
Micro and nanoscale domain engineering in lithium niobate and lithium tantalate
Upload
rwth-aachenCategory
view
0download
0
Nanoscale
REVIEW
Publ
ishe
d on
08
May
201
3. D
ownl
oade
d by
Nat
iona
l Ins
titut
es o
f St
anda
rds
& T
echn
olog
y on
10/
06/2
013
21:2
4:51
.
View Article OnlineView Journal
Molecularly stabili
aRWTH Aachen University, Helmholtz
Biointerface Laboratory, Pauwelsstrasse 30
[email protected]; Fax: +49 241 80-8bRWTH Aachen University, Institute of Ino
Aachen, Germany. E-mail: ulrich.simon@ac
Tel: +49 241 80-94644
AdmRPseaOgTrim
Cite this: DOI: 10.1039/c3nr00916e
Received 21st February 2013Accepted 28th April 2013
DOI: 10.1039/c3nr00916e
www.rsc.org/nanoscale
This journal is ª The Royal Society of
sed ultrasmall gold nanoparticles:synthesis, characterization and bioactivity
Annika Leifert,a Yu Pan-Bartnek,b Ulrich Simona and Willi Jahnen-Dechent*b
Gold nanoparticles (AuNPs) are widely used as contrast agents in electronmicroscopy as well as for diagnostic
tests. Due to their unique optical and electrical properties and their small size, there is also a growing field of
potential applications in medical fields of imaging and therapy, for example as drug carriers or as active
compounds in thermotherapy. Besides their intrinsic optical properties, facile surface decoration with (bio)
functional ligands renders AuNPs ideally suited for many industrial and medical applications. However,
novel AuNPs may have toxicological profiles differing from bulk and therefore a thorough analysis of the
quantitative structure–activity relationship (QSAR) is required. Several mechanisms are proposed that cause
adverse effects of nanoparticles in biological systems. Catalytic generation of reactive species due to the
large and chemically active surface area of nanomaterials is well established. Because nanoparticles
approach the size of biological molecules and subcellular structures, they may overcome natural barriers
by active or passive uptake. Ultrasmall AuNPs with sizes of 2 nm or less may even behave as molecular
ligands. These types of potential interactions would imply a size and ligand-dependent behaviour of any
nanomaterial towards biological systems. Thus, to fully understand their QSAR, AuNPs bioactivity should
be analysed in biological systems of increasing complexity ranging from cell culture to whole animal studies.
Introduction
The main parameters to describe a metal nanoparticle (NP) arechemical composition, size (including both the metal core size
Institute for Biomedical Engineering,
, 52074 Aachen, Germany. E-mail: willi.
2573; Tel: +49 241 80-80163
rganic Chemistry, Landoltweg 1, 52074
.rwth-aachen.de; Fax: +49 241 80-99003;
nnika Leifert received her PhDegree in 2012 from the Depart-ent of Inorganic Chemistry atWTH Aachen University. HerhD project was focused on theynthesis, characterization andvaluation of the biologicalctivity of gold nanoparticles. Inctober 2012 she joined theroup of Prof. Stefan Kaskel atU Dresden as a postdoctoralesearcher. Her current researchs focused on nanoparticle basedaterials with novel properties.
Chemistry 2013
as well as the total size including the ligand shell), bindingstrength between the core and ligand (as this determines theaccessibility of a NP surface), overall charge and stability in thegiven medium (taking potential aggregation into account).These parameters and the respective analytical methods areillustrated in Fig. 1. Only with fully characterized testsubstances, cause–effect relationships may be elucidated.
In this review, well established synthesis methods as well ascharacterization techniques of ultrasmall gold nanoparticles(AuNPs) will be discussed, followed by a summary of analytical
Yu Pan-Bartneck received herPhD degree in 2010 at RWTHAachen University for her workon the toxicity of mono-sulfonated triphenylphosphanegold nanoparticles. She holds aMaster Degree in BiomedicalEngineering from RWTH AachenUniversity and a Bachelor ofMedicine from Shanghai SecondMedical University. Since 2013she has been a counselingscientist for an internationalbiomedical business develop-ment rm.
Nanoscale

Fig. 1 Parameters that determine metal NP properties and most importantanalytical techniques (further discussed in detail below): EA: elemental analysis;NMR: nuclear magnetic resonance (spectroscopy); IR: infrared (spectroscopy); MS:mass spectrometry; AFM: atomic force microscopy; DLS: dynamic light scattering;DSC: differential scanning calorimetry; EM: electron microscopy; SAXS: smallangle X-ray scattering; XRD: X-ray diffractometry.
Nanoscale Review
Publ
ishe
d on
08
May
201
3. D
ownl
oade
d by
Nat
iona
l Ins
titut
es o
f St
anda
rds
& T
echn
olog
y on
10/
06/2
013
21:2
4:51
. View Article Online
methods to determine biological activity, illustrated by recentresults achieved in this eld. The feasibility to compare theresults of different methods will also be discussed. We focus onultrasmall AuNPs as they have revealed several parameters thatdetermine their bioactivity and toxicity, such as size and ligandfunctionalisation. Although there is no commonly accepteddenition for ultrasmall AuNPs, we refer to AuNPs of 2 nm orless, because in this size range size quantization effects as wellas enhanced catalytic activities occur and thus dominate theNPs' properties. Larger colloidal gold nanoparticles are gener-ally considered non-toxic and are in diagnostic and preclinicaluse already and thus are not considered in this review.
Synthesis
Metal NPs can generally be synthesized either in the gas or inthe solution phase, whereas in this work we focus on wetchemical synthesis routes. Gas phase syntheses are describedelsewhere.1 Due to the growing interest in AuNPs for applica-tions in different elds, a huge and still increasing number ofdifferent synthesis routes have been developed.2 Two recent
Ulrich Simon studied chemistryat the University of Essen (Ger-many) and obtained his diplomain 1990, his doctorate in 1992and became an associateprofessor aer having nishedhis habilitation in 1999. Since2000 he has been director of theInstitute of Inorganic Chemistryat the RWTH Aachen University(Germany) and holds the chairof Inorganic Chemistry andElectrochemistry. The main
interest of his current research includes the synthesis, assembly,and electrical properties of metal and semiconducting nano-particles and of nanoporous materials, as well as their applicationin nanoelectronics and biomedicine.
Nanoscale
reviews summarize synthesis strategies of gold clusters invarious media, either stabilized by ligands or polymers,respectively, or immobilized on surfaces and their geometricand electronic properties, for a rational approach to functionalmaterials.3,4 The most common routes are exemplarilydescribed in the following paragraph in order to illustrate theaccess to stable, water soluble AuNPs.
The solution-based synthesis of AuNPs can be performed inpolar and non-polar solvents and typically follows the reduction ofa dissolved Au(III) salt or Au(I) complex to Au(0) by a suitablereducing agent in the presence of ligands, which have to be elec-tron donors (Lewis acid) binding coordinatively to surface atomsof the NPs (Lewis base). The most common ligands are carboxylicacids, amines, phosphines and thiols, whereas the ligand-to-metalbinding strength follows the order O < N < P < S, according toPearson's HSAB concept. Typical reducing agents are, for example,trisodium citrate (Na3C6H5O7), sodium borohydride (NaBH4), ordiborane (B2H6), depending on the polarity of the solvent as wellas on the intended size range, as a strong reducing agent induceda high nucleation rate rather than a high growth rate, thus leadingpreferentially to the formation of ultrasmall AuNPs.5
Fig. 2 summarizes different pathways that can be followed toobtain water soluble AuNPs, which are needed for any kind ofbiological test or application. It illustrates that the synthesis caneither be performed directly in the water phase, or alternativelyin a non-polar solvent, followed by a ligand exchange reaction atthe solvent's interface. The driving force for ligand exchange ina homogeneous phase is the strength of the metal-to-ligandbond, so that carboxylates, such as citrate, amines or chargedphosphines can quantitatively be replaced by thiols, if appliedin huge excess. At the solvent interface, the particles have topass from the non-polar side to the water phase, whereby thenon-polar ligand, initially bound to the particle, is replaced by awater soluble one, making the AuNPs water soluble and thusaccessible for further ligand exchange or e.g. standardbiochemical coupling reactions.
Willi Jahnen-Dechent isProfessor for BiointerfaceScience at the RWTH AachenUniversity. He completed hishabilitation in PhysiologicalChemistry and Pathobiochemis-try from the Medical Faculty ofJohannes Gutenberg UniversityMainz in 1999. He received hisPhD at the University of Colognein 1986 for thesis work at theMax-Planck-Institute. He per-formed postdoctoral research at
Melbourne University, Australia and at the University of Massa-chusetts, Amherst. His work centers on the structure and functionof fetuins, a small family of blood proteins, and on cell–materialinteractions including the clearance and toxicity of nanoparticlesin experimental cell and animal models.
This journal is ª The Royal Society of Chemistry 2013

Fig. 2 Schematic presentation of the reaction pathway for the synthesis of watersoluble AuNPs. Starting in the non-polar phase the precursor is typically a Au–ligand complex, such as [(C6H5)3P]AuCl, which is reduced via a suitable reducingagent, such as B2H6. After AuNPs formation, the cluster is transferred to theaqueous phase via ligand exchange. In the aqueous phase synthesis starts typi-cally with a AuI or AuIII salt, such as HAuCl4, which is reduced to form the NPs.Ligands present during synthesis stabilize the AuNPs in the aqueous environmentand can, if necessary, be replaced by stronger binding ligands, i.e. phosphines (e.g.TPPMS) can be replaced by thiols (e.g. GSH) in the homogeneous phase.
Table 1 Overview of typical ligands for AuNPs, classified by the atom of bindingfunctionality. These determine the ligand donor ability
Ligand Size range/nm Reference
O Diglyme 1.9–8.9a 15Citrate 15–150 6
N Dodecylamine 2.5–7.0 7Lysine 6.5 16
P TPP 1.4 8TPPMS 1.4 10BINAP 1.7 17
S GSH 1.0 13 and 14PEG-SH 1.5–3.2 18Alkylthiols 1.5–5.2 11 and 19DHLA 1.6 20GSH/cysteamine 2.7 21Tiopronin 2.8 22
a Aer capture of primary particles by thiols or amines.
Review Nanoscale
Publ
ishe
d on
08
May
201
3. D
ownl
oade
d by
Nat
iona
l Ins
titut
es o
f St
anda
rds
& T
echn
olog
y on
10/
06/2
013
21:2
4:51
. View Article Online
One of the most commonly applied syntheses for AuNPs isthe reaction published initially by Turkevitch et al. in 1951.6 Inthis reaction, [AuCl4]
� is reduced by trisodium citrate in boilingwater. The citrate which is added in excess acts as a reducingagent as well as a ligand for the formed AuNPs, stabilizing theparticles electrostatically. Depending on the synthesis condi-tions, i.e. the concentrations of the components as well astemperature and reaction time, particle sizes in the range from15 nm up to 150 nm can be obtained. Further renement of thismethod makes particle sizes down to 10 nm accessible.
Leff et al. described a synthesis route using NaBH4 as thereducing agent to obtain amine-stabilized AuNPs in a size rangefrom 2.5 to 7.0 nm, depending on the synthesis conditions.7 Asamines are weakly binding ligands, these AuNPs are of specialinterest for subsequent ligand exchange reactions with strongerbinding ligands.
Very small AuNPs with the chemical formulaAu55[(C6H5)3P]12Cl6 (Au55TPP) and a gold core diameter of1.4 nm can be synthesized by the method developed by Schmidet al. in 1981.8 In this reaction the Au(I) complex [(C6H5)3P]AuClis dissolved in benzene and reduced with diborane at moderateheat. These Au55 clusters are monodisperse and tend to crys-tallize upon solvent evaporation.9 In particular for this clusterSchmid et al. developed a method to transfer these AuNPs intothe aqueous phase.10 For this purpose, a dichloromethanedispersion of the clusters is covered with an aqueous solution ofmono-sulfonated triphenylphosphine (TPPMS) and stirred atroom temperature for three days. An equilibrium of AuNPs inboth phases is reached aer this time. To improve the yield ofwater soluble clusters, the volume of the water phase is largerand TPPMS is added in large excess. Furthermore, TPPMSstabilizes the AuNPs more efficiently compared to triphenyl-phosphine, as the particles are not only stabilized via steric
This journal is ª The Royal Society of Chemistry 2013
repulsion (such as the TPP-stabilized one) but also by an elec-trostatic stabilization of the charged TPPMS which has anegative sulfonate group.
Another break-through in AuNP synthesis was achieved in1994 when Brust et al. developed the so-called Brust–Schiffrinsynthesis.11 In a two-phase system, HAuCl4 is dissolved in H2O,covered with an organic solvent (e.g. toluene) containing analkylthiol, and reduced with an aqueous solution of NaBH4.AuNPs with sizes between 1.5 and 5.2 nm with differentalkylthiol ligands were obtained. One year later, they expandedtheir synthesis concept into a one-phase method, where meth-anol serves as a solvent for all compounds.12
An approach to synthesize thiol-stabilized, water solubleultrasmall AuNPs was introduced by Whetten et al. and renedby Negishi et al.13,14 In this route, HAuCl4 is reduced by NaBH4 inthe presence of glutathione (GSH), a biogenic tripeptide con-sisting of L-cysteine, L-glutamic acid and glycine, and the size ofthe particles reaches down to approximately 1 nm.
Ligands
AuNPs need a stabilizing ligand shell to prevent them fromaggregation.23 As described above, the interaction between theligand and AuNP surface depends on the functional groupwhich binds to the particle and thus inuences the stability andreactivity of the AuNPs. The colloidal stability, which preventsthe particles from aggregation, is achieved either by steric (e.g.alkylthiols) or by electrostatic repulsion (e.g. phosphine sulfo-nates). Typical examples of AuNP ligands are summarized inTable 1 and shown in Fig. 3.
AuNP stability and solubility in different media, such as cellculture media, buffer, etc., mainly depends on the charge andpolarity of the end group that points away from the particlesurface. It determines the surface charge of the particles,experimentally accessible via measurements of the zeta poten-tial, and in biological systems it will be a key parameter, sincethe ligand shell is the entity the cell gets in contact with, whenincubated with AuNP solution.
Nanoscale

Fig. 3 Structures of typical AuNP ligands, classified by the atom of bindingfunctionality.
Nanoscale Review
Publ
ishe
d on
08
May
201
3. D
ownl
oade
d by
Nat
iona
l Ins
titut
es o
f St
anda
rds
& T
echn
olog
y on
10/
06/2
013
21:2
4:51
. View Article Online
Carboxylates such as trisodium citrate are very weak bindingAuNP ligands, which can most easily be replaced by otherligands. The stability of such particles in solution sensitivelydepends on factors such as salt concentration and pH. Incontrast to AuNPs with stronger binding ligands, it is notpossible to redisperse such AuNPs aer drying.
Amines, such as alkylamines or the amino acid lysine, havealso been used as ligands.16 Compared to stronger bindingphosphines and thiols, their affinity is only moderate, but stableAuNP dispersions can be obtained.
Phosphines, which are widely used as AuNP ligands, exhibita lower binding energy than thiols. DFT calculations on Au38and Au39 clusters revealed a binding energy of 0.93 eV for theAu–PH3 bond, while 2.45 eV for Au–SCH3 was obtained.24
Typically, aryl phosphines like triphenyl phosphine (TPP) andits derivatives are used, as their phenyl rings provide high AuNPstability due to sterical bulkiness as well as oxidation resistanceof the phosphine itself.
Thiols have the highest affinity to gold and are therefore themost frequently used class of ligands. For self-assembledmonolayers (SAM) consisting of thiols on a at gold surface, abinding energy of approximately 200 kJ mol�1 (h2.07 eV) wasdetermined.25 However, this value may be slightly different forAuNPs as the surface of NPs differs from a at surface becauseof its curvature and the resulting edge and vertex atoms. Othersulfur containing molecules are e.g. disuldes and thioethers,both forming considerably weaker bonds towards AuNPs.26–32
Dendrimers and polymers such as PAMAM, PVP and PEG arealso used as ligands to stabilize AuNPs. Such AuNPs are oensynthesized by reducing a gold salt or complex in the presenceof the respective polymer, so that the polymer matrix deter-mines the particle growth.2
Nanoscale
Furthermore, a chelate effect can be utilized in ligandbinding to obtain particle–ligand systems of high stability.Analogous to the binding modalities in complex chemistry theenhanced stability is based on a higher probability of recom-bination if one functional group desorbed from the nano-particle surface. A multivalent molecule remains in theproximity of the particle, as opposed to monovalent ligandswhich will desorb completely. Also, entropic effects play a role,since the degree of disorder increases when one chelatingligand replaces two or more monovalent ones.
Perumal et al. investigated binding kinetics of mono-, di- andtrithiols on differently sized AuNPs (2.2, 3.2 and 4.4 nm) viatime resolved uorescence spectroscopy of the leaving ligand (apyrene).33 They found that divalent ligands show the highestexchange rate and explain this by a cooperative activationmodelof vicinal atoms. The lower velocity of trithiols is explained byenhanced sterical hindrance. They also found a particle sizedependence, as the ligand exchange rate increases withincreasing particle size.
The chemical stability of mono-, di- and trithiol-stabilizedAuNPs (2 nm diameter) was also investigated by Srisombat et al.by UV/Vis spectroscopic monitoring of cyanide degradation.34
They found the same trend that dithiols stabilize AuNPs best.Diphosphines can also serve as AuNP ligands. An example is
2,20-bis(diphenylphosphino)-1,10-binaphthyl (BINAP)-stabilizedAuNPs with a mean diameter of 1.7 nm that could be synthe-sized via a direct route.17 Ligand characterization was per-formed by XPS, also showing that the gold atoms are present asAu0.
Furthermore, Au11 clusters, stabilized by four BINAP mole-cules per particle, were reported.35 The formationmechanism ofdiphosphine-stabilized undecagold was investigated in detail byelectrospray ionization-mass spectrometry (ESI-MS).36 It wasfound that ligand lability is an important factor in the equilibriaof relevant gold complex intermediates and that the directformation of diphosphine-stabilized clusters from diphos-phine–gold complexes may be hindered due to high stability ofsuch complexes. A problem in AuNP stabilization with poly-functional ligands may occur by possible network formationdue to cross-linking of AuNPs, resulting in aggregation andprecipitation.
NPs can easily be functionalized via their ligand shell. Thus,targeting molecules can be attached to the NP surface, leading toa specic interaction with cells having suitable receptors.Appropriate target molecules include peptides, proteins,enzymes and antibodies, depending on the aspired application.23
NPs with active targeting functionalities are classied asthird generation NPs and are capable of specically recognizingtheir target. Typically, they interact with receptors present onthe cell surface via peptides, proteins, aptamers and antibodies.Uptake can then occur through receptor-mediatedendocytosis.37
As an example, 2.8 nm sized AuNPs functionalized withtiopronin and then further with a Tat protein derived peptidesequence were tested in a human broblast cell line. In a TEManalysis specic nuclear targeting could successfully bedemonstrated.22
This journal is ª The Royal Society of Chemistry 2013

Review Nanoscale
Publ
ishe
d on
08
May
201
3. D
ownl
oade
d by
Nat
iona
l Ins
titut
es o
f St
anda
rds
& T
echn
olog
y on
10/
06/2
013
21:2
4:51
. View Article Online
Purication
A molecularly dened chemical compound is considered highlypure, when by-products or remaining educts can successfully beseparated by different techniques. In the case of AuNPs, puritywill also depend on the polydispersity of a sample, i.e. the sizedistribution of the particles. Narrow size distribution is a highlydesirable trait of nanoparticle preparations, because chemicaland biological properties are known to vary considerablydepending on size. Several methods can then be applied to purifythe raw material in terms of chemical purity and size selection.
The most frequently used methods for size selection includeltration, selective centrifugation, selective precipitation,column chromatography, and gel electrophoresis. Filtration isuseful if larger NP aggregates have to be removed from the NPsolution. Filters with narrowly dispersed pore sizes down to20 nm are commercially available, fabricated from differentmaterials such as alumina or polymers for minimum interac-tion with the particle to avoid decomposition.
Quantitative centrifugation, relying on the sedimentationprinciple, can also be used to separate particles with a bimodaldistribution of different masses.38 Either the pellet or thesupernatant may contain the desired product. Sample volumesmay vary widely from mL to hundreds of mL, which makescentrifugation especially suitable for larger batches.
Size selective precipitation is based on different solubilitiesof differently sized NPs due to varying numbers of ligands onthe particle surface and therefore differing stabilization. NPsare dispersed in a solvent, and a non-solvent or a salt is addedstepwise, enabling the successive precipitation and removal ofsize-selected NP fractions.39
Column chromatography or size exclusion chromatographyrelies on the same principle, as the retention time of NPsdepends on the particle size and therefore solubility andpolarity.40 Different materials for the stationary phase are usefulfor purication of AuNPs, such as cellulose or Sephadex�(a cross-linked dextran gel). However, not all AuNPs are suitablefor column chromatography, as especially larger particles orweakly stabilized ones tend to aggregate on the adsorbent.
Gel electrophoresis separates species regarding their sizeand charge. It is usually performed in polyacrylamide (PAGE; asmall pore size of 3–6 nm is possible) or agarose gel (larger poresize, typically 100–500 nm). By varying the degree of cross-linking, suitable gels can be generated for different AuNP sizes.Xu et al. used an agarose gel to separate 5, 15 and 20 nm sizedAuNPs.41 Gel electrophoresis can be used for the separation andsubsequent identication of different species in a mixture in avery small scale, or as a purication method of larger batches ina preparative scale. Schaaff et al. isolated GSH-stabilized goldclusters with the chemical formula Au28(GSH)16 by PAGE.13
Depending on the kind of ligand, stability problems of AuNPsmay occur, if these are sensitive to buffer solutions/increasedsalt concentrations. Ackerson et al. investigated various watersoluble, thiol-stabilized AuNP species regarding their ability tobe puried by PAGE.42
Purication methods to eliminate excess of ligand, synthesisby-products, and ionic impurities include centrifugation,
This journal is ª The Royal Society of Chemistry 2013
washing or precipitation, column chromatography and gelelectrophoresis, but also dialysis. Here, different membranesare commercially available with different molecular weight cutoff (MWCO) radii and made of different materials. Althoughdialysis may be a time consumingmethod when performed overseveral days, it is easy to perform.
Depending on particle size and ligand stabilization, AuNPssometimes exhibit reduced stability when kept in solution overlonger periods of time. In such cases the particle size changesdue to ripening effects (Ostwald ripening, digestive ripeningetc.) and partial decomposition and therefore slow enrichmentof impurities is possible. These facts need to be considered forthe preparation, storage and handling of AuNPs. Regularlyrepeated analysis of the NP quality is essential to ensure that theinvestigated material has the intended structure.
Characterization of AuNPsMethods and limits of size determination
In the characterization of AuNPs, or of ligand stabilized NPs ingeneral, besides their chemical composition, the determinationof the particle size and its distribution (core and shell) as well asthe analysis of the ligand shell composition and functionalityhas to be considered. The following paragraph will give anoverview of the most commonly applied methods, oen leadingto complementary information, and their meaning for the NPcharacterization (vide infra Table 2).
UV/Vis spectroscopy
The optical properties of a AuNP sample can easily be investi-gated by UV/Vis spectroscopy. AuNPs above a size of approx.2 nm exhibit metallic properties and show a plasmon resonancepeak in their optical absorbance spectrum. As its shape andspectral position depend on the size, shape and the NPsurrounding, UV/Vis spectroscopy is used for qualitative NPanalysis for dispersions of spherical AuNPs. When parameterssuch as solvent and ligand shell are kept constant for differentsamples, the width of the peak is ameasure of the polydispersityof the sample.
Ultrasmall AuNPs of 2 nm and below are no longer metallic,but exhibit energetically separated electronic states and discreteelectron excitations occur in UV/Vis absorption. The respectivespectra show narrow peaks in the short-wave length range,reecting single electron excitations, as for example shown forthe cluster [Au9[(C6H5)3P]8](NO3)3.43 The positions of theabsorption signals, reecting the energy levels of the clusterorbitals, depend sensitively on the number of metal atoms, onthe atomic conguration, but also on the nature and thebinding mode of the stabilizing ligands.
Electron microscopy
Electron microscopy (EM) is used to determine the size of aAuNP core. The resolution is determined by the wavelength ofthe probe beam and it approaches the sub-nm range.44 By usingaberration correctors, resolution can reach 50 pm in annulardark-eld scanning TEM imaging.45
Nanoscale

Table 2 Different analysis methods for AuNP samples
Method Parameter Resolution/limitation
UV/Vis Plasmon band 2 nmTEM Size Atomic resolutionAFM Size 0.1 nm in the Z direction,
3 nm in the lateral directionXRD Crystallinity/size Atomic resolutionDLS Size 1 nm (solvent dependent)SAXS Size/shape 0.8 nmMS Size Atomic resolutionEA Chemical composition Accuracy typically �0.3%TGA/DSC Chemical composition/
ligand affinityAccuracy typically ng–mg range
NMR Ligand shell Relatively high AuNPconcentrations necessary
IR Ligand shell Relatively high AuNPconcentrations necessary
Fluorescencemicroscopy
Ligand shell/autouorescence
Relatively high AuNPconcentrations necessary
XPS Chemical composition/electronic state
100–1000 ppm
Nanoscale Review
Publ
ishe
d on
08
May
201
3. D
ownl
oade
d by
Nat
iona
l Ins
titut
es o
f St
anda
rds
& T
echn
olog
y on
10/
06/2
013
21:2
4:51
. View Article Online
However, several obstacles have to be overcome when usingTEM for NP size determination. TEM is not a volume integratingmethod, i.e. a relatively small fraction of particles is examinedcompared to other methods. Analysis of a statistically signicant,and hence a representative number of particles is thereforecumbersome, but nevertheless necessary. For ultrasmall NPs, thecontrast between particles and the substrate (usually an amor-phous carbon lm)may be low.46Due to the high electron densityof gold this problem is mostly negligible for AuNPs. The resolu-tion can be improved by measuring in dark-eld imaging modeand even further in high-angle annular dark-eld (HAADF)mode,using a detector operating in a preselected angular range whileexcluding the direct electron beam.
The appropriate choice of magnication and acceleratingvoltage is also crucial. Magnication is especially important forstatistical NP size analysis. With higher magnication, theparticle size can be determined more correctly. On the otherhand, fewer particles per image are available for evaluation.Higher voltage enhances contrast, but may induce severesample damage. Thus typically intermediate energy voltages(80–400 keV) are employed for NP imaging in EM. To reducesample damage by the electron beam, scanning transmissionEM (STEM) is advantageous compared to continuously exposingthe entire sample to the electron beam.
Furthermore, the image analysis is an important parameter.Manual evaluation may be defective, but for samples with lowcontrast, it is preferred compared to soware-assisted or fullyautomated analysis. Smoothing and sharpening processes ofthe images may come along with loss of information andaddition of artefacts.
A state-of-the art example for AuNP EM characterization waspresented by Li et al., who used aberration-corrected HAADF-STEM for the three-dimensional analysis of a Au309 cluster. Theycould achieve atomic resolution when combined with clustersimulations.47
Small angle X-ray scattering
In small angle X-ray scattering (SAXS), elastic scattering ofX-rays is detected at small angles (<10�) to resolve small,mesoscopic structures in solution. For AuNPs, there is a strongscattering contrast of the heavy gold atoms with the solvent,thus, the core radius can be determined. This was for exampleshown by Terrill et al. for 2 nm sized, alkanethiol-stabilizedAuNPs.48 Hostetler et al. used SAXS for the size determination ofseveral AuNP samples, performed two different analyses fromthe data and compared the results with TEM results, ndingthem to be in good agreement.49 The nucleation and growth ofAuNPs during the synthesis were analysed via SAXS by differentgroups, using different ligands, reducing agents and reactionconditions and nding different formation kinetics.50–52 Theresolution limit of common laboratory SAXS systems is around0.8 nm.51
Atomic force microscopy
In atomic forcemicroscopy (AFM), a surface is scanned by a smalltip mounted on a cantilever. The deection of the cantilever is
Nanoscale
detected by a laser beam. The vertical resolution can be as high as0.1 nm,mostly depending on the tip geometry.53 The technique iswell suited to investigate nanostructures and also small NPs onsurfaces, either spatially arranged or randomly dried onto a atAFM sample disc. Thus AFM may be used to determine the sizedistribution of NPs in a statistically meaningful way. Forexample, CTAB-stabilized, differently sized AuNPs ranging from2.6 to 14 nm were analysed and sized together with TEM andSAXS.54 Results were comparable, but AFM slightly overestimatedthe AuNP sizes. Themost likely explanation for this nding is thefact that AFM also detected the ligand shell and not only themetal core like TEM and SAXS do.
X-ray diffractometry
X-ray diffractometry (XRD) is based on elastic scattering of X-rays from the electrons of atoms. Powder diffraction is used toanalyse the crystallographic structure and crystallite size of amaterial. For very small crystallite sizes, reexes in XRD arebroadened, a phenomenon handled by the Scherrer equation.55
Thus XRD can be used to determine the structure of AuNPs.Small angle X-ray diffraction (SAXRD) was applied to determinethe arrangement of Au55[(C6H5)3P]12Cl6 crystallites, showing asimple close packed arrangement of clusters correlating with aneffective cluster distance of 2.3 nm.9 Until now it was notpossible to generate large single crystals of the Au55 cluster.Therefore no single-crystal X-ray structure analysis to solve thecomplete structure could be performed.56
Crystallization of single crystals was however possible forother AuNP species. In 2007, Jadzinsky et al. reported the crys-tallization and X-ray structure determination of a p-mercapto-benzoic acid ( p-MBA)-protected AuNP, consisting of 102 goldatoms and 44 p-MBA molecules.57 For even smaller AuNPs(molecular clusters), single crystal structure analysis is morecommon and established.43,58–60
This journal is ª The Royal Society of Chemistry 2013

Review Nanoscale
Publ
ishe
d on
08
May
201
3. D
ownl
oade
d by
Nat
iona
l Ins
titut
es o
f St
anda
rds
& T
echn
olog
y on
10/
06/2
013
21:2
4:51
. View Article Online
Dynamic light scattering and zeta potential measurement
In dynamic light scattering (DLS) measurements, laser light isused to determine the size distribution prole of a NP disper-sion.61 Photons are scattered by particles, if the particles aresmall compared to the wavelength used. For monochromaticcoherent laser light, time-dependent uctuations of the scat-tering intensity occur, which are induced by the Brownianmotion particles undergo in dispersion. From the intensitytrace, an autocorrelation function can be generated, whichtypically shows exponential decay. This decay is related to thediffusion coefficient of the NP. These data can be used tocalculate the hydrodynamic radius of a sphere through theStokes–Einstein equation.
The zeta potential of a colloidal dispersion describes thepotential difference between the dispersion medium and thestationary layer of uid attached to the dispersed particle.62 It isa measure to approximate the stability of a NP dispersion. If theabsolute value is higher than 30 mV, a dispersion is regarded asstable. Measurement of the zeta potential of a NP dispersion istypically conducted by performing DLS in an electrical eld. Theelectrophoretic mobility thus determined correlates with thezeta potential.
Practical advantages of DLS and zeta potential measure-ments are speed and easy performance. Unlike TEM, theseanalytical methods integrate a large number of particles in onemeasurement, providing statistically relevant data.
There is however a major drawback related to this. Theintensity of scattered light varies with the sixth power of particlediameter. Therefore impurities such as comparatively large dustparticles, which would be immediately spotted and excluded inTEM, are highly disturbing.
Manufacturers of DLS instruments give values around 1 nmor below as lower size detection limits. However, this valuestrongly depends on medium viscosity, temperature and mon-odispersity of the sample. A critical discussion of potentialproblems arising in size determination of AuNPs by DLS wasgiven by Khlebtsov and Khlebtsov.63
As one example, AuNPs with a mean gold core diameter of2.7 nm, stabilized by a mixed ligand shell of GSH and cyste-amine, had a hydrodynamic diameter of 3.1 nmwhenmeasuredby DLS.21
In another study, Rotello and co-workers performed zetapotential measurements of 2 nm sized thiol-stabilized AuNPswith mixed ligand shells of tetra(ethylene glycol) (TEG) and1-pentanethiol, and found differences in the values dependingon the ratio of ligands bound to the particles.64 They found aqualitative correlation of ligand shell ratio and zeta potentials.
Mass spectrometry
Mass spectrometry (MS) can be used for small gold clusters todetermine their molecular formula, i.e. the number of goldatoms and ligands per particle. This was shown by Whetten andcoworkers for AuNPs with core diameters ranging from 1.4 to3.2 nm, stabilized by different thiols,65 and for GSH-stabilizedAuNPs using matrix-assisted laser desorption (MALDI) andelectrospray ionization (ESI) MS, identifying Au28(GSH)16 as the
This journal is ª The Royal Society of Chemistry 2013
most abundant species.13 Chaki et al. investigated the alka-nethiolate-stabilized species Au38 and Au144 in more detail byESI MS and suggested structural models.66 A limiting factor ofMS for AuNP analysis relates to the method of ionization, whichmust mobilize heavy species, but must bemild enough to not bedestructive. Thus, the method is limited (not fundamentally,but practically) by the mass of the respective AuNPs, andspecic measuring parameters have to be adjusted to thecompound to be studied.
Elemental analysis
Elemental analysis (EA) is a standard technique for the deter-mination of elemental composition. Typically, carbon, hydrogenand nitrogen (CHN) amounts are quantied in a sample bycombustion and detection of the resulting combustion products.For AuNPs, EA is useful to characterize the ligand shell compo-sition. If the structure of a cluster and thus the theoreticalelemental composition are known, EA can be used to verify thepurity of the sample. For Au55[(C6H5)3P]12Cl6 the EA result wasinstrumental in determining the chemical composition.8
One intrinsic problem of EA in AuNP characterization is thehigh mass of the gold core and thus an unfavorable mass ratiobetween the core and shell. This makes it difficult to distinguishe.g. samples before and aer a ligand exchange, especially if thechemical compositions of the outgoing and incoming ligandsare similar, and the percentage change of the AuNP species isrelatively small compared to the EA of the pure ligands.
Thermogravimetric analysis/differential scanning calorimetry
Thermogravimetric analysis (TGA) of AuNPs, i.e. heating of adened mass following a temperature program and paralleldetection of the weight loss of the sample, enables to determinethe percentage of organic ligands relative to the amount of goldand other inorganic constituents, such as counter ions.
The TGA of 1.5 nm sized, TPP-stabilized AuNPs resulted in amass loss of 24.5% in a temperature range of 200–250 �C which isassigned to the organic fraction of the AuNPs.67 Hostetler et al.synthesized various alkanethiolate-stabilized AuNPs with sizesranging from 1.5 to 5.2 nm and determined the respectivenumbers of ligands by TGA for each species.49 The decompositiontemperature is characteristic for different ligands but can also bea measure of differing stabilities of the ligand–gold bond.48
In differential scanning calorimetry (DSC), a reference isused to which the same temperature protocol is applied, andenthalpies of endo- and exothermic processes can be quanti-ed. Thus, stabilities of ligand–gold bonds can be determined,7
and intra- and intermolecular interactions of ligand moleculescan be investigated. Terrill et al. showed that alkanethiols withdifferent lengths show varying amounts of interdigitation whenthey are bound as ligands to 2 nm sized AuNPs.48 The degree ofordering of alkanethiol ligands on 3 nm sized AuNPs wasinvestigated with DSC by Badia et al.68
Nuclear magnetic resonance spectroscopy
Nuclear magnetic resonance (NMR) spectroscopy is widely usedin chemical analysis to determine molecular structures. When
Nanoscale

Nanoscale Review
Publ
ishe
d on
08
May
201
3. D
ownl
oade
d by
Nat
iona
l Ins
titut
es o
f St
anda
rds
& T
echn
olog
y on
10/
06/2
013
21:2
4:51
. View Article Online
ligands of a NP species are investigated by NMR spectroscopy,the proximity of the ligand molecules to the metallic coredistorts the resulting spectra, e.g. line broadening may occur.Several reasons account for this phenomenon, the mostimportant being the heterogeneous environments for thebound ligands on the NP, and also exchange broadening, dipolecoupling due to the restricted ligand mobility on the surfaceand other effects.69 Hence, the broadening of NMR signals mayserve as a proof of ligand binding to the AuNP surface, but italso complicates the structural analysis of the respectivemolecules.
NMR spectroscopy can be performed in solution, yieldingaveraged signals as the time required for an NMR spectrumrecording is very long compared to molecular movements. NMRcan also be conducted in the solid state.69 For AuNPs, NMRspectroscopy may yield important information about liganddynamics. This was demonstrated by Schmid forAu55[(C6H5)3P]12Cl6 in interaction with an excess TPP ligand,70
and also for similar TPP-stabilized clusters with a mean diam-eter of 1.8 nm by Sharma et al.69 They found that if excess TPP(in the deuterated form, d15-PPh3) was added, ligand exchangereactions occurred involving the complex TPP-Au(I). Along theseNMR spectroscopy of AuNPs may well allow the study of ligandreplacement reactions by biomolecules.
Infrared spectroscopy
In infrared (IR) spectroscopy, vibrational modes of atoms oratom groups in molecules are excited and the respectiveabsorption is detected. This covers the range from 400 to 4000cm�1 of the electromagnetic spectrum, typically given in wave-numbers. As for NMR spectroscopy, IR spectroscopy of AuNPs ismainly used to analyse the ligand shell. Characteristic bandsfrom functional groups can be identied to ensure particlefunctionalization with the respective molecules. Sometimes,further information can be gained, e.g. for thiols bound toAuNPs. Here, the S–H vibrational mode disappears in AuNPspectra as thiols bind via the sulphur atom to the AuNP surface,as demonstrated by Brust et al.12 Again, the problem of signalbroadening arises and can hinder the identication of weakbands in an IR spectrum.
Vilain et al. used IR spectroscopy to conrm the Janus-typecharacter of 7 nm sized AuNPs, stabilized by a mixed ligandshell consisting of phosphinines and thiols.71 AuNPs with sizesbetween 2.6 and 8.8 nm and a mixed ligand shell of tetraocty-lammonium bromide and tryptophan were characterized byFourier transform infrared reection-absorption spectroscopy(FT-IRRAS), and also here a Janus-type distribution of the twoligands was found by orientation dependent vibrationalchanges in the adsorbed ligand functionalities.72
Fluorescence spectroscopy
Fluorescence spectroscopy uses electromagnetic radiation toexcite uorescent molecules. Typically, emission spectra arerecorded, but absorption can be detected as well. UltrasmallAuNPs can be inherently uorescent. This phenomenon isclearly dependent on size and ligand functionalization. Huang
Nanoscale
and Murray investigated tiopronin-stabilized AuNPs with adiameter of 1.8 nm and found uorescence at 700–800 nm. Theyhypothesized a mechanism of interband transitions betweenthe lled 5d10 band and 6(sp)1 conduction band.73 Furtherexamples for this phenomenon can be found in the litera-ture.74–78 Polymer-stabilized AuNPs with sizes between 1.1 and1.7 nm were found to be uorescent, with 1.1 nm sized AuNPshaving the highest quantum yield of 3%.79,80
The behaviour of uorophores attached to AuNPs was alsoinvestigated. When uorophores are in close proximity toAuNPs, their uorescence is typically quenched e.g. by a Forsterresonance energy transfer (FRET).81 Therefore, uorescencespectroscopy can be a valuable tool to determine ligand releasefrom AuNPs. This was shown for 2 nm sized, uorescein-taggedAuNPs that had a charge dependent ligand release with cationicstabilized AuNPs being more labile than anionic ones.64
Gu et al. investigated the relaxation pathways of 2.2 nmsized, uorophore-labeled AuNPs and found that mainly energytransfer from the uorophore to the gold core plays a signicantrole.82
X-ray photoelectron spectroscopy
In X-ray photoelectron spectroscopy (XPS), X-ray radiation isused as a surface probe to trigger electron emission from thesample surface. As the energies of emitted electrons are elementand oxidation state dependent, valuable information about thechemical composition of the sample surface can be derived.
A limitation of the technique is the necessity of ultrahighvacuum during measurement. Furthermore, radiation candestroy sensitive samples during the measurement.
As a surface analysis method, XPS can be used to charac-terize the ligand shell of NPs, although the relatively low ligandconcentration compared to the metal core mass can be alimiting factor. Also, the oxidation state of the metal core can bedetected because the radiation can sample up to 10 nm depth.Tamura and Fujihara investigated 1.7 nm sized AuNPs, stabi-lized with BINAP, with XPS to conrm the oxidation state of 0 ofthe Au atoms.17 Boyen et al. determined by XPS an unexpectedlyhigh oxidation resistance for Au55 clusters compared to smallerand larger AuNPs, relating this inertness to the closed-shellstructure of the Au55 clusters.83
Bioactivity of ultrasmall AuNPs
Ultrasmall AuNPs are versatile compounds with many potentialapplications in diagnosis and therapy. Safety issues of AuNPshave been widely addressed.84–87 AuNPs are unlikely to causeenvironmental pollution due to the low production compared toother metal oxides (TiO2, SiO2), or carbon nanotubes, which areproduced in kiloton quantities per year worldwide. Nevertheless,several hundred papers are readily retrieved from the Pubmeddatabase using the search term “gold nanoparticle toxicity”.Conicting results are oen reported, and a consensus onexperimental design and standard calibration reagents is direlylacking. The oen quoted “quantitative structure activity rela-tionship” (QSAR) in nanoparticle toxicity is largely a myth,because most nanomaterials are highly polydisperse and have no
This journal is ª The Royal Society of Chemistry 2013

Review Nanoscale
Publ
ishe
d on
08
May
201
3. D
ownl
oade
d by
Nat
iona
l Ins
titut
es o
f St
anda
rds
& T
echn
olog
y on
10/
06/2
013
21:2
4:51
. View Article Online
dened structure. An evaluation guideline was recommended byCard and coworkers to rank the reliability of toxicity studies.88
Briey, the design of the study should be scored according to theKlimisch category89 and the physiochemical properties of theAuNPs including agglomeration, chemical composition, crystalstructure, particle size, purity, shape, surface area, surfacecharge, surface chemistry and the actual status of the AuNPs inthe experimental media should be stated in detail.
Dispersion state of AuNPs in biological uids and the coronaeffect
NPs will only behave as nanomaterials as long as they maintaintheir small size. Aggregating nanoparticles will progressivelybehave like a bulk material. Close attention should therefore begiven to the interaction of NPs and biological uids, whichmight greatly inuence their physicochemical and biologicalactivity. The composition of biological uids was shown to alterthe particle size, surface characters, endocytosis pathway,intracellular trajectory and the toxicity prole of the NPs. On theother hand, NPs can alter the nature of bioactive proteinsdepending on the size, shape, surface charge and curvature.90
Commonly used cell culture media include (with or withoutserum) phosphate buffer and 0.9% sodium chloride. Many NPswill aggregate in saline and in serum free medium because ofthe salt contained in these solutions. Addition of serum can, tosome extent, improve the stability of the NPs in biological uidsby forming a protein halo or corona.90 Serum protein bindingmay alter the biological activity of NPs in more ways thanaggregation. Many proteins will facilitate AuNP endocytosis by aprocess called opsonization.
One of the best studied proteins interacting with NPs is serumalbumin, the most abundant globulin in serum. Albumin isnegatively charged. Binding of albumin to AuNPs can change thepositive surface charge of AuNPs (e.g. cysteine modied AuNPs)into negative, increasing the hydrodynamic diameter of AuNPsand increasing reabsorption of the AuNPs in kidney tubules viathe neonatal Fc receptor–albumin binding.91 HydrophobicAuNPs strongly bind apolipoproteins. This binding can enhancethe clearance of AuNPs by hepatocytes, and apolipoproteins canincrease the penetration of the blood–brain barrier. In principle,AuNPs may also adsorb coagulation factors including freebrinogen, as has been demonstrated for polymeric nano-paricles.92NP protein binding can in fact be engineered to controlthe release of peptides from the NP surface. Thus “intelligentnanomaterials” have been optimized for drug release at sites ofinammation, which have a lower pH than healthy tissue.91
NP binding works both ways in that the particles collectmolecules from the surrounding uid, but also bind tomacromolecular assemblies or cell surfaces. AuNPs approachthe size of typical protein ligands andmay inadvertently activatecell surface receptors. AuNPs are particularly notorious forbinding thiol ligands because of the high binding affinitybetween sulphur and gold. Thus AuNPs will preferentially bindsulfur containing compounds, and may even deplete theglutathione-based redox buffering capacity of biological uidsand perhaps even the cytoplasmic redox buffer inside cells.
This journal is ª The Royal Society of Chemistry 2013
Several methods have been used to study NP–proteinbinding. Certain methods are based on the unique photo-physical properties of AuNPs, either uorescence quenching oruorescence enhancement.80,93 Fluorescence of tryptophan,tyrosine, and phenylalanine residues in proteins was quenched,when they bound to AuNPs, because of the changes in proteinconformation. Fluorescence decay of the AuNPs may also beused as a proxy of AuNP–protein binding. Ultrasmall AuNPshave different protein binding cooperativity and photophysicalproperties than large AuNPs. Shang and colleagues showed thatdihydrolipoic acid modied AuNPs (<2 nm) have strong uo-rescence emission at 684 nm.80 A 6-fold higher signal wasobserved following AuNP binding to human serum albumin,apo-transferrin, lysozyme and apolipoprotein E4. The signalenhancement was directly proportional to the concentration ofthe protein in the test situation. The four proteins are non-uorescent per se. Protein binding of AuNPs led however, to thereduction in the polarity of the local environment and sloweddown the uorescence decay rate of the AuNPs, enhancing theAuNP derived uorescence.80
The examples given reect the fact that our current knowledgemainly relies on indirect methods to probe the interaction ofproteins with NPs in general. Here it is important to note thatprotein adsorption will not only affect the surface functionalityand the state of aggregation of the NPs, but will potentially alsolead to structural changes of the protein and therefore to analtered conformation or even to complete loss of protein function.However, the mechanistic details of this interplay are still ratherunexplored.94 Experimental techniques, which have recently beenapplied to assess the interaction on a molecular level includeFourier transform infrared spectroscopy,95 surface-enhancedRaman spectroscopy,96 surface plasmon resonance spectroscopy,97
and circular dichroism spectroscopy.98 The latter technique hasbeen applied by Treuel et al. to analyse the dissociation constantsfor the interaction of serum albumin with citrate-coated AuNPs incomparison to silver NPs coated with polyvinylpyrrolidone.99
Remarkably, the binding affinities signicantly differed by up tothree orders of magnitude. These studies underscore that thestability of the initial surface functionality is a key parameter incontrolling the NP–protein interaction in biological media. Alto-gether, themechanistic understanding of this complex interplay iselusive and requires more quantitative analyses with hightemporal and spatial resolution on the true molecular level.
Localization and quantication of AuNPs in biologicalsamples
The methods used to track AuNPs with size larger than 5 nm incells and tissues are well established. However, imaging andlocalization of ultrasmall AuNPs remains a big challenge. The useof common contrast agents in e.g. electron microscopy isimpossible, because osmium and silver contrast enhancementboth introduce high background staining caused by metal grainsof comparable nm size like the ultrasmall AuNPs. The detectionof the ultrasmall AuNPs therefore requires uncontrastedsamples. The AuNPs may be imaged using high resolution elec-tron microscopy HR-TEM. However, the high energy electron
Nanoscale

Nanoscale Review
Publ
ishe
d on
08
May
201
3. D
ownl
oade
d by
Nat
iona
l Ins
titut
es o
f St
anda
rds
& T
echn
olog
y on
10/
06/2
013
21:2
4:51
. View Article Online
beam used in this technique destroys all organic matrixsurrounding the AuNPs including biological tissue componentsand indeed the embedding resin. This leaves us with a conun-drum. Conventional TEM cannot image ultrasmall AuNPs inbiological samples because of insufficient resolution,100 and HR-TEM cannot localize ultrasmall AuNPs in any biological contextbecause it destroys the surrounding biological structures.
Exploiting their specic photophysical properties, ultrasmallAuNPs may be traced in biological samples. Ultrasmall AuNPs donot possess plasmon resonance, but may exhibit size dependentuorescence emission. Recording the photoluminescence ofAuNPs or modifying AuNPs with uorescent markers is thus apromising approach to detect AuNPs in biological samples. Linet al. showed that dihydrolipoic acid (DHLA) modied AuNPshave a red photoluminescence aer exposure to 365 nm UV lightalthough either DHLA and AuNPs alone are non-uorescent.Further modifying Au–DHLA with streptavidin specicallylabelled human hepatoma cells, because they are biotin-positive.20
Table 3 lists some key studies on the tracking and imaging ofAuNPs in biological samples.
Biological activity of AuNPs in vitro
The unique biological properties of AuNPs are closely associ-ated with their increased surface activity and penetrationcapacity. Several studies have shown that the toxicity andintracellular distribution of AuNPs are size dependent.112–114
Ultrasmall 1.4 nm TPPMS modied AuNPs (Au1.4MS) had hightoxicity compared to 15 nm AuNPs bearing the same ligand.112
Au1.4MS killed the cells by triggering the production of reactiveoxygen species in the cells.115 Size was not the only parameterdetermining the high toxicity of Au1.4MS. Similar size thiolmodied AuNPs were non-toxic. In addition, the toxicity ofAu1.4MS was abolished by the addition of glutathione.115 Theprotective effect of GSH in Au1.4MS toxicity was due to ligand
Table 3 Methods to quantify nanoparticles in biological samplesa
Method Sample Sensitivity
TEM Cell, tissue HighPhotoluminescence/uorescent marker
Cell, tissue High
ICP-MS Cell, tissue HighICP-AES Cell, tissue HighNAA Cell, tissue 10–100 mg kg�1
AAS Cell, tissue 0.8–1.88 mg kg�1
Silver enhancement Cell, tissue 400 mg Au/mouseSpectroscopicphotoacoustic
Live mouse 400 mg Au/mouse
Photoacoustictomography
Live rat 0.8 � 109 nanocagesper g body weight
CT Live swine 86–99 mg kg�1
Liquid scanningtransmission electronmicroscopy
Live cells 1.8 nM, 2 h
a TEM: transmission electron microscopy; ICP-MS: inductively coupled pemission spectroscopy; NAA: neutron activation analysis; AAS: atomic abscomputed tomography; N.A. not available.
Nanoscale
exchange of GSH for TPPMS resulting in the non-toxiccompound Au1.4GSH.115 Hakkinen and colleagues determinedby density functional theory (DFT) that the binding energy ofthe gold phosphine (Au–PH3) bond is 0.93 eV, compared to2.45 eV for a gold thiolate (Au–SCH3) bond.24 The interaction ofbare, ligand-stripped gold atoms and biological targets, espe-cially thiol containing targets may thus be one prime reason forthe toxicity of Au1.4MS. Once the relatively weak bindingTPPMS was replaced with GSH, the stronger gold thiolate bondwould prevent further interaction between the unprotected,highly electronegative gold atom core and biological targets,thus abrogating toxicity. A genome-wide mRNA expressionanalysis showed that Au1.4MS triggered a robust heat shockand stress-related gene upregulation aer 12 hours in HeLacells115 suggesting extreme cell stress and ultimately apoptosisor necrosis depending on the applied dose of Au1.4MS.
AuNP interaction with DNA
Modeling AuNP interaction with potential intracellular biolog-ical macromolecules demonstrated that gold nanoparticlesbound tightly to the major groove of B-DNA.116,117 The fact thatthese AuNPs were cytotoxic led the authors to hypothesize thatAuNPs inside the cell nucleus may interfere with DNA tran-scription and replication.116 Later work showed that the cellulartarget of the nanoparticles may in fact lie upstream in thecellular trajectory,112,115 but modeling studies, which regardedthe DNA backbone with its multiple phosphate groups as apolydentate ligand, nevertheless conrmed tight DNA bindingof such particles.116
AuNP interaction with ion channels
Prime targets of interaction of ultrasmall NPs are thought to beproteins, the nanoscale machinery of cellular life. Ultrasmall
Data AuNP detection Reference
Localization No 101Localization Yes 20
Quantitative Yes 102Quantitative Yes 103Quantitative Yes 104 and 105Quantitative Yes 106Semi-quantitative No 107Semi- quantitative;localization
N.A. 108
Semi-quantitative;localization
N.A. 109
Semi-quantitative;localization
N.A. 110
Semi-quantitative;localization
N.A. 111
lasma mass spectrometry; ICP-AES: inductively coupled plasma atomicorption spectroscopy; UV-vis: ultraviolet–visible spectroscopy; CT: X-ray
This journal is ª The Royal Society of Chemistry 2013

Review Nanoscale
Publ
ishe
d on
08
May
201
3. D
ownl
oade
d by
Nat
iona
l Ins
titut
es o
f St
anda
rds
& T
echn
olog
y on
10/
06/2
013
21:2
4:51
. View Article Online
AuNPs approach the size of ligands, and could potentiallydirectly interact with cell surface proteins.118 Ion channels areexpressed on the cellular membrane controlling intra- andextra-cellular ion homeostasis and the associated biologicalfunctions. The disturbance of ion channels inevitably changesthe cellular function and causes pathology. The human ether-a-gogo-related potassium channel (hERG) is particularly noto-rious among ion channels to interact promiscuously withmultiple ligands. Blockade of the hERG channel causes severecardiac arrhythmia and associated death. Therefore regulatingbodies have made it mandatory to test hERG function early indrug development.119,120 We recently studied the inuence ofAuNPs by the patch clamp technique in hERG channel trans-fected HEK293 cells.121 Depending on the ligand chemistry 1.4nm diameter AuNPs failed in the electrophysiology based safetytesting. Phosphine-stabilized AuNPs irreversibly blocked hERGchannels, whereas thiol-stabilized AuNPs of similar size had noeffect in vitro. Neither particle blocked the channel in vivo,because serum proteins prevented AuNPs from direct interac-tion with the ion channel. Thus AuNP interaction with ionchannels was clearly demonstrated in, but is probably irrelevantto the observed in vivo toxicity in this particular case. Never-theless our observation suggests that ultrasmall AuNPs shouldbe tested for hERG toxicity before potential in vivo use.
Biological effects and biodistribution ofAuNPs in vivoInteraction with blood components
The majority of NPs designed for in vivo use are administeredintravenously. Blood comprising blood cells and plasma is thusinvariably rst encountered by NPs. A well-studied sequenceoccurs starting with the interactions of bulk serum proteins (e.g.albumin, globulin and brinogen), which are graduallyreplaced by low abundance plasma proteins with high affinity.This so-called Vroman effect122 is the physical basis of theprotein corona,123 which determines the stability and trajectoryof NPs in biological systems.124
Hemolysis
The surface charge of NPs is a major determinant of hemolyticactivity. A study employing lithium–heparin anticoagulatedhuman blood showed that NPs abundant in cationic groupscaused hemolysis.125,126
Platelet activation
Hydrophobic NPs tend to induce aggregation of platelets. Deband co-workers showed that platelet aggregation increased withreduced AuNP size.127 Compared to 68 nm AuNPs, 20 nm AuNPstriggered more platelet aggregation. In addition, a plateletsurface integrin GpIIbIIIa inhibitor stopped AuNP inducedplatelet aggregation as expected, but not CD62P up-regulation,suggesting that the AuNP interaction with platelets triggered afully executed signal transduction cascade, and not merelyhydrophobic cross-linking of AuNPs and platelets. The inter-action of ultrasmall AuNPs and platelets was not studied, but
This journal is ª The Royal Society of Chemistry 2013
the size-dependent pattern suggests that ultrasmall AuNPs maycause severe platelet activation and thus intravascular bloodclotting.128
Complement activation
The complement system is a primitive, yet important part of theimmune defense, which is based on the assembly of activationcomplexes made of “sticky” binding proteins, and subsequentassembly of proteolytic execution cascades. Unwarranted acti-vation of the complement system causes inammation andautoimmune disease. Many interactions between NPs andcomponents of the complement system are possible. A study ofHamad et al. showed that the surface structure of NPs directedthe mode of complement activation.129 Binding of immuno-globins (IgG, IgM), C-reactive protein, and C1q protein all trig-gered the activation of the classical complement pathway. Thebinding of mannose lectin-binding protein and/or colinsinduced the lectin pathway. Surface functionalization of NPswith hydroxyl and amine end groups caused binding ofcomplement C3. The cleavage of a thioester bond in C3 trig-gered by this binding activated the alternative complementarypathway. The elevation of complement activation products inserum including C4d (classical pathway), Bb (alternativepathway) and C5a (common terminal pathway) can be used todifferentiate among the different complementary pathways.
Biodistribution of AuNPs
Small rodents are usually used in pilot studies exploring thepharmacology and biodistribution of NPs in mammals. Unlikecell based assays, these animal studies inform about bloodclearance, organ distribution, and detoxication mechanisms.In addition, the genetics, physiology and behavior of rodentsare well established, and imaging modalities like micro-computed tomography, micro-ultrasound, and uorescencemolecular tomography can all be employed. Mouse102,107,130–136
and rat104,137,138 are the most commonmammal models to assessNP toxicity in vivo. The biodistribution and organ-specictoxicity of NPs can be investigated in normal healthy mice aswell as in genetically engineered or pre-operated micemimicking human diseases. Mouse tumor models facilitate theevaluation of the anti-tumor efficiency of nanomaterials as well.Major disadvantages of these models are that they are cost andlabor intensive and sometimes the results obtained in mice orrats will not apply to humans, because of the differentphysiology.
Doses of AuNPs ranging from 0.1 to 2700 mg kg�1 bodyweight have been injected into mice. The highest dose of 2700mg kg�1 was tested using Aurovist, a commercial AuNP (1.9 nm)designed as an X-ray contrast agent.132 Neither acute toxicity norpathologies regarding histology or blood chemistry werereported at this high dose. Aurovist may therefore serve as aprototype ultrasmall AuNP for biodistribution studies. Fig. 4illustrates a typical experiment when a single bolus of 2000 mgkg�1 body weight of Aurovist was injected intravenously. Fig. 4Ashows that blood and urine samples taken 1 hour post injection(hpi) were dark opaque compared to the clear color of serum
Nanoscale
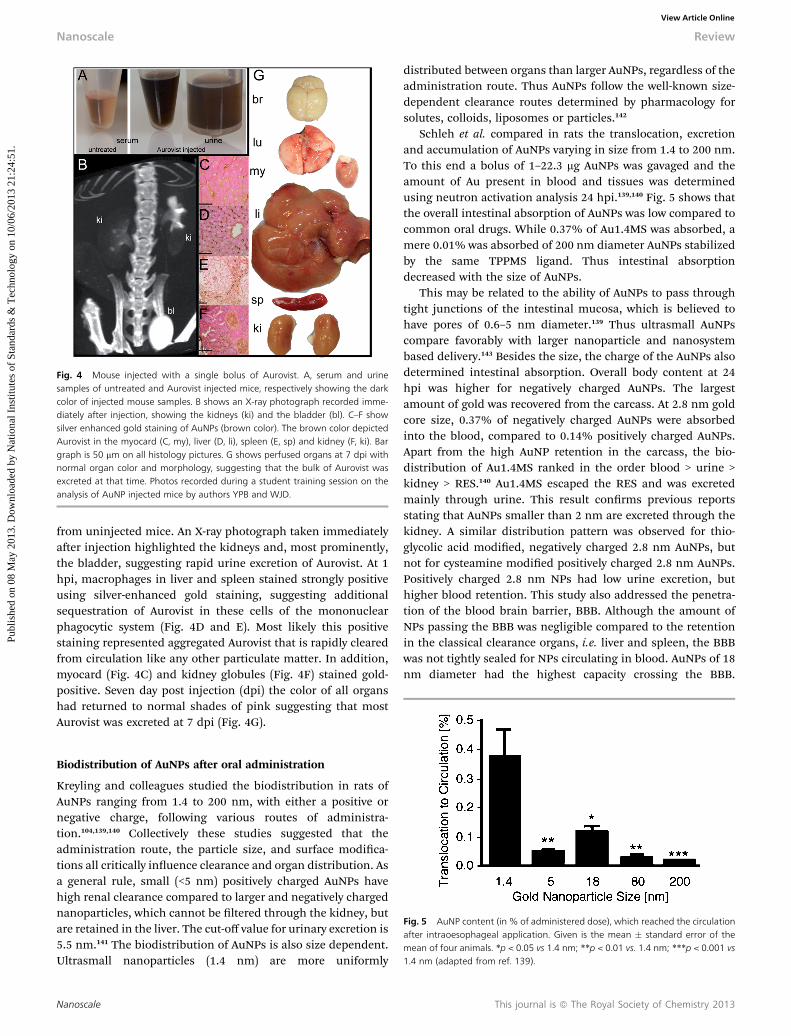
Fig. 4 Mouse injected with a single bolus of Aurovist. A, serum and urinesamples of untreated and Aurovist injected mice, respectively showing the darkcolor of injected mouse samples. B shows an X-ray photograph recorded imme-diately after injection, showing the kidneys (ki) and the bladder (bl). C–F showsilver enhanced gold staining of AuNPs (brown color). The brown color depictedAurovist in the myocard (C, my), liver (D, li), spleen (E, sp) and kidney (F, ki). Bargraph is 50 mm on all histology pictures. G shows perfused organs at 7 dpi withnormal organ color and morphology, suggesting that the bulk of Aurovist wasexcreted at that time. Photos recorded during a student training session on theanalysis of AuNP injected mice by authors YPB and WJD.
Nanoscale Review
Publ
ishe
d on
08
May
201
3. D
ownl
oade
d by
Nat
iona
l Ins
titut
es o
f St
anda
rds
& T
echn
olog
y on
10/
06/2
013
21:2
4:51
. View Article Online
from uninjected mice. An X-ray photograph taken immediatelyaer injection highlighted the kidneys and, most prominently,the bladder, suggesting rapid urine excretion of Aurovist. At 1hpi, macrophages in liver and spleen stained strongly positiveusing silver-enhanced gold staining, suggesting additionalsequestration of Aurovist in these cells of the mononuclearphagocytic system (Fig. 4D and E). Most likely this positivestaining represented aggregated Aurovist that is rapidly clearedfrom circulation like any other particulate matter. In addition,myocard (Fig. 4C) and kidney globules (Fig. 4F) stained gold-positive. Seven day post injection (dpi) the color of all organshad returned to normal shades of pink suggesting that mostAurovist was excreted at 7 dpi (Fig. 4G).
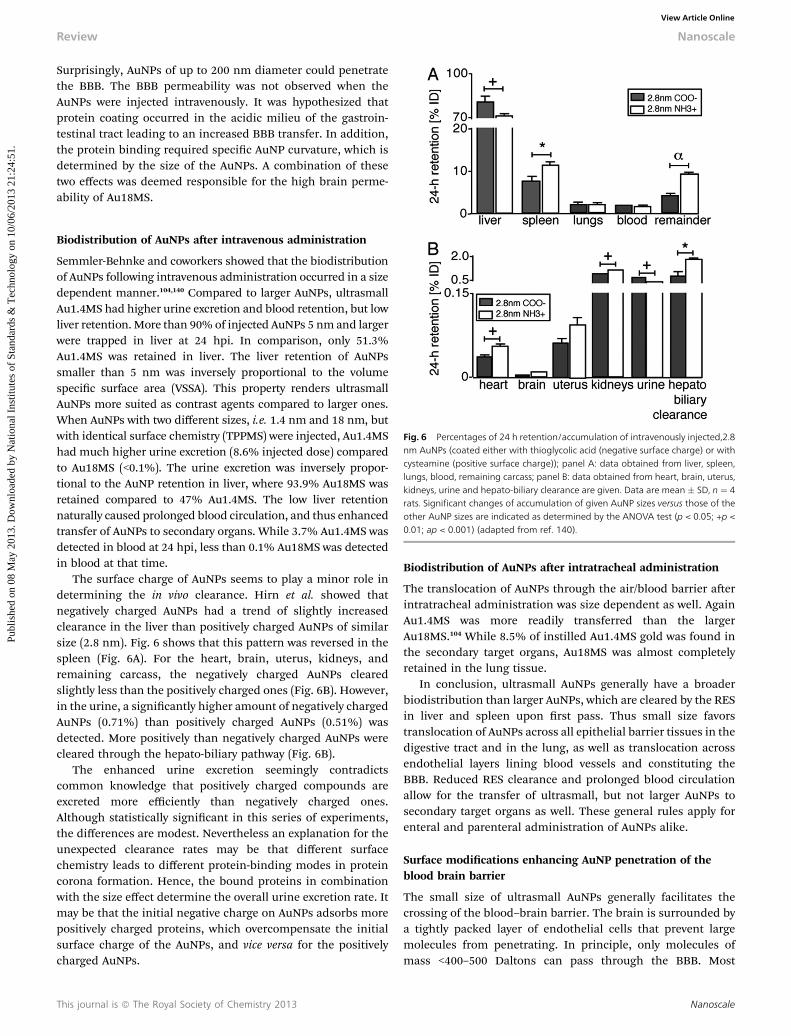
Fig. 5 AuNP content (in % of administered dose), which reached the circulationafter intraoesophageal application. Given is the mean � standard error of themean of four animals. *p < 0.05 vs 1.4 nm; **p < 0.01 vs. 1.4 nm; ***p < 0.001 vs1.4 nm (adapted from ref. 139).
Biodistribution of AuNPs aer oral administration
Kreyling and colleagues studied the biodistribution in rats ofAuNPs ranging from 1.4 to 200 nm, with either a positive ornegative charge, following various routes of administra-tion.104,139,140 Collectively these studies suggested that theadministration route, the particle size, and surface modica-tions all critically inuence clearance and organ distribution. Asa general rule, small (<5 nm) positively charged AuNPs havehigh renal clearance compared to larger and negatively chargednanoparticles, which cannot be ltered through the kidney, butare retained in the liver. The cut-off value for urinary excretion is5.5 nm.141 The biodistribution of AuNPs is also size dependent.Ultrasmall nanoparticles (1.4 nm) are more uniformly
Nanoscale
distributed between organs than larger AuNPs, regardless of theadministration route. Thus AuNPs follow the well-known size-dependent clearance routes determined by pharmacology forsolutes, colloids, liposomes or particles.142
Schleh et al. compared in rats the translocation, excretionand accumulation of AuNPs varying in size from 1.4 to 200 nm.To this end a bolus of 1–22.3 mg AuNPs was gavaged and theamount of Au present in blood and tissues was determinedusing neutron activation analysis 24 hpi.139,140 Fig. 5 shows thatthe overall intestinal absorption of AuNPs was low compared tocommon oral drugs. While 0.37% of Au1.4MS was absorbed, amere 0.01% was absorbed of 200 nm diameter AuNPs stabilizedby the same TPPMS ligand. Thus intestinal absorptiondecreased with the size of AuNPs.
This may be related to the ability of AuNPs to pass throughtight junctions of the intestinal mucosa, which is believed tohave pores of 0.6–5 nm diameter.139 Thus ultrasmall AuNPscompare favorably with larger nanoparticle and nanosystembased delivery.143 Besides the size, the charge of the AuNPs alsodetermined intestinal absorption. Overall body content at 24hpi was higher for negatively charged AuNPs. The largestamount of gold was recovered from the carcass. At 2.8 nm goldcore size, 0.37% of negatively charged AuNPs were absorbedinto the blood, compared to 0.14% positively charged AuNPs.Apart from the high AuNP retention in the carcass, the bio-distribution of Au1.4MS ranked in the order blood > urine >kidney > RES.140 Au1.4MS escaped the RES and was excretedmainly through urine. This result conrms previous reportsstating that AuNPs smaller than 2 nm are excreted through thekidney. A similar distribution pattern was observed for thio-glycolic acid modied, negatively charged 2.8 nm AuNPs, butnot for cysteamine modied positively charged 2.8 nm AuNPs.Positively charged 2.8 nm NPs had low urine excretion, buthigher blood retention. This study also addressed the penetra-tion of the blood brain barrier, BBB. Although the amount ofNPs passing the BBB was negligible compared to the retentionin the classical clearance organs, i.e. liver and spleen, the BBBwas not tightly sealed for NPs circulating in blood. AuNPs of 18nm diameter had the highest capacity crossing the BBB.
This journal is ª The Royal Society of Chemistry 2013
Review Nanoscale
Publ
ishe
d on
08
May
201
3. D
ownl
oade
d by
Nat
iona
l Ins
titut
es o
f St
anda
rds
& T
echn
olog
y on
10/
06/2
013
21:2
4:51
. View Article Online
Surprisingly, AuNPs of up to 200 nm diameter could penetratethe BBB. The BBB permeability was not observed when theAuNPs were injected intravenously. It was hypothesized thatprotein coating occurred in the acidic milieu of the gastroin-testinal tract leading to an increased BBB transfer. In addition,the protein binding required specic AuNP curvature, which isdetermined by the size of the AuNPs. A combination of thesetwo effects was deemed responsible for the high brain perme-ability of Au18MS.
Fig. 6 Percentages of 24 h retention/accumulation of intravenously injected,2.8nm AuNPs (coated either with thioglycolic acid (negative surface charge) or withcysteamine (positive surface charge)); panel A: data obtained from liver, spleen,lungs, blood, remaining carcass; panel B: data obtained from heart, brain, uterus,kidneys, urine and hepato-biliary clearance are given. Data are mean � SD, n ¼ 4rats. Significant changes of accumulation of given AuNP sizes versus those of theother AuNP sizes are indicated as determined by the ANOVA test (p < 0.05; +p <0.01; ap < 0.001) (adapted from ref. 140).
Biodistribution of AuNPs aer intravenous administration
Semmler-Behnke and coworkers showed that the biodistributionof AuNPs following intravenous administration occurred in a sizedependent manner.104,140 Compared to larger AuNPs, ultrasmallAu1.4MS had higher urine excretion and blood retention, but lowliver retention. More than 90% of injected AuNPs 5 nm and largerwere trapped in liver at 24 hpi. In comparison, only 51.3%Au1.4MS was retained in liver. The liver retention of AuNPssmaller than 5 nm was inversely proportional to the volumespecic surface area (VSSA). This property renders ultrasmallAuNPs more suited as contrast agents compared to larger ones.When AuNPs with two different sizes, i.e. 1.4 nm and 18 nm, butwith identical surface chemistry (TPPMS) were injected, Au1.4MShad much higher urine excretion (8.6% injected dose) comparedto Au18MS (<0.1%). The urine excretion was inversely propor-tional to the AuNP retention in liver, where 93.9% Au18MS wasretained compared to 47% Au1.4MS. The low liver retentionnaturally caused prolonged blood circulation, and thus enhancedtransfer of AuNPs to secondary organs. While 3.7% Au1.4MS wasdetected in blood at 24 hpi, less than 0.1% Au18MS was detectedin blood at that time.
The surface charge of AuNPs seems to play a minor role indetermining the in vivo clearance. Hirn et al. showed thatnegatively charged AuNPs had a trend of slightly increasedclearance in the liver than positively charged AuNPs of similarsize (2.8 nm). Fig. 6 shows that this pattern was reversed in thespleen (Fig. 6A). For the heart, brain, uterus, kidneys, andremaining carcass, the negatively charged AuNPs clearedslightly less than the positively charged ones (Fig. 6B). However,in the urine, a signicantly higher amount of negatively chargedAuNPs (0.71%) than positively charged AuNPs (0.51%) wasdetected. More positively than negatively charged AuNPs werecleared through the hepato-biliary pathway (Fig. 6B).
The enhanced urine excretion seemingly contradictscommon knowledge that positively charged compounds areexcreted more efficiently than negatively charged ones.Although statistically signicant in this series of experiments,the differences are modest. Nevertheless an explanation for theunexpected clearance rates may be that different surfacechemistry leads to different protein-binding modes in proteincorona formation. Hence, the bound proteins in combinationwith the size effect determine the overall urine excretion rate. Itmay be that the initial negative charge on AuNPs adsorbs morepositively charged proteins, which overcompensate the initialsurface charge of the AuNPs, and vice versa for the positivelycharged AuNPs.
This journal is ª The Royal Society of Chemistry 2013
Biodistribution of AuNPs aer intratracheal administration
The translocation of AuNPs through the air/blood barrier aerintratracheal administration was size dependent as well. AgainAu1.4MS was more readily transferred than the largerAu18MS.104 While 8.5% of instilled Au1.4MS gold was found inthe secondary target organs, Au18MS was almost completelyretained in the lung tissue.
In conclusion, ultrasmall AuNPs generally have a broaderbiodistribution than larger AuNPs, which are cleared by the RESin liver and spleen upon rst pass. Thus small size favorstranslocation of AuNPs across all epithelial barrier tissues in thedigestive tract and in the lung, as well as translocation acrossendothelial layers lining blood vessels and constituting theBBB. Reduced RES clearance and prolonged blood circulationallow for the transfer of ultrasmall, but not larger AuNPs tosecondary target organs as well. These general rules apply forenteral and parenteral administration of AuNPs alike.
Surface modications enhancing AuNP penetration of theblood brain barrier
The small size of ultrasmall AuNPs generally facilitates thecrossing of the blood–brain barrier. The brain is surrounded bya tightly packed layer of endothelial cells that prevent largemolecules from penetrating. In principle, only molecules ofmass <400–500 Daltons can pass through the BBB. Most
Nanoscale

Nanoscale Review
Publ
ishe
d on
08
May
201
3. D
ownl
oade
d by
Nat
iona
l Ins
titut
es o
f St
anda
rds
& T
echn
olog
y on
10/
06/2
013
21:2
4:51
. View Article Online
medicinal chemistry databases list an average molecular massof CNS active drugs of 357 Daltons.144 Drugs may be transportedby passive diffusion, carrier mediated transport (glucose, aminoacids), receptor mediated transcytosis (insulin, and transferrin),and adsorptive-mediated transcytosis. In addition, the tightendothelial layer, which constitutes the BBB, is known toreverse-transport certain toxic drugs by active eux transportersto protect the CNS from noxious substances. Substances thatreadily pass the BBB by passive diffusion are commonly lipo-philic, non-polar, and have a molecular weight of less than 400Daltons. The transport of larger entities across the BBB can beenhanced by surface modication. Positively charged NPs tendto more easily penetrate BBB through the electrostatic interac-tion with the anionic glycocalyx on the brain endothelial cells.The hydrophilic surfactant polysorbate 80 can improve the BBBpenetration of NPs. Modication with the pH sensitive polymerpoly (N,N-dimethylaminoethyl methacrylate (DMAEMA/2-hydroxyethyl methacrylate (HEMA)) likewise has been shownto improve penetration capacity of NPs.
Importantly, whatever the modication will be, it must bemaintained in blood contact. This brings us back to the allimportant protein corona,90,123,142,145–147 which controls the fateof nanomaterials including AuNPs at least as much as thechemistry used to produce nanomaterials.
Excretion of AuNPs
Urine excretion and RES retention are the major clearancepathways for AuNPs. Several studies showed that the bio-distribution and excretion of ultrasmall AuNPs in vivo is size-and ligand-dependent. Studies from the Kreyling laboratoryshowed that small Au1.4MS has lower RES capture and fasterurine excretion than the larger Au5MS. Zhou et al. showed thatrenal clearance of 2 nm glutathione-coated AuNPs (AuGSH) wasmore than 10 to 100 times faster than that of similarly sizedAuNPs coated by TPPMS and cysteine. AuGSH had high stabilityin physiological medium and low protein binding. Compared toTPPMS modied AuNPs with a similar size, 50% of AuGSH wasexcreted through the urine 24 hpi,148 and only 3.7% AuGSHaccumulated in the liver.
Zebrash embryo as a vertebrate model to test the toxicity ofAuNPs in vivo
AuNP toxicity is still mostly studied using simple cell-basedassays. Translocation of nanomaterials across biologicalbarriers into the intact organism, organ specic toxicity, theinterference of nanoparticles with embryonic development andreproductive toxicity can, however, not be addressed in cell-based experiments. The European Community legislativeframework REACH for Registration, Evaluation, Authorisationand Restriction of Chemicals and its regulatory annex fornanomaterials call for modied test guidelines to determinespecic hazards associated with nanomaterials both in vitro andin vivo.149 However, ethical issues and cost preclude this exces-sive use of higher vertebrates. To this end, the zebrash embryotest may be ideally suited as a complex vertebrate test to studygeneral toxicity, organ toxicity and teratogenicity of NPs in large
Nanoscale
scale. Zebrash have a large number of progeny and are asuccessful renement of conventional rodent animal modelsfor toxicity assessment.150,151 25% of the zebrash genes areknown to be essential for early development and 99% of thesegenes are homologous to human genes, suggesting that resultsobtained in zebrash may be transferable to humans.152
Furthermore, transgenic zebrash may be employed to inter-pret defense or detoxication pathways triggered by NPs in realtime.153 Transgenic zebrash expressing uorescent reporterproteins, e.g. green uorescent protein GFP, under the control ofpromoters that report differential gene regulation followingtoxic insult, are well suited to study the biological response of,e.g., developing embryos in the presence of toxic compounds.Heat shock protein promoters like HSP70, or cytochrome Cdetoxication pathway promoters like CYP1A1 are excellentchoices to interrogate major stress pathways in toxicologyresearch.
We recently showed that the sh embryo test (FET) faithfullyreproduced all important ndings of a previous study in HeLacells and added new important information on teratogenicityand hepatotoxicity that could not be gained from studyingcultured cells.154 Similar sized AuNP carrying ligands with highaffinity to the gold core (glutathione, GSH) were much less toxicthan Au1.4MS, which carries a more labile ligand (TPPMS).Au1.4MS caused embryo coagulation at the lethal dose(400 mM). At the sublethal dose (50 mM), Au1.4MS causedhypopigmentation and pericardial edema. The malformationswere absent in embryos exposed to Au1.4GSH at an even higherdose (1 mM), and, like in HeLa cells, the toxicity of Au1.4MS wasabrogated by the addition of GSH. Fig. 7 shows that toxicitytesting in transgenic hsp70GFP sh embryos was much moresensitive than testing in wildtype sh, in that GFP expression inthe liver clearly commenced at a dose (1/20 LD50) that hadcaused no visible morphological abnormalities whatsoever inthe sh embryo test.
Similar to our work, Lin and coworkers have shown thathsp70GFP transgenic sh report a robust stress responsefollowing exposure to transition metal oxide NPs.153 Theseauthors have also reviewed the use of zebrash in NP toxicologyresearch.155
Application of AuNPs in diagnosis andtherapyAuNP contrast agents
The long blood retention, low liver capture rate and fast urineexcretion render ultrasmall AuNPs well suited as novel contrastagents. AuNPs have higher X-ray contrast than the conventionaliodinated compounds. Xu and colleagues showed that the X-rayattenuation decreased with the size of the nanoparticles.156
Thus 4 nm AuNPs have greater CT attenuation than the 20, 38and 60 nm AuNPs.157
AuNPs in forensic science
Dihydrolipoic acid (DHLA) modied AuNPs were applied inforensic science to detect as little as 0.5 nM intracellular Hg2+.
This journal is ª The Royal Society of Chemistry 2013

Fig. 7 Marked hepatic GFP expression in transgenic Tg(hsp70: GFP) zebrafishtriggered by low doses of Au1.4MS. Untreated embryos kept in embryo mediumdid not express HSP in the liver (A). HSP induction was, however, strongly upre-gulated in the liver in response to the sub-lethal dose Au1.4MS 13 mM (B) and25 mM (C). Note that no morphological malformations were present at this lowconcentration. 200 mM Au1.4MS caused severe malformation and high expres-sion of HSP in liver (D and E). Reproduced with permission.154
Review Nanoscale
Publ
ishe
d on
08
May
201
3. D
ownl
oade
d by
Nat
iona
l Ins
titut
es o
f St
anda
rds
& T
echn
olog
y on
10/
06/2
013
21:2
4:51
. View Article Online
The Au–DHLA complex is uorescent and this uorescence isquenched aer Au–Hg2+ binding.158
AuNPs for percutaneous delivery
Ultrasmall AuNPs have higher stability, and penetrate barrierepithelia better than conventional lipid vehicles, which arelarger.159 The ultrasmall AuNPs penetrate intact skin withoutcausing structural damage, rendering AuNPs an attractivealternative for percutaneous drug delivery.
AuNPs as chemotherapeutic agents
A recent study from Huang et al. showed that the cellularpenetration and tumor accumulation of AuNPs were sizedependent. Ultrasmall AuNPs showed high penetration oftumor tissue compared to 15 nm AuNPs.113
AuNPs for glycoprotein analysis
Hydrazide-functionalized 1.2 nm AuNPs were applied tospecically isolate N-linked glycoproteins from rat kidney withhigh binding capacity, purity and low complexity compared tothe conventional methods like lectin affinity chromatographyand solid-phase extraction.160
First AuNP in clinical testing
Insulin and glucagon-like peptide-1 modied AuNPs with thecore size <2 nm from Midatech (http://midatechgroup.com)
This journal is ª The Royal Society of Chemistry 2013
have received regulatory approval for clinical testing. This is therst ultrasmall AuNP entering the clinical stage. Combined withthe PharmaFilm drug delivery platform, this AuNP is potentiallya novel transbuccal medication for the treatment of diabetes,which overcomes the burden of multiple daily injections.
Conclusions
This review has summarized relevant techniques on the synthesisand characterization of ultrasmall AuNPs in the context of arapidly growing eld of applications, i.e. the application of AuNPsin medicine. Although most of the AuNPs synthesized so far areconsidered non-toxic, some novel ultrasmall AuNPs may in factbe toxic. This requires thorough analysis of QSAR. The examplesgiven in this review shed light on how adverse effects of NPs inbiological systems may occur. Mechanistically, NPs may berendered toxic because of catalytic generation of reactive speciesdue to the large and chemically active surface area of NPs, as wellas by shape complementarity in the sense that NPs approach thesize of biological molecules and subcellular structures and thus(i) may overcome natural barriers by active or passive uptake, or(ii) may even behave as molecular ligands. These types ofpotential interactions underline the need for standardizedmodelsystems, where besides particles size the ligand-dependentproperties can be studied independently in a very systematicmanner and over a broad parameter space, to fully understandtheir QSAR. In view of the high degree of control over particle sizeand ligand chemistry, AuNPs may thus by ideally suited for sucha generalistic approach.
Acknowledgements
This work was supported by the Deutsche For-schungsgemeinscha DFG (investigator grants Si609/8 andJa562/13).
Notes and references
1 F. E. Kruis, H. Fissan and A. Peled, J. Aerosol Sci., 1998, 29,511–535.
2 M.-C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346.3 T. Tsukuda, Bull. Chem. Soc. Jpn., 2012, 85, 151–168.4 P. Maity, S. Xie, M. Yamauchi and T. Tsukuda, Nanoscale,2012, 4, 4027–4037.
5 N. R. Jana, L. Gearheart and C. J. Murphy, Langmuir, 2001,17, 6782–6786.
6 J. Turkevitch, P. C. Stevenson and J. Hillier, Discuss. FaradaySoc., 1951, 11, 55–75.
7 D. V. Leff, L. Brandt and J. R. Heath, Langmuir, 1996, 12,4723–4730.
8 G. Schmid, R. Pfeil, R. Boese, F. Bandermann, S. Meyer,G. H. M. Calis and J. W. A. van der Velden, Chem. Ber.,1981, 114, 3634–3642.
9 G. Schmid, R. Pugin, T. Sawitowski, U. Simon and B. Marler,Chem. Commun., 1999, 1303.
10 G. Schmid, N. Klein, L. Korste, U. Kreibig and D. Schoauer,Polyhedron, 1988, 7, 605–608.
Nanoscale

Nanoscale Review
Publ
ishe
d on
08
May
201
3. D
ownl
oade
d by
Nat
iona
l Ins
titut
es o
f St
anda
rds
& T
echn
olog
y on
10/
06/2
013
21:2
4:51
. View Article Online
11 M. Brust, M. Walker, D. Bethell, D. J. Schiffrin andR. Whyman, J. Chem. Soc., Chem. Commun., 1994, 801.
12 M. Brust, J. Fink, D. Bethell, D. J. Schiffrin and C. Kiely, J.Chem. Soc., Chem. Commun., 1995, 1655.
13 T. G. Schaaff, G. Knight, M. N. Shagullin, R. F. Borkmanand R. L. Whetten, J. Phys. Chem. B, 1998, 102, 10643–10646.
14 Y. Negishi, Y. Takasugi, S. Sato, H. Yao, K. Kimura andT. Tsukuda, J. Am. Chem. Soc., 2004, 126, 6518–6519.
15 M. Schulz-Dobrick, K. V. Sarathy and M. Jansen, J. Am.Chem. Soc., 2005, 127, 12816–12817.
16 P. R. Selvakannan, S. Mandal, S. Phadtare, R. Pasricha andM. Sastry, Langmuir, 2003, 19, 3545–3549.
17 M. Tamura and H. Fujihara, J. Am. Chem. Soc., 2003, 125,15742–15743.
18 R. G. Shimmin, A. B. Schoch and P. V. Braun, Langmuir,2004, 20, 5613–5620.
19 C. M. Goodman, C. D. McCusker, T. Yilmaz andV. M. Rotello, Bioconjugate Chem., 2004, 15, 897–900.
20 C. A. Lin, T. Y. Yang, C. H. Lee, S. H. Huang, R. A. Sperling,M. Zanella, J. K. Li, J. L. Shen, H. H. Wang, H. I. Yeh,W. J. Parak and W. H. Chang, ACS Nano, 2009, 3, 395–401.
21 M. Yu, C. Zhou, J. Liu, J. D. Hankins and J. Zheng, J. Am.Chem. Soc., 2011, 133, 11014–11017.
22 J. M. de la Fuente and C. C. Berry, Bioconjugate Chem., 2005,16, 1176–1180.
23 R. A. Sperling and W. J. Parak, Philos. Trans. R. Soc., A, 2010,368, 1333–1383.
24 H. Hakkinen, M. Walter and H. Gronbeck, J. Phys. Chem. B,2006, 110, 9927–9931.
25 J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo andG. M. Whitesides, Chem. Rev., 2005, 105, 1103–1170.
26 L. A. Porter, D. Ji, S. L. Westcott, M. Graupe,R. S. Czernuszewicz, N. J. Halas and T. R. Lee, Langmuir,1998, 14, 7378–7386.
27 X.-M. Li, M. R. d. Jong, K. Inoue, S. Shinkai, J. Huskens andD. N. Reinhoudt, J. Mater. Chem., 2001, 11, 1919–1923.
28 J. P. Hermes, F. Sander, T. Peterle, C. Cioffi, P. Ringler,T. Pfohl and M. Mayor, Small, 2011, 7, 920–929.
29 J. P. Hermes, F. Sander, U. Fluch, T. Peterle, D. Thompson,R. Urbani, T. Pfohl and M. Mayor, J. Am. Chem. Soc., 2012,134, 14674–14677.
30 T. Peterle, P. Ringler andM.Mayor, Adv. Funct. Mater., 2009,19, 3497–3506.
31 T. Peterle, A. Leifert, J. Timper, A. Sologubenko, U. Simonand M. Mayor, Chem. Commun., 2008, 3438–3440.
32 D. Thompson, J. P. Hermes, A. J. Quinn and M. Mayor, ACSNano, 2012, 6, 3007–3017.
33 S. Perumal, A. Hofmann, N. Scholz, E. Ruhl and C. Graf,Langmuir, 2011, 27, 4456–4464.
34 L.-O. Srisombat, J.-S. Park, S. Zhang and T. R. Lee, Langmuir,2008, 24, 7750–7754.
35 Y. Yanagimoto, Y. Negishi, H. Fujihara and T. Tsukuda, J.Phys. Chem. B, 2006, 110, 11611–11614.
36 D. E. Bergeron, O. Coskuner, J. W. Hudgens andC. A. Gonzalez, J. Phys. Chem. B, 2008, 112, 12808–12814.
37 J. E. A. Barreto, W. O'Malley, M. Kubeil, B. Graham,H. Stephan and L. Spiccia, Adv. Mater., 2011, 23, H18–H40.
Nanoscale
38 J. A. Jamison, K. M. Krueger, C. T. Yavuz, J. T. Mayo,D. LeCrone, J. J. Redden and V. L. Colvin, ACS Nano,2008, 2, 311–319.
39 C.-L. Wang, M. Fang, S.-H. Xu and Y.-P. Cui, Langmuir, 2010,26, 633–638.
40 G.-T. Wei and F.-K. Liu, J. Chromatogr., A, 1999, 836, 253–260.
41 X. Xu, K. K. Caswell, E. Tucker, S. Kabisatpathy,K. L. Brodhacker and W. A. Scrivens, J. Chromatogr., A,2007, 1167, 35–41.
42 C. J. Ackerson, P. D. Jadzinsky and R. D. Kornberg, J. Am.Chem. Soc., 2005, 127, 6550–6551.
43 F. Wen, U. Englert, B. Gutrath and U. Simon, Eur. J. Inorg.Chem., 2008, 2008, 106–111.
44 S. L. Flegler, J. W. Heckman jr and K. L. Klomparens,Elektronenmikroskopie, Spektrum Akademischer Verlag,Heidelberg, 1995.
45 R. Erni, M. D. Rossell, C. Kisielowski and U. Dahmen, Phys.Rev. Lett., 2009, 102, 096101.
46 W. D. Pyrz and D. J. Buttrey, Langmuir, 2008, 24, 11350–11360.
47 Z. Y. Li, N. P. Young, M. D. Vece, S. Palomba, R. E. Palmer,A. L. Bleloch, B. C. Curley, R. L. Johnston, J. Jiang andJ. Yuan, Nature, 2008, 451, 46–48.
48 R. H. Terrill, T. A. Postlethwaite, C.-h. Chen, C.-D. Poon,A. Terzis, A. Chen, J. E. Hutchison, M. R. Clark andG. Wignall, J. Am. Chem. Soc., 1995, 117, 12537–12548.
49 M. J. Hostetler, J. E. Wingate, C.-J. Zhong, J. E. Harris,R. W. Vachet, M. R. Clark, J. D. Londono, S. J. Green,J. J. Stokes, G. D. Wignall, G. L. Glish, M. D. Porter,N. D. Evans and R. W. Murray, Langmuir, 1998, 14, 17–30.
50 B. Abecassis, F. Testard, O. Spalla and P. Barboux, NanoLett., 2007, 7, 1723–1727.
51 J. Polte, R. Erler, A. F. Thuemann, S. Sokolov, T. T. Ahner,K. Rademann, F. Emmerling and R. Kraehnert, ACS Nano,2010, 4, 1076–1082.
52 J. Polte, T. T. Ahner, F. Delissen, S. Sokolov, F. Emmerling,A. F. Thuemann and R. Kraehnert, J. Am. Chem. Soc., 2010,132, 1296–1301.
53 G. Binnig, C. F. Quate and C. Gerber, Phys. Rev. Lett., 1986,56, 930–933.
54 R. Fenger, E. Fertitta, H. Kirmse, A. F. Thunemann andK. Rademann, Phys. Chem. Chem. Phys., 2012, 14, 9343–9349.
55 H. G. Jiang, M. Ruhle and E. J. Lavernia, J. Mater. Res., 1999,14, 549–559.
56 G. Schmid and B. Corain, Eur. J. Inorg. Chem., 2008, 3081.57 P. D. Jadzinsky, G. Calero, C. J. Ackerson, D. A. Bushnell and
R. D. Kornberg, Science, 2007, 318, 430–433.58 B. S. Gutrath, I. M. Oppel, O. Presly, I. Beljakov, V. Meded,
W. Wenzel and U. Simon, Angew. Chem., Int. Ed., 2013,52(12), 3529–3532.
59 Y. Shichibu, Y. Kamei and K. Konishi, Chem. Commun.,2012, 48, 7559–7561.
60 K. Nunokawa, S. Onaka, M. Ito, M. Horibe, T. Yonezawa,H. Nishihara, T. Ozeki, H. Chiba, S. Watase andM. Nakamoto, J. Organomet. Chem., 2006, 691, 638–642.
This journal is ª The Royal Society of Chemistry 2013

Review Nanoscale
Publ
ishe
d on
08
May
201
3. D
ownl
oade
d by
Nat
iona
l Ins
titut
es o
f St
anda
rds
& T
echn
olog
y on
10/
06/2
013
21:2
4:51
. View Article Online
61 B. L. Berne and R. Pecora, Dynamic Light Scattering – WithApplications to Chemistry, Biology and Physics, Wiley, 2000.
62 R. W. O'Brien and L. R. White, J. Chem. Soc., Faraday Trans.,1978, 74, 1607–1626.
63 B. Khlebtsov and N. Khlebtsov, Colloid J., 2011, 73, 118–127.64 A. Chompoosor, G. Han and V. M. Rotello, Bioconjugate
Chem., 2008, 19, 1342–1345.65 M. M. Alvarez, J. T. Khoury, T. G. Schaaff, M. N. Shagullin,
I. Vezmar and R. L. Whetten, J. Phys. Chem. B, 1997, 101,3706–3712.
66 N. K. Chaki, Y. Negishi, H. Tsunoyama, Y. Shichibu andT. Tsukuda, J. Am. Chem. Soc., 2008, 130, 8608–8610.
67 W. W. Weare, S. M. Reed, M. G. Warner and J. E. Hutchison,J. Am. Chem. Soc., 2000, 122, 12890–12891.
68 A. Badia, L. Cuccia, L. Demers, F. Morin and R. B. Lennox, J.Am. Chem. Soc., 1997, 119, 2682–2692.
69 R. Sharma, G. P. Holland, V. C. Solomon, H. Zimmermann,S. Schiffenhaus, S. A. Amin, D. A. Buttry and J. L. Yarger, J.Phys. Chem. C, 2009, 113, 16387–16393.
70 G. Schmid, Struct. Bonding, 1985, 62, 51–85.71 C. Vilain, F. Goettmann, A. Moores, P. Le Floch and
C. Sanchez, J. Mater. Chem., 2007, 17, 3509–3514.72 P. Biji, N. K. Sarangi and A. Patnaik, Langmuir, 2010, 26,
14047–14057.73 T. Huang and R. W. Murray, J. Phys. Chem. B, 2001, 105,
12498–12502.74 W. Guo, J. Yuan and E. Wang, Chem. Commun., 2012, 48,
3076–3078.75 X. Tu, W. Chen and X. Guo,Nanotechnology, 2011, 22, 095701.76 C. Zhou, C. Sun, M. Yu, Y. Qin, J. Wang, M. Kim and
J. Zheng, J. Phys. Chem. C, 2010, 114, 7727–7732.77 W. Chen, X. Tu and X. Guo, Chem. Commun., 2009, 1736–
1738.78 C.-C. Huang, Z. Yang, K.-H. Lee and H.-T. Chang, Angew.
Chem., Int. Ed., 2007, 46, 6824–6828.79 N. Schaeffer, B. Tan, C. Dickinson, M. J. Rosseinsky,
A. Laromaine, D. W. McComb, M. M. Stevens, Y. Wang,L. Petit, C. Barentin, D. G. Spiller, A. I. Cooper andR. Levy, Chem. Commun., 2008, 3986.
80 L. Shang, S. Brandholt, F. Stockmar, V. Trouillet, M. Brunsand G. U. Nienhaus, Small, 2012, 8, 661–665.
81 E. Dulkeith, A. C. Morteani, T. Niedereichholz, T. A. Klar,J. Feldmann, S. A. Levi, F. C. J. M. van Veggel,D. N. Reinhoudt, M. Moller and D. I. Gittins, Phys. Rev.Lett., 2002, 89, 203002.
82 T. Gu, T. Ye, J. D. Simon, J. K. Whitesell and M. A. Fox, J.Phys. Chem. B, 2003, 107, 1765–1771.
83 H. G. Boyen, G. Kastle, F. Weigl, B. Koslowski, C. Dietrich,P. Ziemann, J. P. Spatz, S. Riethmuller, C. Hartmann,M. Moller, G. Schmid, M. G. Garnier and P. Oelhafen,Science, 2002, 297, 1533–1536.
84 Y. Pan, M. Bartneck and W. Jahnen-Dechent, MethodsEnzymol., 2012, 509, 225–242.
85 N. Lewinski, V. Colvin and R. Drezek, Small, 2008, 4, 26–49.86 H. J. Johnston, G. Hutchison, F. M. Christensen, S. Peters,
S. Hankin and V. Stone, Crit. Rev. Toxicol., 2010, 40, 328–346.
This journal is ª The Royal Society of Chemistry 2013
87 A. M. Schrand, M. F. Rahman, S. M. Hussain, J. J. Schlager,D. A. Smith and A. F. Syed,Wiley Interdiscip. Rev.: Nanomed.Nanobiotechnol., 2010, 2, 544–568.
88 J. W. Card and B. A. Magnuson, Int. J. Toxicol., 2010, 29,402–410.
89 H. J. Klimisch, M. Andreae and U. Tillmann, Regul. Toxicol.Pharmacol., 1997, 25, 1–5.
90 A. E. Nel, L. Madler, D. Velegol, T. Xia, E. M. V. Hoek,P. Somasundaran, F. Klaessig, V. Castranova andM. Thompson, Nat. Mater., 2009, 8, 543–557.
91 D. H. Tsai, F. W. Delrio, A. M. Keene, K. M. Tyner,R. I. Maccuspie, T. J. Cho, M. R. Zachariah andV. A. Hackley, Langmuir, 2011, 27, 2464–2477.
92 T. Cedervall, I. Lynch, S. Lindman, T. Berggard, E. Thulin,H. Nilsson, K. A. Dawson and S. Linse, Proc. Natl. Acad.Sci. U. S. A., 2007, 104, 2050–2055.
93 S. H. Lacerda, J. J. Park, C. Meuse, D. Pristinski,M. L. Becker, A. Karim and J. F. Douglas, ACS Nano, 2010,4, 365–379.
94 L. Treuel and G. Nienhaus, Biophys. Rev., 2012, 4, 137–147.95 T. Wang, J. Bai, X. Jiang and G. U. Nienhaus, ACS Nano,
2012, 6, 1251–1259.96 M. Shao, L. Lu, H. Wang, S. Luo and D. Ma,Microchim. Acta,
2009, 164, 157–160.97 Y. Cheng, M. Wang, G. Borghs and H. Chen, Langmuir,
2011, 27, 7884–7891.98 L. Shang, Y. Wang, J. Jiang and S. Dong, Langmuir, 2007, 23,
2714–2721.99 L. Treuel, M. Malissek, J. S. Gebauer and R. Zellner,
ChemPhysChem, 2010, 11, 3093–3099.100 A. A. Sousa, J. T. Morgan, P. H. Brown, A. Adams,
P. Mudiyanselage, G. Zhang, C. J. Ackerson, M. J. Kruhlakand R. D. Leapman, Small, 2012, 8(14), 2277–2286.
101 R. Goel, N. Shah, R. Visaria, G. F. Paciotti and J. C. Bischof,Nanomedicine, 2009, 4, 401–410.
102 C. Lasagna-Reeves, D. Gonzalez-Romero, M. A. Barria,I. Olmedo, A. Clos, V. M. Sadagopa Ramanujam,A. Urayama, L. Vergara, M. J. Kogan and C. Soto, Biochem.Biophys. Res. Commun., 2010, 393, 649–655.
103 B. D. Chithrani, A. A. Ghazani and W. C. W. Chan, NanoLett., 2006, 6, 662–668.
104 M. Semmler-Behnke, W. G. Kreyling, J. Lipka, S. Fertsch,A. Wenk, S. Takenaka, G. E. n. Schmid and W. Brandau,Small, 2008, 4, 2108–2111.
105 J. Lipka, M. Semmler-Behnke, R. A. Sperling, A. Wenk,S. Takenaka, C. Schleh, T. Kissel, W. J. Parak andW. G. Kreyling, Biomaterials, 2010, 31, 6574–6581.
106 V. Kattumuri, K. Katti, S. Bhaskaran, E. J. Boote,S. W. Casteel, G. M. Fent, D. J. Robertson,M. Chandrasekhar, R. Kannan and K. V. Katti, Small,2007, 3, 333–341.
107 J. H. Kim, M. H. Kim, D. H. Jo, Y. S. Yu and T. G. Lee,Biomaterials, 2011, 32, 1865–1871.
108 S. Kim, Y. Chen, G. Luke and S. Emelianov, Biomed. Opt.Express, 2011, 2, 2540–2550.
109 X. Yang, S. E. Skrabalak, Z. Y. Li, Y. Xia and L. V. Wang,Nano Lett., 2007, 7, 3798–3802.
Nanoscale

Nanoscale Review
Publ
ishe
d on
08
May
201
3. D
ownl
oade
d by
Nat
iona
l Ins
titut
es o
f St
anda
rds
& T
echn
olog
y on
10/
06/2
013
21:2
4:51
. View Article Online
110 E. Boote, G. Fent, V. Kattumuri, S. Casteel, K. Katti,N. Chanda, R. Kannan and R. Churchill, Acad. Radiol.,2010, 17, 410–417.
111 D. B. Peckys and N. de Jonge, Nano Lett., 2011, 11, 1733–1738.
112 Y. Pan, S. Neuss, A. Leifert, M. Fischler, F. Wen, U. Simon,G. Schmid, W. Brandau and W. Jahnen-Dechent, Small,2007, 3, 1941–1949.
113 K. Huang, H. Ma, J. Liu, S. Huo, A. Kumar, T. Wei, X. Zhang,S. Jin, Y. Gan, P. C. Wang, S. He and X. J. Liang, ACS Nano,2012, 6(5), 4483–4493.
114 E. Oh, J. B. Delehanty, K. E. Sapsford, K. Susumu,R. Goswami, J. B. Blanco-Canosa, P. E. Dawson, J. Granek,M. Shoff, Q. Zhang, P. L. Goering, A. Huston andI. L. Medintz, ACS Nano, 2011, 5, 6434–6448.
115 Y. Pan, A. Leifert, D. Ruau, S. Neuss, J. Bornemann,G. Schmid, W. Brandau, U. Simon and W. Jahnen-Dechent, Small, 2009, 5, 2067–2076.
116 M. Tsoli, H. Kuhn, W. Brandau, H. Esche and G. Schmid,Small, 2005, 1, 841–844.
117 Y. Liu, W. Meyer-Zaika, S. Franzka, G. Schmid, M. Tsoli andH. Kuhn, Angew. Chem., 2003, 42, 2853–2857.
118 W. Jahnen-Dechent and U. Simon, Nanomedicine, 2008, 3,601–603.
119 EMA, in Report # CPMP/986/96, European Agency for theEvaluation of Medicinal Products, London, 1997.
120 FDA, in ICH (Expert Working Group (Safety) of theInternational Conference on Harmonisation of TechnicalRequirements for Registration of Pharmaceuticals for HumanUse), FDA (Food and Drug Administration, USA),Rockville, Maryland, USA, 2001.
121 A. Leifert, Y. Pan, A. Kinkeldey, F. Schiefer, J. Setzler,O. Scheel, H. Lichtenbeld, G. Schmid, W. Wenzel,W. Jahnen-Dechent and U. Simon, Proc. Natl. Acad. Sci.U. S. A., 2013, 110(20), 8004–8009.
122 L. Vroman, Ann. N. Y. Acad. Sci., 1987, 516, 300–305.123 M. Lundqvist, J. Stigler, T. Cedervall, T. Berggad,
M. B. Flanagan, I. Lynch, G. Elia and K. Dawson, ACSNano, 2011, 5, 7503–7509.
124 R. B. Sim and R. Wallis, Nat. Nanotechnol., 2011, 6, 80–81.
125 M. A. Dobrovolskaia, P. Aggarwal, J. B. Hall andS. E. McNeil, Mol. Pharmacol., 2008, 5, 487–495.
126 M. A. Dobrovolskaia, J. D. Clogston, B. W. Neun, J. B. Hall,A. K. Patri and S. E. McNeil, Nano Lett., 2008, 8, 2180–2187.
127 S. Deb, H. K. Patra, P. Lahiri, A. K. Dasgupta, K. Chakrabartiand U. Chaudhuri, Nanomedicine, 2011, 7, 376–384.
128 A. Radomski, P. Jurasz, D. Alonso-Escolano, M. Drews,M. Morandi, T. Malinski and M. W. Radomski, Br. J.Pharmacol., 2005, 146, 882–893.
129 I. Hamad, O. Al-Hanbali, A. C. Hunter, K. J. Rutt,T. L. Andresen and S. M. Moghimi, ACS Nano, 2010, 4,6629–6638.
130 E. Sadauskas, N. R. Jacobsen, G. Danscher, M. Stoltenberg,U. Vogel, A. Larsen, W. Kreyling and H. Wallin, Chem. Cent.J., 2009, 3, 16.
Nanoscale
131 X. D. Zhang, H. Y. Wu, D. Wu, Y. Y. Wang, J. H. Chang,Z. B. Zhai, A. M. Meng, P. X. Liu, L. A. Zhang andF. Y. Fan, Int. J. Nanomed., 2010, 5, 771–781.
132 J. F. Hainfeld, D. N. Slatkin, T. M. Focella andH. M. Smilowitz, Br. J. Radiol., 2006, 79, 248–253.
133 E. Hutter, S. Boridy, S. Labrecque, M. Lalancette-Hebert,J. Kriz, F. M. Winnik and D. Maysinger, ACS Nano, 2010,4, 2595–2606.
134 Z. Ji, D. Zhang, L. Li, X. Shen, X. Deng, L. Dong, M. Wu andY. Liu, Nanotechnology, 2009, 20, 445101.
135 S. Hussain, J. A. Vanoirbeek, K. Luyts, V. De Vooght,E. Verbeken, L. C. Thomassen, J. A. Martens, D. Dinsdale,S. Boland, F. Marano, B. Nemery and P. H. Hoet, Eur.Respir. J., 2011, 37, 299–309.
136 K. Yamashita, Y. Yoshioka, K. Higashisaka, K. Mimura,Y. Morishita, M. Nozaki, T. Yoshida, T. Ogura,H. Nabeshi, K. Nagano, Y. Abe, H. Kamada, Y. Monobe,T. Imazawa, H. Aoshima, K. Shishido, Y. Kawai,T. Mayumi, S. Tsunoda, N. Itoh, T. Yoshikawa,I. Yanagihara, S. Saito and Y. Tsutsumi, Nat.Nanotechnol., 2011, 6, 321–328.
137 W. H. De Jong, W. I. Hagens, P. Krystek, M. C. Burger,A. J. Sips and R. E. Geertsma, Biomaterials, 2008, 29,1912–1919.
138 S. K. Balasubramanian, J. Jittiwat, J. Manikandan,C. N. Ong, L. E. Yu and W. Y. Ong, Biomaterials, 2010, 31,2034–2042.
139 C. Schleh, M. Semmler-Behnke, J. Lipka, A. Wenk, S. Hirn,M. Schaffler, G. Schmid, U. Simon and W. G. Kreyling,Nanotoxicology, 2012, 6, 36–46.
140 S. Hirn, M. Semmler-Behnke, C. Schleh, A. Wenk, J. Lipka,M. Schaffler, S. Takenaka, W. Moller, G. Schmid, U. Simonand W. G. Kreyling, Eur. J. Pharm. Biopharm., 2011, 77, 407–416.
141 H. S. Choi, W. Liu, P. Misra, E. Tanaka, J. P. Zimmer, B. IttyIpe, M. G. Bawendi and J. V. Frangioni, Nat. Biotechnol.,2007, 25, 1165–1170.
142 N. Bertrand and J.-C. Leroux, J. Controlled Release, 2011.143 P. Ruenraroengsak, J. M. Cook and A. T. Florence, J.
Controlled Release, 2010, 141, 265–276.144 A. K. Ghose, V. N. Viswanadhan and J. J. Wendoloski, J.
Comb. Chem., 1999, 1, 55–68.145 M. Lundqvist, J. Stigler, G. Elia, I. Lynch, T. Cedervall and
K. A. Dawson, Proc. Natl. Acad. Sci. U. S. A., 2008, 105,14265–14270.
146 M. P. Monopoli, D. Walczyk, A. Campbell, G. Elia, I. Lynch,F. B. Bombelli and K. A. Dawson, J. Am. Chem. Soc., 2011,133, 2525–2534.
147 C. Rocker, M. Potzl, F. Zhang, W. J. Parak andG. U. Nienhaus, Nat. Nanotechnol., 2009, 4, 577–580.
148 C. Zhou, M. Long, Y. Qin, X. Sun and J. Zheng, Angew.Chem., Int. Ed., 2011, 50, 3168–3172.
149 http://guidance.echa.europa.eu/.150 S. Scholz, S. Fischer, U. Gundel, E. Kuster, T. Luckenbach
and D. Voelker, Environ. Sci. Pollut. Res., 2008, 15, 394–404.151 T. Kosmehl, J. C. Otte, L. Yang, J. Legradi, K. Bluhm,
C. Zinsmeister, S. H. Keiter, G. Reifferscheid, W. Manz,
This journal is ª The Royal Society of Chemistry 2013

Review Nanoscale
Publ
ishe
d on
08
May
201
3. D
ownl
oade
d by
Nat
iona
l Ins
titut
es o
f St
anda
rds
& T
echn
olog
y on
10/
06/2
013
21:2
4:51
. View Article Online
T. Braunbeck, U. Strahle and H. Hollert, Reprod. Toxicol.,2012, 33, 245–253.
152 A. Amsterdam, R. M. Nissen, Z. Sun, E. C. Swindell,S. Farrington and N. Hopkins, Proc. Natl. Acad. Sci.U. S. A., 2004, 101, 12792–12797.
153 S. Lin, Y. Zhao, T. Xia, H. Meng, Z. Ji, R. Liu, S. George,S. Xiong, X. Wang, H. Zhang, S. Pokhrel, L. Madler,R. Damoiseaux and A. E. Nel, ACS Nano, 2011, 5, 7284–7295.
154 Y. Pan, A. Leifert, M. Graf, F. Schiefer, S. Thoroe-Boveleth,J. Broda, M. C. Halloran, H. Hollert, D. Laaf, U. Simonand W. Jahnen-Dechent, Small, 2013, 9(6), 863–869.
155 S. Lin, Y. Zhao, A. E. Nel and S. Lin, Small, 2013, 9, 1608–1618.
This journal is ª The Royal Society of Chemistry 2013
156 C. Xu, G. A. Tung and S. Sun, Chem. Mater., 2008, 20, 4167–4169.
157 A. Zhang, Y. Tu, S. Qin, Y. Li, J. Zhou, N. Chen, Q. Lu andB. Zhang, J. Colloid Interface Sci., 2012, 372, 239–244.
158 L. Shang, L. Yang, F. Stockmar, R. Popescu, V. Trouillet,M. Bruns, D. Gerthsen and G. U. Nienhaus, Nanoscale,2012, 4, 4155–4160.
159 Y. Huang, F. Yu, Y. S. Park, J. Wang, M. C. Shin,H. S. Chung and V. C. Yang, Biomaterials, 2010, 31,9086–9091.
160 T. H. Tran, S. Park, H. Lee, B. Kim, O. H. Kim, B. C. Oh andD. Lee, Analyst, 2012, 137, 991–998.
Nanoscale
Copyright © 2022 FDOKUMEN




















