
LIGAND-BASED PHARMACOPHORE DETECTION AND SCREENING OF POTENTIAL GLITAZONES
38
TITLE: LIGAND-BASED PHARMACOPHORE DETECTION AND SCREENING OF POTENTIAL GLITAZONES Ritesh Agrawal*, Pratima Jain and S. N. Dikshit Department of Chemistry, Shrimant Madhavrao Scindia, Government Model Science College, Jhansi Road, Gwalior 474001, Madhya Pradesh, India; Email: [email protected]
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of LIGAND-BASED PHARMACOPHORE DETECTION AND SCREENING OF POTENTIAL GLITAZONES

TITLE:
LIGAND-BASED PHARMACOPHORE DETECTION AND SCREENING OF POTENTIAL GLITAZONES
Ritesh Agrawal*, Pratima Jain and S. N. Dikshit
Department of Chemistry, Shrimant Madhavrao Scindia, Government Model Science College, Jhansi Road, Gwalior 474001, Madhya Pradesh, India; Email: [email protected]

ABSTRACT
Three-dimensional pharmacophore hypothesis was built based on a set of knownProtein tyrosine Phosphatase 1B (PTP1B) agonists using PharmaGist program tounderstand the essential structural features for Protein Tyrosine Phosphatase1B (PTP1B) agonists. The various marketed or under development potentialglitazones have been opted to build a pharmacophore model. PharmaGist webbased program is employed for pharmacophore development. Four pointspharmacophore with the hydrogen bond acceptor (A), hydrophobic group (H),Spatial Features and aromatic rings (R) have been considered to developpharmacophoric features by PharmaGist program. The best pharmacophore modelhaving the Score 30.547, which has been opted to screen on ZincPharmerdatabase to derive the novel potential antidiabetic ligands. The bestpharmacophore having various Pharmacophore features, including GeneralFeatures 6, Spatial Features 6, Aromatic 2, Hydrophobic 1, Donors 1, andAcceptors 2. The algorithm identifies the best pharmacophores by computingmultiple flexible alignments between the input ligands. The multiplealignments are generated by combining pairwise alignments between one of theglitazone input ligand, which acts as pivot and the other glitazones asligand. The resulting multiple alignments reveal spatial arrangements ofconsensus features shared by different subsets of input ligands. The bestpharmacophore model has been derived using both pairwise and multiplealignment methods, which have been weighted in Pharmacophore Generationprocess. The highest-scoring pharmacophore model selected as potentialpharmacophore model. Finally, 3D structure search have been performed on the“ZincPharmer Database” to identify potential compounds that have been matchedwith the proposed pharmacophoric features. The 3D ZincPharmer Database hasbeen matched with 553 ligands hits. The physicochemical properties of 553ligands hits have been calculated by PaDEL-Descriptor software, which have

been filtered based on the Lipinski’s rule‐of‐five criteria (i.e. MW <500, H‐bond acceptor ≤ 10, H‐bond donor ≤ 5, Log P ≤ 5) by to get the potentialantidiabetic ligands. We have been found various substituted“pyrido[3',2':4,5]thieno[3,2-d]pyrimidin-4(3H)-one” as potential antidiabeticligands, which can be used for further development of antidiabetic agents. Inthe present research work, we have covered rational of Thiazolidinedione's nucleus based on MaximalCommon Substructure (MCS) as well as Ligand-Based Pharmacophore.
Keywords: Pharmacophore; Glitazones; Protein tyrosine Phosphatase 1B (PTP1B); PharmaGist; PPAR-γ; Selective PPAR-γ modulators; PPARs; Thiazolidinediones; Novel Emerging Targets for Diabetes; Diabetes; Diabetes Type-II; Non-Insulin-Dependent Diabetes Mellitus; NIDDM.
Introduction:
Type II Diabetes is a major cause of morbidity and mortality in theindustrialized world, with cardiovascular disease as the leading cause ofdeath, accounting for almost 50% of all Type II Diabetes deaths. The numbersof type 2 diabetes patients are increasing rapidly, and the number of patientsis expected to reach between 300 and 380 million by 2025, thereby placing anenormous economic burden on global healthcare. The worldwide epidemic of Type

II Diabetes (NIDDM) has been stimulating the search for new concepts andtargets for the treatment of this incurable disease. Most current therapieswere developed in the absence of defined molecular targets. Increasingknowledge on the biochemical and cellular alterations occurring in NIDDM hasled to the development of novel and potentially more effective therapeuticapproaches to treat the disease. The role of peroxisome proliferator activatedreceptors (PPARs) in the regulation of lipid metabolism, insulin andtriglycerides leads to the rational design of several PPAR agonists. [1]
Computational screening of databases has become increasingly popular in thepharmaceutical research. Virtual screening uses computer-based methods todiscover new ligands based on biological structures. Virtual screening isdivided into structural based screening (docking) and screening using activecompounds as templates (ligand based virtual screening). Ligand basedscreening techniques mainly focus on comparing molecular similarity analysesof compounds with known and unknown moiety, regardless of the methods of theused algorithm. Virtual screening approach based on structural similaritybetween known and potential active ligands. The rationale is that moleculesthat share some structural similarity may have a similar activity. Somemethods use a known active ligand as a query to extract structurally similarcompounds from large databases. [2]
Pharmacophore is the spatial arrangement of features that is essential for amolecule to interact with a specific target receptor. These selected featuresare important to achieving best Pharmacophore Model. In the present work, weused PharmaGist a webserver to build ligand based best Pharmacophore model.PharmaGist employed ligand-based pharmacophore detection method.
PPAR-γ receptor interacts with diverse range of ligands selected from thenatural ligands, such as prostaglandin PGJ2, Linolenic, Eicosapentaenoic,Docohexaenoic, Arachidonic Acids, and synthetic ligands, such as theThiazolidinediones (TZDs), L-tyrosine-based compounds, 7-Hydroxy-benzopyran-4-one several nonsteroidal anti-inflammatory drugs (NSAIDs), and a variety ofnew chemical classes. [3,4]
The basic difference between the PPAR-γ full agonist and PPAR-γ partialagonist is that the PPAR-γ partial agonist selectively modifies geneexpression needed for insulin sensitization without activating the genesresponsible for weight gain and edema, which are most side effects in PPAR-γfull agonist.
World major Pharma companies are developing partial agonists, with the goal ofretaining the beneficial effects of PPAR-γ full agonist, while diminishingtheir adverse effects. In vitro studies by researchers from Astellas and Rochealso indicate that their PPARγ partial agonists (FK614 and PA-082,respectively) activate pathways that ameliorate insulin resistance withoutstimulating fat accumulation in adiposities (Fujimura, Kimura, Oe et al. 2006;

Burgermeister, Schnoebelen, Flament et al. 2006). Studies with partialagonists of PPARγ suggest that a focus on partial PPAR agonists may be a wayof developing agents that have the desired efficacy of PPAR agonists withoutat least some of their potential adverse effects. However, development of PPARagonists remains a challenging endeavour.[5,6]
Common side effects associated with PPARγ receptor agonists are weight gain,oedema and adipogenesis. Balaglitazone is a selective partial PPARγ agonistcompound, which possess better safety profile comparative to full agonists.Unlike the other marketed PPAR gamma agonists, Phase II clinical trialsBalaglitazone shown weight neutral change and in preclinical experiments tocause less fluid retention than full PPAR gamma (γ) agonists. Based on theabove facts we included Balaglitazone in the present study and limited theexperiment on Thiazolidinediones or Glitazones or PPAR agonists. [7]
The present work elaborates pharmacophore generation via Ligand basedpharmacophore generation method. The present involves various marketed orunder developmental Thiazolidinediones or Glitazones or PPAR agonistsmolecules and followed by virtual screening on the ZincPharmer database tofind novel potential Protein tyrosine Phosphatase 1B (PTP1B) agonists.
Novel Emerging targets for diabetes include Peroxisome Proliferator-ActivatedReceptors agonist (PPARA) is nuclear receptors - ligand-activatedtranscription factors that bind to response elements in the promoter region oftarget genes to regulate gene expression. 2,4-Thiazolidinedione’s are well known agonistactivity with PPARA receptor.
Thiazolidinediones or TZDs act by binding to PPARs gamma and alpha receptors), agroup of receptor molecules inside the cell nucleus, specifically PPAR gamma(γ). The ligands for these receptors are free fatty acids (FFAs) andeicosanoids.[8]
PPARs are known to act via two distinct prominent mechanism transrepressionand transcription. The former is a DNA independent mechanism and is associatedwith disruption of other transcription factors pathways while the latter isDNA dependent and involves binding to PPAR response element (PPRE) of thetarget gene. [9]
The traditional approach is to search Pharmacophore is based on Maximal CommonSubstructure (MCS), namely the largest set of features shared by all inputligands. However, since some features are less frequent than others, thelargest common pattern can be sometimes misleading as a pharmacophore. A betterapproach is to assign weights to the features based on their infrequency of appearance and to look forthe highest weighted sum of common features. Another drawback of the MCS approach isthat it is based on the assumption that there is a single pharmacophore, andall ligands possess all its features. This is not always the case. The input

ligands can bind to different binding sites; they may have different bindingaffinities, and in a noisy input some of them might be outliers. [10,11]
The wonderful example of Maximal Common Substructure (MCS), we have been noticed thatthe discovery of Thiazolidinediones was started from Citaglitazone, which wasa classical prototype lead discovery of novel series of 5-(4-alkoxybenzyl)-2,4-thiazolidinediones as novel antidiabetic agent. Citaglitazone has beendiscovered as prototypical compound of the Thiazolidinediones class, which wasobserved to normalize hyperglycemia, hyperinsulinemia and hypertriglyceridemiain various insulin-resistant animal models. [12]
Ciglitazone was developed by Takeda Pharmaceuticals in the early 1980s.Ciglitazone was never used as a medication, but it sparked interest in theeffects of Thiazolidinedione class. Several analogues were later developed,e.g. pioglitazone and Troglitazone made it to the market. [6,12-14]
One of the studies before 1980 reported the structure activity relation (SAR)on Thiazolidinedione class, e.g. pioglitazone and its related compound. Thestudy revealed that the presence of pyridine nitrogen to oxyethyl chain was avery important factor for antidiabetic activity. These finding led to otherheterocyclic analogs such as Pioglitazone having nitrogen at the same relativeposition. The finding is significant in case of Balaglitazone as it possessednitrogen at the same relative position. [15]
Thiazolidinedione's increases the synthesis of certain proteins involved inmetabolism of fat as well as glucose, which reduces levels of certain types oflipids, and circulating free fatty acids. Thiazolidinediones generallydecrease triglycerides and increase high-density lipoprotein cholesterol (HDL-C) and low-density lipoprotein cholesterol (LDL-C). [16]
Several basic and clinical studies have exemplified the beneficial effects ofPPARα and PPARγ ligands in preventing the cardiovascular risks. The PPARα/γdual agonists are developed to increase insulin sensitivity and simultaneouslyprevent diabetic cardiovascular complications. Such compounds are underclinical trials and proposed for treatment of Type II diabetes with secondarycardiovascular complications. [17]
The present work we have selected 9 potential 2,4-Thiazolidinedione, formallyknown as Glitazone. Some of them selected from USFDA or EMEA approvedGlitazone, and some are still undergoing in clinical trial development.Approved Glitazone have been marketed globally for the treatment of Type-IIDiabetes as depicted in Table 1.

SN Molecule Chemical Structure Originator Current Market Status1.
PioglitazoneS
OO
O
HN
N
TakedaPharmaceuticals.
Approved in USA on Jul15, 1999 [18]
2.
Rosiglitazone (BRL-49653)S
OOO
NHNN
GlaxoSmithKline
Approved in USA on May25, 1999 [19]
In Europe, suspendedSeptember 2010 [20]
3.Rivoglitazone (CS-011) S
O
OOON
N
NH
Daiichi SankyoCo undergoing research [21]
4.
Darglitazone
S
O
O
O
O
NH
N
Pfizer Inc. undergoing research [5]
5.
Ciglitazone
S
O
O
ONH
TakedaPharmaceuticals
considered theprototypical compound
for thethiazolidinedione class
[6,12-14]
6.
Englitazone
S
O O
ONH
Pfizer Discontinued [22]
7.Netoglitazone
(MCC555/RWJ241947) S
F
O
O
O
HNSorbera et al. Discontinued [23]
8.
Balaglitazone (DRF-2593)
S
OO
O
O
NHN N Dr. Reddy's
LaboratoriesLtd.
Phase III clinical trialin United States and
Europe [24]
9.
Troglitazone
S
OO
OH
O
O
HN
Daiichi SankyoCo. Withdrawn [25,26]

Table 1, Various Protein Tyrosine Phosphatase 1B (PTP1B) agonists,which have been involved as an input query for the instant study. Research Design and method:
The present study comprises two modules. The first module is for detectingpharmacophore candidates from a set of input ligands. The other module is topharmacophore-based virtual screening on the ZincPharmer. Since we have beenevaluated potential ligands information, the identification has been doneusing indirect in other means ligand based drug-design (LBDD) (refer toFigure-1). The ligand based Pharmacophore based. [2,27,28]
Figure 1: Schematic diagram of pharmacophore development process. LBDD—ligandbased drug design.
PharmaGist pharmacophore generation employs an indirect approach requiring a3D training set input. PharmaGist is very fast requiring seconds to minute togenerate new pharmacophore. PharmaGist, accessible through the web interface,detects new pharmacophore using multiple flexible alignment of the trainingset. Each ligand in the training set serves as a pivot point for alignment. Toensure efficiency, the method requires the choice of highly active molecule orligands as to generate the best pharmacophore. [2,27,28]

The input is a set of chemical structures of drug-like molecules that areknown to bind to the PPAR receptor agonists. The output consists of candidatepharmacophores that are computed by multiple flexible alignments of the inputligands. The method handles the flexibility of the input ligands explicitlyand in a deterministic manner within the alignment process.
The PharmaGist pharmacophore generation method comprises four major stages:(i) ligand representation, (ii) pairwise alignment, (iii) multiple alignmentsand (iv) solution clustering and output. In the initial process of PharmaGistpharmacophore generation method involves each input ligand, which has beenprocessed separately. The PharmaGist method detects the rotatable bonds of theligand and divides it into rigid groups accordingly. In addition, the ligandis assigned to a set of physicochemical features (hydrogen bonddonor/acceptor, anion/cation, aromatic ring, hydrophobic group or, optionally,other features defined by the user). In the second stage, given a pivot(treated as rigid) and one target ligand (treated as flexible), pairwisealignment are computed as follows. First, for each rigid group of the targetligand, the method generates a set of transformations for superimposing thetarget rigid group onto the pivot. The result for each target rigid group is aset of a candidate new poses on the pivot. Then, these poses are reassembledinto new conformations of the target ligand aligned on the pivot. [2,27,28]

Figure 2: PharmaGist method flow. [19][]
The score of a resulting pairwise alignment is a weighted sum of the matchedpivot features. Two features, one from the pivot and one from a confirmationof the target ligand, can be matched if they are of the same type, and thedistance between them is below a predefined threshold (1A˚ by default).[2,27,28]
The algorithm uses default weight values for each feature type (0.3 forhydrophobicity and 1 for the rest), unless other values are supplied by theuser. The output of the stage is a large number of high-scoring pairwisealignments between the pivot and the target ligand. The third stage also workswith a selected pivot. [2,27,28]
Pairwise alignments between the pivot and the target ligands are combined intomultiple alignments. The goal is to find significant subsets of pivot featuresthat are matched by as many pairwise alignments for different target ligandsas possible. However, maximizing the number of aligned ligands can becontradictory to maximizing the score of the matched features. Thus, themethod produces multiple alignments for each subset size of input ligands. Due

to efficiency considerations, this is achieved by enumerating all the possiblesubsets of pivot features and selecting the ones that can be aligned by asmany ligands as possible. Subsets of pivot features with a significant scoreare candidate pharmacophores and will be reported to the user. By default, acandidate pharmacophore must consist of at least three spatially distinctfeatures, but this parameter is user-defined. In the last fourth stage,possible pharmacophores derived from different pivot iterations are clusteredand the highest scoring non-redundant ones for each number of molecules arereported. [2,27,28]
A pharmacophoric feature is defined as a set of atoms in the same rigid groupwith a physico-chemical property important for binding. Namely, a feature is(i–ii) a hydrogen-bond acceptor/donor atom; (iii–iv) an anion/cation atom; (v)a set of atoms of an aromatic ring; or (vi) a pair of adjacent hydrophobicatoms (Table 3). In addition, users can define other feature types of theirown. [27]
The performance of PharmaGist was successfully evaluated on different testcases (7). The whole dataset consists of almost 80 crystal structures ofreceptor–ligand complexes taken mainly from the FlexS benchmark dataset (27).The complexes are classified into 12 different cases, where each case includescomplexes of the same receptor with different ligands. For validation, weproduced a reference pharmacophore for each test case. The referencepharmacophore was extracted from a 3D alignment of the bounded ligands. Thisalignment was derived from a superposition of the receptor in the differentcomplexes. In all cases, the highest scoring pharmacophore candidate detectedby PharmaGist was similar to the reference pharmacophore. The runtime rangesfrom seconds to a few minutes on a typical set of ligands. [2,27]
Input Query:
The basic input query comprises the 9 glitazone molecules namely Pioglitazone,Rosiglitazone (BRL-49653), Rivoglitazone (CS-011), Darglitazone,Citaglitazone, Englitazone, Netoglitazone (MCC-555), Balaglitazone (DRF-2593)and Troglitazone. Glitazone is having common the thaizolidine di one nucleusas depicted in Table 1.
The chemical structures have been imported from Pubmed in the SDF format. TheOpen Babel GUI format converter software was used to convert .Mol2 format(Mol2—Sylbyl Mol2 format). The 9 glitazone molecules were put as input queryon the PharmaGist webserver to build ligand based best Pharmacophore model.PharmaGist employed ligand-based pharmacophore detection method.
Pharmacophore feature Generation:

PharmaGist employed 7 glitazones molecules out of 9 glitazones to build ligandbased best Pharmacophore model. The goal is to detect pharmacophorecandidates, namely the highest scoring 3D configurations of featuresresponsible for binding that are common to a significant number of inputligands. The various possible scored pharmacophore have been arranged based onscores. Table-2, presents the highest scoring pharmacophores, which have been sharedby seven glitazone molecules, including pivot. The Table-3 presents sortedpossible pharmacophoric features in descending order. The candidature ofpharmacophores was automatic detected by PharmaGist using the algorithm.
The algorithm identifies pharmacophores by computing multiple flexiblealignments between the input ligands. The multiple alignments are generated bycombining pairwise alignments between the pivot and the other ligands.However, the optimal multiple alignment does not necessarily consist of theoptimal pairwise alignments. Therefore, to ensure that we detect the optimalsolution, a large number of pairwise alignments are generated and used by themultiple alignment stage. The resulting multiple alignments reveal spatialarrangements of consensus features shared by different subsets of inputligands. The highest-scoring ones are potential pharmacophores.
Pairwise Alignment:
In the pairwise alignment build the pair of a pivot ligand and a single targetligand in present case one of the 9 Glitazones. The pivot is considered to berigid, whereas the target ligand is treated as flexible. The goal is togenerate the K highest-scoring pairwise alignments, where a pairwise alignmentis a superposition of a feasible conformation of the target ligand onto thepivot. The PharmaGist works by via a scoring method for pairwise alignmentbased on the pivot features matched by the superimposed conformation of thetarget ligand. Two features, one from the pivot and one from a conformation ofthe target ligand, can be matched if they are of the same type, and theirdistance is below a predefined threshold. [2,27]
The task is tackled by a two-tier procedure. First, for each rigid group ofthe target ligand, a set of rigid transformations for superimposing it on thepivot is computed. Then, the resulting candidate new poses of the target rigidgroups are assembled to form the K highest-scoring pairwise alignments. Theprocedure is highly efficient. A hybrid technique of Pose-Clustering andGeometric Hashing is employed for generating candidate new poses for thetarget rigid groups. Their best assemblies are computed by dynamicprogramming. [2,27]
Multiple Alignments:

The input is a pivot ligand, a collection of M target ligands and a largenumber (K) of pairwise alignments between the pivot and each target ligand.The goal is to compute the highest scoring multiple alignments.
A multiple alignment is defined by a subset of target ligands and exactly onepairwise alignment for each one of them. The score of a multiple alignment isa function of the number of participating target ligands, and the consensussubset of pivot features matched by them. Aim is to find multiple alignmentswith significant subsets of pivot features matched by as many target ligandsas possible. However, there is an inherent tradeoff between the tworequirements. The larger the number of participating ligands (m), the smalleris their consensus subset of pivot features.
Given K pairwise alignments for each of the M target ligands, there is anexponential number of (K + 1)M combinations for a multiple alignment. Anenumeration over all these combinations is impractical since K is a largenumber (1500 by default), and M is unbounded. Instead, we apply an exhaustivesearch on the possible subsets of matched pivot features. If n is the numberof pivot features, then there are 2n subsets to enumerate, in the worst case.This enumeration is practical since the number of atoms, and thus the numberof features (n), in a typical drug-like molecule is small. The algorithm worksin a bottom-up manner. It starts with subsets of one matched pivot feature andincrementally extends them until no larger subsets are matched. Theimplementation is efficient using bitwise representation.[2,27]
Weighted Pharmacophore Generation:
The pharmacophore derived from such an ensemble of multiple alignments iscalled a weighted pharmacophore. The input is a list of multiple alignmentsbetween the pivot and different subsets of target ligands. In principle,pharmacophore models can be derived from the highest scoring multiplealignments. However, due to the tradeoff between the number of ligandsparticipating in a multiple alignment and its consensus set of features, theinput multiple alignments are not disjoint and some of them can be combined togain more information on the candidate pharmacophores. Specifically,PharmaGist employs the combining parent multiple alignments with otherchildren multiple alignments for which the consensus is partial to theconsensus of the parent due to additional target ligands. [2,27]
Pharmacophore Clustering:
The input Glitazone molecules build the Pharmacophore cluster based on thethree stages described above (Pairwise Alignment, Multiple Alignment andWeighted Pharmacophore Generation) are repeated for each selected pivot.

Different pivots may lead to the same or similar weighted pharmacophores.Therefore, after iterating over all the possible pivots, we cluster aroundweighted pharmacophores with a similar 3D pattern of features. [2,27]
Pharmacophore Candidates Selection:
The algorithm produces a large set of weighted Protein tyrosine Phosphatase 1B(PTP1B) inhibitor pharmacophores and the task of selecting the mostappropriate ones for virtual screening is based on scored pharmacophore. Theinput Glitazone molecules may bind different binding sites or belong todifferent classes of chemical molecules. Based on these observations, we havedeveloped a strategy for selecting a small set of weighted pharmacophores thatcover as much as possible input molecules. The weighted pharmacophores areselected iteratively as follows. The first selected weighted pharmacophore isthe top-scoring one. The ligands of the parent multiple alignments of thisweighted pharmacophore and these only, are marked as covered. Next, we iterateover the remaining weighted pharmacophores and select the next-best scoringweighted pharmacophore that has no more than two molecules in common with theset of already covered ones. The process is repeated until all molecules arecovered or no more weighted pharmacophores can be selected. These results in aranked list of weighted pharmacophores associated with different classes ofbinding ligands.
The Table-2 represents the names of the input molecules along with theirnumber of atoms, and their assigned physico-chemical features. In addition,the user can click on "view details" and the Jmol window with molecules, andtheir features will be shown. The user can select/unselect each input moleculeand the associated features by clicking the relevant checkbox.
# Molecule AtomsFeatures
Spatial Features
Aromatic
Hydrophobic
Donors
Acceptors
Negatives
Positives
1 FromOBAll_1.mol2 58 21 20 3 10 2 6 0 0
2 FromOBAll_2.mol2 44 11 11 3 2 1 5 0 0
3 FromOBAll_3.mol2 47 15 15 4 3 1 6 0 1
4 FromOBAll_4.mol2 45 13 13 3 4 1 5 0 0
5 FromOBAll_5.mol2 43 10 10 4 1 1 4 0 0
6 FromOBAll_6.mol2 44 12 12 3 4 1 4 0 0
7 FromOBAll_7.mol2 3 0 0 0 0 0 0 0 0
Table 2, Various molecular feature of seven input Molecules to find out best Pharmacophore
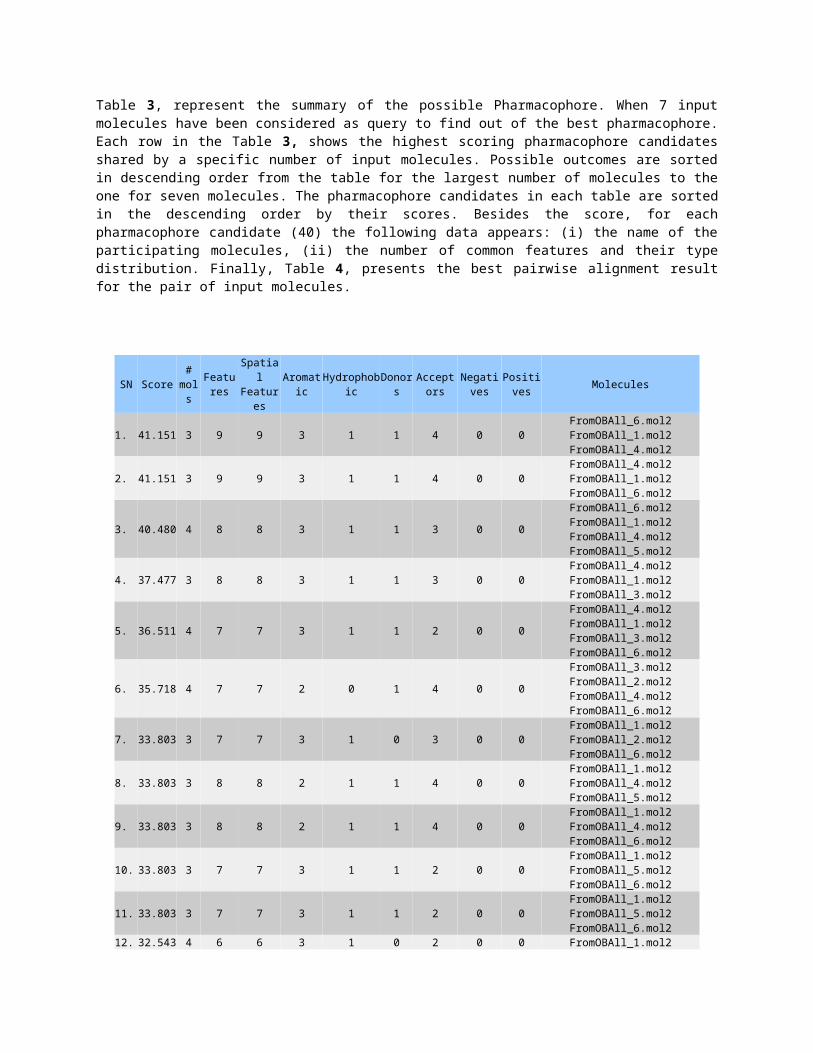
Table 3, represent the summary of the possible Pharmacophore. When 7 inputmolecules have been considered as query to find out of the best pharmacophore.Each row in the Table 3, shows the highest scoring pharmacophore candidatesshared by a specific number of input molecules. Possible outcomes are sortedin descending order from the table for the largest number of molecules to theone for seven molecules. The pharmacophore candidates in each table are sortedin the descending order by their scores. Besides the score, for eachpharmacophore candidate (40) the following data appears: (i) the name of theparticipating molecules, (ii) the number of common features and their typedistribution. Finally, Table 4, presents the best pairwise alignment resultfor the pair of input molecules.
SN Score#
mols
Features
Spatial
Features
Aromatic
Hydrophobic
Donors
Acceptors
Negatives
Positives Molecules
1. 41.151 3 9 9 3 1 1 4 0 0FromOBAll_6.mol2FromOBAll_1.mol2FromOBAll_4.mol2
2. 41.151 3 9 9 3 1 1 4 0 0FromOBAll_4.mol2FromOBAll_1.mol2FromOBAll_6.mol2
3. 40.480 4 8 8 3 1 1 3 0 0
FromOBAll_6.mol2FromOBAll_1.mol2FromOBAll_4.mol2FromOBAll_5.mol2
4. 37.477 3 8 8 3 1 1 3 0 0FromOBAll_4.mol2FromOBAll_1.mol2FromOBAll_3.mol2
5. 36.511 4 7 7 3 1 1 2 0 0
FromOBAll_4.mol2FromOBAll_1.mol2FromOBAll_3.mol2FromOBAll_6.mol2
6. 35.718 4 7 7 2 0 1 4 0 0
FromOBAll_3.mol2FromOBAll_2.mol2FromOBAll_4.mol2FromOBAll_6.mol2
7. 33.803 3 7 7 3 1 0 3 0 0FromOBAll_1.mol2FromOBAll_2.mol2FromOBAll_6.mol2
8. 33.803 3 8 8 2 1 1 4 0 0FromOBAll_1.mol2FromOBAll_4.mol2FromOBAll_5.mol2
9. 33.803 3 8 8 2 1 1 4 0 0FromOBAll_1.mol2FromOBAll_4.mol2FromOBAll_6.mol2
10. 33.803 3 7 7 3 1 1 2 0 0FromOBAll_1.mol2FromOBAll_5.mol2FromOBAll_6.mol2
11. 33.803 3 7 7 3 1 1 2 0 0FromOBAll_1.mol2FromOBAll_5.mol2FromOBAll_6.mol2
12. 32.543 4 6 6 3 1 0 2 0 0 FromOBAll_1.mol2

FromOBAll_2.mol2FromOBAll_5.mol2FromOBAll_6.mol2
13. 32.543 4 7 7 2 1 1 3 0 0
FromOBAll_5.mol2FromOBAll_1.mol2FromOBAll_4.mol2FromOBAll_6.mol2
14. 32.543 4 7 7 2 1 1 3 0 0
FromOBAll_4.mol2FromOBAll_1.mol2FromOBAll_2.mol2FromOBAll_6.mol2
15. 32.543 4 7 7 2 1 1 3 0 0
FromOBAll_4.mol2FromOBAll_1.mol2FromOBAll_5.mol2FromOBAll_6.mol2
16. 31.749 4 6 6 2 0 1 3 0 0
FromOBAll_6.mol2FromOBAll_1.mol2FromOBAll_2.mol2FromOBAll_4.mol2
17. 30.547 6 6 6 2 1 1 2 0 0
FromOBAll_4.mol2FromOBAll_1.mol2FromOBAll_2.mol2FromOBAll_5.mol2FromOBAll_6.mol2FromOBAll_3.mol2
18. 30.547 5 6 6 2 1 1 2 0 0
FromOBAll_4.mol2FromOBAll_1.mol2FromOBAll_2.mol2FromOBAll_3.mol2FromOBAll_6.mol2
19. 30.547 5 6 6 2 1 1 2 0 0
FromOBAll_4.mol2FromOBAll_1.mol2FromOBAll_2.mol2FromOBAll_3.mol2FromOBAll_6.mol2
20. 30.547 5 6 6 2 1 1 2 0 0
FromOBAll_2.mol2FromOBAll_1.mol2FromOBAll_3.mol2FromOBAll_4.mol2FromOBAll_6.mol2
21. 30.547 5 6 6 2 1 0 3 0 0
FromOBAll_6.mol2FromOBAll_1.mol2FromOBAll_2.mol2FromOBAll_3.mol2FromOBAll_4.mol2
22. 30.129 3 6 6 3 1 0 2 0 0FromOBAll_2.mol2FromOBAll_1.mol2FromOBAll_5.mol2
23. 30.129 3 6 6 3 1 0 2 0 0FromOBAll_1.mol2FromOBAll_2.mol2FromOBAll_6.mol2
24. 29.698 5 5 5 2 0 1 2 0 0
FromOBAll_6.mol2FromOBAll_1.mol2FromOBAll_2.mol2FromOBAll_3.mol2FromOBAll_4.mol2
25. 29.698 5 5 5 2 0 1 2 0 0 FromOBAll_6.mol2FromOBAll_1.mol2FromOBAll_2.mol2

FromOBAll_4.mol2FromOBAll_5.mol2
26. 29.698 5 5 5 2 0 1 2 0 0
FromOBAll_1.mol2FromOBAll_3.mol2FromOBAll_4.mol2FromOBAll_5.mol2FromOBAll_6.mol2
27. 29.368 4 7 7 2 2 0 3 0 0
FromOBAll_1.mol2FromOBAll_2.mol2FromOBAll_3.mol2FromOBAll_6.mol2
28. 28.574 5 6 6 2 1 1 2 0 0
FromOBAll_6.mol2FromOBAll_1.mol2FromOBAll_3.mol2FromOBAll_4.mol2FromOBAll_2.mol2
29. 28.574 4 5 5 3 1 0 1 0 0
FromOBAll_1.mol2FromOBAll_2.mol2FromOBAll_5.mol2FromOBAll_6.mol2
30. 27.900 6 5 5 2 1 1 1 0 0
FromOBAll_4.mol2FromOBAll_1.mol2FromOBAll_2.mol2FromOBAll_3.mol2FromOBAll_5.mol2FromOBAll_6.mol2
31. 27.900 6 5 5 2 1 0 2 0 0
FromOBAll_6.mol2FromOBAll_1.mol2FromOBAll_2.mol2FromOBAll_3.mol2FromOBAll_4.mol2FromOBAll_5.mol2
32. 27.780 6 6 6 1 0 1 4 0 0
FromOBAll_6.mol2FromOBAll_1.mol2FromOBAll_2.mol2FromOBAll_4.mol2FromOBAll_3.mol2FromOBAll_5.mol2
33. 27.000 6 4 4 2 0 1 1 0 0
FromOBAll_6.mol2FromOBAll_1.mol2FromOBAll_2.mol2FromOBAll_3.mol2FromOBAll_4.mol2FromOBAll_5.mol2
34. 27.000 6 4 4 2 0 1 1 0 0
FromOBAll_6.mol2FromOBAll_1.mol2FromOBAll_2.mol2FromOBAll_3.mol2FromOBAll_4.mol2FromOBAll_5.mol2
35. 27.000 6 4 4 2 0 0 2 0 0
FromOBAll_4.mol2FromOBAll_1.mol2FromOBAll_2.mol2FromOBAll_3.mol2FromOBAll_5.mol2FromOBAll_6.mol2
36. 27.000 6 4 4 2 0 0 2 0 0 FromOBAll_1.mol2FromOBAll_2.mol2FromOBAll_3.mol2

FromOBAll_4.mol2FromOBAll_5.mol2FromOBAll_6.mol2
37. 27.000 6 5 5 1 0 1 3 0 0
FromOBAll_6.mol2FromOBAll_1.mol2FromOBAll_2.mol2FromOBAll_3.mol2FromOBAll_4.mol2FromOBAll_5.mol2
38. 26.304 5 4 4 3 1 0 0 0 0
FromOBAll_1.mol2FromOBAll_2.mol2FromOBAll_4.mol2FromOBAll_5.mol2FromOBAll_6.mol2
39. 26.304 5 5 5 2 1 1 1 0 0
FromOBAll_2.mol2FromOBAll_1.mol2FromOBAll_3.mol2FromOBAll_4.mol2FromOBAll_5.mol2
40. 23.400 6 4 4 2 1 1 0 0 0
FromOBAll_3.mol2FromOBAll_1.mol2FromOBAll_2.mol2FromOBAll_4.mol2FromOBAll_5.mol2FromOBAll_6.mol2
Table 3, Various possible pharmacophore outcome when sort by scores
Score Features SpatialFeatures Aromatic Hydrophobic Donor
sAccepto
rs Negatives Positives
30.547 6 6 2 1 1 2 0 0
Table 4, Best Pharmacophore features selected via best Pairwise Alignments,which have been considered to find out potential ligands in the present study.
Virtual Screening:
ZINCPharmer – A Free an online interface for Commercially Available Compoundsfor Virtual Screening. A Web-based query tool incorporating a moleculardrawing interface enables the database to be searched and browsed and subsetsto be created. Users can process their own molecules by uploading them to aserver. Our hope is that this database will bring virtual screening librariesto a wide community of structural biologists and medicinal chemists.[27,29,29]

The ZINCPharmer tool having various filers to limit screen the databasecompounds according to pharmacophoric features like
Max Hits per Conf to reduce the number of orientations (resulting fromdifferent mappings of query features to ligand features) of the sameconformer returned. Highly symmetric queries may result in acombinatorial number of poses, necessitating this filer.
Max Hits per Molecule: Limit the number of hits returned of the same molecule(in different conformations or poses). Hits are chosen based on thespecified priority sort order. We have put 1 per molecule.
Max Total Hits: Limit the total number of hits returned. Hits are chosenbased on the specified sort order in the result's window.
Max RMSD: Screen by the optimal RMSD between the query features and theresult molecule. Statically, significance of RMSD: If the RMSD is below2 Aº, it is generally considered a successful prediction. The obviousgoal is that such a "near native" solution is ranked best of the set ofligand poses generated. In the present study, we have filtered the 553-Ligand via the rendering the maximum RMSD value more than 0.42. [30]
Molecular Weight: Screen by the molecular weight in Daltons (valueincludes hydrogens). In the present study, we have filtered the 553-Ligand via the rendering the Molecular Weight value not more than 500Dalton.
Rotable Bonds: Screen by the number of rotatable bonds. Rotatable bondsare identified using the SMARTS expression:
Drug likeness screening:
Many drug candidate fail in the clinical trials reason is unrelated in thepotency against the intended drug target. Pharmacokinetic and toxicity issuesare blamed for more than half of all failure in the clinical trials. Thephysicochemical properties anticipate potential in-vivo effect andbioavailability of active pharmaceutical ingredient or Drug-likeliness.Therefore, first part of the virtual screening evaluates drug likeness ofsmall molecules, drug like molecule's exhibit favorable absorption,distribution, metabolism, excretion, toxicological (ADMET) parameters. Usingfollowing types of method currently assesses drug likeness.
Counting method Functional group filter Topological filter Pharmacophore filter
In the present work, we have been followed simple counting method, which isbased on 2D-physicochemical parameters, e.g. molecular mass, high

lipophilicity (expressed as cLogP greater than 5), more than 5 hydrogen bonddonors and more than 10 hydrogen bond acceptors.
In Silico Prediction of ADME Properties would be possible to evaluate throughLipinski’s rule-of-five analysis, The simplest ADME-concerned filters may be“rule of 5” proposed by Lipinski et al. in 1997. Lipinski and coworkers analyzeda subset of 2245 drugs from the World Drug Index (WDI). They found that poorabsorption and permeation are more likely to occur when the molecular weightis over 500, the octanol/water partition coefficient is over 5 (CLOGP) or 4.15(MLOGP); the number of hydrogen-bond donors (OH and NH groups) is more than 5,and the number of hydrogen-bond acceptors (N and O atoms) is more than 10. Thefast estimations of logP allow the “rule of 5” screening of library prior toenumeration.
The guidelines were quickly adopted by the pharmaceutical industry as ithelped apply ADME considerations early in preclinical development and couldhelp avoid costly late-stage preclinical and clinical failures. The guidelinespredict that poor absorption or permeation of an orally administered compoundis more likely if the compound meets the following criteria:
Conclusion:
The best candidate pharmacophore have been detected by multiple flexiblealignments of the input ligands, where the flexibility of the ligands istreated explicitly and in a deterministic manner in the alignment process.Another key advantage of the method is the ability to detect pharmacophorescommon to subsets of input ligands, a characteristic that makes PharmaGisttolerant of outliers and to several binding modes.
The best pharmacophore model having the Score 30.547, which has been opted toscreen on ZincPharmer database to derive the novel potential antidiabeticligands. The best pharmacophore having various Pharmacophore features,including General Features 6, Spatial Features 6, Aromatic 2, Hydrophobic 1,Donors 1, and Acceptors 2.
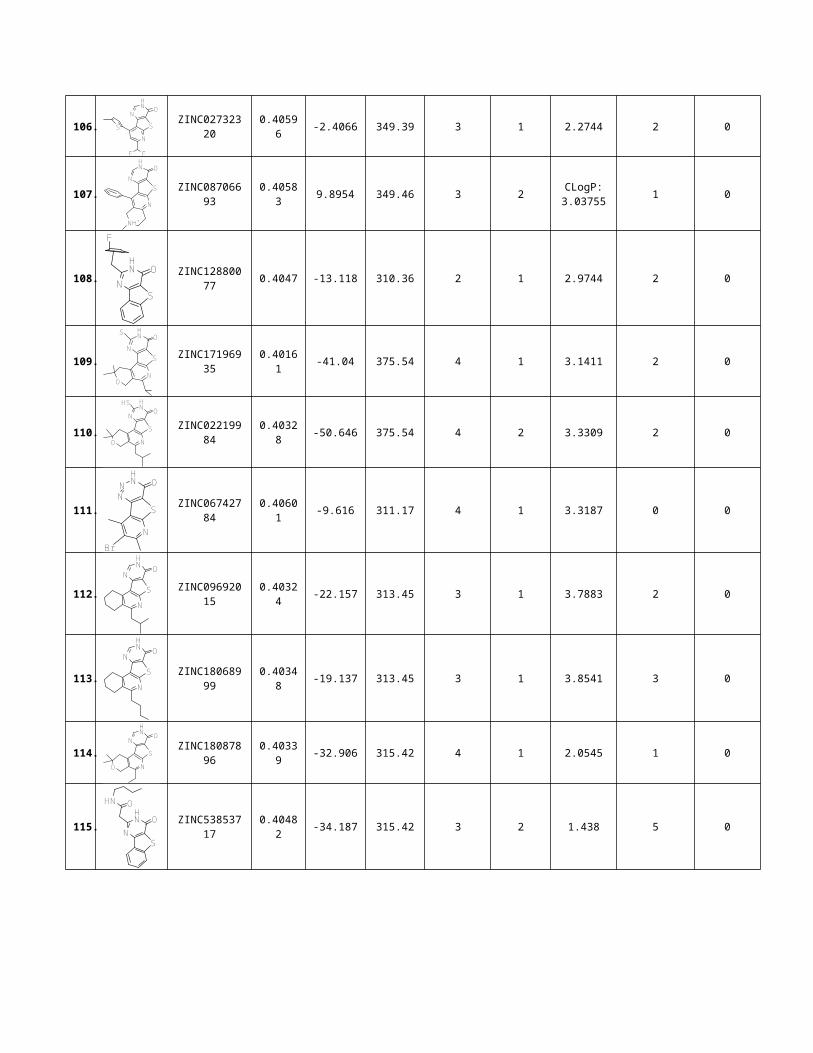
We have found 553 potential candidates for the Protein tyrosine Phosphatase 1B(PTP1B) targets, which were reduced to 116 potential ligands(Pyrido[3',2':4,5]thieno[3,2-d]pyrimidin-4(3H)-one Nucleus), when ZINCPharmer query wasfiltered by RMSD values 4.42 and molecular weight more than 500 Dalton. Table-5 represents 116- potential candidates for the Protein tyrosine Phosphatase 1B(PTP1B) targets.
We have found almost 116- potential ligands, which qualify the Lipinski’srule-of-five theory for ADME. Based on the above observation, we can concludethat these ligands may be used for further studies to evaluate its in-vivoeffect in the biological system.

The screened best pair aligned pharmacophore on the ZincPharmer databaseprovides various substituted “pyrido[3',2':4,5]thieno[3,2-d]pyrimidin-4(3H)-one” ligands. All 116 ligands generated ligands possessed the common nucleus“pyrido[3',2':4,5]thieno[3,2-d]pyrimidin-4(3H)-one”.
In the year 2010, the pyrido[3',2':4,5]thieno[3,2-d]pyrimidin compounds, whichis closely related pharmacophore screed nucleus “pyrido[3',2':4,5]thieno[3,2-d]pyrimidin-4(3H)-one”, found to have phosphodiesterase IV inhibitors (PDE4)activity, a target for the treatment of asthma and chronic obstructivepulmonary disease (COPD). [33]
In the same year 2010, synthesis, in vitro and in vivo evaluation of [O-methyl-(11)C]dimethylamino-3(4-methoxyphenyl)-3H-pyrido[3',2':4,5]thieno[3,2-d]pyrimidin-4-one , as a potential imaging agent for mGluR1 receptors (type ofglutamate receptor) have been described by J. Prabhakaran et.al, which suggestthat the “pyrido[3',2':4,5]thieno[3,2-d]pyrimidin-4(3H)-one” nucleus bindswith mGluR1. [31]
In one another independent study well before the J. Prabhakaran et.al study,have been found that the possibility of glutamate receptor ligands to inducediabetes. The study determined that binding properties of glutamate receptorsin streptozotocin-induced diabetes in rats. The study was published in theyear 1997 by J. Gagné et.al. The study determined the effects of streptozotocin(STZ) administration on the binding properties of alpha-amino-3-hydroxy-5-methylisoxazolepropionic acid (AMPA) and N-methyl-D-aspartate (NMDA) subtypesof glutamate receptors in rats, using quantitative autoradiographic analysisof (3)H-AMPA and [(3)H]glutamate binding on brain tissue sections. The STZinjection produced a reduction of (3)H-AMPA binding in various brain regions,an effect that is due to a decrease in receptor affinity. The effect of STZ-induced diabetes appeared to be specific to the AMPA subtype of glutamatereceptors, as the same treatment did not modify [(3)H]glutamate binding toNMDA receptors. These changes in AMPA receptor properties may have importantimplications for understanding the biochemical mechanisms underlying cognitiveimpairment in diabetes. [32]
These finding suggests that the mGluR1 receptors may involve in the inductiondiabetes, which may be rational for the antidiabetic activity of“pyrido[3',2':4,5]thieno[3,2-d]pyrimidin-4(3H)-one”. Hence, based abovefinding and developed pharmacophore model. We would conclude that“pyrido[3',2':4,5]thieno[3,2-d]pyrimidin-4(3H)-one” nucleus has been potentialto produce novel series antidiabetic ligands. The exact mechanistic activityhypothesis remains need to be draw by in vivo study.

SN WholeMolecule
Name(Whole
Molecule)rmsd
CosmicTotalEnergy(WholeMolecul
e)
ADMEWeight(WholeMolecul
e)
ADMEH-bondAcceptor
s(WholeMolecule
)
ADMEH-bondDonors(WholeMolecul
e)
ADMElogP(WholeMolecule
)
ADMERotatable
Bonds(Whole
Molecule)
ADMEViolatio
ns(WholeMolecule
)
1. N
NH
HN O
O
OS
ZINC06721865
0.40435 -39.607 401.46 4 2 2.3198 3 0
2.N
N
HN O
OF
F
FF F
S
ZINC36611497
0.40716 -10.236 427.37 4 1 4.6992 3 0
3.N
N
HN O
O
O
FF
S
ZINC20608411
0.40709 107.95 427.45 5 1 2.9187 3 0
4.N
N
HNN O
OO
F F
S
S
ZINC06150618
0.40555 87.204 454.55 6 1 1.2862 4 0
5.N
N
HNN OO
O
O
FF
S
S
ZINC02731144
0.40521 -12.892 480.54 7 1 1.229 4 0
6.N
N
HN O
F F
S
Cl
ZINC08419174
0.40719 91.583 403.85 3 1 4.1199 3 0
7. N
HN O
SZINC182177
700.4055
3 -7.1071 202.24 2 1 1.2743 0 0
8.N
N
HN O
O
S ZINC09327367
0.40322 0.54673 323.39 4 1 2.9754 1 0

9.
N
N
HN O
SZINC001718
10 0.4061 -20.977 203.23 3 1 1.1854 0 0
10.
N N
N
HN O
SZINC038470
100.4049
1 -12.955 204.22 4 1 0.2688 0 0
11.N
N
HN O
OO
S ZINC12437012
0.40195 -11.927 353.42 5 1 1.9684 1 0
12.N
HN O
F SZINC002607
710.4056
9 -4.6613 220.23 2 1 1.4138 0 0
13.
N
N
HN O
S
S ZINC08670378
0.40606 -34.807 353.48 3 1 4.2975 3 0
14.N
N
HN O
SCl ZINC131088
870.4068
1 -9.924 353.84 3 1 4.0041 1 0
15.
N
N
HN O
S ZINC00061359 0.4061 -28.003 231.29 3 1 1.0851 0 0
16.
N
NN
HN O
SZINC092351
390.4058
5 -18.391 232.28 4 1 2.5269 0 0
17.N
N
HN O
FF
SZINC356780
490.4073
1 -11.384 433.5 3 1 5.5307 3 1
18.N
HN O
SClZINC573523
980.4055
7 -4.1125 236.68 2 1 1.7923 0 0

19.N
N
HN O
S ZINC00888373
0.40605 -6.5185 355.43 3 1 4.4884 2 0
20.
N
N
HN O
O
FF
FF
S
ZINC06275829
0.40744 -12.541 409.38 4 1 3.9806 4 0
21.N
NH+
HN
N
O
O
O
S
S
ZINC01865796 0.4022 -21.471 461.67 4 2 2.6497 5 0
22.N
N
HN O
OH
FF
S SZINC027308
110.4071
6 -18.115 491.55 4 2 4.6203 3 0
23.
N
N
N
N HN O
O
F F
S
Br
ZINC02731618
0.40741 21.921 492.32 5 1 3.652 4 0
24.
N
N
HN O
SZINC002091
280.4058
5 -23.039 245.32 3 1 1.5523 0 0
25.N
NNHN O
S ZINC06742786
0.40594 -2.9015 356.42 4 1 5.1782 2 1
26.N
N
HN
N
O
O
S ZINC18187795
0.40395 -11.989 328.42 4 1 1.6952 1 0
27.N
NH+
HN
N
O
SZINC358969
660.4052
2 -125.88 247.32 2 2 0.6514 1 0
28. N
HN O
S
SZINC064118
190.4054
4 -16.836 248.33 2 1 1.9581 1 0
29.N
N
HN O
FF
S ZINC08436368
0.40614 -6.0157 329.34 3 1 2.9682 2 0

30.N
HN O
SClZINC404787
710.4050
8 -12.597 250.71 2 1 1.5075 0 0
31.N
NH+
HN
N
O
O
O
S
S
ZINC13512136
0.40192 -35.131 495.68 4 2 3.2189 4 0
32.N
N
HN O
O
SZINC066642
520.4022
2 -31.909 329.45 4 1 2.4573 1 0
33.N
N
HN O
O
S ZINC18068875
0.40333 -32.737 329.45 4 1 2.4508 2 0
34.N
N
HN O
OO
F F
F
F
S
ZINC36611509
0.40744
-0.90689 439.41 5 1 3.7279 5 0
35.
N
NH+
HN
N
O
O
S ZINC18165184
0.40209 -6.2495 357.49 3 2 1.4807 1 0
36.N
N
HN
H+N
O
S ZINC06897265
0.40987 -5.8006 357.54 3 2 2.2447 2 0
37.N
N
NN H
N
N+-O
O
O
O
FF
S
ZINC22818729
0.40732 2.4804 498.5 7 1 3.4305 5 0
38.N
N
HN O
S
S
ZINC09353886
0.40349 -9.865 441.59 3 1 6.0911 4 1
39.N
N
HN O
OS
ZINC18202598 0.4058 -13.243 261.32 4 1 1.1129 2 0

40.N
N
HN O
O
FF
F
F
S
ZINC36611344 0.4073 -7.1128 471.45 4 1 5.1978 5 1
41.
N
N
N
N
HN O
O
FF F
S
ZINC22707513
0.40721 12.849 471.49 5 1 4.1955 4 0
42.N
N
HN O
S
S
ZINC08770596
0.40264 -32.872 331.48 3 1 2.497 1 0
43.N
NN
HN O
OS
ZINC00085659
0.40603 -16.401 262.31 5 1 1.8027 2 0
44.
N
N
HN OO
S
Br
ZINC09235261
0.40731 -29.531 416.31 4 1 3.5328 3 0
45.N
N NN
HN O
F F
S
SZINC196382
270.4073
1 -6.6551 443.52 4 1 3.6896 4 0
46.N
N
HN O
S
S
ZINC04785862
0.40266 -35.4 359.54 3 1 3.2238 2 0
47.N
NN
HN O
S
Cl
ZINC00091867
0.40602 -10.877 266.72 4 1 3.0449 0 0
48.N
N
HN O
FF
S ZINC08409902 0.4058 0.46827 267.27 3 1 1.751 1 0
49.N
N
HN O
S ZINC05218732
0.40308 -3.1425 333.43 3 1 4.027 1 0
50.N
N
HN O
FF F
S ZINC32117011
0.40569 -23.247 361.36 3 1 3.402 1 0

51.
N
N
N
NHN O
F F
SZINC068714
010.4074
1 10.678 361.4 4 1 2.6946 3 0
52.N
N
HN O
SZINC056463
930.4031
7 -23.943 271.36 3 1 2.5929 0 0
53.
N
N
N
HN O
F F
SZINC027856
060.4073
1 -12.779 420.46 4 1 4.2165 3 0
54.N
N
HN
NH+
O
S ZINC06368217
0.40585 12.462 273.36 3 2 CLogP:
1.14955 0 0
55. N
N
HN O
S
SCl
ZINC08411754
0.40585 -9.9996 421.93 3 1 4.5839 1 0
56.N
N
HN O
OS
ZINC03157512
0.40585 -22.004 275.35 4 1 0.8281 2 0
57.N
N
HN O
FF F
S
S
SZINC206084
400.4073
1 18.33 449.5 3 1 4.3655 2 0
58.
N
N
N
NHN O
FF
SZINC028002
510.4072
9 4.1053 423.47 4 1 3.9118 4 0
59. N
N
HNH2N O
S
ClCl
ZINC16386001
0.40661 -49.441 363.23 3 2 3.4555 1 0
60. N
N
HNH2N O
S
Cl
Cl
ZINC06402293 0.4064 -54.113 363.23 3 2 3.4555 1 0

61. N
N
HN
N
O
O
O
S ZINC17056917
0.40263 -26.251 386.51 5 1 1.5531 2 0
62.N
N
HN
N
O
O
O
S ZINC18187751 0.4022 -26.114 386.51 5 1 1.5531 2 0
63.N
N
HN O
O
FF F
S ZINC12395069
0.40596 16.015 337.29 4 1 2.6352 1 0
64.N
N
HN O
S ZINC12284935
0.40605 -11.292 279.33 3 1 2.804 1 0
65.N
N
HN O
O
S ZINC02296449
0.40224 -15.705 363.46 4 1 3.02 1 0
66.N
N
HN O
OS
ZINC12445626
0.40704 -16 337.42 4 1 2.741 3 0
67.N
NH+
HN
N
O
O
O
S ZINC02283144
0.40214 -19.976 387.52 4 2 0.4737 1 0
68.N
NH+
HN
N
O
O
O
S ZINC04625885
0.40176 -8.0794 387.52 4 2 1.2271 2 0
69.N
N
HN O
FF F
S ZINC18162196
0.40568 10.06 285.26 3 1 2.4696 0 0

70.N
N
HN O
S
S ZINC15224322
0.40624 -15.749 285.35 3 1 2.0957 1 0
71.N
N
HN O
SZINC182120
050.4038
8 -31.213 285.39 3 1 2.3081 0 0
72.N
N
HN O
SZINC179922
310.4031
7 -18.979 285.39 3 1 3.0615 1 0
73.N
N
HN O
SZINC086917
540.4039
6 -14.649 285.39 3 1 3.068 1 0
74.N
N
HN
N
O
O
S
S
ZINC13115826
0.40074 -30.731 388.54 4 1 1.527 1 0
75.N
N
HN O
S
S
ZINC09311867
0.40343 -10.721 365.49 3 1 4.3145 2 0
76.N
NNHN O
S
S ZINC16430737
0.40622 -2.7387 286.34 4 1 2.7855 1 0
77.N
N
HN O
O
S ZINC00085455
0.40561 -19.539 287.36 4 1 1.136 0 0
78.N
N
HN
+HN
O
S ZINC08706717
0.40513 3.7695 287.39 3 2 CLogP:
1.64855 0 0

79.N
N
HN
N
O
S ZINC18086360
0.40309 -17.988 340.48 3 1 3.156 1 0
80.N
NN
HN O
OS
Br
ZINC06742782
0.40601 -8.9572 341.2 5 1 2.5945 2 0
81.N
N
HN O
O
S
S
ZINC02231897
0.40156 -44.824 389.57 4 1 3.4651 3 0
82.N
NN
HN
N
O
S ZINC31811269
0.40338 -3.4201 341.47 4 1 3.8458 1 0
83.N
N
HN O
O
O SZINC124456
250.4071
3 -31.617 367.45 5 1 2.4883 4 0
84.N
N
HN O
S
S
ZINC13143816
0.40606 -23.835 291.41 3 1 2.8634 2 0
85.
N
N
HN O
S
S ZINC05646468
0.40604 -38.117 367.51 3 1 4.7647 3 0
86.N
N
HN
N
O
O
S ZINC19144440
0.40315 -16.485 342.45 4 1 2.0915 1 0
87.N
N
HN O
SZINC124150
360.4061
4 -20.433 293.36 3 1 3.0543 1 0
88.N
N
HN O
FF
SZINC181623
150.4063
3 -10.715 343.37 3 1 3.4354 2 0
89.N
N
HN O
O
S ZINC18144679
0.40339 -35.684 343.48 4 1 2.7813 2 0

90.N
N
HN O
FF F
SZINC001036
770.4058
5 -26.123 299.29 3 1 1.9679 0 0
91.N
N
HN O
S
S ZINC05634512 0.4057 -23.183 299.38 3 1 1.8109 1 0
92.N
N
HN O
S ZINC09692013
0.40305 -18.896 299.42 3 1 3.4578 2 0
93.N
N
HN O
S ZINC09019787
0.40423 -15.693 299.42 3 1 3.4578 3 0
94.N
N
HN O
S ZINC04922947
0.40423 -18.24 299.42 3 1 3.392 2 0
95.N
N
HN O
SZINC180849
520.4031
7 -17.886 299.42 3 1 3.4643 1 0
96.N
NNHN O
O S ZINC06742788
0.40601 4.1622 346.38 5 1 4.1266 2 0
97.N
N
HNH2N O
S
S ZINC16430734
0.40671 -48.358 300.37 3 2 1.7112 1 0

98. N
N
HN O
O
SZINC171123
530.4060
9 -11.746 301.39 4 1 1.9227 2 0
99. N
N
HN O
O
S ZINC08856477
0.40537 -11.746 301.39 4 1 1.9227 2 0
100.N
N
HN O
O
SZINC170458
680.4056
8 -12.414 301.39 4 1 1.9292 1 0
101. N
N
HN O
O
SZINC170458
700.4058
2 -11.833 301.39 4 1 1.9292 1 0
102.N
N
HN O
O
SZINC124369
090.4022
3 -37.422 301.39 4 1 1.5859 0 0
103.N
N
HN
N
O
O
O
S ZINC19144435
0.40207 -30.246 372.48 5 1 1.0845 1 0
104.N
N
HN O
O
O
O
S ZINC13108885
0.40554 -21.34 395.46 6 1 2.8742 5 0
105.N
N
HN O
S
S
ZINC08622872
0.40474 -20.488 303.42 3 1 1.5785 0 0
106.N
N
HN O
FF
S SZINC027323
200.4059
6 -2.4066 349.39 3 1 2.2744 2 0
107.N
N
HN
NH+
O
S ZINC08706693
0.40583 9.8954 349.46 3 2 CLogP:
3.03755 1 0
108. N
HN O
F
S
ZINC12880077 0.4047 -13.118 310.36 2 1 2.9744 2 0
109.N
N
HN O
O
S
S
ZINC17196935
0.40161 -41.04 375.54 4 1 3.1411 2 0
110.N
N
HN O
O
S
HS
ZINC02219984
0.40328 -50.646 375.54 4 2 3.3309 2 0
111.N
NN
HN O
S
Br
ZINC06742784
0.40601 -9.616 311.17 4 1 3.3187 0 0
112.N
N
HN O
S ZINC09692015
0.40324 -22.157 313.45 3 1 3.7883 2 0
113.N
N
HN O
S ZINC18068999
0.40348 -19.137 313.45 3 1 3.8541 3 0
114.N
N
HN O
O
SZINC180878
960.4033
9 -32.906 315.42 4 1 2.0545 1 0
115. N
HN
HN
OO
S
ZINC53853717
0.40482 -34.187 315.42 3 2 1.438 5 0

116.N
N
HN O
S ZINC09379812
0.40426
-0.65276 319.4 3 1 3.6307 1 0
Table 5, Zincpharmer sorted all potential Protein Tyrosine Phosphatase1B (PTP1B) agonists [33]
CONFLICT OF INTEREST:
Authors declare that they have no conflict of interest by any means withrespect to the instant research manuscript.
ACKNOWLEDGEMENTS
We would like to thanks to the ChemAxon software team to providing us suchuseful software. The prepared chemical structures were imported from pubmedand make it as drawn by ChemAxon MarvinSketch.
ABBREVIATIONS:
Metabotropic glutamate receptor 1 : mGluR1TZD : Thiazolidinedione), FDA : Food and Drug AdministrationSPPARγMs : Selective PPAR-γ modulators PTP1B : Protein tyrosine Phosphatase 1B

Journal Reference List
[1] Santilli, A. A.; Scotese, A. C.; Tomarelli, R. M. A potentantihypercholesterolemic agent: (4-chloro-6-(2,3-xylidino)-2-pyrimidinylthio) acetic acid (Wy-14643). Experientia 1974, 30 (10), 1110-1111.
[2] Dror, O.; Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H. J.Novel approach for efficient pharmacophore-based virtual screening:method and applications. J. Chem. Inf. Model. 2009, 49 (10), 2333-2343.
[3] Zhang, F.; Lavan, B. E.; Gregoire, F. M. Selective Modulators of PPAR-gamma Activity: Molecular Aspects Related to Obesity and Side-Effects.PPAR. Res. 2007, 2007, 32696.
[4] Matin, A.; Gavande, N.; Kim, M. S.; Yang, N. X.; Salam, N. K.; Hanrahan,J. R.; Roubin, R. H.; Hibbs, D. E. 7-Hydroxy-benzopyran-4-onederivatives: a novel pharmacophore of peroxisome proliferator-activatedreceptor alpha and -gamma (PPARalpha and gamma) dual agonists. J. Med. Chem.2009, 52 (21), 6835-6850.
[5] Hulin, B.; Clark, D. A.; Goldstein, S. W.; McDermott, R. E.; Dambek, P.J.; Kappeler, W. H.; Lamphere, C. H.; Lewis, D. M.; Rizzi, J. P. Novelthiazolidine-2,4-diones as potent euglycemic agents. J. Med. Chem. 1992, 35(10), 1853-1864.
[6] Pershadsingh, H. A.; Szollosi, J.; Benson, S.; Hyun, W. C.; Feuerstein,B. G.; Kurtz, T. W. Effects of ciglitazone on blood pressure andintracellular calcium metabolism. Hypertension 1993, 21 (6 Pt 2), 1020-1023.
[7] New Drugs Online Report for Balaglitazone.http://www.ukmi.nhs.uk/applications /ndo / record_view_open.asp?newDrugID=3940 (Accessed Aug 06, 2011).

[8] U S Food and Drug Administration (USFDA). http://www.fda.gov/ (AccessedMar 07, 2011).
[9] Matin, A.; Gavande, N.; Kim, M. S.; Yang, N. X.; Salam, N. K.; Hanrahan,J. R.; Roubin, R. H.; Hibbs, D. E. 7-Hydroxy-benzopyran-4-onederivatives: a novel pharmacophore of peroxisome proliferator-activatedreceptor alpha and -gamma (PPARalpha and gamma) dual agonists. J. Med. Chem.2009, 52 (21), 6835-6850.
[10] Shatsky, M.; Shulman-Peleg, A.; Nussinov, R.; Wolfson, H. J. The multiplecommon point set problem and its application to molecule binding patterndetection. J. Comput. Biol. 2006, 13 (2), 407-428.
[11] Brint, A.; Willett, P. Algorithms for the identification of three-dimensional maximal common substructures. J Chem Inf Comput Sci, 1987, 27,152-158.
[12] Sohda, T.; Kawamatsu, Y.; Fujita, T.; Meguro, K.; Ikeda, H. [Discoveryand development of a new insulin sensitizing agent, pioglitazone].Yakugaku Zasshi 2002, 122 (11), 909-918.
[13] Hulin, B.; McCarthy, P.A.; Gibbs, E.M. The glitazone family ofantidiabetic agents. Current Pharmaceutical Design, 1996, 2, 85-102.
[14] Imoto, H.; Imamiya, E.; Momose, Y.; Sugiyama, Y.; Kimura, H.; Sohda, T.Studies on non-thiazolidinedione antidiabetic agents. 1. Discovery ofnovel oxyiminoacetic acid derivatives. Chem. Pharm. Bull. (Tokyo) 2002, 50(10), 1349-1357.
[15] Sohda, T.; Mizuno, K.; Momose, Y.; Ikeda, H.; Fujita, T.; Meguro, K.Studies on antidiabetic agents. 11. Novel thiazolidinedione derivativesas potent hypoglycemic and hypolipidemic agents. J. Med. Chem. 1992, 35(14), 2617-2626.
[16] NHS: Avandia diabetes drug suspended.http://www.nhs.uk/news/2010/09September/Pages/ rosiglitazone-avandia-drug-suspended-heart-risk.aspx (Accessed Aug 06, 2011).
[17] Balakumar, P.; Rose, M.; Ganti, S. S.; Krishan, P.; Singh, M. PPAR dualagonists: are they opening Pandora's Box? Pharmacol. Res. 2007, 56 (2), 91-98.
[18] Orange Book: Approved Drug Products with Therapeutic EquivalenceEvaluations.http://www.accessdata.fda.gov/scripts/cder/ob/docs/obdetail.cfm?Appl_No=021073&TABLE1=OB_Rx (Accessed Sep 23, 2011).

[19] Schneidman-Duhovny, D.; Dror, O.; Inbar, Y.; Nussinov, R.; Wolfson, H. J.PharmaGist: a webserver for ligand-based pharmacophore detection. NucleicAcids Res. 2008, 36 (Web Server issue), W223-W228.
[20] European Medicines Agency recommends suspension of Avandia, Avandamet andAvaglim.http://www.ema.europa.eu/ema/index.jsp?curl=pages/news_and_events/news/2010/09/news_detail_001119.jsp&murl=menus/news_and_events/news_and_events.jsp&mid=WC0b01ac058004d5c1&jsenabled=false (Accessed Sep23, 2011).
[21] Kong, A. P.; Yamasaki, A.; Ozaki, R.; Saito, H.; Asami, T.; Ohwada, S.;Ko, G. T.; Wong, C. K.; Leung, G. T.; Lee, K. F.; Yeung, C. Y.; Chan, J.C. A randomized-controlled trial to investigate the effects ofrivoglitazone, a novel PPAR gamma agonist on glucose-lipid control intype 2 diabetes. Diabetes Obes. Metab 2011, 13 (9), 806-813.
[22] Clark, D. A.; Goldstein, S. W.; Volkmann, R. A.; Eggler, J. F.; Holland,G. F.; Hulin, B.; Stevenson, R. W.; Kreutter, D. K.; Gibbs, E. M.; Krupp,M. N.; . Substituted dihydrobenzopyran and dihydrobenzofuranthiazolidine-2,4-diones as hypoglycemic agents. J. Med. Chem. 1991, 34 (1),319-325.
[23] Sorbera, L.A., Castaner, J., del Fresno, M., Silvestre, J. Netoglitazone.Drugs Fut., 2002, 27(2), 132.
[24] Agrawal, R.;Jain, P.;N. Dikshit, S. Balaglitazone: A Second GenerationPeroxisome Proliferator-Activated Receptor (PPAR) Gamma (ã) Agonist. MiniReviews in Medicinal Chemistry, 2012, 12(2), 87-97.
[25] Retired Drugs: Failed Blockbusters, Homicidal Tampering, FatalOversights.http://www.wired.com/medtech/drugs/multimedia/2008/10/gallery_retired_drugs?slide=6&slideView=6 (Accessed Sep 29, 2011).
[26] Cohen, J. S. Risks of troglitazone apparent before approval in USA.Diabetologia 2006, 49 (6), 1454-1455.
[27] Sun, H. Pharmacophore-based virtual screening. Curr. Med. Chem. 2008, 15(10), 1018-1024.
[28] Schneidman-Duhovny, D.; Dror, O.; Inbar, Y.; Nussinov, R.; Wolfson, H. J.Deterministic pharmacophore detection via multiple flexible alignment ofdrug-like molecules. J. Comput. Biol. 2008, 15 (7), 737-754.
[29] Irwin, J. J.; Shoichet, B. K. ZINC--a free database of commerciallyavailable compounds for virtual screening. J. Chem. Inf. Model. 2005, 45 (1),177-182.

[30] Kamau, Edwin. Pharmacophore Model Development for the Identification of NovelAcetylcholinesterase Inhibitors.Kennesaw State University, June 2007.
[31] Prabhakaran, J.; Majo, V. J.; Milak, M. S.; Kassir, S. A.; Palner, M.;Savenkova, L.; Mali, P.; Arango, V.; Mann, J. J.; Parsey, R. V.; Kumar,J. S. Synthesis, in vitro and in vivo evaluation of [11C]MMTP: apotential PET ligand for mGluR1 receptors. Bioorg. Med. Chem. Lett. 2010, 20(12), 3499-3501.
[32] Gagne, J.; Milot, M.; Gelinas, S.; Lahsaini, A.; Trudeau, F.; Martinoli,M. G.; Massicotte, G. Binding properties of glutamate receptors instreptozotocin-induced diabetes in rats. Diabetes 1997, 46 (5), 841-846.
[33] ZINCPharmer (http://zincpharmer.csb.pitt.edu), an online interface to thePharmer pharmacophore search software for the ZINC database.




















