Unfolding Various Concepts of Junctional Epithelium - Thieme ...
Upload
independentCategory
view
1download
0
doi:10.1006/smim.2001.0347, available online at http://www.idealibrary.com onseminars in IMMUNOLOGY, Vol. 14, 2002: pp. 105–113
Leukocyte transendothelial migration: A junctional affair
Francis W. Luscinskas a,∗, Shuo Ma a, Asma Nusrat b, Charles A. Parkos b andSunil K. Shaw a
A critical function of the inflammatory response is deliveryof leukocytes to a site of injury, immune reaction orinfection. Considerable information is available concerningthe molecular mechanisms that capture flowing leukocytesand initiate their stable arrest on the lumenal surface of theblood vessel wall. In comparison, much less is known aboutthe subsequent step(s) in migration of circulating bloodleukocytes across endothelial cell-to-cell lateral borders tounderlying tissues. This article will focus on the endothelial-dependent processes that coordinate transmigrations inperipheral vasculature during the inflammatory response.
Key words: endothelium / leukocytes / inflammation /transmigration / cell–cell adhesion
c© 2002 Elsevier Science Ltd. All rights reserved.
Introduction
The vascular endothelium lines the entire cardio-vascular system and acts as a nonthrombogenicand selectively permeable boundary betweenthe bloodstream and extravascular space. Thus,the endothelium by virtue of its anatomicallocation is an enormous organ that possesses aunique and influential position during normal andpathophysiological processes.
Much progress has been made in understandingthe mechanisms responsible for inducible surface
From the aVascular Research Division, Departments of Pathology,Brigham and Women’s Hospital and Harvard Medical School, Boston,MA 02115, USA and bDivision of Gastrointestinal Pathology,Department of Pathology and Laboratory Medicine, Emory University,Atlanta, GA 30322, USA. *Corresponding author. Brigham and Women’sHospital, 221 Longwood Avenue LMRC Bldg. Rm 420b, Boston, MA02115, USA. E-mail: [email protected]
c©2002 Elsevier Science Ltd. All rights reserved.1044–5323/02/$ - see front matter
expression of adhesion molecules and chemoat-tractants/chemokines by endothelium, identifyingrelevant constitutively expressed surface adhesionmolecules on leukocytes and confirming the multi-step process (i.e. multi-step adhesion moleculecascade) involved in leukocyte—endothelial adhesiveinteractions. Likewise, a more cohesive picture isavailable regarding the orchestration of the leukocytedependent side of transendothelial migration (TEM)(reviewed in Reference 1). However, much less isknown about later steps in recruitment includingthe endothelial-dependent molecular mechanismsunderlying leukocyte penetration through lateraljunctions and the signal(s) that control this process.This article will discuss the molecules that localizeat endothelial cell contacts emphasizing recentadvances in identifying members of the JAM genefamily. The article will then discuss how thesemolecules may participate in leukocyte TEM anddiscuss the approaches our laboratory has taken tobetter understand the role of endothelial cell-to-celljunctions during TEM using real time imaging andgreen fluorescent protein (GFP) labelled lateraljunction proteins.
Leukocyte TEM is a junctional affair
Leukocyte TEM across peripheral vascular endothe-lium during inflammation usually occurs at the levelof the small diameter postcapillary and collectingvenules, where the endothelium is specialized forexpression of adhesion molecules for leukocytes(reviewed in Reference 2). The lumenal surfaceof these vessels is lined with endothelium, andthe outer surface intersects intimately with muralcells (i.e. pericytes or smooth muscle cells). Thedegree of investment of vessels with mural cellsvaries with the tissue and its matrix compositionand microenvironment. The pioneering studies byFlorey and Marchesi using electron microscopy
105

F. W. Luscinskas et al.
techniques3,4 gave rise to the paradigm thatleukocytes migrate out of vessels at intercellularjunctions.5 More recently, however, very specificexperimental systems have documented thatleukocytes also can migrate through an endothelialcell body (transcytosis).6
Original reports using electron microscopyelegantly described PMN migration at intercellularjunctions of venules.5 Leukocytes were observedextending pseudopods into the interendothelialjunctions, followed by the use of mechanicalleverage gained by pseudopods to squeeze throughthe now widened junctions. The appearance ofstricture points in migrating leukocytes suggestedleukocytes could loosen the endothelial junctionalinterconnections to a limited extent. In thesesituations, the leukocyte and endothelial cellmembranes appeared in close contact and showedprominent cytoskeletal structures and membranevesicles. These features preceded the apparentmovement of endothelial-derived membrane overthe trailing end of the leukocyte, which ultimatelysurrounded and then enveloped the lumenal portionof the leukocyte. The molecular mechanisms orintracellular signals underlying these steps are notcompletely understood although recent reportsstrongly suggest that the endothelium and leukocytecommunicate with one another during TEM andthat preventing certain of these signals impairsTEM (the reader is referred to detailed reviews ofthis literature1,7). Most available evidence pointsto mobilization of intracellular Ca2+ ([Ca2+]i ) andreorganization of actin and myosin and associatedmolecules8–12 in endothelium as critical signals andresponses, respectively, for leukocyte TEM. Multiplereports (reviewed in Reference 7) have documentedthat leukocyte adhesion triggers a rise in endothelial[Ca2+]i and pretreatment with intracellular Ca2+
chelators dampens this rise in [Ca2+]i , essentiallypreventing leukocyte TEM.11 A recent refinement12
using fluorescence imaging of single endothelialcells under defined flow conditions showed thatneutrophil TEM, but not adhesion or rolling events,triggered necessary rises in endothelial [Ca2+]i
that were localized in EC adjacent to the migratingleukocyte. Taken together, this line of investigationhas provided evidence for active participation by theendothelium during leukocyte TEM and also raisesseveral new questions. How does the endotheliumaccommodate passage of one or many leukocytesthrough their lateral junctions without exhibitingfrank signs of damage or gross changes in vessel
Figure 1. Endothelial lateral junctional proteins.Endothelial cells (purple) are schematically shownon a subcellular matrix (blue). Lateral junctionaltransmembrane components are depicted, with theirmembrane topology, ligands, and (some) cytoplasmiccomponents. The peri-junctional actin cytoskeleton linksto both tight junctional and adherens junction complexes.
permeability? Do ‘gaps’ in endothelial cell junctionsform prior to leukocyte TEM? What triggers thesegaps and how do they reseal? Does the passage of oneleukocyte favor the passage of subsequent leukocytes?Answers to such questions remain elusive, butsignificant progress has been made in determiningthe individual molecules that preferentially localizeto the endothelial lateral junctions.
Molecular structure of endothelial cell-to-celllateral junction components
Endothelial cell–cell lateral junctions are less easilydifferentiated into tight and adherens junctionsby light or fluorescence microscopy techniques. Incontrast, epithelial cell junctions are easily distin-guishable. In spite of this appearance, at least threedistinct zones based on morphological and functionalcharacterization are found (1) tight junctions(zona occludens, TJ), (2) adherens junctions (zonaadherens, AJ) and (3) gap junctions (Figure 1). Thesezones are dynamic and are composed of transmem-brane and cytoplasmic molecules that associate withthe cytoskeleton in mature vessels. The tight junctionsare composed of multiple transmembrane proteins(JAM1, occludin, and claudins) and adherensjunctions contain VE-cadherin and its associatedcytosolic molecules, the catenins. The actual numberof tight junctions in the vasculature decreases asvessel diameter narrows. Gap junctions are importantfor inter-endothelial cell communication and are notthought to participate in leukocyte TEM.
106

Endothelial cell junctions and leukocyte diapedesis
Table 1. Identity and expressions of JAM family members
Families Members (GenBank #) Cellular Expression
JAM1 (JAM; F11 receptor; Mouse (U89915) EpithelialMurine Antigen 106) Human (AF111713) Endothlial
Cow (AF111714) WBC, RBCRat (AF276998) Platelet
JAM2 (VE-JAM; hJAM2) Mouse (AF255911) EndothelialHuman (AF255910)
JAM3 (**mJAM-2; hJAM3) Mouse (AJ300304) EndothelialHuman (AF356518) Activated T cells
mJAM-2 was initially identified and cloned in murine tissues but is the murine homolog of hJAM3.
The JAM family of adhesion proteins
JAM1, JAM2 and JAM3 are recently identifiedadhesion molecules belonging to the Ig superfamilyand are type I integral membrane proteins thatpossess an extracellular domain with two Ig loops intandem, a single transmembrane domain and a shortcytoplasmic tail. The cellular expression patternsof JAM members are summarized in Table 1. JAM1is expressed in both epithelium and endotheliumwhere it is concentrated at the tight junctions.13
JAM1 is also expressed by blood cells includingerythrocytes, PMNs, monocytes, lymphocytes andplatelets.14 JAM2 (VE-JAM, hJAM2) is expressed inendothelial cells of high endothelial cell venules15,16
and in venules at certain other tissues. JAM3 isexpressed in endothelium and in activated T cellsubsets.17,18
It has been suggested that JAM1 can formhomodimers and multimers in solution.19 A recentstudy of the crystal structure of JAM1 extracellulardomain revealed a novel dimerization motif.20 Thismotif is conserved among the extracellular domainsof the JAM family members, suggesting a structuralbasis for homodimer formation in other JAMs,although there is no evidence that such structure isrequired for JAM1 function. The short cytoplasmictails of JAMs are conserved, with a PDZ bindingmotif and a protein kinase C consensus site near theC-terminus. This PDZ binding motif is implicatedin JAM1 association with several PDZ domain-containing proteins at the intercellular junctions,including ZO-1, AF-6, ASIP/PAR-3 and CASK.21–25
ZO-1 and AF-6 are important components of theTJ. A mutant JAM1 lacking the PDZ-binding motiffailed to locate to cell–cell contacts, suggesting arole of AF-6 and ZO-1 in recruiting and stabilizing
JAM1 to site of junction formation.21 PAR proteinsare determinants of asymmetric cell division andpolarized cell growth. The interaction of JAM1 withthe PAR-3 signaling complex suggests a potential roleof JAM in generating and maintaining cell polarity inepithelial cells.23,25 As JAM2 and JAM3 also containthe conserved PDZ-binding motif, it is conceivablethat they might also have similar associations with thePDZ domain-containing proteins and thus junctionallocalization. At this point is it important to note thatthe study of JAM is evolving and further study of JAMfamily members in a variety of models is crucial to ourunderstanding their roles in physiological processes.
JAM1 in regulation of tight junction assembly andparacellular permeability
All three JAM members are targeted to cell–cell contacts and decrease cell permeability (i.e.enhance barrier function) in CHO transfectionexperiments. Because JAM1 localizes to TJ ofepithelium and endothelium and associates withseveral proteins containing the PDZ binding motif,it is not surprising that JAM1 has been implicatedin regulation of paracellular barrier function and TJformation in epithelial cells. Monoclonal antibodiesto JAM1 markedly inhibited transepithelial resistancerecovery of epithelial monolayers after disruptionof intercellular junctions by transient calciumdepletion,14 or by trypsin-EDTA treatment.26
Consistent with this inhibiting effect, JAM1 mAb alsoinhibited the redistribution of JAM1 and occludinto tight junction following recovery, suggesting arole for JAM1 in the regulation of tight junctionreassembly.14 Interestingly, ZO-1 and E-cadherinwere shown to be redistributed normally, despiteJAM1 and occludin disruption, suggesting different
107

F. W. Luscinskas et al.
mechanisms of JAM1 regulation of TJ reassemblywithin confluent monolayers.
JAM1 regulation of leukocyte TEM
JAM1 was first described as a junctional adhesionmolecule involved in regulating leukocyte TEMin murine models of inflammation. mAb BV11, amonoclonal antibody to murine JAM1, inhibitedmonocyte TEM in a chemotaxis assay in vitro andblocked monocyte infiltration in vivo in subcutaneousair pouches in mice.13 mAb BV11 also inhibitedrecruitment of both PMN and monocytes in amurine model of cytokine-induced experimentalmeningitis.27 However, the same mAb failed toprevent leukocyte influx into the meninges in aninfectious meningitis model in mice after viral orbacterial infection,28 suggesting the involvementof JAM1 in leukocyte TEM is not universal. Theregulatory effect of JAM1 on leukocyte TEM hasbeen examined recently in a human system. Thecombined treatment of TNF-α and IFN-γ wereshown to cause reduced expression of JAM1 as wellas PECAM-1 and redistribution away from the celljunctions in HUVEC29 and hence led to decreasedTEM under static assay conditions. However, ourlab has shown that this reduced level of JAM1 andPECAM-1 did not affect the efficiency of monocyte orPMN TEM when assayed under flow conditions.30 Inaddition, several different monoclonal and polyclonalantibodies against human JAM1 had no significanteffect on leukocyte migration across epithelialmonolayers in the basolateral to apical direction.14
Nor did they have any effect on PMN TEM underflow.30 On the other hand, a recent article55 hasimplicated leukocyte LFA-1 (CD11a/CD18) as abinding partner for endothelial JAM1. A blockingmAb to JAM1 reduced LFA-1 dependent T celland PMN transmigration and LFA-1 dependentT cell adhesion under flow. Clearly more work isnecessary to determine the role of JAM molecules ininflammation in human tissues.
In another context, JAM1 binds directly toreovirus σ1 head domain with high affinity andserves as a reovirus receptor capable of mediatingvirus attachment, infection, and intracellularsignaling.31 JAM1 is also the platelet receptorfor F11, an activating antibody that induced granularsecretion and aggregation in human platelets.32
Platelet activation by various stimuli causes JAM1phosphorylation via protein kinase C, indicating arole of JAM1 in platelet signaling.33
JAM2–JAM3 heterophilic interactions
One of the most interesting aspects of JAM2 andJAM3 is that they appear to be binding partners.Immobilized recombinant JAM2 is able to captureseveral T cell lines in static adhesion assays. Clearlythe adhesion is not through homophilic interactions,because JAM2 itself is not present on these T cells.16
It was later discovered that JAM3 is the counter-receptor that mediates JAM2 adhesion to T cells.17
This is the first example of a heterophilic interactionbetween different members of the JAM family. Thephysiological significance of JAM2–JAM3 interactionis as yet unknown and requires further study.Because JAM2 is expressed on endothelium in highendothelial venules in secondary lymphoid organs,one intriguing possibility is that JAM2 participatesin lymphocyte subset homing via interaction withJAM3.
Occludin
Occludin is a 65 kDa protein that is present attight junctions of epithelial and endothelial cells.34
Sequence analysis indicates four transmembraneregions, suggesting a topology of two extracellularloops, with a short cytoplasmic N-terminus, and along cytoplasmic C-terminal tail. The C-terminaltail associates with several cytoplasmic componentsincluding zona occludens-(ZO-)1, -2 and -3,35 whichin turn link to the actin cytoskeleton. The role ofoccludin in regulation of paracellular permeability isnot yet completely understood. A dominant mutantof occludin disrupts tight junction structure andfunction.36 On the other hand, in mice lackingoccludin,37 impedance analysis of cultured intestinalepithelium monolayers revealed no significant differ-ence in occludin−/− mice compared to normal mice.However, the baseline resistance values were low andparacellular flux studies were not performed. Furtherstudies are needed to define its exact contribution tolateral junction structure and function.
Claudins
Claudins are a family of proteins (18 members andgrowing) that contain a similar membrane topologybut share little sequence homology with occludin.Claudin-5 is expressed primarily in endothelialcells.38 It is expressed in brain and lung endothelialcells, but only in some segments of blood vessels inother tissues. Like occludin, the C-terminal tail of the
108

Endothelial cell junctions and leukocyte diapedesis
claudins associate with ZO-1, -2 and -3.39 Claudinshave been shown to reconstitute tight junctionalstrands by transfection experiments,40 and arethought to play an important role in permeability tomacromolecules. Surprisingly, in specific instances,transfection of a claudin increases monolayerpermeability in epithelial cell monolayers.41 Itmay be a result of heterophilic adhesion betweendifferent claudins, possibly leading to pore formationand poor barrier function.
VE-cadherin
VE-cadherin is a member of the cadherin superfamily(Cadherin-5).42 Classical cadherins are transmem-brane proteins with a conserved cytoplasmic domainand an extracellular domain consisting of five repeatmotifs. The extracellular domain is responsiblefor Ca2+ dependent homophilic adhesion, whilethe cytoplasmic tail provides a linkage to the actincytoskeleton via α- and β-catenins, plakoglobin, andp120 in endothelium.43 This linkage is critical forstable adhesion because deletion of the cadherincytoplasmic tail results in greatly diminishedadhesive properties, while overexpression results incompetition for cytosolic proteins with endogenousfull-length cadherin and disruption of their functionvia a dominant negative mechanism.44 VE-cadherinis expressed at endothelial lateral junctions, andplays a major role in maintaining barrier function tomacromolecules. Antibodies to VE-cadherin cause anincrease in permeability in vitro and in vivo.42,45,46
Transfection of the cDNA into heterologous celltypes likewise increases barrier function.
Platelet/endothelial cell adhesion molecule-1
Platelet/endothelial cell adhesion molecule-1(PECAM-1, CD31) is a type I transmembrane proteinof the Ig gene superfamily. PECAM-1 is localizedto endothelial cell lateral junctions and has beendemonstrated to be involved in leukocyte TEM. Itsrole in TEM has been covered in many reviews (seeRef. 7 and articles therein) and will not be covered infurther detail here.
Leukocyte TEM triggers alterations in AJcomponents
The components of the AJ are of particular interestbecause they appear to be the predominant mech-
anisms regulating permeability of macromoleculesin microvascular endothelium. Thus, we initiallyconsidered the hypothesis that leukocyte TEMtransiently alters the VE-cadherin complex, causing atransient and localized ‘gap’, and thus accommodateleukocyte passage. Indeed, the first studies (DelMaschio and coworkers47 and subsequently byAllport and colleagues48) to examine this conceptfound dramatic alterations in the VE-cadherincomplex as a result of PMN leukocyte adhesion,namely loss of VE-cadherin and its associated cateninsas assessed by immunofluorescence microscopyand protein gel electrophoresis/blotting. Furthersupporting evidence came from both in vivo andin vitro studies that found that antibodies to VE-cadherin led to increases in permeability andgreater leukocyte TEM.45,46 These data led to amodel of VE-cadherin functioning as a gatekeeperfor leukocyte TEM. Hence, leukocyte adhesiontriggered disassembly of VE-cadherin complex,allowing adherent leukocytes to migrate throughthe newly created gap. Subsequently, Moll andcolleagues49 reported that adhesion dependentloss of catenins was not be due to specific signalingevents, but rather due to leukocyte proteases releasedduring post-assay incubations (i.e. non-physiologicalproteolysis). PMN contain abundant proteases andCarden and colleagues50 had shown that activatedPMN or purified PMN elastase cleaves VE-cadherinin vitro. Nonetheless, we considered the possibilitythat some form of disruption of the VE-cadherincomplex occurs and constitutes a physiologicalstep during TEM. Our lab51 examined TEM ofmonocytes and the U937 leukocyte cell line, bothof which lack the PMN serine protease elastase andalso performed experiments under defined flowconditions rather than static conditions as had beenused before. We found that adherent/transmigratingmonocytes or U937 cells induced focal and transientloss of the VE-cadherin complex, that antibodiesthat inhibit TEM, but not adhesion, blocked lossof VE-cadherin and that the gaps appeared toreseal after TEM. However, like earlier studies,post-assay degradation of junctional proteinsremained a possibility and the technique couldnot establish accurate kinetics of the exact cellularevents or kinetics of leukocyte TEM. Meanwhile,alternative hypotheses of leukocyte TEM wereforwarded by other groups. In an in vivo model,chemoattractant induced PMN TEM occurredvia transcellular migration, rather than at lateraljunctions (paracellular), obviating the need to
109
F. W. Luscinskas et al.
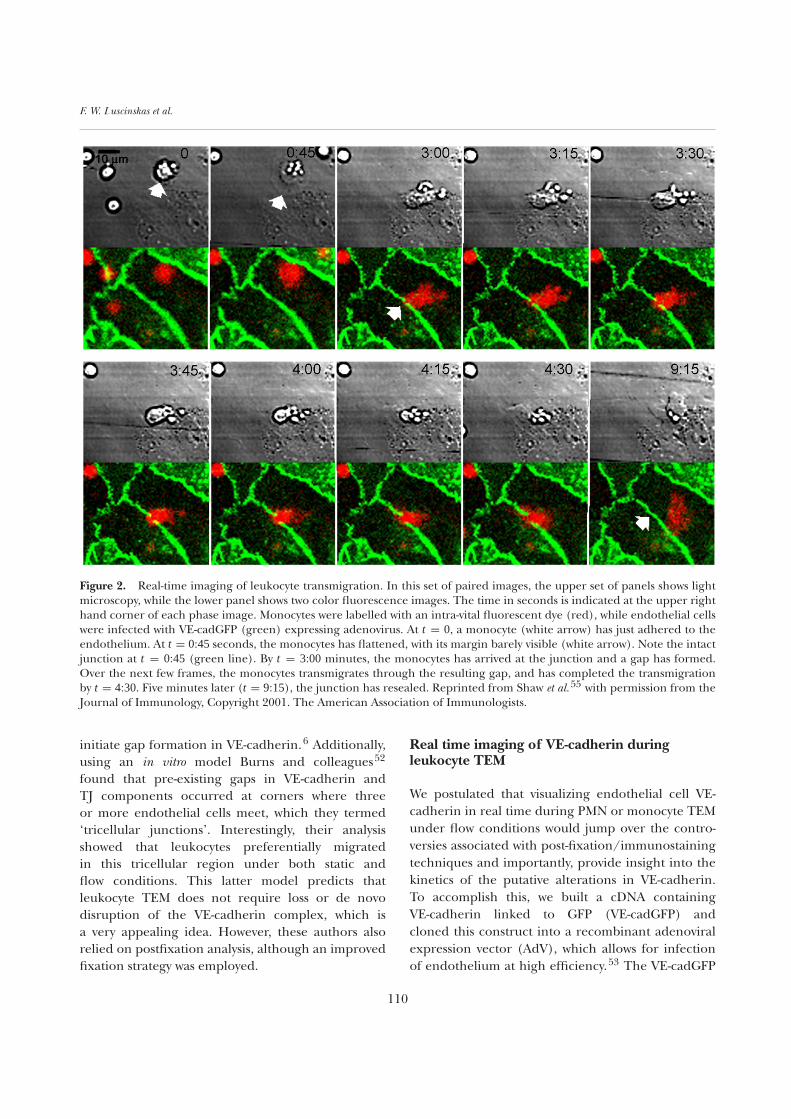
Figure 2. Real-time imaging of leukocyte transmigration. In this set of paired images, the upper set of panels shows lightmicroscopy, while the lower panel shows two color fluorescence images. The time in seconds is indicated at the upper righthand corner of each phase image. Monocytes were labelled with an intra-vital fluorescent dye (red), while endothelial cellswere infected with VE-cadGFP (green) expressing adenovirus. At t = 0, a monocyte (white arrow) has just adhered to theendothelium. At t = 0:45 seconds, the monocytes has flattened, with its margin barely visible (white arrow). Note the intactjunction at t = 0:45 (green line). By t = 3:00 minutes, the monocytes has arrived at the junction and a gap has formed.Over the next few frames, the monocytes transmigrates through the resulting gap, and has completed the transmigrationby t = 4:30. Five minutes later (t = 9:15), the junction has resealed. Reprinted from Shaw et al.55 with permission from theJournal of Immunology, Copyright 2001. The American Association of Immunologists.
initiate gap formation in VE-cadherin.6 Additionally,using an in vitro model Burns and colleagues52
found that pre-existing gaps in VE-cadherin andTJ components occurred at corners where threeor more endothelial cells meet, which they termed‘tricellular junctions’. Interestingly, their analysisshowed that leukocytes preferentially migratedin this tricellular region under both static andflow conditions. This latter model predicts thatleukocyte TEM does not require loss or de novodisruption of the VE-cadherin complex, which isa very appealing idea. However, these authors alsorelied on postfixation analysis, although an improvedfixation strategy was employed.
Real time imaging of VE-cadherin duringleukocyte TEM
We postulated that visualizing endothelial cell VE-cadherin in real time during PMN or monocyte TEMunder flow conditions would jump over the contro-versies associated with post-fixation/immunostainingtechniques and importantly, provide insight into thekinetics of the putative alterations in VE-cadherin.To accomplish this, we built a cDNA containingVE-cadherin linked to GFP (VE-cadGFP) andcloned this construct into a recombinant adenoviralexpression vector (AdV), which allows for infectionof endothelium at high efficiency.53 The VE-cadGFP
110

Endothelial cell junctions and leukocyte diapedesis
fusion protein was shown by biochemical andfunctional criteria (e.g. localization to cell–celljunctions and enhanced permeability function inendothelium) to mimic the behavior of nativeendothelial cell VE-cadherin.54 The cassette oftime stamped confocal images shown in Figure 2is representative of many experiments wheremonocytes (labelled in red) crawled to endothelialcell–cell junctions bearing a continuous junctionof VE-cadGFP, triggered gaps in VE-cadherin andthen migrated through so formed gaps. As depictedin the last two panels, the resealing required ∼5minutes. The companion differential interferencecontrast microscopy images (Figure 2, top panels)provide vivid details showing the multistep processof monocyte arrest, spreading and extension oflamellipodia into cell–cell junctions and TEM (seeFigure 2 legend). Monocytes and PMNs behavedessentially the same in this system. Typically, thegaps formed in VE-cadGFP were on the order of4–6 µm. Interestingly, 36% of the time PMNs wereassociated with de novo gap formation while inthe remainder of the observations 64% of PMNsmigrated through pre-existing gaps. This figureis consistent with the previous reports of Burnset al. using postfixation staining techniques.52 Formonocytes, the result was just the opposite (60%de novo gaps, 40% pre-existing gaps). However, forpre-existing gaps, transmigrating monocytes andPMNs caused significant widening of the gaps inVE-cadGFP. It is also worth noting that gaps presentbefore the approach of a leukocyte were still able toreseal, suggesting that the VE-cadherin complex isvery dynamic and appears to be actively regulated.In this system we did not observe transcellularmigration of PMNs or monocytes. Thus, the greensignal for VE-cadherin is lost only at sites of activeleukocyte migration (Figure 3) while mere adhesionof leukocytes, even at a lateral junction, does notdisrupt the VE-cadGFP signal.
There are several conclusions that follow from thislatter series of experiments using VE-cadGFP. First,for leukocyte TEM a pre-existing gap or de novogap formed as a result of leukocyte adhesion wasnecessary for TEM. Second, de novo gap formation orwidening of gaps occurred only when the leukocytearrived at the junction. Third, that leukocyte TEMalways occurred at cell–cell junctions (paracellular)and was not observed to proceed by a transcellularroute in this system. Lastly, that specific leukocytetypes may prefer multicellular (PMN) versus bicel-lular (monocytes) endothelial cell borders, and in
Figure 3. Schematic of VE-cadherin distribution duringleukocyte transmigration. In the upper half of thisimage, a endothelial monolayer is shown en face. Twoleukocytes are shown, one is transmigrating, while theother leukocyte is not. The transmigrating leukocyte hasmade a gap in junctional VE-cadherin, while an intactVE-cadherin junction is visible through the body of thenon-transmigrating leukocyte. A section of this monolayerthrough the dotted line is shown in the bottom half. Thenon-transmigrating leukocyte is shown resting above thejunction, while the transmigrating leukocyte has piercedthe junction, and VE-cadherin is no longer present in theplane of this section.
regard to the type of gap formed, monocytes prefer-entially initiated de novo gap formation as comparedto PMN. Future studies are needed to assess the kinet-ics of gap formation and resealing under differentlevels of defined fluid shear stress. Certainly it will beimportant to investigate the processes that drive VE-cadherin gap formation and resealing in the presenceor absence of leukocyte TEM. Our approach usingGFP-tagged lateral junction proteins should allowfor determination of changes in other junctionalproteins during leukocyte TEM. One would also liketo monitor simultaneously a junctional molecule anda cytoskeletal component to gain insight into howthese two components may be coordinate in a spatialand temporal fashion during TEM events.
Conclusions
The studies reviewed above indicate that endothe-lium actively participates in the process of leukocyteTEM. Here we have described our laboratory’s recentwork on the dynamics of endothelial cell adherensjunction component VE-cadGFP during leukocyteTEM under defined fluid shear conditions. Theformation of de novo gaps or pre-existing gaps inVE-cadherin provides one mechanism for traffickingof leukocytes through postcapillary vessels and laysthe technical ground work for determining whether
111

F. W. Luscinskas et al.
leukocytes can modify tight junctions to gain passageor whether leukocytes seek out areas in the vesselwall that have preexisting gaps. Lastly we assume thatapplications of new techniques or novel strategies willallow a finer resolution of the process of leukocyteTEM across endothelium and no doubt a betterunderstanding of the mechanisms underlying thiscritical component of innate immunity.
Acknowledgements
The authors would like to thank our colleagues inthe Department of Pathology, Brigham and Women’sHospital for their helpful discussions. The authors’laboratories were supported by grants KO1-DK02398,PO1-HL-36028, RO-1 HL-53993, RO-1 HL65090, RO-1 HL54229, and PO-1 HL-56985 from the N.H.L.B.I.,National Institutes of Health, Washington, DC, andresearch grants from the Arthritis Foundation andCrohns and Colitis Foundation of America.
References
1. Worthylake RA, Burridge K (2001) Leukocyte transendothe-lial migration: orchestrating the underlying molecularmachinery. Curr Opin Cell Biol 13:569–577
2. Cotran RS (1999) Robbins Pathological Basis of Disease,pp. 50–89. WB Saunders Co, Philadelphia
3. Marchesi VT (1961) The site of leukocyte emigration duringinflammation. Quart J Exp Physiol 46:115–118
4. Marchesi VT, Florey HW (1960) Electron microscopicobservation on the emigration of leukocytes. Quart J ExpPhysiol 45:343–348
5. Granger HJ, Yuan Y, Zawieja DC (1995) Ultrastructural basisof leukocyte migration through the microvascular membrane,in Physiology and Pathophysiology of Leukocyte Adhesion(Granger DN, Schmid-Schoenbien GW, eds) pp. 185–195.Oxford University Press, New York
6. Feng D, Nagy JA, Pyne K, Dvorak HF, Dvorak AM (1998)Neutrophils emigrate from venules by a transendothelial cellpathway in response to FMLP. J Exp Med 187:903–915
7. Johnson-Leger C, Aurrand-Lions M, Imhof BA (2000) Theparting of the endothelium: miracle, or simply a junctionalaffair? J Cell Sci 113:921–933
8. Lorenzon P, Vecile E, Nardon E, Ferrero E, Harlan JM,Tedesco F, Dobrina A (1998) Endothelial cell E- and P-selectinand vascular cell adhesion molecule-1 function as signalingreceptors. J Cell Biol 142:1381–1391
9. Yoshida M, Szente BE, Kiely JM, Rosenzweig A, Gim-brone MA (1998) Phosphorylation of the cytoplasmic domainof E-selectin is regulated during leukocyte-endothelial adhe-sion. J Immunol 161:933–941
10. Yoshida M, Westlin WF, Wang N, Ingber DE, Rosenzweig A,Resnick N, Gimbrone MA Jr (1996) Leukocyte adhesion tovascular endothelium induces E-selectin linkage to the actincytoskeleton. J Cell Biol 133:445–455
11. Huang AJ, Manning JE, Bandak TM, Ratau MC, Hanser KR,
Silverstein SC (1993) Endothelial cell cytosolic free calciumregulates neutrophil migration across monolayers of endothe-lial cells. J Cell Biol 120:1371–1380
12. Su WH, Chen HI, Huang JP, Jen CJ (2000) Endothelial[Ca2+]i signaling during transmigration of polymorphonu-clear leukocytes. Blood 96:3816–3822
13. Martin-Padura I et al. (1998) Junctional adhesion molecule,a novel member of the immunoglobulin superfamily thatdistributes at intercellular junctions and modulates monocytetransmigration. J Cell Biol 142:117–127
14. Liu Y, Nusrat A, Schnell FJ, Reaves TA, Walsh S, Pochet M,Parkos CA (2000) Human junction adhesion moleculeregulates tight junction resealing in epithelia. J Cell Sci113:2363–2374
15. Palmeri D, van Zante A, Huang CC, Hemmerich S,Rosen SD (2000) Vascular endothelial junction-associatedmolecule, a novel member of the immunoglobulin superfam-ily, is localized to intercellular boundaries of endothelial cells.J Biol Chem 275:19139–19144
16. Cunningham SA, Arrate MP, Rodriguez JM, Bjercke RJ,Vanderslice P, Morris AP, Brock TA (2000) A novelprotein with homology to the junctional adhesion molecule.Characterization of leukocyte interactions. J Biol Chem275:34750–34756
17. Arrate MP, Rodriguez JM, Tran TM, Brock TA, Cunning-ham SA (2001) Cloning of human junctional adhesionmolecule 3 (JAM3) and its identification as the JAM2 counter-receptor. J Biol Chem 276:45826–45832
18. Aurrand-Lions MA, Duncan L, Ballestrem C,Imhof BA (2000) JAM-2, a novel immunoglobulin superfamilymolecule, expressed by endothelial and lymphatic cells. J BiolChem 276:2733–2741
19. Bazzoni G, Martinez-Estrada OM, Mueller F, Nelboeck P,Schmid G, Bartfai T, Dejana E, Brockhaus M (2000)Homophilic interaction of junctional adhesion molecule.J Biol Chem 275:30970–30976
20. Kostrewa D et al. (2001) X-ray structure of junctional adhesionmolecule: structural basis for homophilic adhesion via anovel dimerization motif. EMBO J 20:4391–4398
21. Ebnet K, Schulz CU, Meyer ZBM, Pendl GG, Vestwe-ber D (2000) Junctional adhesion molecule interacts with thePDZ domain-containing proteins AF-6 and ZO-1. J Biol Chem275:27979–27988
22. Bazzoni G, Martinez-Estrada OM, Orsenigo F, Cordenonsi M,Citi S, Dejana E (2000) Interaction of junctional adhesionmolecule with the tight junction components ZO-1, cingulin,and occludin. J Biol Chem 275:20520–20526
23. Ebnet K, Suzuki A, Horikoshi Y, Hirose T, Meyer ZBM,Ohno S, Vestweber D (2001) The cell polarity proteinASIP/PAR-3 directly associates with junctional adhesionmolecule (JAM). EMBO J 20:3738–3748
24. Martinez-Estrada OM, Villa A, Breviario F, Orsenigo F,Dejana E, Bazzoni G (2000) Association of junctional adhe-sion molecule with calcium/calmodulin-dependent serineprotein kinase (CASK/LIN-2) in human epithelial Caco-2cells. J Biol Chem 276:9291–9296
25. Itoh M, Sasaki H, Furuse M, Ozaki H, Kita T, Tsukita S (2001)Junctional adhesion molecule (JAM) binds to PAR-3:a possible mechanism for the recruitment of PAR-3 totight junctions. J Cell Biol 154:491–497
26. Liang TW et al. (2000) Characterization of huJAM: evidencefor involvement in cell–cell contact and tight junctionregulation. Am J Physiol 279:C1733–C1743
27. Del Maschio A et al. (1999) Leukocyte recruitment in thecerebrospinal fluid of mice with experimental meningitis is
112

Endothelial cell junctions and leukocyte diapedesis
inhibited by an antibody to junctional adhesion molecule(JAM). J Exp Med 190:1351–1356
28. Lechner F, Sahrbacher U, Suter T, Frei K, Brockhaus M,Koedel U, Fontana A (2000) Antibodies to the junctionaladhesion molecule cause disruption of endothelial cells anddo not prevent leukocyte influx into the meninges after viralor bacterial infection. J Infect Dis 182:978–982
29. Ozaki H, Ishii K, Horiuchi H, Arai H, Kawamoto T,Okawa K, Iwamatsu A, Kita T (1999) Cutting edge: Combinedtreatment of TNF-alpha and IFN-gamma causes redistributionof junctional adhesion molecule in human endothelial cells.J Immunol 163:553–557
30. Shaw SK, Perkins BA, Lim Y-C, Liu Y, Nusrat A, Schnell FJ,Parkos CA, Luscinskas FW (2001) Reduced expression ofjunctional adhesion molecule and platelet/endothelial celladhesion molecule-1 (CD31) at human vascular endothelialjunctions by cytokines tumor necrosis factor-α plus interferon-γ does not reduce leukocyte transmigration under flow. Am JPath 159:2281–2291
31. Barton ES, Forrest JC, Connolly JL, Chappell JD, Liu Y,Schnell FJ, Nusrat A, Parkos CA, Dermody TS (2001)Junction Adhesion Molecule is a receptor for reovirus. Cell104:441–451
32. Sobocka MB et al. (2000) Cloning of the human plateletF11 receptor: a cell adhesion molecule member of theimmunoglobulin superfamily involved in platelet aggrega-tion. Blood 95:2600–2609
33. Ozaki H, Ishii K, Arai H, Horiuchi H, Kawamoto T, Suzuki H,Kita T (2000) Junctional adhesion molecule (JAM) isphosphorylated by protein kinase C upon platelet activation.Biochem Biophys Res Commun 276:873–878
34. Furuse M, Hirase T, Itoh M, Nagafuchi A, Yonemura S,Tsukita S (1993) Occludin: a novel integral membraneprotein localizing at tight junctions. J Cell Biol 123:1777–1788
35. Tsukita S, Furuse M, Itoh M Structural and signalingmolecules come together at tight junctions. Curr Opin CellBiol 11:628–633
36. Bamforth SD, Kniesel U, Wolburg H, Engelhardt B,Risau W (1999) A dominant mutant of occludin disrupts tightjunction structure and function. J Cell Sci 112:1879–1888
37. Saitou M, Furuse M, Sasaki H, Schulzke JD, Fromm M,Takano H, Noda T, Tsukita S (2000) Complex phenotype ofmice lacking occludin, a component of tight junction strands.Mol Biol Cell 11:4131–4142
38. Morita K, Sasaki H, Furuse M, Tsukita S (1999) EndothelialClaudin: claudin-5/TMVCF constitutes tight junction strandsin endothelial cells. J Cell Biol 147:185–194
39. Itoh M, Furuse M, Morita K, Kubota K, Saitou M,Tsukita S (1999) Direct binding of three tight junction-associated MAGUKs, ZO-1, ZO-2, and ZO-3, with the COOHtermini of claudins. J Cell Biol 147:1351–1363
40. Furuse M, Sasaki H, Fujimoto K, Tsukita S (1998) A singlegene product, claudin-1 or -2, reconstitutes tight junctionstrands and recruits occludin in fibroblasts. J Cell Biol 143:391
41. Furuse M, Furuse K, Sasaki H, Tsukita S (2001) Conversionof zonulae occludentes from tight to leaky strand type byintroducing claudin-2 into Madin-Darby canine kidney I cells.J Cell Biol 153:263–272
42. Lampugnani MG, Resnati M, Raiteri M, Pigott R, Pisacane A,Houen G, Ruco LP, Dejana E (1992) A novel endothelial-specific membrane protein is a marker of cell–cell contacts.J Cell Biol 118:1511–1522
43. Lampugnani MG, Corada M, Caveda L, Breviario F, Ayalon O,Geiger B, Dejana E (1995) The molecular organizationof endothelial cell to cell junctions: differential associationof plakoglobin, β -catenin, and α-catenin with vascularendothelial cadherin (VE-cadherin). J Cell Biol 129:203–214
44. Navarro P, Caveda L, Breviario F, Mandoteanu I, Lam-pugnani MG, Dejana E (1995) Catenin-dependent and-independent functions of vascular endothelial cadherin.J Biol Chem 270:30965–30972
45. Gotsch U, Borges E, Bosse R, Boggemeyer E, Simon M,Mossmann H, Vestweber D (1997) VE-cadherin antibodyaccelerates neutrophil recruitment in vivo. J Cell Sci110:583–588
46. Hordijk PL, Anthony E, Mul FP, Rientsma R, Oomen LC,Roos D (1999) Vascular-endothelial-cadherin modulatesendothelial monolayer permeability. J Cell Sci 112:1915–1923
47. Del Maschio A, Zanetti A, Corada M, Rival Y, Ruco L,Lampugnani MG, Dejana E (1996) Polymorphonuclearleukocyte adhesion triggers the disorganization of endothelialcell-to-cell adherens junctions. J Cell Biol 135:497–509
48. Allport JR, Ding H, Collins T, Gerritsen ME, Luscin-skas FW (1997) Endothelial-dependent mechanisms regulateleukocyte transmigration: a process involving the proteasomeand disruption of the vascular endothelial-cadherin complexat endothelial cell- to-cell junctions. J Exp Med 186:517–528
49. Moll T, Dejana E, Vestweber D (1998) In vitro degradationof endothelial catenins by a neutrophil protease. J Cell Biol140:403–407
50. Carden D, Xiao F, Moak C, Willis BH, Robinson-Jackson S,Alexander S (1998) Neutrophil elastase promotes lungmicrovascular injury and proteolysis of endothelial cadherins.Am J Physiol 275:H385–H392
51. Allport JR, Ding H, Muller WA, Luscinskas FW (2000) Mono-cytes induce reversible focal changes in VE-cadherin complexduring transendothelial migration under flow. J Cell Biol147:203–216
52. Burns AR, Bowden RA, MacDonell SD, Walker DC,Odebunmi TO, Donnachie EM, Simon SI, Entman ML,Smith CW (2000) Analysis of tight junctions during neu-trophil transendothelial migration. J Cell Sci 113:45–57
53. Gerszten RE, Luscinskas FW, Ding HT, Dichek DA, Stool-man LM, Gimbrone MA Jr, Rosenzweig A (1996) Adhesionof memory lymphocytes to vascular cell adhesion molecule-1- transduced human vascular endothelial cells under sim-ulated physiological flow conditions in vitro. Circ Res 79:1205–1215
54. Shaw SK, Bamba PS, Perkins BN, Luscinskas FW (2001)Real-time imaging of vascular endothelial-cadherin duringleukocyte transmigration across endothelium. J Immunol167:2323–2330
55. Ostermann G, Weber KSC, Zernecke A, Schroder A,Weber C (2002) JAM-1 is a ligand of the β2 integrin LFA-1 involved in transendothelial migration of leukocytes. NatureImmunol 3:151–158
113
Copyright © 2022 FDOKUMEN