
West Nile virus-induced disruption of the blood-brain barrier in mice is characterized by the...
11
Downloaded from www.microbiologyresearch.org by IP: 54.160.113.5 On: Tue, 07 Jun 2016 05:53:10 West Nile virus-induced disruption of the blood– brain barrier in mice is characterized by the degradation of the junctional complex proteins and increase in multiple matrix metalloproteinases Kelsey Roe,3 Mukesh Kumar,3 Stephanie Lum, Beverly Orillo, Vivek R. Nerurkar and Saguna Verma Correspondence Saguna Verma [email protected] Received 3 January 2012 Accepted 2 March 2012 Department of Tropical Medicine, Medical Microbiology and Pharmacology, Pacific Center for Emerging Infectious Diseases Research, John A. Burns School of Medicine, University of Hawaii at Manoa, Honolulu, Hawaii 96813, USA West Nile virus (WNV) encephalitis is characterized by neuroinflammation, neuronal loss and blood–brain barrier (BBB) disruption. However, the mechanisms associated with the BBB disruption are unclear. Complex interactions between the tight junction proteins (TJP) and the adherens junction proteins (AJP) of the brain microvascular endothelial cells are responsible for maintaining the BBB integrity. Herein, we characterized the relationship between the BBB disruption and expression kinetics of key TJP, AJP and matrix metalloproteinases (MMPs) in the mice brain. A dramatic increase in the BBB permeability and extravasation of IgG was observed at later time points of the central nervous system (CNS) infection and did not precede virus–CNS entry. WNV-infected mice exhibited significant reduction in the protein levels of the TJP ZO-1, claudin-1, occludin and JAM-A, and AJP b-catenin and vascular endothelial cadherin, which correlated with increased levels of MMP-1, -3 and -9 and infiltrated leukocytes in the brain. Further, intracranial inoculation of WNV also demonstrated increased extravasation of IgG in the brain, suggesting the role of virus replication in the CNS in BBB disruption. These data suggest that altered expression of junction proteins is a pathological event associated with WNV infection and may explain the molecular basis of BBB disruption. We propose that WNV initially enters CNS without altering the BBB integrity and later virus replication in the brain initiates BBB disruption, allowing enhanced infiltration of immune cells and contribute to virus neuroinvasion via the ‘Trojan-horse’ route. These data further implicate roles of multiple MMPs in the BBB disruption and strategies to interrupt this process may influence the WNV disease outcome. INTRODUCTION West Nile virus (WNV) is a mosquito-borne neurotropic flavivirus responsible for the largest outbreak of viral meningoencephalitis in the Western Hemisphere since its appearance in New York in 1999. A subset of WNV patients with febrile illness progress to encephalitis (Davis et al., 2006; Gubler, 2007) and neurological disease syndrome is observed in approximately 30 % of confirmed WNV cases with a higher frequency in the elderly and immunocompromised (Campbell et al., 2002; Hayes & Gubler, 2006). No specific therapy for WNV is currently approved for use in humans. In the mouse model, WNV- associated neurological disease is characterized by disrup- tion of the blood–brain barrier (BBB), increased infiltra- tion of immune cells into the central nervous system (CNS), production of inflammatory cytokines, microglia activation and eventual loss of neurons (Glass et al., 2005; Klein et al., 2005; Samuel & Diamond, 2006). WNV entry into the CNS represents an important event in the clinical outcome of WNV. The proposed routes of entry are by crossing the BBB and by retrograde axonal transport via peripheral nervous system (Samuel et al., 2007). Since high viraemia correlates with early WNV entry into the CNS, haematogenous route via crossing the BBB is suggested to be one of the major routes by which WNV enters the CNS (King et al., 2007) although the associated mechanisms are yet unclear. We previously demonstrated in vitro that WNV can infect and efficiently replicate in human brain microvascular endothelial cells, which line the BBB, and can be one of the routes of virus entry into the CNS without affecting the BBB integrity (Verma et al., 2009). The BBB, which is the interface between circulating blood and the CNS, is composed of specialized microvascular 3These authors contributed equally to this paper. A supplementary table is available with the online version of this paper. Journal of General Virology (2012), 93, 1193–1203 DOI 10.1099/vir.0.040899-0 040899 G 2012 SGM Printed in Great Britain 1193
-
Upload
independent -
Category
Documents
-
view
8 -
download
0
Transcript of West Nile virus-induced disruption of the blood-brain barrier in mice is characterized by the...

Downloaded from www.microbiologyresearch.org by
IP: 54.160.113.5
On: Tue, 07 Jun 2016 05:53:10
West Nile virus-induced disruption of the blood–brain barrier in mice is characterized by thedegradation of the junctional complex proteins andincrease in multiple matrix metalloproteinases
Kelsey Roe,3 Mukesh Kumar,3 Stephanie Lum, Beverly Orillo,Vivek R. Nerurkar and Saguna Verma
Correspondence
Saguna Verma
Received 3 January 2012
Accepted 2 March 2012
Department of Tropical Medicine, Medical Microbiology and Pharmacology, Pacific Center forEmerging Infectious Diseases Research, John A. Burns School of Medicine, University of Hawaii atManoa, Honolulu, Hawaii 96813, USA
West Nile virus (WNV) encephalitis is characterized by neuroinflammation, neuronal loss and
blood–brain barrier (BBB) disruption. However, the mechanisms associated with the BBB
disruption are unclear. Complex interactions between the tight junction proteins (TJP) and the
adherens junction proteins (AJP) of the brain microvascular endothelial cells are responsible for
maintaining the BBB integrity. Herein, we characterized the relationship between the BBB
disruption and expression kinetics of key TJP, AJP and matrix metalloproteinases (MMPs) in the
mice brain. A dramatic increase in the BBB permeability and extravasation of IgG was observed at
later time points of the central nervous system (CNS) infection and did not precede virus–CNS
entry. WNV-infected mice exhibited significant reduction in the protein levels of the TJP ZO-1,
claudin-1, occludin and JAM-A, and AJP b-catenin and vascular endothelial cadherin, which
correlated with increased levels of MMP-1, -3 and -9 and infiltrated leukocytes in the brain.
Further, intracranial inoculation of WNV also demonstrated increased extravasation of IgG in the
brain, suggesting the role of virus replication in the CNS in BBB disruption. These data suggest
that altered expression of junction proteins is a pathological event associated with WNV infection
and may explain the molecular basis of BBB disruption. We propose that WNV initially enters
CNS without altering the BBB integrity and later virus replication in the brain initiates BBB
disruption, allowing enhanced infiltration of immune cells and contribute to virus neuroinvasion via
the ‘Trojan-horse’ route. These data further implicate roles of multiple MMPs in the BBB disruption
and strategies to interrupt this process may influence the WNV disease outcome.
INTRODUCTION
West Nile virus (WNV) is a mosquito-borne neurotropicflavivirus responsible for the largest outbreak of viralmeningoencephalitis in the Western Hemisphere since itsappearance in New York in 1999. A subset of WNVpatients with febrile illness progress to encephalitis (Daviset al., 2006; Gubler, 2007) and neurological diseasesyndrome is observed in approximately 30 % of confirmedWNV cases with a higher frequency in the elderly andimmunocompromised (Campbell et al., 2002; Hayes &Gubler, 2006). No specific therapy for WNV is currentlyapproved for use in humans. In the mouse model, WNV-associated neurological disease is characterized by disrup-tion of the blood–brain barrier (BBB), increased infiltra-tion of immune cells into the central nervous system
(CNS), production of inflammatory cytokines, microgliaactivation and eventual loss of neurons (Glass et al., 2005;Klein et al., 2005; Samuel & Diamond, 2006). WNV entryinto the CNS represents an important event in the clinicaloutcome of WNV. The proposed routes of entry are bycrossing the BBB and by retrograde axonal transport viaperipheral nervous system (Samuel et al., 2007). Since highviraemia correlates with early WNV entry into the CNS,haematogenous route via crossing the BBB is suggested tobe one of the major routes by which WNV enters the CNS(King et al., 2007) although the associated mechanisms areyet unclear. We previously demonstrated in vitro thatWNV can infect and efficiently replicate in human brainmicrovascular endothelial cells, which line the BBB, andcan be one of the routes of virus entry into the CNSwithout affecting the BBB integrity (Verma et al., 2009).
The BBB, which is the interface between circulating bloodand the CNS, is composed of specialized microvascular
3These authors contributed equally to this paper.
A supplementary table is available with the online version of this paper.
Journal of General Virology (2012), 93, 1193–1203 DOI 10.1099/vir.0.040899-0
040899 G 2012 SGM Printed in Great Britain 1193

Downloaded from www.microbiologyresearch.org by
IP: 54.160.113.5
On: Tue, 07 Jun 2016 05:53:10
endothelial cells and perivascular astrocytes separated bythe basement membrane (Abbott, 2005; Persidsky et al.,2006). The BBB-endothelial cells are unique because of thepresence of specific junctional complex proteins that sealthe intracellular gaps between adjacent endothelial cells,resulting in the formation of a selective barrier thatregulates the entrance of circulating leukocytes andpathogens into the brain (Persidsky et al., 2006). Thejunctional complex includes two groups of proteins, thetight junction proteins (TJP), located at the apical side andthe adherens junction proteins (AJP), that form acontinuous belt on the basolateral side of the membrane(Zlokovic, 2008). The TJP complex comprises transmem-brane proteins, including occludin and claudins thatinteract with cytosolic adaptor proteins, the zonulaoccludins (ZO), which connect to apical actin filamentsforming a tight seal between adjacent cells. The keyproteins in the AJ complex are the transmembrane vascularendothelial cadherin (VE-cadherin) and cytosolic catenins(Bazzoni et al., 2000; Zlokovic, 2008). This continuous AJcomplex holds neighbouring cells together and assists instabilizing the interactions between TJP, thereby support-ing the integrity of the BBB and regulating paracellularpermeability (Bazzoni et al., 2000). Junctional complexproteins are highly sensitive to the microenvironment andrespond to inflammatory molecules, resulting in alterationsin their subcellular distribution and/or dissociation of thetransmembrane-adaptor protein complexes.
Several neurotropic pathogens have evolved strategies tocross the BBB to gain access to the CNS, including directinfection of BBB cells, paracellular entry through thecompromised BBB and transmigration within infectedleukocytes via the Trojan-horse mechanism. Increasedpermeability of the BBB is a pathological hallmark inseveral neurological disorders such as multiple sclerosis,bacterial meningitis (Huber et al., 2001) and neurotropicvirus infections including human immunodeficiency virus(HIV) (Dallasta et al., 1999), measles virus (Cosby &Brankin, 1995), mouse adenovirus (Gralinski et al., 2009)and arthropod-borne viruses such as Japanese encephalitisvirus (JEV) (Mishra et al., 2009), WNV (Morrey et al., 2008;Wang et al., 2004) and Venezuelan equine encephalitis virus(VEEV) (Schafer et al., 2011). BBB disruption is associatedwith the degradation of specific junction proteins, whichcontributes to virus entry (Afonso et al., 2007; Luabeya et al.,2000) and enhanced transmigration of activated immunecells into the brain (Boven et al., 2000). These eventscorrelate with simultaneous production of matrix-degradingmetalloproteinases (MMPs), a large family of endopepti-dases in the region of injury (Rosenberg, 1995). In CNSinfections, MMPs are thought to play a major role inpromoting destructive neuroinflammatory processes includ-ing BBB disruption via TJP degradation, oedema formationand disintegration of the neurovascular unit (Liu &Rosenberg, 2005; Rosenberg, 2002).
Previous studies have demonstrated that BBB integrity iscompromised in WNV-infected mice; however, the underlying
mechanisms are not well defined. It is hypothesized that thecascade of the production of proinflammatory mediatorssuch as tumour necrosis factor alpha, MMP-9 andmacrophage migration inhibitory factor may alter the BBBintegrity (Arjona et al., 2007; Wang et al., 2004, 2008a). Ourprevious in vitro studies provide evidence for the role ofWNV-induced MMPs in human astrocytes in comprom-ising the integrity of the in vitro BBB model (Verma et al.,2010). Additionally, Wang et al. (2008a) demonstrated thatMMP-92/2 mice were resistant to WNV infection andmarkers of BBB disruption such as IgG extravasation andEvans blue leakage into the brain were markedly reduced inthe MMP92/2 mice compared with controls. Although thesestudies though collectively suggest the pathogenic role ofBBB disruption and MMPs in WNV neuropathogenesis, theprecise mechanism, specifically the effect of WNV on theexpression kinetics of key BBB junction proteins has notbeen delineated in vivo. Therefore, in this study wecharacterized the expression pattern of multiple TJP andAJP in the mouse brains and correlated it with the kinetics ofBBB disruption, MMPs production and infiltration ofleukocytes during the course of infection.
RESULTS
In vivo WNV-induced BBB disruption correlateswith peak viral titres in the brain
To understand the specific in vivo mechanisms by whichWNV mediates BBB disruption, we examined the kineticsof BBB disruption during the course of WNV infection bymeasuring the leakage of Evans blue dye and systemic IgGproteins into mice brains. Evans blue is a cationic dye thatbinds to the albumin present in the sera and forms acomplex that cannot pass through the intact BBB.Extravasation of this complex into the brain is indicativeof increased BBB permeability. Our results demonstratethat in the mock-infected animals the Evans blue dyeperfused only into the peripheral tissues such as spleen andkidney but not into the brain (Fig. 1a) thereby suggestingan intact BBB. In WNV-infected mice, the BBB wasobserved to be intact at days 2 and 4 after infection, similarto the control brains. However, detection of leakage of thedye was first observed at day 6 after infection, which becamemore prominent at day 8 after infection (Fig. 1a). In thosemice that survived WNV infection, no leakage of Evans bluedye was observed in the brain at day 21 after infection,suggesting restoration of the BBB integrity (data not shown).Similarly, Fig. 1(b) indicates that IgG (heavy and lightchains) levels in the perfused brains increased significantly inthe brains at day 8 after infection in comparison to themock-infected brains, further validating that compromisedBBB leads to increased transmigration of systemic proteinsin the brain. The WNV titres in the sera displayed peakviraemia in the sera at days 3 and 4 after infection (Fig. 1c).In the brains, while WNV was not detected at day 4 afterinfection, the titres reached 104 p.f.u. mg21 RNA at day 6 and
K. Roe and others
1194 Journal of General Virology 93

Downloaded from www.microbiologyresearch.org by
IP: 54.160.113.5
On: Tue, 07 Jun 2016 05:53:10
further increased at day 8 after infection (Fig. 1d). Overall,the kinetics of BBB disruption demonstrating compromisedintegrity at day 8 after infection correlated positively withthe peak virus titres in the brain.
BBB disruption is accompanied by decreasedprotein levels of TJP and AJP
To further understand the relationship between BBBdisruption and the levels of junctional proteins, weanalysed the mRNA and protein levels of multiple TJPand AJP in the infected-brain tissue. As determined byquantitative real-time PCR (qRT-PCR), the mRNA levelsof the TJP claudin-1, occludin, ZO-1 and JAM-A revealedno significant change at any time points following WNVinfection (Fig. 2a). However, the protein levels of theseTJP, as determined by Western blot analysis, weresignificantly altered in the brains of infected mice. Exceptfor JAM-A, the protein levels of claudin-1, occludin andZO-1 did not change significantly in the brain at day 4 afterinfection; however, by day 6 after infection, the decrease inthese TJP was approximately 20–50 % as compared withcontrols (Fig. 2b). This decrease became even morepronounced at day 8 after infection. As seen in Fig. 2(c),decrease in the expression of claudin-1 and occludin was70–80 % (P,0.05) and JAM-A was approximately 85 %(P,0.001) as compared with controls at day 8 afterinfection. Since in vivo AJP are also critical for maintainingthe paracellular permeability across the BBB, the expressionprofile of b-catenin and VE-cadherin was also analysed. Asdepicted in Fig. 3(a), compared to controls no significant
changes were observed in the mRNA expression of AJP, b-catenin and VE-cadherin in WNV-infected brains. Similarto the TJP, while WNV infection did not alter protein levelsof these AJP at day 4 after infection, they decreased byapproximately 40 and 60 % at days 6 and 8 after infection,respectively (Fig. 3b and c).
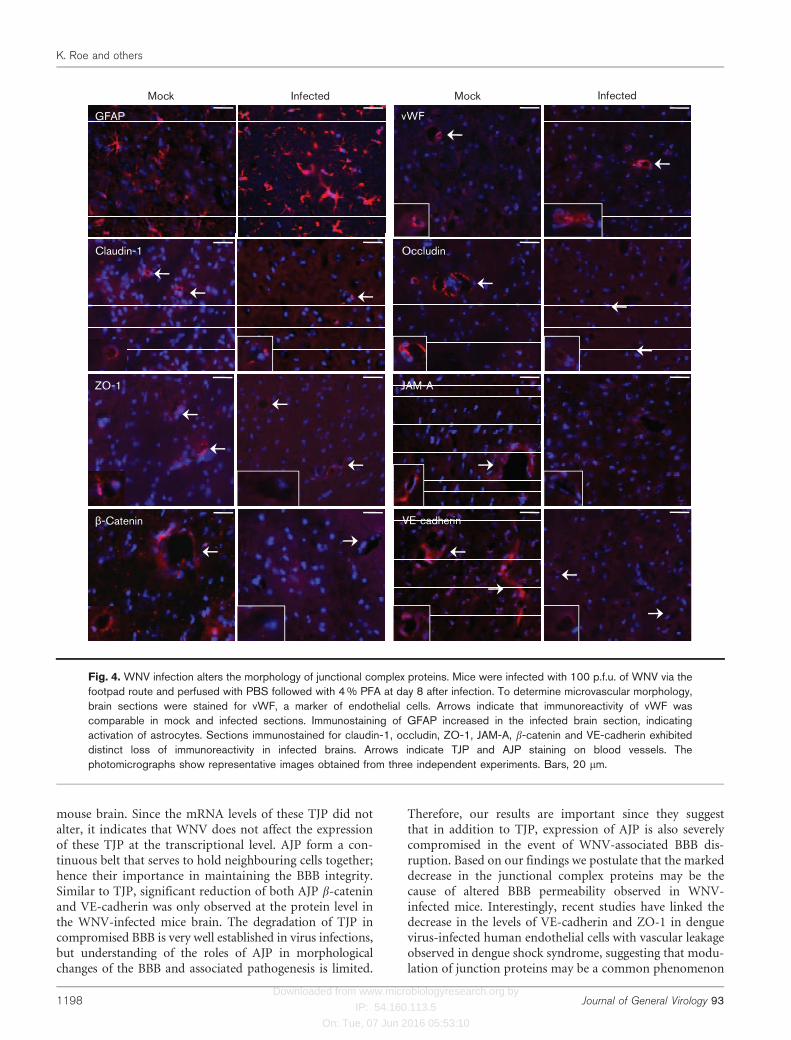
Alterations in the expression of brain TJP and AJP werefurther validated by immunostaining of the brain sectionsharvested at day 8 after infection. We also examined thebrain sections for the expression of glial fibrillary acidicprotein (GFAP), a marker of astrocytes activation and vonWillebrand Factor (vWF), a specific marker of micro-vascular endothelial cells. As seen in Fig. 4, increasedimmunostaining of GFAP was observed in infected brainand indicates activation of astrocytes, a well characterizedfeature of WNV-associated pathology. However, the fluor-escence intensity of vWF staining was similar in both mock-and WNV-infected brain sections, suggesting similarcapillary morphology. In brain sections from mock-infectedmice, distinct immunostaining of TJP and AJP surroundingthe capillaries was evident. However, the fluorescent signalof all the TJP and AJP examined decreased dramaticallyin WNV-infected brain sections. In contrast to linear andcontinuous staining pattern in control tissue, immuno-reactivity of claudin-1 and ZO-1 in infected tissue resembledspot-like structures. Immunostaining of occludin, JAM-Aand b-catenin in infected tissue also demonstrated either lossof immunoreactivity or areas of fragmented staining (Fig. 4).These results are consistent with the Western blot data (Fig.3b) and collectively indicate that the severe loss of both TJP
lgG-heavy chain
M
Peripheraltissues
(b) (c) (d)
(a)
D4 D6 D8
M D2 D4 D6 D8
Days after inoculation
101
102
103
104
100
105
101
102
103
104
105
100
106
2 3 4 6 2 4 6 8
Sera Brain
Days after inoculation
p.f.u
. ml–
1 se
ra
p.f.u
. µg–
1 R
NA
lgG-light chain
-Actin
Fig. 1. WNV infection induces BBB disruption in vivo. Mice were inoculated with 100 p.f.u. of WNV via footpad, and sera andbrain were harvested at different time points after infection. (a) Measurement of the BBB permeability using Evans blue dye atdays 2, 4, 6 and 8 after infection. Mice were injected i.p. with 1 ml Evans blue dye (1 % w/v) and after 1 h were cardiac perfusedwith PBS. The dye diffused into the peripheral tissues and turned them blue. The extravasation of the dye was evident in thebrains harvested at days 6 and 8 after infection. (b) Brain lysates were resolved by SDS-PAGE and Western blotting wasconducted to detect heavy and light chains of IgG. (c) WNV titres in the serum were determined by plaque assay using Verocells and were expressed as p.f.u. ml”1 serum and (d) WNV copy numbers in the brain of WNV-infected mice were determinedby qRT-PCR and were expressed as p.f.u. mg”1 RNA. M, Mock infected.
WNV-induced changes in the markers of BBB integrity
http://vir.sgmjournals.org 1195

Downloaded from www.microbiologyresearch.org by
IP: 54.160.113.5
On: Tue, 07 Jun 2016 05:53:10
and AJP protein in the brain correlates with the BBBdisruption (Fig. 1a).
We next assessed the correlation of BBB disruption with theinfiltration of leukocytes in the brain. Haematoxylin andeosin stained sections demonstrated the presence ofinfiltrating leukocytes in the meninges of the brain at day8 after infection (Fig. 5a). Further, while infiltratingleukocytes represented by CD11b+ cells (monocytes andneutrophils) in the brains did not increase at day 6 (data notshown), the increase was significant at day 8 after infectionand correlated positively with BBB disruption (Fig. 5b).
Multiple MMPs are induced by WNV in vivo
To determine if the decreased TJP and AJP levels (Figs 2, 3,4) could be explained through the actions of MMPs, weanalysed the expression levels of multiple MMPs in the brainfollowing WNV infection. A six- to eightfold increase in themRNA transcripts of MMP-1 and -3 were observed in thebrain at day 6 after infection, which further increased at day8 after infection (Fig. 6a). While the mRNA expressions ofMMP-9 did not increase at day 6 after infection, a modesttwofold increase of MMP-9 was observed in the infectedbrain tissue at day 8 after infection (Fig. 6a). The expressionof tissue inhibitor of metalloproteinases 2 (TIMP-2) andMMP-2 in the brain did not alter at any time pointsfollowing WNV infection (data not shown).
Western blotting further demonstrated the increase ofMMP-3 and -9 at days 6 and 8 after infection (Fig. 6b).Since MMPs are secretory proteins, their levels were alsoexamined in the serum and brain using ELISA. Surprisingly,as compared to mock, MMP-3 levels did not change in theserum although a significant increase in MMP-9 levels wasobserved at day 3 after infection (P,0.05), which returnedto the basal level by day 6 and remained low at day 8 afterinfection (Fig. 6c). However, in the brain, levels of MMP-3and -9 increased significantly at day 8 as compared withcontrols, thus coinciding with the increase in their mRNAtranscripts at the same time points (Fig. 6d). The positivecorrelation between the BBB disruption and increasedMMPs in the brain and not in the serum, suggest thatinflammatory processes within the CNS might be critical inTJP and AJP degradation.
WNV infection via intracranial route also inducesBBB disruption
As we observed that the time points of BBB disruptioncoincided with high MMP levels and virus titres in thebrain and not in the serum, we hypothesized that decreasedTJP and AJP expression could be a result of increased virusreplication-induced inflammation in the brain. To validatethis hypothesis, we inoculated mice with similar infectiousdose of WNV (100 p.f.u.) directly into the cranium and
3
2
1
0
0.0
0.1
0.2
0.3
(c)
(a) (b)
0.0
0.2
0.4
0.6
0.0
0.5
1.0
1.5
2.0
0.0
0.4
0.2
0.6
0.8
Claudin-1
Days after inoculation
M 4 6 8
*
Days after inoculation
M 4 6 8
**
Days after inoculation
M 4 6 8
* *
Days after inoculation
M 4 6 8
** *
mR
NA
rela
tive
fold
-cha
nge
as c
ompa
red
to c
ontr
ols
Cla
udin
-1/
-act
in
Occ
ludi
n/ -
actin
ZO
-1/
-act
in
JAM
-A/
-act
in
Occludin ZO-1 JAM-A
D2D4D6D8
M
Claudin-1
Occludin
ZO-1
JAM-A
-Actin
D4 D6 D8
Fig. 2. BBB disruption is characterized by decrease in TJP protein. (a) qRT-PCR was conducted on RNA extracted from brainsharvested from mice inoculated via footpad to determine mRNA fold-change of claudin-1, occludin, ZO-1 and JAM-Aexpression at days 2, 4, 6 and 8 after WNV infection. Data represents the means±SEM of at least five mice conducted induplicate. (b) Total brain lysates were separated by SDS-PAGE, transferred onto PVDF membranes and immunoblotted withantibodies specific to claudin-1, occludin, ZO-1 and JAM-A. Equal loading was confirmed by reprobing with anti-b-actinantibody and the bands were detected using the Li-Cor Odyssey infrared method. (c) Quantitative analysis of Western blotresults represented as a ratio between TJP and b-actin in control and infected brain tissue. Mean comparisons were based ondata from at least four mice. *P,0.05 as compared to mock controls (M).
K. Roe and others
1196 Journal of General Virology 93

Downloaded from www.microbiologyresearch.org by
IP: 54.160.113.5
On: Tue, 07 Jun 2016 05:53:10
analysed extravasation of IgG as a marker of BBBdisruption. As observed in Fig. 7, intracranially infectedmice demonstrated high WNV titres in the brains at days 4and 6 after infection, which correlated with the concom-itant increase in the mRNA levels of multiple MMPs.Increased levels of IgG were observed at day 6 afterinfection, suggesting compromised BBB (Fig. 7b).Intracranially infected mice did not survive after day 6 ofinfection and virus was not detected in the sera of thesemice at days 2, 4 and 6 after infection (data not shown).
DISCUSSION
Impaired function of the brain vasculature can contributeto the trafficking of pathogens and activated immune cells
into the CNS causing death of neurons. Previous studieshave demonstrated that BBB permeability is compromisedin WNV-infected mice; however, the associated mechan-isms have not been elucidated so far in vivo. In this report,we demonstrate that WNV-induced BBB disruption occursgradually during the course of infection but does notprecede WNV-CNS entry. Further, BBB disruption resultsin significant alterations in the expression of TJP and AJP,which correlate with peak WNV titres, increased produc-tion of MMPs and infiltrated leukocytes in the brain. Insummary, these data suggest that initially, WNV enters intothe CNS without disrupting the BBB, and initiates virusreplication and inflammation, which leads to the openingof the BBB allowing unrestricted entry of virus andimmune cells into the CNS.
WNV infection disrupts the BBB
Inflammation of the CNS accompanied by migration ofactivated immune cells into the CNS and increased BBBpermeability are vital components of encephalitis-associatedwith neurotropic viruses including HIV, human T-celllymphotropic virus (HTLV) and flaviviruses such as JEV,WNV and Murray Valley encephalitis virus (Afonso et al.,2007; Dallasta et al., 1999; King et al., 2007; Mishra et al.,2009). Previous studies demonstrating increased mortality byadministration of lipopolysaccharide, which is known toenhance BBB permeability, in mice infected with non-neuroinvasive strain of WNV supports the fact that BBBdisruption contributes to virus–CNS entry and diseaseoutcome (Lustig et al., 1992). Our data using two well-established methods of BBB permeability assay, suggest thatchanges in BBB permeability develop slowly during the courseof infection and the most dramatic decrease in its integrity isobserved when virus infection is already established in theCNS. Though intrathecal antibody production can alsocontribute to the IgG in the brain, the presence of systemicproteins including IgG is an established indicator ofcompromised BBB integrity. The BBB disruption we observedconcurs with previous reports wherein IgG extravasation wasdetected in the brain at day 7 after infection (Wang et al.,2008a). Further, Wang et al. (2004) also established the role ofthe BBB disruption in WNV pathogenesis; however, theyobserved BBB disruption as early as day 3 after infection,which may be because of either higher infectious dose (LD100)inoculated via intraperitoneal route or a different strain of theWNV. On the other hand, Morrey et al. (2008) did not reportcorrelation between increased BBB permeability and lethalityof WNV-infected rodents. These differences could beattributed to the virus dose used for inoculation (105
p.f.u.), which resulted in early mortality, starting at day 6after infection (Morrey et al., 2008).
Degradation of junction proteins is a pathologicalhallmark of WNV infection
In this report, for the first time we demonstrate that WNVinfection results in the degradation of multiple TJP in the
mR
NA
rela
tive
fold
-cha
nge
as c
ompa
red
to c
ontr
ols
D2D4D6D8
M D4 D6 D8
VE-cadherin-Catenin
VE-cadherin
-Actin
2.5(a)
(b)
(c)
2.0
1.5
1.0
0.5
0.0
Days after inoculationM
0
5
10
15
0.0
0.1
0.2
0.4
0.3
4 6 8
* *
Days after inoculationM 4 6 8
**
VE
-cad
herin
/ -a
ctin
-Cat
enin
/ -a
ctin
-Catenin
Fig. 3. WNV infection induces alterations in the expression of AJP.(a) As compared to mock brains, the mRNA expression of b-catenin and VE-cadherin did not change in the brains at days 2, 4,6 and 8 after infection via the footpad route as determined by qRT-PCR. Data represent the means±SEM of at least five miceconducted in duplicate. (b) Protein expression of b-catenin andVE-cadherin as determined by Western blot analysis wassignificantly reduced in the brains at days 6 and 8 after infection.(c) Quantitative analysis of Western blot results. All values arerelative to b-actin and represent at least four samples per timepoint. *P,0.05 as compared to mock controls (M).
WNV-induced changes in the markers of BBB integrity
http://vir.sgmjournals.org 1197
Downloaded from www.microbiologyresearch.org by
IP: 54.160.113.5
On: Tue, 07 Jun 2016 05:53:10
mouse brain. Since the mRNA levels of these TJP did notalter, it indicates that WNV does not affect the expressionof these TJP at the transcriptional level. AJP form a con-tinuous belt that serves to hold neighbouring cells together;hence their importance in maintaining the BBB integrity.Similar to TJP, significant reduction of both AJP b-cateninand VE-cadherin was only observed at the protein level inthe WNV-infected mice brain. The degradation of TJP incompromised BBB is very well established in virus infections,but understanding of the roles of AJP in morphologicalchanges of the BBB and associated pathogenesis is limited.
Therefore, our results are important since they suggestthat in addition to TJP, expression of AJP is also severelycompromised in the event of WNV-associated BBB dis-ruption. Based on our findings we postulate that the markeddecrease in the junctional complex proteins may be thecause of altered BBB permeability observed in WNV-infected mice. Interestingly, recent studies have linked thedecrease in the levels of VE-cadherin and ZO-1 in denguevirus-infected human endothelial cells with vascular leakageobserved in dengue shock syndrome, suggesting that modu-lation of junction proteins may be a common phenomenon
*
GFAP
VE-cadherin
ZO-1 JAM-A
β-Catenin
OccludinClaudin-1
vWF
kcoMkcoM Infected Infected
Fig. 4. WNV infection alters the morphology of junctional complex proteins. Mice were infected with 100 p.f.u. of WNV via thefootpad route and perfused with PBS followed with 4 % PFA at day 8 after infection. To determine microvascular morphology,brain sections were stained for vWF, a marker of endothelial cells. Arrows indicate that immunoreactivity of vWF wascomparable in mock and infected sections. Immunostaining of GFAP increased in the infected brain section, indicatingactivation of astrocytes. Sections immunostained for claudin-1, occludin, ZO-1, JAM-A, b-catenin and VE-cadherin exhibiteddistinct loss of immunoreactivity in infected brains. Arrows indicate TJP and AJP staining on blood vessels. Thephotomicrographs show representative images obtained from three independent experiments. Bars, 20 mm.
K. Roe and others
1198 Journal of General Virology 93

Downloaded from www.microbiologyresearch.org by
IP: 54.160.113.5
On: Tue, 07 Jun 2016 05:53:10
in flavivirus infection (Kanlaya et al., 2009). Correlation ofbreakdown of TJP with BBB disruption and leukocyteinfiltration has been well established in infection with virusessuch as HIV, HTLV and mouse adenovirus (Afonso et al.,2007; Gralinski et al., 2009; Kanmogne et al., 2007).
MMPs are widely implicated in modulating BBB integrityand affect the entry of peripheral immune cells into the CNS.Therefore, the expression of MMPs is tightly regulated at thelevel of gene transcription, conversion of pro-enzyme toactive MMPs and by the action of TIMP (Rosenberg, 2002).While the increase in MMP-9 levels as observed by ussupports the previous studies, demonstrating its increase byWNV, JEV and VEEV (Mishra et al., 2009; Schafer et al.,2011; Wang et al., 2008a), the unique aspect of our data isthe increase of MMP-1 and -3 in the brains of WNV-infected mice. The increase of MMP-3 was at a greater extentcompared with MMP-9 in the brain. This result is inagreement with our previous in vitro studies, demonstratingupregulation of MMP-1, -3 and -9 in WNV-infected humanastrocytes (Verma et al., 2010). Based on several novel rolesof MMP-1 and -3 in mediating neuroinflammation andapoptosis (Kim et al., 2005; Mun-Bryce et al., 2002; Suzukiet al., 2007), this observation warrants further delineation ofthe role of other MMPs in WNV neurological disease. In ourstudy, the increased MMP levels coincided with increasedvirus titres in the CNS (Fig. 1), activation of astrocytes (Fig.3), BBB disruption and decreased TJP expression (Fig. 2).Based on our previous observation (Verma et al., 2010) aswell as recent report of MMP-9 production by JEV-infectedastrocytes (Tung et al., 2010), it seems likely that activatedastrocytes might be one of the potential sources of theseMMPs in the brain, which participate in the remodelling ofthe BBB by degrading junctional complex proteins.However, the possibility of infiltrating immune cells asanother source of MMPs production at day 8 after infectioncannot be ruled out.
WNV replication in the CNS plays a critical role inmediating BBB disruption
In the mouse model of WNV disease, it is believed thatWNV invades the CNS when viraemia is high and is firstdetected in the CNS between days 4 and 6 after infection(Klein & Diamond, 2008). During this period, WNV canenter the CNS via multiple routes, including by crossingthe BBB. Our results indicate that in spite of high viraemiaand increased MMP-9 levels in the serum there were nodetectable changes in the BBB permeability between days 2and 4 after infection. On the other hand, the most dramaticchange in BBB integrity was observed at day 8 afterinfection when the virus titres and MMP levels weresignificantly elevated in the brain. These results suggest thatthe initial entry of infectious West Nile virions into theCNS between days 4 and 6 after infection might occurwithout altering the BBB integrity by one or more of theproposed routes of virus entry such as directly infectingBBB-endothelial cells (Samuel et al., 2007; Verma et al.,2009). Virus replication in the CNS then inducesinflammation, including production of multiple MMPsand cytokines, which causes BBB disruption. To furtherconfirm the hypothesis of BBB disruption being driven byinflammatory mediators produced in the brain, we infectedmice intracranially with WNV. In favour of this notion, weobserved a similar increase in BBB disruption as measuredby the presence of IgG and MMPs in the brain at day 6after infection (Fig. 7). The pathophysiological importanceof BBB disruption at later time points supports thehypothesis that enhanced infiltration of activated immunecells into the brain, some of which may be infected, mayfacilitate a second wave of virus neuroinvasion via the‘Trojan-horse’ route. Recent studies using Drak22/2 micehave emphasized this phenomenon, by demonstrating thatinfected infiltrating T-cells can be potential carriers forWNV entry into the CNS (Wang et al., 2008b). As observed
Day 7 Day 80
5
1020
40
60
80
Days after infection
CD
11b+
cel
ls p
er b
rain
(×
104 )
(a) (b)
Fig. 5. Analysis of infiltrating leukocytes in the brains. (a) Brain sections were stained with haematoxylin and eosin and examinedby microscopy. Multiple cellular infiltrates (black arrows) were observed in the meninges region of the brain infected via thefootpad route. (b) At days 7 and 8 after infection, brains were analysed for the presence of infiltrating CD11b+ cells using flowcytometry. Each group represents the number of cells per brain from analysis of three brains that were pooled for staining.
WNV-induced changes in the markers of BBB integrity
http://vir.sgmjournals.org 1199

Downloaded from www.microbiologyresearch.org by
IP: 54.160.113.5
On: Tue, 07 Jun 2016 05:53:10
by us (Fig. 5) and others (Lim et al., 2011), increasedleukocyte infiltration in the infected brain beginning at day8 after infection further supports the fact that BBBdisruption might facilitate unrestricted entry of peripheralimmune cells into the CNS. A similar argument, that virusreplication in the brain induces BBB opening allowing asecond wave of invading virus, has been recently proposedfor VEEV (Schafer et al., 2011). Our data do not explainthe relationship of BBB disruption with overall diseaseoutcome; however, they do indicate an important role ofBBB disruption in the increased transmigration ofactivated infected and/or naıve immune cells into thebrain. Inhibition of BBB opening may not block the initialinvasion of the virus, but it is likely to reduce the
neuropathology associated with unrestricted entry of virusand activated infected and/or naıve immune cells in theCNS. Although virus specific T-cells are critical for WNVclearance from the brain, enhanced infiltration of immunecells as a result of BBB disruption may exacerbate neuronaldamage by potentiating neuroinflammation, resulting inpoor disease prognosis.
In conclusion, this first in vivo study demonstratesdramatic alterations in the expression kinetics of junctionproteins of the brain vascular endothelial cells followingWNV infection in mice. The kinetics of TJP and AJPdegradation follow closely the pattern of BBB disruptionduring the course of WNV infection. Furthermore,
12.5
10.0
7.5
5.0
2.5
0.0
2.0
1.5
1.0
0.5
0.0
150
50
100
15
10
5
0
0.4
0.6
0.8
1.0
20
0
40
60
80
5
0
10
15
20
0.5
0.0
1.0
1.5
2.0
0
2
4
6
0.2
0.3
0.4
1
2
3
2 4 6 8
Days after inoculation
6M 8
Days after inoculation
3M 6Days after inoculation
3
*
M 6 8Days after inoculation
M 6 8
*
*
Days after inoculation
M 6 8
*
Days after inoculation
6M 8
*
Days after inoculation
6M 8
*
Days after inoculation
2 4 6 8
*
*
Days after inoculation2 4 6 8
Days after inoculation
MMP-1 MMP-3
MMP-3
(c) (d)
(b)
(a)
MMP-9 MMP-3 MMP-9
MMP-9
MMP-1
MMP-3
MMP-9
mR
NA
rela
tive
fold
-cha
nge
as c
ompa
red
to c
ontr
ols
ng m
l–1
sera
ng/g
Bra
in
-Actin
M D4 D6 D8
MM
P-1
/ -a
ctin
MM
P-3
/ -a
ctin
MM
P-9
/ -a
ctin
Fig. 6. Multiple MMPs are induced in the sera and brains of WNV-infected mice. (a) qRT-PCR was conducted on RNAextracted from mock- and WNV-infected brains to determine fold-change in MMP-1, -3 and -9 gene expression. Datarepresents the mean±SEM of at least five mice conducted in duplicate. (b) The protein levels of MMP-1, -3 and -9 wereanalysed using Western blot analysis. Quantitative analysis of Western blot results are represented as a ratio between MMPand b-actin in mock and infected brain tissue at days 6 and 8 after infection. Mean comparisons were based on data from atleast four mice. *P,0.05 as compared to mock controls (M). Total MMP-3 and -9 levels were measured in the (c) sera and (d)brain homogenates at different time points after infection using ELISA. The data expressed are the mean concentration of theamount of MMP-3 and -9 in sera (ng ml”1)±SEM and brains (ng g”1)±SEM. Sera (n56 per time point) and brains (n57 per timepoint). *P,0.05.
K. Roe and others
1200 Journal of General Virology 93

Downloaded from www.microbiologyresearch.org by
IP: 54.160.113.5
On: Tue, 07 Jun 2016 05:53:10
concomitant increase of multiple MMPs in the CNS ofmice inoculated either in the footpad or directly into thecranium indicates that MMPs derived from the brain play acritical role in TJP degradation. Overall these findingssuggest that BBB-tight junction disruption is a criticalpathological event in WNV neuropathogenesis that maypermit the paracellular transit of virus and immune cellsinto the CNS. Future studies of blocking BBB disruptionare warranted to further delineate the regulatory mechan-isms underlying WNV-induced BBB disruption, to developtherapeutic modalities to ameliorate the pathology asso-ciated with WNV neuropathogenesis.
METHODS
Virus and mouse experiments. Wild-type C57BL/6 mice boughtfrom the Jackson Laboratory were bred in the animal facility at theJohn A. Burns School of Medicine. All experiments were approvedand conducted in accordance with the University of Hawaii at Manoaanimal study guidelines. All infections were conducted on 8–10-week-old-mice by inoculation of 100 p.f.u. (LD50) of virus (NY99 isolatedfrom crow brain passaged once in Vero cells) either in the footpad orcranium as described previously (Verma et al., 2011). Blood wascollected by cardiac puncture and serum was separated bycentrifugation and was stored at 280 uC. Mice were then perfusedwith 20 ml cold PBS under anaesthesia and harvested organs, afterflash freezing, were stored at 280 uC.
Assay of BBB integrity. BBB integrity was evaluated by using theEvans blue dye exclusion test and by measuring IgG extravasation inthe brain (Wang et al., 2008a). Control and infected mice were
injected in the peritoneal cavity with 1 ml Evans blue dye [1 % (w/v)in PBS]. One hour later, mice were perfused with 20 ml PBS,euthanized under anaesthesia and the brains and peripheral tissueswere harvested and photographed. Western blotting was conductedby using total protein extract of the brain to detect heavy and lightchains of IgG.
Quantification of viral load in blood and tissues. Virus titreswere analysed by plaque assays and qRT-PCR in the serum and brain,respectively, as described previously (Verma et al., 2011). qRT-PCRwas conducted using primers and FAM- and TAMRA-labelled probesspecific for WNV env region and the standard curve was generated byusing RNA extracted from previously titrated WNV dilutions (10 000to 0.1 p.f.u.) as described by Lanciotti et al. (2000). The data areexpressed as WNV p.f.u. equivalents mg21 of RNA.
qRT-PCR determination of host genes. The mRNA levels ofmultiple TJP, AJP and MMPs were determined by using qRT-PCR,and the fold change in infected brains as compared to controls wascalculated after normalizing to the GAPDH gene (Verma et al., 2010).The primer sequences and annealing temperatures used for qRT-PCRare listed in Table S1 (available in JGV Online).
Western blot analysis. Total cellular protein was extracted bysonicating the brain in chilled Cell Lytic buffer (Sigma) followed bycentrifugation at 11 000 g. for 20 min and the soluble supernatantfraction was collected. An equal amount of total protein (25–40 mg)was separated by SDS-PAGE, transferred onto nitrocellulose mem-brane and incubated overnight with polyclonal antibodies againstclaudin-1, occludin, ZO-1 (Zymed), VE-cadherin, JAM-A (SantaCruz), b-catenin (Abcam) and b-actin (Sigma) as describedpreviously (Verma et al., 2010). Following incubation with secondaryantibodies conjugated with IRDye 800 and IRDye 680 (Li-CorBiosciences), the membranes were scanned by using the Odyssey
Days after inoculation
20
5
10
15
20
25
109
108
107
106
105
104
103
420
6
2
1
0
3
100200300400
4 6
Days after inoculation
2 4 6
Days after inoculation
2 4 6
Days after inoculation
MMP-1
(c)
(a) (b)
MMP-3 MMP-9
M D2 D4 D6
2 4 6
lgG-heavy chain
lgG-light chain
-Actin
p.f.u
. µg–
1 R
NA
mR
NA
rela
tive
fold
-cha
nge
as c
ompa
red
to c
ontr
ols
Fig. 7. Intracranial inoculation of WNV results in BBB disruption. (a) WNV titres in the brain of mice intracranially inoculated with100 p.f.u. WNV. Brain titres were determined by qRT-PCR and expressed as p.f.u. mg”1 RNA. (b) Leakage of IgG in the brainwas determined by Western blotting. (c) MMP-1, -3 and -9 mRNA transcripts increased in the brains of intracranially inoculatedmice as determined by qRT-PCR. Data represents at least three mice per time point.
WNV-induced changes in the markers of BBB integrity
http://vir.sgmjournals.org 1201

Downloaded from www.microbiologyresearch.org by
IP: 54.160.113.5
On: Tue, 07 Jun 2016 05:53:10
infrared imager (Li-Cor Biosciences). Intensity of bands for densito-
metric analysis was determined by using the Odyssey program
software.
Measurement of MMPs using ELISA. The protein levels of MMP-3
and -9 in mouse brain homogenates and sera were measured using
commercial ELISA kits (R&D) following the instruction manual as
described previously (Verma et al., 2010).
Immunohistochemistry. Mice were transcardially perfused with
20 ml PBS followed by 20 ml of 4 % paraformaldehyde (PFA), brains
were harvested, cryoprotected in 30 % sucrose (Sigma) for 3 days at
4 uC and frozen in Optimal Cutting Temperature solution (Tissue-
Tek). Horizontal sections of 10 mm thickness were fixed in 2 % PFA
for 15 min, permeablized by incubating 0.3 % Triton X-100 (Sigma)
in PBS for 1 h at room temperature and then blocked for 1 h using
5 % goat serum (Jackson ImmunoResearch) in PBS. The staining with
various primary antibodies (GFAP, vWF, claudin-1, occludin, ZO-1,
JAM-A, VE-cadherin and b-catenin) was conducted overnight at
4 uC, followed by incubation with biotinylated secondary antibodies
for 2 h at room temperature and developed by using fluorescently
labelled streptavidin conjugate. Images were acquired by using the
fluorescent microscope (Zeiss Axiovert 200). Tissue sections were also
stained with haematoxylin and eosin and examined for pathological
changes by using a Nikon E600 phase-contrast microscope.
Preparation of cells and staining for flow cytometry. Single-cell
suspensions of the brains were prepared by using MACS Neural
Tissue Dissociation kit and GentleMACS cell dissociator system
(Miltenyi Biotec). The suspension was brought to 10 ml in 30 %
Percoll with FACS buffer and overlaid on 1 ml 70 % Percoll. After
centrifuging at 700 g for 30 min, leukocytes at the interphase were
isolated, washed, blocked in 200 ml Fc block (CD16/32) and stained
with anti-CD11b-PE-Cy7 for 30 min and analysed using FACS Aria
(BD) and FlowJo software.
Statistical analysis. All mRNA and protein quantification data are
reported as SEM. Either unpaired Student’s t-test or Mann–Whitney U
test for single-mean comparison was conducted using GraphPad
Prism 5.0 (GraphPad software). P,0.05 was considered as statistically
significant for all analyses.
ACKNOWLEDGEMENTS
This work was partially supported by grants from the Hawaii
Community Foundation (20050405), Centers of Biomedical Research
Excellence (P20RR018727 and 8P20GM1033S16-08), Research
Centers in Minority Institutions Program (G12RR003061), National
Center for Research Resources, National Institutes of Health, and
institutional funds. We thank Dr Duane J. Gubler for the generous
gift of the WNV strain NY99, Ms. Janet Meeks for assistance in the
JABSOM Biocontainment Facility and Ms. Alexandra Gurary for
assistance with the flow cytometry. We also thank Ms. Miyoko
Bellinger, Dr Abby Collier and Mr Arjun Raman for assistance with
the sectioning of the brains and immunostaining, and Ms. Melissa
Valdez for assistance with the manuscript preparation.
REFERENCES
Abbott, N. J. (2005). Dynamics of CNS barriers: evolution,
differentiation, and modulation. Cell Mol Neurobiol 25, 5–23.
Afonso, P. V., Ozden, S., Prevost, M. C., Schmitt, C., Seilhean, D.,Weksler, B., Couraud, P. O., Gessain, A., Romero, I. A. & Ceccaldi, P.E. (2007). Human blood-brain barrier disruption by retroviral-infected
lymphocytes: role of myosin light chain kinase in endothelial tight-
junction disorganization. J Immunol 179, 2576–2583.
Arjona, A., Foellmer, H. G., Town, T., Leng, L., McDonald, C., Wang, T.,Wong, S. J., Montgomery, R. R., Fikrig, E. & Bucala, R. (2007). Abrogationof macrophage migration inhibitory factor decreases West Nile virus
lethality by limiting viral neuroinvasion. J Clin Invest 117, 3059–3066.
Bazzoni, G., Martinez-Estrada, O. M., Orsenigo, F., Cordenonsi, M.,Citi, S. & Dejana, E. (2000). Interaction of junctional adhesion
molecule with the tight junction components ZO-1, cingulin, and
occludin. J Biol Chem 275, 20520–20526.
Boven, L. A., Middel, J., Verhoef, J., De Groot, C. J. & Nottet, H. S.(2000). Monocyte infiltration is highly associated with loss of the tightjunction protein zonula occludens in HIV-1-associated dementia.
Neuropathol Appl Neurobiol 26, 356–360.
Campbell, G. L., Marfin, A. A., Lanciotti, R. S. & Gubler, D. J. (2002).West Nile virus. Lancet Infect Dis 2, 519–529.
Cosby, S. L. & Brankin, B. (1995). Measles virus infection of cerebralendothelial cells and effect on their adhesive properties. Vet Microbiol
44, 135–139.
Dallasta, L. M., Pisarov, L. A., Esplen, J. E., Werley, J. V., Moses, A. V.,Nelson, J. A. & Achim, C. L. (1999). Blood-brain barrier tight junction
disruption in human immunodeficiency virus-1 encephalitis. Am J
Pathol 155, 1915–1927.
Davis, L. E., DeBiasi, R., Goade, D. E., Haaland, K. Y., Harrington, J. A.,Harnar, J. B., Pergam, S. A., King, M. K., DeMasters, B. K. & Tyler, K. L.(2006). West Nile virus neuroinvasive disease. Ann Neurol 60, 286–300.
Glass, W. G., Lim, J. K., Cholera, R., Pletnev, A. G., Gao, J. L. &Murphy, P. M. (2005). Chemokine receptor CCR5 promotes leukocytetrafficking to the brain and survival in West Nile virus infection. J Exp
Med 202, 1087–1098.
Gralinski, L. E., Ashley, S. L., Dixon, S. D. & Spindler, K. R. (2009).Mouse adenovirus type 1-induced breakdown of the blood-brain
barrier. J Virol 83, 9398–9410.
Gubler, D. J. (2007). The continuing spread of West Nile virus in the
western hemisphere. Clin Infect Dis 45, 1039–1046.
Hayes, E. B. & Gubler, D. J. (2006). West Nile virus: epidemiologyand clinical features of an emerging epidemic in the United States.
Annu Rev Med 57, 181–194.
Huber, J. D., Egleton, R. D. & Davis, T. P. (2001). Molecular
physiology and pathophysiology of tight junctions in the blood-brain
barrier. Trends Neurosci 24, 719–725.
Kanlaya, R., Pattanakitsakul, S. N., Sinchaikul, S., Chen, S. T. &Thongboonkerd, V. (2009). Alterations in actin cytoskeletal assembly
and junctional protein complexes in human endothelial cells inducedby dengue virus infection and mimicry of leukocyte transendothelial
migration. J Proteome Res 8, 2551–2562.
Kanmogne, G. D., Schall, K., Leibhart, J., Knipe, B., Gendelman, H. E.& Persidsky, Y. (2007). HIV-1 gp120 compromises blood-brain
barrier integrity and enhances monocyte migration across blood-brain barrier: implication for viral neuropathogenesis. J Cereb Blood
Flow Metab 27, 123–134.
Kim, Y. S., Kim, S. S., Cho, J. J., Choi, D. H., Hwang, O., Shin, D. H.,Chun, H. S., Beal, M. F. & Joh, T. H. (2005). Matrix metalloproteinase-
3: a novel signaling proteinase from apoptotic neuronal cells that
activates microglia. J Neurosci 25, 3701–3711.
King, N. J., Getts, D. R., Getts, M. T., Rana, S., Shrestha, B. & Kesson,A. M. (2007). Immunopathology of flavivirus infections. Immunol CellBiol 85, 33–42.
Klein, R. S. & Diamond, M. S. (2008). Immunological headgear:
antiviral immune responses protect against neuroinvasive West Nilevirus. Trends Mol Med 14, 286–294.
K. Roe and others
1202 Journal of General Virology 93

Downloaded from www.microbiologyresearch.org by
IP: 54.160.113.5
On: Tue, 07 Jun 2016 05:53:10
Klein, R. S., Lin, E., Zhang, B., Luster, A. D., Tollett, J., Samuel, M. A.,Engle, M. & Diamond, M. S. (2005). Neuronal CXCL10 directs CD8+
T-cell recruitment and control of West Nile virus encephalitis. J Virol
79, 11457–11466.
Lanciotti, R. S., Kerst, A. J., Nasci, R. S., Godsey, M. S., Mitchell, C. J.,Savage, H. M., Komar, N., Panella, N. A., Allen, B. C. & other authors(2000). Rapid detection of West Nile virus from human clinical
specimens, field-collected mosquitoes, and avian samples by a
TaqMan reverse transcriptase-PCR assay. J Clin Microbiol 38, 4066–
4071.
Lim, J. K., Obara, C. J., Rivollier, A., Pletnev, A. G., Kelsall, B. L. &Murphy, P. M. (2011). Chemokine receptor Ccr2 is critical for
monocyte accumulation and survival in West Nile virus encephalitis.
J Immunol 186, 471–478.
Liu, K. J. & Rosenberg, G. A. (2005). Matrix metalloproteinases and
free radicals in cerebral ischemia. Free Radic Biol Med 39, 71–80.
Luabeya, M. K., Dallasta, L. M., Achim, C. L., Pauza, C. D. & Hamilton,R. L. (2000). Blood-brain barrier disruption in simian immunodefi-
ciency virus encephalitis. Neuropathol Appl Neurobiol 26, 454–462.
Lustig, S., Danenberg, H. D., Kafri, Y., Kobiler, D. & Ben-Nathan, D.(1992). Viral neuroinvasion and encephalitis induced by lipopoly-
saccharide and its mediators. J Exp Med 176, 707–712.
Mishra, M. K., Dutta, K., Saheb, S. K. & Basu, A. (2009).Understanding the molecular mechanism of blood-brain barrier
damage in an experimental model of Japanese encephalitis:
correlation with minocycline administration as a therapeutic agent.
Neurochem Int 55, 717–723.
Morrey, J. D., Olsen, A. L., Siddharthan, V., Motter, N. E., Wang, H.,Taro, B. S., Chen, D., Ruffner, D. & Hall, J. O. (2008). Increased blood-
brain barrier permeability is not a primary determinant for lethality of
West Nile virus infection in rodents. J Gen Virol 89, 467–473.
Mun-Bryce, S., Lukes, A., Wallace, J., Lukes-Marx, M. & Rosenberg,G. A. (2002). Stromelysin-1 and gelatinase A are upregulated before
TNF-a in LPS-stimulated neuroinflammation. Brain Res 933, 42–49.
Persidsky, Y., Ramirez, S. H., Haorah, J. & Kanmogne, G. D. (2006).Blood-brain barrier: structural components and function under
physiologic and pathologic conditions. J Neuroimmune Pharmacol
1, 223–236.
Rosenberg, G. A. (1995). Matrix metalloproteinases in brain injury.
J Neurotrauma 12, 833–842.
Rosenberg, G. A. (2002). Matrix metalloproteinases in neuroin-
flammation. Glia 39, 279–291.
Samuel, M. A. & Diamond, M. S. (2006). Pathogenesis of West Nilevirus infection: a balance between virulence, innate and adaptiveimmunity, and viral evasion. J Virol 80, 9349–9360.
Samuel, M. A., Wang, H., Siddharthan, V., Morrey, J. D. & Diamond,M. S. (2007). Axonal transport mediates West Nile virus entry into thecentral nervous system and induces acute flaccid paralysis. Proc NatlAcad Sci U S A 104, 17140–17145.
Schafer, A., Brooke, C. B., Whitmore, A. C. & Johnston, R. E. (2011).The role of the blood-brain barrier during Venezuelan equineencephalitis virus infection. J Virol 85, 10682–10690.
Suzuki, Y., Nagai, N., Umemura, K., Collen, D. & Lijnen, H. R. (2007).Stromelysin-1 (MMP-3) is critical for intracranial bleeding after t-PAtreatment of stroke in mice. J Thromb Haemost 5, 1732–1739.
Tung, W. H., Tsai, H. W., Lee, I. T., Hsieh, H. L., Chen, W. J., Chen, Y. L.& Yang, C. M. (2010). Japanese encephalitis virus induces matrixmetalloproteinase-9 in rat brain astrocytes via NF-kB signallingdependent on MAPKs and reactive oxygen species. Br J Pharmacol161, 1566–1583.
Verma, S., Lo, Y., Chapagain, M., Lum, S., Kumar, M., Gurjav, U., Luo,H., Nakatsuka, A. & Nerurkar, V. R. (2009). West Nile virus infectionmodulates human brain microvascular endothelial cells tight junctionproteins and cell adhesion molecules: transmigration across the invitro blood-brain barrier. Virology 385, 425–433.
Verma, S., Kumar, M., Gurjav, U., Lum, S. & Nerurkar, V. R. (2010).Reversal of West Nile virus-induced blood-brain barrier disruptionand tight junction proteins degradation by matrix metalloproteinasesinhibitor. Virology 397, 130–138.
Verma, S., Hoffmann, F. W., Kumar, M., Huang, Z., Roe, K., Nguyen-Wu, E., Hashimoto, A. S. & Hoffmann, P. R. (2011). Selenoprotein Kknockout mice exhibit deficient calcium flux in immune cells andimpaired immune responses. J Immunol 186, 2127–2137.
Wang, T., Town, T., Alexopoulou, L., Anderson, J. F., Fikrig, E. & Flavell,R. A. (2004). Toll-like receptor 3 mediates West Nile virus entry into thebrain causing lethal encephalitis. Nat Med 10, 1366–1373.
Wang, P., Dai, J., Bai, F., Kong, K. F., Wong, S. J., Montgomery, R. R.,Madri, J. A. & Fikrig, E. (2008a). Matrix metalloproteinase 9 facilitatesWest Nile virus entry into the brain. J Virol 82, 8978–8985.
Wang, S., Welte, T., McGargill, M., Town, T., Thompson, J., Anderson,J. F., Flavell, R. A., Fikrig, E., Hedrick, S. M. & Wang, T. (2008b). Drak2contributes to West Nile virus entry into the brain and lethalencephalitis. J Immunol 181, 2084–2091.
Zlokovic, B. V. (2008). The blood-brain barrier in health and chronicneurodegenerative disorders. Neuron 57, 178–201.
WNV-induced changes in the markers of BBB integrity
http://vir.sgmjournals.org 1203




















