies [92,105-110].
Therapy of neurodegenerative diseases, which progressover an extended time, demands a prolonged therapy.Ideally, a gene therapy approach should provide a life-long
The local delivery in the CNS, together with the absenceof genes encoding viral proteins, should decrease theinflammatory and/or immune response following lentiviral

460 Current Drug Targets - CNS & Neurological Disorders, 2005, Vol. 4, No. 4 de Lima et al.
vector administration. Kordower and co-workers [111]described minor CD45 and CD8 staining in 2 out of 18monkeys injected with lentiviral vectors. In rats, infiltratinglymphocytes and macrophages were observed 2 weeks aftersaline or vector injection, but no CD4, OX8 or ED-1positive cells were detected for 6 to 12 weeks post-injection[92]. Deglon and Aebischer have reported some tissuedamage and perivascular cuffing at the needle tract level, butonly when a very high number of viral particles wereinjected into the rat striatum [112]. Moreover, transductionof the rat striatum with lentiviral vectors coding for β-galactosidase did not interfere with subsequent transductionmediated by these vectors coding for green fluorescentprotein (GFP), 6 weeks later. These studies suggested thatno immune reaction, which would prevent repeatedtransduction, developed after the first administration of thelentiviral vectors.
This may interact with the displacement of the pre-integration complex, allowing translocation across thenuclear pore or acting as a docking site for the cellularmachinery involved in intracellular trafficking and nucleartransport [122].Enhancement of Gene Expression
Enhancement of transgene expression delivered bylentiviral vectors was obtained upon introduction of theWoodchuck post-transcriptional regulatory element (WPRE)from the hepatitis virus [125]. Apparently, the WPREstimulates gene expression by facilitating RNA exportand/or processing at the level of 3’ cleavage andpolyadenylation. Moreover, it has been suggested that itcould assist the generation of RNA-protein complexes thatwould protect the newly synthesized transcripts fromdegradation within the nucleus, resulting in an increasedgene expression [108,125].3.4.2. Improvement of the Vector DesignRegulation - Tet and Cre-Lox SystemsBesides vector biosafety, other strategies aiming at
improving the in vivo performance of lentiviral vectors,particularly their tropism, nuclear translocation, geneexpression, regulation and reproducibility of production havebeen explored.
An important feature for successful gene therapy,particularly when working with vectors that integrate theirgenetic cargo into host chromosomes, is to be able tocontrol gene expression. Regulated systems are necessary toallow turning the transgene expression off in case of sideeffects, or to control transgene expression levels andtherefore achieve optimal therapeutic effect. Among thedifferent regulated systems that have been described, the tetsystem is the best characterized [2,126,127] and was firstintroduced in lentiviral vectors by Kafri and co-workers[128]. This system regulates expression of GFP, both invitro and in the rat brain, under control of doxycycline. Inthe absence of doxycycline, GFP is expressed but uponaddition of doxycycline in the drinking water, transgeneexpression is blocked. Another regulated system, the cre-loxsystem, was also associated with the lentiviral vector,allowing conditional but irreversible gene modification[129].
TropismThe original envelope glycoproteins from HIV-1
assemble a fragile envelope that restricts the infection mainlyto CD4+ cells. In order to adapt the vector to gene therapy,lentiviral vectors have been pseudotyped with a wide rangeof heterologous envelope proteins of different origins:Mokola virus (classified as rabies envelope), Moloneymurine leukemia virus, gibbon ape leukemia virus, Ebolavirus and (VSV-G) [101,113-116].
The envelope glycoprotein from the VSV-G, used in theoriginal paper of Naldini and colaborators [95], is still themost widely used, because it confers high physical stabilityto the viral particles allowing concentration byultracentrifugation. Furthermore, VSV-G pseudotypedvectors have a broad host range and are able to transduceboth mammalian and non-mammalian cells [117].
Reproducibility of Viral ProductionThe stringent requirements of clinical applications
demand a consistent and reproducible production of viralvectors. Packaging cell lines are more suited for this purposethan transient transfection. Nevertheless, the development ofpackaging cell lines was somewhat difficult for HIV-derivedvectors due to the toxicity of VSV-G envelope and of somelentiviral proteins (vpr, gag and tat) [129]. Nevertheless,different studies have reported the successful development ofvarious packaging cell lines [130-133]. Farson and co-workers [134] generated a stable packaging cell line carryingthe coding sequences of gag, pol, tat and rev under controlof a tet promoter. The authors claim that followingtransfection with the transfer vectors, production of vectorsis sustained for several days, resulting in titers identical tothose obtained by transient transfection.
Retrograde vector transport had been described for Herpessimplex and adenoviruses but only recently for lentiviruses[118-120]. It has been shown that equine lentiviral vectorspseudotyped with the rabies glycoprotein are transportedretrogradely from neurites to the cell body, particularly fromthe rat striatum to substantia nigra and, from thegastrocnemius and facial muscles to spinal motoneurons.This interesting feature could allow non-invasiveneurological gene therapy to be established, particularly inmotoneuron disease, chronic pain and spinal injury[104,121].Nuclear Translocation
A significant increase in transduction efficiency (3- to 5-fold in vitro) was observed when the central polypurine tract(cPPT), a 118 bp sequence, from HIV-1 pol gene wasincluded in lentiviral transfer vectors [122-124]. This effectwas attributed to an increased nuclear translocation of theviral genome. Although the mechanism is still unclear, ithas been suggested that cPPT acts as a second origin of plusstrand synthesis, introducing a discontinuity in viral DNA.
3.4.3. Therapeutic Applications of Lentiviral GeneTransfer for the CNS
Blömer and co-workers [135] were the first to describe atherapeutic effect using lentiviral vectors in a model ofneurodegenerative disease. The authors showed that braininjection of lentiviral vectors coding for either the Bcl-xLanti-apoptotic factor, or hNGF neurotrophin rescues 73 and

Liposomal and Viral Vectors for Gene Therapy Current Drug Targets - CNS & Neurological Disorders, 2005, Vol. 4, No. 4 461
53% septal cholinergic neurons compared to a 38% controlsurvival, thus demonstrating a neuroprotective effect of thesegenes in the fimbria-fornix rat model of Alzheimer’s disease.
strategies of gene therapy for PD takes advantage of thestrong neuroprotective effect of GDNF in dopaminergicneurons [144]. Lentiviral vectors-mediated delivery ofGDNF in rodent and monkey brains allowed significantneuropathological and behavioral ameliorations in models ofPD [108,145-147]. This strategy is particularly suited forearly stages of the disease when a large number of neuronscan still be rescued with trophic support.
In lysosomal storage diseases, the various defectiveenzymes lead to metabolite accumulation within thelysosomes, progressive neurological pathologies andpremature death. In type VII β-glucuronidase deficient mice,a mouse model of mucopolysaccharidose, lentiviral vectorshave been used to supply the missing enzyme and to reversethe phenotype [136]. Similarly, multiple lentiviral injectionswere used to replace the missing arylsulphatase in the AS2-/-mouse model of metachromatic leukodystrophy (MLD),which resulted in reversal of learning disabilities andcorrection of neuropathology [137]. More recently,transplantation of autologous hematopoietic stem cellstransduced with lentiviral vectors encoding thearylsulphatase A gene restored enzyme activity in thehematopoietic system of the MLD mouse model, andprevented the development of behavioral andneuropathological abnormalities, which are typical of thedisease [138].
An alternative strategy based on the replacement of themissing striatal dopamine has shown potential for genetherapy of late-stage PD. Several studies using AAV inducedoverexpression of the enzymes involved in dopaminebiosynthesis by co-transduction with mixtures of vectorsseparately encoding each of the enzymes [80,148-150].Lentiviral vectors have a higher loading capability, whichallowed Azzouz and co-workers [151] to engineer equinelentiviral vectors simultaneously encoding the three rate-limiting enzymes involved in dopamine biosynthese TH,aromatic amino acid dopa decarboxylase , and GTP CH1, ina single multicistronic transcription unit. Stereotacticinjection of the vectors into the dopamine-denervatedstriatum of 6-hydroxydopamine (6-OHDA)-lesioned animalshas been shown to ensure sustained striatal expression ofeach enzyme and production of dopamine, resulting infunctional improvement in this rat model of PD.
A recessive mutation in the gene coding for the rodphotoreceptor cGMPP phosphodiesterase β (PDEβ) subunitleads to retinal degeneration. Lentiviral delivery of PDEβgene into the subretinal space of the retinal degenerationmouse model resulted in rescue of the photoreceptors [139],thus paving the way for a treatment of retinitis pigmentosa.Recently, delivery of ciliary neurotrophic factor (CNTF) vialentiviral vectors has been shown to protect axotomizedretinal ganglion cells for an extended period of time,suggesting that lentiviral vectors-mediated delivery of CNTFcould serve as a potential treatment for retinal disordersinvolving optic neuropathy and retinal ganglion cell injurysuch as in glaucoma [140].
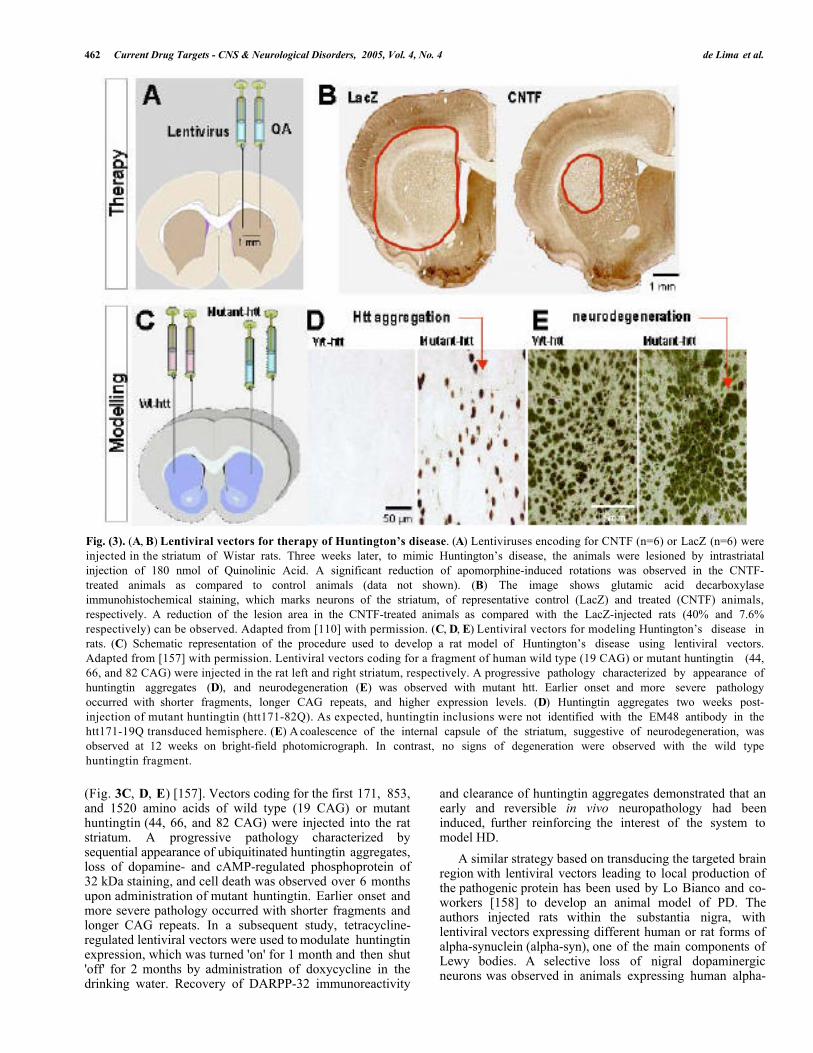
The genetic etiology of Huntington’s disease (HD), amember of the polyglutamine (polyQ) group ofneurodegenerative disorders, makes gene therapy a naturaltherapeutic approach for this disease. The first report on theuse of lentiviral vectors for gene therapy of HD was based onneuroprotection by the local striatal production of CNTF inthe quinolinic acid rat model of HD (Fig. 3A, B) [110].This strategy was extended by development of tetracycline(Tet)-regulated lentiviral vectors in order to investigate thedose-dependent neuroprotection. Neuroprotective effects wereassociated with the production of CNTF in the “on” state,while residual CNTF expression in the “off” state did notprotect against quinolinic acid toxicity, demonstrating thefeasibility of lentiviral-mediated tetracycline-regulated genetransfer in the brain [152]. Recently, the effects of CNTFwere also evaluated in a genetic model of HD, the YAC72transgenic mice, expressing full-length huntingtin with 72CAG repeats. CNTF expression reduced the behavioralactivity levels and the number of striatal dark cells ofYAC72 mice compared to controls. Moreover, in both WTand YAC72 mice, CNTF expression was demonstrated up to1 year after lentiviral injection [153].
The progressive loss of spinal cord motor neurons inALS leads to muscular atrophy, paralysis and death byasphyxiation 5 to 10 years after onset of the disease.Axotomy of the facial nerves in 4-month old BALB/c miceinduces the degeneration or atrophy of approximately 50% ofthe neurons. Hottinger and co-workers [141] have shown thatlentiviral delivery of GDNF was able to rescue the totality ofthe motor neurons in this model of ALS. Nevertheless, theuse of the same local lentiviral-mediated delivery of (GDNF)in a transgenic model of ALS, the SOD1(G93A) transgenicmice protected facial nerve motoneurons but not spinalmotoneurons [142]. More recently, in another study usingthis model, Azzouz and co-workers [143] have shown thatinjection of vascular endothelial growth factor (VEGF)-expressing equine lentiviral vectors into various musclesdelayed onset, slowed disease progression and increased lifeexpectancy without causing toxic side effects.
The dominant underlying cause of polyglutaminediseases is associated with a gain of function of thepolyglutamine expanded proteins, which suggests thatknocking down the pathogenic gene should result in abeneficial effect. Lentiviral vectors encoding short hairpinRNAs have recently been shown to efficiently mediate genesilencing in vitro and in vivo, particularly in the rodentbrain, suggesting that this strategy may be useful for therapyof neurodegenerative diseases [154-156].
Parkinson’s disease (PD) is a pathology that isparticularly amenable to gene therapy approaches due to thesmall discrete size of the degenerating region, the substantiapars compacta (SNc). Degeneration of dopaminergic neuronswithin this region results in depletion of theneurotransmitter dopamine (DA) in the striatum and leads torigidity, resting tremor and motor function impairment.Injection of an efficient vector with stereotactic guidanceshould allow a precise local delivery, enabling transductionof most neurons within CNS. One of the most promising
Since lentiviral vectors have a reasonable loadingcapability allowing cloning of a mutant gene; they constitutea natural choice for modeling genetic disorders. To ourknowledge, HD was the first neurodegenerative disorder forwhich a lentiviral-mediated genetic model was developed
462 Current Drug Targets - CNS & Neurological Disorders, 2005, Vol. 4, No. 4 de Lima et al.
Fig. (3). (A, B) Lentiviral vectors for therapy of Huntington’s disease. (A) Lentiviruses encoding for CNTF (n=6) or LacZ (n=6) wereinjected in the striatum of Wistar rats. Three weeks later, to mimic Huntington’s disease, the animals were lesioned by intrastriatalinjection of 180 nmol of Quinolinic Acid. A significant reduction of apomorphine-induced rotations was observed in the CNTF-treated animals as compared to control animals (data not shown). (B) The image shows glutamic acid decarboxylaseimmunohistochemical staining, which marks neurons of the striatum, of representative control (LacZ) and treated (CNTF) animals,respectively. A reduction of the lesion area in the CNTF-treated animals as compared with the LacZ-injected rats (40% and 7.6%respectively) can be observed. Adapted from [110] with permission. (C, D, E) Lentiviral vectors for modeling Huntington’s disease inrats. (C) Schematic representation of the procedure used to develop a rat model of Huntington’s disease using lentiviral vectors.Adapted from [157] with permission. Lentiviral vectors coding for a fragment of human wild type (19 CAG) or mutant huntingtin (44,66, and 82 CAG) were injected in the rat left and right striatum, respectively. A progressive pathology characterized by appearance ofhuntingtin aggregates (D), and neurodegeneration (E) was observed with mutant htt. Earlier onset and more severe pathologyoccurred with shorter fragments, longer CAG repeats, and higher expression levels. (D) Huntingtin aggregates two weeks post-injection of mutant huntingtin (htt171-82Q). As expected, huntingtin inclusions were not identified with the EM48 antibody in thehtt171-19Q transduced hemisphere. (E) A coalescence of the internal capsule of the striatum, suggestive of neurodegeneration, wasobserved at 12 weeks on bright-field photomicrograph. In contrast, no signs of degeneration were observed with the wild typehuntingtin fragment.
(Fig. 3C, D, E) [157]. Vectors coding for the first 171, 853,and 1520 amino acids of wild type (19 CAG) or mutanthuntingtin (44, 66, and 82 CAG) were injected into the ratstriatum. A progressive pathology characterized bysequential appearance of ubiquitinated huntingtin aggregates,loss of dopamine- and cAMP-regulated phosphoprotein of32 kDa staining, and cell death was observed over 6 monthsupon administration of mutant huntingtin. Earlier onset andmore severe pathology occurred with shorter fragments andlonger CAG repeats. In a subsequent study, tetracycline-regulated lentiviral vectors were used to modulate huntingtinexpression, which was turned 'on' for 1 month and then shut'off' for 2 months by administration of doxycycline in thedrinking water. Recovery of DARPP-32 immunoreactivity
and clearance of huntingtin aggregates demonstrated that anearly and reversible in vivo neuropathology had beeninduced, further reinforcing the interest of the system tomodel HD.
A similar strategy based on transducing the targeted brainregion with lentiviral vectors leading to local production ofthe pathogenic protein has been used by Lo Bianco and co-workers [158] to develop an animal model of PD. Theauthors injected rats within the substantia nigra, withlentiviral vectors expressing different human or rat forms ofalpha-synuclein (alpha-syn), one of the main components ofLewy bodies. A selective loss of nigral dopaminergicneurons was observed in animals expressing human alpha-

Liposomal and Viral Vectors for Gene Therapy Current Drug Targets - CNS & Neurological Disorders, 2005, Vol. 4, No. 4 463
syn, together with appearance of abundant alpha-syn-positiveinclusions and extensive neuritic pathology. Similarly, ratalpha-syn led to protein aggregation but without cell loss,suggesting that inclusions are not the primary cause of celldegeneration in PD. This viral-mediated genetic model ofPD recapitulates the essential neuropathological features ofPD [158]. In summary, lentiviral vector-based modelsrepresent a new valuable approach in biomedicine as theymay contribute to elucidate mechanisms of disease and allowscreening of candidate therapies.
MLD = Metachromatic leukodystrophyPDEβ = PhosphodiesteraseCNTF = Cilliary neurotrophic factorRGC = Retinal ganglion cellHD = Huntington’s disease
REFERENCES[1] Kay, M.A.; Liu, D.; Hoogerbrugge, P.M. Proc. Natl. Acad. Sci.
USA, 1997, 94, 12744.4. FUTURE CHALLENGES / PROSPECTS /CONCLUSION
[2] Blau, H.M.; Rossi, F.M. Proc. Natl. Acad. Sci. USA, 1999, 96, 797.[3] Cavazzana-Calvo, M.; Hacein-Bey, S.; de Saint Basile, S.; Gross,
F.; Yvon, E.; Nusbaum, P.; Selz, F.; Hue, C.; Certain, S.; Casanova,J.L.; Bousso, P.; Deist, F.L.; Fischer, A. Science, 2000, 288, 669.A large number of animal studies have been performed in
order to demonstrate the feasibility of gene therapy inmodels of neurodegeneration, trauma, pain, ischemia andbrain tumors. The therapeutic approaches used for CNSrecovery have involved four primary strategies: (1) todecrease the action of a mutant protein by antisensetechnology; (2) to replace a missing protein in recessiveconditions; (3) to promote neuronal survival with trophicfactors, anti-oxidative enzymes, heat shock proteins, andanti-apoptotic factors; and (4) to prevent brain tumor growthby activation of anti-cancer drugs, inhibition of tumor cellmigration, increase of the immune response and induction ofapoptosis. Before gene therapy to CNS disorders becomes areality, significant improvements need to be addressed,namely regarding efficacy and safety of gene deliverysystems and the regulation of gene expression being two ofthe major goals. Accordingly, the increasing knowledge ofgenomics and proteomics, and of cell and molecular biologyof neurological disorders, may help to generate animalmodels with an etiology and a phenotype similar to thehuman disease, which will allow a more efficient and safeapplicability of gene delivery systems to gene therapyclinical trials.
[4] Fischer, D.; Pavlidis, M.; Thanos, S. Invest. Ophthalmol. Vis. Sci.,2000, 41, 3943.
[5] Aiuti, A.; Slavin, S.; Aker, M.; Ficara, F.; Deola, S.; Mortellaro,A.; Morecki, S.; Andolfi, G.; Tabucchi, A.; Carlucci, F.;Marinello, E.; Cattaneo, F.; Vai, S.; Servida, P.; Miniero, R.;Roncarolo, M.G.; Bordignon, C. Science, 2002, 296, 2410.
[6] Tseng, J.L.; Aebischer, P. Prog. Brain Res., 2000, 127, 189.[7] Lysaght, M.J.; Aebischer, P. Sci. Am., 1999, 280, 76.[8] Geisert, E.E.; Delmar, N.A.; Owens, J.L.; Holmberg, E.G.
Neurosci. Lett., 1995, 184, 40.[9] Zhang, Y.; Zhu, C.; Pardridge, W.M. Mol. Ther., 2002, 6, 67.[10] Zhang, Y.; Schlachetzki, F.; Pardridge, W.M. Mol. Ther. , 2003, 7,
11.[11] Zhang, Y.; Boado, R.J.; Pardridge, W.M. J. Gene Med., 2003, 5,
157.[12] Zhang, Y.; Schlachetzki, F.; Zhang, Y.F.; Boado, R.J.; Pardridge,
W.M. Hum. Gene Ther., 2004, 15, 339.[13] Schwartz, B.; Benoist, C.; Abdallah, B.; Rangara, R.; Hassan, A.;
Scherman, D.; Demeneix, B.A. Gene Ther., 1996, 3, 405.[14] Jiao, S.; Cheng, L.; Wolff, J.A.; Yang, N.S. Biotechnology (N. Y. ) ,
1993, 11, 497.[15] Sato, H.; Hattori, S.; Kawamoto, S.; Kudoh, I.; Hayashi, A.;
Yamamoto, I.; Yoshinari, M.; Minami, M.; Kanno, H. Biochem.Biophys. Res. Commun. , 2000, 270, 163.
[16] Hui, K.M.; Sabapathy, T.K.; Oei, A.A.; Chia, T.F. J. Immunol.Meth., 1994, 171, 147.
[17] Teruel, M.N.; Blanpied, T.A.; Shen, K.; Augustine, G.J.; Meyer,T. J. Neurosci. Methods, 1999, 93, 37.
ABBREVIATIONS[18] Ghosh, C.; Song, W.; Lahiri, D.K. Mol. Biol. Rep., 2000, 27, 113.[19] Washbourne, P.; McAllister, A.K. Curr. Opin. Neurobiol., 2002,
12, 566.[20] Abdallah, B.; Hassan, A.; Benoist, C.; Goula, D.; Behr, J.P.;
Demeneix, B.A. Hum. Gene. Ther., 1996, 7, 1947.CNS = Central nervous systemIONP-PLL = Iron oxide nanoparticles [21] Goula, D.; Remy, J.S.; Erbacher, P.; Wasowicz, M.; Levi, G.;
Abdallah, B.; Demeneix, B.A. Gene. Ther., 1998, 5, 712.TH = Tyrosine hydroxylase [22] Lemkine, G.F.; Goula, D.; Becker, N.; Paleari, L.; Levi, G.;
Demeneix, B.A. J. Drug Target., 1999, 7, 305.PD = Parkinson’s disease [23] Li, Y.; Wang, J.; Lee, C.G.; Wang, C.Y.; Gao, S.J.; Tang, G.P.;Ma, Y.X.; Yu, H.; Mao, H.Q.; Leong, K.W.; Wang, S. GeneTher., 2004, 11, 109.NGF = Nerve growth factor
PEG = Polyethyleneglycol [24] Xiang, J.J.; Tang, J.Q.; Zhu, S.G.; Nie, X.M.; Lu, H.B.; Shen, S.R.;Li, X.L.; Tang, K.; Zhou, M.; Li, G.Y. J. Gene. Med. , 2003, 5,803.GTPCH1 = GTP cyclohydrolase 1
[25] Aris, A.; Feliu, J.X.; Knight, A.; Coutelle, C.; Villaverde, A.Biotechnol. Bioeng. , 2000, 68, 689.GDNF = Glial cell line-derived growth factor
[26] Peluffo, H.; Aris, A.; Acarin, L.; Gonzalez, B.; Villaverde, A.;Castellano, B. Hum. Gene. Ther., 2003, 14, 1215.ALS = Amyotrophic lateral sclerosis
[27] Gershon, H.; Ghirlando, R.; Guttman, S.B.; Minsky, A.Biochemistry , 1993, 32, 7143.
VSV-G = Vesicular stomatitis virusEIAV = Either equine lentivirus [28] Sternberg, B.; Sorgi, F.L.; Huang, L. FEBS Lett., 1994, 356, 361.
[29] Gustafsson, J.; Arvidson, G.; Karlsson, G.; Almgren, M. Biochim.Biophys. Acta, 1995, 1235, 305.FIV = Feline lentivirus
[30] Tomlinson, E.; Rolland, A.P. J. Control. Release , 1996, 39, 357.GFP = Green fluorescent protein [31] Radler, J.O.; Koltover, I.; Salditt, T.; Safinya, C.R. Science, 1997,275, 810.cPPT = Central polypurine tract [32] Zhou, X.; Huang, L. Biochim. Biophys. Acta , 1994, 1189, 195.
WPRE = Woodchuck post-transcriptional regulatory element
[33] Wrobel, I.; Collins, D. Biochim. Biophys. Acta , 1995, 1235, 296.

464 Current Drug Targets - CNS & Neurological Disorders, 2005, Vol. 4, No. 4 de Lima et al.
[34] Zabner, J.; Fasbender, A.J.; Moninger, T.; Poellinger, K.A.;Welsh, M.J. J. Biol. Chem., 1995, 270, 18997.
[71] Bajocchi, G.; Feldman, S.H.; Crystal, R.G.; Mastrangeli, A. Nat.Genet. , 1993, 3, 229.
[35] El Ouahabi, A.; Thiry, M.; Schiffmann, S.; Fuks, R.; Nguyen-Tran,H.; Ruysschaert, J.M.; Vandenbranden, M. J. Histochem.Cytochem., 1999, 47, 1159.
[72] Davidson, B.L.; Allen, E.D.; Kozarsky, K.F.; Wilson, J.M.;Roessler, B.J. Nat. Genet., 1993, 3, 219.
[73] Le Gal, L.S.; Robert, J.J.; Berrard, S.; Ridoux, V.; Stratford-Perricaudet, L.D.; Perricaudet, M.; Mallet, J. Science, 1993, 259,988.
[36] Friend, D.S.; Papahadjopoulos, D.; Debs, R.J. Biochim. Biophys.Acta, 1996, 1278, 41.
[37] Ajmani, P.S.; Hughes, J.A. Neurochem. Res., 1999, 24, 699. [74] Wu, N.; Ataai, M.M. Curr. Opin. Biotechnol., 2000, 11, 205.[38] Wiesenhofer, B.; Kaufmann, W.A.; Humpel, C. J. Neurosci.
Methods, 1999, 92, 145.[75] Blacklow, N.R.; Hoggan, M.D.; Rowe, W.P. J. Natl. Cancer Inst.,
1968, 40, 319.[39] Wiesenhofer, B.; Humpel, C. Exp. Neurol., 2000, 164, 38. [76] Carter, B.J. Contrib. Microbiol., 2000, 4, 85.[40] Wangerek, L.A.; Dahl, H.H.; Senden, T.J.; Carlin, J.B.; Jans,
D.A.; Dunstan, D.E.; Ioannou, P.A.; Williamson, R.; Forrest, S.M.J. Gene. Med., 2001, 3, 72.
[77] Kaplitt, M.G.; Leone, P.; Samulski, R.J.; Xiao, X.; Pfaff, D.W.;O'Malley, K.L.; During, M.J. Nat. Genet., 1994, 8, 148.
[78] Matsushita, T.; Elliger, S.; Elliger, C.; Podsakoff, G.; Villarreal, L.;Kurtzman, G.J.; Iwaki, Y.; Colosi, P. Gene. Ther., 1998, 5, 938.[41] Murray, K.D.; Mcquillin, A.; Stewart, L.; Etheridge, C.J.; Cooper,
R.G.; Miller, A.D.; Gurling, H.M. Gene. Ther., 1999, 6, 190. [79] Szczypka, M.S.; Mandel, R.J.; Donahue, B.A.; Snyder, R.O.; Leff,S.E.; Palmiter, R.D. Neuron, 1999, 22, 167.[42] Imaoka, T.; Date, I.; Ohmoto, T.; Nagatsu, T. Hum. Gene. Ther.,
1998, 9, 1093. [80] Shen, Y.; Muramatsu, S.I.; Ikeguchi, K.; Fujimoto, K.I.; Fan, D.S.;Ogawa, M.; Mizukami, H.; Urabe, M.; Kume, A.; Nagatsu, I.;Urano, F.; Suzuki, T.; Ichinose, H.; Nagatsu, T.; Monahan, J.;Nakano, I.; Ozawa, K. Hum. Gene. Ther., 2000, 11, 1509.
[43] Zou, L.L.; Huang, L.; Hayes, R.L.; Black, C.; Qiu, Y.H.; Perez-Polo, J.R.; Le, W.; Clifton, G.L.; Yang, K. Gene. Ther., 1999, 6,994.
[44] Hagihara, Y.; Saitoh, Y.; Kaneda, Y.; Kohmura, E.; Yoshimine, T.Gene. Ther., 2000, 7, 759.
[81] Kirik, D.; Rosenblad, C.; Bjorklund, A.; Mandel, R.J. J. Neurosci.,2000, 20, 4686.
[45] Hayashi, K.; Morishita, R.; Nakagami, H.; Yoshimura, S.; Hara,A.; Matsumoto, K.; Nakamura, T.; Ogihara, T.; Kaneda, Y.;Sakai, N. Gene. Ther., 2001, 8, 1167.
[82] McBride, J.L.; During, M.J.; Wuu, J.; Chen, E.Y.; Leurgans, S.E.;Kordower, J.H. Exp. Neurol., 2003, 181, 213.
[83] Azzouz, M.; Hottinger, A.; Paterna, J.C.; Zurn, A.D.; Aebischer,P.; Bueler, H. Hum. Mol. Genet., 2000, 9, 803.[46] da Cruz, M.T.; Simoes, S.; de Lima, M.C. Exp. Neurol., 2004, 187,
65. [84] Bosch, A.; Perret, E.; Desmaris, N.; Heard, J.M. Mol. Ther. , 2000,1, 63.[47] da Cruz, M.T.; Cardoso, A.L.C.; de Almeida, L.P.; Simoes, S.; de
Lima, M.C. Gene. Ther., 2005 (accepted for publication). [85] Haberman, R.P.; Samulski, R.J.; McCown, T.J. Nat. Med. , 2003, 9,1076.[48] Huwyler, J.; Wu, D.; Pardridge, W.M. Proc. Natl. Acad. Sci. USA,
1996, 93, 14164. [86] Richichi, C.; Lin, E.J.; Stefanin, D.; Colella, D.; Ravizza, T.;Grignaschi, G.; Veglianese, P.; Sperk, G.; During, M.J.; Vezzani,A. J. Neurosci., 2004, 24, 3051.
[49] Shi, N.Y.; Pardridge, W.M. Proc. Natl. Acad. Sci. USA, 2000, 97,7567.
[50] Shi, N.; Zhang, Y.; Zhu, C.; Boado, R.J.; Pardridge, W.M. Proc.Natl. Acad. Sci. USA, 2001, 98, 12754.
[87] Xia, H.; Mao, Q.; Eliason, S.L.; Harper, S.Q.; Martins, I.H.; Orr,H.T.; Paulson, H.L.; Yang, L.; Kotin, R.M.; Davidson, B.L. Nat.Med. , 2004, 10, 816.[51] Hermens, W.T.; Verhaagen, J. Prog. Neurobiol., 1998, 55, 399.
[52] Pfeifer, A.; Verma, I.M. Annu. Rev. Genomics Hum. Genet.,2001, 2, 177.
[88] Nishimura, I.; Uetsuki, T.; Dani, S.U.; Ohsawa, Y.; Saito, I.;Okamura, H.; Uchiyama, Y.; Yoshikawa, K. J. Neurosci., 1998,18, 2387.[53] Ho, D.Y.; Mocarski, E.S. Virology, 1988, 167, 279.
[54] Palella, T.D.; Hidaka, Y.; Silverman, L.J.; Levine, M.; Glorioso, J.;Kelley, W.N. Gene, 1989, 80, 137.
[89] Senut, M.C.; Suhr, S.T.; Kaspar, B.; Gage, F.H. J. Neurosci., 2000,20, 219.
[55] Costantini, L.C.; Bakowska, J.C.; Breakefield, X.O.; Isacson, O.Gene. Ther., 2000, 7, 93.
[90] Telfeian, A.E.; Federoff, H.J.; Leone, P.; During, M.J.;Williamson, A. Neurobiol. Dis., 2000, 7, 362.
[56] Walther, W.; Stein, U. Drugs, 2000, 60, 249. [91] Kirik, D.; Rosenblad, C.; Burger, C.; Lundberg, C.; Johansen, T.E.;Muzyczka, N.; Mandel, R.J.; Bjorklund, A. J. Neurosci., 2002, 22,2780.
[57] Andersen, J.K.; Frim, D.M.; Isacson, O.; Breakefield, X.O. CellMol. Neurobiol., 1993, 13, 503.
[58] During, M.J.; Naegele, J.R.; O'Malley, K.L.; Geller, A.I. Science,1994, 266, 1399.
[92] Blomer, U.; Naldini, L.; Kafri, T.; Trono, D.; Verma, I.M.; Gage,F.H. J. Virol., 1997, 71, 6641.
[59] Fink, D.J.; DeLuca, N.A.; Goins, W.F.; Glorioso, J.C. Annu. Rev.Neurosci., 1996, 19, 265.
[93] Bukrinsky, M.I.; Sharova, N.; Dempsey, M.P.; Stanwick, T.L.;Bukrinskaya, A.G.; Haggerty, S.; Stevenson, M. Proc. Natl. Acad.Sci. USA, 1992, 89, 6580.[60] Sodeik, B.; Ebersold, M.W.; Helenius, A. J. Cell Biol., 1997, 136,
1007. [94] Bukrinsky, M.I.; Haggerty, S.; Dempsey, M.P.; Sharova, N.;Adzhubel, A.; Spitz, L.; Lewis, P.; Goldfarb, D.; Emerman, M.;Stevenson, M. Nature, 1993, 365, 666.
[61] Bearer, E.L.; Schlief, M.L.; Breakefield, X.O.; Schuback, D.E.;Reese, T.S.; LaVail, J.H. Biol. Bull., 1999, 197, 257.
[62] Ballay, A.; Levrero, M.; Buendia, M.A.; Tiollais, P.; Perricaudet,M. EMBO J., 1985, 4, 3861.
[95] Naldini, L.; Blomer, U.; Gallay, P.; Ory, D.; Mulligan, R.; Gage,F.H.; Verma, I.M.; Trono, D. Science, 1996, 272, 263.
[63] Chattopadhyay, M.; Goss, J.; Wolfe, D.; Goins, W.C.; Huang, S.;Glorioso, J.C.; Mata, M.; Fink, D.J. Brain, 2004, 127, 929.
[96] Zufferey, R.; Nagy, D.; Mandel, R.J.; Naldini, L.; Trono, D. Nat.Biotechnol., 1997, 15, 871.
[64] Kochanek, S.; Clemens, P.R.; Mitani, K.; Chen, H.H.; Chan, S.;Caskey, C.T. Proc. Natl. Acad. Sci. USA, 1996, 93, 5731.
[97] Dull, T.; Zufferey, R.; Kelly, M.; Mandel, R.J.; Nguyen, M.;Trono, D.; Naldini, L. J. Virol., 1998, 72, 8463.
[65] Chen, H.H.; Mack, L.M.; Kelly, R.; Ontell, M.; Kochanek, S.;Clemens, P.R. Proc. Natl. Acad. Sci. USA, 1997, 94, 1645.
[98] Miyoshi, H.; Takahashi, M.; Gage, F.H.; Verma, I.M. Proc. Natl.Acad. Sci. USA, 1997, 94, 10319.
[66] Hardy, S.; Kitamura, M.; Harris-Stansil, T.; Dai, Y.; Phipps, M.L.J. Virol., 1997, 71, 1842.
[99] Zufferey, R.; Dull, T.; Mandel, R.J.; Bukovsky, A.; Quiroz, D.;Naldini, L.; Trono, D. J. Virol., 1998, 72, 9873.
[67] Morsy, M.A.; Gu, M.; Motzel, S.; Zhao, J.; Lin, J.; Su, Q.; Allen,H.; Franlin, L.; Parks, R.J.; Graham, F.L.; Kochanek, S.; Bett, A.J.;Caskey, C.T. Proc. Natl. Acad. Sci. USA, 1998, 95, 7866.
[100] Olsen, J.C. Gene. Ther., 1998, 5, 1481.[101] Mitrophanous, K.A.; Yoon, S.; Rohll, J.B.; Patil, D.; Wilkes, F.J.;
Kim, V.N.; Kingsman, S.M.; Kingsman, A.J.; Mazarakis, N.D.Gene. Ther., 1999, 6, 1808.[68] Schiedner, G.; Morral, N.; Parks, R.J.; Wu, Y.; Koopmans, S.C.;
Langston, C.; Graham, F.L.; Beaudet, A.L.; Kochanek, S. Nat.Genet. , 1998, 18, 180.
[102] Poeschla, E.M.; Wong-Staal, F.; Looney, D.J. Nat. Med., 1998, 4,354.
[69] Thomas, C.E.; Edwards, P.; Wickham, T.J.; Castro, M.G.;Lowenstein, P.R. J. Virol., 2002, 76, 3452.
[103] Johnston, J.C.; Gasmi, M.; Lim, L.E.; Elder, J.H.; Yee, J.K.; Jolly,D.J.; Campbell, K.P.; Davidson, B.L.; Sauter, S.L. J. Virol., 1999,73, 4991.[70] Akli, S.; Caillaud, C.; Vigne, E.; Stratford-Perricaudet, L.D.;
Poenaru, L.; Perricaudet, M.; Kahn, A.; Peschanski, M.R. Nat.Genet. , 1993, 3, 224.
[104] Mazarakis, N.D.; Azzouz, M.; Rohll, J.B.; Ellard, F.M.; Wilkes,F.J.; Olsen, A.L.; Carter, E.E.; Barber, R.D.; Baban, D.F.;

Liposomal and Viral Vectors for Gene Therapy Current Drug Targets - CNS & Neurological Disorders, 2005, Vol. 4, No. 4 465
Kingsman, S.M.; Kingsman, A.J.; O'Malley, K.; Mitrophanous,K.A. Hum. Mol. Genet., 2001, 10, 2109.
[133] Xu, K.; Ma, H.; McCown, T.J.; Verma, I.M.; Kafri, T. Mol. Ther. ,2001, 3, 97.
[105] Naldini, L.; Blomer, U.; Gage, F.H.; Trono, D.; Verma, I.M. Proc.Natl. Acad. Sci. USA, 1996, 93, 11382.
[134] Farson, D.; Witt, R.; McGuinness, R.; Dull, T.; Kelly, M.; Song, J.;Radeke, R.; Bukovsky, A.; Consiglio, A.; Naldini, L. Hum. Gene.Ther., 2001, 12, 981.[106] Naldini, L. Curr. Opin. Biotechnol., 1998, 9, 457.
[107] Kordower, J.H.; Bloch, J.; Ma, S.Y.; Chu, Y.; Palfi, S.; Roitberg,B.Z.; Emborg, M.; Hantraye, P.; Deglon, N.; Aebischer, P. Exp.Neurol., 1999, 160, 1.
[135] Blomer, U.; Kafri, T.; Randolph-Moore, L.; Verma, I.M.; Gage,F.H. Proc. Natl. Acad. Sci. USA, 1998, 95, 2603.
[136] Bosch, A.; Perret, E.; Desmaris, N.; Trono, D.; Heard, J.M. Hum.Gene. Ther., 2000, 11, 1139.[108] Deglon, N.; Tseng, J.L.; Bensadoun, J.C.; Zurn, A.D.; Arsenijevic,
Y.; Pereira, d.A.; Zufferey, R.; Trono, D.; Aebischer, P. Hum.Gene. Ther., 2000, 11, 179.
[137] Consiglio, A.; Quattrini, A.; Martino, S.; Bensadoun, J.C.; Dolcetta,D.; Trojani, A.; Benaglia, G.; Marchesini, S.; Cestari, V.; Oliverio,A.; Bordignon, C.; Naldini, L. Nat. Med., 2001, 7, 310.[109] Bjorklund, A.; Kirik, D.; Rosenblad, C.; Georgievska, B.;
Lundberg, C.; Mandel, R.J. Brain Res., 2000, 886, 82. [138] Biffi, A.; De Palma, M.; Quattrini, A.; Del Carro, U.; Amadio, S.;Visigalli, I.; Sessa, M.; Fasano, S.; Brambilla, R.; Marchesini, S.;Bordignon, C.; Naldini, L. J. Clin. Invest., 2004, 113, 1118.
[110] de Almeida, L.P.; Zala, D.; Aebischer, P.; Deglon, N. Neurobiol.Dis., 2001, 8, 433.
[111] Kordower, J.H.; Emborg, M.E.; Bloch, J.; Ma, S.Y.; Chu, Y.;Leventhal, L.; McBride, J.; Chen, E.Y.; Palfi, S.; Roitberg, B.Z.;Brown, W.D.; Holden, J.E.; Pyzalski, R.; Taylor, M.D.; Carvey, P.;Ling, Z.; Trono, D.; Hantraye, P.; Deglon, N.; Aebischer, P.Science, 2000, 290, 767.
[139] Koide, R.; Kobayashi, S.; Shimohata, T.; Ikeuchi, T.; Maruyama,M.; Saito, M.; Yamada, M.; Takahashi, H.; Tsuji, S. Hum. Mol.Genet. , 1999, 8, 2047.
[140] van Adel, B.A.; Kostic, C.; Deglon, N.; Ball, A.K.; Arsenijevic, Y.Hum. Gene. Ther., 2003, 14, 103.
[112] Deglon, N.; Aebischer, P. Curr. Top. Microbiol. Immunol., 2002,261, 191.
[141] Hottinger, A.F.; Azzouz, M.; Deglon, N.; Aebischer, P.; Zurn,A.D. J. Neurosci., 2000, 20, 5587.
[113] Mochizuki, H.; Schwartz, J.P.; Tanaka, K.; Brady, R.O.; Reiser, J.J. Virol., 1998, 72, 8873.
[142] Guillot, S.; Azzouz, M.; Deglon, N.; Zurn, A.; Aebischer, P.Neurobiol. Dis., 2004, 16, 139.
[114] Reiser, J. Gene. Ther., 2000, 7, 910. [143] Azzouz, M.; Ralph, G.S.; Storkebaum, E.; Walmsley, L.E.;Mitrophanous, K.A.; Kingsman, S.M.; Carmeliet, P.; Mazarakis,N.D. Nature, 2004, 429, 413.
[115] Stitz, J.; Buchholz, C.J.; Engelstadter, M.; Uckert, W.; Bloemer,U.; Schmitt, I.; Cichutek, K. Virology, 2000, 273, 16.
[116] Kobinger, G.P.; Weiner, D.J.; Yu, Q.C.; Wilson, J.M. Nat.Biotechnol., 2001, 19, 225.
[144] Lin, L.F.; Doherty, D.H.; Lile, J.D.; Bektesh, S.; Collins, F.Science, 1993, 260, 1130.
[117] Burns, J.C.; Friedmann, T.; Driever, W.; Burrascano, M.; Yee,J.K. Proc. Natl. Acad. Sci. USA, 1993, 90, 8033.
[145] Bensadoun, J.C.; Deglon, N.; Tseng, J.L.; Ridet, J.L.; Zurn, A.D.;Aebischer, P. Exp. Neurol., 2000, 164, 15.
[118] Wood, M.J.; Byrnes, A.P.; Kaplitt, M.G.; Pfaff, D.W.; Rabkin,S.D.; Charlton, H.M. Exp. Neurol., 1994, 130, 127.
[146] Georgievska, B.; Kirik, D.; Rosenblad, C.; Lundberg, C.;Bjorklund, A. Neuroreport, 2002, 13, 75.
[119] Ghadge, G.D.; Roos, R.P.; Kang, U.J.; Wollmann, R.; Fishman,P.S.; Kalynych, A.M.; Barr, E.; Leiden, J.M. Gene. Ther., 1995, 2,132.
[147] Azzouz, M.; Ralph, S.; Wong, L.F.; Day, D.; Askham, Z.; Barber,R.D.; Mitrophanous, K.A.; Kingsman, S.M.; Mazarakis, N.D.Neuroreport, 2004, 15, 985.
[120] Lilley, C.E.; Groutsi, F.; Han, Z.; Palmer, J.A.; Anderson, P.N.;Latchman, D.S.; Coffin, R.S. J. Virol., 2001, 75, 4343.
[148] Leff, S.E.; Spratt, S.K.; Snyder, R.O.; Mandel, R.J. Neuroscience,1999, 92, 185.
[121] Wong, L.F.; Azzouz, M.; Walmsley, L.E.; Askham, Z.; Wilkes,F.J.; Mitrophanous, K.A.; Kingsman, S.M.; Mazarakis, N.D. Mol.Ther., 2004, 9, 101.
[149] Bankiewicz, K.S.; Eberling, J.L.; Kohutnicka, M.; Jagust, W.;Pivirotto, P.; Bringas, J.; Cunningham, J.; Budinger, T.F.; Harvey-White, J. Exp. Neurol., 2000, 164, 2.
[122] Follenzi, A.; Ailles, L.E.; Bakovic, S.; Geuna, M.; Naldini, L. Nat.Genet. , 2000, 25, 217.
[150] Sanchez-Pernaute, R.; Harvey-White, J.; Cunningham, J.;Bankiewicz, K.S. Mol. Ther., 2001, 4, 324.
[123] Sirven, A.; Pflumio, F.; Zennou, V.; Titeux, M.; Vainchenker, W.;Coulombel, L.; Dubart-Kupperschmitt, A.; Charneau, P. Blood,2000, 96, 4103.
[151] Azzouz, M.; Martin-Rendon, E.; Barber, R.D.; Mitrophanous,K.A.; Carter, E.E.; Rohll, J.B.; Kingsman, S.M.; Kingsman, A.J.;Mazarakis, N.D. J. Neurosci., 2002, 22, 10302.
[124] Zennou, V.; Petit, C.; Guetard, D.; Nerhbass, U.; Montagnier, L.;Charneau, P. Cell, 2000, 101, 173.
[152] Regulier, E.; Pereira, d.A.; Sommer, B.; Aebischer, P.; Deglon, N.Hum. Gene. Ther., 2002, 13, 1981.
[125] Zufferey, R.; Donello, J.E.; Trono, D.; Hope, T.J. J. Virol., 1999,73, 2886.
[153] Zala, D.; Bensadoun, J.C.; Pereira, d.A.; Leavitt, B.R.; Gutekunst,C.A.; Aebischer, P.; Hayden, M.R.; Deglon, N. Exp. Neurol.,2004, 185, 26.[126] Gossen, M.; Freundlieb, S.; Bender, G.; Muller, G.; Hillen, W.;
Bujard, H. Science, 1995, 268, 1766. [154] Abbas-Terki, T.; Briand, P.A.; Donze, O.; Picard, D. Biol. Chem.,2002, 383, 1335.[127] Baron, U.; Gossen, M.; Bujard, H. Nucleic Acids Res., 1997, 25,
2723. [155] Rubinson, D.A.; Dillon, C.P.; Kwiatkowski, A.V.; Sievers, C.;Yang, L.; Kopinja, J.; Rooney, D.L.; Ihrig, M.M.; McManus, M.T.;Gertler, F.B.; Scott, M.L.; Van Parijs, L. Nat. Genet., 2003, 33,401.
[128] Kafri, T.; van Praag, H.; Gage, F.H.; Verma, I.M. Mol. Ther.,2000, 1, 516.
[129] Pfeifer, A.; Brandon, E.P.; Kootstra, N.; Gage, F.H.; Verma, I.M.Proc. Natl. Acad. Sci. USA, 2001, 98, 11450. [156] Van den, H.C.; Eggermont, K.; Nuttin, B.; Debyser, Z.;
Baekelandt, V. Hum. Gene. Ther., 2003, 14, 1799.[130] Yu, H.; Rabson, A.B.; Kaul, M.; Ron, Y.; Dougherty, J.P. J. Virol.,1996, 70, 4530. [157] de Almeida, L.P.; Ross, C.A.; Zala, D.; Aebischer, P.; Deglon, N.
J. Neurosci., 2002, 22, 3473.[131] Kafri, T.; van Praag, H.; Ouyang, L.; Gage, F.H.; Verma, I.M. J.Virol., 1999, 73, 576. [158] Lo, B.C.; Ridet, J.L.; Schneider, B.L.; Deglon, N.; Aebischer, P.
Proc. Natl. Acad. Sci. USA, 2002, 99, 10813.[132] Klages, N.; Zufferey, R.; Trono, D. Mol. Ther., 2000, 2, 170.




















